

We propose nanomedicines able to target cells phenotypically that bind when a precise combination of receptors is expressed.
Abstract
Tight control on the selectivity of nanoparticles’ interaction with biological systems is paramount for the development of targeted therapies. However, the large number of tunable parameters makes it difficult to identify optimal design “sweet spots” without guiding principles. Here, we combine superselectivity theory with soft matter physics into a unified theoretical framework and we prove its validity using blood brain barrier cells as target. We apply our approach to polymersomes functionalized with targeting ligands to identify the most selective combination of parameters in terms of particle size, brush length and density, as well as tether length, affinity, and ligand number. We show that the combination of multivalent interactions into multiplexed systems enable interaction as a function of the cell phenotype, that is, which receptors are expressed. We thus propose the design of a “bar-coding” targeting approach that can be tailor-made to unique cell populations enabling personalized therapies.
INTRODUCTION
Possibly the most defining feature of a drug is its ability to interact with its biological target as selectively as possible, and indeed, most drug discovery tools are fined to identify those molecules that bind the strongest. Such a concept goes back to the 19th century, when Nobel laureate Paul Ehrlich postulated the side-chain theory proposing the existence of receptors and ligands (1). Selective drugging, popularized as the “magic bullet,” made the fortune of Ehrlich, and indeed, it is still the cornerstone of modern medicine. Today, drug discovery is a highly rigorous process that spans across structural and cell biology, bioinformatics, and computational and medicinal chemistry. It is now evolving and merging with -omic technologies to promise personalized therapies (2). Alongside drug development, we have also advanced our ability to deliver drugs combining molecular recognition with nanoscopic carriers equipped with the necessary attributes to navigate biological environments (3). Here, selectivity is bestowed decorating the carrier with ligands that enable targeting and crossing biological barriers. Drug discovery is thus now extending to target biological macromolecules that are not accessible via simple passive diffusion such as the inside of cells (4) or the central nervous system (5).
Today, our ability to create ligands is well advanced and can be extended to almost any biological unit. When the targeted receptor is exogenous to the host, such as the case of infections and poisoning, ligands can be made with selectivity close to “Ehlrich’s magic bullet.” However, in most diseases, with cancer being the most exemplary one, the malfunction is often associated with receptors that are endogenous and hence expressed by both healthy and diseased cells. Such a promiscuity is the major reason why most drugs come with side effects, and many failed to go through the clinical pipeline. Yet, such a promiscuous nature is managed with exquisite precision within a living system with molecules, proteins, nucleic acids, and cells interacting with one another with extremely high selectivity. Historically, the strength of interaction between a given ligand, L, and it receptor, R, is measured by its affinity, and this is defined by the same thermodynamic principles that apply to a reversible reaction. The reaction association constant , where kon and koff being the rates of binding and unbinding, respectively, is defined as the ligand affinity. The higher the ligand affinity, the lower the ligand concentration required to saturate its receptor. Affinity can be augmented by combining different ligands into multivalent scaffolds (6), and in these cases, the binding is defined by the term avidity, which represents the total effect of the bound units collectively (7). Multivalent interactions are critical in most biological processes as they allow the translation of weak bonds into strong ones, enabling clustering and signal transduction (8). Similarly, multivalent interaction is the bread and butter of supramolecular chemistry and often at the core of the design of nanoscale devices (9). From a theoretical standpoint, the probability of a single ligand to bind to a receptor-coated target can be expressed roughly as
| (1) |
where NR is the number of receptors on the target and ρ is the number concentration of ligands and can be written as ρ = [L]NA, where [L] is the molar concentration of the ligands and NA is the Avogadro number (see the Supplementary Materials for derivation). Thus, probability saturates to 1 for either large number of receptors NR or high binding strength. For this reason, high affinity means that a large proportion of ligands will bind to any cells that express the targeted receptors, not just those overexpressing them. This inevitably leads to unwanted interactions, which in the case of anticancer therapy, where the final aim is often to kill the abnormal cells, can lead to reactions that outweigh the clinical benefits. For multivalent systems, the binding affinity has a strong contribution from combinatorial entropy (7), which can be exploited for targeting. In 2007, Carlson et al. showed that multivalent targeting was more selective when multivalent low-affinity ligands were used (10). Noting such a peculiar feature of multivalency, in 2011, Martinez-Veracoechea and Frenkel came up with a very interesting approach proposing, based on a statistical mechanical description, the superselectivity theory (SST) (11). They show that, in contrast to what happens with high-affinity ligands, the combination of multiple low-affinity ligands creates on-off association profiles, where the multivalent scaffold saturates the receptors only above a given onset receptor density, while it does not bind at all at lower densities. Such a scenario is indeed what is required to target cancer cells, which often overexpress receptors otherwise present in several healthy tissues. SST was proven experimentally in model systems such as supramolecular polymers (12) and multivalent polymers (13, 14). However, a major limitation to the applicability of SST is that the affinity required to create superselective profiles is rather low, corresponding to binding energies of order of few kBTs, where kB is the Boltzmann’s constant and T is the absolute temperature. The receptor and its ligand interact via supramolecular forces emerging such as Coulombic forces, hydrogen bonds, aromatic interaction, hydrophilic and hydrophobic effects, and van der Waals interactions. Although, these are usually weak forces, the range of realistic binding energy is much higher than that required by SST with the lowest limit being the water hydrogen bond of about 8kBT (15) to the strongest biological supramolecular bond known so far, the avidin/biotin complex, with association energy of c.a. 30kBT (16). In addition to this, as recently demonstrated by Angioletti-Uberti, multivalent systems are strongly affected by unspecific interactions induced by the presence of ligands other than the targeted ones, and this is exacerbated by the use of low-affinity ligands (17). In the following, we show how these problems can be solved by combining the general concept of SST theory with principles from soft matter and polymer physics that allow to concurrently modulate the bond-mediated–specific interaction and avoid nonspecific ones. In doing that, we also show how to achieve multiplexed targeting based on multiple receptor types.
THEORY
Rules of engagement in a biological environment
From blood plasma to interstitial fluid to the cell cytosol, biological liquids are crowded aqueous oversaturated solutions with high molecular diversity. Typical protein volume fractions range from 10% in the blood plasma to up to 40% in cellular organelles. Assuming hexagonal packing, the ratio between the protein radius, rP, and the interprotein distance, rij, is a function of the volume fraction, . Hence, each protein is packed with average interparticle distance ranging from 6 to 0.3 times its own radius. Macromolecular crowding means that protein diffusion is considerably decreased (18, 19), while metabolites and ions diffusing through the protein network exhibit enhanced percolation (18). The same water that bathes the proteins is confined in a thin interface and hence exhibits properties different from bulk behavior (20). In addition to the constitutive bonds that characterizes each unit, biological molecules and macromolecules interact via weaker supramolecular forces. These interactions are either electrostatic such as ionic, hydrogen bonding, π-π, metal complexation, and van der Waals or emerging from the interaction with the solvent (i.e., the water) including attractive hydrophobic and repulsive hydrophilic interaction. Supramolecular forces combine into isotropic nonspecific potentials that, for the single unit to be stable, ought to be repulsive and stronger than the thermal fluctuations at very short distances (i.e., U(r → 0) > 1kBT). At larger distances, the word fluid already indicates that these forces are weaker and of similar magnitude to thermal fluctuations. Any attractive net potential stronger than the thermal fluctuations will lead to association and aggregation. A good example of such a scenario is when an exogenous element placed within a biological fluid quickly attracts all the proteins around. Such a fouling process, known as opsonization, is a critical step of immunological surveillance and correlates with fast riddance (21). Many biological structures are also capable of creating unique chemical combinations that make supramolecular forces between two complementary molecular arrangements very specific, directional, and stronger than thermal fluctuations. Molecular recognition processes such as ligand/receptor binding are critical to control interaction as well as to serve as a template for drug design. It is also important to notice that in complex multicellular organisms, opsonization acts together with preprogrammed proteins that recognize nonself or abnormal self-species and hence bestowed with the ability to detect chemical signatures classified by the adaptive immune system as non gratae (22).
