

Abstract
The effective therapeutic treatment and the disease-modifying therapy for spinocerebellar ataxia type 2 (SCA2) (a progressive hereditary disease caused by an expansion of polyglutamine in the ataxin-2 protein) is not available yet. At present, only symptomatic treatment and methods of palliative care are prescribed to the patients. Many attempts were made to study the physiological, molecular, and biochemical changes in SCA2 patients and in a variety of the model systems to find new therapeutic targets for SCA2 treatment. A better understanding of the uncovered molecular mechanisms of the disease allowed the scientific community to develop strategies of potential therapy and helped to create some promising therapeutic approaches for SCA2 treatment. Recent progress in this field will be discussed in this review article.
Key Words: Spinocerebellar ataxia type 2, polyglutamine disorders, cerebellum, calcium signaling, aggregation.
Introduction
Spinocerebellar ataxia type 2 (SCA2) represents a genetic disorder with an autosomal dominant inheritance caused by a CAG expansion in the ubiquitously expressed ATXN2 gene encoding the polyglutamine (polyQ)-expanded ataxin-2 protein [1–5]. The pathophysiological features of SCA2 include the progressive atrophy of the cerebellum and the subsequent gradually evolving ataxia [2]. At present, there is no disease-modifying therapy for SCA2, only symptomatic treatment, and the methods of palliative care are used to support SCA2 patients. The modern methods of neuroimaging, electrophysiology, and the tracking of the different biomarkers in the patient’s body fluids together with the motor and cognitive assessments are used for the evaluation of the disease progression. The disease-causing protein, ataxin-2, exhibits different cellular functions and is involved in RNA metabolism [6]. The different modeling systems on cells, yeast, worms, flies, mice, and induced pluripotent stem cell (iPSC)-derived neurons from SCA2 patients were developed to study the basics of SCA2 pathology. Cerebellar Purkinje cells (PCs) and the cerebellar pathways are primarily affected in SCA2 mice and patients. Studies with these model systems revealed the disturbances in the morphology, biochemistry, and neurophysiology of the PCs and the cerebellar circuits [7, 8] and helped to identify the possible molecular mechanisms of the disease. Aggregation, oxidative stress, disturbed cell signaling, dysregulation of calcium homeostasis, abnormal autophagy, and impaired DNA processing all seem to be involved in SCA2 molecular pathogenesis [9–16]. The promising data obtained in these studies gave rise to some perspective therapeutic approaches that may give decisive results in the treatment of SCA2 patients. The encouraging data have been obtained in preclinical studies on the antisense oligonucleotide (ASO) therapy [17, 18] and in some early clinical trials on iPSCs [19] and riluzole [20] treatment. The disease-modifying therapy with calcium stabilizer approach also exhibited some promising results in the preclinical studies on SCA2 transgenic mice [21–23].
Epidemiology
Spinocerebellar ataxias (SCAs) represent a group of autosomal dominant cerebellar ataxias (ADCAs) characterized first of all by a progressive ataxia because of the degeneration of the cerebellum and the cerebellar pathways [1–5]. To date, about 47 SCA subtypes have been described, and 35 causal genes have been identified [24]. The prevalence of SCAs is estimated to be 1 to 5 per 100,000 [2, 5], but can vary markedly depending on the geography and ethnicity [2, 3, 5]. The most frequent and most extensively studied are the polyglutamine (polyQ) expansion disorders including SCA1 to SCA3, SCA6, SCA7, SCA17, and dentatorubral–pallidoluysian atrophy [1, 3, 24]. These diseases are all caused by a CAG repeat expansion, which is different for each gene [5, 24]. Based on the meta-analysis of the global epidemiological data, SCA2 has large worldwide geographical distribution and represents the most prevalent type of the polyQ ataxias in Cuba, India, Mexico, and southern Italy [25]. SCA2 also represents the second most common subtype of ADCA across the world in many regions (after SCA3) [25, 26]. A large epidemiological study conducted recently to analyze the frequencies of various SCAs in a population of South Korea revealed SCA3 as the most common subtype of SCA (34.8%), followed by SCA2 (23.0%) and SCA6 (13.7%) [27]. The largest prevalence rates of SCA2 are found in Cuba because of a prominent founder effect, and the highest case frequency is found in the Holguin province of Cuba, with 40.18 cases and 182.75 carriers per 100,000 inhabitants [26]. Survival studies in SCAs have shown that survival rate in SCA2, as well as prevalence, depends on the geographical location and ethnical groups. Thus, longer survival is found in southern Italy—67 years, whereas in Spanish and Cuban groups, the survival rate is considerably less—54 and 52 years accordingly [25].
In SCA2, as well as in other polyQ diseases, the age of onset depends on the length of the CAG repeat expansion: the longer is the polyQ expansion, the earlier is the age of the disease onset. Average age at onset of symptoms in SCA2 is about 35 years [28, 29]. A recent study on the Cuban SCA2 founder population has shown that the age of onset also depends on its modifiers such as the DNA repair, mitochondrial fission, and oxidoreductase activity [30]. The Scale for the Assessment and Rating of Ataxia (SARA) has revealed that the longer disease duration in SCA2 patients is correlated with a greater disease severity, worse balance, and higher functional dependency [28]. The studies on the severity of ataxia via the SARA scale in SCA patients have shown that the natural progression of SCA2, as well as SCA3 and SCA6, seems to be similar between the Chinese and Caucasians [31]. The analysis of the long-term evolution of the patient-reported outcome measures in SCA patients revealed that functional capacity, health-related quality of life, and mood deteriorated over the 8-year observation period in SCA2 [32].
ATXN2 Mutation
Genetically, SCA2 is caused by an expansion of the CAG DNA repeat in the coding region of the ATXN2 gene localized to chromosome 12q24.1 [33]. SCA2 pathology manifests in patients with ≥ 33 CAG repeats [34]. Consistent with a dominant inheritance in human pedigrees, the CAG repeat expansion acts as a gain-of-function mutation [35]. Generally, the intergenerational instability at ATXN2 locus in SCA2 is influenced by the sex, the CAG expansion length, and the age at conception of the transmitting parent [36, 37].
Ataxin-2 is a 140-kDa cytoplasmic protein, product of the ATXN2 gene, localized at the rough endoplasmic reticulum (ER) [38] and other cellular compartments including Golgi, stress granules, inclusion bodies, and cytoplasm. Ataxin-2 deficiency leads to obesity, dyslipidemia, insulin resistance [39, 40], and changes in circadian system in mice [41] and flies [42, 43]. The studies on a polyQ-expanded ataxin-2 via the granular cytoplasmic staining and the analysis of the frequency of neuronal inclusions in the brainstem areas in SCA2 patients revealed the aggregation of mutant ataxin-2 in the SCA2 tissue [44]. The protein structure of ataxin-2 contains PAM2 motif (Fig. 1a) which takes part in the association with the poly(A)-binding protein (PABP) [45], that is likely to lead to ataxin-2 association with the polyribosomes [6]. Ataxin-2 also contains 2 globular Lsm domains and the Lsm-associated domain (Fig. 1a) that were shown to interact directly with RNA [6]; thus, ataxin-2 most likely is involved in the RNA metabolism (Fig. 1b). Indeed, in vitro studies on neurons and the mouse embryonic fibroblasts have shown the rise of the ataxin-2 expression levels in response to the nutrient and trophic stress in a mammalian target of rapamycin-signaling dependent manner, whereas the absence of ataxin-2 leads to adaptive changes in the phosphoinositide 3-kinase/mammalian target of rapamycin-mediated phosphorylation of the ribosomal protein S6 and the eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1) as regulatory components of the 48S pre-initiation complex of the mRNA translation [46].
Fig. 1.
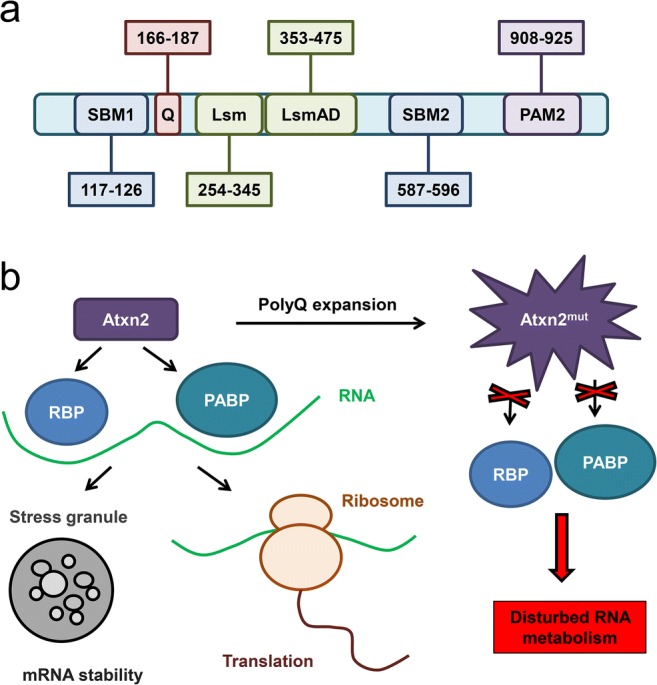
Domain structure and function of Atxn2. (a) Known functional motifs are diagrammed for ataxin-2 (Atxn2) protein with their amino acid (aa) residue numbering. Identified domains include polyglutamine tract (Q; aa 166–187), SRC homology 3 domain binding motifs 1 (SBM1; aa 117–126) and 2 (SBM2; aa 587–596), Like sm domain (Lsm; aa 254–345), Lsm-associated domain (LsmAD; aa 353–475), and poly(A)-binding protein (PABP) interacting motif 2 (PAM2; aa 908–925). (b) Atxn2 interacts directly or indirectly with various RNA-binding proteins (RBP), such as STAU1 protein, and also with PABP. These proteins are involved in translation and stress granule formation processes. Polyglutamine expansion in Atxn2 protein may interrupt these interactions and lead to the disturbed RNA metabolism. Modified from Paulson et al. [3] and Scoles and Pulst [4]
Another study has shown that the intracellular levels of ataxin-2 affect the stress granules (SGs) and P-bodies formation (Fig. 1b), thus regulating the mRNA metabolism including translation, stability, and degradation [47]. It was shown that the Lsm/Lsm-associated domain of ataxin-2 interacts with the DEAD/H-Box RNA helicase DDX6 which is involved in the formation of SGs and P-bodies. Reduction of the ataxin-2 level led to the abnormal SG assembly and to the elevation of the level of PABP, another SG component [47]. Recently, it was shown that the disordered domains of ataxin-2 led to the formation of the granules containing mRNA and RNA-binding proteins as well as the long-term memory and cytotoxicity [48].
It was recently reported that the Pbp1, the yeast ortholog of the human ataxin-2, regulated the retrotransposon activity because the changes in its expression repressed transposons of yeast retrotransposons via distinct mechanisms [49]. The analysis of the SCA2 patient blood global transcriptome has indicated an ataxin-2 as a cytosolic RNA processing factor and displayed its role in the transcript regulation of the mitochondrial stress response and quality control factor PINK1 [50]. The mentioned physiological functions of ataxin-2 most likely are compromised in SCA2 and the increase in the concentration of not-expanded ataxin-2 could probably have the therapeutic potential for SCA2.
Studies on the human postmortem SCA2 brain tissues, SCA2 fibroblasts, SCA2 iPSCs, and SCA2 mice model have shown that the ATXN2 locus is bidirectionally transcribed and the antisense transcript ATXN2-AS with a CUG repeat expansion exhibits neurotoxic activity in an SCA2 cell model and forms RNA foci in SCA2 PCs [51]. Thus, the antisense transcript ATXN2-AS may be involved into the additional mechanism of SCA2 pathogenesis and can be considered as a new therapeutic target in SCA2.
The discussion as to whether the ataxin-2 mutation in SCA2 represents a loss-of-function or gain-of-function mutation seems to be a controversial issue. Many studies have shown that the lack of ataxin-2 led to the disturbances in the mRNA metabolism including translation, stability, and degradation [47]; indirectly provoked the impairment in the pathways of the ribosomal biogenesis, translation initiation, ER secretion, and lipid trafficking [52]; and altered the nutrition and metabolism [53], as well as negatively affected the gene expression in a calcium homeostasis pathway [54]. Thus, the physiological reduction in the ataxin-2 levels caused by the mutation in the ATXN2 gene and the further production of the dysfunctional mutant protein indeed can be considered as a loss-of-function mutation (Fig. 1b). However, the display of the mutant ataxin-2 features as a gain-of-function mutation is also remarkable. The observation of the mutant ataxin-2 aggregation in the cerebellar tissue from SCA2 patients [44] together with the experimental results on the planar lipid bilayers model system, which demonstrated that a polyQ-expanded ataxin-2 protein binds to the inositol 1,4,5-trisphosphate receptor (IP3R) and significantly enhances its activation by the IP3 molecules [21], suggests that the SCA2 causing mutation leads to the gain of a new abnormal function. We can only presume that the truth about the mutation type lies somewhere in between: whereas the loss of the normal physiological function of the ataxin-2 is observed in SCA2, the mutant polyQ-expanded protein reveals new cytotoxic signs.
Molecular Changes Underlying the PC Loss
On the cellular level, the PCs are primarily affected in SCA2 patients. Thus, the postmortem studies on SCA2 patients detected the heterotopic PCs whose cell bodies were dislocated in the molecular layer of the cerebellum [55]. The same pathology was observed in the subjects with an essential tremor [55].
Recent research via a complex approach combining the methods of the whole exome sequencing, targeted resequencing, and gene network analysis in 20 SCA families has identified the 5 novel disease genes for SCAs, involved in the central nervous system development, axon guidance, transcription regulation, mitochondrial function control, autophagy, and synaptic transmission [56]. The disturbances in these important physiological features may explain the cerebellar PC elimination in SCA2 pathology and will be discussed further in the section related to the molecular mechanism of SCA2.
