

Abstract
The success of RNA interference (RNAi) as a research tool and potential therapeutic approach has reinvigorated interest in chemical modifications of RNA. Replacement of the negatively charged phosphates with neutral amides may be expected to improve bioavailability and cellular uptake of small interfering RNAs (siRNAs) critical for in vivo applications. In this study, we introduced up to seven consecutive amide linkages at the 3´-end of the guide strand of an siRNA duplex. Modified guide strands having four consecutive amide linkages retained high RNAi activity when paired with a passenger strand having one amide modification between its first and second nucleosides at the 5´-end. Further increase in the number of modifications decreased the RNAi activity; however, siRNAs with six and seven amide linkages still showed useful target silencing. While an siRNA duplex having nine amide linkages retained some silencing activity, the partial reduction of the negative charge did not enable passive uptake in HeLa cells. Our results suggest that further chemical modifications, in addition to amide linkages, are needed to enable cellular uptake of siRNAs in the absence of transfection agents.
An siRNA having four consecutive phosphates of its guide strand replaced by amide linkages showed high silencing activity when the passenger strand had an amide linkage between its first and second nucleosides. Further increase in amide modification decreased the RNAi activity; however, siRNAs having up to nine amide linkages retained some silencing activity at higher concentrations.
Keywords: RNA interference, chemical modifications, backbone modified RNA, internucleoside amide linkage, non-ionic RNA analogues
Graphical Abstract
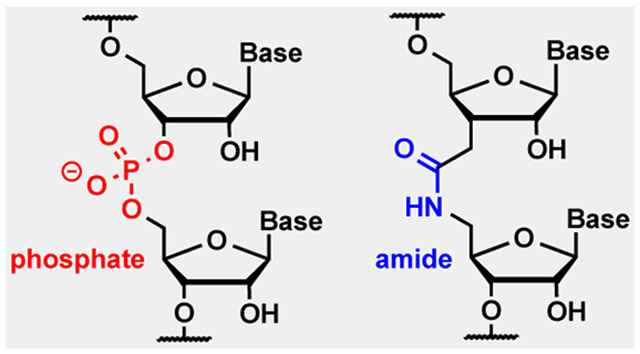
Introduction
RNA interference (RNAi) has become a well-established and powerful tool in the broad fields of biochemistry and molecular biology,[1] and has an intriguing potential to become a new therapeutic approach, with more than 20 RNAi-based clinical trials currently being pursued.[2] Natural RNAi is driven by external (invading virus) or endogenous (microRNA) double-stranded RNAs (dsRNA) that are processed by cellular endonucleases (Drosha and Dicer) into 21-23 nucleotide-long short interfering RNAs (siRNAs) that are loaded into Argonaute 2 (Ago2), the principal protein of RNAi.[3] For research or therapeutic applications, the initial endonuclease-processing step may be bypassed by directly delivering synthetic siRNAs.[4] During loading, an siRNA duplex is unwound and its guide strand (complimentary to the target mRNA) is incorporated into Ago2. The loaded guide strand directs Ago2 to cleave complementary target-mRNA, preventing the mRNA from being translated into protein, thus silencing the target gene. Ideally, only the guide strand of an siRNA would be loaded into Ago2. However, in real systems, the passenger strand may be loaded instead, causing its own mRNA targets to be cleaved, and resulting in off-target effects. The success of in vivo applications of RNAi, including clinical trials, has been limited by the siRNA-associated issues of challenging systemic delivery, inadequate cellular uptake, and off-target microRNA-like activity of both guide and passenger strands.[2a]
The large size and negatively charged phosphate backbone of siRNAs cause the problems of poor bioavailability, short half-life in plasma, and poor cellular uptake. Complexation with lipids and liposomes improves bioavailability and cellular uptake[5] and enables in vivo delivery of siRNAs.[6] However, lipid nanoparticles are not an ideal solution due to limited diffusion and poor pharmacokinetics caused by their large size (∼100 nm and 100 megaD). Conjugation with spermine[7] and cholesterol[8] has also been used to deliver siRNAs in cultured cells and mice. Conjugation with N-acetylgalactosamine (GalNac) enables efficient delivery of siRNAs to hepatocytes via the asialoglycoprotein receptor,[5b] and achieves 80% reduction of transthyretin, a protein implicated in amyloidosis, in non-human primates.[9] Chemical modifications of RNA sugar-phosphate backbone have also been used extensively to improve the properties of siRNAs.[10] Patisiran, the recently approved first RNAi drug, combines strategically-placed chemical modifications with liposome formulation for delivery.[2d, 11] Despite these promising results, current systemic delivery is limited mostly to the liver and spleen. Off target effects, poor biodistribution, and poor cellular uptake remain bottlenecks for the broader in vivo application of siRNAs.