Proteins control their repulsive potential via their surface charge and structure. For large objects such as cells and viruses, their surfaces are often coated with long polysaccharides also known as glycans (23), either chemically linked to proteins forming glycoproteins (e.g., syndecan, mucins, etc.) or bound to dedicated receptors (e.g., CD44/hyaluronan) (24, 25). Glycans are often packed densely, and this in combination with their strong affinity with water drives the chain to stretch forming brush-like profiles. Such an arrangement creates a very strong repulsive steric repulsive potential that prevents nonspecific interaction (26, 27).
Synthetic hydrophilic polymer brushes are the most common strategy in biomaterial design to prevent unspecific interaction and protein fouling so as to ensure long-term compatibility (28–30). Experimental observations have shown that unspecific interaction are best controlled using nonimmunogenic moieties that have either neutral polar or zwitterionic functional groups (30–32). These are known to interact with water orienting it in their close proximity creating repulsive potentials relatively insensitive to other species (30, 32). The most common polymer that fulfills such requirements is the poly(ethylene oxide) (PEO) also known as poly(ethylene glycol) (28–30). This is one of the very few synthetic polymers that are generally recognized as safe for most medical applications, and it is used routinely in the clinic as adjuvant/coating for several devices and drugs (28). Alternatives to the PEO include poly(vinyl pyrrolidone), poly(2-methacryloyloxyethyl phosphorylcholine) (PMPC), poly(glycerol)s, poly(amino acid)s, polysarcosine, poly(2-oxazoline)s, and poly(N-(2-hydroxypropyl)methacrylamide) (28, 29).
The case study
To help the discussion and facilitate theory derivation and calculations, while at the same time studying a relevant system, we selected brain endothelial cells (BECs) as biological target. BECs are the most important actors in the blood-brain barrier and hence, the most critical target to devise brain delivery strategies (5). BECs are very much like any other endothelial cells in our body, and their major function is forming the vessels that carry blood supply. Yet, every organ, and brain in particular, conditions endothelial cells to control the crossing of metabolites and immune cells according to their specific needs. BECs are programmed to be very “permeable” to glucose and several other small metabolites but almost impermeable to most drugs and to immune cells (5). This has created a considerable hurdle to any neurological pharmaceutical development (5). We demonstrated that BECs can be targeted and crossed using multivalent polymersomes bearing ligands for the low-density lipoprotein receptor–related protein 1 (LRP1) (33, 34). We also demonstrated that the PMPC polymer chains target the scavenger receptor B1 (SRB1) (35), also expressed by BECs (33). We know from the literature (36, 37) that endothelial cells (38, 39), and BECs (37) in particular, express high levels of the glycocalyx syndecan 4. On the basis of this information, we thus reconstructed a possible arrangement of LRP1, SRB1, and syndecan 4 proteins on a stereotypical BEC membrane. The resulting scheme is shown in Fig. 1A, where all the molecules are drawn to scale. The structure of the LRP1 was reconstructed using iterative threading assembly refinement (see the Supplementary Materials), while the glycocalyx structure was used as reported by Cruz-Chu et al. (40). For the SRB1, we used the Protein Data Bank–deposited crystal structure (4F7B) as reported by Neculai et al. (41). The syndecan 4 is decorated with four heparan sulfate (HS) chains with a polymerization degree of 100 and placed in the membrane with density estimated by experimental reconstructions (36, 37). The resulting arrangement clearly shows that anything interacting with either LRP1 or SRB1 will be also affected by the steric hindrance of the HS chains and suggests that only by analyzing the holistic interaction we can disclose the most effective targeting strategy.
Fig. 1. Examples of polymer brushes.
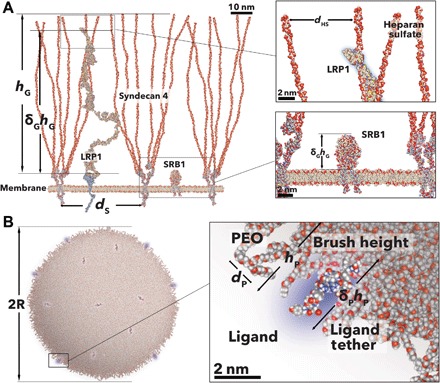
Schemes of glycocalyx syndecan 4 LRP1 and SRB1 receptor. Proteins were reconstructed with atomic resolution using computational methods and minimized for a stretched brush conformation. Both insets show the details of the end part of the LRP1 next to the four HS chains and the SRB1 size with respect to the syndecans (A). Scheme of a POEGMA-PDPA polymersome decorated with angiopep peptides. The polymersomes were reconstructed using a minimized atomistic model of the single blocks that, in turn, were assembled into a 50-nm vesicle. The inset details show that the peptide is well embedded in the PEO brush.
As targeting units, we propose synthetic vesicles, known as polymersome, made of the self-assembly in water of amphiphilic poly[oligo(ethylene glycol) methyl methacrylate]-poly(2-(diisopropylamino)ethyl methacrylate) (P[(OEG)10MA]20-PDPA100), which we will refer to as EP for simplicity. Their structure is shown in Fig. 1B, where some of the POEGMA chains are functionalized with a peptide ligand. We have studied this particular system in large detail and demonstrated its use for the in vivo targeting of the blood-brain barrier (33, 34) and peritoneal metastasis (42, 43). As shown in Fig. 1B, the oligo(ethylene glycol) chains cluster at high density on the surface, creating a brush-like layer that protects polymersomes from unspecific interaction and modulates the ligand binding as it will be discussed in the following section.
Steric potentials as interference effect
Now that we have established the “rules of engagement” and the model study, the quest is to define how either glycans or antifouling polymers contort the ligand/receptor interaction. We thus define three possible scenarios: In Fig. 2A, the EP polymersome is decorated with angiopep-2 peptides, known to target the LRP1 receptor (44); in Fig. 2B, the EP polymersome is decorated with PMPC polymer chains to target the SRB1 receptor (35); lastly, in Fig. 2C, the two ligands are combined together (Fig. 2E). In all three scenarios, the EP polymersomes interact with LRP1 (Fig. 2D) and/or SRB1 receptors dispersed in a matrix of syndecan 4. This particular glycocalyx is known to be overexpressed by endothelial cells (38, 39) and the brain endothelial in particular (37), and its role is to control both the blood fluid dynamics and the transport across (36, 45). We use structural data available from the literature and inferred that each receptor is associated with at least two syndecan 4 (38, 39). In each scenario, the receptor/ligand interaction drives the association, and this is opposed by two steric potentials: one arising from the glycocalyx brush and one arising from the polymer brush that coats the nanoparticle. The magnitude of both depends on how accessible ligands and receptors are. This, in turn, depends on (i) the relative height of the receptor with respect to the HS chains, which we define as δGhG where hG is the HS length and (ii) the tether length of the ligands, which we define here as δPhP where hP is the PEO chains length. Both, δG and δP are defined as the interference parameters, with δP, δG ∈ [0,1]. For simplicity, we neglect the protein component of the syndecan and consider only the HS chains. For receptors shorter than the HS chains, binding requires that the nanoparticle inserts into the HS brush and hence feels a steric potential arising from both the osmotic pressure due to its volume being depleted and an elastic component due to the compressions of the chains. According to Halperin (46), the two components depend on the relative ratio between the object entering the brush (here, the nanoparticle) and the brush height. If we assume that the nanoparticle radius is smaller than the brush height, i.e., R < hG, then, as in (46), the compression component can be neglected. Considering that HS chains are as long as 100 nm (38, 39), we believe this condition applies to most nanomedicines whose optimal radius is always around 50 nm. We can thus calculate the potential as a function of the glycol interference parameter, δG, considering only the osmotic pressure component as
| (2) |
where β = (kBT)−1, R is the particle radius, σHS is the area per HS chain, which can be derived as , with dS being the interchain distance between two syndecans each bearing six HS chains (see Fig. 1A). Note that the glycocalyx potential is invariant with the HS chain polymerization degree as long as we do not consider the compression of the chains, i.e., R < hG.
