

Abstract
Genetic and functional studies have confirmed an important role for the voltage-gated sodium channel Nav1.9 in human pain disorders. However, low functional expression of Nav1.9 in heterologous systems (e.g. in human embryonic kidney 293 (HEK293) cells) has hampered studies of its biophysical and pharmacological properties and the development of high-throughput assays for drug development targeting this channel. The mechanistic basis for the low level of Nav1.9 currents in heterologous expression systems is not understood. Here, we implemented a multidisciplinary approach to investigate the mechanisms that govern functional Nav1.9 expression. Recombinant expression of a series of Nav1.9-Nav1.7 C-terminal chimeras in HEK293 cells identified a 49-amino-acid-long motif in the C terminus of the two channels that regulates expression levels of these chimeras. We confirmed the critical role of this motif in the context of a full-length channel chimera, Nav1.9-Ct49aaNav1.7, which displayed significantly increased current density in HEK293 cells while largely retaining the characteristic Nav1.9-gating properties. High-resolution live microscopy indicated that the newly identified C-terminal motif dramatically increases the number of channels on the plasma membrane of HEK293 cells. Molecular modeling results suggested that this motif is exposed on the cytoplasmic face of the folded C terminus, where it might interact with other channel partners. These findings reveal that a 49-residue-long motif in Nav1.9 regulates channel trafficking to the plasma membrane.
Keywords: sodium channel, pain, electrophysiology, structural model, trafficking, functional expression, live imaging, Nav1.9, nociception, sensory neurons, voltage clamp, channel trafficking
Introduction
Voltage-gated sodium channel 1.9 (Nav1.9),3 first reported to be preferentially expressed in small nociceptive neurons, displays the most divergent amino acid (aa) sequence within the Nav channel family (1) and has unique gating properties (2). Nav1.9 produces a tetrodotoxin-resistant (TTX-R) current, which is activated at voltages considerably more hyperpolarized compared with other Nav channels and inactivates with ultra-slow kinetics. This results in a large overlap window and a significant persistent current, suggesting a threshold channel role for Nav1.9 in action potential generation (3, 4).
Nav1.9 is preferentially expressed in peripheral sensory and myenteric neurons and is linked to human pain disorders, including rare familial diseases and more common disorders, such as some forms of small fiber neuropathy (5, 6). Gain-of-function mutations in NaV1.9 have been reported in patients with early- and late-onset pain symptoms (7–9) and in syndromes that include painless injuries (10–12). Patients carrying Nav1.9 mutations with pain syndromes and with painless injuries reported gastrointestinal (GI) disturbances, consistent with animal studies showing a role of Nav1.9 in intestinal contraction (13). NaV1.9 has been found in submucosal plexuses in normal human colon, and its expression is reduced in ganglionic tissue from patients with Hirschsprung's disease (14), supporting a contribution of NaV1.9 in human GI tract function. These findings have triggered a rise of interest in Nav1.9 as a potential target for new medications for pain and GI disturbances.
Efforts to understand the molecular basis for Nav1.9 gating properties and to develop high-throughput assays for drug development have been hampered by the exceedingly low levels of Nav1.9 current in heterologous expression systems (e.g. the human embryonic kidney 293 (HEK293) cell line). Although successful expression of Nav1.9 in DRG-derived ND7/23 (8, 10, 15) and HEK293 (16) cell lines has been recently reported, the currents were still relatively small (≤1 nA) and required special cell culture and recording conditions that included co-transfection with sodium channel β-subunits, cell incubation at 30 °C, and inclusion of GTPγS in the recording pipette. Another strategy for boosting the expression of Nav1.9 was the use of chimeric channels in which the C terminus of Nav1.9 was exchanged with that of the Nav1.4 channel, which resulted in enhanced expression in ND7/23 and HEK293 cells as well as in Xenopus oocytes; however, the biophysical properties of this chimeric channel were dramatically altered compared with native Nav1.9 with significantly accelerated inactivation (17). The mechanisms underlying the small currents produced by Nav1.9 channels in these heterologous expression systems are not well-understood and could include instability of the channel protein or impaired trafficking to the plasma membrane in these cell types. To unravel what limits the functional expression of Nav1.9 sodium channels in heterologous expression systems, we implemented a multidisciplinary study designed to identify molecular determinants governing functional expression of Nav1.9. We applied molecular analysis, voltage-clamp recordings, high-resolution live imaging of channel trafficking, and structural modeling and have identified a 49-aa motif in the Nav1.9 C terminus that regulates functional expression of the channel in HEK293 cells.
Results
Low expression levels of the C terminus of Nav1.9 in HEK293 cells
To look for molecular determinants that regulate Nav1.9 expression, we focused on the C terminus because its exchange with that of Nav1.4 increases current amplitude (17), similarly to what we have previously shown for a Nav1.8-Nav1.4 chimera (18). Our initial attempts were designed to use channel C terminus proteins for affinity purification of cytosolic proteins that might play a role in the delivery of the channels to the cell surface. The C terminus of Nav1.9, Nav1.7, Nav 1.6, or a full GFP sequence (as a control) was subcloned between a myristoylation signal and a Strep/FLAG tag within a piggyBac shuttle vector (Fig. 1A). The myristoylation signal (19) was added to target these channel segments to the plasma membrane where they are normally present. A Strep/FLAG tag was chosen because it was previously shown to be very efficient in tandem affinity purification (TAP) of protein complexes from mammalian cells under native conditions (20, 21). A piggyBac transposon system was reported to provide a highly efficient nonrandom integration of large inserts with no overproduction inhibition observed (22, 23), making this approach a method of choice for our purpose, to purify enough protein partners to be able to identify them by MS.
Figure 1.
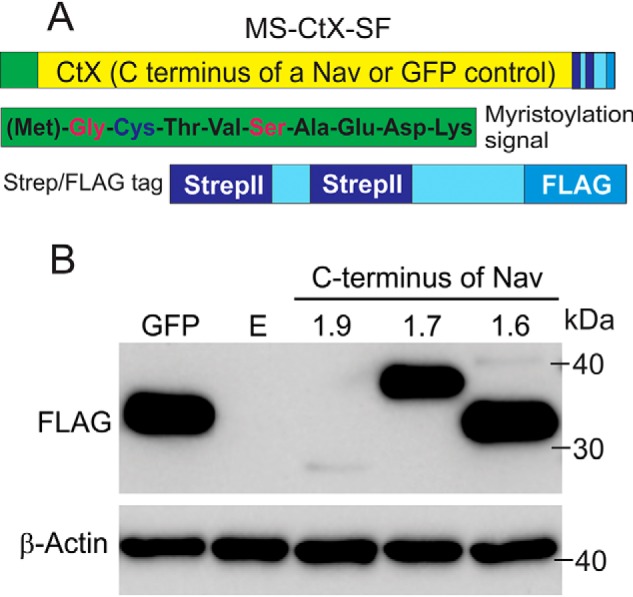
C terminus of Nav1.9 is expressed at low levels in transiently transfected HEK293 cells. A, schematic representation of the MS-CtX-SF construct showing location and composition of the myristoylation signal (green box, full sequence included) and streptavidin-FLAG tag (light blue box with stripes). The C terminus of a sodium channel (CtX) or a control full GFP sequence was inserted in-frame between the myristoylation and SF tags (yellow). B, representative Western blotting showing comparative expression levels of MS-CtNav1.9-SF, MS-CtNav1.7-SF, and MS-CtNav1.6-SF constructs as well as a control MS-GFP-SF. Total protein extracts were run on a 4–12% polyacrylamide gel, transferred to a nitrocellulose membrane, and probed with FLAG (top) or β-actin (bottom) antibodies. Lane E, a control transfection with an empty vector.
The DNA constructs (MS-CtNav1.9-SF, MS-CtNav1.7-SF, MS-CtNav1.6-SF, and MS-GFP-SF) were transfected into HEK293 cells, and total protein lysates were collected and subjected to Western blotting using a FLAG antibody. Surprisingly, only trace levels of Nav1.9 C terminus protein were detected, whereas the other C termini as well as a control GFP were robustly expressed (Fig. 1B). This result was reproducible using either transient or stable transfection in HEK293 cells. Probing for β-actin as a control confirmed comparable amounts of the lysates that were loaded (Fig. 1B). A similarly low expression of Nav1.9 C terminus was observed when ND7/23 cells were used as an alternative heterologous expression system (data not shown), indicating that this is not a cell background effect. The exceedingly low levels of the Nav1.9 C terminus in the HEK293 cells preclude downstream implementation of a TAP/MS approach to identify protein partners that might regulate Nav1.9 channel trafficking. The C terminus of Nav1.9 (184 aa) is 47.8% identical to that of Nav1.6 (211 aa) and 51.1% identical to Nav1.7 (225 aa). A closer look at the aligned sequences shows segments that are highly conserved and other sequences that are isoform-specific, which suggests that the difference in protein stability might be regulated by the divergent sequences in Nav1.9.
A swap of a 49-amino acid motif between Nav1.7 and Nav1.9 is sufficient for rescuing expression of the Nav1.9 C terminus
To identify molecular determinants in the Nav1.9 C terminus that are responsible for its low expression in HEK293 cells, we created three C-terminal Nav1.9-Nav1.7 chimeras by using the MS-CtNav1.9-SF construct as a template and successively replacing fragments of Nav1.9 with those of Nav1.7. Sequences of Nav1.9 and Nav1.7 C termini were aligned and divided into three regions, proximal (Pr), middle (Md), and distal (Ds) (Fig. 2, A and B). The Pr (indigo rectangle; 52 amino acid residues) and Md (red rectangle; 49 amino acid residues) regions were almost identical in length and degree of homology between Nav1.9 and Nav1.7 (the resulting constructs were named constructs ChimPr and ChimMd, respectively), whereas the Ds region (green rectangle; 74 amino acid residues for Nav1.9 and 116 amino acid residues for Nav1.7) was considerably less conserved and included isoform-specific sequences (the resulting construct was named ChimDs). The three C-terminal short chimera constructs were transiently transfected into HEK293 cells, and the protein levels were analyzed by Western blotting. Although both the ChimPr and ChimDs showed improved protein levels, the ChimMd shows a dramatic increase in expression compared with the native Nav1.9 C terminus and the two other chimera proteins (Fig. 2C). The 49-amino acid motif is 63% similar between Nav1.7 and Nav1.9, with 31 identical amino acid residues.
Figure 2.

Chimeric construct with a 49-amino acid swap between Nav1.7 and Nav1.9 is sufficient for rescuing expression of the Nav1.9 C terminus. A, aa sequences of Nav1.7 and Nav1.9 C termini are aligned using DNASTAR Lasergene software; nonconserved residues in Nav1.9 are highlighted in red. Aligned sequences were divided into three parts that contained divergent sequences separated by stretches of identical residues (region Pr (proximal) in blue, region Md (middle) in pink, and region Ds (distal) in green). B, three chimeric constructs were created by replacing the three regions of the Nav1.9 C terminus (yellow) with the corresponding sequences from the Nav1.7 C terminus. The 1.7Pr sequence (proximal 52 aa residues from Nav1.7 C terminus) is represented by a blue rectangle; 1.7Md (middle 49 aa from Nav1.7 C terminus) is represented by a pink rectangle; and 1.7Ds (distal 116 aa from Nav1.7 C terminus) is represented by a green rectangle. C, representative Western blotting showing comparative expression of ChimPr, ChimMd, and ChimDs constructs. Total protein extracts were probed with FLAG (top) or β-actin (bottom) antibodies. D, three chimeric constructs named ChimMd#1, ChimMd#2, and ChimMd#3 were created by replacing three subregions of the 49-aa middle region of the Nav1.9 C terminus (yellow) with the corresponding sequences from Nav1.7 (Md#1, Md#2, and Md#3, red, purple, and orange, respectively). E, representative Western blotting showing protein levels of ChimMd#1, ChimMd#2, and ChimMd#3 chimera. Total protein extracts were probed with FLAG (top) or β-actin (bottom) antibodies.
