

ABSTRACT
Vascular endothelial cells (VECs) that form the inner wall of blood vessels can be injured by high glucose-induced autophagy and apoptosis. Although the role of long noncoding RNA in regulating cell fate has received widespread attention, long noncoding RNAs (lncRNAs) that can both regulate autophagy and apoptosis need to be discovered. In this study, we identified that a small chemical molecule, 3-benzyl-5-([2-nitrophenoxy] methyl)-dihydrofuran-2(3H)-one (3BDO), synthesized by us, could inhibit VEC autophagy and apoptosis induced by a high concentration of glucose. To find new lncRNAs that regulate autophagy and apoptosis in VECs, we performed lncRNA microarray analysis. We found and verified an upregulated lncRNA named CA7-4 that was induced by a high concentration of glucose could be downregulated by 3BDO most obviously among all of the detected lncRNAs. Meanwhile, we investigated the mechanism of CA7-4 in regulating VEC autophagy and apoptosis. The results showed that CA7-4 facilitated endothelial autophagy and apoptosis as a competing endogenous RNA (ceRNA) by decoying MIR877-3P and MIR5680. Further study elucidated that MIR877-3P could trigger the decrease of CTNNBIP1 (catenin beta interacting protein 1) by combining with its 3ʹ UTR and then upregulating CTNNB1 (catenin beta 1); MIR5680 inhibited the phosphorylation of AMP-activated protein kinase (AMPK) by targeting and decreasing DPP4 (dipeptidyl peptidase 4). Therefore, CA7-4, MIR877-3P and MIR5680 represent new signal pathways that regulate VEC autophagy and apoptosis under the high-glucose condition.
Abbreviations: 3BDO: 3-benzyl-5-([2-nitrophenoxy] methyl)-dihydrofuran-2(3H)-one; 3ʹ UTR: 3ʹ untranslated region; AGO2: argonaute RISC catalytic component 2; AMPK: AMP-activated protein kinase/protein kinase AMP-activated; BAX/BCL2L4: BCL2 associated X, apoptosis regulator; BCL2: BCL2 apoptosis regulator; CASP3: caspase 3; ceRNA: competing endogenous RNA; CTNNB1: catenin beta 1; CTNNBIP1/ICAT: catenin beta interacting protein 1; DPP4: dipeptidyl peptidase 4; FGF2/FGF-2: fibroblast growth factor 2; HG: high concentration glucose (30 mM glucose); lncRNA: long noncoding RNA; MAP1LC3B/LC3B: microtubule associated protein 1 light chain 3 beta; miRNA: microRNA; MIR4778-3P: microRNA 4778-3p; MIR561-3P: microRNA 561-3p; MIR5680: microRNA 5680; MIR877-3P: microRNA 877-3p; MTOR: mechanistic target of rapamycin kinase; Mut: mutant; NC: negative control; NG: normal concentration glucose (5.5 mM glucose); PARP1: poly(ADP-ribose) polymerase 1; qPCR: quantitative real-time PCR; RNA-FISH: RNA-fluorescence in situ hybridization; ROS: reactive oxygen species; RT-PCR: reverse transcription polymerase chain reaction; siRNA: small interfering RNA; SQSTM1: sequestosome 1; TGFB2-OT1: TGFB2 overlapping transcript 1; TUNEL: terminal deoxynucleotidyl transferase dUTP nick end labeling; VECs: vascular endothelial cells; WT: wild type
KEYWORDS: Apoptosis, autophagy, high glucose, lncRNA, miRNA, vascular endothelial cells
Introduction
A high concentration of glucose in the blood is harmful to vascular endothelial cells (VECs) [1]. Under hyperglycemia, the synthesis of nitric oxide (NO) decreases and reactive oxygen species (ROS) levels notably increase due to the inactivation of endothelial NO synthase. High glucose-induced oxidative stress induces cell death, impairs angiogenesis, and then leads to vascular diseases [1,2]. In parallel, studies have focused on investigating the mechanism of endothelial cell autophagy, apoptosis, inflammation and senescence induced by high glucose. For instance, the production of ROS induced by hyperglycemia contributes to caspase-dependent apoptosis of endothelial cells by triggering CYCS (cytochrome c, somatic) release [3]. The chronic elevation of glucose level promotes the expression of various inflammatory factors in endothelial cells, including ICAM1 (intercellular adhesion molecule 1), IL6 (interleukin 6) [4], and VCAM1 (vascular cell adhesion molecule 1) [5]. In addition, high glucose accelerates endothelial cell senescence, apoptosis and autophagy by inducing renin-angiotensin system-mitochondrial damage [6]. Consequently, exploring novel regulatory factors to reduce the injury caused by hyperglycemia on endothelial cells is of great significance.
Only about 2% of sequences of human genome can be translated into protein. Transcripts that do not encode a protein can be divided according to length, including short non-coding RNAs (<200 nt) and long non-coding RNAs (lncRNAs, >200 nt) [7]. However, these noncoding transcripts do not simply exist as trash or transcriptional noise [8]. LncRNAs have been extensively considered critical players in regulating dosage compensation, genomic imprinting, chromatin remodeling, DNA methylation, cell differentiation, organogenesis, and the physiological and pathological processes of organisms [9]. Beyond that, many reports demonstrate that lncRNAs are emerging as central regulators in controlling cell autophagy or apoptosis of various diseases, including cancers [10–12], cardiovascular diseases [13–15], and diabetic vascular complications [16].
Nevertheless, only a few studies have investigated lncRNA regulation in endothelium injury caused by hyperglycemia. For example, under high glucose, MALAT1 (metastasis associated lung adenocarcinoma transcript 1) knockdown ameliorates retinal vessel apoptosis and capillary impairment [17]; MEG3 (maternally expressed 3) knockdown aggravates retinal vascular dysfunction [18]; MIAT (myocardial infarction associated transcript) is involved in regulating retinal vessel dysfunction as the decoy of MIR150-5P [19]; and CDKN2B-AS1 (CDKN2B antisense RNA 1) promotes angiogenesis by upregulating VEGFA/VEGF (vascular endothelial growth factor A) and activating NFKB1 (nuclear factor kappa B subunit 1) signaling [20].
MicroRNAs (miRNAs) also play important roles in endothelial injury induced by hyperglycemia. In general, these are a class of evolutionarily conserved non-coding RNAs approximately 20–22 nt in length. Accumulating evidence has revealed that miRNAs, alone or in combination with lncRNAs, are involved in regulating specific gene expression at the transcription or translation level, then changing cell signaling pathways associated with different physiological and pathological processes [21]. For the past few years, studies have elevated our understanding of miRNA regulation in hyperglycemia-induced endothelial dysfunction. For example, MIR126 inhibits inflammation and ROS production by targeting HMGB1 (high-mobility group box 1) in diabetic vascular endothelium [22]; overexpression of MIR149-5P improves high glucose-induced VEC dysfunction and apoptosis by targeting and downregulating TNF/TNFα (tumor necrosis factor) [23]. Consequently, noncoding RNAs, including lncRNAs and miRNAs, are emerging as diagnostic biomarkers or therapeutic targets of diabetes-associated vascular complications because of their regulation of endothelial injury induced by high glucose [24].
Our previous study indicates that a small chemical molecule, 3BDO, inhibits serum and FGF2 (fibroblast growth factor 2) deprivation-induced VEC apoptosis [25]. In 2014, we have identified that 3BDO inhibits autophagy as a novel activator of MTOR (mechanistic target of rapamycin kinase) and also found a new lncRNA, TGFB2-OT1 (TGFB2 overlapping transcript 1), that regulates autophagy in VECs by decoying MIR4459 targeting ATG13 (autophagy-related 13), and 3BDO reduces TGFB2-OT1 by promoting the phosphorylation of TIA1 (TIA1 cytotoxic granule associated RNA binding protein) [26]. Furthermore, 3BDO ameliorates autophagy and inflammation of VECs caused by lipopolysaccharide and oxidized low-density lipoprotein through diminishing TGFB2-OT1 [27]. Thus, according to the available evidence, we speculated that 3BDO may both suppress endothelial autophagy and apoptosis induced by high glucose.
In this study, we first demonstrated that 3BDO could alleviate high glucose-induced VEC autophagy and apoptosis and also identified a new lncRNA named CA7-4 by high-throughput sequencing. Here, 3BDO could decrease the level of CA7-4. Additionally, our findings highlighted novel miRNA signal pathways regulated by CA7-4 in autophagy and apoptosis of VECs.
Results
3BDO restrained the upregulation of CA7-4 induced by high glucose in VECs
First, we found that 3BDO inhibited high glucose-induced VEC autophagy and apoptosis (Figure S1). However, the data revealed that 3BDO decreased high glucose-induced autophagy in VECs without a remarkable effect on the expression of SQSTM1 (sequestosome 1) (Figure S1(c)). To investigate novel lncRNAs participating in VEC autophagy and apoptosis under high glucose, we performed lncRNA microarray analysis after treating VECs with normal concentration glucose (5.5 mM glucose) (NG), high concentration glucose (30 mM glucose) (HG), and HG+3BDO (60 μM) (Figure 1(a)) and found a new lncRNA, CA7-4, prominently regulated by HG+3BDO (Figure 1(b)). We examined the expression of CA7-4 by quantitative real-time PCR (qPCR) after treatment with high glucose or 3BDO. 3BDO inhibited the upregulated CA7-4 caused by high glucose (Figure 1(d,e)). Under normal glucose, 3BDO still could downregulate CA7-4 (Figure 1(f)).
Figure 1.