Fig. 2. Repulsive steric potentials.
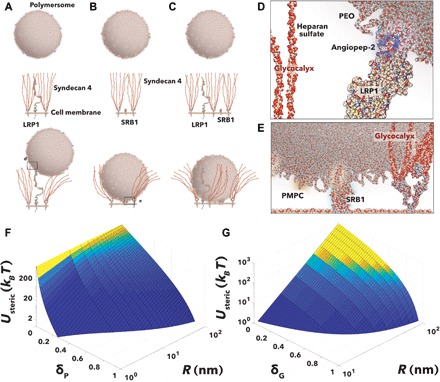
Schematics of the binding of a multivalent POEGMA-PDPA polymersome decorated with angiopep peptide and targeting LRP1 (A) with PMPC chains and targeting SRB1 receptors (B) and with both ligands and targeting both receptors (C). The detail of the interaction between angiopep and LRP1 (D) and PMPC and SRB1 (E) modulated by both the PEO and glycocalyx brushes. The corresponding repulsive steric potentials exerted on the LRP1 insertion in the PEO brush (F) and the polymersome inserting in the glycocalyx brush (G). These are calculated as a function of the polymersome radius, R, and insertion parameter for the PEO chains, δP, and for the glycocalyx HS chains, δG, respectively.
If we now consider the polymer brush made by the PEO chains and expressed on the nanoparticle surface, as the receptor binds to its ligand, the chains apply a steric repulsion to the receptor tip. For simplicity, we assume the receptor tip with volume VP and , we can write, using the same model for Eq. 2
| (3) |
Because of the nonnegligible curvature of the nanoparticle, here, σP, i.e., the area per PEO chain, changes along the brush height, hP, as a function of the nanoparticle size R. Using the model proposed by Zhulina et al. (47), we derive the area per chain σP as a function the interference parameter, δP as
| (4) |
with σ0 being the grafting density at the surface (i.e., δP = 0) and γ being a geometrical parameter to represent the packing of the chains on the surface, which is for and γ = 3 for any other values, where is the value of the brush height on a planar surface. According to Zhulina et al. (47), we can write the brush height as
| (5) |
where aPEO is the PEO monomer size, is the monomer excluded volume parameter and NPEO is the PEO degree of polymerization. Note that . If we substitute Eq. 5 in Eq. 4 we can write
| (6) |
Both Eqs. 3 and 6 can be used to calculate the steric potentials (Fig. 2, F and G) as a function of the particle core radius R and two insertion parameters δG and δP. In both cases, we use experimental values using the polymersome model and the syndecan 4 brush expressed by BECs. The resulting curves show considerable energetic barriers for both brushes. As shown in Fig. 2F potentials up to ∼200kBT are required to overcome completely the PEO brush repulsion at δP = 0. The graph in Fig. 2G shows that the syndecan 4 brush act as an effective filter imposing prohibitive (∼1000kBT) potentials for particles closer to 100 nm. However, it is important to notice that for larger radii, the steric potential is no longer due to osmotic displacement of the chain but to their compression.
Multivalent interactions
Now that we have shown how both the polymer brush and the glycocalyx brush tune the binding strength of a given ligand-receptor pair, we need to describe how the possibility to form multiple bonds at the same time affects the overall binding energy of the nanoparticle. In other words, we need a general model to describe multivalent effects. The latter arise from the fact that nanoparticles can use their ligands to bind the cell surface forming many distinct bond arrangements. Each of these constitute a possible microstate of the system that should be taken into account when calculating the free energy due to bond formation (7). As first shown by Kitov and Bundle, there is a degeneracy, Ω, associated to each microstate that can strongly contribute to its weight in determining the overall binding free energy. This is simply due to the fact that this degeneracy translates in an associated entropy, typically named “avidity entropy” Savidity = kBlogΩ. In calculating the binding energy of a nanoparticle to a receptor-bearing surface, each microstate should be properly taken into account, including its entropic contribution. Angioletti-Uberti et al. showed (48) that when this is done, a general analytical formula arises for the free energy due to bond formation
| (7) |
where pi is the probability that a ligand or receptor i is unbound and the sum is all possible ligand and receptors. The values of pi are given by the solution of the following set of self-consistent equations
| (8) |
one for each ligand or receptor in the system, all coupled together. The sum in the left-hand side of Eq. 8 runs over all possible neighbors j of a binder i (i being either a ligand or a receptor) and χij = e−βΔGij is the bond strength for that specific ligand-receptor pair (46). In the case where all ligands bind to a single type of receptor only, and vice-versa, and considering that receptors are mobile on the cell surface, one can take an average over all receptors’ positions and substitute χij with its average value 〈χ〉, which would only depend on the type of ligand and receptor (49, 50). In this case, the equations leading to Ebond can be solved analytically (see the Supplementary Materials). At this point, it is important to discuss what the various contributions to the bond energy are, since this is crucial to understand how to engineer/design our targeting system. As shown in (49) the bond energy can be written as with being the binding energy or affinity from association of ligand i and receptor j in solution, as measured by the ligand/receptor equilibrium dissociation constant, KD, that can be measured experimentally and ρ° = 1 M is the standard concentration. is a configurational contribution due the constraints imposed by binding (49). In our system, there are two contributions that we need to include in . The first arises because of the mobility of the receptors (50)
| (9) |
This contribution accounts for the fact that to interact and bind to a ligand, receptors need to be in its proximity. This limits their position within an area Abind < Afree, where Afree is the area that they can span in the free, unbound state. To estimate , hence, we take Abind = 2π(δhP)2, i.e., approximately the area spanned by a rigid ligand, whereas for Afree, we take the surface area exposed by a typical cell, of about 400 μm2. The second important contribution to comes from the fact that to bind a ligand, receptors need to penetrate the PEO brush. We calculate this latter contribution assuming that the equilibrium adsorption distance between nanoparticles surface and receptors is the average ligand length, i.e., we set
| (10) |
Given the free energy for adsorption defined by Eqs. 7 to 10, as in (11), we use a simple Langmuir-Hill model to describe the binding of the nanoparticles to a cell, considered as a multivalent surface. By using this model, we implicitly assume that (i) we have a fixed number of adsorption sites and the number of receptors on each is given by a Poisson distribution with average <NR>, (ii) different nanoparticles do not compete for the same receptors, and (iii) a surface can only be occupied by one nanoparticle at a time. Hence, we can write the fraction of bound nanoparticles, θ, as
| (11) |
where 〈〉P is an average over all possible distributions of receptors on the adsorption sites, weighted by their Poisson probability. In Eq. 11, q is the ratio between the bound versus unbound partition function for a single nanoparticle on an adsorption site, while a is the nanoparticle activity, which under the experimentally relevant dilute conditions can be approximated as
| (12) |
where [P] is the molar concentration of the nanoparticles in the bulk solution. The volume vB can be derived for a spherical particle with radius R and ligand tether length, d = δphP, approaching a surface as
| (13) |
The single-site bound state partition function is related to the adsorption free energy by (17)
| (14) |
where Ebond is the free energy due to bond formation, properly summed up over all possible bonding combinations given by Eq. 7. The additional −1 takes into account the fact that the nanoparticle is considered bound only in the case where at least one bond is present (16), and the factor exp(−βUG) accounts for the fact that in the bound state, the particle gain an energy contribution of UG due to the repulsion caused by the glycocalyx. We can thus combine Eqs. 12 to 14 to obtain
| (15) |
Equation 15 associates binding to several design parameters and hence allows to identify the most effective combinations. To facilitate the identification of superselective regimes we use, from now on, the same selectivity function defined by Martinez-Veracoechea and Frenkel (11) as
| (16) |
For monovalent binding the selectivity α ≤ 1 and under superselective conditions for multivalent interactions. Equation 11 can be also expressed as an Hill function with n being the Hill coefficient that defines the binding cooperativity (51) and indeed at very low surface coverage α ∼ n. However, here, we consider all possible bonds making α not constant. We thus define the receptor (or ligand) number, where α takes its maximum value as the onset density Nonset (note that this number is actually the average value per site that controls the Poisson distribution) and the corresponding value of α(Nonset) ≡ αmax as the super selectivity parameter. As discussed previously (11), superselective binding corresponds to quasi-step–like θ(NR) or θ(NL) functions where the fraction of bound particles rapidly grows from c.a. zero to c.a. 1 as the number of receptors (or ligands) goes above the onset density. Across this threshold value, a minimal change in NR corresponds to changes of . Therefore, nonselective binding corresponds to αmax < 1, whereas superselective profiles will have αmax > 1.