It was reported in the literature that the developing cerebellum is highly vulnerable to the damaging effects of the reacting oxygen species, and the structural changes in the PCs, their dendrites, granule cells, astrocytes, microglia, neuronal axons, and oligodendroglia are observed in the induced oxidative stress [57]. The cerebellar oxidative stress-related alterations typical for SCA2 patients can be related to the altered homeostasis of the different elements in the serum or blood samples [58, 59]. Thus, it was reported that the levels of copper, manganese, zinc, and vanadium are disturbed in the blood of SCA2 patients [58]. The reduced levels of the superoxide dismutase (SOD3) enzymatic activity were detected in SCA2 patients with no significant impact on the clinical phenotype [60]. Recent studies have shown the association of the omega 2 glutathione-S-transferase polymorphism and SCA2 [61]. Another polymorphism, A10398G at the mitochondrial DNA, was noted to be involved in the cognitive decline during the earlier ages at onset in SCA2 [62]. The significant increase in the global 5-hydroxymethylcytosine levels in the blood samples of SCA2 patients was observed indicating global DNA methylation as a biomarker of the disease [63]. It was reported that the serum neurofilament light might be another promising peripheral biomarker in SCAs [64].
Not only cerebellar PCs reflect the SCA2 pathology signs. Recently, the examination of the frequencies of the micronuclei formations (MNi) and binucleated cells (BN) in the buccal cells, collected by an oral mucosa scraping from SCA2 patients, revealed the significant increase in the MNi and BN number in buccal cells from SCA2 patients compared with healthy control, progressing with time [65]. The authors suggested that the MNi and BN cell frequencies could be useful as the peripheral biomarkers for SCA2 [65]. In SCA2 patients, the significant increase in the markers of gliosis including myoinositol and glutamine is observed [66] together with the significant rise in the level of the tau protein compared with control [67]. Generally, disease biomarkers seem to be useful for disease progression evaluation, whereas diagnosing SCA2 may be done simply by a genetic test using PCR and capillary electrophoresis [68].
Cerebellar Atrophy in SCA2
The neuropathological analysis reveals an extensive atrophy of the cerebellum, basal ganglia, and frontal lobes in SCA2 patients [69, 70]. Recent fractal analysis on the index of the structural complexity of the cortical gray matter and white matter demonstrated the reduction of the structural complexity of the cerebellum and cerebral cortex in SCA2 patients in vivo [71]. The recent whole-brain voxel-based morphometry (VBM) in vivo studies on SCA2 showed that the gray matter volume reductions in SCA2 patients were mainly observed in the cerebellum and the part of the cerebral cortex in the supratentorial regions responsible for the regulation of cognitive functions and emotions, whereas significant white matter volume reductions widely existed in the cerebellum along with its afferent pathways and pyramidal system [72]. Another recent VBM study has shown the significant gray matter loss in the bilateral regions of the anterior cerebellar hemisphere (I-V), the posterior lobe (VI-IX), and the posterior vermis (VI-IX). In this research, it was proposed that there is a selective topography between the cerebellar lobules and the specific attentional impairments, namely, the attention deficits observed in SCA2 patients are caused by the dysfunction of cerebrocerebellar circuitry and the specific site of the cerebellar degeneration is also involved [73]. The VBM-based quantitative analysis of the degeneration patterns revealed that the degeneration was clearly localized to the lobules VII to IX in SCA2 patients representing a unique degeneration signature in the cerebellar cortex that determines the unique symptomology in SCA2 [74].
Magnetic resonance imaging (MRI) represents 1 of the main tools for neuroimaging study of SCA patients and can be used for diagnosis, pathophysiological studies, disease progression evaluation, and even as an instrument for the assessment of the efficacy of new treatments [75]. In SCA2 patients, MRI reveals olivopontocerebellar atrophy, pontine brainstem volume loss before clinical onset, diffuse spin–spin relaxation time (T2) high signal in pons, middle cerebellar peduncles and cerebellar white matter with “hot cross bun” sign, and thalamus and parietal cortical atrophy in advanced phases, whereas in terms of the volume of the pontine brainstem and cerebellum, diffusion tensor imaging indexes of the brainstem and cerebellum are the most promising imaging measures to monitor disease progression in SCA2 [76, 77]. Recent MRI studies have shown the hypertrophy of the inferior olivary nuclei in SCA2 patients that represents a unique feature of SCA2 compared with other hereditary SCAs [78]. A special kind of diffusion-weighted MRI, diffusion-tensor imaging (DTI), has revealed the microstructural changes associated with the pontocerebellar degeneration in SCA2 and has shown that the median value of the mean diffusivity might be a marker for SCA2 progression [79] and may account for the specificity of the cognitive symptomatology observed in SCA2 patients [80]. DTI studies together with a novel tractography method, called a fixel-based analysis, have indicated the increased cerebellar and pontine atrophy at the early stages of SCA2, which slowed with disease progression [81]. Recently, diffusion kurtosis imaging, an extension of DTI, has been shown to exhibit better sensitivity to subtle microstructural changes in various neurological disorders, and can be used for the differential diagnosis of SCA2 [82]. The dysfunction of the corticospinal tract is also observed in SCA2 patients before clinical onset [83]. Recent studies via the transcranial magnetic stimulation have shown that the transcranial magnetic stimulation markers of corticospinal tract dysfunction progress significantly during the pre-ataxic stage of SCA2 and may serve as a biomarker of SCA2 progression before the clinical onset [84, 85], that can also be detected via the corticomuscular (electroencephalography—electromyography (EMG)) [86] and intermuscular (EMG–EMG) coherence approach [87].
A Cerebellar–Thalamic–Cortical Circuit Dysregulation
The differences in the cerebellothalamic circuit functions were detected in SCA2 patients via the method of the polysomnographic recordings, an electrophysiological tool used in sleep research. Thus, the sleep spindle densities were significantly decreased in SCA2 patients, most likely because of the effect of the thalamic disruption observed in SCA2 [88]. The rapid eye movement (REM) sleep density was absent in the majority of the studied SCA2 patients and showed a marked reduction in the remaining patients [89]. Most likely, the progression of the REM sleep pathology is affected by the thalamic degeneration in the later course of SCA2, as the REM sleep is known to correlate with the increased metabolic activities in both the pons and the thalamus [89]. Another recent study on the sleep research in SCA2 has demonstrated that the reduction in the sleep spindles and K-complex activity caused by the thalamocortical circuit impairment seems to be corresponded with the abnormal neuronal plasticity and affected verbal memory in SCA2 patients [90]. SCA2 patients can also suffer from such sleep disorders as restless leg syndrome and periodic leg movement (PLM). The study on SCA patients has shown that restless leg syndrome developed more frequently in aged patients, but did not depend on the CAG tract length or the age at onset [91]. SCA2 patients also exhibited PLM during the REM sleep that might be caused by the pathological alterations in the dopaminergic, brainstem, and cerebellar circuits [92]. The PLM frequency depended on the ataxia severity and the disease duration but did not depend on the CAG repeat length [93]. An antiparkinson agent lisuride exerted a beneficial effect in the sleep-related disorders in SCA2 patients [94].
In SCA2 patients, cognitive decline is also observed as a common nonmotor clinical feature during the pre-ataxic stage. Recent studies on the P300 wave component revealed the impaired processes related to attention, decision-making, and memory updating in SCA2 patients and the preclinical carriers that tend to appear some years before the ataxia onset and may serve as a biomarker for the cognitive deterioration and disease onset in SCA2 [95]. A new VBM study on the correlation between the cerebellar structures and the cognitive dysfunction in SCA2 patients has demonstrated that gray matter loss in the cognitive posterior lobules (VI, crus I, crus II, VIIB, IX) correlated with the visuospatial, verbal memory, and executive tasks, whereas additional correlations with the motor anterior (V) and posterior (VIIIA, VIIIB) lobules were observed for the tasks involving the motor and planning components [96]. Recent functional MRI studies on the functional connectivity via the network-based statistics approach have shown that the cerebellar disturbances may affect the long-distance regions in the cerebral cortex through the cerebellar projections resulting in various motor, cognitive, and emotional dysfunction in SCA2 patients [97].
Ataxia
The initial clinical symptom observed in the majority of SCA2 patients is a gait abnormality. SCA2 patients can also have slow saccades, postural and action tremor, polyneuropathy, upper motor neuron signs, and parkinsonism, which could be a predominant feature in some cases. The recent study on SCA patients from the Clinical Research Consortium for SCAs has shown that the initial symptom in SCA2 had a different influence on the age of onset and motor progression [98]. Another clinical feature of SCA2 is postural tremor, which could be associated with the different rates of an ataxia progression and might be affected by the brain circuitry influenced by the genetic interactions between the ataxia genes [99]. It was recently found that both postural and rest tremors occur more frequently in the later stage of the cerebellar degeneration in SCAs patients and can independently contribute to worse functional status in SCA2 patients [100]. Some of SCA2 patients have lower cranial dystonia as a clinical feature, especially jaw and tongue dystonia [101]. The prevalence of dystonia has been reported to be 14% in SCA2 [102]. It also has been reported that dystonia was associated with more severe ataxia in SCA2 [103]. The chronic symptoms of sensory neuropathy are also observed in SCA2 patients and upper limb ultrasound can be used for detecting this medical condition [104].
Eye movements are frequently impaired in SCA2; thus, reduced saccade velocity or “slow saccades” are observed in many SCA2 patients [105]. It was reported that the vestibulo-ocular reflex gain was reduced in 30% of SCA2 patients and the caloric responses were reduced in 75% of patients [106]. However, a more recent study on a small cohort of SCA2 patients reported that the vestibulo-ocular reflex gain was not significantly affected in these patients [107]. The oculomotor dysfunction in SCA2 can be quantified by using the optokinetic nystagmus testing [108] and the susceptibility-weighted imaging of the dentate nuclei hypointensity [109] because the dentate nuclei are involved in a human oculomotor control [110].
Together with neuroimaging studies, electrophysiological examination might be a potential marker of disease progression and subtype identification. Thus, a motor nerve conduction study shows a prolonged distal latency, reduced nerve conduction velocity, and reduced compound muscle action potential (AP) amplitudes in SCA2 patients; a sensory nerve conduction study reveals absent or reduced sensory nerve AP amplitudes and reduced nerve conduction velocity; the needle EMG discovers fasciculations, giant motor unit APs, and reduced requirement; the brainstem auditory evoked potentials (EPs) demonstrate prolonged I to III and III to V interpeak intervals; the motor EPs are characterized by a delay or absence; the somatosensory EPs observe prolonged P40 latencies or absence of P40 wave; the visual EPs have prolonged latencies and reduced amplitudes of P100; and finally, electrooculography reveals the lack of the gaze-evoked nystagmus and dysmetric saccades [111]. The other important oculomotor feature revealed by electrooculography is the severe slowing of horizontal saccades in a horizontal plane which is significantly correlated with the expanded CAG repeats [112], precedes the ataxia onset [113], progresses notably along time [114], and shows a high familiar aggregation leading to its conceptualization as a disease endophenotype marker [115].
Although SCA2 is mostly manifested with a cerebellar ataxia, slow saccades, and polyneuropathy, in some cases, parkinsonism is also reported as a clinical feature of SCA2. Thus, a Korean SCA2 family with a long-duration (up to 34 years) levodopa-responsive parkinsonism without cerebellar ataxia was reported [116]. Later, a member of this family who developed a unique intrafamilial jaw-opening and lingual-protrusion dystonia as a main symptom got reported demonstrating the heterogeneity within the single SCA2 kindred [117]. The link between SCA2 and Parkinson’s disease (PD) might be explained by the findings of a recently conducted transcriptome study, that has revealed a distinctive ataxin-2-mediated dysfunction of pathways related to cell contact, proliferation, and differentiation in PD patients, together with the disturbances of the RNA metabolism with the involvements of the poly(A) RNA-binding and mRNA-binding in SCA2 and PD patients revealing a putative continuum from PD to SCA2 [118]. Interestingly, ataxin-2 with an intermediate-length polyQ expansion has been reported as a risk factor for amyotrophic lateral sclerosis [119].
At present, there is no disease-modifying therapy for SCAs, only the symptomatic treatment and the methods of a palliative care are used to support SCA patients [120]. A recent 24-week, rater-blinded, randomized, controlled trial on SCA2 patients revealed that the tested rehabilitation program significantly improved the SARA scores of SCA2 patients likely as a result of the partial preservation of the motor learning and neural plasticity mechanisms suggesting that palliative treatment does have an actual effect in the treatment of SCA2 patients [121]. Current clinical studies show that some ataxia signs can be partly improved pharmacologically by treatment with 4-aminopyridine, riluzole, valproic acid, and thyrotropin-releasing hormone, whereas lithium and deferiprone have a negative effect or its absence [122]. Low-frequency deep cerebellar stimulation has been suggested as a potential strategy for treating cerebellum-related motor symptoms, but no clinical studies on SCA patients have been carried out yet [123].
Review of SCA2 Model Systems
Different modeling systems were developed to study the basics of SCA2 pathology. Several models of yeast, worms, flies, and mice were created to investigate the molecular mechanisms and to test the potential therapeutic strategies. The first developed SCA2 models were simple models like yeast, Caenorhabditis elegans, and Drosophila melanogaster, whereas they allowed to identify the basic functions of ataxin-2, and the further created murine models of SCA2 enabled researchers to test the therapeutic strategies that might be translated into the clinic [7]. The reviewed murine models of SCA2 are summarized in Table 1.
Table 1.