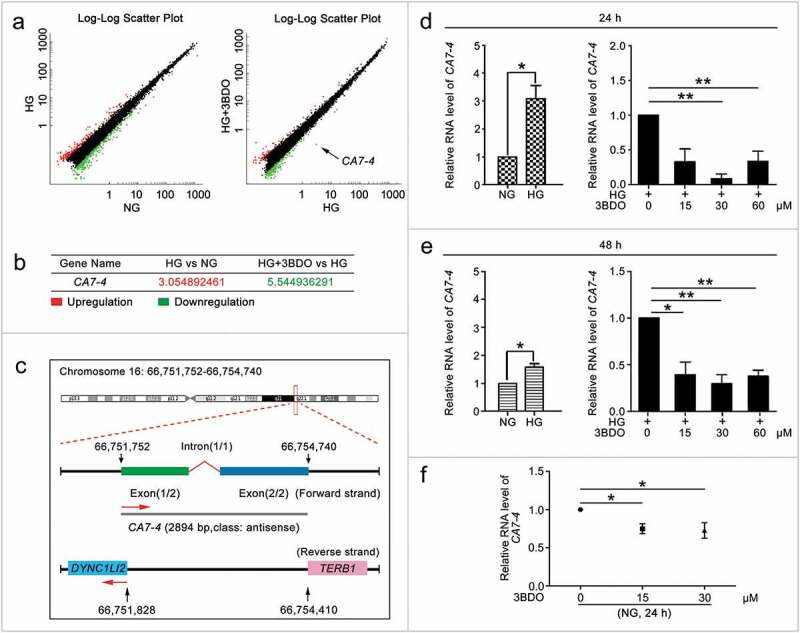
3BDO could suppress the increase in CA7-4 expression caused by high glucose. (a) The scatter plot of microarray analysis depicts genes upregulated (red) and downregulated (green). CA7-4 level was decreased obviously, as indicated by the arrow. (b) Changes in CA7-4 level with treatment. (c) Information on CA7-4 in the human genome is available at https://lncipedia.org/db/transcript/lnc-CA7-4:1. (d,e) VECs were treated with 5.5 mM glucose (NG), 30 mM glucose (HG), and HG+3BDO (15, 30, 60 μM) for 24 h and 48 h. (f) VECs were treated with NG, 3BDO (0, 15, 30 μM) for 24 h. The level of CA7-4 was detected by qPCR. (*, p < 0.05; **, p < 0.01; n = 3.).
In the LNCipedia database (version 5.2), CA7-4 has been identified as an lncRNA (https://lncipedia.org/db/transcript/lnc-CA7-4:1). It is located in the forward strand of chromosome 16 (hg38), is 2894 bp in full length, consists of two exons and one intron, and is antisense to DYNC1LI2 (dynein cytoplasmic 1 light intermediate chain 2) and TERB1 (telomere repeat binding bouquet formation protein 1) (Figure 1(c)). However, no one has studied the function of CA7-4. First, we analyzed the expression pattern of CA7-4 in different human cell lines (Figure S2(a)) and found CA7-4 expressed highly in VEC, Hela and A549 cells. Then we predicted the subcellular localization of CA7-4 via http://www.csbio.sjtu.edu.cn/bioinf/lncLocator (Figure S2(b)). RNA-fluorescence in situ hybridization (RNA-FISH) and nuclear/cytoplasmic RNA separation analysis revealed that CA7-4 distributed in both nucleus and cytoplasm of VECs (Figure S2(c,d)). Moreover, HG increased the RNA level of CA7-4 in the cytoplasm instead of the nucleus (Figure S2(e)). After treatment with 3BDO, the distribution of CA7-4 in the cytoplasm was significantly reduced(Figure S2(f,g)).
Overexpression CA7-4 promoted high glucose-induced VEC autophagy
To clarify the function of CA7-4 in VECs, we cloned full-length CA7-4 into the pcDNA3.1 plasmid (pcDNA3.1-CA7-4) and synthesized specific siRNAs against CA7-4. Then the overexpression and interference efficiency of CA7-4 was assessed by qPCR analysis (Figure 2(a,b)). In VECs, after overexpression of CA7-4 and treatment with high glucose, we detected the distribution of LC3B (microtubule associated protein 1 light chain 3 beta) by immunofluorescence. CA7-4 substantially increased LC3B puncta and the protein level of LC3B-II in VECs with high glucose treatment (Figure 2(c,d)) and without high glucose treatment (Figure 2(e)). 3BDO inhibited the upregulation of LC3B-II induced by CA7-4 (Figure 2(f)). Thus, CA7-4 aggravated VEC autophagy. We found that cells containing a large number of autophagosomes were also accompanied with nuclear shrinkage, so CA7-4 may also regulate VEC apoptosis.
Figure 2.
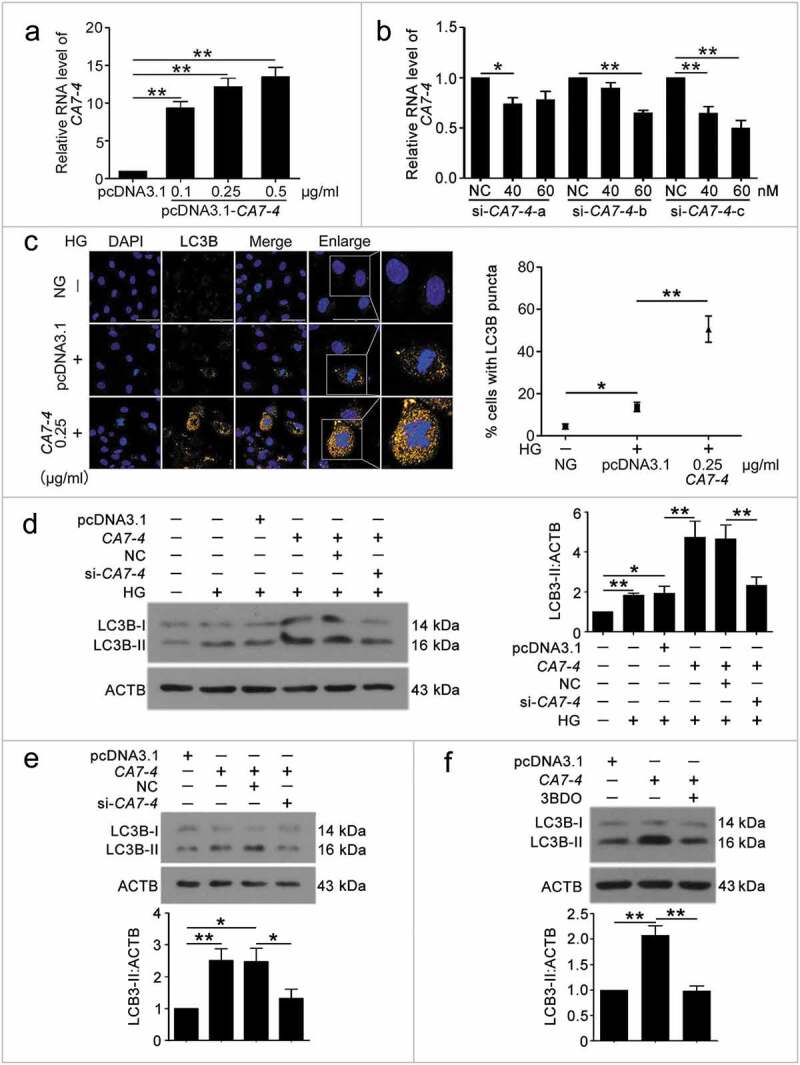
CA7-4 encouraged VEC autophagy. (a) VECs were transfected with empty vector pcDNA3.1 and CA7-4 (0.1, 0.25, 0.5 μg/ml) for 48 h. qPCR analysis of CA7-4 overexpression efficiency. (b) VECs were transfected with NC or siRNA-CA7-4-a, siRNA-CA7-4-b, and siRNA-CA7-4-c for 48 h. qPCR analysis of the RNA level of CA7-4. (c) After transfection with pcDNA3.1 or CA7-4 (0.25 μg/ml) for 24 h, VECs were treated with HG for 48 h. The distribution of LC3B in VECs was detected by immunofluorescence, and the proportion of cells containing > 5 LC3B puncta was investigated. Scale bar: 50 μm. Rescue experiment: VECs were transfected with pcDNA3.1 or CA7-4, then co-transfected with CA7-4 (0.1 μg/ml) and NC; CA7-4 (0.1 μg/ml) and si-CA7-4 (60 nM) for 24 h (d) or 48 h (e), then treated with HG (d) for 48 h. (f) VECs were treated with 3BDO (15 μM) for 48 h after transfection with CA7-4 (0.1 μg/ml) overnight. Western blot analysis of LC3B-II level. (*, p < 0.05; **, p < 0.01; n = 3.).
High glucose-induced VEC apoptosis was aggravated by CA7-4
To detect whether CA7-4 affected VEC apoptosis, we treated cells with high glucose after transfection with CA7-4 and si-CA7-4. CA7-4 heightened the levels of BAX and cleaved-PARP1 (Figure 3(a,b)); these observations could be reversed by CA7-4 knockdown (Figure 3(c,d)). Then we used Hoechst 33258 staining analysis after overexpression of CA7-4 and found that CA7-4 favored VEC apoptosis (Figure 3(e)). Also, CA7-4 increased the level of cleaved-CASP3 in VECs, whereas CA7-4 knockdown reversed the result (Figure 3(f)). Hence, CA7-4 increased VEC apoptosis directly.
Figure 3.
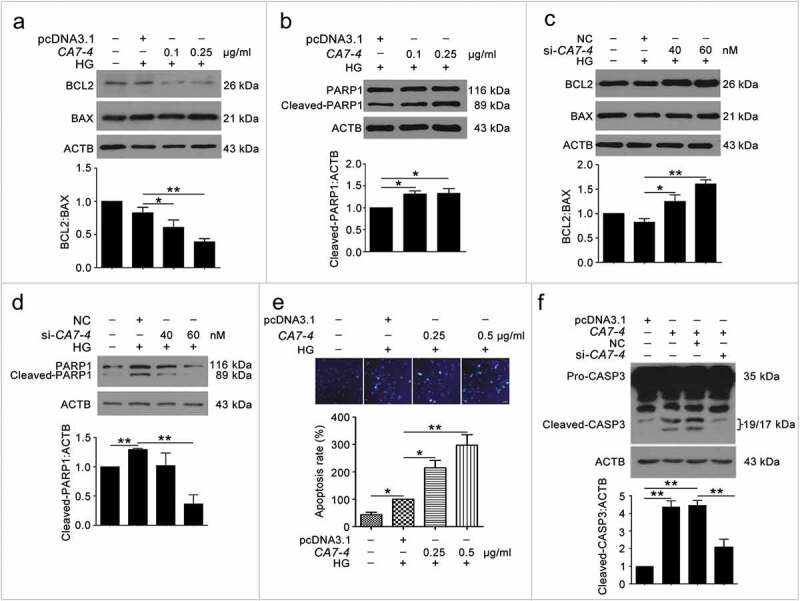
Overexpressing CA7-4 increased VEC apoptosis. (a–d) After being transfected with pcDNA3.1 or CA7-4; NC or si-CA7-4 overnight, VECs were treated with HG for 48 h, and the protein levels of BCL2, BAX and cleaved-PARP1 were detected by western blot. (e) VECs were treated with HG for 48 h after overexpression of CA7-4. Hoechst 33258 staining analysis of cell apoptosis. Scale bar: 30 μm. (f) VECs were transfected with pcDNA3.1 or CA7-4; co-transfected with CA7-4 (0.1 μg/ml) and NC or si-CA7-4 (60 nM) for 48 h. The level of cleaved-CASP3 was detected by western blot. (*, p < 0.05; **, p < 0.01; n = 3.).