In an attempt to further delineate the region responsible for the improved expression of the Nav1.9 C terminus in HEK293 cells, we created a new set of C-terminal chimeras in which the least conserved sequences within the 49-aa motif were swapped with the corresponding sequences from the Nav1.7 C terminus. Three small amino acid sequence stretches in the 49-aa Md segment with the lowest homology between Nav1.9 and Nav1.7 were labeled Md#1, Md#2, and Md#3 (Fig. 2A). These three sequences were individually replaced in the MS-CtNav1.9-SF and were named ChimMd#1, ChimMd#2, and ChimMd#3, respectively (Fig. 2D). Western blot analysis of the protein levels of each of these chimera constructs showed improved expression levels compared with the parent Nav1.9 C terminus construct, but unlike the ChimMd construct (Fig. 2C), none recovered expression of the Nav1.9 C terminus to a level comparable with that of the Nav1.7 C terminus (Fig. 2E). This result suggests that the entire 49-aa motif may be necessary for Nav1.9 C terminus expression.
Increased levels of full-length Nav1.9–1.7Ct49aa chimera
We found that the exchange of the 49-amino acid motif within the C-terminal part of Nav1.9 with the corresponding sequence from Nav1.7 substantially increased the protein level of the C terminus chimera protein in HEK293 cells. We tested whether this same motif enhances the functional expression of the channel. To answer this question, we created two new chimeras where the 49-amino acid motif was swapped between full-length Nav1.9 and Nav1.7 channels. The resulting constructs were called Nav1.9-Ct49aaNav1.7 and Nav1.7-Ct49aaNav1.9, respectively (Fig. 3A).
Figure 3.
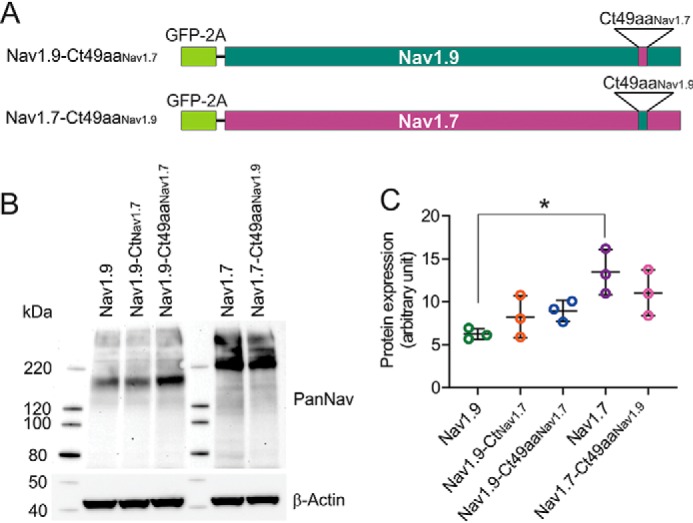
The reciprocal swapping of the 49-amino acid motif between Nav1.7 and Nav1.9 has only a minor effect on steady-state protein level. A, schematic representation of the Nav1.9-Ct49aaNav1.7 and Nav1.7-Ct49aaNav1.9 chimeric constructs showing approximate location of the 49-aa residue swap between Nav1.9 and Nav1.7 in the context of full channel sequences (green for Nav1.9, purple for Nav1.7) as well as the N-terminal GFP-2A sequence (bright green rectangle). B, representative Western blotting image showing protein levels of Nav1.9, Nav1.9-CtNav1.7, Nav1.9-Ct49aaNav1.7, Nav1.7, and Nav1.7-Ct49aaNav1.9 in transiently transfected HEK293 cells. Total protein extracts were probed with pan-sodium channel (top) or β-actin (bottom) antibodies. C, protein expression levels from three independent transfections for each construct were quantified using ImageLab software and presented as mean ± S.D. (error bars). A 2.2-fold higher protein level of Nav1.7 channel compared with Nav1.9 channel was observed (Nav1.9: 6.24 ± 0.64 AU, n = 3; Nav1.7: 13.46 ± 2.62 AU, n = 3, p < 0.05), by one-way ANOVA (F(4, 10) = 5.220, p = 0.0156), followed by Tukey's post hoc analysis. Differences in protein levels of all of the other constructs were statistically insignificant.
The level of protein expression was assessed for both chimera constructs and compared with that of the parent Nav1.9 and Nav1.7 channels. Western blotting of total protein lysates from transiently transfected HEK293 cells (Fig. 3B) showed that protein level of the parent Nav1.7 channel is higher than that of all of the other constructs. Quantitation of the protein signal from three independent transfections shows a 2.2-fold higher protein level of Nav1.7 channel compared with Nav1.9 channel (Nav1.9: 6.24 ± 0.64 AU, n = 3; Nav1.7: 13.46 ± 2.62 AU, n = 3, by one-way ANOVA (F(4, 10) = 5.220, p = 0.0156, followed by Tukey's post hoc analysis). Differences in protein levels of all of the other constructs were insignificant. However, a trend of increased expression in the case of Nav1.9-Ct49aaNav1.7 compared with Nav1.9 and decreased expression in the case of Nav1.7-Ct49aaNav1.9 compared with Nav1.7 is worth noting.
Using voltage-clamp recordings, we compared biophysical properties of the chimeric channels with the corresponding parent channel. HEK293 cells were transiently transfected with human Nav1.9 channel (Fig. 4A) or its C-terminal chimera constructs: Nav1.9-CtNav1.7 (Fig. 4B) and Nav1.9-Ct49aaNav1.7 (Fig. 4C, representative traces of recordings). Fig. 4D shows traces with Nav1.9-characteristic ultra-slow inactivation that were recorded from ∼20% of HEK293 cells transfected with the Nav1.9-Ct49aaNav1.7 chimera. Voltage-clamp characterization was performed after expressing the channels at 30 °C for 96 h post-transfection. In addition, Nav1.7 channel (Fig. 4E) and the chimera in which the reciprocal 49-amino acid motif of Nav1.7 was replaced by that of Nav1.9 (Nav1.7-Ct49aaNav1.9) (Fig. 4F) were also examined. The human Nav1.9 channel produces a very small current in HEK293 cells, compared with human Nav1.7 (Fig. 4G). The current density of Nav1.9 channel is 130-fold lower than that of Nav1.7 channels (Nav1.9: 4.07 ± 0.66 pA/pF, n = 15; Nav1.7: 542 ± 148 pA/pF, n = 11; p < 0.001, one-way ANOVA, followed by Tukey's post hoc analysis) (Fig. 4G). Replacing the entire C terminus of Nav1.9 with the C terminus of Nav1.7 channel increased current density 6-fold (Nav1.9-CtNav1.7: 24.8 ± 4.5 pA/pF, n = 8; p < 0.0001, one-way ANOVA, followed by Tukey's post hoc analysis) (Fig. 4H). More importantly, replacing the 49-amino acid stretch alone is sufficient to increase the expression of Nav1.9 (Nav1.9-Ct49aaNav1.7: 16.8 ± 3.8 pA/pF, n = 7; p < 0.01, one-way ANOVA, followed by Tukey's post hoc analysis) (Fig. 4H); there was no statistically significant difference in current density between Nav1.9-CtNav1.7 and Nav1.9-Ct49aaNav1.7 chimera (p = 0.160) (Fig. 4H). By contrast, the reciprocal chimera of Nav1.7 channel in which the 49-amino acid motif in the C terminus was substituted by that of Nav1.9 channel produces smaller currents than the parent Nav1.7 channel (Nav1.7: 542 ± 148 pA/pF, n = 11; Nav1.7-Ct49aaNav1.9: 199 ± 22 pA/pF, n = 10; p < 0.05) (Fig. 4, E–G).
Figure 4.
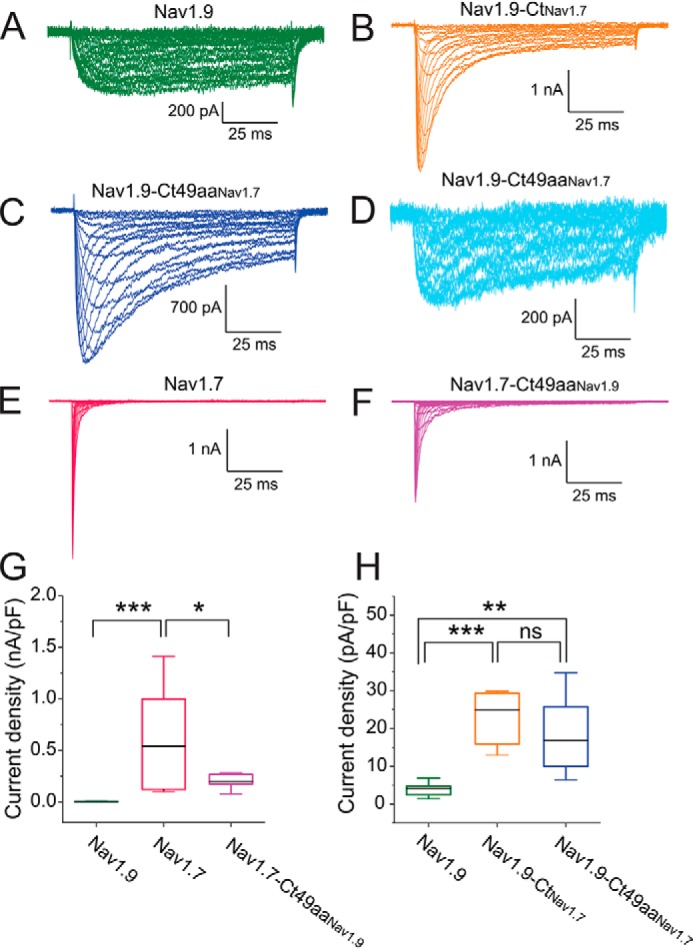
The 49-aa motif in the C terminus increases Nav1.9 current amplitude in HEK293 cells. A–F, representative current traces recorded from HEK293 cells expressing either Nav1.9 (A, green), Nav1.9-CtNav1.7 (B, orange), Nav1.9-Ct49aaNav1.7 (C, navy blue), Nav1.7 (E, red), or Nav1.7-Ct49aaNav1.9 (F, magenta). Current traces recorded from HEK293 cells expressing Nav1.9-Ct49aaNav1.7 and demonstrating ultra-slow inactivation kinetics and large persistent current (in ∼20% HEK293 cells) similar to the parent Nav1.9 current are shown separately (D, light blue). G, comparison of current density among Nav1.9 (n = 15), Nav1.7 (n = 11), and chimera Nav1.7-Ct49aaNav1.9 (n = 10). H, comparison of current density among Nav1.9 (n = 15), Nav1.9-CtNav1.7 (n = 8), and Nav1.9-Ct49aaNav1.7 (n = 7) channels. Error bars, S.E. Statistical significance is indicated by asterisks: *, p < 0.05; **, p < 0.01; ***, p < 0.001, by one-way ANOVA followed by Tukey's multiple-comparison test. ns, not significant.
Voltage dependence of activation (Fig. 5A) and steady-state fast inactivation (Fig. 5B) were analyzed by fitting data to a Boltzmann function (Fig. 5, A and B). Neither of the two Nav1.9 chimeras, Nav1.9-CtNav1.7 or Nav1.9-Ct49aaNav1.7, showed altered midpoint voltage of activation (V½Act), compared with the parent Nav1.9 channel (Nav1.9: −56.6 ± 2.5 mV, n = 4; Nav1.9-CtNav1.7: −63.2 ± 2.9 mV, n = 8, p = 0.256; Nav1.9-Ct49aaNav1.7: −65.3 ± 1.9 mV, n = 7, p = 0.118) (Fig. 5C). The voltage dependence of activation is not statistically significantly different between the two Nav1.9 chimeras either (p = 0.813) (Fig. 5C). Swapping the 49-amino acid stretch in the Nav1.7 channel also preserves the channel's voltage dependence of activation (Nav1.7: −26.2 ± 2.1 mV, n = 10; Nav1.7-Ct49aaNav1.9: −22.7 ± 1.6 mV, n = 10, p = 0.207) (Fig. 5C).