We envisioned that the replacement of phosphates with neutral backbone modifications, such as amide linkages, might complement currently-used modifications and provide additional improvements of bioavailability and cellular uptake of siRNAs. Our hypothesis was inspired by the recent work of Dowdy and co-workers,[12] who showed that bioreversible phosphate neutralization by phosphotriesters enabled efficient siRNA delivery after conjugation with cell-penetrating peptides. In the present study, we tested the limits of consecutive amide linkages that could be placed in an siRNA duplex while retaining useful RNAi activity. Earlier, we showed[13] that the replacement of isolated phosphates with amide linkages did not change the thermal stability and overall A-type conformation of short RNA duplexes. In more recent studies,[14] we discovered that amides were well tolerated at most positions of siRNA guide strands, except between the first and second nucleosides, and actually increased the RNAi activity when placed in the middle of a guide strand. Most remarkably, an amide linkage between the first and second nucleosides of the passenger strand completely abolished its undesired activity and increased the desired activity of the complementary guide strand.[14b] Taken together, these results suggested that amides were promising modifications for the optimization of siRNAs, and prompted us to explore the limits of amide modification in an siRNA duplex.
Herein we report that up to five amide linkages were well tolerated in an siRNA duplex. An siRNA having four consecutive amide linkages in the guide strand maintained high silencing activity (~80% at 10 nM) when the passenger strand had an amide linkage between its first and second nucleosides. A similar result was obtained for an siRNA having three and two consecutive amide linkages in the guide and passenger strands, respectively. Further increases of amide modifications in either the guide or passenger strand decreased RNAi activity; however, siRNAs with six and seven amide linkages still showed useful silencing of target gene expression. An siRNA duplex with nine amide linkages did not show any notable RNAi activity in the absence of lipid transfection agent, suggesting that further modification is needed to explore if reducing charge may have a beneficial effect on the cellular uptake of siRNAs.
Results and Discussion
Overall design.
In an earlier preliminary study,[15] we used uridine amino acid 2a (Figure 1) along with chemistry-switching (switch) monomers 1a and 4 to incorporate short stretches of amide-linked uridines in the middle of a short RNA duplex and near the 5´-end of the passenger strand of an siRNA. We found that three consecutive amides did not interfere with normal base-pairing and A-type conformation of dsRNA, and caused little change in RNAi activity.[15] In the present study, we tested how many amide linkages can be introduced at the 3´-end of the guide strand and 5´-end of the passenger strand using both modified uridine and modified adenosine nucleosides (Figure 1). Our goal was to create an siRNA duplex having a stretch of non-ionic backbone at one of the ends. Previous studies by Iwase et al.[16] showed that siRNAs having two consecutive amide linkages at the 3´-ends of both guide and passenger strands maintained full RNAi activity. In the present study, we started with three and introduced up to seven consecutive amides at the 3´-end of the guide strand. The passenger strand had either a single amide or two consecutive amides at its 5’-end. The synthesis of 3´-amide-modified guide strands (G3-G7, Table 1) started with attachment of the first monomer, 5´-amino 2´-succinyluridine 3 (Figure 1), to the solid support. This was followed by peptide-like couplings of uridine 2a and adenosine 2b amino acids to assemble the amide-linked RNA portion. Amide-to-phosphate switch monomer 1a or 1b was then introduced, allowing for the syntheses of G3-G7 to be completed using standard RNA 2´-O-TOM phosphoramidites (Glen Research). Preparation of the passenger strand having two 5’-amide-modifications (P2, Table 1) started with standard automated RNA synthesis. Phosphate-to-amide switch monomer 4 was then introduced, allowing for assembly of the amide-linked RNA portion using the adenosine 2b and uridine 2a amino acids.
Figure 1.

Structures of 3´-end and 5´-end amide-modified RNAs and the building blocks required for their preparation.
Table 1.