CA7-4 decoyed MIR877-3P and MIR5680 as a sponge RNA
Studies have shown that lncRNAs affect mRNA stability by competing endogenous RNAs (ceRNAs) [28]. To determine the mechanism of CA7-4 boosting autophagy and apoptosis in VECs, we predicted miRNAs that may combine with CA7-4 by bioinformatics analysis (http://starbase.sysu.edu.cn/mirLncRNA.php) and found MIR877-3P, MIR5680, MIR4778-3P, MIR561-3P with higher binding scores. CA7-4 overexpression decreased the levels of these miRNAs (Figures 4(a,c), S3(a,c)), whereas CA7-4 knockdown elevated the levels (Figures 4(b,d), S3(b,d)), and 3BDO promoted the expression of MIR877-3P and MIR5680 especially (Figure 4(e–h)). Furthermore, we predicted the possible sites for MIR877-3P, MIR5680, MIR4778-3P, and MIR561-3P bonding with CA7-4, with putative sites at positions 1302 to 1322, 863 to 884, 2316 to 2337 and 2323 to 2344, respectively. Luciferase activity was reduced after co-transfection with Luc-CA7-4-WT plasmid and MIR877-3P or MIR5680 mimics into HEK293T cells as compared with the negative control or plasmids containing the mutant sequences of presumptive binding sites (Figure 4(i,j)). However, luciferase activity did not differ after co-transfection with Luc-CA7-4-WT plasmid and MIR4778-3P or MIR561-3P mimics as compared with the negative control (Figure S3(e,f)). As we know, microRNAs fail to modulate target mRNAs when ceRNAs bind to microRNA-induced silencing complex (RISC) as microRNA sponges [29]. AGO2 (argonaute RISC catalytic component 2) is the core component of RISC, RNA-binding protein immunoprecipitation (RIP) experiment demonstrated the existence of CA7-4 in the anti-AGO2 group under the treatments of NG, 3BDO, HG, HG+3BDO (Figure 4(k–m)), and 3BDO conspicuously decreased the enrichment of CA7-4 in AGO2 (Figure 4(l,m)). So, CA7-4 directly combined with MIR877-3P and MIR5680 as a miRNA decoy.
Figure 4.
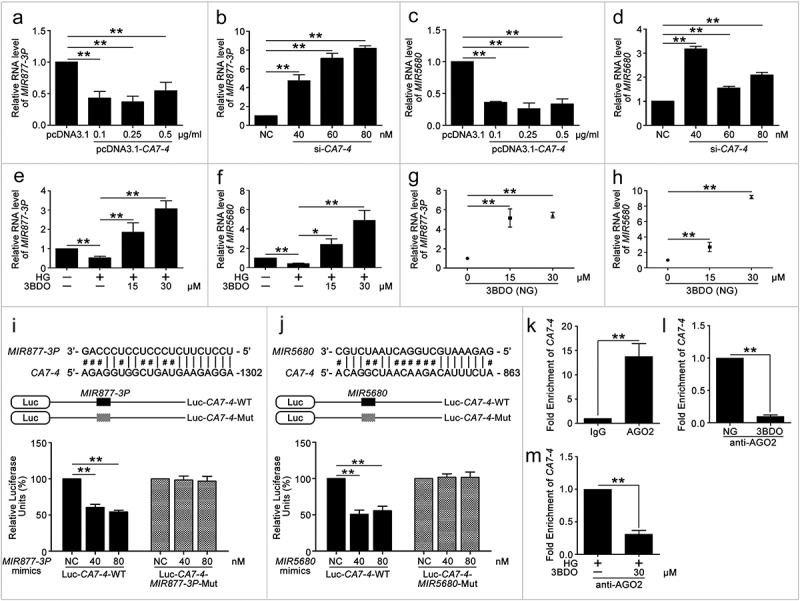
CA7-4 directly bonded with MIR877-3P and MIR5680. (a–d) qPCR analysis of MIR877-3P and MIR5680 in VECs after overexpression or knockdown CA7-4 for 48 h. (e–h) VECs were treated with NG, HG, HG+3BDO, or 3BDO (NG) for 24 h, qPCR analysis of MIR877-3P and MIR5680. (i,j) Luc-CA7-WT, Luc-CA7-4-MIR877-3P-Mut, and Luc-CA7-4- MIR5680-Mut plasmids were separately co-transfected into HEK293T cells with NC, MIR877-3P mimics or MIR5680 mimics for 24 h. Detecting luciferase activity. (k–m) RNA binding protein immunoprecipitation experiments were performed by using AGO2 antibody in VECs after treatment with NG, 3BDO (30 μM), HG and HG+3BDO (30 μM) for 24 h. IgG was used as a negative control. The fold enrichment of pulled-down CA7-4 was detected by qPCR analysis. (*, p < 0.05; **, p < 0.01; n = 3.).
MIR877-3P and MIR5680 suppressed VEC autophagy and apoptosis
To understand the mechanism of CA7-4 regulating VEC autophagy and apoptosis, we considered MIR877-3P and MIR5680 as the point of penetration. First, RNA-FISH revealed that MIR877-3P and MIR5680 distributed in both nucleus and cytoplasm of VECs (Figure S4). Next, we transfected VECs with MIR877-3P and MIR5680 mimics or inhibitor overnight and then incubated cells with high glucose. LC3B-II level and LC3B immunofluorescence indicated that MIR877-3P (Figure 5(a,b)) and MIR5680 (Figure 6(a–c)) protected VECs against high glucose-caused autophagy. Simultaneously, according to TUNEL assay and Hoechst 33258 staining, MIR877-3P (Figure 5(c–e)) and MIR5680 (Figure 6(d)) alleviated VEC apoptosis. Furthermore, in normal cultured VECs, MIR877-3P (Figure S6) and MIR5680 (Figure S7) had a parallel effect on autophagy and apoptosis. Therefore, MIR877-3P and MIR5680 weakened VEC autophagy and apoptosis directly.
Figure 5.
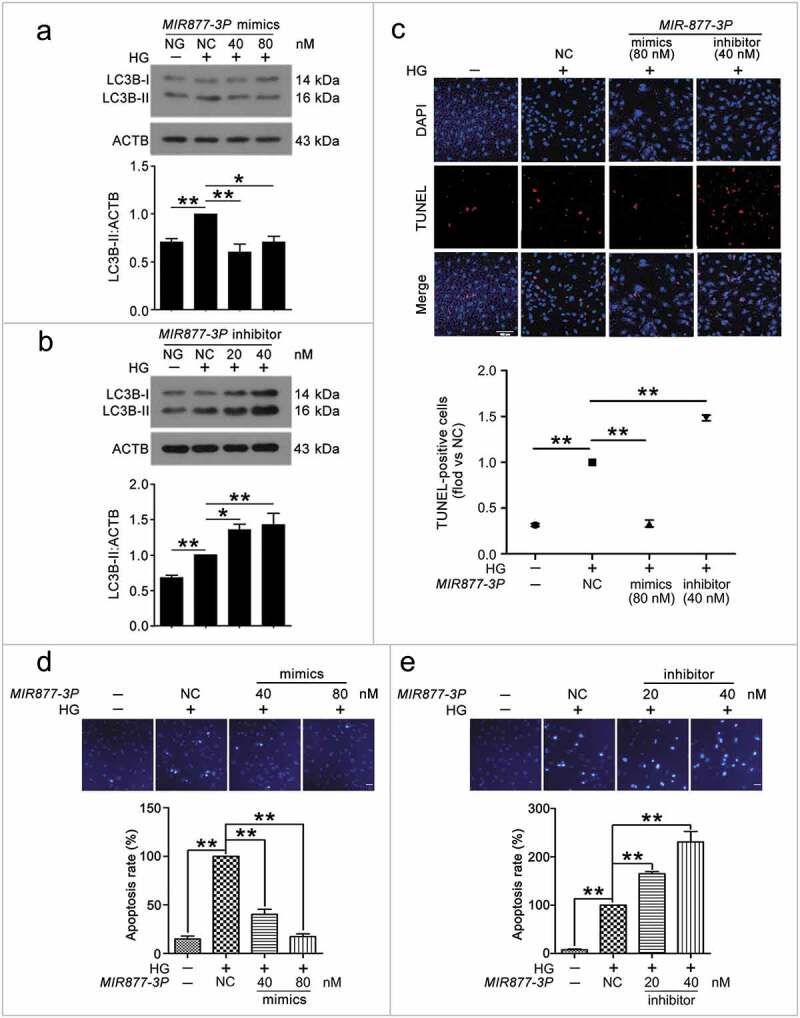
MIR877-3P inhibited VEC autophagy and apoptosis. After transfection with MIR877-3P mimics or inhibitor overnight, VECs were treated with HG for 48 h. (a,b) Western blot analysis of LC3B-II level. (c) TUNEL staining analysis of cell apoptosis. Scale bar: 100 μm. (d,e) Cell apoptosis was detected by Hoechst 33258 staining. Scale bar: 30 μm. (*, p < 0.05; **, p < 0.01; n = 3.).
Figure 6.