Figure 5.
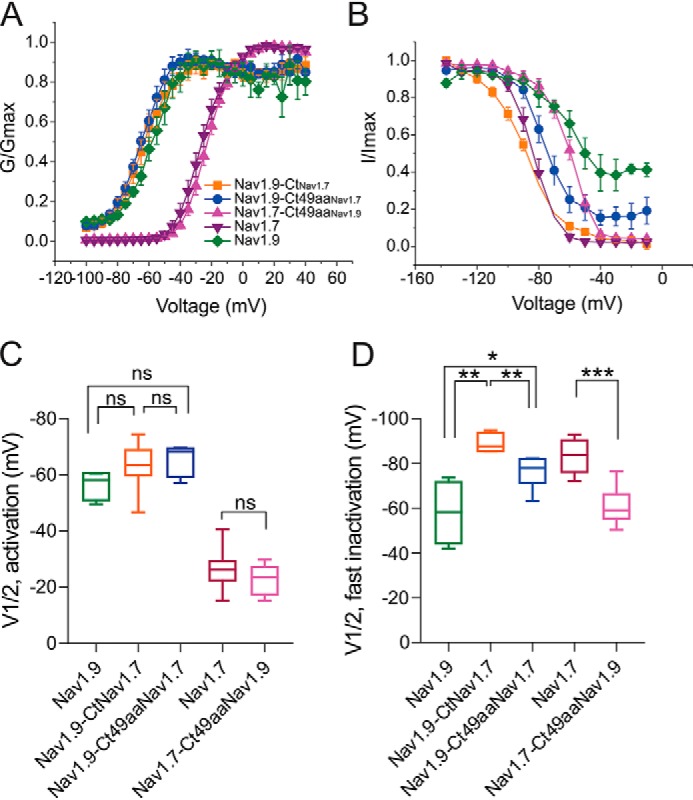
The C terminus of Nav1.9 modulates voltage-dependence of steady-state fast-inactivation but not activation. Boltzmann fits of voltage dependence of activation (A) and steady-state fast inactivation (B) for WT Nav1.9 and Nav1.7 channels and their chimeras. C, comparison of midpoint voltage of activation (V½Act) among all tested channels showing no difference in chimeric channels Nav1.9-CtNav1.7 (n = 8), Nav1.9-Ct49aaNav1.7 (n = 7), and Nav1.7-Ct49aaNav1.9 (n = 10), compared with parent channels Nav1.9 (n = 4) and Nav1.7 (n = 10). D, comparison of midpoint voltage of fast inactivation (V½Inact) among all tested channels (Nav1.9-CtNav1.7 (n = 8); Nav1.9-Ct49aaNav1.7 (n = 7); Nav1.7-Ct49aaNav1.9 (n = 10); Nav1.9 (n = 4); Nav1.7 (n = 10)). Error bars, S.E. Statistical significance is indicated by asterisks: *, p < 0.05; **, p < 0.01; ***, p < 0.001, by one-way ANOVA followed by Tukey's multiple-comparison test. ns, not significant.
The C termini of Nav1.9 and Nav1.7 channels have a more profound impact on steady-state fast inactivation. When the entire C terminus of the Nav1.9 channel was replaced by that from the Nav1.7 channel (Nav1.9-CtNav1.7), the V½Inact voltage dependence of fast inactivation as reflected by the voltage for half-inactivation (V½Inact) was hyperpolarized by 31.2 mV (Nav1.9: −58.1 ± 6.8 mV, n = 4; Nav1.9-CtNav1.7: −89.3 ± 1.8 mV, n = 5; p < 0.01, one-way ANOVA, followed by Tukey's post hoc analysis) (Fig. 5D), bringing it close to that of parent Nav1.7 channels (Nav1.7: −83.3 ± 2.4 mV, n = 9). This deep hyperpolarization of the V½Inact was partially rescued when only the 49-amino acid motif was swapped (Nav1.9-CtNav1.7: −89.3 ± 1.8 mV, n = 5; Nav1.9-Ct49aaNav1.7: −76.3 ± 2.8 mV, n = 6, p < 0.01) (Fig. 5D); note that recordings that are represented in Fig. 4C were used for this analysis. Similarly, the 49-aa motif of Nav1.9 channel significantly depolarized V½Inact of Nav1.7 channel (Nav1.7: −83.3 ± 2.4 mV, n = 9; Nav1.7-Ct49aaNav1.9: −60.9 ± 2.4 mV, n = 10, p < 0.0001) (Fig. 5D).
The WT Nav1.9 channel is known for its characteristic ultra-slow inactivation, which results in a marked persistent current. We therefore examined the effect of the 49-amino acid stretch on the kinetics of open-state inactivation as well as the persistent current. Nav1.9-Ct49aaNav1.7 decelerated inactivation, compared with Nav1.9-CtNav1.7 (Fig. 6A). The 49-aa motif from Nav1.9 markedly slowed down the inactivation of the Nav1.749aaNav1.9 channel (Fig. 6B). The normalized level of persistent current (percentage of total current) of Nav1.9 channel was significantly reduced after its C terminus was replaced by that from Nav1.7 channel (Nav1.9: 57.1 ± 7.1%, n = 4; Nav1.9-CtNav1.7: 11.8 ± 2.6%, n = 8; p < 0.0001) (Fig. 6C). In the Nav1.9 chimeric channel where only the 49-aa motif was altered, the magnitude of the persistent current was partially restored toward that of the WT Nav1.9 channel (Nav1.9-Ct49aaNav1.7: 20.1 ± 6.0 mV, n = 7) (Fig. 6C). However, the average persistent current in Nav1.9-Ct49aaNav1.7 chimeric channel is less than that of the WT Nav1.9 channel (p = 5.95E−7) (Fig. 6C). It is notable that we found that Nav1.9-Ct49aaNav1.7 produced currents in some HEK293 cells (∼20%) with ultra-slow inactivation and with persistent currents as large as the WT Nav1.9 channel (Fig. 4D). As for Nav1.7 channel, the exchange of the 49-aa motif with that of Nav1.9 has little effect on the persistent current (Nav1.7: 2.36 ± 0.74%, n = 11; Nav1.7-Ct49aaNav1.9: 3.82 ± 0.69%, n = 10; p = 0.995) (Fig. 6C).
Figure 6.
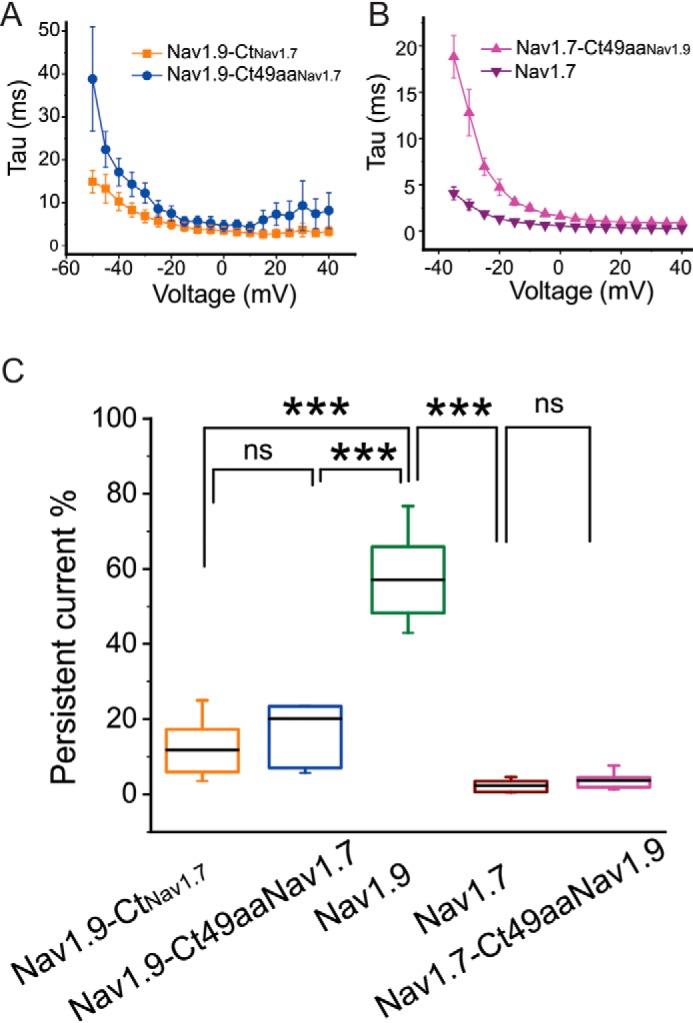
The effect of the 49-aa motif on kinetics of open-state fast inactivation and persistent current. A, time constants of open-state fast inactivation for Nav1.9-CtNav1.7 (orange, n = 8) and Nav1.9-Ct49aaNav1.7 (blue, n = 7). B, time constants of open-state fast inactivation were compared between Nav1.7 (n = 11) and Nav1.7-Ct49aaNav1.9 (n = 10). C, comparison of persistent current, normalized to the peak current, among all channels tested (Nav1.9-CtNav1.7, n = 8; Nav1.9-Ct49aaNav1.7, n = 7; Nav1.9 (n = 4); Nav1.7 (n = 11); Nav1.7-Ct49aaNav1.9 (n = 10)). Error bars, S.E. ***, statistical significance with p < 0.001. ns, not significant.
The 49-aa motif from Nav1.7 enhances the number of Nav1.9 channels at the cell surface
The data in Fig. 4 show that the Nav1.9 current density is 130-fold lower than that of Nav1.7 channels in HEK293 cells, which cannot be explained by the 2.5-fold reduction in the Nav1.9 protein levels, compared with Nav1.7 channels (Fig. 3), in these cells. Alternative explanations include impaired delivery of Nav1.9 channels to the cell surface or an alteration of single channel conductance, which could be regulated by post-translational modification that may occur at the plasma membrane, including an interaction with channel partners. To assess the number of Nav1.9 channels on the plasma membrane in transiently transfected HEK293 cells, we used a signal quantification approach based on total internal reflection fluorescence (TIRF) microscopy of Nav channels (24).
We engineered a Nav1.9 construct with a biotin acceptor domain (BAD) sequence inserted within the S1-S2 extracellular loop of the domain IV (Fig. 7A). This location is not known to substantially alter properties of Nav1.6 (24) and Nav1.7 (25) channels that have been tagged in a similar manner. The BAD is recognized by a bacterial biotin ligase that is co-transfected with the channel and results in biotinylation of the acceptor lysine residue within this sequence (26). Because the BAD domain is in an extracellular loop, we can selectively label channels on the cell surface with a far-red streptavidin-conjugated fluorophore (SA-C640F fluorophore). We also added a GFP tag to the C terminus of the Nav1.9-BAD construct to monitor total protein levels within the cell. This allowed us to observe total expression of Nav1.9 in the green fluorescence channel, whereas the far-red fluorescence was used to monitor channel presence at the cell surface.
Figure 7.