Overview of all reviewed murine models of SCA2
| Transgenic line | Transgene | The age at onset of the motor dysfunction (weeks) | Reported pathological display | Reference |
|---|---|---|---|---|
| SCA2-58Q | Human ATXN2 gene with 58 CAG repeats | 32 | Significant loss of cerebellar PCs; progressive loss of calbindin-28k; disturbances of the electrophysiological functions | [21, 22, 124–127] |
| SCA2-75Q | Human ATXN2 gene with 75 CAG repeats | 12 | Specific cerebellar PC degeneration | [128] |
| SCA2-42KI | Murine ATXN2 gene with 42 CAG repeats | 72 | Reduced body weight; neuropathological mild and late-onset alterations | [129] |
| SCA2-127Q | Human ATXN2 gene with 127 CAG repeats | 8 | Ataxin-2 aggregates in the PCs; electrophysiological changes of the PCs; PC neuronal loss; abnormalities in the cellular gene expression | [130, 131] |
| BAC-SCA2-72Q | Human ATXN2 gene with 72 CAG repeats | 16 | Body weight loss; PC dendritic trees thinning; reduction of the calbindin and Pcp2 levels in the cerebellum | [132] |
The first created mouse model of SCA2 was a transgenic line SCA2-58Q. In these mice, a full-length human ATXN2 gene with 58 CAG repeats was expressed under Purkinje cell protein 2 (Pcp2) promoter specifically in the cerebellar PCs of the mice [124]. These mice are characterized by a progressive general motor coordination impairment and the age at onset is 32 weeks of age, as scored by the beam walk and rotarod tests, that showed longer latencies to cross the beam and an increased number of foot slips compared with wild-type (WT) mice [21]. At 24 weeks of age, significant loss of the cerebellar PCs was observed in these mice together with a progressive loss of calbindin-28k that represents a marker for neuronal dysfunction [21]. Disturbances of the electrophysiological functions have been reported in these mice in the studies on the cerebellar slices [22, 125] and also in in vivo studies [126, 127].
Another SCA2 mouse model was a transgenic line SCA2-75Q that expresses a human full-length version of ATXN2 under the control of the endogenous SCA2 promoter. Although the transgene expression was ubiquitous, the neuropathological analysis revealed that only cerebellar PCs were degenerated. Motor coordination assessments, namely, the rotarod assay, demonstrated a significant decline in the motor functions of the heterozygous SCA2-75Q mice at 12 weeks and the homozygous at 6 weeks of age when compared with the WT littermates [128].
At present, there is only 1 reported knock-in (KI) mouse model of SCA2 [129]. In these SCA2-42KI mice, the 42 CAG repeat expansion was introduced into the murine ATXN2 gene. The SCA2-42KI mice showed phenotypes with a reduced weight and subtle motor incoordination. A significant difference in the motor performance on the accelerated rotarod was detected for the homozygous mice only at 72 weeks of age, whereas the heterozygous mice have not shown any differences at any age compared with the WT littermates. The neuropathological examination also discovered very mild and late-onset alterations, consistent with the results on the motor coordination assessments [129].
Another mouse model of SCA2 is a transgenic line SCA2-127Q with 127 CAG repeats under the Pcp2 promoter [130]. Because of the longer polyQ expansion, this mouse model exhibits a much more severe pathology. Thus, the presence of ataxin-2 aggregates in the PC cell bodies was observed at 4 weeks; further, the statistically significant changes in the electrophysiological behavior of PCs were detected at 6 weeks, and finally, the progressive motor dysfunction in the accelerating rotarod performance became significant at 8 weeks. The biochemical, electrophysiological, and behavioral disturbances worsened over the period between 8 and 40 weeks, but the PC cell neuronal loss was not detected until 40 weeks of age in this model [130]. The weighted gene co-expression network analysis has revealed early and progressive abnormalities in the cellular gene expression primarily in genes associated with the GTPase signaling, calcium signaling, and cell death, but temporal mRNA changes in expression as early as day 1 have shown that the upregulated genes were associated with the histone acetylation and chromatin remodeling, whereas the downregulated genes were associated with cell adhesion and extracellular matrix terms [131].
The most recent transgenic mouse model of SCA2, the BAC-72Q line, was created with a bacterial artificial chromosome (BAC), including the entire ATXN2 gene in which an exon 1 was edited to include an expanded CAG repeat under the endogenous human promoter [132]. The BAC-SCA2-72Q mice were characterized by weight loss, progressive impairment in the accelerating rotarod performance starting at 16 week of age, and also by thinning of the PC dendritic trees and the reduction of the calbindin and the Pcp2 expression levels in the cerebellum [132].
Regarding the studies on the murine models of SCA2, ataxin-2 knockout (KO) mice should be mentioned. The first generated ataxin-2 KO mutant mice were obtained via the method of electroporation of the targeting construct into the embryonic stem cells [39]. The morphological analysis of these KO mice did not reveal any significant impairment in the histological features; only adult-onset obesity was determined. The authors concluded that the ataxin-2 protein is not required for the development or survival in mice [39]. Although the morphology and overall well-being of ataxin-2 KO mice were not significantly affected, the disturbances in their biochemical and cellular machinery were observed. Thus, the phosphorylation of the ribosomal protein S6 was enhanced and global protein synthesis was pathologically altered in ataxin-2 KO mice [52]. Recently, it was also reported that ataxin-2 KO mice demonstrate the fatty acid, leucine, valine, isoleucine amino acid [53], and calcium homeostasis [54] pathway alterations.
Mouse transgenic SCA2 lines are very useful in basic research on the biochemical, electrophysiological, and especially motor behavior onset of the disease, but even though most of the mouse lines contain human ataxin-2, the murine organism is very different from human organism, and the therapeutic approaches that show promising results in mice often fail in human trials. Recently, the success in the pluripotency reprogramming technology allowed the researchers to extract the disease-specific iPSCs from SCA2 patients. These cells can be differentiated into neurons giving the opportunity to study the SCA2 disease mechanisms and pathological progression in vitro on the actual patient-derived neurons [7, 133]. The iPSC lines were successfully generated from the human skin fibroblasts of SCA2 patients [134], from the peripheral blood mononuclear cells [135], or both [133]. The SCA2-iPSC-derived neurons exhibited SCA2-associated pathological phenotype in vitro, namely, the polyQ aggregation and distorted mitochondrial microstructures [133].
Molecular Mechanism of SCA2
The molecular mechanism of SCA2 has been discussed for years now, many research groups have made an important contribution to the understanding of the SCA2 pathology, and many different approaches have been used as attempts to build a clear picture of SCA2 molecular pathogenesis. Only a clear comprehension of the molecular bases of the disease can give us a chance to modify the pathological pathways and, finally, find the treatment for the disease.
Aggregation
A polyQ-expanded ataxin-2 is characterized by a conformational shift to the β-sheet–rich structure, which forms the insoluble aggregates with the β-sheet–rich amyloid fibrillar structures and accumulates as inclusion bodies in neurons [9]. The role of these aggregates and inclusions into the degenerative processes in the SCA2 cerebellum is still not clear [136]. The examination of the aggregation pathology in the brainstem of SCA2 patients revealed a significant positive correlation between the presence of granular cytoplasmic staining and more severe pathological processes, whereas the neuronal nuclear inclusions displayed a protective role [44]. In SCA2 patients, via the method of 1C2-immunoreactive typing, it was shown that the granular cytoplasmic pattern was observed at the early stage of the disease, the cytoplasmic and nuclear pattern at the active stage, and the nuclear with inclusion pattern at the final stage [137]. Studies on the polyQ oligomer behavior in the mouse brain have demonstrated that the presence of polyQ-expanded oligomers leads to the disruption of the ER membrane through the insertion of the apoptosis regulator Bax protein into the ER membrane and the following caspase-7 activation [138].
Oxidative Stress
The examination of the mitochondrial oxidative stress in fibroblasts from SCA2 patients has revealed the increased SOD expression and the decreased catalase expression at both the transcript and protein levels [139]. The increased levels of the intracellular hydrogen peroxide, produced by the increased SOD from a superoxide, could not be decomposed by the reduced levels of catalase, thus leading to increased oxidative stress, disturbances in the antioxidant system, changes in the oxidative phosphorylation system, and abnormalities in the mitochondrial activity that were detected in the fibroblasts from SCA2 patients. Treatment with the antioxidant coenzyme Q10 alleviated the increased oxidative stress in the fibroblasts from SCA2 patients [139]. In SCA patients themselves, the coenzyme Q10 treatment led to improvements indexed by the SARA and the Unified Huntington’s Disease Rating scales within a cross-sectional 2-year study in SCA1 and SCA3, whereas improvement was not statistically significant in SCA2 patients most likely because of too small size of the experimental group [140]. Thus, the cellular antioxidant system may be involved in the SCA2 pathogenesis and its regulation might be a potential therapeutic target.
Disturbed Cell Signaling
The communication processes between cells are very important for normal development, neurogenesis, reparation, homeostasis, immune response, and other intrinsic cell actions. Abnormalities in the cell signaling can lead to the diverse pathological processes and neurodegeneration. The Src family of the nonreceptor tyrosine kinases (SFK) is essential for the nervous system homeostasis and may be involved in the neurodegenerative disease. Recent studies have discovered the involvement of the SFK inhibitor Missing-in-metastasis (MTSS1) in SCA molecular pathology [141]. The lack of MTSS1 in mice led to the increased SFK enzyme activity, accompanied by the PC firing dysfunction and, eventually, to PC cell death. Treatment of MSST1 mutant mice with the FDA-approved SFK inhibitor dasatinib prevented the SFK-dependent firing defects and delayed the ataxia progression in these mice. Interestingly, the reduced MTSS1 protein level and elevated SFK activity was observed in SCA2-127Q transgenic mice [141].
Another kinase, protein kinase C (PKC), is especially important for the cerebellar PCs to maintain their homeostasis as mutations in the gene encoding this kinase cause the abnormal PC dendritic development and cerebellar ataxia, namely, SCA14 [142]. Carbonic anhydrase-related protein 8 (Car8) encoding CAR8 and Itpr1 encoding IP3R1 were identified as the upregulated genes in SCA14 mice [143]. Recent experimental findings have revealed the increased PKC substrate phosphorylation in SCA2-127Q mice that serve as a protective modifier of PC degeneration because normalizing of the PKC activity accelerated neurodegeneration in these mice [144]. PKC activation in PCs is required for the long-term depression (LTD) at the parallel fiber (PF)–PC synapse, and these studies also demonstrated that the increased PKC activity may mediate its protective effect on the PC degeneration by limiting the membrane hyperexcitability of the PCs [144].
Not only kinases are implicated in the neuronal cell signaling and are responsible for the proper function of the cellular machinery, different transcription factors are also important in regulating cellular responses. Thus, NF-kB (nuclear factor kappa-light-chain-enhancer of activated B cells) is involved in the control of the DNA transcription, cytokine production, and cell survival and has also been implicated in the processes of synaptic plasticity and memory [145]. Recently, it was demonstrated that the NF-kB signaling is required for the increase in microglial numbers and tumor necrosis factor alpha (TNF-α) production in SCA1 mice cerebellum, but also is involved in the synaptical processing during cerebellar development [146]. In the SCA3 fly model, NF-kB was identified as an enhancer of degeneration that mediated the activation of astrocytes leading to neurodegeneration, whereas the astrocyte-specific inhibition of the NF-kB enhanced vitality and extended lifespan [147]. Although so far there are no publications on NF-kB involvement in SCA2 pathogenesis, downregulation of the NF-kB pathway was observed in the gene co-expression network analysis in SCA2-127Q mice [131].
Calcium Dysregulation
Calcium ions represent an essential part of cell signaling and govern an allosteric regulatory effect on many enzymes and regulatory proteins. Calcium can activate the ion channels and influence the AP shape and frequency, and changes in the calcium concentration levels can control the important physiological processes including learning, memory, and behavior. High levels of calcium in the cytoplasm provoke apoptosis. Deranged neuronal calcium signaling may result in disturbances of the cell homeostasis, synaptic loss and dysfunction, and eventually in cell death. Calcium dysfunction was observed in the models for Huntington’s disease (HD), SCAs, and Alzheimer’s disease [11–14].
Calcium signaling plays a special role in the cerebellar PC functioning as PCs express a very big amount of various calcium-dependent proteins and enzymes to maintain the intracellular calcium homeostasis. Thus, the cerebellar PCs have high concentrations of the dendritic calbindin D-28k and somatic parvalbumin. These proteins belong to the large family of EF-hand calcium-binding proteins [148], and their loss leads to the alterations in the function of Cav2.1 channels (P/Q-type voltage-dependent calcium channels (VDCCs)), encoded by the CACNA1A gene [149].
The regulation of the calcium influx to PCs through VDCCs is very important for the proper formation of a climbing fiber (CF)–PC synapse during postnatal development [150]. The abnormalities in the CF–PC circuitry contribute to the neuronal impairment in SCA [151]. The PCs also highly express the calmodulin-binding transcription activator 1 (CAMTA1) and deletion of the CAMTA1 gene in mice causes a severe ataxia with the PC degeneration and cerebellar atrophy [152]. The LTD at PF on a PC is considered to be the main basis for motor learning. PCs express the calcium/calmodulin-dependent protein kinase II whose activation leads to a prolonged increase of cGMP, supporting the signaling mechanism of the LTD induction by calcium/calmodulin-dependent protein kinase II [153].
Inside the cell, calcium is stored in the ER and mitochondria. It has been shown that mitochondrial fission and mitochondrial transport are involved in the development of the dendritic arborization in the cerebellar PCs [154]. Mutations in the mitochondrial protease AFG3L2 implicated in the mitochondrial proteome support cause SCA28 [155].
There are 2 general ways of calcium entry into the cytoplasm of a PC. In both cases, glutamate, an excitatory neurotransmitter, is required. The first way is the calcium influx through the VGCCs from the extracellular space. These channels are activated by the membrane depolarization, caused by the activation of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors by glutamate, released from the presynaptic terminals of the PF and CF to the synaptic clefs. The second way is the activation of metabotropic glutamate receptor (mGluR) which leads to a calcium release from the ER through activation of the IP3R, and this calcium influx is called the IP3-induced calcium release (IICR). The activation of mGluR or other G protein-coupled receptors leads to the activation of a phospholipase C on the cell membrane that catalyzes the hydrolysis of the phospholipid phosphatidylinositol 4,5-bisphosphate into a diacylglycerol (DAG) and IP3 that are secondary messengers implicated in the signal transduction and lipid signaling in different cell types. DAG stays bound to the membrane, whereas soluble IP3 is released into the cytosol. Next, IP3 binds to the IP3R provoking the IICR leading to the increase of the intracellular calcium level that starts a cascade of signaling changes and affects the activity of various proteins and enzymes [156]. It was recently shown that the IICR initiates at numerous immobile sites and the more intense stimulation increases the probability of a site initiating a calcium puff without increasing the underlying number of the competent calcium release sites [157].