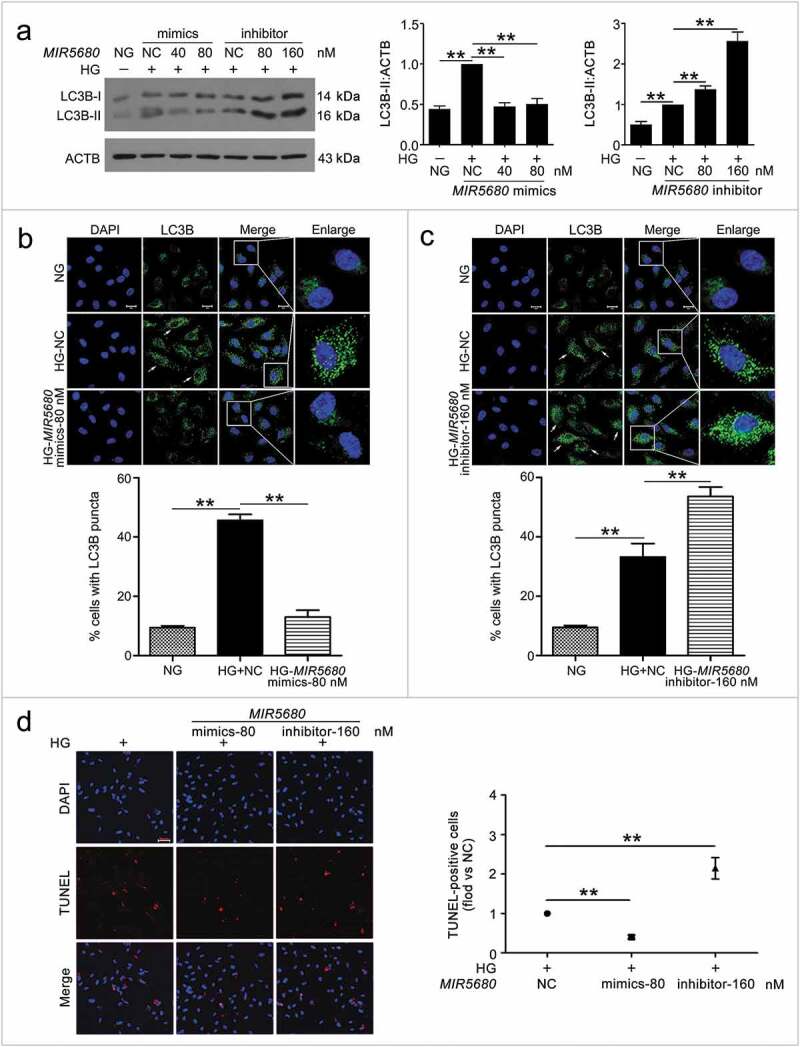
MIR5680 prevented VEC autophagy and apoptosis. VECs were treated with HG for 48 h after transfection with MIR5680 mimics or inhibitor overnight. (a) Western blot analysis of LC3B-II level. (b,c) Immunofluorescence analysis of the distribution of LC3B in VECs, and the proportion of cells containing > 5 LC3B puncta was analyzed. Scale bar: 20 μm. (d) TUNEL staining analysis of cell apoptosis. Scale bar: 50 μm. (*, p < 0.05; **, p < 0.01; n = 3.).
MIR877-3P and MIR5680 targeted the 3ʹ UTRs of CTNNBIP1 and DPP4 respectively
Because CA7-4 increased VEC autophagy and apoptosis by sponging MIR877-3P and MIR5680, we predicted the possible targets of MIR877-3P and MIR5680 via miRDB-MicroRNA Target Prediction and Functional Study Database (http://www.mirdb.org/). Among the predicted target genes of MIR877-3P and MIR5680, CTNNBIP1 (catenin beta interacting protein 1) and DPP4 (dipeptidyl peptidase 4) strongly drew our attention (Table S1). CTNNBIP1 injures T-cell development by contributing to T-cell apoptosis and destroying the interactions between CTNNB1 (catenin beta 1) and T-cell factor [30]. However, we have no data on the association between CTNNBIP1 and autophagy. Research has illuminated that inhibitors of DPP4 show excellent anti-hyperglycemic performance, superior to traditional treatment, especially for older people and women [31]. Moreover, as one of the DPP4 inhibitors, sitagliptin contributes to protect mesenchymal stem cells against hypoxia-induced apoptosis and autophagy [32]. Therefore, we explored whether MIR877-3P and MIR5680 targeted and affected CTNNBIP1 and DPP4, respectively.
We found that positions 2368 to 2388 contained the binding sites of MIR877-3P and CTNNBIP1 3ʹ UTR (Figure 7(a)). Likewise, according to the prediction results, MIR5680 has two putative DPP4 3ʹ UTR binding sites at positions 225 to 246 and 845 to 866 (Figure 8(a)). Luciferase assays showed that MIR877-3P mimics co-transfection with Luc-CTNNBIP1 3ʹ UTR-WT plasmid repressing luciferase activity significantly as compared with transfection of the negative control or Luc-CTNNBIP1 3ʹ UTR-Mut (Figure 7(b)). Also, MIR5680 mimics weakened luciferase activity of Luc-DPP4 3ʹUTR-WT instead of Luc-DPP4 3ʹ UTR-Mut (Figure 8(b)). Hence, MIR877-3P and MIR5680 directly targeted CTNNBIP1 and DPP4 3ʹ UTRs, respectively.
Figure 7.
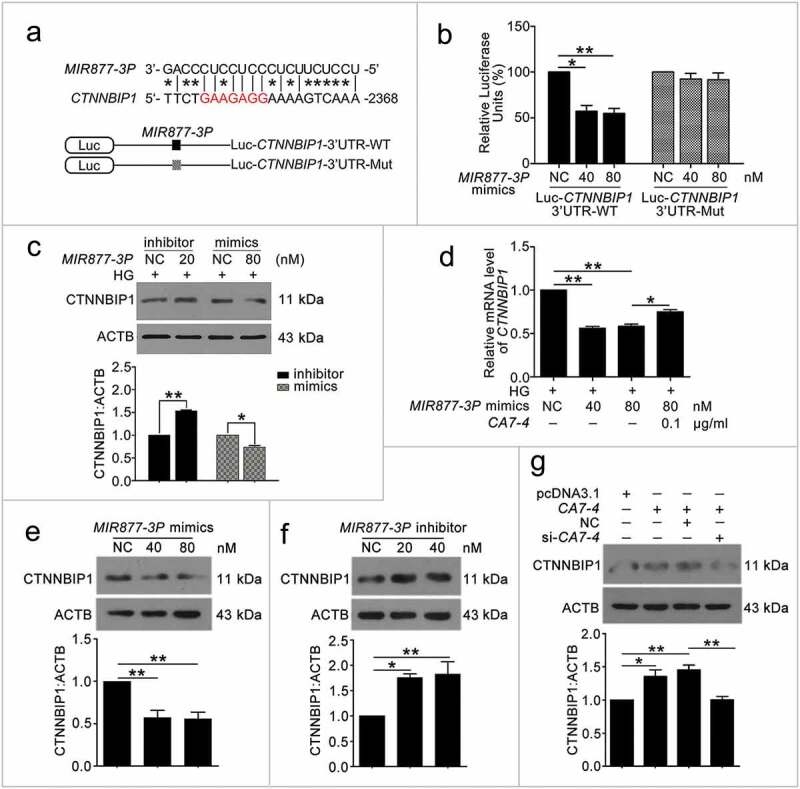
MIR877-3P targeted and inhibited CTNNBIP1. (a) Potential bind sites of MIR877-3P in CTNNBIP1 3ʹ UTR. (b) Luc-CTNNBIP1 3ʹ UTR-WT or Luc-CTNNBIP1 3ʹ UTR-Mut plasmid was co-transfected with NC or MIR877-3P mimics into HEK293T cells for 24 h. Luciferase activity was determined. (c) After transfection with NC, MIR877-3P inhibitor or mimics overnight, VECs were treated with HG for 48 h. Western blot analysis of CTNNBIP1. (d) VECs were treated with HG for 48 h after transfection with MIR877-3P mimics or co-transfection with MIR877-3P mimics and CA7-4. qPCR analysis of CTNNBIP1 mRNA level. (e,f) VECs were transfected with NC, MIR877-3P mimics or inhibitor for 48 h. (g) VECs were transfected with pcDNA3.1 or CA7-4 (0.1 μg/ml); co-transfected with CA7-4 (0.1 μg/ml) and NC or si-CA7-4 (60 nM) for 48 h. Western blot analysis of CTNNBIP1. (*, p < 0.05; **, p < 0.01; n = 3.).
Figure 8.
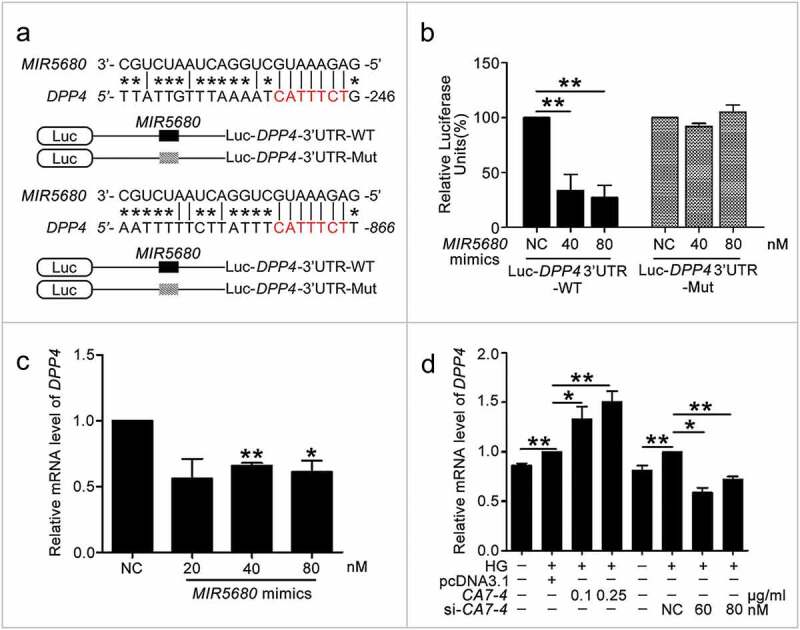
MIR5680 targeted DPP4 and decreased the mRNA level of it. (a) Possible binding sites of MIR5680 in DPP4 3ʹ UTR. (b) Luc-DPP4 3ʹ UTR-WT or Luc-DPP4 3ʹ UTR-Mut plasmids were co-transfected with NC or MIR5680 mimics into HEK293T cells for 24 h. Luciferase activity was determined. (c) After transfection with NC, MIR5680 mimics for 48 h, detecting the mRNA level of DPP4 by qPCR analysis. (d) VECs were treated with HG for 48 h after overexpression or knockdown of CA7-4. qPCR analysis of DPP4 mRNA level. (*, p < 0.05; **, p < 0.01; n = 3.).