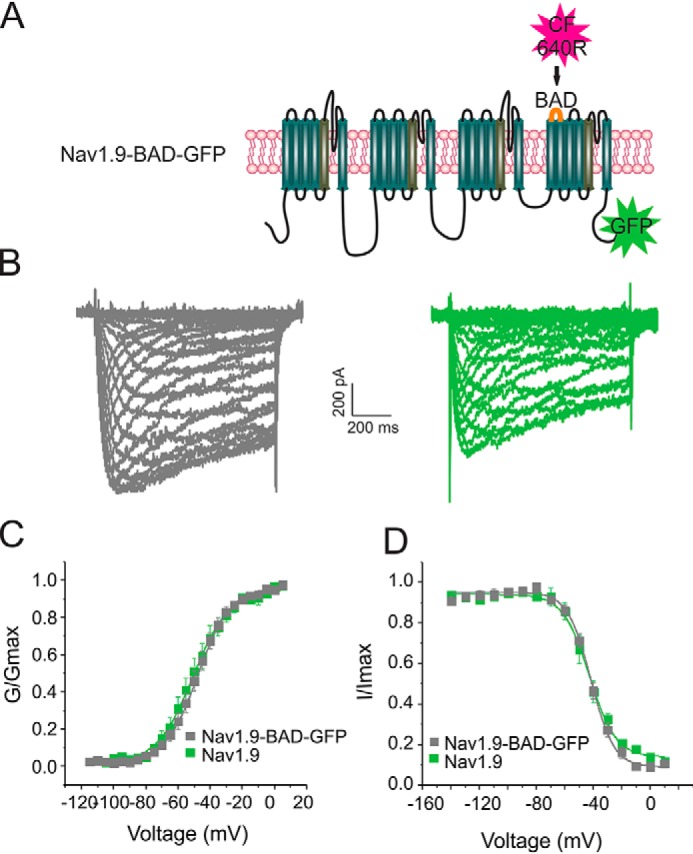
Nav1.9-BAD-GFP channels produce currents comparable with untagged Nav1.9 channels. A, schematic representation of the Nav1.9-BAD-GFP construct showing locations of the BAD (orange loop) and the C-terminal GFP tag (green star). The BAD domain is biotinylated by a co-transfected bacterial biotin ligase, BirA, which allows channel detection using a streptavidin-conjugated fluorophore CF-640R (red star). B, representative current traces recorded from superior cervical ganglion neurons expressing either Nav1.9 (green) or Nav1.9-BAD-GFP (gray). C, comparison of voltage dependence of activation for Nav1.9 (green, n = 9) and Nav1.9-BAD-GFP (gray, n = 8) channels. D, comparison of voltage dependence of steady-state fast inactivation for Nav1.9 (green, n = 9) or Nav1.9-BAD-GFP (gray, n = 8) channels. Error bars, S.E.
We have previously shown that Venus-Nav1.7-BAD channels produce currents that have properties similar to those of untagged channels and that they are trafficked to the cell surface in HEK293 cells and in DRG neuron (25). We characterized properties of the Nav1.9-BAD-Venus channel construct in superior cervical ganglion (SCG) neurons by voltage clamp. SCG neurons were isolated from 0–5-day postnatal Sprague–Dawley rats, transfected with either Nav1.9 or Nav1.9-BAD-GFP constructs, and cultured at 37 °C for 48–72 h post-transfection before voltage-clamp recordings. SCG neurons were used because they do not express endogenous TTX-R channels (27); thus, the properties of Nav1.9 channels can be studied in isolation by blocking the TTX-S channels with 1 μm TTX. Recordings of the Nav1.9 current in SCG neurons (Fig. 7) show that voltage dependence of activation and inactivation are comparable for untagged Nav1.9 channels and the Nav1.9-BAD-GFP channels. Although the current density of the Nav1.9-BAD-GFP channel was reduced compared with the untagged channel, this difference did not reach statistical significance (Fig. 7, B–E) (Table 1). Thus, the Nav1.9-BAD-GFP was considered to be suitable for use in trafficking studies.
Table 1.
Summary of Nav1.9 and Nav1.9-BAD-GFP electrophysiological properties
| Peak current density | Voltage dependence of activation |
Voltage dependence of inactivation |
|||
|---|---|---|---|---|---|
| V½ | k | V½ | k | ||
| pA/pF | mV | mV | |||
| Nav1.9 | 101.95 ± 37.78 | −47.46 ± 0.58 | 10.12 ± 0.35 (n = 14) | −41.75 ± 1.24 | 8.14 ± 0.69 (n = 9) |
| Nav1.9-BAD-GFP | 46.81 ± 12.67 | −50.60 ± 0.47 | 11.27 ± 0.44 (n = 7) | −40.98 ± 0.87 | 8.45 ± 0.74 (n = 8) |
Venus-Nav1.7-BAD and Nav1.9-BAD-GFP constructs were transiently transfected into HEK293 cells, and the protein level was assessed by Western blotting (Fig. 8A). Three independent transfections of different batches of HEK293 cells were performed to control for batch-to-batch differences in cultured cells. Similar to what we observed in the case of unmodified channels, a 3.1-fold difference in steady-state protein level was observed between Venus-Nav1.7-BAD and Nav1.9-BAD-GFP constructs (Nav1.9-BAD-GFP: 0.32 ± 0.10 AU, n = 3; Venus-Nav1.7-BAD constructs: 1.00 ± 0.36 AU, p < 0.05, by unpaired t test). Thus, no marked influence of BAD domain insertion on channel expression was observed. To clarify the role of the 49-aa motif in increasing Nav1.9 current density, a chimeric construct Nav1.9-BAD-Ct49aaNav1.7-GFP was created.
Figure 8.
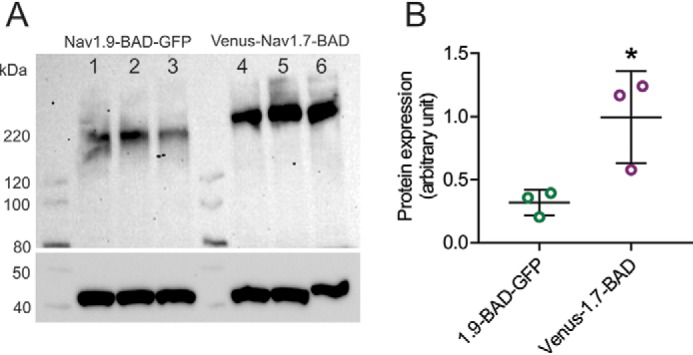
Steady-state protein levels of Nav1.9-BAD-GFP and Venus-Nav1.7-BAD in transiently transfected HEK293 cells. A, Western blotting of cell lysate of HEK293 cells transfected with either Nav1.9-BAD-GFP or Venus-Nav1.7-BAD. The blot was probed with pan-sodium channel (top) or β-actin (bottom) antibodies. Three independent transfections of different batches of HEK293 cells were analyzed: lanes 1–3 (Nav1.9-BAD-GFP) and lanes 4–6 (Venus-Nav1.7-BAD). B, protein expression levels were quantified using ImageLab software and presented as mean ± S.D. (error bars). A 3.1-fold difference was observed in protein expression between Nav1.9-BAD-GFP (0.32 ± 0.10 AU; n = 3) and Venus-Nav1.7-BAD constructs (1.00 ± 0.36 AU; n = 3), p < 0.05. Significance was determined using a two-tailed unpaired t test.
To measure surface expression, all three BAD-carrying constructs (Nav1.9-BAD-GFP, Venus-Nav1.7-BAD, and Nav1.9-BAD-Ct49aaNav1.7-GFP) were individually transfected into HEK293 cells, and resulting cells were incubated at 30 °C for 48–72 h post-transfection. Cells were surface-labeled with the far-red SA-CF640R ligand and then imaged using TIRF microscopy. Cells were selected for imaging by confirming that their basal membrane was within the TIRF evanescence field based on the Venus or GFP fluorescence. The surface expression was determined by quantifying the SA-CF640 signal on the basal membrane of each cell (Fig. 9A). A cell outline was determined using a green fluorescence image, whereas cell quality (single cells lying flat with well-defined borders) was assessed using the bright field image. In the far-red fluorescence image, the signal was measured within the determined cell outline (main signal, white dotted shape) as well as immediately outside of it (background). Cell surface fluorescence intensity was quantified by subtracting background from the main signal. Time-lapse imaging was used to confirm the characteristic movement of single channels diffusing within the plasma membrane, which allows labeled channels to be distinguished from background artifacts (Movies S1–S3). Data from 30 cells per construct were used for quantitative analysis of surface channels.
Figure 9.
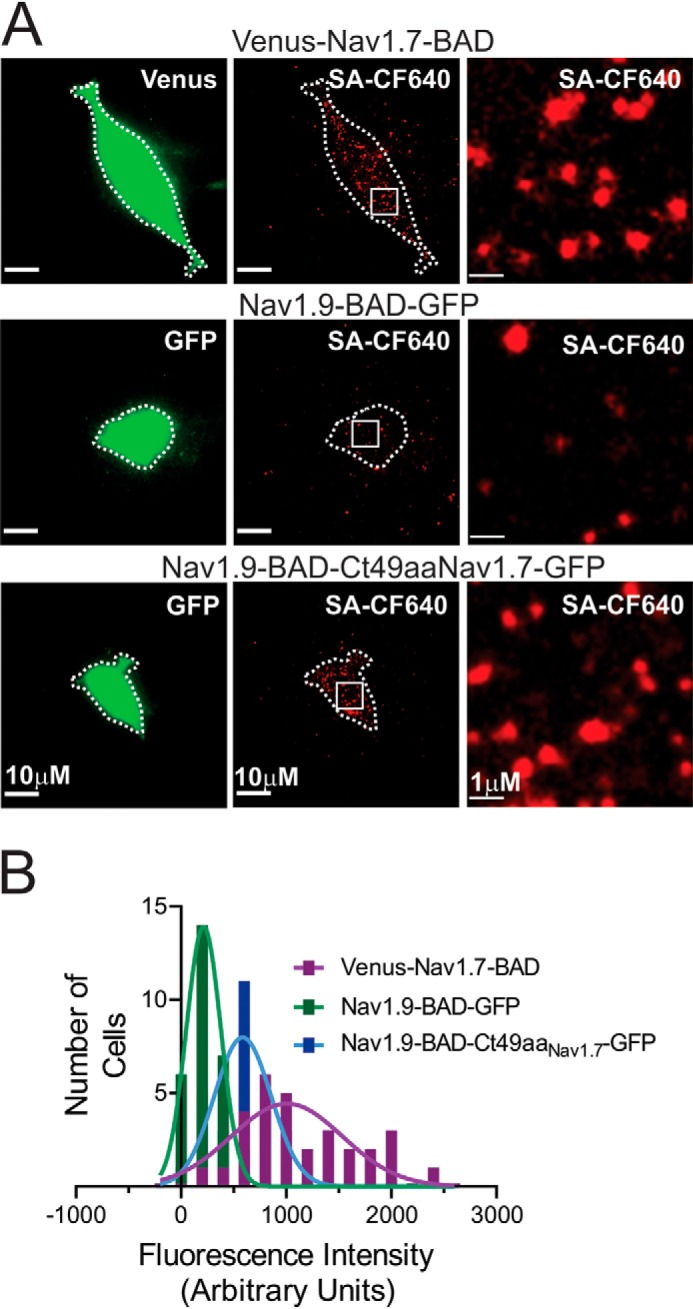
Imaging of Nav1.9 and Nav1.7 channels at the cell surface in HEK293 cells. A, representative TIRF images (green, 488-nm laser, first column of images; red, 647-nm laser, second column) of HEK293 cells transfected with any of the three constructs Venus-Nav1.7-BAD, Nav1.9-BAD-GFP, and Nav1.9-BAD-Ct49aaNav1.7-GFP. The third column shows ×10 magnified areas (insets marked by a square) within images from the second column. B, histograms of surface fluorescence intensity of SA-CF-640R–labeled Nav1.9-BAD-GFP (green), Nav1.9-BAD-Ct49aaNav1.7-GFP (blue), and Venus-Nav1.7-BAD (purple) channels on HEK293 cell surface. Solid green, blue, and purple lines are fits to the histogram distribution of the Nav1.9-BAD-GFP and Venus-Nav1.7-BAD constructs, respectively, with a mean value of 249 ± 38 AU, n = 30 for Nav1.9-BAD-GFP; 658 ± 64 AU, n = 30 for Nav1.9-BAD-Ct49aaNav1.7-GFP; and 1156 ± 100 AU, n = 30 for the Venus-Nav1.7-BAD construct. Differences between these values are statistically significant (one-way ANOVA (F(2, 87) = 9.769, p = 0.0001), followed by Tukey's multiple-comparison test).