Sequences of on-target siRNA (G0-P0), amide-modified passenger and guide strands, and non-target control siRNA (NTC) used in the RNAi experiments.
| Code[a] | N[b] | RNA sequence[c] |
|---|---|---|
| G0-P0 | 0 | 3´-UU GUU AAG CAU CCA GUU UUA U-5´ (passenger) 5´-CAA UUC GUA GGU CAA AAU AUU-3´ (guide) |
| P0 | 0 | 3´-UUG UUA AGC AUC CAG UUU UAU-5´ |
| P1 | 1 | 3´-UUG UUA AGC AUC CAG UUU UAamU-5´ |
| P2 | 2 | 3´-UUG UUA AGC AUC CAG UUU UamAamU-5´ |
| G0 | 0 | 5´-CAA UUC GUA GGU CAA AAU AUU-3´ |
| G3 | 3 | 5´-pCAA UUC GUA GGU CAA AAUamAamUamU-3´ |
| G4 | 4 | 5´-pCAA UUC GUA GGU CAA AAamUamAamUamU-3´ |
| G5 | 5 | 5´-pCAA UUC GUA GGU CAA AamAamUamAamUamU-3´ |
| G6 | 6 | 5´-pCAAUUCGUAGGU CAAamAamAamUamAamUamU-3´ |
| G7 | 7 | 5´-pCAAUUCGUAGGU CAamAamAamAamUamAamUamU-3´ |
| NTC | 0 | 3´-UU AUC GCU GAU UUG UGU AGU U-5´ (passenger) 5´-UAG CGA CUA AAC ACA UCA A UU-3´ (guide) |
P and G denote the passenger and guide strands, respectively; NTC denotes non-target control siRNA.
N is number of amide modifications.
In RNA sequences, p denotes a phosphate monoester at the 5´-end and each am denotes an internucleoside amide linkage. U or A denotes either a phosphate-to-amide (4) or amide-to-phosphate (1a or 1b) switch monomer being used during solid phase synthesis.
Main Text Paragraph.
Synthesis of monomeric building blocks.
The key monomers for preparation of amide-modified RNA were the nucleoside amino acids 2a and 2b (boxed in Figure 1). In earlier work,[15] we synthesized the uridine monomer 2a starting from d-xylose.[17] However, such a route required 18 steps and gave 2a in only 4% overall yield. Moreover, we were unable to make adenosine monomer 2b following this route because of elimination of the adenine heterocycle during attempts to introduce the 2´-O-TBS protection. In the present study, we used our recently developed and more efficient synthesis route that gave 2a in only six steps and 31% overall yield and 2b in eight steps and 16% overall yield.[18] Switch monomers 1a and 1b were prepared as previously reported.[14a] The synthesis of monomers 3 and 4 (for details, see Supporting Information) started with a non-selective TOM protection of 5´-azidouridine (Scheme 1) that gave a separable mixture of 3´- and 2´-O-TOM isomers 5 and 6, respectively. Reduction of the azide was followed by 5´-tritylation to give 7 and 8, which were converted into succinylated monomer 3 and switch monomer 4 using standard nucleoside chemistry procedures.
Scheme 1.

Synthesis of monomers 3 and 4. Steps: (a) TOM-Cl, Bu2SnCl2, DIPEA, ClCH2CH2Cl, reflux, 1 h, 26% of 5 and 41% of 6; (b) H2S (gas), pyridine:water (4:1), rt, overnight; (c) p-methoxytrityl chloride, DMAP, pyridine, rt, overnight, 65% of 7 and 65% of 8; (d) succinic anhydride, DBU, CH2Cl2, rt, 30 min, 80%; (e) P(OCH2CH2CN)(N(iPr)2)2, tetrazole, N-methylimidazole, CH2Cl2, rt, 24 h, 35%.
Design and synthesis of siRNAs containing consecutive amide linkages.
For the current study, we chose an siRNA (G0-P0, Table 1) that targets the mRNA of cyclophilin B (a housekeeping gene, peptidylprolyl isomerase B, PPIB) which we have previously used[14] to study the effect of amide modifications in RNAi. The G0-P0 duplex was rich in Us and As at the 3´-end of the guide and 5´-end of the passenger strand, which made synthesis of amide-linked RNAs easier because only modified U and A monomers were needed. Our previous studies[14a, 14b] showed that both guide and passenger strands of this siRNA were highly active (the passenger was also loaded in Ago2) and that isolated amide modifications in the guide strand strongly reduced its activity when paired with an unmodified passenger strand. Hence, G0-P0 was a good model system due to its relatively easy synthesis and high sensitivity to amide modification, which would provide a rigorous test of the limits of consecutive amide modifications that may be tolerated in an siRNA.