MIR877-3P, MIR5680 and 3BDO separately downregulated CTNNBIP1 and DPP4; CA7-4 upregulated CTNNBIP1 and DPP4 by sponging MIR877-3P and MIR5680
We investigated whether MIR877-3P and MIR5680 affected CTNNBIP1 and DPP4 expression. The protein level of CTNNBIP1 was promoted by MIR877-3P inhibitor but decreased by MIR877-3P mimics under high glucose (Figure 7(c)). Additionally, MIR877-3P mimics downregulated the mRNA level of CTNNBIP1, whereas overexpression CA7-4 abolished this downregulation (Figure 7(d)). In normal VECs, MIR877-3P mimics (Figure 7(e)) inhibited the protein level of CTNNBIP1, whereas the MIR877-3P inhibitor (Figure 7(f)) and CA7-4 increased it (Figure 7(g)). Additionally, MIR5680 mimics decreased the expression of DPP4 at the levels of transcription (Figure 8(c)) and translation with or without high glucose treatment (Figure 9(a,d)). MIR5680 inhibitor (Figure 9(b,e)) or CA7-4 (Figures 8(d), 9(c)) transfection had the opposite results. As well, elevated CTNNBIP1 and DPP4 levels caused by high glucose decreased after treatment with siRNA-CA7-4 or 3BDO (Figure S8). Therefore, we verified that MIR877-3P, MIR5680, 3BDO negatively modulated and CA7-4 promoted the expression of CTNNBIP1 and DPP4.
Figure 9.
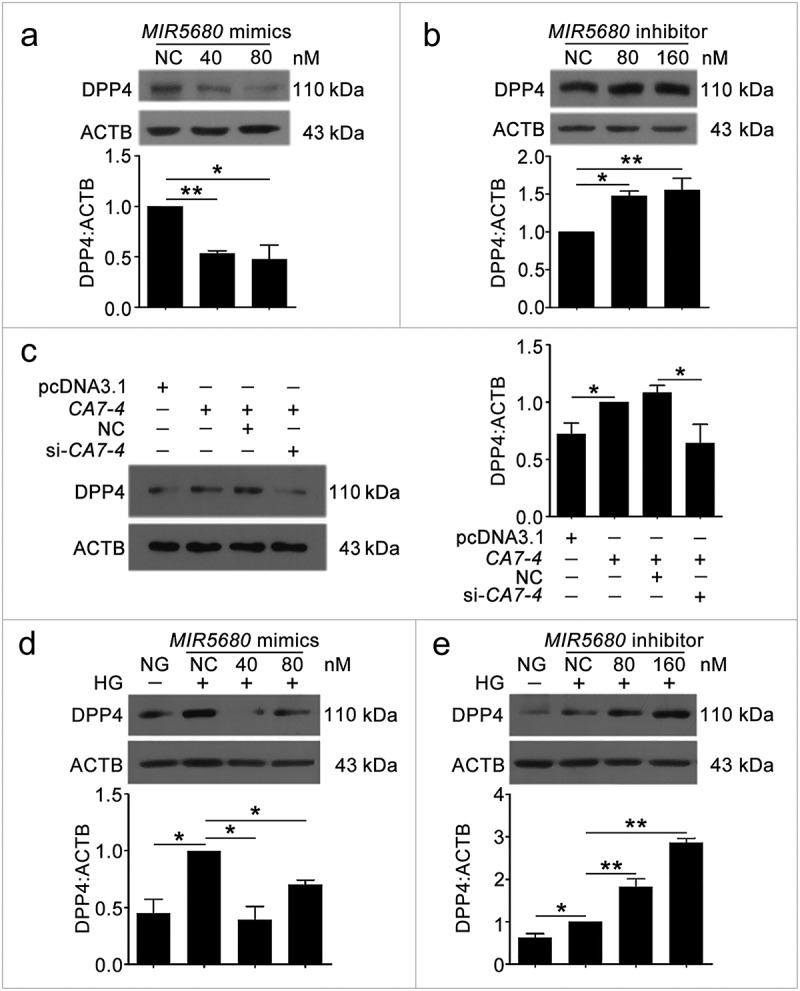
The protein level of DPP4 was negatively regulated by MIR5680. (a,b) VECs were transfected with MIR5680 mimics or inhibitor for 48 h. (c) VECs were transfected with pcDNA3.1 or CA7-4 (0.1 μg/ml); co-transfected with CA7-4 (0.1 μg/ml) and NC or si-CA7-4 (60 nM) for 48 h. (d,e) After transfection with NC, MIR5680 mimics or inhibitor overnight, VECs were treated with HG for 48 h. Western blot analysis of DPP4 level. (*, p < 0.05; **, p < 0.01; n = 3.).
CTNNBIP1 and DPP4 aggravated VEC autophagy and apoptosis under high glucose by inhibiting CTNNB1 expression and improving AMPK phosphorylation, respectively
CTNNBIP1 works as a key negative regulator of CTNNB1 and affects cell fate [33]. Thus, we analyzed whether MIR877-3P regulated the level of CTNNB1. After transfecting VECs with MIR877-3P mimics or inhibitor, we detected the protein level of CTNNB1 (Figure S9(a)) and found that MIR877-3P enhanced CTNNB1 expression. We also conducted a rescue experiment to explore whether CA7-4 affected the expression of CTNNB1. High glucose and overexpression of CA7-4 negatively regulated CTNNB1, but the protein level of CTNNB1 recovered after knockdown CA7-4, transfection with MIR877-3P mimics or treatment with 3BDO (Figure S9(b–d)).
AMPK regulates cellular metabolism as an energy sensor, and its activation leads to β-cell apoptosis [34]. In addition, AMPK is famous for activating autophagy to control cellular energy homeostasis through the AMPK/MTOR pathway [35]. Here, we hypothesized that MIR5680 may have an impact on AMPK phosphorylation by restraining the expression of DPP4, then regulating autophagy in VECs. The data revealed that MIR5680 suppressed AMPK phosphorylation (Figure S10). Also, we found that CA7-4 overexpression-upregulated AMPK phosphorylation could be abolished by 3BDO (Figure S10(c)).
To verify the roles of CTNNBIP1 and DPP4 in high glucose-induced VEC autophagy and apoptosis, we used siRNA knockdown and examined the protein levels of CTNNBIP1, CTNNB1, DPP4, p-AMPK, cleaved-CASP3, and LC3B-II (Figure 10). The results suggested that CTNNBIP1 reduced CTNNB1 expression and DPP4 promoted AMPK phosphorylation to enhance VEC autophagy and apoptosis.
In conclusion, high glucose-induced CA7-4 upregulation, VEC autophagy and apoptosis could be inhibited by the small chemical molecule 3BDO. As a competing endogenous RNA, CA7-4 decoyed MIR877-3P and MIR5680. MIR877-3P and MIR5680 protected VECs against high glucose-caused autophagy and apoptosis by targeting the 3ʹ UTRs of CTNNBIP1 and DPP4, negatively regulating their levels, as well as promoting the expression of CTNNB1 and downregulating AMPK phosphorylation (Figure 11).
Figure 11.
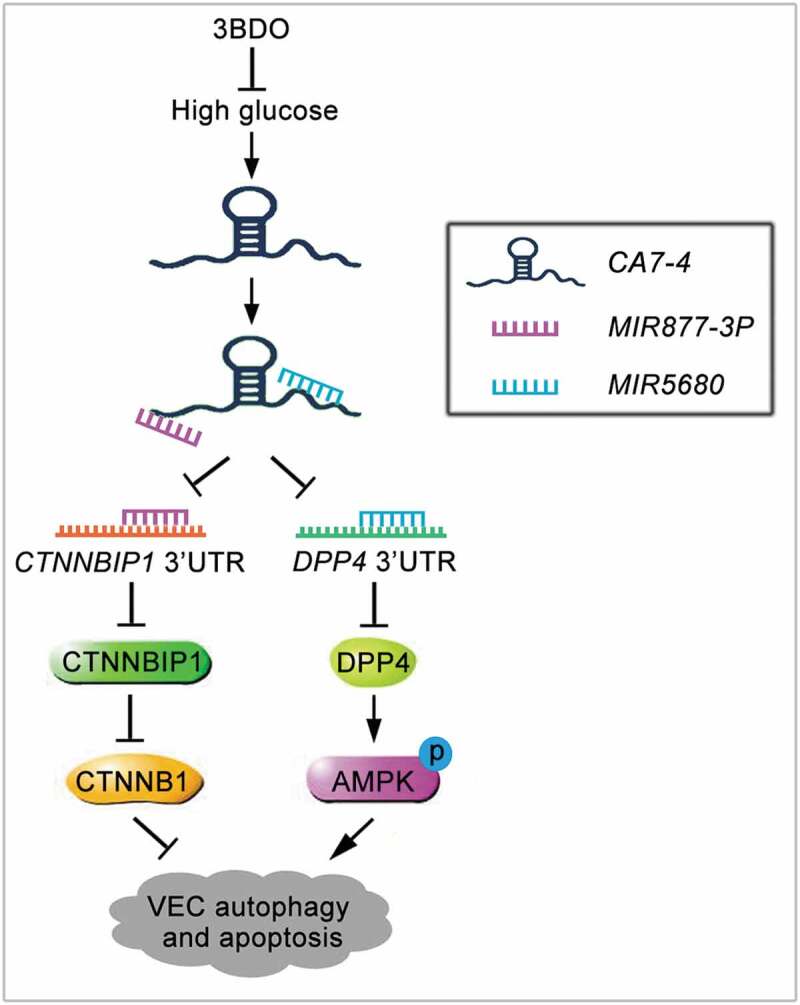
Schematic of CA7-4 regulating VEC autophagy and apoptosis caused by high glucose. High glucose induces VEC autophagy and apoptosis. In this progress, upregulated CA7-4 as a miRNA sponge to decoy MIR877-3P and MIR5680, promote the expression of CTNNBIP1 and DPP4, decrease the level of CTNNB1 and increase AMPK phosphorylation. 3BDO can attenuate high glucose-induced VEC autophagy and apoptosis by weakening the upregulation of CA7-4.