Histograms of the fluorescence intensity of SA-CF640R–labeled channels Venus-Nav1.7-BAD, Nav1.9-BAD-GFP, and Nav1.9-BAD-Ct49aaNav1.7-GFP on the surface of transiently transfected HEK293 cells are presented in Fig. 9B. Sparse Nav1.9 channels (Nav1.9-BAD-GFP) were registered on the surface of most HEK293 cells, although there were a few rare cells (<10%) that showed considerable surface expression. This observation is in agreement with a very small amplitude of Nav1.9 current in this heterologous system. In contrast, large numbers of Nav1.7 channels (Venus-Nav1.7-BAD construct) were observed on the surface of HEK293, in agreement with the big amplitude of this channel (Fig. 4, compare A and E). Signal from the chimera Nav1.9-BAD-Ct49aaNav1.7-GFP reflects an intermediate level of surface expression compared with the two other constructs, showing that the 49-aa motif from Nav1.7 significantly improves Nav1.9 channel surface expression. The distributions have a mean of 249 ± 38 AU, n = 30 for Nav1.9-BAD-GFP, 658 ± 64 AU, n = 30 for Nav1.9-BAD-Ct49aaNav1.7-GFP, and 1156 ± 100 AU, n = 30 for Venus-Nav1.7-BAD construct (one-way ANOVA (F(2, 87) = 9.769, p = 0.0001) followed by Tukey's multiple-comparison test: p < 0.0001 for Venus-Nav1.7-BAD versus Nav1.9-BAD-GFP; p < 0.0001 for Venus-Nav1.7-BAD versus Nav1.9-BAD-Ct49aaNav1.7-GFP comparison; and p < 0.001 for Nav1.9-BAD-GFP versus Nav1.9-BAD-Ct49aaNav1.7-GFP comparison).
The 49-aa motif is exposed at the cytosolic interface of the C terminus within the Nav1.9-Ct49aaNav1.7 chimeric channel
The increase in the number of Nav1.9-BAD-Ct49aaNav1.7-GFP channels at the cell surface, compared with the parent Nav1.9-BAD-GFP channel, suggests that the 49-aa motif might be engaged in channel trafficking to the plasma membrane or in increasing the stability of the channels at the cell surface. Knowing the extent of exposure of a particular amino acid motif at the surface of a protein structure is important to infer the probability for this particular motif to engage with protein partners. We utilized the homology-modeling tool in Molecular Operating Environment (MOE) software to generate energetically stable structures of human Nav1.7 and Nav1.9 intracellular loops. For this study, we have created homology models for four different channels to look at a specific region of the C-terminal domain. The structures of the C-terminal domain for Nav1.9 and Nav1.7 are illustrated in (Fig. 10, A and C), and the 49-aa motif is colored green for the Nav1.9 and cyan for the Nav1.7 channels. The C termini of the parent Nav1.9 and Nav1.7 channels show divergent overall structures and specifically different structure of the 49-aa motif (Fig. 10, A and C). The chimeric versions of the channels are shown in Fig. 10 (B and D) with the 49-aa motif colored as in the donor channel. In all models, the 49 aa form α-helical secondary structures that appear to be exposed to the intracellular aqueous environment and are thus available for interactions with other cytosolic proteins. No significant change in Nav1.9 C terminus structure is observed when the 49-aa motif is replaced with the corresponding Nav1.7 motif. This result suggests that specific amino acids of this motif, rather than the location of the whole motif within the channel, are responsible for the strong effect that this region is exerting on Nav1.9 functional expression. However, a marked change in the conformation of the Nav1.7 C terminus is observed when a reciprocal swap with the Nav1.9 49-aa motif is performed. These results suggest that the Nav1.9 49-aa motif may have a dominant effect on the folding of the C terminus of the both Nav1.7 and Nav1.9.
Figure 10.
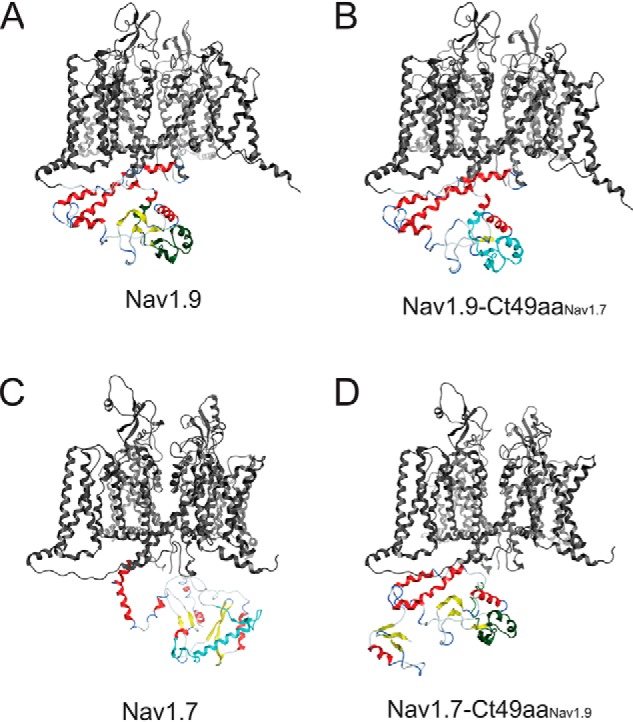
Structures for the C-terminal domains of Nav1.7 and Nav1.9 and their chimeric versions. All channel structures were created using the homology modeling tool in the MOE program using the recently published core structure of hNav1.7 (Protein Data Bank entry 6J8I) to create energetically stable conformations of the intracellular loops of the channel constructs used in the current study. The highly constrained structure of the core membrane-spanning segments are colored in gray. Although all of the intracellular sequences were modeled, only the C-terminal structure is shown for clarity. The default color scheme is red for α-helix and yellow for β-sheet. A, view of the Nav1.9 homology model illustrating the C-terminal domain. The 49-aa motif in Nav1.9 is colored green. B, view of the Nav1.9-Ct49aaNav1.7 chimera homology model illustrating the C-terminal domain. The 49-aa motif from Nav1.7 is colored cyan. C, view of the Nav1.7 homology model illustrating the C-terminal domain. The 49-aa motif in Nav1.7 is colored cyan. D, view of the Nav1.7-Ct49aaNav1.9 chimera homology model illustrating the C-terminal domain. The 49-aa motif from Nav1.9 is colored green.
Discussion
We have identified a 49-aa motif within the C terminus of Nav1.9 that regulates channel trafficking to the plasma membrane. We show here that the small currents that are produced by Nav1.9 in HEK293 cells are not due to the instability of the channel protein; rather, they are caused by altered channel trafficking. The number of channels on the cell surface of a chimeric Nav1.9 channel carrying the corresponding 49-amino acid motif from Nav1.7 C terminus was significantly increased, and the chimera channels produced significantly larger currents with Nav1.9-like activation properties in HEK293 cells, but inactivation properties and persistent current that are intermediate between the parent Nav1.7 and Nav1.9 channels. Our data have validated a chimera that might be used as a new tool for studying Nav1.9 in isolation in a heterologous system, and, importantly, our observations identified a motif within a channel that regulates channel trafficking.
Until recently, mechanisms underlying sodium channel trafficking to the cell surface have remained elusive due to a lack of imaging techniques that allow monitoring of channels that are specifically located on the cell surface. Novel methods for live imaging of labeled sodium channels in cultured neurons have now begun to unravel these mechanisms (24, 28). For example, the TIRF microscopy in combination with the use of GFP-tagged BAD-carrying Nav1.6 channel constructs allowed studies of channel trafficking to the soma and axon initial segment in hippocampal neurons (24, 25), and the optical pulse-chase axonal long-distance (OPAL) imaging method using Halo-Nav1.7 and spinning-disc microscopy allowed the analysis of channel trafficking in DRG axons (28). Using the Nav1.9-BAD-GFP channel and TIRF microscopy, in the present study we simultaneously observed the total population of channels (green fluorescence) and surface channels (red fluorescence) (Fig. 9), which allowed a comparison of surface channels in HEK293 cells expressing Nav1.9, Nav1.7, and chimeric Nav1.9-Ct49aaNav1.7 channels. This analysis confirmed a marked increase in the number of cells with greater Nav1.9-Ct49aaNav1.7 surface channels, compared with cells expressing the parent Nav1.9 channels in HEK293 cells, and supports the conclusion that impaired trafficking causes low Nav1.9 current amplitude in HEK293 cells.
The C terminus of sodium channels has been previously shown to play an important role in regulating sodium channel expression and gating properties. Plasma membrane targeting of sodium channel Nav1.8, another Nav channel with low functional expression in nonneuronal cells, improved dramatically upon replacement of its C terminus with that from Nav1.4 (18) or Nav1.7 (29) and altered gating and kinetic properties in an isoform-dependent manner. For Nav1.9, replacing the C terminus with that of Nav1.4 increased current amplitude in both mammalian cells and Xenopus oocyte and caused faster inactivation of the chimera channel (17), abrogating the characteristic ultra-slow inactivation of native Nav1.9 channels (2, 30). We observed a similar effect on channel inactivation in the Nav1.9-CtNav1.7 chimera. The Nav1.9-CtNav1.7 channels produced bigger currents than the parent Nav1.9 channel and retained the hyperpolarized voltage dependence of activation but manifested a voltage-dependence and kinetics of fast-inactivation that are comparable with those of the native Nav1.7 (Figs. 5 and 6).
The small Nav1.9 current in HEK293 cells could be due to instability of the channel protein in these cells. Our data show robust levels of Nav1.6 and Nav1.7 C terminus proteins, but for unknown reasons, the Nav1.9 C terminus protein is not stable in HEK293 cells, with levels barely detectable above background (Fig. 1). The instability of the Nav1.9 C terminus could have led to a precipitous reduction of the full-length Nav1.9 channel in these cells. However, we detected stable full-length Nav1.9 protein in HEK293 cells, suggesting that within the context of the full-length channel, the C terminus is protected from degradation. Although the 2.5-fold reduction in total protein levels of Nav1.9 compared with Nav1.7 is significant, it is not enough to explain the 130-fold greater amplitude of the Nav1.7 current in HEK293 cells. We interpret these data to suggest that additional mechanisms, including impaired trafficking of the Nav1.9 channels to the cell surface, contribute to this difference.
The 4-fold increase in the current density produced by Nav1.9-Ct49aaNav1.7 channels, compared with the parent Nav1.9 channels, while comparable levels of the protein are produced by these two channels in HEK293 cells, suggests that the 49-aa motif might increase channel trafficking rather than enhancing protein stability. This conclusion is supported by our data from live imaging of surface channels showing a marked increase in the fluorescence intensity at the surface of HEK293 expressing the Nav1.9-Ct49aaNav1.7 channels (Fig. 9). The reciprocal chimera Nav1.7-Ct49aaNav1.9 channels produced a current that is less than half of the parent Nav1.7 channels (Fig. 4). The fact that the exchange of this motif between Nav1.9 and Nav1.7 increased the current amplitude of the Nav1.9 chimera and reduced the amplitude of the Nav1.7 chimera further supports the conclusion that the 49 amino acids act as an independent trafficking motif within the C termini of sodium channels. A caveat is that swapping this motif alone does not increase the Nav1.9 chimera current to comparable levels of Nav1.7 current or reduce the Nav1.7 chimera current to that of Nav1.9, which suggests that additional motifs elsewhere in these channels are also necessary for full surface delivery. A closer look at the 49-amino acid motif in the Nav1.9 C terminus reveals an axonal targeting motif. A dileucine peptide (Ile-Leu, p.1685–1686) has been mapped to the axonal targeting motif in several axonal proteins, including sodium channels (31). The dileucine motif could also act as a selective endocytosis signal for Nav1.2 in the somatodendritic compartment of hippocampal neurons while retaining this channel in the axonal compartment (32). Although both Nav1.9 and Nav1.7 channels carry this motif, a number of adjacent amino acids are not conserved, suggesting that the local environment may alter the effectiveness of the motif in enhancing Nav1.7 but not Nav1.9 trafficking to the plasma membrane of HEK293 cells.
