

ABSTRACT
Macroautophagy/autophagy is implicated in age-dependent neurodegenerative diseases, including amyotrophic lateral sclerosis and Parkinson, Huntington and Alzheimer diseases, suggesting that an age-related decline in neuronal autophagy may contribute to the onset of neurodegeneration. We identified a significant decline in the rate of axonal autophagosome formation in neurons cultured from aged mice, accompanied by a striking increase in the accumulation of autophagic structures with aberrant morphologies. Using live-cell microscopy, we identified the specific step in autophagosome formation that becomes impaired with age, focusing on the role of the phosphoinositide binding protein WIPI2. We determined that the dynamic and local phosphorylation of WIPI2 is a critical regulatory step in autophagosome biogenesis in neurons and that this step is specifically affected by aging. Together, these results provide new insights into the regulation of autophagosome biogenesis in neurons and delineate how autophagosome formation is affected by age. These observations also point to a potential new target for therapeutic intervention.
KEYWORDS: Aging, autophagosome biogenesis, autophagy, macroautophagy, neurodegeneration, neuronal autophagy, WIPI2
Autophagy functions as part of a stress response in many cell types and tissues, induced by environmental factors such as nutrient deprivation or acute organelle damage. In neurons, accumulating evidence indicates that nonselective macroautophagy is a constitutive pathway that contributes to cellular homeostasis. Neurons are long-lived, highly polarized cells, with axons and dendrites that can extend up to a meter from the cell body, the primary site of anabolic and catabolic processing. Both the spatial and temporal challenges of maintaining healthy neurons with long processes over decades of life are considerable. But how neuronal autophagy is affected by aging has remained unclear.
Axonal autophagosomes are generated at a constant rate in primary neurons isolated from young adult mice. We wondered if the rate of autophagosome biogenesis in neurons changes with age. We isolated dorsal root ganglion (DRG) neurons from GFP-LC3B transgenic mice at 4 different ages: young mice (1 mo), young adult mice (3 mo), aged mice (16–17 mo) and advanced age mice (24 mo). Using GFP-LC3B to track the formation of autophagosomes, we observed a striking 94% decrease in the rate of autophagosome biogenesis with age. Further, using electron microscopy, we identified aberrant, multilamellar structures in neurons from aged mice that we did not observe in neurons from young adult mice. These multilamellar structures are similar to those seen in pathological analysis of tissue from aged patients with Alzheimer disease. Remarkably, in aged mice only 34% of autophagic vesicles found in axon terminals have normal, double-membrane morphologies, compared to 80% in young adult mice. We confirmed that the ultrastructural differences between neurons from young and aged mice are not unique to primary neurons, as similar observations were made in vivo at the neuromuscular junctions of motor neurons [1].
To identify which step in the autophagosome biogenesis pathway was altered with age, we used live-cell, multi-color confocal microscopy to examine components of each autophagy complex. We determined that the induction (marked with ATG13) and nucleation (phosphatidylinositol 3-phosphate [PtdIns3P] generation visualized with ZFYVE1/DFCP1) steps do not display altered kinetics across the 4 ages. However, neurons from aged mice frequently display “stalled” events, in which ATG13 or ATG5 remain as stable puncta whereas LC3B is not recruited. Additionally, we observed abnormalities with ATG9 localization; ATG9 remains persistently associated with stalled autophagic vesicles. Lipidated LC3B is not recruited to stalled phagophore structures, although the elongation complex (marked by ATG5) required for lipidation is present at stalled autophagic vesicles and is able to lipidate other LC3 homologs, indicating that the lipidation machinery is intact and functional and that the defect is specific to a failure to recruit LC3B.
These observations led us to focus on WIPI2, a PtdIns3P effector that links PtdIns3P generation to LC3 lipidation. Strikingly, we found that overexpression of WIPI2 is sufficient to fully restore the rate of autophagosome formation in neurons from aged mice. This observation suggested that there might be an age-dependent decline in WIPI2 expression levels, but quantitative analysis of Wipi2 mRNA and WIPI2 protein levels does not reveal a decrease in Wipi2/WIPI2 expression levels with age.
We next hypothesized that post-translational modification of WIPI2 affects autophagosome biogenesis and may change with age. We found that WIPI2 is reversibly phosphorylated at S395 in neurons. Interestingly, only a WIPI2 construct mimicking the dephosphorylated form is able to reverse the age-induced stalling observed during autophagosome formation, suggesting that dephosphorylation of WIPI2 is required for productive autophagosome biogenesis. We further demonstrated that phosphorylated WIPI2 is required for subsequent extension of the phagophore membrane. Thus, our data suggest that WIPI2 is first dephosphorylated to facilitate recruitment to the nascent phagophore, then is subsequently rephosphorylated to allow the phagophore membrane to expand. Finally, we determined that phosphorylation of WIPI2 decreases its affinity for membranes, suggesting that phosphorylation enhances dissociation of WIPI2, allowing autophagosome biogenesis to proceed. Thus, we propose that there is a dynamic phosphorylation cycle for WIPI2 S395 during autophagosome biogenesis that is locally regulated at the forming autophagosome.