The graphs showed in Fig. 3 shows heat maps of θ as a function of the receptor average number per adsorption site <NR> and different functional parameters. The maps were used to calculate the selectivity αmax, and the onset density Nonset as a function of the same parameters. We used the angiopep-2 affinity to LRP1 as reported in literature (52) and used the size of the LRP1 receptor and its insertion in the glycocalyx matrix as shown in Fig. 1A.
Fig. 3. Scaling principles in superselectivity.
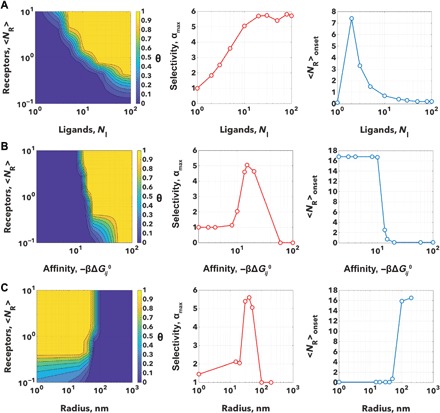
Heat maps showing the fraction of bound particle θ as a function of the numbers of receptors <NR> and number of ligands NL (A), the additive inverse ligand affinity −βΔGij (B) and particle radius, R (C). Each map was analyzed to calculate the selectivity αmax and the corresponding <NR> onset, and the graphs of these as a function of the varying parameter are reported alongside.
Each variable was optimized to achieve high selectivity and tunable onset density. As shown in Fig. 3A, θ is extremely sensitive to the ligand number, NL, with selectivities always larger than one and even approaching 6. However, the higher the number of ligands, the higher the selectivity and the lower is the onset density. This varies with a normal-like trend with a peak at around two ligands and decaying to zero as NL → ∞. The hyperbolic decay allows for precise tuning only between 2 and 10 ligands, while for larger NL, the onset density goes to zero. For the ligand affinity (Fig. 3B), we observe no interaction above −8 kBT where the binding energy is too low to overcome the steric potentials. For lower values, the selectivity parameter αmax peaks between −10kBT and −20kBT to values close to 5 to then decay to nonselective values. In the similar range, the high selectivity corresponds to a high-onset receptor concentration with a fast decay in few kBT units. A very similar trend is observed with the polymer insertion parameter, δP and the glycocalyx insertion, δG (fig. S1A). It is also very important to note that the glycocalyx spacing, dS needs to be large enough to allow the nanoparticle to access the receptor and indeed if this is too tight or the receptor is well hidden within the glycocalyx network, no interaction takes place. However (as shown in fig. S1B), the extra steric potential considerably enhances the selectivity to 6 units while allowing for a good control on the onset receptor concentration. A similar argument can be made for the particle radius where small particles lack selectivity and large particles cannot penetrate the glycocalyx network (see Fig. 3C). The two counteracting parameters thus lead to well-defined size-controlled selectivity. Similarly, we can state that particles with larger radii have denser brushes and thus stronger repulsive contribution to the bond strength, which effectively decreases with increasing particle size. At the same time, however, the activity coefficient a increases with particle size. However, it is important to emphasize that the radius dependence effectively is mixed with the dependence on the number of active ligands as well as details relating to the ligand tether length. If we increase the radius but keep NL fixed, then we reduce the overall grafting density and/or we decrease the ligand length so that overall, the same number can bind (fig. S1A).
In our discussion, we also emphasize that the exact values of αmax and Nonset, as well as the sweet spots in terms of the optimal parameter range to tune these quantities, depend on the choice of the values for all other parameters involved (we have six, and we fix five of them in each graph). However, the trends observed are not qualitatively affected by this choice.
Multiplexing
As shown in Fig. 3, we can vary different parameters to achieve the selectivity required for the targeted receptor ensuring that binding occurs above a certain therapeutic threshold. However, the very same physics determining the conditions for superselectivity makes multivalent targeting of extremely high sensitivity and small changes of some of these parameters can lead to very different outcomes. In turn, this can lead to evolutionary responses that might simply render the system ineffective. For example, a small mutation in a receptor might make its binding energy toward targeting ligands weaker, shifting the required expression threshold at higher values than those experimentally achievable. We thus propose that to make multivalent targeting more robust and indeed precise toward these changes is to target more than one receptor type using different ligands. In this way, one can make sure that more complex evolutionary adaptation responses must occur before targeting is made ineffective. In doing this, we can make use of the growing amount of bioinformatic data available about cancer-related receptors and their expression in different cancer lines, making a step closer to fulfilling the “big data” revolution expected in the treatment of cancer (53).
We thus propose the design of nanoparticles comprising ζ > 1 type of multiple ligands, where each i type is expressed at numbers (Nl)i > 1 on the surface with tether length (δPhp)i. Each ligand is supposed to target a specific receptor type among those expressed on the surface. Considering that ligands are specific for one type of receptor only, hence competition between different ligands for the same receptor does not occur, Eqs. 7 and 8 show that the corresponding free energy of multiplexed and multivalent binding, (Ebond)MP can be expressed as
| (17) |
where ζ is an index running over all possible ligand-receptor pairs in the system. The values of is given simply by solving Eqs. 7 and 8 considering a subsystem where only that specific ligand-receptor pairs are present (and hence even in this case, an analytical solution is available; see the Supplementary Materials). In this multiplexed case, one should also account for the different volumes of the various receptors, leading to , where we explicitly indicated that the two steric penalties UP and UG are different for different ligand-receptor pairs ζ. We can hence rewrite Eq. 15 as
| (18) |
where we assumed that dζ = δPhP(1) = δPhP(2) = … = δPhP(ζ) or in other words that ligand tethers are equal for all the ζ receptor/ligand couples. If we had ligands with tethers of different lengths, then the situation would be more complicated, as the system would preferably stay at an intermediate distance from the surface. While multiplexing affects the single binding shifting it toward lower receptor densities, the clear advantage comes from the fact that we can engineer holistic binding profiles where nanoparticles bind to surfaces only if they express all the targeted receptors at densities above a given threshold as shown in Fig. 4 for ζ = 2, 3, and n. This means that nanoparticles can be designed to target specific cell populations that overexpress unique combinations and compositions of receptors. In other words, we can “bar-code” targeting to information gathered from -omic screenings on the specific biological target, hence potentially focusing interaction on a single-cell population. Note that this is different than the approach developed by Curk et al. (54), where it is shown to design nanoparticles so as to target a specific distribution of receptors in terms of its relative composition, at a fixed number of receptors. In their case, any change around the targeted distribution would decrease the binding probability. In our case, we look for the design conditions where binding would occur when more than one receptor is expressed above a certain threshold, but anything above that number would still lead to binding, making the system robust toward any biological fluctuations among the different cell populations.