The activation of mGluR1 results in the DAG-mediated positive nodulation of a transient receptor potential canonical 3 (TRPC3), a nonselective cation channel, that forms the slow excitatory postsynaptic currents. Loss of the TRPC3-mediated slow excitatory postsynaptic currents leads to cerebellar ataxia caused by mGluR dysregulation [158]. It has been shown that the transcriptional factor retinoid-related orphan receptor α (RORα) is involved in mGluR1 expression [159] and also that it upregulates the expression of the multiple genes involved in the calcium-dependent pathways including the SLC1A6 encoding the excitatory amino-acid transporter 4 (EAAT4), ITPR1 encoding IP3R1, and PCP2/L7 encoding PcP2/L7 protein [160]. To achieve this upregulation, RORα interacts with the ataxin-1, but the polyQ-expanded ataxin-1 fails to make a transcription complex with RORα, so the expression of the important genes of calcium homeostasis is disturbed in SCA1 mice leading to neurodegeneration [161].
Microarray transcriptome studies on ataxin-2 KO mice and SCA2-42KI mice cerebellum have revealed in these mice similar dysregulation of several factors involved in calcium homeostasis pathways together with the multiply factors of RNA processing, bioenergetics, cell adhesion and growth, and lipid signaling [54]. In particular, a decrease was observed in the mRNA levels of the genes, encoding important calcium homeostasis proteins, such as sarco/endoplasmic reticulum calcium ATPase, IP3-phosphatase (5PP), IP3R, and transcription factor RORα [54].
In the experiments on the planar lipid bilayer model system via the single channel recordings of a IP3R1 co-expressed with a polyQ-expanded ataxin-2, it was demonstrated that the mutant ataxin-2 protein binds to the C-terminal domain of the IP3R1 (Fig. 2) and that finding was not observed for the WT ataxin-2. The expression of the mutant ataxin-2 significantly enhanced the IP3R activation by IP3 (Fig. 2) [21]. Further studies on calcium imaging carried out in the primary PC cultures from the SCA2-58Q transgenic and WT mice have determined the significantly increased IICR via the IP3R1 in SCA2-58Q PCs, but not in WT PCs. To reduce the IICR through the inhibition of the ryanodine receptors (RyRs), other intracellular calcium channels, ryanodine, or dantrolene was applied in the PC cultures (Fig. 2). The pathological effect of the presence of the mutant ataxin-2 was abolished by these treatments as the IICR converted to WT levels. It was suggested that dantrolene and ryanodine improved the altered calcium signaling by a reduction of the calcium release from the ER [21]. Long-term feeding of SCA2-58Q mice with dantrolene rescued the motor coordination defects and alleviated the PC loss [21].
Fig. 2.
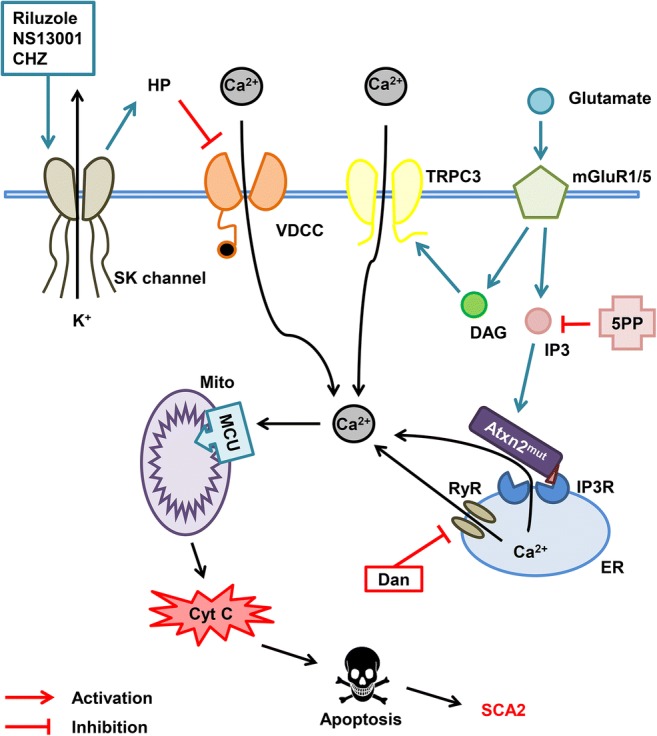
Calcium hypothesis of SCA2 pathogenesis. Molecules of glutamate released into the synaptic cleft activate metabotropic glutamate receptor (mGluR) promoting the release of inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG) molecules into the cytoplasm and further activation of the IP3 receptor (IP3R) on the endoplasmic reticulum (ER) membrane leading to the calcium entry from the ER to the cytoplasm. This process is called IP3-induced calcium release (IICR). DAG directly activates transient receptor potential channel 3 (TRPC3) and more calcium ions pass to the cytoplasm from extracellular space. It was shown that mutant ataxin-2 protein (Atxn2mut) with pathological polyglutamine expansion, but not wild-type Atxn2, associates with IP3R and increases its sensitivity to IP3. Hyperactivation of IP3R results in the abnormal calcium signaling in cerebellar Purkinje cells. Overloaded calcium (Ca2+) ions are pumped into the mitochondria (Mito) through the mitochondrial calcium uniporter (MCU) leading to the mitochondrial swelling, followed later by the rupture of outer mitochondrial membrane and further release of pro-apoptotic factors like cytochrome C (Cyt C) into the cytoplasm thus initiating apoptosis. Dramatically increased IICR can be suppressed by adeno-associated virus-mediated expression of the IP3-5-phosphatase enzyme (5PP) which converts IP3 into nonactive form IP2. Another way to reduce calcium release from ER may be the inhibition of ryanodine receptors (RyRs) with dantrolene (Dan). Small-conductance calcium-activated potassium channels (SK channels) are required for proper PC pacemaker activity. SK channel activation with riluzole, NS13001, and chlorzoxazone (CHZ) increases hyperpolarization (HP) of the PC membrane thus inhibiting the voltage-dependent calcium channels (VDCC) leading to the decrease of calcium influx from extracellular space
In further experiments, the chronic suppression of IICR via the adeno-associated virus–mediated expression of the 5PP enzyme was achieved in SCA2-58Q PCs to reduce the IP3 level in SCA2 PCs (Fig. 2) [22]. The patch-clamp experiments on the cerebellar slices showed improvement in the age-dependent abnormal firing of SCA2 PCs. Examination of the motor coordination and the subsequent neuropathological analysis have shown that the chronic 5PP overexpression also alleviated the age-dependent motor decline and PC death in SCA2-58Q mice [22]. The obtained results support the idea that the disturbed calcium signaling may play an important role in SCA2 pathogenesis. Thus, the partial suppression of the IICR could provide a therapeutic benefit for SCA2 patients and possibly for patients with other types of SCAs [11, 22].
Recent experiments on the cerebellar slices from SCA2-127Q transgenic mice via the combined method of the whole-cell current clump recordings and 2 photon calcium imaging have shown that the somatic calcium levels were elevated over a physiological range of firing frequencies in the SCA2-127Q PCs and the synaptic activation of mGluR1 by the PFs’ electric stimulation has revealed the significant increase in the slow EPSPs and the larger intracellular calcium release in SCA2-127Q PCs [162]. The enhanced mGluR signaling was also observed in these mice. The authors suggested that the SCA2 pathology is relying on the positive feedback mechanisms based on the enhanced calcium level on mGluR1 coupling to TRPC3 channels and to IICR (Fig. 2) [162].
The recent mechanistic studies on the SCA2-iPSC-derived neurons have demonstrated that overexposure to glutamate alters the expression of the mGluR genes and negatively affects the calcium signaling. Treatment of the SCA2-iPSC-derived neurons with the anti-glutamate drugs such as riluzole, NMDA receptor antagonist dizocilpine, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor antagonist NBQX, and the calcium stabilizer dantrolene reduced cell death and improved mitochondrial dysfunction, thus supporting the idea of mGluR involvement in SCA2 molecular pathology [133].
Changes in the PC spontaneous firing, observed in various models of SCA2 mice [22, 126, 163], might also be explained by the altered calcium and mGluR-mediated signaling. Calcium-activated potassium SK channels are responsible for the PC pacemaker activity; thus, the slowed pacemaking of SCA2-PCs occurs because the constantly enhanced level of the intracellular calcium [164]. It was also suggested that the expression and/or post-translational modulation of the various ion channels required for the normal electrophysiological function of PCs might be disturbed because of the chronically increased calcium levels [164].
Autophagy
The polyQ-expanded mutant proteins tend to aggregate and form toxic inclusions and autophagy is required for their clearance. Autophagy is a selective lysosomal-mediated degradation process when the accumulated and aggregated ubiquitin-labeled proteins are destroyed. Proteins essential for autophagy include the microtubule-associated protein 1 light-chain 3 (MAP1LC3, gene MAP1LC3), p62 (gene SQSTM1), and autophagy-linked FYVE domain protein (Alfy, gene WDFY3). The examination of the peripheral markers of autophagy in polyQ patients has revealed the increased expression levels of the MAP1LC3B, SQSTM1, and WDFY3 in HD patients, whereas in SCA2 patients, only the WDFY3 level was elevated [165]. The authors suggested that such a difference may be explained by the assumption on much less aggregation formation in SCA2 compared to HD which is also supported by the rare occurrence or absence of the nuclear inclusions in SCA2 mice [124].
In the experiments on SCA2 patient-derived fibroblasts and SCA2 mice, it has been demonstrated that the polyQ expansion in ataxin-2 also leads to abnormal autophagy, accompanied by the enhanced expression of staufen1 (STAU1), a double-stranded RNA-binding protein required for the formation of the cytoplasmic inclusions in neuroglia and cultured cells and involved in the modulation of SG functions, co-localized with ataxin-2 in SG-like structures [166]. It has been shown that the increased STAU1 levels induced the aberrant processing of the RNA targets. Although more studies on the role of ataxin-2 in the abnormal autophagy are still required, it has been shown that the reduction of the STAU1 levels improved the impaired motor coordination in SCA2 mice and may be used as a potent therapeutic approach for SCA2 treatment [166].
Role of Microglia
The role of the cerebellar microglia in SCA pathogenesis was recently described in a comprehensive review by Ferro et al. [167]. The microglia is a type of neuroglia representing the macrophage cells of the central nervous system. These cells take part in the processes of development and aging; their main functions include immune response formation and maintaining homeostasis, including scavenging, phagocytosis, and extracellular signaling. The development of a microglia is strictly regulated by the distinct transcriptional factors [168]. Microglia in its turn plays an important role in cerebellar development. Thus, it takes part in the pruning of the excessive and dysfunctional CF–PC synapses during postnatal development that is required for the proper operation of the cerebellar circuits in the adult life [169]. Studies on SCA1 mice, indeed, have demonstrated that CF development is affected in these mice [170]. It was shown that at early stages, each PC is innervated by multiple CFs, but as development proceeds, only 1 strengthened CF becomes the dominant maintaining its synapses with the PC dendritic tree. In SCA1-82Q-S776 mice, the translocation of the dominant CF terminals up the PC dendritic tree was delayed in time, and some of its terminals remained on the PC soma, whereas in SCA1-30Q-D776, the impaired pruning led to the much more severe consequences [170].
Microglial activation in the cerebellum has been detected in SCA patients and several SCA mice models. In different SCA1 mice models, the early increase in the microglial number and TNF-α was observed before neuronal cell death and this glial activation tightly correlated with pathological progression, suggesting a possible mechanism of neurodegeneration based on the microglia-driven neuroinflammation [171]. Pharmacological inhibition of the colony-stimulating factor receptor 1, a protein that is responsible on the microglia production, differentiation, and function, resulted in the depletion by 69% in the cerebellar microglia leading to the amelioration of the motor deficits in SCA1 mice [172].
Neurophysiology of the PCs and Changes in the Cerebellar Circuit Function in SCA2
The cerebellum represents a part of the brain that plays a fundamental role in the regulation of muscular activity and motor control. Cerebellar pathways are the key players in the coordination of movements. The PCs are considered to be the main dynamic element in the cerebellum because their axons represent the unique output coming from the cerebellar cortex to the cerebellar nuclei and other deep structures of the brain (Fig. 3). The PCs spontaneously fire the action potentials at a constant frequency [173–180]. This tonic pacemaking activity of the PCs is assumed to be crucial for the correct encoding of the cortical cerebellar information [125, 180, 181]. In studies with the cerebellar slices from ataxia mouse models, it has been reported that the neuronal activity of the aged PCs in the various models of ataxia is abnormal when compared with the PCs from the age-matched WT mice [22, 125, 163, 182–186]. These results suggested an idea that the early ataxic symptoms can be caused by a neuronal dysfunction, i.e., disturbances in firing patterns of PCs, and not by PC death. It was assumed that in different SCAs a common pattern of the PC electrophysiological dysfunction exists, and addressing these abnormalities might have a therapeutic potential [187].
Fig. 3.