Discussion
Normal endothelium function is essential for vascular homeostasis and remodeling. For decades, hyperglycaemia induced by diabetes has been reported to destroy the function of endothelial cells and result in vascular complications [36]. As a consequence, avoiding endothelial cell injury is the purpose of diabetic vascular complications therapy. A spate of reports show that lncRNAs involved in the pathogenesis of various human diseases can work as decoys, guides, signals, or scaffolds to combine DNA, RNA, or protein and consequently exert diverse biological functions [16]. According to our study, 3BDO inhibited high glucose-induced VEC autophagy and apoptosis (Figure S1). LncRNA CA7-4, which was upregulated by high glucose, could be supressed by 3BDO (Figure 1). The function of CA7-4 lacks evidence, particularly in endothelial cells during the development of hyperglycemia. We found that high glucose-induced autophagy and apoptosis of VECs sharpened with overexpression of CA7-4 and its knockdown had the reverse effect (Figures 2, 3).
A number of lncRNAs are dysregulated in vascular cells under the circumstance of high glucose [37]. However, most have been identified to participate in regulating vascular dysfunction, as well as vascular apoptosis and inflammation, with no study about lncRNA regulating high glucose-induced VEC autophagy. Therefore, CA7-4 discovered to promote VEC autophagy and apoptosis is of significance.
LncRNAs can function as ceRNAs. The crosstalk between lncRNAs, miRNAs and mRNA targets represents a complex network of gene expression regulation [38]. Hence, to explore the mechanism of CA7-4 regulation in high glucose-induced VEC autophagy and apoptosis, we used bioinformatics prediction and searched for miRNAs that CA7-4 might combine with. Further study showed that CA7-4 could decoy MIR877-3P and MIR5680, and the levels of MIR877-3P and MIR5680 were increased after treatment with 3BDO (Figure 4).
Previous study indicates that according to miRNA profiling, MIR877-3P is upregulated in earlier type II diabetic kidney disease [39]. MIR877-3P also plays important roles in else several diseases, including nephropathy [40], lung fibrosis [41], bladder cancer [42] and inflammatory [43]. However, the effect of MIR877-3P on VEC damage induced by high glucose had not been studied. Additionally, there were no reports on MIR5680 functions. Here, MIR877-3P and MIR5680 prevented VEC autophagy and apoptosis with or without high glucose treatment (Figures 5, 6, S6, S7). Then we predicted and detected the target genes of MIR877-3P and MIR5680 (Table S1). The data suggested that MIR877-3P and MIR5680 targeted CTNNBIP1 (Figure 7) and DPP4 (Figures 8, 9), respectively, and inhibited their expression at the transcription and translation levels, whereas CA7-4 weakened the functions of MIR877-3P (Figure 7(d,g)) and MIR5680 (Figures 8(d), 9(c)). In addition, treatment with si-CA7-4 and 3BDO decreased the upregulation of CTNNBIP1 and DPP4 induced by high glucose (Figure S8).
CTNNBIP1 induces cell apoptosis by suppressing CTNNB1 activity [44], but no study has investigated the relationship between CTNNBIP1 and autophagy. DPP4, also known as CD26, can be expressed as a soluble enzyme in vascular beds including endothelial cells [45]. GLP1 (Glucagon like peptide-1), a substrate that belongs to DPP4, can promote insulin secretion and reduce glucagon production as a kind of incretin hormone. The increased DPP4 level leads to hyperglycemia by suppressing GLP1 [46]. As a kind of DPP4 inhibitor, sitagliptin reduces high glucose-caused apoptosis by activating AMPK in endothelial cells [47]. As well, sitagliptin alleviates hypoxia-induced autophagy and apoptosis by suppressing DPP4 to protect mesenchymal stem cells [32]. So, we investigated how DPP4 affected AMPK activity during VEC autophagy and apoptosis caused by high glucose.
First, we analyzed whether MIR877-3P affected the expression of CTNNB1 and whether MIR5680 affected AMPK phosphorylation. MIR877-3P or 3BDO increased the level of CTNNB1, and CA7-4 downregulated it (Figure S9). The phosphorylation of AMPK was inhibited by MIR5680 or 3BDO, and overexpression CA7-4 reversed the result (Figure S10). To understand how CTNNBIP1 and DPP4 affect high glucose-induced VEC autophagy and apoptosis, we transfected cells with si-CTNNBIP1 and si-DPP4. CTNNBIP1 and DPP4 knockdown reduced VEC autophagy and apoptosis under HG (Figure 10). Hence, we found novel pathways regulating VEC autophagy and apoptosis: MIR877-3P/CTNNBIP1/CTNNB1 and MIR5680/DPP4/AMPK.
Figure 10.
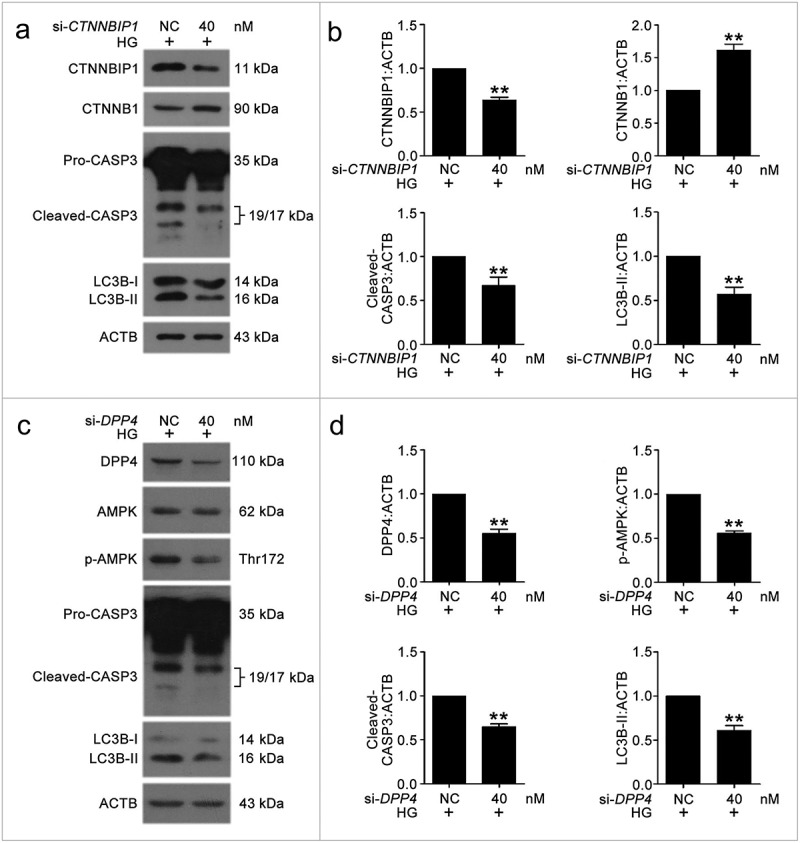
The knockdown of CTNNBIP1 and DPP4 reduced VEC autophagy and apoptosis caused by high glucose. (a–d) After transfection with NC, si-CTNNBIP1 or si-DPP4 overnight, VECs were treated with high glucose for 48 h. The protein levels of CTNNBIP1, CTNNB1, cleaved-CASP3, LC3B-II, DPP4, p-AMPK were analyzed by western blot. (*, p < 0.05; **, p < 0.01; n = 3.).
In summary, under high glucose, 3BDO attenuates VEC autophagy and apoptosis by weakening the upregulation of CA7-4. As a ceRNA, CA7-4 decoys MIR877-3P and MIR5680, promotes the expression of CTNNBIP1 and DPP4, decreases the level of CTNNB1 and increases AMPK phosphorylation, thereby aggravating VEC autophagy and apoptosis (Figure 11). Therefore, CA7-4 might become an attractive target against autophagy and apoptosis of vascular endothelial cells.
Materials and methods
Cell culture
Source of VECs from human umbilical vein was consistent with the previous description [48]. The VECs were grown in M199 medium (Gibco, 31100-035) with 10% (V/V) bovine calf serum (Sigma-Aldrich, 13063C) and 8.4 IU/mL FGF2. HEK293T cells stemed from the Cell Bank of the Chinese Academy of Sciences (Shanghai) and grew in DMEM-H medium (Gibco, 12800017) with 10% fetal bovine serum (Gibco, 10270-106). H9C2, HepG2, PC3 and U87 cells were grown in DMEM-H medium with 10% fetal bovine serum. Hela cells were grown in DMEM-H medium with 10% bovine calf serum. A549 cells were grown in RPMI-1640 (Gibco, 31800089) medium with 10% bovine calf serum. HCT116 and THP-1 cells were grown in RPMI-1640 medium with 10% fetal bovine serum. All cell lines were cultured in a humidified incubator at 37°C with 5% CO2.
Immunofluorescence assay
Immunofluorescence assay was performed as described previously [49]. First, VECs were fixed with 4% paraformaldehyde for 15 min and washed 3 times with 0.1 M phosphate-buffered saline (PBS: 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, pH 7.4), then permeabilized with 0.1% Triton X-100 (Sangon Biotech, A600198). Cells were washed 3 times with 0.1 M PBS. At room temperature, 10% normal donkey serum (Solarbio, SL050) was applied to block cells for 20 min. Then VECs were incubated with LC3B primary antibody (1:100) (Cell Signaling Technology, 2775S) at 4°C overnight and corresponding secondary antibody (1:200) at 37°C for 1 h. Cells were washed 3 times with 0.1 M PBS. After treatment with DAPI (4’, 6-diamidino-2-phenylindole) (Sigma-Aldrich, D8417) for 10 min, cells were washed 3 times with 0.1 M PBS, then photographed by confocal fluorescence microscopy Zeiss LSM700 (Germany).