Synthesis of amide-modified guide strands G3-G7 was done on long chain alkylamine controlled pore glass (CPG, standard solid support for DNA/RNA synthesis, Glen Research) derivatized with monomer 3 through its succinamide linker (for details, see Supporting Information). Synthesis of amide-modified passenger strand P2 was performed on standard U-derivatized CPG RNA support (Glen Research). Amide couplings were done using either PyAOP (for G3-G7) or HATU (for P2) as activators for carboxylic acid monomers 2a and 2b (Figure 1); coupling yields were in the 80-85% range. The unmodified RNA parts of all sequences were synthesized by standard phosphoramidite chemistry using 2´-O-TOM protected monomers (Glen Research) on an Expedite 8909 DNA/RNA synthesizer. The amide-modified guide and passenger strands were cleaved from the solid support, deprotected, purified, and analyzed (MALDI-TOF MS) using standard RNA synthesis protocols, as recommended by Glen Research and previously reported by us.[14a, 15] Overall, the modest amide coupling yields enabled synthesis of sufficient amounts of modified RNA strands having up to six consecutive amide linkages (for yields, see Table S1 and Figures S1-S12). We obtained only a small amount of G7 (0.11 nmols).
RNAi activity of siRNAs having consecutive amide linkages.
The effect of amide modifications on RNAi silencing of the target, PPIB, was studied in HeLa cells (CCL2 variant) using a bDNA assay (QuantiGene 2.0 Reagent System, Figures S13) with GAPD (glyceraldehyde-3-phosphate dehydrogenase) serving as an internal reference. Initial experiments showed that RNAi activities of amide-modified guides G3 and G4, each paired with the unmodified passenger P0, were both <20% at 1 nM duplex concentration. For comparison, unmodified guide G0 yielded an activity of ~85% under the same conditions. Increasing siRNA duplex concentrations to 100 nM somewhat improved the silencing activity to 35%, 25%, and 90% for G3, G4, and G0, respectively (Figures S14). This result was consistent with our earlier study[14a] that showed that isolated amide linkages at most internal positions of this guide strand strongly reduce its RNAi activity. We later showed[14b] that the decreased silencing activity was due to preferential loading of the unmodified passenger P0 (instead of the modified guides) into Ago 2, and that the activity could be restored by using passenger strand P1, having a single amide linkage between its first and second nucleosides. Consistent with these earlier findings, pairing either G3 or G4 with P1 resulted in >80% silencing activity at both 100 and at 10 nm duplex concentrations (Figure 2).
Figure 2.

Concentration-dependent silencing activity of amide-modified guide strands G3, G4, G5, and G6 (for sequences, see Table 1) paired with passenger strand P1 having a single amide linkage between its first and second nucleosides. Activity is presented as a PPIB-to-GAPD mRNA ratio in HeLa (CCL2) cells, normalized to a non-target control (NTC). G0-P1 is the positive control. The data are averages of biological duplicate experiments, each performed as a technical triplicate.
Overall, all amide-modified guide strands (G3 through G6) showed the expected concentration-dependent silencing of the target, PPIB. While the activity of G3-P1 was similar to that of the positive control G0-P1 at both 100 and at 10 nm, increasing the number of consecutive guide-strand amide linkages from three to six resulted in progressively weaker silencing (Figure 2). Due to this trend and the limited amount of G7 synthesized, G7 was not included in these experiments. The results in Figure 2 showed that siRNA having five total amide linkages (four in guide strand G4 and one in passenger strand P1) had high silencing activity (~80%) at 10 nM siRNA concentration; siRNAs with higher number of amide modifications retained useful, albeit lower silencing activity at 100 nM.
Similar results were observed when the amide-modified guide strands were paired with passenger strand P2 having two amide linkages at its 5´-end (Figure 3). While the activity of G3-P2 was similar to that of the positive control G0-P2 at both 100 and at 10 nM, increasing the number of consecutive guide strand amide linkages lead to progressively weaker silencing. Similar to results obtained with P1-containing siRNAs in Figure 2, the results in Figure 3 showed that G3-P2 having five total amide linkages (three in guide strand G3 and two in passenger strand P2) had high silencing activity (~80%) at 10 nM siRNA concentration, while G4-P2 and G5-P2 retained useful, albeit lower silencing activity at 100 nM.
Figure 3.