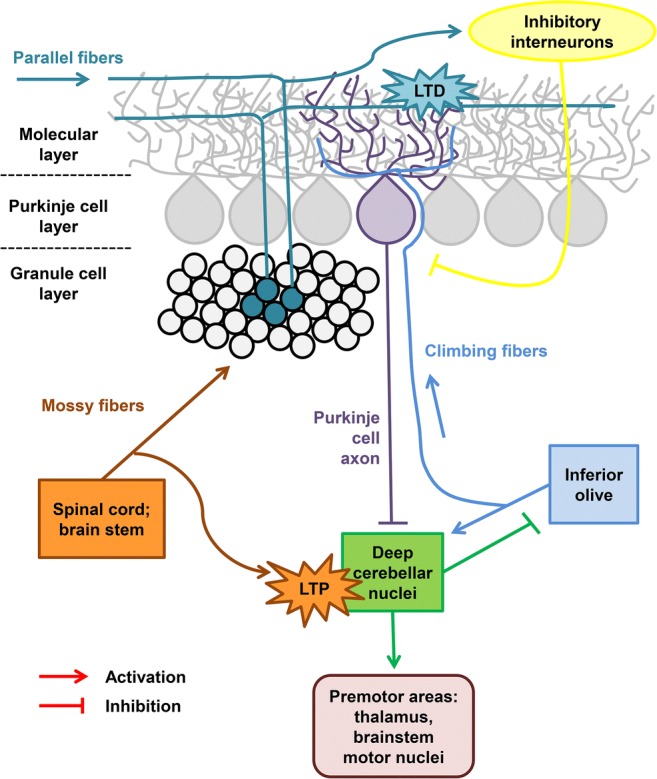
A schematic overview of the cerebellar circuit. The cerebellar Purkinje cells (PCs) organize the Purkinje cell layer in the cerebellar cortex and their axons form the unique output coming from the cerebellar cortex to the cerebellar nuclei. The PCs receive inhibitory and excitatory impulses through multiple synapses formed on their dendritic tree, which is located in the molecular layer. Climbing fibers (CF, in blue) originate from neurons in the inferior olive, climb to the cerebellum, and form between 250 and 1500 synapses upon a single PC dendritic tree, in a unique 1:1 ratio. Parallel fibers (PF, in pine) originate from granule cells in the granule cell layer of the cerebellar cortex and form a few synapses upon the distal dendritic tree of a single PC. Each PC dendritic tree forms as many PF–PC synapses as 200,000 parallel fibers contacting them. Mossy fibers (MF, in orange) originate from neurons in the spinal cord and brain stem and transport information from the periphery and cerebral cortex to PCs through granular cell axons forming PF. The pine and orange starbursts depict supposed sites of CF-mediated, associative forms of synaptic plasticity required for associative motor learning (LTD of PF inputs and LTP of MF inputs, respectively). These forms of plasticity may be impaired in SCA2. Modified from Smeets and Verbeek [8] and Meera et al. [164]
The disturbance of the PCs’ electrophysiological functions was firstly detected in SCA2-127Q transgenic mice: the PCs demonstrated a progressive, age-dependent decrease in the firing frequency together with a progressive decline in the motor performance, the beginning of which was observed before the PCs’ degeneration [130]. Further, it was demonstrated that in the SCA2-58Q transgenic mouse model, the PCs’ firing frequency was also significantly decreased with time, but additionally, significant reduction in firing precision and abnormal burst pattern of firing were observed [22, 125].
Recent studies on SCA2-127Q transgenic mice have revealed that reduced expression of the Kv3.3 voltage-gated potassium channels and large-conductance calcium-activated potassium channels (BK channels) followed the reduction in the firing frequency in SCA2-127Q PCs and the development of a newly discovered afterhyperpolarization with slow kinetics [188]. It was shown that the diverse ion-channel mutations lead to SCA. In polyQ ataxias, the altered ion-channel function is observed because of the changes in the ion-channel transcript levels [189]. A recent panel study on patients with a dominantly inherited cerebellar ataxia discovered the prominent implication of the point mutations in the genes of the calcium voltage-gated channel subunit alpha 1 A (CACNA1A) and in a gene known to cause the hereditary spastic paraplegia (SPG7) in the progressive ataxias [190]. Mutations in the other ion channels also can lead to ataxia and movement disorder phenotype. Thus, a new mutation in the voltage-gated sodium channel type VIII alpha subunit was recently found in mice with a chronic movement disorder with the early-onset tremor and adult-onset dystonia characterized by a shortened lifespan and the impaired motor functions. The PCs from these mice also displayed the lack of the spontaneous and induced firing activity [191].
Although the PCs are primarily affected in SCAs, the impairment of the cerebellar pathways is also observed. Normal functionality of the excitatory PC inputs, such as CFs, parallel fibers, and their synaptic plasticity, is required for the normal cerebellar function and motor behavior (Fig. 3) [8]. It was demonstrated that the dysfunction of the PCs and cerebellar fibers provokes the ataxic phenotype before the PC loss, and the CFs control the proper functioning of the PCs [8]. Proper PC inhibition in the cerebellar circuit is necessary for cerebellar memory formation [192]. There are lines of evidence that CFs participate in both motor learning and motor control functions of the cerebellum, and might also play a role in cerebellar development [193]. The study on the CF–PC synaptic pathology in the cerebellum of patients with different degenerative movement disorders revealed the distinct CF pathological features across these disorders, reflecting most likely the different mechanisms of the movement pathologies [194].
The PC generates 2 types of spikes: the simple spikes and complex spikes (CS). These different types of spikes are produced via the 2 main types of the afferent fibers in the cerebellum: the mossy fibers (MF) and CF (Fig. 3). MF originate from neurons in the spinal cord and brain stem and transport the information from the periphery and cerebral cortex to PCs through the granular cells axons forming the PF. CF originate in the inferior olive nucleus and send the information from the cortex to PCs by making the numerous synaptic contacts with the proximal dendrites of a PC. CF carry an excitation that makes PC generate a CS: an initial large-amplitude action potential followed by a high-frequency burst of potentials with smaller amplitude (also called spikelets). At the same time, the PC generates the simple spikes responding to the excitatory potentials produced by the PF [176, 195, 196]. The pharmacological stimulation of the olivocerebellar circuit by harmaline, an alkaloid that acts directly on the inferior olive neurons, uncovered the disturbances in the SCA2-58Q PC activity pattern in vivo and in the CS shape when compared with the age-matched WT cells [127]. The abnormalities in the CF–PC circuitry were aggravated with age. Thus, it was proposed that the alterations in the CF–PC circuitry represent 1 of the potential causes of ataxic symptoms in SCA2 and in other SCAs [127].
Therapeutic Approaches to SCA2
The effective therapeutic treatment and disease-modifying therapy for SCA2 is not available yet, only the symptomatic treatment is prescribed to patients, but it is incapable of preventing pathology development and brings only a temporary relief of symptoms at the early stage of the disease. Many attempts were made in studying the physiological, biochemical, and functional brain characteristics in the pathology of SCA2 to find new therapeutic targets for SCA2 treatment in the early stages and preventing the development of further symptoms. The modern understanding of the uncovered molecular mechanisms of the disease allowed the scientific community to underlie the strategies of a potential therapy and helped to create some promising therapeutic approaches.
Antisense Oligonucleotide Therapy
One of the most promising therapeutic approaches for SCA2 to date is the ASO therapy (Fig. 4a) as this technique showed promise in clinical trials with patients suffering from HD, another polyQ disease [197]. In the ASO therapy, the oligonucleotide sequence complementary to a target mRNA is used to suppress its expression and, thus, reduce the target protein level (Fig. 4a). A recently performed in vitro screening of the 152 ASOs designed in silico as targets for human ataxin-2 identified ASO7 as a most promising oligonucleotide for SCA2 ASO therapy [17]. In the subsequent studies, ASO7 was delivered to mice by intracerebroventricular injection. The SCA2-127Q and BAC-72Q transgenic SCA2 mice models were used in this study [17]. ASO7 injections led to the reduction of the human ATXN2 expression in mice cerebellum by 75%. Both mouse models exhibited significant improvement in motor functions after treatment with ASO7 according to a rotarod assay [17]. ASO7 also restored the slow firing frequency of the PCs from both transgenic SCA2 lines. Interestingly, in both mouse models, the treatment with ASO7 normalized the protein levels of several SCA2-related proteins expressed in PCs, including a regulator of G protein signaling (Rgs8) that suppresses the mGluR1 activity, a regulator of G-coupled receptors including the P-type calcium channel involved in the sensorimotor control (Pcp2), Pcp4 required for the normal development of the PF–PC synapses, another mGluR1 regulator Homer3, and Cep76 protein that is necessary for normal centriole function [17]. Therefore, these findings support the idea that ASO therapy is promising as a potential treatment for SCA2 and other polyQ diseases [17, 18]. Interestingly, the lowering ataxin-2 via ASOs also showed promising results in a mouse model of amyotrophic lateral sclerosis in which ataxin-2 with an intermediate-length polyQ expansion has been reported as a risk factor [198].
Fig. 4.

Strategies for SCA2 therapeutic development. (a) Antisense oligonucleotide (ASO) therapy. The oligonucleotide sequence complementary to a target mRNA is used to prevent the translation of the ataxin-2 mutant (Atxn2mut) protein. (b) Stem cell therapy. Mesenchymal stem cells are extracted from healthy donors and intravenously administrated into SCA patients. Stem cells act as immunomodulators and neuroprotectors and regenerate affected neurons in SCA patients. (c) Calcium stabilizers, such as ryanodine, dantrolene, riluzole, inositol-1,4,5-trisphosphate-5-phosphatase (5PP), NS13001, and chlorzoxazone (CHZ), normalize calcium signaling disturbed in SCA2 cerebellar Purkinje cells (PCs), improving PC firing and morphology and ameliorating motor performance of SCA2 mice. (d) Prevention of mutant ataxin-2 aggregation can be achieved by the polyQ-binding proteins such as polyQ-binding protein 1 (QBP1) or huntingtin polyQ-binding peptoid 09 (HQP09) that inhibit polyQ aggregation. Another way to suppress mutant ataxin-2 aggregation is the overexpression of the co-chaperone/E3 ligase C-terminal constitutive Hsp70 (Hsc70)-interacting protein (CHIP) that links the protein folding machinery with the ubiquitin–proteasome system (UPS) and lead to the mutant ataxin-2 degradation in the proteasome. Molecular chaperones such as heat shock protein (HSP) 70 kDa (Hsp70) and heat shock transcription factor (HSF1) that regulates the expression of the HSPs also provoke significant suppression of aggregation formation
Anionic oligonucleotides penetrate the blood–brain barrier very poorly or do not cross it at all; this gives rise to a consistent concern about the simple methods of ASO delivery to the cerebellar cells of SCA2 patients. To address this issue, a novel liposome formulation, DCL64, was developed [199]. Confocal microscopy studies have revealed that this vehicle was selectively absorbed by the brain microvascular endothelial cells through the selective interaction with the low-density lipoprotein receptor family members on the cell membrane which are abundantly localized on the cerebellar PCs. Intravenous administration of the DCL64 in mice confirmed the prevalent distribution of oligonucleotides in the cerebellar PCs compared with other brain regions. No pathological side effects were revealed in the use of DCL64. This property potentially makes the DCL64 particularly attractive as a promising delivery vehicle in the treatments of SCAs [199].
Stem Cells
Another promising therapeutic approach for SCA2 treatment involves the implication of the mesenchymal stem cells (MSCs) (Fig. 4b). The MSCs represent the multipotent stromal fibroblast-like cells that can differentiate into different cell types and also exhibit some immune-modulatory functions as release of the neurotrophic factors. The study on the transgenic SCA2 mice has shown that the intravenous delivery of the human MSCs led to significant improvement in the motor activity assessed by a rotarod performance in SCA2 mice and also delayed the onset of motor coordination dysfunction by preventing PC loss in the SCA2 cerebellum [200]. Previous experiments, conducted on Lurcher mice, which have a mutation in the delta2 glutamate receptor that lead to the degeneration of the cerebellar PCs, olivary neurons, and granule cells [201], have shown that the transplanted MSCs migrate throughout the cerebellum toward the PC layer and express the neurotrophic factors such as a brain-derived neurotrophic factor, neurotrophin-3, and glial cell-derived neurotrophic factor, thus improving PC survival [202].
Following the promising results of the MSC treatment observed in the SCA2 mouse model, a study on the effect of MSCs was carried out on SCA3 patients. The pilot, open-labeled, phase I/IIa clinical study was conducted in Taiwan to check the safety, tolerability, and efficacy of the intravenous injections with the allogeneic adipose tissue-derived MSCs from healthy donors [19]. The experimental group consisted of 6 SCA3 patients. Patients were aged between 21 and 66 years, the disease duration was in between 3 to 13 years, and SARA scores at baseline were between 12 and 16 points. SCA3 patients were injected with 106 MSCs cells/kg body weight. Following the 1-year monitoring revealed no significant side effects: all subjects tolerated the procedures well and successfully completed the study. Efficacy evaluations included clinical assessment with SARA, sensory organization testing, oculomotor testing, magnetic resonance spectroscopy, and 18F-fluorodeoxyglucose positron emission tomography of the brain. The SARA scores assessment showed insignificant improvement in SCA patients most likely because of the limited number of subjects that was the limitation of this phase I/IIa study. Thus, the difference in SARA units was − 0.25 at 0.5 months, 0 at 6 months, and 0 at 12 months. In 1, 3, and 6 months after the MSC injection, 83% of the SCA3 patients had a significantly better performance in the sensory organization testing. In 9 months after the injection, the significant improvement of the cerebral and cerebellar glucose consumption was observed in 5 of 6 SCA3 patients. The obtained results suggest the tolerability and safety of the treatment with MSCs, but a larger-sized, randomized, double-blind, placebo-controlled phase II clinical trial has to be conducted to confirm the possible therapeutic use of MSC therapy for ataxia treatment [19]. However, it remains unclear if the MSC-derived neurons are able to properly integrate into the cerebellar circuit.
Calcium Stabilizers
Many SCAs share the same features of the molecular mechanism pathology. The disturbed calcium signaling and downregulation of several calcium homeostasis proteins have been reported in the different types of ADCA. The positive modulation of the calcium-binding protein expression and the suppression of the calcium entry from the intracellular stores may be a useful strategy for PC functional improvement by blockage of the enhanced calcium level neurotoxicity [203].
Indeed, the experiments on SCA2-58Q transgenic mice have shown that the inhibition of the ER calcium channels RyRs with ryanodine or dantrolene normalized the altered calcium signaling in the SCA2 PCs, and a long-term feeding of SCA2-58Q mice with dantrolene rescued the motor coordination defects and alleviated the PC loss in SCA2 mice (Figs. 2 and 4c) [21]. The chronic suppression of the IICR via the adeno-associated virus–mediated expression of the 5PP reduced the IP3 level in the SCA2 PCs and alleviated the age-dependent motor decline and PC death in SCA2-58Q mice (Figs. 2 and 4c) [22]. Thus, the partial suppression of the IICR could provide a therapeutic benefit for SCA2 patients and possibly for patients with other types of SCAs [11, 22].