Neither the full-length C terminus nor the 49-aa motif of Nav1.7 altered voltage dependence of activation of the chimera channels, compared with their parent channels. However, voltage dependence of fast inactivation of the Nav1.9 and Nav1.7 channels was shifted in the direction of the source channel of the 49-aa motif. Kinetics of inactivation were altered by the replacement of the full-length C terminus; however, the Nav1.9-Ct49aaNav1.7 channels retained slower inactivation kinetics compared with Nav1.9-CtNav1.7 channels. There was a significant reduction in the percentage of the persistent current produced by Nav1.9-CtNav1.7 and Nav1.9-Ct49aaNav1.7 channels, compared with the parent Nav1.9 channels. By contrast, fast inactivation of Nav1.7-Ct49aaNav1.9 channels was shifted to more depolarized potentials; however, the persistent current was not increased. These data indicate that the 49-aa motif contributes to modulation of channel inactivation properties in an isoform-dependent manner. It was surprising, however, to record Nav1.9-Ct49aaNav1.7 currents that are comparable with those of native Nav1.9 channels in 20% of the HEK293 cells, which suggests that voltage dependence and ultra-slow kinetics of inactivation (Fig. 4D) may not be entirely channel-intrinsic, but rather may be modifiable by cellular factors that vary even among the clonal HEK293 cells.
Visualization of the 49-aa motif within the folded channel structure offers an opportunity for a better understanding of its role in regulating channel properties. We have focused on the C terminus structure with special emphasis on a 49-aa motif from Nav1.7, which when swapped onto Nav1.9 confers a greater surface delivery of the channel. Although we do not understand the contribution of the tertiary structure adopted by the Nav1.9 49 aa motif, shown in green, to the altered trafficking and gating changes in the chimera channels, we note that this motif is located at the exterior cytosolic surface of the folded C terminus structure, making it accessible to interact with the cellular trafficking machinery. The Nav1.7 49-aa motif adopted a tertiary structure similar to that of the Nav1.9 motif in the Nav1.9-Ct49aaNav1.7 chimeric channel, which was different from that in its native Nav1.7 environment (Fig. 10), suggesting constraints on folding of this motif that are exerted by other parts of the Nav1.9 C terminus. However, this motif in the middle of the channel C terminus appears to remain widely exposed to external interactions in both WT and chimeric channels. Easy accessibility to the cytosol for protein-protein interaction shown by molecular structure modeling for the 49-aa motif confirms once again the potential importance of this region not just for channel trafficking, but also for forming contacts that are isoform-selective, which could explain the gating and kinetic changes that we observed in the chimera channels.
Our multidisciplinary approach identified a 49-aa motif in the Nav1.9 C terminus that regulates surface delivery and current amplitude of the channel in HEK293 cells. Our findings describe chimeric channels that may be useful for investigating mechanisms underlying sodium channel trafficking.
Experimental procedures
DNA constructs
The pcDNA3mod-GFP-2A-Navx plasmids carrying a complete sequence of human sodium channel 1.6, 1.7, and 1.9 α subunits (Nav1.6, Nav1.7, and Nav1.9 or Navx as a general designation for all three channels), subcloned into a pcDNA3 vector modified to become a low-copy number plasmid, were previously created in the laboratory (see Refs. 7 and 34) for details. The GFP-2A part of these plasmids is encoding GFP and a “StopGo” 33-amino acid 2A linker upstream of the human sodium channel ATG codon. As a result, GFP and sodium channel are produced as independent proteins from the same mRNA (35–37). The pcDNA3mod-GFP-2A-Navx plasmids were used as a template for creating all DNA constructs described below. All DNA modifications were introduced by a site-directed mutagenesis protocol adapted for large insert introduction (38, 39) using the QuikChange Lightning site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA). Insert identity was confirmed by Sanger sequencing at the Keck facility, Yale University. To create the MS-CtNavx-SF and MS-GFP-SF constructs, either the full C-terminal part of either Nav1.6 (aa 1770–1980 of human Nav1.6 sequence (hNav1.6)), Nav1.7 (aa 1764–1988 of hNav1.7), or Nav1.9 (aa 1608–1791 of hNav1.9) or a coding sequence of GFP (GFP control) was subcloned into a piggyBac shuttle vector (gift from Alfred L. George, Northwestern University). Strep-FLAG tag was added to each construct C-terminally to allow easier protein identification by Western blotting and the possibility to purify corresponding protein complexes using a TAP approach (20). An N-terminal myristoylation signal, GCTVSAEDK, was added to target these constructs to the plasma membrane. The ChimNt, ChimMd, and ChimCt Nav1.9-Nav1.7 chimeras were created by replacing sequences of Nav1.9 within the MS-CtNav1.9-SF construct with corresponding sequences of Nav1.7 for region Nt (aa 1608–1659 of hNav1.9 sequence), Md (aa 1665–1713), or Ct (aa 1717–1791) respectively (see Fig. 3, A and B). The ChimMd#1, ChimMd#2, and ChimMd#3 chimeras were created by successively replacing Md#1 (aa 1665–1669), Md#2 (aa 1677–1683), and Md#3 (aa 1696–1706) sequences of Nav1.9 with corresponding sequences from Nav1.7 (Fig. 4, A and B). To generate the Nav1.9-Ct49aaNav1.7 and Nav1.7-Ct49aaNav1.9 constructs (Fig. 5A), corresponding sequences of a 49-amino acid stretch (Md), aa 1665–1713 of hNav1.9 and aa 1821–1869 of hNav1.7, were swapped between pcDNA3mod-GFP-2A-Nav1.9 and pcDNA3mod-GFP-2A-Nav1.7 constructs. Finally, the Nav1.9-BAD-GFP construct was created by successively adding a C-terminal GFP tag and introducing a 50-amino acid insertion that includes the 17-amino acid BAD (biotin-accepting lysine: GLNDIFEAQKIEWH) between S1 and S2 of channel domain IV; the lysine residue (boldface type) is the biotin acceptor residue within the BAD. The Nav1.9-BAD-Ct49aaNav1.7-GFP construct was created by replacing the C-terminal 49-amino acid stretch in question (aa 1665–1713 of hNav1.9) with corresponding sequences from Nav1.7 (aa 1821–1869).
Cell culture and transfection
HEK293 cells were grown at 37 °C with 5% CO2 in Dulbecco's modified Eagle's medium/F12 medium (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (Hyclone Laboratories, Logan, UT) and penicillin-streptomycin antibiotics (Thermo Fisher Scientific). Unless otherwise stated, cell transfection was performed using LipoJet (SignaGen Laboratories, Rockville, MD) by following a standard protocol. When no GFP was present as a terminal tag or a part of a GFP-2A construct, HEK cells were co-transfected with a plasmid pEGFP (Takara Bio, Mountain View, CA) to monitor for transfection efficiency. For electrophysiology experiments, HEK293 cells were allowed to recover at 37 ºC for 18 h after transfection and then seeded onto 10 poly-l-lysine–coated glass coverslips (BD Biosciences) in a 24-well plate, allowed to attach at 37 ºC, and transferred to 30 °C for 48–72 h, as incubation at lower temperature (28–30 °C) was shown to improve Nav1.9 functional expression. For imaging experiments, HEK293 cells were plated on poly-d-lysine–coated 35-mm dishes with 14-mm glass coverslips (MatTek, Ashland, MA) that were precoated with Matrigel (Corning, Inc.). Cells for imaging were transfected using Lipofectamine 2000 (Invitrogen) as described previously (24). Briefly, cells were plated at low density on glass coverslip plates and incubated at 37 °C for 4 h before transfection with 1 μg of the construct DNA. To allow biotinylation of the BAD constructs, the latter were co-transfected with 0.5 μg of bacterial biotin ligase plasmid pSec-BirA. Imaging was performed after incubation for 48–72 h at 30 °C post-transfection. Isolation (40) and transfection (41) of SCG neurons was performed as described. All animal care and experimental procedures were approved by the Veterans Administration Connecticut Healthcare System Institutional Animal Care and Use Committee.
Protein lysate preparation and Western blotting
HEK293 cells (nontransfected as a control as well as transiently or stably transfected with DNA constructs described above) were lysed and subjected to Western blotting as described (42, 43). Briefly, cells were trypsinized following standard procedure and collected by centrifugation at 500 × g for 5 min at 4 °C. Cell pellets were washed twice with ice-cold PBS and lysed in a buffer made of 20 mm Tris-Cl (pH 7.4), 150 mm NaCl, 1% Triton X-100, 1 mm DTT, 10 mm EGTA and 2× Complete protease inhibitor mixture (Roche Applied Science) at 4 °C for 1 h with rotation. Supernatant containing total protein was separated from cell pellet by centrifugation at 15,000 × g for 10 min at 4 °C, aliquoted, and stored at −80 °C. Protein concentrations in cell lysates were measured using the Bradford reagent (Bio-Rad). For Western blotting, about 30 μg of lysate were denatured in NuPAGE® LDS sample buffer, resolved on NuPAGE® Novex® 4–12% BisTris gels (1.0 mm, 12 wells), and transferred to a nitrocellulose membrane. Membranes were incubated in a blocking solution (5% nonfat dry milk and 1% BSA in TBS with 0.1% of Tween 20, TBST) for 1 h, washed, and incubated overnight at 4 °C with the desired primary antibodies diluted in the blocking buffer. Either monoclonal antibodies against Nav subunits (pan-Navα; 1:1000; Sigma), FLAG tag (FLAG; 1:1000; Sigma), Strep tag (Strep; 1:1000; Sigma), or β-actin (1:20,000; Sigma) were used. Blots were washed with TBST and incubated with secondary antibodies (horseradish peroxidase–conjugated goat anti-mouse (1:10,000; Dako, Santa Clara, CA) or goat anti-rabbit (1:10,000; Dako)) for 1 h. Finally, blots were washed with PBST and developed with the Western Lightning Plus-ECL enhanced chemiluminescence kit (PerkinElmer Life Sciences) using a ChemiDoc Imaging System (Bio-Rad).