Western blot analysis
Cells were lysed in RIPA lysis buffer (Beyotime, P0013B) containing protease inhibitor cocktail (Sigma-Aldrich, P8340) after being washed twice with 0.1 M PBS. Lysates were centrifuged at 12,000 × g, 4°C for 15 min, collecting supernatant. Bicinchoninic acid (BCA) protein assay kit (Beyotime, P0011) was used for measuring protein concentration. After 15% or 12% SDS-PAGE at 4°C, protein was transferred to polyvinylidene difluoride (PVDF) membrane (Millipore, IPFL00010). The membrane was blocked with 5% non-fat milk for 1 h at room temperature, incubated with primary antibodies: PARP1 (Cell Signaling Technology, 9542S); LC3B (Cell Signaling Technology, 2775S); BCL2 (Proteintech, 12789-1-AP); BAX (Proteintech, 50599-2-Ig); CASP3 (Cell Signaling Technology, 9662S); ACTB (Sigma-Aldrich, A-5441); DPP4 (Abcam, ab215711); AMPK (Cell Signaling Technology, 5832S); p-AMPK (Cell Signaling Technology, 2535S); SQSTM1 (BD, 610833); CTNNBIP1 (Abcam, ab129011); CTNNB1 (Proteintech, 51067-2-AP), 1:1000 in TBS-Tween 20 (0.1%) at 4°C overnight (0.1 M TBS: 3 g Tris, 0.2 g KCl, 8 g NaCl, pH 7.4), washed TBST three times, then incubated with horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch, goat anti-rabbit: 111-035-003, goat anti-mouse: 115-035-003) for 1 h at room temperature. Fluorescence signals were detected by X-ray films after being incubated with HRP substrate for 3 min. The expression of protein was quantified by ImageJ software.
Quantitative real-time PCR
Total RNA was purified by means of Trizol reagent (TAKARA, 9109). PARIS™ Kit (Invitrogen, AM1921) was utilized to isolate nuclear and cytoplasmic RNA according to the manufacturer’s instructions. Following the PrimeScript RT reagent kit’s (TAKARA, DRR047) protocol, RNA (1 μg) was reverse-transcribed to cDNA. Quantitative real-time PCR was implemented in a 20 μl volume containing 10 μl EvaGreen 2 × qPCR MasterMix (Applied Biological Materials, MasterMix-S), forward primer, reverse primer, template DNA, and nuclease-free H2O. Relative gene expression was normalized to that of actin beta. Additionally, the levels of MIR877-3P, MIR5680, MIR4778-3P and MIR561-3P were analyzed by Hairpin-it microRNA and RNU6 snRNA Normalization RT-PCR Quantitation Kit (GenePharma, E22001-E22010). Relative gene expression was normalized to that of RNU6 snRNA. Primer information is provided in the supplemental material.
Transient transfection
CA7-4 overexpression full-length plasmid (pcDNA3.1-CA7-4) was obtained by inserting the full length of CA7-4 into pcDNA3.1 vector. Control: pcDNA3.1-empty vector. Specific small interfering RNAs, including CA7-4, CTNNBIP1, DPP4, scramble siRNA (negative control); miRNA mimics and inhibitors for MIR877-3P, MIR5680, MIR4778-3P, and MIR561-3P were designed and synthesized by Invitrogen. Plasmids were transfected by using Lipofectamine 2000 (Invitrogen, 11668-019) according to protocols. In addition, siRNAs, miRNA mimics and inhibitor were transfected by using HiPerFect Transfection Reagent (QIAGEN, 301705) according to the manufacturer’s instructions.
Hoechst 33258 staining
After treatment, VECs grown on 24 well plates were stained with 10 mg/ml Hoechst 33258 (Sangon Biotech, E607329) at 37°C for 15 min, avoid light. Cells were washed with PBS twice and photographed by using a fluorescence microscope (Japan).
Luciferase activity assay
Luciferase reporter plasmids were reconstructed by inserting target fragments into pmirGLO Dual-Luciferase miRNA Target Expression Vectors (dual-luciferase: firefly luciferase and renilla luciferase). Dual luciferase reporter plasmids of Luc-CA7-4-WT, Luc-CA7-4-MIR877-3P or MIR5680 or MIR4778-3P or MIR561-3P-Mut, Luc-CTNNBIP1 or DPP4-3ʹ UTR-WT, Luc-CTNNBIP1 or DPP4-3ʹ UTR-Mut, were used in the experiments. HEK293T cells were cultured onto 96-well plates at 10000 cells per well overnight in DMEM-H medium (Gibco, 12800017) with 10% fetal bovine serum, then co-transfected with plasmids of dual-luciferase reporters and miRNA mimics or NC, detecting dual-luciferase activity by using the Dual-Glo Luciferase Assay System Kit (Promega, E2920) according to the instructions. Firefly luciferase activity was normalized to renilla luciferase activity.
TUNEL assay
TUNEL assay was performed by using In Situ Cell Detection Kit (Roche Applied Science, 11767291910) according to the manufacturer’s instructions and photographed by laser scanning confocal microscopy (Zeiss LSM700, Germany).
RNA-binding protein immunoprecipitation (RIP) assay
RIP assay was operated with the Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Millipore, 17-701). Briefly, VECs were lysed, and incubated with magnetic beads conjugated with human anti-AGO2 antibody (Millipore, 03-110) or anti-mouse IgG (Millipore, 03-110). The immunoprecipitated RNA was detected by qPCR.
Statistical analysis
Experimental data are shown as means ± SEM from at least three independent experiments and were analyzed by using SPSS 17.0 (SPSS Inc., Chicago, IL, USA). P < 0.05 was considered statistically significant.
Funding Statement
This work was supported by the National Natural Science Foundation of China (No. 91539105, 81321061, 91313303).
Highlights
A small chemical molecule — 3 - benzyl −5 - ((2-nitrophenoxy) methyl) - dihydrofuran-2 (3H) - one (3BDO) — synthesized by us could simultaneously inhibit autophagy and apoptosis induced by a high concentration of glucose in vascular endothelial cells (VECs).
A novel long noncoding RNA called CA7-4 could promote VEC autophagy and apoptosis triggered by high glucose; 3BDO inhibited the role of CA7-4.
CA7-4 directly bonded with MIR877-3P and MIR5680 as a decoy.
We confirmed that MIR877-3P and MIR5680 targeted 3ʹUTRs of CTNNBIP1 and DPP4, respectively.
MIR877-3P inhibited high glucose-induced VEC autophagy and apoptosis by decreasing the expression of CTNNBIP1 and upregulating CTNNB1.
MIR5680 protected VECs against high glucose-induced autophagy and apoptosis by targeting DPP4 and weakening PRKAA1 phosphorylation.
Therefore, CA7-4, MIR877-3P and MIR5680 represent novel signal pathways regulating high glucose-induced autophagy and apoptosis in VECs.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Eelen G, de Zeeuw P, Simons M, et al. Endothelial cell metabolism in normal and diseased vasculature. Circ Res. 2015. March 27;116(7):1231–1244. PubMed PMID: 25814684; PubMed Central PMCID: PMC4380230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Zaitseva II, Berggren PO, Zaitsev SV.. Insulinotropic compounds decrease endothelial cell survival. Toxicol In Vitro. 2016. June;33:1–8. PubMed PMID: 26883446. [DOI] [PubMed] [Google Scholar]
- [3].Zhu M, Wen M, Sun X, et al. Propofol protects against high glucose-induced endothelial apoptosis and dysfunction in human umbilical vein endothelial cells. Anesth Analg. 2015. April;120(4):781–789. PubMed PMID: 25793913. [DOI] [PubMed] [Google Scholar]
- [4].Zhao Q, Gao C, Cui Z.. Ginkgolide A reduces inflammatory response in high-glucose-stimulated human umbilical vein endothelial cells through STAT3-mediated pathway. Int Immunopharmacol. 2015. April;25(2):242–248. PubMed PMID: 25681539. [DOI] [PubMed] [Google Scholar]
- [5].Jayakumar T, Chang CC, Lin SL, et al. Brazilin ameliorates high glucose-induced vascular inflammation via inhibiting ROS and CAMs production in human umbilical vein endothelial cells. Biomed Res Int. 2014;2014:403703. PubMed PMID: 24716195; PubMed Central PMCID: PMC3955648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chen F, Chen B, Xiao FQ, et al. Autophagy protects against senescence and apoptosis via the RAS-mitochondria in high-glucose-induced endothelial cells. Cell Physiol Biochem. 2014;33(4):1058–1074. PubMed PMID: 24732710. [DOI] [PubMed] [Google Scholar]
- [7].Sallam T, Sandhu J, Tontonoz P. Long noncoding RNA discovery in cardiovascular disease: decoding form to function. Circ Res. 2018. January 5;122(1):155–166. PubMed PMID: 29301847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jandura A, Krause HM. The new RNA world: growing evidence for long noncoding RNA functionality. Trends Genet. 2017. October;33(10):665–676. PubMed PMID: 28870653. [DOI] [PubMed] [Google Scholar]
- [9].Fok ET, Scholefield J, Fanucchi S, et al. The emerging molecular biology toolbox for the study of long noncoding RNA biology. Epigenomics. 2017. October;9(10):1317–1327. PubMed PMID: 28875715. [DOI] [PubMed] [Google Scholar]
- [10].Li L, Chen H, Gao Y, et al. Long noncoding RNA MALAT1 promotes aggressive pancreatic cancer proliferation and metastasis via the stimulation of autophagy. Mol Cancer Ther. 2016. September;15(9):2232–2243. PubMed PMID: 27371730. [DOI] [PubMed] [Google Scholar]
- [11].Chen CL, Tseng YW, Wu JC, et al. Suppression of hepatocellular carcinoma by baculovirus-mediated expression of long non-coding RNA PTENP1 and MicroRNA regulation. Biomaterials. 2015. March;44:71–81. PubMed PMID: 25617127. [DOI] [PubMed] [Google Scholar]
- [12].Schmitz SU, Grote P, Herrmann BG. Mechanisms of long noncoding RNA function in development and disease. Cell Mol Life Sci. 2016. July;73(13):2491–2509. PubMed PMID: 27007508; PubMed Central PMCID: PMC4894931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ballantyne MD, McDonald RA, Baker AH. lncRNA/MicroRNA interactions in the vasculature. Clin Pharmacol Ther. 2016. May;99(5):494–501. PubMed PMID: 26910520; PubMed Central PMCID: PMC4881297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wang K, Liu CY, Zhou LY, et al. APF lncRNA regulates autophagy and myocardial infarction by targeting miR-188-3p. Nat Commun. 2015. April 10;6:6779. PubMed PMID: 25858075. [DOI] [PubMed] [Google Scholar]