Concentration-dependent silencing activity of amide-modified guide strands G3, G4, G5, and G6 (for sequences, see Table 1) paired with passenger strand P2 having two consecutive amide linkages at its 5´-end. Activity is presented as a PPIB-to-GAPD mRNA ratio in HeLa (CCL2) cells, normalized to a non-target control (NTC). G0-P2 is the positive control. The data are averages of biological duplicate experiments, each performed as a technical triplicate.
We have previously reported that isolated amide linkages did not significantly change the structure and thermal stability of RNA duplexes[13] and caused relatively small decreases in RNAi activity when incorporated in an siRNA guide strand.[14a] Three consecutive amide linkages caused relatively small changes in the structure of an A-type duplex; however, the melting temperature of the duplex was significantly lowered.[15] To test if the observed decrease in RNAi activity in Figures 2 and 3 correlated with systematic changes in thermal stability, we recorded the melting temperatures of the corresponding siRNA duplexes (Table S2). A single amide modification caused a drop in melting temperature from ~65 °C for G0-P0 to ~60 °C for G0-P1. However, further increasing the number of amide linkages in either guide or passenger strand did not significantly change the thermal stability of siRNA duplexes, with melting temperatures ranging from 60.1 to 61.6 °C. We concluded that decreases in RNAi activity observed when increasing the number of amide modifications did not correlate with changes in thermal stability of the modified siRNA duplexes.
To test if the decreased negative charge due to multiple amide modifications enabled passive uptake of siRNAs, we compared the silencing activities of G7-P1 (8-amides), G7-P2 (9-amides), and the unmodified G0-P0 (no amides) in the presence and in the absence of transfection reagent (RNAiMAX) at 100 nM siRNA duplex concentrations (Figures S15). As expected from the results in Figures 2 and 3, in the presence of RNAiMAX the strong RNAi activity of G0-P0 decreased with the increasing number of amide linkages in G7-P1 and G7-P2. Nevertheless, G7-P2 achieved ~50% silencing of PPIB at 100 nM. There was no detectable knockdown for any of the duplexes in the absence of RNAiMAX, suggesting that elimination of up to nine phosphates (~22% of all siRNA phosphates) was not sufficient to enable passive uptake of amide-modified siRNAs.
Nuclease stability of amide-modified RNAs.
Previous studies by Iwase et al.[16] showed that two consecutive amide linkages at the 3´-ends of both guide and passenger strands enhanced the resistance of siRNAs against degradation by nuclease S1 and 50% mouse plasma. We observed similar enhancement of stability of the 3´-amide-modified guide strands against 3´-specific snake venom phosphodiesterase (SVPD) and the 5´-amide-modified passenger strands against 5´-specific bovine spleen phosphodiesterase (BSPD). For example, >90% of G3-G5 remained intact (Figures S20) under conditions where more than 50% of the unmodified G0 was digested by SVPD. Similarly, P1 and P2 where digested slower by BSPD than P0 or G3 that lacked the 5´-amide modifications (Figures S21). Overall, these results were consistent with Iwase et al.[16] and confirmed the expected trend that amide-linkages increase the nuclease stability of RNA.
Conclusions
In this study, we developed methods that allowed us for the first time to introduce up to seven consecutive amide linkages between uridine and adenosine nucleosides in RNA. Limitations currently remain due to challenges associated with the synthesis of cytosine and guanosine nucleoside amino acids, as well as modest amide-coupling efficiencies. Synthesizing cytosine and guanosine monomers and optimizing amide couplings remain future tasks whose accomplishment will expand the sequences that can be linked with amides and allow for the preparation of longer stretches of amide-linked RNA. Nevertheless, the current methodology allowed us to explore the limits of amide modification in a model siRNA duplex. We found that up to five amide linkages were tolerated without significant loss of RNAi activity. An siRNA having four consecutive amide linkages in the guide strand maintained high silencing activity (~80% of target silencing) at 10 nM when paired with a passenger strand having one amide modification between its first and second nucleosides. A similar result was observed for an siRNA having three and two consecutive amide linkages in the guide and passenger strands, respectively. Further increases in the number of amide modifications in either the guide or passenger strand decreased RNAi activity; however, siRNA duplexes with up to nine amide linkages retained useful RNAi activity at higher (100 nM) siRNA concentrations. It should be noted that the siRNA duplex studied herein showed high sensitivity to amide modifications in one of our previous studies.[14a] It is conceivable that other siRNA sequences may be more tolerant to multiple amide substitutions. While an siRNA duplex having nine amide linkages retained some silencing activity in the presence of lipid transfecting agent, we did not detect any activity in the absence of transfection agent, indicating that the partial reduction of the negative charge did not enable passive uptake in HeLa cells. Taken together our results suggest that multiple consecutive amide linkages may be used to optimize biological properties of siRNAs. However, additional chemical modifications, such as conjugation with cell-penetrating peptides, are needed to enable cellular uptake of siRNAs in the absence of transfection agents.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health (R01 GM071461 and R35 GM130207 to E.R.). The Regional NMR Facility (600 MHz instrument) at Binghamton University is supported by NSF (CHE-0922815).