Recent experiments on the SCA2 and SCA3 patients’ iPSC-derived neurons have shown that the glutamate toxicity alters the expression of the mGluR-related genes and develop the SCA phenotype in these neurons [133]. The studies on the measuring of the intracellular calcium concentration with Fluo-4 showed the reduction in the calcium levels after treatment with dantrolene and riluzole in the SCA-iPSC-derived neurons. Overall, the results of the experiments on the iPSCs from SCA patients suggested that molecular targeting of the mGluR or calcium-mediated signaling and treatment with calcium stabilizers may have beneficial effects on the SCA pathological phenotypes [133].
It was previously demonstrated that the PC pacemaker activity is regulated by a small-conductance calcium-activated potassium channel (SK channel) activity [204]. Constantly increased concentration of the intracellular calcium ions slows down the pacemaker activity of the cerebellar PCs through the SK channels [164]. Targeting SK and BK channels for the therapeutic modulation of the SCA2 pathology had some promising results. The combination of the calcium-activated potassium channels activator chlorzoxazone (CHZ) and GABAB agonist baclofen, that potentiates a subthreshold-activated potassium channel current, restored the spiking in SCA1 PCs and improved the dendritic hyperexcitability in SCA1-82Q mice and also sustained the improvement in the motor dysfunction in these mice [23]. It has also been demonstrated that the subtype nonselective SK channel positive modulator 1-ethyl-2-benzimidazolinone and CHZ normalize the PC firing and exert beneficial effects in a mouse model of episodic ataxia type 2 [182, 186, 205]. Exposure to SKA-31, a riluzole analogue optimized for SK channel activation, provided benefit in a mouse model of SCA3 by correcting the abnormal PC firing and improving motor function in SCA3 mice [185].
Studies on SCA2-58Q transgenic mice demonstrated that a novel specific positive modulator of SK2/3 channels (NS13001) stabilized the PC firing rates and exerted beneficial effects in SCA2 mice [125]. Thus, the application of NS13001 reverted the abnormal bursting pattern into the normal tonic rate of a repetitive spiking, and a significant improvement in motor performance was also observed [125]. Using an extracellular single-unit in vivo recording method to compare the spontaneous activity of the PCs in the age-matched WT mice and SCA2-58Q transgenic mice, it was discovered that the fraction of the PCs with a bursting and irregular pattern dramatically increased in the aged SCA2-58Q mice compared with the WT mice at the same age [126]. Intraperitoneal injections with CHZ normalized the firing activity of the cerebellar PCs from the aging SCA2-58Q mice [126]. These results provided further mechanistic support to the hypothesis that positive modulators of SK channels may help to stabilize the abnormal firing of ataxic PCs and hold promise as potential therapeutic agents for ataxia (Fig. 2).
Another small molecule riluzole binds and allosterically modulates the SK2 channels [206]. A randomized, double-blind, placebo-controlled trial on SCA patients revealed that treatment with 100 mg/day riluzole improved the SARA scores in subjects and did not have any significant side effects [20]. The experimental groups included 19 SCA patients and 9 patients with Friedreich’s ataxia in the riluzole group and 19 SCA and 8 Friedreich’s ataxia patients in the placebo group. The average age of the patients was 46.2 years in the riluzole group and 43.1 years in the placebo group. The average SARA score at baseline was 15.3 points in the riluzole group and 16.5 points in the placebo group. Patients were given 100 mg riluzole per day during a 12-month period. The following procedures were applied over the study period: clinical assessments, ECG, and laboratory tests every 3 months; SARA at months 3 and 12; and Short Form Health Survey Questionnaire and Beck Depression Inventory after 12 months. The significant improvement in SARA score of 5.5 points or lower was observed in 50% patients in the riluzole group at month 12, whereas in the placebo group, this was only 11%. It was revealed that there was no significant difference in the Beck Depression Inventory between the riluzole and placebo groups. The mean score for the quality of life was measured by the Health Survey Questionnaire and was significantly higher in the riluzole group compared with the placebo group [20].
Thus, studies on the animal models and clinical trials on SCA patients demonstrated the potential beneficial effect of riluzole and other SK and BK channel activators (Fig. 2).
Aggregation as a Therapeutic Target
The inclusion bodies and aggregates have been found postmortem in the tissues from SCA2 patients. Misfolded polyQ-expanded ataxin-2 organizes into insoluble aggregates with β-sheet–rich amyloid fibrillar structures gaining thus the toxicity. Prevention of the aggregate formation and accumulation of the misfolded polyQ proteins has been studied as a promising therapeutic approach for the development of the disease-modifying therapies for the polyQ diseases (Fig. 4d) [9].
The screening of a combinational peptide library expressed on the M13 phage pIII protein was performed to identify the peptides that bind the polyQ-expanded proteins preventing their aggregation and the polyQ-binding protein 1 (QBP1) was chosen as a best candidate [207]. In vitro studies on the transfected fibroblast-like cell line COS-7 have shown that QBP1 potently suppresses the polyQ-mediated cell death in these cells (Fig. 4d) [207]. Consequent experiments on the fly polyQ disease model which expresses an expanded polyQ specifically in the eyes have demonstrated that treatment with QBP1 inhibited the polyQ aggregation and rescued the degeneration in the compound eye in these flies, whereas on the fly model with a polyQ expression in the nervous system, the application of QBP1 significantly increased the average lifespan from 5.5 to 52 days [208]. Long-term intraperitoneal administration of QBP1 fused with a protein transduction domain to polyQ mice ameliorated the weight loss phenotype of these mice, whereas the rotarod performance remained the same between polyQ and WT mice probably because of the limited blood–brain barrier permeability of the QBP1-protein transduction domain [209]. The single chain Fv antibodies specific for the 17 N-terminal residues of a mutant huntingtin prevented the huntingtin aggregation and suppressed the neuropathology in the cell [210], fly [211], and mouse [212] models of HD.
Another screening study selected the huntingtin polyQ-binding peptoid 09 as a specific ligand of the N-terminal fragment of a mutant huntingtin protein (Htt-N-82Q) and ataxin3-77Q mutant protein that exhibited the specific polyQ-binding activity (Fig. 4d) [213]. Huntingtin polyQ-binding peptoid 09 inhibited the mutant Htt aggregation and stabilized the glutamate-induced calcium signals in the striatal neurons from HD mice and also rescued the HD neurons from the glutamate-induced apoptosis. The intracerebroventricular delivery of HPQ09 into HD mice brain suppressed the mutant Htt aggregation and ameliorated the beam walk performance in these mice [213].
Misfolding and consequent aggregation of the polyQ-expanded mutant proteins can be controlled by the molecular chaperones, the proteins that assist the folding and conformational maintenance of other proteins via their affinity for the hydrophobic regions of the misfolded proteins. Most of the molecular chaperones have a historical name of heat shock proteins (HSPs) and consist of members with different molecular weights—Hsp40, Hsp60, Hsp70, Hsp90, Hsp100, and the small Hsp [214]. The enhanced functional capacity of HSPs could help to prevent the initiation of the aggregate formation or disaggregation of the aggregating core and might be a promising therapeutic strategy for polyQ diseases [215]. Thus, the co-chaperone/E3 ligase C-terminal constitutive Hsp70-interacting protein (CHIP) that links the protein folding machinery with the ubiquitin–proteasome system was co-localized in the nuclear inclusions in both cell culture and SCA1 postmortem neurons, and the CHIP overexpression decreased both mutant and normal ataxin-1 levels in fly SCA1 model, suppressing the neurotoxicity of a mutant ataxin-1 (Fig. 4d) [216]. Genetic reduction of CHIP accelerated the disease in SCA3 mice and increased the mutant ataxin-3 aggregation, whereas studies on the cell model showed that CHIP selectively reduced the level of mutant ataxin-3 [217]. The overexpression of the inducible Hsp70 suppressed the neuropathological phenotype and improved the motor performance in SCA1 mice (Fig. 4d) [218]. Active forms of the heat shock transcription factor (HSF1) provoked much more significant suppression of the aggregation formation in HD mice (Fig. 4d) [219]. Although these results seemed promising, the contradictory data were obtained. Thus, the Hsp70 overexpression had no effect on the mutant ataxin-7-induced toxicity in SCA7 mice [220].
A humanized yeast-based high-throughput screening revealed a small molecule HSF1A as a potent HSF1 activator in mammalian and fly cells, which provoked the enhanced protein chaperone expression and decreased the protein misfolding and neuronal cell death in the cells and reduced cytotoxicity in a fly model of the polyQ-induced neurodegeneration [221]. Another new HSF activator, azadiradione, also exhibited promising results in the cell and fly models [222]. Although the measurement of the chaperone-mediated effects on polyQ protein aggregation seems a promising tool for research of potential therapeutic strategies and although new methods are developing [223], no articles on preclinical trials in SCA2 models were published so far.
Modifiers of Transcription, Translation, and DNA Repair
The effective DNA repair is absolutely required for the proper development of the nervous system. The disturbances in DNA reparation in the adult brain are associated with the processes of aging and neurodegeneration. Oxidative metabolism is very important for neuronal functioning, but oxidative stress can cause the single-strand breaks in DNA, leading to alterations in gene expression and even cell death [16]. Cerebellar PCs have been reported to be highly affected by oxidative stress [224]. Impaired DNA repair was observed in patients with ataxia telangiectasia [225] and some other diseases, suggesting that the functional activity of the proteins involved in DNA repair suffers in these cerebellar disturbances. Studies on the SCA3-78Q fly model revealed CAG repeat instability accompanied by declined activity of histone acetyltransferase, a regulator of DNA repair. Treating SCA3 flies with histone deacetylase inhibitors normalized the acetylation and suppressed the increased rate of repeat instability [226]. Further search for histone deacetylase inhibitors with better characteristics via a pharmacophore modeling and structural analysis defined 2 lead compounds as the potential agents for SCA2 treatment [227]. In the SCA1 mouse transgenic model, the deficiency in the Xpa null allele, which eliminates the nucleotide excision repair, had a tissue-specific effect: the CAG repeat instability was selectively reduced in the neuronal tissues, but not in the kidney or liver [228]. A more recent study on the SCA3-135Q transgenic mice, postmortem brain tissue from SCA3 patients, and SCA3 cell model has revealed the inactivation of the polynucleotide kinase 3′-phosphatase, an enzyme required for the DNA strand break repair, through the negative modulation by a mutant ataxin-3, leading to the inefficient DNA repair and constant increase of DNA damage/strand breaks in SCA3. Polynucleotide kinase 3′-phosphatase overexpression immediately stopped the mutant ataxin-3-induced cell death [229]. Thus, the mechanisms of DNA repair are also involved in the polyQ SCA pathology and their modulation could have a possible therapeutic effect in SCA treatment.
The central dogma of molecular biology states that DNA and RNA make proteins describing a 2-step process: transcription and translation. Although these 2 steps are complicated enough and require high precision of actions of the intracellular machinery, the final version of a new protein requires a few more steps. Intrinsic protein functions highly depend on the post-translational modifications (PTMs) such as phosphorylation, acetylation, ubiquitination, SUMOylation, and many others, thus affecting the protein localization, functions, stability, aggregation, and interactions with other molecules. The majority of SCAs have disease-causing mutant proteins, in which modulation by the proper PTMs might have a potential disease-modifying effect in SCA pathology [230]. Regarding SCA2 pathology in particular, the studies on COS-7 cells expressing different fragments of both mutant and normal ataxin-2 have shown the importance of the phosphorylation of ataxin-2 by cyclin-dependent kinase 5 (Cdk5) [231]. The Cdk5-mediated phosphorylation induced the degradation of the N-terminal fragment remarkably and middle fragment moderately, and the mutant ataxin-2 with 41 CAG repeats was exposed to the proteasomal degradation after the phosphorylation by Cdk5 suggesting the increased activation of Cdk5 as a therapeutic strategy for SCA2 [231]. Although the involvement of other PTMs in the modulation of SCA2 pathology has not been described yet, the field is wide open for extensive research on the role of PTMs in SCA2 pathology.
Studies on cell and fly models of the polyQ-expanded diseases have shown that the expanded CAG RNA interacts with the nucleolin, a nucleolar protein implicated in the synthesis and maturation of ribosomes, thus preventing the preribosomal RNA transcription and leading to a nucleolar stress-induced apoptosis in polyQ diseases [232]. Further experiments demonstrated that a new synthetized nucleolin-derived beta-structured inhibitor for neurodegenerative diseases fused together with a TAT peptide, a cell-penetrating peptide that facilitate cellular uptake of different cargos, directly interacted with a CAG-expanded RNA, suppressed the nucleolar stress in cells, inhibited the mutant RNA-induced neurodegeneration in polyQ flies, improved the climbing ability and extended a lifespan of these flies, and finally, suppressed the neurotoxicity in SCA2 and SCA3 cell and fly models [233]. Thus, the exclusion of the mutant polyQ protein from the transcription process and suppression of its altered functions might have a potential therapeutic effect in SCA2.
Conclusion
Although SCA2 still remains an incurable progressive neurodegenerative disorder, there are some promising therapeutic approaches that may give decisive results in the treatment of SCA2 patients. Encouraging data have been obtained in preclinical studies with ASO therapy, Ca2+ stabilizers, and SK-positive allosteric modulators and in some pilot clinical trials with MSCs and riluzole. The abolishment of the mutant ataxin-2 neurotoxic functions on the protein level might be also a perspective strategy, although more studies are required. Thus, clinical trials using ataxin-2–targeted ASO therapy, stem cell treatment, Ca2+ stabilizers, and SK channel modulators should be carried out to give us the chance of breaking the vital problem of incurable SCA and other polyQ expansion diseases.