Whole-cell patch-clamp recordings
Voltage-clamp recordings in HEK293 cells were performed using an EPC-10 amplifier and the Patchmaster program (version 69; HEKA Elektronik, Lambrecht/Pfalz, Germany) at room temperature (22 ± 1 °C). Fire-polished electrodes were fabricated from 1.6-mm outer diameter borosilicate glass micropipettes (World Precision Instruments, Sarasota, FL) using a Sutter Instruments (Novato, CA) P-97 puller and had a resistance of 0.7–1.5 megaohms. Pipette potential was adjusted to zero before seal formation. Liquid junction potential was not corrected. To reduce voltage errors, 70–80% series resistance compensation was applied. Cells were excluded from analysis if the predicted voltage error exceeded 3 mV. Linear leak currents were subtracted out using the P/6 method. Sodium current recordings were initiated after a 10-min equilibration period once whole-cell configuration was achieved. Current traces were sampled at 50 kHz and filtered with a low-pass Bessel setting of 10 kHz. The pipette solution contained the following: 140 mm CsF, 10 mm NaCl, 1 mm EGTA, 10 mm Hepes, 10 mm dextrose, pH 7.30, with CsOH (adjusted to 310 mosmol/liter with sucrose). The extracellular bath solution contained the following: 140 mm NaCl, 3 mm KCl, 1 mm MgCl2, 1 mm CaCl2, 10 mm Hepes, 10 mm dextrose, pH 7.30, with NaOH (adjusted to 320 mosmol/liter with sucrose). TTX (300 nm) was included in the bath to block the endogenous sodium currents in HEK293 cells (41, 44, 45). Transfected HEK293 cells were held at −120 mV for all parameters examined. Current-voltage relationships were measured using a series of 100-ms step depolarizations (−100 to +40 mV in 5-mV increments at 5-s intervals) from holding potential. Current density was calculated by normalizing maximal peak currents with cell capacitance. Peak inward currents obtained from activation protocols were converted to conductance values using the equation, G = I/(Vm − ENa), where G is the conductance, I is the peak inward current, Vm is the membrane potential step used to elicit the response, and ENa is the reversal potential for sodium channel, which is determined for each cell using the x axis intercept of a linear fit of the peak inward current responses to the last six voltage steps of the activation protocol. Conductance data were normalized by the maximum conductance value and fit with a Boltzmann equation of the form, G = Gmax + (Gmin − Gmax)/(1 + exp ((Vm − V½)/k)), where V½ is the midpoint of activation and k is a slope factor. Steady-state fast inactivation was assessed with a series of 500-ms prepulses (−140 to −10 mV in 10-mV increments); the remaining noninactivated channels were activated by a 50-ms step depolarization to −45 mV. Peak inward currents obtained from steady-state fast inactivation were normalized by the maximum current amplitude and fit with a Boltzmann equation of the form, I/Imax = 1/(1 + exp((Vm − V½)/k)), where Vm represents the inactivating prepulse membrane potential and V½ represents the midpoint of fast inactivation. To measure open-state inactivation, decaying currents after the peak during the depolarizing steps of the activation protocol were fit with a single-exponential equation of the form, I = A × exp(−t/τ) + Ic, where A is the amplitude of the fit, t is time, τ is the time constant of decay, and Ic is the asymptotic minimum to which the tail currents decay. Persistent currents were assessed as mean amplitudes of inward currents between 93 and 98 ms after the onset of depolarization steps in the activation protocol and are presented as a percentage of the peak current. Voltage-clamp recordings in SCG cells were performed as described (40) using 1 μm TTX.
Live-cell surface labeling and microscopy
Labeling of Nav1.9-BAD-GFP, Venus-Nav1.7-BAD, and Nav1.9-BAD-49aa1.7-GFP was performed immediately prior to imaging as described (24, 46). In brief, cells were rinsed with HEK imaging saline (146 mm NaCl, 4.7 mm KCl, 2.5 mm CaCl2, 0.6 mm MgSO4, 0.15 mm NaH2PO4, 0.1 mm ascorbic acid, 8 mm glucose, and 20 mm HEPES (pH 7.4)) and then incubated for 10 min at 37 °C with CF640R-streptavidin (Biotium, Hayward, CA) diluted 1:1000 in HEK imaging saline. Excess label was removed by rinsing with HEK imaging saline. Visualization of surface expression was performed on a Nikon TIRF microscope equipped with a 1024 × 1024 Andor iXon Ultra 888 EMCCD camera attached to a Nikon TiE inverted microscope base with Perfect Focus. Images were acquired using a ×100 Plan Apo-TIRF 1.49 numeric aperture objective. Illumination was with 100-mW 488-nm and 300-mW 639-nm solid-state lasers through the TIRF illumination path. All imaging was performed at 37 °C using a heated stage. Exactly the same settings for laser power (10% for 488 nm, 15% for 647 nm), camera (200-ms exposure for both fluorescence channels), and TIRF angles were used for imaging of cells carrying each of the three constructs. Thirty cells per construct from 3–5 independent cultures were recorded during the same day. Due to the broad range of fluorescence intensities, representative images shown in Fig. 8A could not be displayed with identical contrasts and were adjusted using Volocity software. Overall, Nav1.9-BAD-GFP was brightened to allow visualization, and the displayed intensity of Venus-Nav1.7-BAD was reduced so that it did not appear saturated. The following settings were used for black and white points, respectively: Nav1.9-BAD-GFP (2300 and 5500 for green channel; 1200 and 3300 for red channel), Venus-Nav1.7-BAD (3900 and 11,000 for green channel; 3300 and 9000 for red channel), Nav1.9-BAD-49aa1.7-GFP (3000 and 12,000 for green channel; 1000 and 6000 for red channel). To quantify signal corresponding to surface channel expression, a cell outline was drawn on the green (488-nm) image and then moved to the red (647-nm) image, where the resulting signal was detected as a difference between average fluorescence intensity inside the cell outline and outside of it (background) (Fig. 9A). Data were analyzed using Prism software (GraphPad, Inc., San Diego, CA), and data from 30 cells per construct type are presented as histograms of fluorescence intensity detected on the surface of HEK293 cells transfected with each of the constructs.
Structural modeling and statistical analysis
Ion channel structural visualization and homology model generation were performed using Molecular Operating Environment (MOE) software, version 2018.01 (Chemical Computing Group ULC, Montreal, Canada). All residue homology models including C-terminal sequences of hNav1.7 and hNav1.9 were created using the recently published structure of hNav1.7 (Protein Data Bank entry 6J8I) as a template (33). The homology-modeling module was run using default settings. Homology modeling uses minimization of energy algorithms to find possible allowed structural conformations. Although algorithms and protocols for de novo protein folding are not available to date, probable structural conformations can be generated based on homology to template structures. The highly constrained template structures are colored gray in our homology models, leaving the calculated energetically possible intracellular sequence structures in default display color conventions. Note that the solved structure of Nav1.7 that was used as the template (33) is presumed to represent an inactivated conformation, and the IFM motif is docked. All of our homology models maintained the docked location of the IFM motif, and thus our modeling does not provide structural insights into the swapped inactivation kinetics observed in the chimera channels. The ability to focus on particular regions of the structures was aided by the use of selective hiding and/or coloring of stretches of sequence.
Voltage-clamp and current-clamp data were analyzed using FitMaster (HEKA Electronics, Bellmore, NY) and OriginPro (OriginLab Corp., Northampton, MA). We carried out head-to-head comparisons of cells transfected with WT controls and corresponding chimeric channels from cells transfected by the same researcher to minimize culture-to-culture variability. Imaging data were analyzed and graphed using Prism (GraphPad) software. Statistical significance was determined using either an independent t test (two samples) or one-way ANOVA followed by Tukey's multiple-comparison test (more than two samples) where appropriate. All data are presented as the mean value ± S.E. p < 0.05 was considered statistically significant.
Author contributions
D. V. S., S. G. W., and S. D. D.-H. conceptualization; D. V. S., J. H., E. J. A., M. E., C. G.-P., and S. D. D.-H. data curation; D. V. S. software; D. V. S., J. H., E. J. A., M. E., and C. G.-P. formal analysis; D. V. S. and E. J. A. validation; D. V. S., E. J. A., S. G. W., and S. D. D.-H. investigation; D. V. S., J. H., E. J. A., and M. E. visualization; D. V. S., J. H., E. J. A., M. E., C. G.-P., and S. D. D.-H. methodology; D. V. S., J. H., M. E., and S. D. D.-H. writing-original draft; D. V. S., S. G. W., and S. D. D.-H. project administration; D. V. S., J. H., E. J. A., M. E., C. G.-P., S. G. W., and S. D. D.-H. writing-review and editing; S. G. W. and S. D. D.-H. resources; S. G. W. and S. D. D.-H. supervision; S. G. W. and S. D. D.-H. funding acquisition.
Supplementary Material
Acknowledgments
We thank Peng Zhao and Shujun Liu for preparation and transfection of SCG cultures.
This work was supported by Center Grant B9253-C from the United States Department of Veterans Affairs Rehabilitation Research and Development Service. The Center for Neuroscience and Regeneration Research is a Collaboration of the Paralyzed Veterans of America with Yale University. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Movies S1–S3.