Fig. 4. Multiplexing.
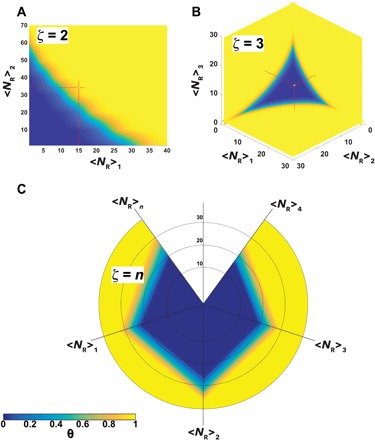
Heat maps showing the fraction of bound particle θ calculated for multiplexed multivalent nanoparticles targeting ζ = 2 (A) and ζ = 3 (B) for ζ > 3. The data are shown using a radar plot with a heat map (C) where multiple receptors can be combined in a potentially infinite number of permutations.
EXPERIMENTAL VALIDATION
As already anticipated, we decided to use EP polymersomes decorated with angiopep-2 peptides to target the LRP1 receptor (44) and responsible for the transport across the endothelial cells that make the blood-brain barrier (33, 34) and with PMPC chains to target the SRB1 (35). We prepared several polymersome formulations, which were all purified and characterized by transmission electron microscopy and dynamic light scattering (see fig. S2). The angiopep peptide was conjugated to POEGMA-PDPA copolymers, and these were mixed at different concentrations with pristine POEGMA-PDPA. The resulting arrangement of peptide expressed on the surface and immersed in the oligoethylene oxide chain (NP = 10) with an average interference parameter of δP = 0.8 as shown in Figs. 1 and 2. The PMPC chains were copolymerized with DPA to form PMPC24-PDPA70, and these were mixed with pristine POEGMA-PDPA chains at different concentrations. It is important to point out that while POEGMA-PDPA and PMPC-PDPA chains can undergo phase separation forming patchy polymersomes (55), the cellular experiments were performed right after preparation and hence without giving the sufficient time to separate (3 to 5 days). All the formulations had an average radius of 50 nm (±10), and the addition of the ligand did not alter the final structure as confirmed by both transmission electron microscopy and dynamic light scattering (DLS). We also labeled about 5% of the POEGMA-PDPA chains with Cy5 dye to allow fluorescence quantification. We incubated different polymersome formulations for an hour with BECs (BEnd3), and macrophages (LADMAC). The total fluorescence per cell was measured using automated imaging analysis on the two cell types by confocal microscopy. We opted for short incubation times to ensure the nanoparticle per cell interaction is kinetically controlled by the binding, and while endocytosis is present, such a process accounts only for a negligible component of the overall process. In fig. S3, an example of micrograph used for quantification is shown to illustrate the effective binding of ligand-modified polymersomes to BECs. In Fig. 5, A to C, the average fluorescence per cell was measured after 1-hour incubation with BECs and macrophages. To assess the ability of polymersomes to selectively target a given cell phenotype, we define the selectivity index, s, as
| (19) |
where FBE is the average fluorescence per cell in BECs, here, considered as target, and FS is the average fluorescence per cell in macrophages, here, considered as sentinel cells. Formulations with s > 1 interact preferentially with target cells than with the sentinels while s < 0 do the actual opposite, while those with selectivity index 1 ≥ s ≥ 0 are considered indifferent and incapable of distinguish between targets and sentinels. As shown in the data in Fig. 5A, the angiopep EP polymersomes interact preferentially with BECs compared to the sentinel cells with selectivity peaking to 2.5 at around 30 of average ligand numbers. As expected at higher ligand numbers, the selectivity is lost and polymersomes interact equally with both cell populations. A very different outcome is observed for PMPC chains where the macrophages show the highest uptake and with the BECs a lower value (albeit with magnitude similar to the angiopep polymersomes). Here, the selectivity is negative with regard to the BECs. It is worth mentioning that this is also due to the choice of target cells; if macrophages are considered as a target, then the PMPC chains will show high selectivity as we also demonstrated recently (56). In Fig. 5C, we report the cellular uptake of mixed angiopep and PMPC EP polymersomes where we varied the average number of angiopep ligands and fixed the PMPC number at 200. The data show a similar trend to the angiopep alone but with a shift of the selectivity from 30 of ligand average number down to 20, indicating that the PMPC chains increases the selectivity of the angiopep, making the binding more synergistic with the selectivity of the formulation. However, the same can also be said about the macrophages where an increase of angiopep ligands shifts the binding at 200 PMPC chains, a value that did not show detectable uptake with PMPC-alone formulations (see Fig. 5B), and the multiplexed polymersomes expand the selectivity range to lower numbers of ligands.
Fig. 5. Selective cellular uptake.
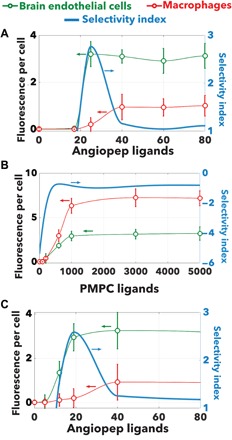
Average fluorescence per cell measured after 1-hour incubation of polymersomes with BECs and macrophages as a function of ligand numbers for the angiopep peptides (A) and PMPC chains (B) and angiopep peptides mixed with 200 PMPC chains (C). The blue line shows the selectivity index calculated using the BECs as target and the macrophages as sentinel cells.
If we normalized the data in Fig. 5 for the maximum value, then we can reinterpret the data using a Hill binding assay measuring cell receptor saturations as a function of the number of ligands. This allows us to fit the experimental data using Eq. 15 and 18 fixing the number of receptors 〈NR〉P and varying the average number of ligands NL. As shown in Fig. 6A, the angiopep functionalization results in binding in both cell models with uptake of following a sigmoidal trend with the number of ligands confirming the expected high selectivity. The BECs reached saturation at a lower number of ligands than macrophages, suggesting that they express a higher number of LRP1 receptors. Experimentally, we have access to the polymersome radius and the average number of ligands NL. Using the reported dissociation constant of the angiopep with LRP1 (44), i.e., KD = 313 nM, we also know that NPEO = 10, aPEO = 0.35 nm, σ0 = 3.14 nm2, and R = 50 nm (experimentally measured); from Fig. 1, we can estimate d = 5 nm, δP = 0.25, δG = 0.7, LRP1 tip volume VP = 188.4 nm3. Last, we assume for both cell types a syndecan interchain distance dS = 20 nm, in agreement with previously reported data (37). We can thus estimate the LRP1 density for BECs <NR>BEC = 18 molecules μm−2 and macrophages <NR>M = 13 molecules μm−2. We repeated the same experiment using PMPC-functionalized EP polymersomes, and the result here is rather different with macrophages expressing the highest numbers of SRB1 and the other cells requiring higher numbers of ligands to reach saturation (Fig. 6B). Similarly to angiopep, for PMPC, we can assume δP = 0.1, δG = 0.1, dS = 20 nm, SRB1 volume VP = 68.4 nm3, to estimate the PMPC/SRB1 dissociation constant KD = 350 nM, and the SRB1 for BECs <NR>BEC = 17 molecules μm−2 and macrophages <NR>M = 25 molecules μm−2.