Footnotes
Experimental Section
Detailed methods are described in the Supporting Information.
Supporting information for this article is given via a link at the end of the document.
References
- [1].Tripp Ralph A., Karpilow JM, Frontiers in RNAi, Vol. 1, Bentham Science, 2014. [Google Scholar]
- [2] a).Bobbin ML, Rossi JJ, Annu. Rev. Pharmacol. Toxicol 2016, 56, 103–122 [DOI] [PubMed] [Google Scholar]; b) Zuckerman JE, Davis ME, Nat. Rev. Drug Discovery 2015, 14, 843–856 [DOI] [PubMed] [Google Scholar]; c) Wittrup A, Lieberman J, Nat. Rev. Genet 2015, 16, 543–552 [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Garber K, Nat. Biotechnol 2018, 36, 777–778 [DOI] [PubMed] [Google Scholar]; e) Setten RL, Rossi JJ, Han S.-p., Nat. Rev. Drug Discovery 2019, 18, 421–446. [DOI] [PubMed] [Google Scholar]
- [3].Wilson RC, Doudna JA, Annu. Rev. Biophys 2013, 42, 217–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T, Nature 2001, 411, 494–498. [DOI] [PubMed] [Google Scholar]
- [5] a).Whitehead KA, Langer R, Anderson DG, Nat. Rev. Drug Discovery 2009, 8, 129–138 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kanasty R, Dorkin JR, Vegas A, Anderson D, Nat. Mater 2013, 12, 967–977. [DOI] [PubMed] [Google Scholar]
- [6] a).Morrissey DV, Lockridge JA, Shaw L, Blanchard K, Jensen K, Breen W, Hartsough K, Machemer L, Radka S, Jadhav V, Vaish N, Zinnen S, Vargeese C, Bowman K, Shaffer CS, Jeffs LB, Judge A, MacLachlan I, Polisky B, Nat. Biotechnol 2005, 23, 1002–1007 [DOI] [PubMed] [Google Scholar]; b) Zimmermann TS, Lee ACH, Akinc A, Bramlage B, Bumcrot D, Fedoruk MN, Harborth J, Heyes JA, Jeffs LB, John M, Judge AD, Lam K, McClintock K, Nechev LV, Palmer LR, Racie T, Roehl I, Seiffert S, Shanmugam S, Sood V, Soutschek J, Toudjarska I, Wheat AJ, Yaworski E, Zedalis W, Koteliansky V, Manoharan M, Vornlocher H-P, MacLachlan I, Nature 2006, 441, 111–114 [DOI] [PubMed] [Google Scholar]; c) Akinc A, Zumbuehl A, Goldberg M, Leshchiner ES, Busini V, Hossain N, Bacallado SA, Nguyen DN, Fuller J, Alvarez R, Borodovsky A, Borland T, Constien R, de Fougerolles A, Dorkin JR, Narayanannair Jayaprakash K, Jayaraman M, John M, Koteliansky V, Manoharan M, Nechev L, Qin J, Racie T, Raitcheva D, Rajeev KG, Sah DWY, Soutschek J, Toudjarska I, Vornlocher H-P, Zimmermann TS, Langer R, Anderson DG, Nat. Biotechnol 2008, 26, 561–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Nothisen M, Kotera M, Voirin E, Remy J-S, Behr J-P, J. Am. Chem. Soc 2009, 131, 17730–17731. [DOI] [PubMed] [Google Scholar]
- [8].Soutschek J, Akinc A, Bramlage B, Charisse K, Constien R, Donoghue M, Elbashir S, Geick A, Hadwiger P, Harborth J, John M, Kesavan V, Lavine G, Pandey RK, Racie T, Rajeev KG, Rohl I, Toudjarska I, Wang G, Wuschko S, Bumcrot D, Koteliansky V, Limmer S, Manoharan M, Vornlocher H, Nature 2004, 432, 173–178. [DOI] [PubMed] [Google Scholar]