- [15].Viereck J, Kumarswamy R, Foinquinos A, et al. Long noncoding RNA chast promotes cardiac remodeling. Sci Transl Med. 2016. February 17;8(326):326ra22. PubMed PMID: 26888430. [DOI] [PubMed] [Google Scholar]
- [16].Leti F, DiStefano JK. Long noncoding RNAs as diagnostic and therapeutic targets in type 2 diabetes and related complications. Genes. 2017. August 22;8(8):207. PubMed PMID: 28829354; PubMed Central PMCID: PMC5575670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Liu JY, Yao J, Li XM, et al. Pathogenic role of lncRNA-MALAT1 in endothelial cell dysfunction in diabetes mellitus. Cell Death Dis. 2014. October 30;5:e1506. PubMed PMID: 25356875; PubMed Central PMCID: PMC4649539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Qiu GZ, Tian W, Fu HT, et al. Long noncoding RNA-MEG3 is involved in diabetes mellitus-related microvascular dysfunction. Biochem Biophys Res Commun. 2016. February 26;471(1):135–141. PubMed PMID: 26845358. [DOI] [PubMed] [Google Scholar]
- [19].Yan B, Yao J, Liu JY, et al. lncRNA-MIAT regulates microvascular dysfunction by functioning as a competing endogenous RNA. Circ Res. 2015. March 27;116(7):1143–1156. PubMed PMID: 25587098. [DOI] [PubMed] [Google Scholar]
- [20].Zhang B, Wang D, Ji TF, et al. Overexpression of lncRNA ANRIL up-regulates VEGF expression and promotes angiogenesis of diabetes mellitus combined with cerebral infarction by activating NF-kappaB signaling pathway in a rat model. Oncotarget. 2017. March 7;8(10):17347–17359. PubMed PMID: 28060742; PubMed Central PMCID: PMC5370045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Nemecz M, Alexandru N, Tanko G, et al. Role of MicroRNA in endothelial dysfunction and hypertension. Curr Hypertens Rep. 2016. December;18(12):87. PubMed PMID: 27837398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tang ST, Wang F, Shao M, et al. MicroRNA-126 suppresses inflammation in endothelial cells under hyperglycemic condition by targeting HMGB1. Vascul Pharmacol. 2017. January;88:48–55. PubMed PMID: 27993686. [DOI] [PubMed] [Google Scholar]
- [23].Yuan J, Chen M, Xu Q, et al. Effect of the diabetic environment on the expression of MiRNAs in endothelial cells: mir-149-5p restoration ameliorates the high glucose-induced expression of TNF-α and ER stress markers. Cell Physiol Biochem. 2017;43(1):120–135. PubMed PMID: 28848152. [DOI] [PubMed] [Google Scholar]
- [24].Zhang Y, Sun X, Icli B, et al. Emerging roles for MicroRNAs in diabetic microvascular disease: novel targets for therapy. Endocr Rev. 2017. April 1;2017(1):1–22. PubMed PMID: 28520942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wang WW, Liu X, Zhao J, et al. A novel butyrolactone derivative inhibited apoptosis and depressed integrin β4 expression in vascular endothelial cells. Bioorg Med Chem Lett. 2007. January 15;17(2):482–485. PubMed PMID: WOS:000244012200038; English. [DOI] [PubMed] [Google Scholar]
- [26].Ge D, Han L, Huang S, et al. Identification of a novel MTOR activator and discovery of a competing endogenous RNA regulating autophagy in vascular endothelial cells. Autophagy. 2014. June;10(6):957–971. PubMed PMID: 24879147; PubMed Central PMCID: PMC4091179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Huang S, Lu W, Ge D, et al. A new microRNA signal pathway regulated by long noncoding RNA TGFB2-OT1 in autophagy and inflammation of vascular endothelial cells. Autophagy. 2015;11(12):2172–2183. PubMed PMID: 26565952; PubMed Central PMCID: PMC4835209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Gong Q, Su G. Roles of miRNAs and long noncoding RNAs in the progression of diabetic retinopathy. Biosci Rep. 2017. December 22;37(6):BSR20171157. PubMed PMID: 29074557; PubMed Central PMCID: PMC5705777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Noh JH, Kim KM, McClusky WG, et al. Cytoplasmic functions of long noncoding RNAs. Wiley Interdiscip Rev RNA. 2018. May–Jun;9(3):e1471. PubMed PMID: WOS:000430472400007; English. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hossain MZ, Yu Q, Xu M, et al. ICAT expression disrupts beta-catenin-TCF interactions and impairs survival of thymocytes and activated mature T cells. Int Immunol. 2008. July;20(7):925–935. PubMed PMID: 18511409; PubMed Central PMCID: PMC2556852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Deacon CF. A review of dipeptidyl peptidase-4 inhibitors. Hot topics from randomized controlled trials. Diabetes Obes Metab. 2018. February;20 Suppl 1:34–46. PubMed PMID: 29364584. [DOI] [PubMed] [Google Scholar]
- [32].Wang XM, Yang YJ, Wu YJ, et al. Attenuating hypoxia-induced apoptosis and autophagy of mesenchymal stem cells: the potential of sitagliptin in stem cell-based therapy. Cell Physiol Biochem. 2015;37(5):1914–1926. PubMed PMID: 26584290. [DOI] [PubMed] [Google Scholar]
- [33].Fu X, Zhu X, Qin F, et al. Linc00210 drives Wnt/β-catenin signaling activation and liver tumor progression through CTNNBIP1-dependent manner. Mol Cancer. 2018. March 14;17(1):73. PubMed PMID: 29540185; PubMed Central PMCID: PMC5853034. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [34].Huang CN, Wang CJ, Lee YJ, et al. Active subfractions of Abelmoschus esculentus substantially prevent free fatty acid-induced β cell apoptosis via inhibiting dipeptidyl peptidase-4. PloS one. 2017;12(7):e0180285. PubMed PMID: 28715446; PubMed Central PMCID: PMC5513409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Yang F, Zhang L, Gao Z, et al. Exogenous H2S protects against diabetic cardiomyopathy by activating autophagy via the AMPK/mTOR pathway. Cell Physiol Biochem. 2017;43(3):1168–1187. PubMed PMID: 28977784. [DOI] [PubMed] [Google Scholar]
- [36].Chen X, Duong MN, Psaltis PJ, et al. High-density lipoproteins attenuate high glucose-impaired endothelial cell signaling and functions: potential implications for improved vascular repair in diabetes. Cardiovasc Diabetol. 2017. September 29;16(1):121. PubMed PMID: 28962618; PubMed Central PMCID: PMC5622442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Leung A, Amaram V, Natarajan R. Linking diabetic vascular complications with LncRNAs. Vascul Pharmacol. 2018. February 1;114:139–144. PubMed PMID: 29398367; PubMed Central PMCID: PMC6070440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014. January 16;505(7483):344–352. PubMed PMID: 24429633; PubMed Central PMCID: PMC4113481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Xie Y, Jia Y, Cuihua X, et al. Urinary exosomal MicroRNA profiling in incipient type 2 diabetic kidney disease. J Diabetes Res. 2017;2017:6978984. PubMed PMID: 29038788; PubMed Central PMCID: PMC5605810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Liang Y, Zhao G, Tang L, et al. Corrigendum to “MiR-100-3p and miR-877-3p regulate overproduction of IL-8 and IL-1beta in mesangial cells activated by secretory IgA from IgA nephropathy patients” [Exp. Cell Res. 347 (2016) 312-321]. Exp Cell Res. 2016. November 15;349(1):198. PubMed PMID: 27745865. [DOI] [PubMed] [Google Scholar]
- [41].Wang C, Gu S, Cao H, et al. miR-877-3p targets Smad7 and is associated with myofibroblast differentiation and bleomycin-induced lung fibrosis. Sci Rep. 2016. July 22;6:30122. PubMed PMID: 27444321; PubMed Central PMCID: PMC4957095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Li S, Zhu Y, Liang Z, et al. Up-regulation of p16 by miR-877-3p inhibits proliferation of bladder cancer. Oncotarget. 2016. August 9;7(32):51773–51783. PubMed PMID: 27429046; PubMed Central PMCID: PMC5239514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Williams AE, Choi K, Chan AL, et al. Sjogren’s syndrome-associated microRNAs in CD14+ monocytes unveils targeted TGFβ signaling. Arthritis Res Ther. 2016. May 3;18(1):95. PubMed PMID: 27142093; PubMed Central PMCID: PMC4855899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zhang K, Zhu S, Liu Y, et al. ICAT inhibits glioblastoma cell proliferation by suppressing Wnt/β-catenin activity. Cancer Lett. 2015. February 1;357(1):404–411. PubMed PMID: 25434796. [DOI] [PubMed] [Google Scholar]
- [45].Mulvihill EE, Drucker DJ. Pharmacology, physiology, and mechanisms of action of dipeptidyl peptidase-4 inhibitors. Endocr Rev. 2014. December;35(6):992–1019. PubMed PMID: 25216328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Rahimi N, Samavati Sharif MA, Goharian AR, et al. The effects of aerobic exercises and 25(OH) D supplementation on GLP1 and DPP4 level in type II diabetic patients. Int J Prev Med. 2017;8:56. PubMed PMID: 28900535; PubMed Central PMCID: PMC5582496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Wu C, Hu S, Wang N, et al. Dipeptidyl peptidase4 inhibitor sitagliptin prevents high glucoseinduced apoptosis via activation of AMPactivated protein kinase in endothelial cells. Mol Med Rep. 2017. June;15(6):4346–4351. PubMed PMID: 28440488. [DOI] [PubMed] [Google Scholar]
- [48].Jaffe EA, Nachman RL, Becker CG, et al. Culture of human endothelial cells derived from umbilical veins. Identification by morphologic and immunologic criteria. J Clin Invest. 1973. November;52(11):2745–2756. PubMed PMID: 4355998; PubMed Central PMCID: PMC302542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Zhao X, Dong W, Gao Y, et al. Novel indolyl-chalcone derivatives inhibit A549 lung cancer cell growth through activating Nrf-2/HO-1 and inducing apoptosis in vitro and in vivo. Sci Rep. 2017. June 20;7(1):3919. PubMed PMID: 28634389; PubMed Central PMCID: PMC5478673. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.