Fig. 6. Superselectivity validation on BECs.
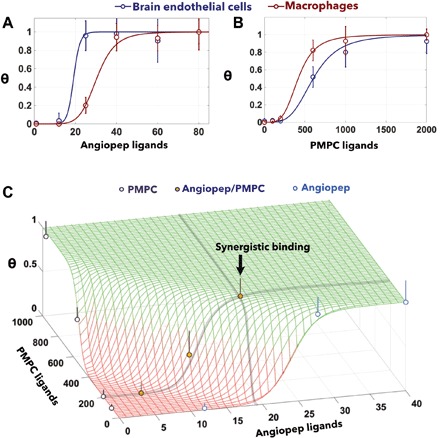
Binding of polymersomes to BECs and macrophages decorated with angiopep peptides (A) and PMPC chains (B). The experimental data are fitted assuming the geometries shown in Fig. 2 and using Eq. 16. The two ligands are combined to form multiplexed polymersomes, and their binding is measured in BECs (C). The data are thus fitted using Eq. 19. Fitting parameters for angiopep: δP = 0.25, δG = 0.7, NPEO = 10, aPEO = 0.35 nm, σ0 = 3.14 nm2, R = 50 nm, d = 5 nm, LRP1 tip volume VP = 188.4 nm3, and angiopep KD = 313 nM. Best fit from monovalent: syndecan interchain distance dS = 20 nm, receptor density for BECs <NR>BEC = 18 molecules μm−2, and macrophages <NR>M = 13 molecules μm−2. Fitting parameters for PMPC: δP = 0.1, δG = 0.1, NPEO = 10, aPEO = 0.35 nm, σ0 = 3.14 nm2, R = 50 nm, d = 0 nm, SRB1 volume VP = 68.4 nm3, and syndecan interchain distance dS = 20 nm. Best fit from monovalent: PMPC KD = 350 nM, receptor density for BECs <NR >BEC = 17 molecules μm−2, and macrophages <NR>M = 25 molecules μm−2.
This result confirms our previous observations that macrophages (as well as other antigen presenting cells) express a high level of SRB1 receptors and that PMPC-functionalized polymersomes target them with high selectivity (56). Last, we formulated three EP polymersomes with both ligands expressed and incubate them with BECs. The results, plotted in Fig. 6C, show a good agreement between Eq. 19 and the experimental data. We show that the two ligands act synergistically allowing targeting using ligand numbers that alone will not correspond to any interactions. This result clearly show that, albeit at short incubation times, our theoretical model well fits experimental data and indeed proposed an effective way to design nanoparticles.
DISCUSSION
We present here a general discussion on how exogenous material interacts with a complex biological system presenting a very simple potential term that accounts for specific and unspecific interactions. We use this as rules of engagement for the design of selective targeting, and we thus derive a model adapting the SST theory to a defined multivalent nanoparticle (see Fig. 1) equipped with realistic binding energies, introducing a nonspecific repulsive potential by inserting the ligand within a PEO polymer brush and from the insertion of the nanoparticle into the endogenous expressed glycoprotein network that characterizes most cells. Such a strategy was partially validated by Wang and Dormidontova using Monte Carlo simulation where it was shown that the shielding ligands by long chains lead to an extra loss of entropy at the onset density (57). Here, we build on this and show, using established models for polymer brush steric repulsion to proteins, that we can tune the interaction so as to create the low affinity necessary for superselectivity as showed in Fig. 3. We show that particle size, ligand number, and polymer brush length can be computed together with ligand affinity and receptor volume to identify the most efficient formulations to achieve superselectivity. Last, we show that the combination of multiple ligands into a multiplexed system can indeed create purely superselective targeting where multiple overexpressed receptors would be required for binding, increasing the robustness of the proposed targeting platform. We thus use cellular uptake of multivalent and multiplexed polymersomes to validate our theory, showing a good agreement between our model and the experimental data. Overall, the model that we present provides not only a very powerful tool to design personalized nanomedicines but also gives important insights into how biological systems can achieve such high selectivity. One can easily extrapolate from the theory here presented as “rules of thumb” to how cells, viruses, bacteria, protein, and nucleic acid interact with each other, hence adding a powerful tool to the existing system biology approaches.
MATERIALS AND METHODS
Polymersome preparation
P[(OEG)10MA]20-PDPA100, Cy5-P[(OEG)10MA]20-PDPA100) angiopep-P[(OEG)10MA]20-PDPA100 and PMPC]25-PDPA70 copolymers were synthesized as reported in (33). To make polymersomes (10 mg/ml), the copolymers were weighed and dissolved using pH 2 phosphate-buffered saline. Once the film dissolved, the pH was increased to 5.0. Peptide-functionalized copolymers can be added at this point to avoid acidic degradation. The pH was gradually increased to pH 6.8 to pH 7.0, eventually stopping at pH 7.4 to pH 7.5. Prolonged stirring at pH 6.8 to pH 7.0 allowed polymersomes to form. Polymersomes were then ultrasound sonicated for 15 to 30 min, at 4°C. The purification of polymersomes was lastly performed by passing through a gel permeation chromatography column prepacked with Sepharose 4B (Sigma-Aldrich). For long-term storage, polymersomes can be kept at 4°C and when conjugated to dyes protected from light. The peptide-functionalized polymersomes were freshly made just before use. Polymersomes were characterized by transmission electron microscopy (FEI Tecnai G2) using phosphotungstenic acid as a staining agent and dynamic light scattering (Malvern Nanosizer).
Cell cultures
BEC bEND.3 cells (ATCC CRL-2299) were seeded on a rat tail collagen type I (Sigma-Aldrich, C3867) precoated T-75 flask, maintained in Dulbecco’s modified Eagle’s medium (DMEM) (DMEM high glucose, D5671, Sigma-Aldrich) supplemented with 2 mM l-glutamine, penicillin (100 IU/ml), streptomycin (100 mg/ml), and 10% fetal calf serum (FCS). Cultures were maintained at 37°C in an atmosphere of 5% CO2 and 95% air and subcultured routinely using 0.02% (w/v) EDTA trypsin (5 ml, 5 min 37°C, 5% CO2 incubation) once 100% confluence was reached. LADMAC macrophages were purchased from the American Type Culture Collection (Manassas, VA, USA) and were cultured in Eagle’s minimal essential medium supplemented with 10% (v/v) FCS and 2 mM l-glutamine.
Confocal laser scanning microscopy
Cells were placed on 96 plates with image-read bottom plastic; they were treated for 1 hour with the different polymersome formulations, and subsequently, their medium was replenished. The cells were imaged using confocal laser scanning microscopy equipped with an incubation chamber connected to the ZEISS temperature control unit 37-2 and a CO2 controller (1 to 2 hours before the experiment was allowed for stabilization of the temperature and CO2 concentration). All the confocal laser scanning microscopy was performed on a ZEISS LSM 510 microscope, equipped with the following lasers: Ar laser, 30 mW; HeNe laser, 1 mW; and HeNe laser, 5 mW. The laser excitation wavelengths used were 405 nm [4′,6-diamidino-2-phenylindole (DAPI)], and 548 nm (Cy5-polymersomes). The quantification of the fluorescence intensity was performed on normally 30 micrographs per formulation over triplicate experiments. The images were analyzed with ImageJ by creating an ad hoc region of interest around the nuclei (prestained with DAPI) and measuring the intensity in the Cy5 channel.