- [9] a).Akinc A, Querbes W, De S, Qin J, Frank-Kamenetsky M, Jayaprakash KN, Jayaraman M, Rajeev Kallanthottathil G, Cantley William L, Dorkin JR, Butler James S, Qin L, Racie T, Sprague A, Fava E, Zeigerer A, Hope Michael J, Zerial M, Sah Dinah WY, Fitzgerald K, Tracy Mark A, Manoharan M, Koteliansky V, Fougerolles Antonin d., Maier Martin A, Mol. Ther 2010, 18, 1357–1364 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Falsini S, Ciani L, Ristori S, Fortunato A, Arcangeli A, J. Med. Chem 2014, 57, 1138–1146. [DOI] [PubMed] [Google Scholar]
- [10] a).Watts JK, Corey DR, J. Pathol 2012, 226, 365–379 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Deleavey GF, Damha MJ, Chem. Biol 2012, 19, 937–954 [DOI] [PubMed] [Google Scholar]; c) Bramsen JB, Corey DRJ. Kjems, in Methods Mol. Biol, Vol. 942, Springer, New York, 2013, pp. 87–109 [DOI] [PubMed] [Google Scholar]; d) Bramsen JB, Grunweller A, Hartmann RK, Kjems J, in Handbook of RNA Biochemistry, Second Edition (Eds.: Hartmann ABRK, Schon A, and Westhof E), Wiley-VCH, Weinheim, Germany, 2014, pp. 1243–1277 [Google Scholar]; e) Bramsen JB, Kjems J, Frontiers in Genetics: Non-Coding RNA 2012, 3, Article 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Mullard A, Nat. Rev. Drug Discovery 2018, 17, 613. [DOI] [PubMed] [Google Scholar]
- [12].Meade BR, Gogoi K, Hamil AS, Palm-Apergi C, Berg A. v. d., Hagopian JC, Springer AD, Eguchi A, Kacsinta AD, Dowdy CF, Presente A, Lonn P, Kaulich M, Yoshioka N, Gros E, Cui X-S, Dowdy SF, Nat. Biotechnol 2014, 32, 1256–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13] a).Rozners E, Katkevica D, Bizdena E, Strömberg R, J. Am. Chem. Soc 2003, 125, 12125–12136 [DOI] [PubMed] [Google Scholar]; b) Selvam C, Thomas S, Abbott J, Kennedy SD, Rozners E Angew. Chem., Int. Ed 2011, 50, 2068–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14] a).Mutisya D, Hardcastle T, Cheruiyot SK, Pallan PS, Kennedy SD, Egli M, Kelley ML, Smith Anja van B., Rozners E, Nucleic Acids Res. 2017, 45, 8142–8155 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hardcastle T, Novosjolova I, Kotikam V, Cheruiyot SK, Mutisya D, Kennedy SD, Egli M, Kelley ML, van Brabant Smith A, Rozners E, ACS Chem. Biol 2018, 13, 533–536 [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Mutisya D, Selvam C, Lunstad BD, Pallan PS, Haas A, Leake D, Egli M, Rozners E, Nucleic Acids Res. 2014, 42, 6542–6551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Tanui P, Kennedy SD, Lunstad BD, Haas A, Leake D, Rozners E, Org. Biomol. Chem 2014, 12, 1207–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16] a).Iwase R, Miyao H, Toyama T, Nishimori K, Nucleic Acids Symposium Series 2006, 175–176 [DOI] [PubMed] [Google Scholar]; b) Iwase R, Toyama T, Nishimori K, Nucleosides, Nucleotides, Nucleic Acids 2007, 26, 1451–1454. [DOI] [PubMed] [Google Scholar]
- [17] a).Tanui P, Kullberg M, Song N, Chivate Y, Rozners E, Tetrahedron 2010, 66, 4961–4964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- b).Rozners E, Liu Y, Org. Lett 2003, 5, 181–184 [DOI] [PubMed] [Google Scholar]; c) Rozners E, Liu Y, J.Org.Chem 2005, 70, 9841–9848. [DOI] [PubMed] [Google Scholar]
- [18].Kotikam V, Rozners E, Org. Lett 2017, 19, 4122–4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.