Acknowledgments
Ilya B. Bezprozvanny is a holder of the Carl J. and Hortense M. Thomsen Chair in Alzheimer’s Disease Research. This work was supported by the National Institutes of Health Grant R01NS056224 (Ilya B. Bezprozvanny), Russian State Grant 17.991.2017/4.6 (Ilya B. Bezprozvanny), Russian Science Foundation Grant 18-75-00025 (Polina A. Egorova), Presidential Grant МК-1299.2019.4 (Polina A. Egorova), and Russian Science Foundation Grant 19-15-00184 (Ilya B. Bezprozvanny). The financial support was divided in the following way: research work related to Figure 1 was supported by the Russian State Grant 17.991.2017/4.6, research work related to Figure 2 was supported by the Presidential Grant МК-1299.2019.4, research work related to Figure 3 was supported by the Russian Science Foundation Grant 18-75-00025, and research work related to Figure 4 was supported by the Russian Science Foundation Grant 19-15-00184.
Abbreviations
- ADCA
Autosomal dominant cerebellar ataxia
- AP
Action potential
- ASO
Antisense oligonucleotide
- BAC
Bacterial artificial chromosome
- BK channel
Large-conductance calcium-activated potassium channel
- BN
Binucleated cell
- Cdk5
Cyclin-dependent kinase 5
- CF
Climbing fiber
- CHIP
C-terminal constitutive Hsc70-interacting protein
- CHZ
Chlorzoxazone
- CS
Complex spike
- DAG
Diacylglycerol
- DTI
Diffusion-tensor imaging
- EMG
Electromyography
- EP
Evoked potential
- ER
Endoplasmic reticulum
- HD
Huntington’s disease
- HSP
Heat shock protein
- Htt
Huntingtin
- IICR
IP3-induced calcium release
- iPSCs
Induced pluripotent stem cells
- IP3R
Inositol 1,4,5-trisphosphate receptor
- KI
Knock-in
- KO
Knockout
- LTD
Long-term depression
- MF
Mossy fiber
- mGluR
Metabotropic glutamate receptor
- MNi
Micronuclei formation
- MRI
Magnetic resonance imaging
- mRNA
Messenger RNA
- MSCs
Mesenchymal stem cells
- PABP
Poly(A)-binding protein
- PC
Purkinje cell
- Pcp2
Purkinje cell protein 2
- PD
Parkinson’s disease
- PF
Parallel fiber
- PKC
Protein kinase C
- PLM
Periodic leg movement
- PolyQ
Polyglutamine
- PTMs
Post-translational modifications
- QBP1
PolyQ-binding protein 1
- REM
Rapid eye movement
- RORα
Retinoid-related orphan receptor α
- RyR
Ryanodine receptor
- SARA
Scale for the Assessment and Rating of Ataxia
- SCA
Spinocerebellar ataxia
- SCA2
Spinocerebellar ataxia type 2
- SGs
Stress granules
- SK channel
Small-conductance calcium-activated potassium channel
- SOD
Superoxide dismutase
- TRPC3
Transient receptor potential canonical 3
- VBM
Voxel-based morphometry
- VDCCs
Voltage-dependent calcium channels
- WT
Wild type
- 5PP
IP3-phosphatase
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Ashizawa T, Oz G, Paulson HL. Spinocerebellar ataxias: prospects and challenges for therapy development. Nat Rev Neurol. 2018;14(10):590–605. doi: 10.1038/s41582-018-0051-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Magana JJ, Velazquez-Perez L, Cisneros B. Spinocerebellar ataxia type 2: clinical presentation, molecular mechanisms, and therapeutic perspectives. Mol Neurobiol. 2013;47(1):90–104. doi: 10.1007/s12035-012-8348-8. [DOI] [PubMed] [Google Scholar]
- 3.Paulson HL, Shakkottai VG, Clark HB, Orr HT. Polyglutamine spinocerebellar ataxias—from genes to potential treatments. Nat Rev Neurosci. 2017;18(10):613–26. doi: 10.1038/nrn.2017.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scoles DR, Pulst SM. Spinocerebellar ataxia type 2. Adv Exp Med Biol. 2018;1049:175–95. doi: 10.1007/978-3-319-71779-1_8. [DOI] [PubMed] [Google Scholar]
- 5.Buijsen RAM, Toonen LJA, Gardiner SL, van Roon-Mom WMC. Genetics, mechanisms, and therapeutic progress in polyglutamine spinocerebellar ataxias. Neurotherapeutics. 2019. [DOI] [PMC free article] [PubMed]
- 6.Satterfield TF, Pallanck LJ. Ataxin-2 and its Drosophila homolog, ATX2, physically assemble with polyribosomes. Hum Mol Genet. 2006;15(16):2523–32. doi: 10.1093/hmg/ddl173. [DOI] [PubMed] [Google Scholar]
- 7.Alves-Cruzeiro JM, Mendonca L, Pereira de Almeida L, Nobrega C. Motor dysfunctions and neuropathology in mouse models of spinocerebellar ataxia type 2: a comprehensive review. Front Neurosci. 2016;10:572. doi: 10.3389/fnins.2016.00572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smeets CJ, Verbeek DS. Climbing fibers in spinocerebellar ataxia: a mechanism for the loss of motor control. Neurobiol Dis. 2016;88:96–106. doi: 10.1016/j.nbd.2016.01.009. [DOI] [PubMed] [Google Scholar]
- 9.Takeuchi T, Nagai Y. Protein misfolding and aggregation as a therapeutic target for polyglutamine diseases. Brain Sci. 2017;7(10). [DOI] [PMC free article] [PubMed]
- 10.Massey TH, Jones L. The central role of DNA damage and repair in CAG repeat diseases. Dis Model Mech. 2018;11(1). [DOI] [PMC free article] [PubMed]
- 11.Egorova P, Popugaeva E, Bezprozvanny I. Disturbed calcium signaling in spinocerebellar ataxias and Alzheimer’s disease. Semin Cell Dev Biol. 2015;40:127–33. doi: 10.1016/j.semcdb.2015.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Egorova PA, Bezprozvanny IB. Inositol 1,4,5-trisphosphate receptors and neurodegenerative disorders. FEBS J. 2018;285(19):3547–65. doi: 10.1111/febs.14366. [DOI] [PubMed] [Google Scholar]
- 13.Hisatsune C, Hamada K, Mikoshiba K. Ca(2+) signaling and spinocerebellar ataxia. Biochim Biophys Acta Mol Cell Res 2018. [DOI] [PubMed]
- 14.Mark MD, Schwitalla JC, Groemmke M, Herlitze S. Keeping our calcium in balance to maintain our balance. Biochem Biophys Res Commun. 2017;483(4):1040–50. doi: 10.1016/j.bbrc.2016.07.020. [DOI] [PubMed] [Google Scholar]
- 15.Ashkenazi A, Bento CF, Ricketts T, et al. Polyglutamine tracts regulate autophagy. Autophagy. 2017;13(9):1613–4. doi: 10.1080/15548627.2017.1336278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yau WY, O’Connor E, Sullivan R, Akijian L, Wood NW. DNA repair in trinucleotide repeat ataxias. FEBS J. 2018;285(19):3669–82. doi: 10.1111/febs.14644. [DOI] [PubMed] [Google Scholar]
- 17.Scoles DR, Meera P, Schneider MD, et al. Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nature. 2017;544(7650):362–6. doi: 10.1038/nature22044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teive HAG, Camargo CHF, Munhoz RP. Antisense oligonucleotide therapy for spinocerebellar ataxias: good news for terrible diseases. Mov Disord Clin Pract. 2018;5(4):402–3. doi: 10.1002/mdc3.12627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsai YA, Liu RS, Lirng JF, et al. Treatment of spinocerebellar ataxia with mesenchymal stem cells: a phase I/IIa clinical study. Cell Transplant. 2017;26(3):503–12. doi: 10.3727/096368916X694373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Romano S, Coarelli G, Marcotulli C, et al. Riluzole in patients with hereditary cerebellar ataxia: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2015;14(10):985–91. doi: 10.1016/S1474-4422(15)00201-X. [DOI] [PubMed] [Google Scholar]
- 21.Liu J, Tang TS, Tu H, et al. Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 2. J Neurosci. 2009;29(29):9148–62. doi: 10.1523/JNEUROSCI.0660-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kasumu AW, Liang X, Egorova P, Vorontsova D, Bezprozvanny I. Chronic suppression of inositol 1,4,5-triphosphate receptor-mediated calcium signaling in cerebellar purkinje cells alleviates pathological phenotype in spinocerebellar ataxia 2 mice. J Neurosci. 2012;32(37):12786–96. doi: 10.1523/JNEUROSCI.1643-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bushart DD, Chopra R, Singh V, Murphy GG, Wulff H, Shakkottai VG. Targeting potassium channels to treat cerebellar ataxia. Ann Clin Transl Neurol. 2018;5(3):297–314. doi: 10.1002/acn3.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coarelli G, Brice A, Durr A. Recent advances in understanding dominant spinocerebellar ataxias from clinical and genetic points of view. F1000Research. 2018;7. [DOI] [PMC free article] [PubMed]
- 25.Antenora A, Bruzzese D, Lieto M, et al. Predictors of survival in spinocerebellar ataxia type 2 population from Southern Italy. Neurol Sci. 2018;39(11):1857–60. doi: 10.1007/s10072-018-3504-1. [DOI] [PubMed] [Google Scholar]
- 26.Velazquez-Perez LC, Rodriguez-Labrada R, Fernandez-Ruiz J. Spinocerebellar ataxia type 2: clinicogenetic aspects, mechanistic insights, and management approaches. Front Neurol. 2017;8:472. doi: 10.3389/fneur.2017.00472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim JS, Kwon S, Ki CS, Youn J, Cho JW. The etiologies of chronic progressive cerebellar ataxia in a Korean population. J Clin Neurol. 2018;14(3):374–80. doi: 10.3988/jcn.2018.14.3.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Amarante TRP, Takeda SYM, Teive HAG, Zonta MB. Impact of disease duration on functional status of patients with spinocerebellar ataxia type 2. Arq Neuropsiquiatr. 2017;75(11):773–7. doi: 10.1590/0004-282X20170146. [DOI] [PubMed] [Google Scholar]
- 29.Diallo A, Jacobi H, Cook A, et al. Survival in patients with spinocerebellar ataxia types 1, 2, 3, and 6 (EUROSCA): a longitudinal cohort study. Lancet Neurol. 2018;17(4):327–34. doi: 10.1016/S1474-4422(18)30042-5. [DOI] [PubMed] [Google Scholar]
- 30.Figueroa KP, Coon H, Santos N, Velazquez L, Mederos LA, Pulst SM. Genetic analysis of age at onset variation in spinocerebellar ataxia type 2. Neurol Genet. 2017;3(3):e155. doi: 10.1212/NXG.0000000000000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin YC, Lee YC, Hsu TY, Liao YC, Soong BW. Comparable progression of spinocerebellar ataxias between Caucasians and Chinese. Parkinsonism Relat Disord. 2018. [DOI] [PubMed]
- 32.Jacobi H, du Montcel ST, et al. Long-term evolution of patient-reported outcome measures in spinocerebellar ataxias. J Neurol. 2018;265(9):2040–51. doi: 10.1007/s00415-018-8954-0. [DOI] [PubMed] [Google Scholar]
- 33.Gispert S, Twells R, Orozco G, et al. Chromosomal assignment of the second locus for autosomal dominant cerebellar ataxia (SCA2) to chromosome 12q23-24.1. Nat Genet. 1993;4(3):295–9. doi: 10.1038/ng0793-295. [DOI] [PubMed] [Google Scholar]
- 34.Fernandez M, McClain ME, Martinez RA, et al. Late-onset SCA2: 33 CAG repeats are sufficient to cause disease. Neurology. 2000;55(4):569–72. doi: 10.1212/wnl.55.4.569. [DOI] [PubMed] [Google Scholar]
- 35.Pulst SM. The complex structure of ATXN2 genetic variation. Neurol Genet. 2018;4(6):e299. doi: 10.1212/NXG.0000000000000299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Almaguer-Mederos LE, Mesa JML, Gonzalez-Zaldivar Y, et al. Factors associated with ATXN2 CAG/CAA repeat intergenerational instability in spinocerebellar ataxia type 2. Clin Genet. 2018;94(3–4):346–50. doi: 10.1111/cge.13380. [DOI] [PubMed] [Google Scholar]
- 37.Sena LS, Castilhos RM, Mattos EP, et al. Selective forces related to spinocerebellar ataxia type 2. Cerebellum. 2019;18(2):188–94. doi: 10.1007/s12311-018-0977-7. [DOI] [PubMed] [Google Scholar]
- 38.van de Loo S, Eich F, Nonis D, Auburger G, Nowock J. Ataxin-2 associates with rough endoplasmic reticulum. Exp Neurol. 2009;215(1):110–8. doi: 10.1016/j.expneurol.2008.09.020. [DOI] [PubMed] [Google Scholar]
- 39.Kiehl TR, Nechiporuk A, Figueroa KP, Keating MT, Huynh DP, Pulst SM. Generation and characterization of Sca2 (ataxin-2) knockout mice. Biochem Biophys Res Commun. 2006;339(1):17–24. doi: 10.1016/j.bbrc.2005.10.186. [DOI] [PubMed] [Google Scholar]
- 40.Lastres-Becker I, Brodesser S, Lutjohann D, et al. Insulin receptor and lipid metabolism pathology in ataxin-2 knock-out mice. Hum Mol Genet. 2008;17(10):1465–81. doi: 10.1093/hmg/ddn035. [DOI] [PubMed] [Google Scholar]
- 41.Pfeffer M, Gispert S, Auburger G, Wicht H, Korf HW. Impact of Ataxin-2 knock out on circadian locomotor behavior and PER immunoreaction in the SCN of mice. Chronobiol Int. 2017;34(1):129–37. doi: 10.1080/07420528.2016.1245666. [DOI] [PubMed] [Google Scholar]
- 42.Lim C, Allada R. ATAXIN-2 activates PERIOD translation to sustain circadian rhythms in Drosophila. Science. 2013;340(6134):875–9. doi: 10.1126/science.1234785. [DOI] [PubMed] [Google Scholar]