Supplementary Material
Acknowledgments
Funding: We thank the EPSRC (grants EP/G062137/1 and EP/I001697/1) for sponsoring part of the experimental work and salary of X.T. G.B. thanks the EPSRC (EP/N026322/1) for funding part of his salary via an Established Career Fellowship and the ERC (CheSSTag 769798) for this consolidator award. S.A.-U. thanks the Beijing Advanced Innovation Centre for Soft Matter Science (BAIC-SM) for funding. X.T. thanks the National Natural Science Foundation of China (NNSFC 21602003), Anhui Provincial Returnees Innovation and Entrepreneurship key support program, and Open fund for Discipline Construction, Institute of Physical Science and Information Technology, Anhui University. G.B. and X.T. thank the “Distinguished Expert” Anhui province 100 talent program. S.A.-U. acknowledges funding via the Presidential International Fellowship Initiative (PIFI) of the Chinese Academy of Sciences. Author contributions: G.B. ideated the work, designed the experimental and computational models, derived the steric potentials, and combined them with the SST to produce the numerical simulations for the multiplexing, as well as supervised the experimental work. S.A.-U. derived the binding potential equations and produced the numerical simulations for the scaling rules. X.T. produced and analyzed all the experimental data. All authors wrote and reviewed the manuscript. Competing interests: G.B. is an inventor on a patent application (United Kingdom Patent Application No. 1900185.8) related to this work filed by UCLB. The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials (United Kingdom Patent Application No. 1900185.8). Additional data related to this paper may be requested directly from the authors. Requests for all the copolymers synthetized in-house used in the study should be submitted to G.B. The copolymers can be provided pending scientific review and a completed material transfer agreement.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/4/eaat0919/DC1
Supporting Information
Fig. S1. Scaling principles in superselectivity continued.
Fig. S2. Polymersome characterization.
Fig. S3. Polymersome cellular uptake.
REFERENCES AND NOTES
- 1.Ehrlich P., Croonian Lecture: On immunity with special reference to cell life. Proc. R. Soc. London 66, 424–433 (1899). [Google Scholar]
- 2.Aronson S. J., Rehm H. L., Building the foundation for genomics in precision medicine. Nature 526, 336–342 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng Z., Al Zaki A., Hui J. Z., Muzykantov V. R., Tsourkas A., Multifunctional nanoparticles: Cost versus benefit of adding targeting and imaging capabilities. Science 338, 903–910 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Akinc A., Battaglia G., Exploiting endocytosis for nanomedicines. Cold Spring Harb. Perspect. Biol. 5, a016980 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fullstone G., Nyberg S., Tian X., Battaglia G., From the blood to the central nervous system: A nanoparticle’s journey through the blood-brain barrier by transcytosis. Int. Rev. Neurobiol. 130, 41–72 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Mammen M., Choi S. K., Whitesides G. M., Polyvalent interactions in biological systems: Implications for design and use of multivalent ligands and inhibitors. Angew. Chem. Int. Ed. Engl. 37, 2754–2794 (1998). [DOI] [PubMed] [Google Scholar]
- 7.Kitov P. I., Bundle D. R., On the nature of the multivalency effect: A thermodynamic model. J. Am. Chem. Soc. 125, 16271–16284 (2003). [DOI] [PubMed] [Google Scholar]
- 8.Kiessling L. L., Lamanna A. C., Multivalency in biological systems. Chem. Probes Biol. 129, 345–357 (2003). [Google Scholar]
- 9.Badjic J. D., Nelson A., Cantrill S. J., Turnbull W. B., Stoddart J. F., Multivalency and cooperativity in supramolecular chemistry. Acc. Chem. Res. 38, 723–732 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Carlson C. B., Mowery P., Owen R. M., Dykhuizen E. C., Kiessling L. L., Selective tumor cell targeting using low-affinity, multivalent interactions. ACS Chem. Biol. 2, 119–127 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Martinez-Veracoechea F. J., Frenkel D., Designing super selectivity in multivalent nano-particle binding. Proc. Natl. Acad. Sci. U.S.A. 108, 10963–10968 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Albertazzi L., Martinez-Veracoechea F. J., Leenders C. M. A., Voets I. K., Frenkel D., Meijer E. W., Spatiotemporal control and superselectivity in supramolecular polymers using multivalency. Proc. Natl. Acad. Sci. U.S.A. 110, 12203–12208 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dubacheva G. V., Curk T., Mognetti B. M., Auzély-Velty R., Frenkel D., Richter R. P., Superselective targeting using multivalent polymers. J. Am. Chem. Soc. 136, 1722–1725 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dubacheva G. V., Curk T., Auzély-Velty R., Frenkel D., Richter R. P., Designing multivalent probes for tunable superselective targeting. Proc. Natl. Acad. Sci. U.S.A. 112, 5579–5584 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Israelachvili J., Differences between non-specific and bio-specific, and between equilibrium and non-equilibrium, interactions in biological systems. Q. Rev. Biophys. 38, 331–337 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Merkel R., Nassoy P., Leung A., Ritchie K., Evans E., Energy landscapes of receptor-ligand bonds explored with dynamic force spectroscopy. Nature 397, 50–53 (1999). [DOI] [PubMed] [Google Scholar]
- 17.Angioletti-Uberti S., Exploiting receptor competition to enhance nanoparticle binding selectivity. Phys. Rev. Lett. 118, 068001 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Yu I., Mori T., Ando T., Harada R., Jung J., Sugita Y., Feig M., Biomolecular interactions modulate macromolecular structure and dynamics in atomistic model of a bacterial cytoplasm. eLife 5, e19274 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gura Sadovsky R., Brielle S., Kaganovich D., England J. L., Measurement of rapid protein diffusion in the cytoplasm by photo-converted intensity profile expansion. Cell Rep. 18, 2795–2806 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harada R., Sugita Y., Feig M., Protein crowding affects hydration structure and dynamics. J. Am. Chem. Soc. 134, 4842–4849 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilhelm S., Tavares A. J., Dai Q., Ohta S., Audet J., Dvorak H. F., Chan W. C. W., Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 1, 16014 (2016). [Google Scholar]
- 22.Medzhitov R., Janeway C. A. Jr., Decoding the patterns of self and nonself by the innate immune system. Science 296, 298–300 (2002). [DOI] [PubMed] [Google Scholar]
- 23.A. Varki, R. D. Cummings, J. D. Esko, P. Stanley, G. W. Hart, M. Aebi, A. G. Darvill, T. Kinoshita, N. H. Packer, J. H. Prestegard, R. L. Schnaar, P. H. Seeberger, Essentials of Glycobiology (Cold Spring Harbor Laboratory Press, ed. 3, 2017), pp. 823. [PubMed] [Google Scholar]
- 24.Mitchell M. J., King M. R., Physical biology in cancer. 3. The role of cell glycocalyx in vascular transport of circulating tumor cells. Am. J. Physiol. Cell Physiol. 306, C89–C97 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuo J. C.-H., Gandhi J. G., Zia R. N., Paszek M. J., Physical biology of the cancer cell glycocalyx. Nat. Phys. 14, 658–669 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.P. G. de-Gennes, Scaling Concepts in Polymer Physics (Cornell Univ. Press, 1980). [Google Scholar]
- 27.Morra M., On the molecular basis of fouling resistance. J. Biomater. Sci. Polym. Ed. 11, 547–569 (2000). [DOI] [PubMed] [Google Scholar]