Together, our data provide a new understanding of neuronal autophagy during aging, indicating that dynamic phosphorylation of WIPI2 is critical for autophagosome biogenesis; further, this step becomes locally misregulated with age in neurons. We speculate that a phagophore-targeted phosphatase initially dephosphorylates WIPI2 to allow productive autophagosome biogenesis. Subsequently, a targeted kinase phosphorylates WIPI2, decreasing WIPI2 membrane association and enabling phagophore membrane expansion (Figure 1). We hypothesize that defects in the dephosphorylation of WIPI2 with increasing age may occur due to an age-related mislocalization of the phosphatase. With age, failure to dephosphorylate WIPI2 at the phagophore membrane might result in the multilamellar structures observed, which are reminiscent of those found in AD patients. We hope that increased mechanistic insights into this age-induced pathway will result in more effective therapies to stave off degeneration by enhancing autophagy at the neuronal level.
Figure 1.
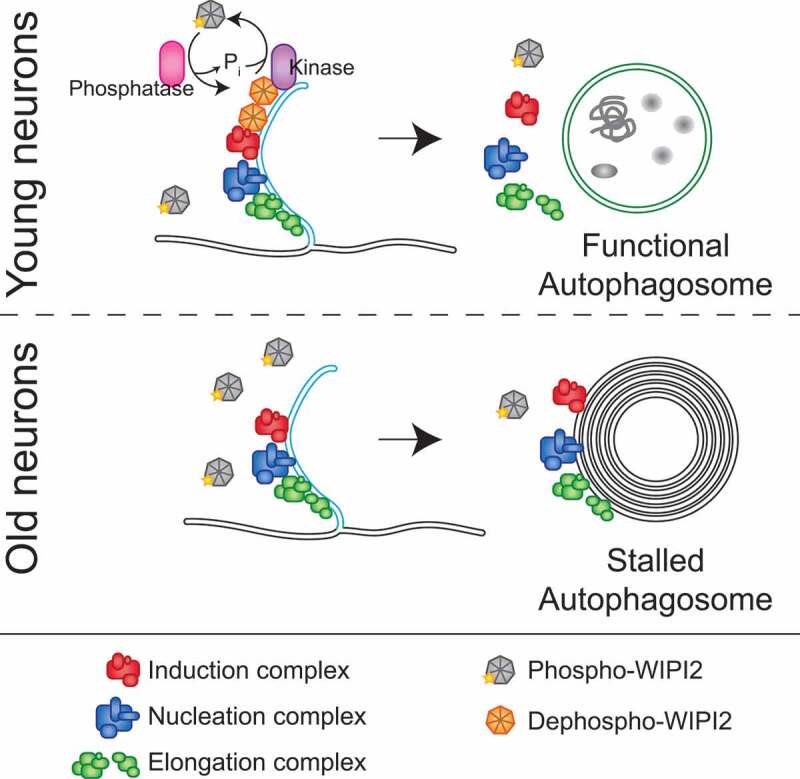
Model of neuronal autophagosome biogenesis changes during aging. In neurons from young adult mice (top panel), dephosphorylation of WIPI2B at the phagophore allows autophagosome biogenesis to continue, ultimately yielding a stereotypical autophagosome, marked with LC3B (green membranes), enclosing bulk cytoplasm. Autophagosome biogenesis complexes are as differently colored protein complexes (see legend at bottom of figure). In neurons from aged mice (lower panel), localized dephosphorylation of WIPI2B does not occur, resulting in autophagic multilamellar vesicles that retain the components of the autophagosome biogenesis complexes, but do not recruit LC3B. These multilamellar structures do not appear to contain bulk cytoplasm.
Funding Statement
This work was supported by the National Institutes of Health (K99 NS109286 to AKHS and R37 NS060698 to ELFH).
Disclosure statement
No potential conflict of interest was reported by the authors.
Reference
- [1].Stavoe AKH, Gopal PP, Gubas A, et al. Expression of WIPI2B counteracts age-related decline in autophagosome biogenesis in neurons. eLife. 2019;8(pii):e44219. PMID: 31309927. [DOI] [PMC free article] [PubMed] [Google Scholar]