- Nav1.9
- voltage gated sodium channel 1.9
- Nav1.7
- voltage gated sodium channel 1.7
- TTX
- tetrodotoxin
- HEK293 cells
- human embryonic kidney 293 cells
- aa
- amino acid(s)
- GI
- gastrointestinal
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate
- TAP
- tandem affinity purification
- Pr
- proximal
- Md
- middle
- Ds
- distal
- AU
- arbitrary units
- ANOVA
- analysis of variance
- pF
- picofarads
- TIRF
- total internal reflection fluorescence
- BAD
- biotin acceptor domain
- DRG
- dorsal root ganglion
- SCG
- superior cervical ganglion
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
References
- 1. Dib-Hajj S. D., Tyrrell L., Black J. A., and Waxman S. G. (1998) NaN, a novel voltage-gated Na channel, is expressed preferentially in peripheral sensory neurons and down-regulated after axotomy. Proc. Natl. Acad. Sci. U.S.A. 95, 8963–8968 10.1073/pnas.95.15.8963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cummins T. R., Dib-Hajj S. D., Black J. A., Akopian A. N., Wood J. N., and Waxman S. G. (1999) A novel persistent tetrodotoxin-resistant sodium current in SNS-null and wild-type small primary sensory neurons. J. Neurosci. 19, RC43 10.1523/JNEUROSCI.19-24-j0001.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rush A. M., Cummins T. R., and Waxman S. G. (2007) Multiple sodium channels and their roles in electrogenesis within dorsal root ganglion neurons. J. Physiol. 579, 1–14 10.1113/jphysiol.2006.121483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dib-Hajj S. D., Black J. A., and Waxman S. G. (2015) NaV1.9: a sodium channel linked to human pain. Nat. Rev. Neurosci. 16, 511–519 10.1038/nrn3977 [DOI] [PubMed] [Google Scholar]
- 5. Bennett D. L., Clark A. J., Huang J., Waxman S. G., and Dib-Hajj S. D. (2019) The role of voltage-gated sodium channels in pain signaling. Physiol. Rev. 99, 1079–1151 10.1152/physrev.00052.2017 [DOI] [PubMed] [Google Scholar]
- 6. Dib-Hajj S. D., and Waxman S. G. (2019) Sodium channels in human pain disorders: genetics and pharmacogenomics. Annu. Rev. Neurosci. 42, 87–106 10.1146/annurev-neuro-070918-050144 [DOI] [PubMed] [Google Scholar]
- 7. Huang J., Han C., Estacion M., Vasylyev D., Hoeijmakers J. G., Gerrits M. M., Tyrrell L., Lauria G., Faber C. G., Dib-Hajj S. D., Merkies I. S., Waxman S. G., and PROPANE Study Group (2014) Gain-of-function mutations in sodium channel Na(v)1.9 in painful neuropathy. Brain 137, 1627–1642 10.1093/brain/awu079 [DOI] [PubMed] [Google Scholar]
- 8. Leipold E., Hanson-Kahn A., Frick M., Gong P., Bernstein J. A., Voigt M., Katona I., Oliver Goral R., Altmüller J., Nürnberg P., Weis J., Hübner C. A., Heinemann S. H., and Kurth I. (2015) Cold-aggravated pain in humans caused by a hyperactive NaV1.9 channel mutant. Nat. Commun. 6, 10049 10.1038/ncomms10049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Han C., Yang Y., Te Morsche R. H., Drenth J. P., Politei J. M., Waxman S. G., and Dib-Hajj S. D. (2017) Familial gain-of-function Nav1.9 mutation in a painful channelopathy. J. Neurol. Neurosurg. Psychiatry 88, 233–240 10.1136/jnnp-2016-313804 [DOI] [PubMed] [Google Scholar]
- 10. Leipold E., Liebmann L., Korenke G. C., Heinrich T., Giesselmann S., Baets J., Ebbinghaus M., Goral R. O., Stodberg T., Hennings J. C., Bergmann M., Altmuller J., Thiele H., Wetzel A., Nurnberg P., et al. (2013) A de novo gain-of-function mutation in SCN11A causes loss of pain perception. Nat. Genet. 45, 1399–1404 10.1038/ng.2767 [DOI] [PubMed] [Google Scholar]
- 11. Woods C. G., Babiker M. O., Horrocks I., Tolmie J., and Kurth I. (2015) The phenotype of congenital insensitivity to pain due to the NaV1.9 variant p.L811P. Eur. J. Hum. Genet. 23, 561–563 10.1038/ejhg.2014.166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huang J., Vanoye C. G., Cutts A., Goldberg Y. P., Dib-Hajj S. D., Cohen C. J., Waxman S. G., and George A. L. Jr. (2017) Sodium channel NaV1.9 mutations associated with insensitivity to pain dampen neuronal excitability. J. Clin. Invest. 127, 2805–2814 10.1172/JCI92373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hockley J. R., Boundouki G., Cibert-Goton V., McGuire C., Yip P. K., Chan C., Tranter M., Wood J. N., Nassar M. A., Blackshaw L. A., Aziz Q., Michael G. J., Baker M. D., Winchester W. J., Knowles C. H., and Bulmer D. C. (2014) Multiple roles for NaV1.9 in the activation of visceral afferents by noxious inflammatory, mechanical, and human disease-derived stimuli. Pain 155, 1962–1975 10.1016/j.pain.2014.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. O'Donnell A. M., Coyle D., and Puri P. (2016) Decreased Nav1.9 channel expression in Hirschsprung's disease. J. Pediatr. Surg. 51, 1458–1461 10.1016/j.jpedsurg.2016.05.007 [DOI] [PubMed] [Google Scholar]
- 15. Vanoye C. G., Kunic J. D., Ehring G. R., and George A. L. Jr. (2013) Mechanism of sodium channel NaV1.9 potentiation by G-protein signaling. J. Gen. Physiol. 141, 193–202 10.1085/jgp.201210919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lin Z., Santos S., Padilla K., Printzenhoff D., and Castle N. A. (2016) Biophysical and pharmacological characterization of Nav1.9 voltage dependent sodium channels stably expressed in HEK-293 cells. PLoS One 11, e0161450 10.1371/journal.pone.0161450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Goral R. O., Leipold E., Nematian-Ardestani E., and Heinemann S. H. (2015) Heterologous expression of NaV1.9 chimeras in various cell systems. Pflugers Arch. 467, 2423–2435 10.1007/s00424-015-1709-1 [DOI] [PubMed] [Google Scholar]
- 18. Choi J. S., Tyrrell L., Waxman S. G., and Dib-Hajj S. D. (2004) Functional role of the C-terminus of voltage-gated sodium channel Na(v)1.8. FEBS Lett. 572, 256–260 10.1016/j.febslet.2004.07.047 [DOI] [PubMed] [Google Scholar]
- 19. Gorfe A. A., and Hocker H. J. (2012) Membrane targeting: methods. eLS 10.1002/9780470015902.a0002615.pub2 10.1002/9780470015902.a0002615.pub2 [DOI] [Google Scholar]
- 20. Gloeckner C. J., Boldt K., and Ueffing M. (2009) Strep/FLAG tandem affinity purification (SF-TAP) to study protein interactions. Curr. Protoc. Protein Sci. 57, 19.20.1–19.20.19 10.1002/0471140864.ps1920s57 [DOI] [PubMed] [Google Scholar]
- 21. Gloeckner C. J., Boldt K., Schumacher A., and Ueffing M. (2009) Tandem affinity purification of protein complexes from mammalian cells by the Strep/FLAG (SF)-TAP tag. Methods Mol. Biol. 564, 359–372 10.1007/978-1-60761-157-8_21 [DOI] [PubMed] [Google Scholar]
- 22. Kahlig K. M., Saridey S. K., Kaja A., Daniels M. A., George A. L. Jr., and Wilson M. H. (2010) Multiplexed transposon-mediated stable gene transfer in human cells. Proc. Natl. Acad. Sci. U.S.A. 107, 1343–1348 10.1073/pnas.0910383107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wilson M. H., Coates C. J., and George A. L. Jr. (2007) PiggyBac transposon-mediated gene transfer in human cells. Mol. Ther. 15, 139–145 10.1038/sj.mt.6300028 [DOI] [PubMed] [Google Scholar]
- 24. Akin E. J., Solé L., Dib-Hajj S. D., Waxman S. G., and Tamkun M. M. (2015) Preferential targeting of Nav1.6 voltage-gated Na+ channels to the axon initial segment during development. PLoS One 10, e0124397 10.1371/journal.pone.0124397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Solé L., Wagnon J. L., Akin E. J., Meisler M. H., and Tamkun M. M. (2019) The MAP1B binding domain of Nav1.6 is required for stable expression at the axon initial segment. J. Neurosci. 39, 4238–4251 10.1523/JNEUROSCI.2771-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen I., Howarth M., Lin W., and Ting A. Y. (2005) Site-specific labeling of cell surface proteins with biophysical probes using biotin ligase. Nat. Methods 2, 99–104 10.1038/nmeth735 [DOI] [PubMed] [Google Scholar]
- 27. Rush A. M., Dib-Hajj S. D., Liu S., Cummins T. R., Black J. A., and Waxman S. G. (2006) A single sodium channel mutation produces hyper- or hypoexcitability in different types of neurons. Proc. Natl. Acad. Sci. U.S.A. 103, 8245–8250 10.1073/pnas.0602813103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Akin E. J., Higerd G. P., Mis M. A., Tanaka B. S., Adi T., Liu S., Dib-Hajj F. B., Waxman S. G., and Dib-Hajj S. D. (2019) Building sensory axons: delivery and distribution of NaV1.7 channels and 2 effects of inflammatory mediators. Sci. Adv. 5, eaax4755 10.1126/sciadv.aax4755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vijayaragavan K., Acharfi S., and Chahine M. (2004) The C-terminal region as a modulator of rNa(v)1.7 and rNa(v)1.8 expression levels. FEBS Lett. 559, 39–44 10.1016/S0014-5793(04)00019-5 [DOI] [PubMed] [Google Scholar]
- 30. Dib-Hajj S. D., Tyrrell L., Cummins T. R., Black J. A., Wood P. M., and Waxman S. G. (1999) Two tetrodotoxin-resistant sodium channels in human dorsal root ganglion neurons. FEBS Lett. 462, 117–120 10.1016/S0014-5793(99)01519-7 [DOI] [PubMed] [Google Scholar]
- 31. Lasiecka Z. M., and Winckler B. (2011) Mechanisms of polarized membrane trafficking in neurons—focusing in on endosomes. Mol. Cell Neurosci. 48, 278–287 10.1016/j.mcn.2011.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Garrido J. J., Fernandes F., Giraud P., Mouret I., Pasqualini E., Fache M. P., Jullien F., and Dargent B. (2001) Identification of an axonal determinant in the C-terminus of the sodium channel Na(v)1.2. EMBO J. 20, 5950–5961 10.1093/emboj/20.21.5950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shen H., Liu D., Wu K., Lei J., and Yan N. (2019) Structures of human Nav1.7 channel in complex with auxiliary subunits and animal toxins. Science 363, 1303–1308 10.1126/science.aaw2493 [DOI] [PubMed] [Google Scholar]
- 34. Yang Y., Huang J., Mis M. A., Estacion M., Macala L., Shah P., Schulman B. R., Horton D. B., Dib-Hajj S. D., and Waxman S. G. (2016) Nav1.7-A1632G mutation from a family with inherited erythromelalgia: enhanced firing of dorsal root ganglia neurons evoked by thermal stimuli. J. Neurosci. 36, 7511–7522 10.1523/JNEUROSCI.0462-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ryan M. D., and Drew J. (1994) Foot-and-mouth disease virus 2A oligopeptide mediated cleavage of an artificial polyprotein. EMBO J. 13, 928–933 10.1002/j.1460-2075.1994.tb06337.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Luke G. A., de Felipe P., Lukashev A., Kallioinen S. E., Bruno E. A., and Ryan M. D. (2008) Occurrence, function and evolutionary origins of “2A-like” sequences in virus genomes. J. Gen. Virol. 89, 1036–1042 10.1099/vir.0.83428-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Atkins J. F., Wills N. M., Loughran G., Wu C. Y., Parsawar K., Ryan M. D., Wang C. H., and Nelson C. C. (2007) A case for “StopGo”: reprogramming translation to augment codon meaning of GGN by promoting unconventional termination (Stop) after addition of glycine and then allowing continued translation (Go). RNA 13, 803–810 10.1261/rna.487907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Geiser M., Cèbe R., Drewello D., and Schmitz R. (2001) Integration of PCR fragments at any specific site within cloning vectors without the use of restriction enzymes and DNA ligase. BioTechniques 31, 88–90, 92 10.2144/01311st05 [DOI] [PubMed] [Google Scholar]
- 39. Wang W., and Malcolm B. A. (1999) Two-stage PCR protocol allowing introduction of multiple mutations, deletions and insertions using QuikChange Site-Directed Mutagenesis. BioTechniques 26, 680–682 10.2144/99264st03 [DOI] [PubMed] [Google Scholar]
- 40. Han C., Yang Y., de Greef B. T., Hoeijmakers J. G., Gerrits M. M., Verhamme C., Qu J., Lauria G., Merkies I. S., Faber C. G., Dib-Hajj S. D., and Waxman S. G. (2015) The domain II S4-S5 linker in Nav1.9: a missense mutation enhances activation, impairs fast inactivation, and produces human painful neuropathy. Neuromolecular Med. 17, 158–169 10.1007/s12017-015-8347-9 [DOI] [PubMed] [Google Scholar]
- 41. Dib-Hajj S. D., Choi J. S., Macala L. J., Tyrrell L., Black J. A., Cummins T. R., and Waxman S. G. (2009) Transfection of rat or mouse neurons by biolistics or electroporation. Nat. Protoc. 4, 1118–1126 10.1038/nprot.2009.90 [DOI] [PubMed] [Google Scholar]
- 42. Dash B., Dib-Hajj S. D., and Waxman S. G. (2018) Multiple myosin motors interact with sodium/potassium-ATPase α1 subunits. Mol. Brain 11, 45 10.1186/s13041-018-0388-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dash B., Han C., Waxman S. G., and Dib-Hajj S. D. (2018) Nonmuscle myosin II isoforms interact with sodium channel α subunits. Mol. Pain 14, 1744806918788638 10.1177/1744806918788638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. He B., and Soderlund D. M. (2010) Human embryonic kidney (HEK293) cells express endogenous voltage-gated sodium currents and Na v 1.7 sodium channels. Neurosci. Lett. 469, 268–272 10.1016/j.neulet.2009.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cummins T. R., Rush A. M., Estacion M., Dib-Hajj S. D., and Waxman S. G. (2009) Voltage-clamp and current-clamp recordings from mammalian DRG neurons. Nat. Protoc. 4, 1103–1112 10.1038/nprot.2009.91 [DOI] [PubMed] [Google Scholar]
- 46. Akin E. J., Solé L., Johnson B., Beheiry M. E., Masson J. B., Krapf D., and Tamkun M. M. (2016) Single-molecule imaging of Nav1.6 on the surface of hippocampal neurons reveals somatic nanoclusters. Biophys. J. 111, 1235–1247 10.1016/j.bpj.2016.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.