- 28.Knop K., Hoogenboom R., Fischer D., Schubert U. S., Poly(ethylene glycol) in drug delivery: Pros and cons as well as potential alternatives. Angew. Chem. Int. Ed. Engl. 49, 6288–6308 (2010). [DOI] [PubMed] [Google Scholar]
- 29.Barz M., Luxenhofer R., Zentel R., Vicent M. J., Overcoming the PEG-addiction: Well-defined alternatives to PEG, from structure–property relationships to better defined therapeutics. Polym. Chem. 2, 1900–1918 (2011). [Google Scholar]
- 30.Wei Q., Becherer T., Angioletti-Uberti S., Dzubiella J., Wischke C., Neffe A. T., Lendlein A., Ballauff M., Haag R., Protein interactions with polymer coatings and biomaterials. Angew. Chem. Int. Ed. 53, 8004–8031 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Chapman R. G., Ostuni E., Takayama S., Holmlin R. E., Yan L., Whitesides G. M., Surveying for surfaces that resist the adsorption of proteins. J. Am. Chem. Soc. 122, 8303–8304 (2000). [Google Scholar]
- 32.Shao Q., Jiang S., Molecular understanding and design of zwitterionic materials. Adv. Mater. 27, 15–26 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Tian X., Nyberg S., Sharp P. S., Madsen J., Daneshpour N., Armes S. P., Berwick J., Azzouz M., Shaw P., Abbott N. J., Battaglia G., LRP-1-mediated intracellular antibody delivery to the Central Nervous System. Sci. Rep. 5, 11990 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Joseph A., Contini C., Cecchin D., Nyberg S., Ruiz-Perez L., Gaitzsch J., Fullstone G., Tian X., Azizi J., Preston J., Volpe G., Battaglia G., Chemotactic synthetic vesicles: Design and applications in blood-brain barrier crossing. Sci. Adv. 3, e1700362 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Colley H. E., Hearnden V., Avila-Olias M., Cecchin D., Canton I., Madsen J., MacNeil S., Warren N., Hu K., McKeating J. A., Armes S. P., Murdoch C., Thornhill M. H., Battaglia G., Polymersome-mediated delivery of combination anticancer therapy to head and neck cancer cells: 2D and 3D in vitro evaluation. Mol. Pharm. 11, 1176–1188 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Arkill K. P., Knupp C., Michel C. C., Neal C. R., Qvortrup K., Rostgaard J., Squire J. M., Similar endothelial glycocalyx structures in microvessels from a range of mammalian tissues: Evidence for a common filtering mechanism? Biophys. J. 101, 1046–1056 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu J., Li X., Yin J., Hu Y., Gu Y., Pan S., Glycocalyx degradation leads to blood–brain barrier dysfunction and brain edema after asphyxia cardiac arrest in rats. J. Cereb. Blood Flow Metab. 38, 1979–1992 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weinbaum S., Tarbell J. M., Damiano E. R., The structure and function of the endothelial glycocalyx layer. Annu. Rev. Biomed. Eng. 9, 121–167 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Curry F. E., Adamson R. H., Endothelial glycocalyx: Permeability barrier and mechanosensor. Ann. Biomed. Eng. 40, 828–839 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cruz-Chu E. R., Malafeev A., Pajarskas T., Pivkin I. V., Koumoutsakos P., Structure and response to flow of the glycocalyx layer. Biophys. J. 106, 232–243 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Neculai D., Schwake M., Ravichandran M., Zunke F., Collins R. F., Peters J., Neculai M., Plumb J., Loppnau P., Pizarro J. C., Seitova A., Trimble W. S., Saftig P., Grinstein S., Dhe-Paganon S., Structure of LIMP-2 provides functional insights with implications for SR-BI and CD36. Nature 504, 172–176 (2013). [DOI] [PubMed] [Google Scholar]
- 42.Simon-Gracia L., Hunt H., Scodeller P. D., Gaitzsch J., Braun G. B., Willmore A. M. A., Ruoslahti E., Battaglia G., Teesalu T., Paclitaxel-loaded polymersomes for enhanced intraperitoneal chemotherapy. Mol. Cancer Ther. 15, 670–679 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Simón-Gracia L., Hunt H., Scodeller P. D., Gaitzsch J., Kotamraju V. R., Sugahara K. N., Ruoslahti E., Battaglia G., Teesalu T., iRGD peptide conjugation potentiates intraperitoneal tumor delivery of paclitaxel with polymersomes. Biomaterials 104, 247–257 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Demeule M., Currie J. C., Bertrand Y., Ché C., Nguyen T., Régina A., Gabathuler R., Castaigne J. P., Béliveau R., Involvement of the low-density lipoprotein receptor-related protein in the transcytosis of the brain delivery vector angiopep-2. J. Neurochem. 106, 1534–1544 (2008). [DOI] [PubMed] [Google Scholar]
- 45.Gao L., Lipowsky H. H., Composition of the endothelial glycocalyx and its relation to its thickness and diffusion of small solutes. Microvasc. Res. 80, 394–401 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Halperin A., Polymer brushes that resist adsorption of model proteins: Design parameters. Langmuir 15, 2525–2533 (1999). [Google Scholar]
- 47.Zhulina E. B., Birshtein T. M., Borisov O. V., Curved polymer and polyelectrolyte brushes beyond the Daoud-Cotton model. Eur. Phys. J. E Soft Matter 20, 243–256 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Angioletti-Uberti S., Varilly P., Mognetti B. M., Tkachenko A. V., Frenkel D., Communication: A simple analytical formula for the free energy of ligand–receptor-mediated interactions. J. Chem. Phys. 138, 021102 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Varilly P., Angioletti-Uberti S., Mognetti B. M., Frenkel D., A general theory of DNA-mediated and other valence-limited colloidal interactions. J. Chem. Phys. 137, 094108 (2012). [DOI] [PubMed] [Google Scholar]
- 50.Angioletti-Uberti S., Varilly P., Mognetti B. M., Frenkel D., Mobile linkers on DNA-coated colloids: Valency without patches. Phys. Rev. Lett. 113, (2014). [DOI] [PubMed] [Google Scholar]
- 51.Localisation B., PROCEEDINGS OF THE PHYSIOLOGICAL SOCIETY: January 22, 1910. J. Physiol. 40, i–vii (1910). [Google Scholar]
- 52.Demeule M., Régina A., Ché C., Poirier J., Nguyen T., Gabathuler R., Castaigne J. P., Béliveau R., Identification and design of peptides as a new drug delivery system for the brain. J. Pharmacol. Exp. Ther. 324, 1064–1072 (2008). [DOI] [PubMed] [Google Scholar]
- 53.Senft D., Leiserson M. D. M., Ruppin E., Ronai Z. A., Precision oncology: The road ahead. Trends Mol. Med. 23, 874–898 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Curk T., Dobnikar J., Frenkel D., Optimal multivalent targeting of membranes with many distinct receptors. Proc. Natl. Acad. Sci. U.S.A. 114, 7210–7215 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.LoPresti C., Massignani M., Fernyhough C., Blanazs A., Ryan A. J., Madsen J., Warren N. J., Armes S. P., Lewis A. L., Chirasatitsin S., Engler A. J., Battaglia G., Controlling polymersome surface topology at the nanoscale by membrane confined polymer/polymer phase separation. ACS Nano 5, 1775–1784 (2011). [DOI] [PubMed] [Google Scholar]
- 56.Rizzello L., Robertson J. D., Elks P. M., Poma A., Daneshpour N., Prajsnar T. K., Evangelopoulos D., Canseco J. O., Yona S., Marriott H. M., Dockrell D. H., Foster S. J., De Geest B., De Koker S., Hugh T. M., Renshaw S. A., Battaglia G., Targeting mononuclear phagocytes for eradicating intracellular parasites. BioRxiv 2017, 119297 (2017). [Google Scholar]
- 57.Wang S., Dormidontova E. E., Selectivity of ligand-receptor interactions between nanoparticle and cell surfaces. Phys. Rev. Lett. 109, 238102 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/4/eaat0919/DC1
Supporting Information
Fig. S1. Scaling principles in superselectivity continued.
Fig. S2. Polymersome characterization.
Fig. S3. Polymersome cellular uptake.