- 43.Zhang Y, Ling J, Yuan C, Dubruille R, Emery P. A role for Drosophila ATX2 in activation of PER translation and circadian behavior. Science. 2013;340(6134):879–82. doi: 10.1126/science.1234746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seidel K, Siswanto S, Fredrich M, et al. On the distribution of intranuclear and cytoplasmic aggregates in the brainstem of patients with spinocerebellar ataxia type 2 and 3. Brain Pathol. 2017;27(3):345–55. doi: 10.1111/bpa.12412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lim NS, Kozlov G, Chang TC, et al. Comparative peptide binding studies of the PABC domains from the ubiquitin-protein isopeptide ligase HYD and poly(A)-binding protein. Implications for HYD function. J Biol Chem. 2006;281(20):14376–82. doi: 10.1074/jbc.M600307200. [DOI] [PubMed] [Google Scholar]
- 46.Lastres-Becker I, Nonis D, Eich F, et al. Mammalian ataxin-2 modulates translation control at the pre-initiation complex via PI3K/mTOR and is induced by starvation. Biochim Biophys Acta. 2016;1862(9):1558–69. doi: 10.1016/j.bbadis.2016.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nonhoff U, Ralser M, Welzel F, et al. Ataxin-2 interacts with the DEAD/H-box RNA helicase DDX6 and interferes with P-bodies and stress granules. Mol Biol Cell. 2007;18(4):1385–96. doi: 10.1091/mbc.E06-12-1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bakthavachalu B, Huelsmeier J, Sudhakaran IP, et al. RNP-granule assembly via Ataxin-2 disordered domains is required for long-term memory and neurodegeneration. Neuron. 2018;98(4):754–66. doi: 10.1016/j.neuron.2018.04.032. [DOI] [PubMed] [Google Scholar]
- 49.Ostrowski LA, Hall AC, Szafranski KJ, et al. Conserved Pbp1/Ataxin-2 regulates retrotransposon activity and connects polyglutamine expansion-driven protein aggregation to lifespan-controlling rDNA repeats. Commun Biol. 2018;1:187. doi: 10.1038/s42003-018-0187-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sen NE, Drost J, Gispert S, et al. Search for SCA2 blood RNA biomarkers highlights Ataxin-2 as strong modifier of the mitochondrial factor PINK1 levels. Neurobiol Dis. 2016;96:115–26. doi: 10.1016/j.nbd.2016.09.002. [DOI] [PubMed] [Google Scholar]
- 51.Li PP, Sun X, Xia G, et al. ATXN2-AS, a gene antisense to ATXN2, is associated with spinocerebellar ataxia type 2 and amyotrophic lateral sclerosis. Ann Neurol. 2016;80(4):600–15. doi: 10.1002/ana.24761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fittschen M, Lastres-Becker I, Halbach MV, et al. Genetic ablation of ataxin-2 increases several global translation factors in their transcript abundance but decreases translation rate. Neurogenetics. 2015;16(3):181–92. doi: 10.1007/s10048-015-0441-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meierhofer D, Halbach M, Sen NE, Gispert S, Auburger G. Ataxin-2 (Atxn2)-knock-out mice show branched chain amino acids and fatty acids pathway alterations. Mol Cell Proteomics. 2016;15(5):1728–39. doi: 10.1074/mcp.M115.056770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Halbach MV, Gispert S, Stehning T, Damrath E, Walter M, Auburger G. Atxn2 knockout and CAG42-knock-in cerebellum shows similarly dysregulated expression in calcium homeostasis pathway. Cerebellum. 2017;16(1):68–81. doi: 10.1007/s12311-016-0762-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Louis ED, Kuo SH, Tate WJ, et al. Heterotopic Purkinje cells: a comparative postmortem study of essential tremor and spinocerebellar ataxias 1, 2, 3, and 6. Cerebellum. 2018;17(2):104–10. doi: 10.1007/s12311-017-0876-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nibbeling EAR, Duarri A, Verschuuren-Bemelmans CC, et al. Exome sequencing and network analysis identifies shared mechanisms underlying spinocerebellar ataxia. Brain. 2017;140(11):2860–78. doi: 10.1093/brain/awx251. [DOI] [PubMed] [Google Scholar]
- 57.Aboulhoda BE, Hassan SS. Effect of prenatal tramadol on postnatal cerebellar development: role of oxidative stress. J Chem Neuroanat. 2018;94:102–18. doi: 10.1016/j.jchemneu.2018.10.002. [DOI] [PubMed] [Google Scholar]
- 58.Squadrone S, Brizio P, Mancini C, Abete MC, Brusco A. Altered homeostasis of trace elements in the blood of SCA2 patients. J Trace Elem Med Biol. 2018;47:111–4. doi: 10.1016/j.jtemb.2018.02.011. [DOI] [PubMed] [Google Scholar]
- 59.Guevara-Garcia M, Gil-del Valle L, Velasquez-Perez L, Garcia-Rodriguez JC. Oxidative stress as a cofactor in spinocerebellar ataxia type 2. Redox Rep. 2012;17(2):84–9. doi: 10.1179/1351000212Y.0000000005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Almaguer-Gotay D, Almaguer-Mederos LE, et al. Spinocerebellar ataxia type 2 is associated with the extracellular loss of superoxide dismutase but not catalase activity. Front Neurol. 2017;8:276. doi: 10.3389/fneur.2017.00276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Almaguer-Mederos LE, Almaguer-Gotay D, Aguilera-Rodriguez R, et al. Association of glutathione S-transferase omega polymorphism and spinocerebellar ataxia type 2. J Neurol Sci. 2017;372:324–8. doi: 10.1016/j.jns.2016.11.075. [DOI] [PubMed] [Google Scholar]
- 62.Monte TL, Pereira FS, Reckziegel EDR, et al. Neurological phenotypes in spinocerebellar ataxia type 2: role of mitochondrial polymorphism A10398G and other risk factors. Parkinsonism Relat Disord. 2017;42:54–60. doi: 10.1016/j.parkreldis.2017.06.010. [DOI] [PubMed] [Google Scholar]
- 63.Hamzeiy H, Savas D, Tunca C, et al. Elevated global DNA methylation is not exclusive to amyotrophic lateral sclerosis and is also observed in spinocerebellar ataxia types 1 and 2. Neurodegener Dis. 2018;18(1):38–48. doi: 10.1159/000486201. [DOI] [PubMed] [Google Scholar]
- 64.Wilke C, Bender F, Hayer SN, et al. Serum neurofilament light is increased in multiple system atrophy of cerebellar type and in repeat-expansion spinocerebellar ataxias: a pilot study. J Neurol. 2018;265(7):1618–24. doi: 10.1007/s00415-018-8893-9. [DOI] [PubMed] [Google Scholar]
- 65.Cuello-Almarales DA, Almaguer-Mederos LE, Vazquez-Mojena Y, et al. Buccal cell micronucleus frequency is significantly elevated in patients with spinocerebellar ataxia type 2. Arch Med Res. 2017;48(3):297–302. doi: 10.1016/j.arcmed.2017.06.008. [DOI] [PubMed] [Google Scholar]
- 66.Oz G, Iltis I, Hutter D, Thomas W, Bushara KO, Gomez CM. Distinct neurochemical profiles of spinocerebellar ataxias 1, 2, 6, and cerebellar multiple system atrophy. Cerebellum. 2011;10(2):208–17. doi: 10.1007/s12311-010-0213-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brouillette AM, Oz G, Gomez CM. Cerebrospinal fluid biomarkers in spinocerebellar ataxia: a pilot study. Dis Markers. 2015;2015:413098. doi: 10.1155/2015/413098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cagnoli C, Brussino A, Mancini C, et al. Spinocerebellar ataxia tethering PCR: a rapid genetic test for the diagnosis of spinocerebellar ataxia types 1, 2, 3, 6, and 7 by PCR and capillary electrophoresis. J Mol Diagn. 2018;20(3):289–97. doi: 10.1016/j.jmoldx.2017.12.006. [DOI] [PubMed] [Google Scholar]
- 69.Estrada R, Galarraga J, Orozco G, Nodarse A, Auburger G. Spinocerebellar ataxia 2 (SCA2): morphometric analyses in 11 autopsies. Acta Neuropathol. 1999;97(3):306–10. doi: 10.1007/s004010050989. [DOI] [PubMed] [Google Scholar]
- 70.Martin JJ, Van Regemorter N, Krols L, et al. On an autosomal dominant form of retinal-cerebellar degeneration: an autopsy study of five patients in one family. Acta Neuropathol. 1994;88(4):277–86. doi: 10.1007/BF00310370. [DOI] [PubMed] [Google Scholar]
- 71.Marzi C, Ciulli S, Giannelli M, et al. Structural complexity of the cerebellum and cerebral cortex is reduced in spinocerebellar ataxia type 2. J Neuroimaging. 2018;28(6):688–93. doi: 10.1111/jon.12534. [DOI] [PubMed] [Google Scholar]
- 72.Han Q, Yang J, Xiong H, Shang H. Voxel-based meta-analysis of gray and white matter volume abnormalities in spinocerebellar ataxia type 2. Brain Behav. 2018;8(9):e01099. doi: 10.1002/brb3.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lupo M, Olivito G, Iacobacci C, et al. The cerebellar topography of attention sub-components in spinocerebellar ataxia type 2. Cortex. 2018;108:35–49. doi: 10.1016/j.cortex.2018.07.011. [DOI] [PubMed] [Google Scholar]
- 74.Hernandez-Castillo CR, King M, Diedrichsen J, Fernandez-Ruiz J. Unique degeneration signatures in the cerebellar cortex for spinocerebellar ataxias 2, 3, and 7. Neuroimage Clin. 2018;20:931–8. doi: 10.1016/j.nicl.2018.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Baldarcara L, Currie S, Hadjivassiliou M, et al. Consensus paper: radiological biomarkers of cerebellar diseases. Cerebellum. 2015;14(2):175–96. doi: 10.1007/s12311-014-0610-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mascalchi M, Vella A. Neuroimaging applications in chronic ataxias. Int Rev Neurobiol. 2018;143:109–62. doi: 10.1016/bs.irn.2018.09.011. [DOI] [PubMed] [Google Scholar]
- 77.Reetz K, Rodriguez-Labrada R, Dogan I, et al. Brain atrophy measures in preclinical and manifest spinocerebellar ataxia type 2. Ann Clin Transl Neurol. 2018;5(2):128–37. doi: 10.1002/acn3.504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yoshii F, Tomiyasu H, Watanabe R, Ryo M. MRI signal abnormalities of the inferior olivary nuclei in spinocerebellar ataxia type 2. Case Rep Neurol. 2017;9(3):267–71. doi: 10.1159/000481303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mascalchi M, Marzi C, Giannelli M, et al. Histogram analysis of DTI-derived indices reveals pontocerebellar degeneration and its progression in SCA2. PLoS One. 2018;13(7):e0200258. doi: 10.1371/journal.pone.0200258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Olivito G, Lupo M, Iacobacci C, et al. Microstructural MRI basis of the cognitive functions in patients with spinocerebellar ataxia type 2. Neuroscience. 2017;366:44–53. doi: 10.1016/j.neuroscience.2017.10.007. [DOI] [PubMed] [Google Scholar]
- 81.Adanyeguh IM, Perlbarg V, Henry PG, et al. Autosomal dominant cerebellar ataxias: imaging biomarkers with high effect sizes. Neuroimage Clin. 2018;19:858–67. doi: 10.1016/j.nicl.2018.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ito K, Ohtsuka C, Yoshioka K, et al. Differentiation between multiple system atrophy and other spinocerebellar degenerations using diffusion kurtosis imaging. Acad Radiol. 2019. [DOI] [PubMed]
- 83.Velazquez-Perez L, Rodriguez-Labrada R, Torres-Vega R, et al. Central motor conduction time as prodromal biomarker in spinocerebellar ataxia type 2. Mov Disord. 2016;31(4):603–4. doi: 10.1002/mds.26555. [DOI] [PubMed] [Google Scholar]
- 84.Velazquez-Perez L, Rodriguez-Labrada R, Torres-Vega R, et al. Progression of corticospinal tract dysfunction in pre-ataxic spinocerebellar ataxia type 2: a two-years follow-up TMS study. Clin Neurophysiol. 2018;129(5):895–900. doi: 10.1016/j.clinph.2018.01.066. [DOI] [PubMed] [Google Scholar]
- 85.Velazquez-Perez L, Rodriguez-Labrada R, Torres-Vega R, et al. Abnormal corticospinal tract function and motor cortex excitability in non-ataxic SCA2 mutation carriers: a TMS study. Clin Neurophysiol. 2016;127(8):2713–9. doi: 10.1016/j.clinph.2016.05.003. [DOI] [PubMed] [Google Scholar]
- 86.Velazquez-Perez L, Tunnerhoff J, Rodriguez-Labrada R, et al. Corticomuscular coherence: a novel tool to assess the pyramidal tract dysfunction in spinocerebellar ataxia type 2. Cerebellum. 2017;16(2):602–6. doi: 10.1007/s12311-016-0827-4. [DOI] [PubMed] [Google Scholar]
- 87.Velazquez-Perez L, Tunnerhoff J, Rodriguez-Labrada R, et al. Early corticospinal tract damage in prodromal SCA2 revealed by EEG-EMG and EMG-EMG coherence. Clin Neurophysiol. 2017;128(12):2493–502. doi: 10.1016/j.clinph.2017.10.009. [DOI] [PubMed] [Google Scholar]
- 88.Seshagiri DV, Botta R, Sasidharan A, et al. Assessment of sleep spindle density among genetically positive spinocerebellar ataxias types 1, 2, and 3 patients. Ann Neurosci. 2018;25(2):106–11. doi: 10.1159/000484516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zanatta A, Camargo CHF, Germiniani FMB, Raskin S, de Souza Crippa AC, Teive HAG. Abnormal findings in polysomnographic recordings of patients with spinocerebellar ataxia type 2 (SCA2) Cerebellum. 2019;18(2):196–202. doi: 10.1007/s12311-018-0982-x. [DOI] [PubMed] [Google Scholar]
- 90.Rodriguez-Labrada R, Galicia-Polo L, Canales-Ochoa N, et al. Sleep spindles and K-complex activities are decreased in spinocerebellar ataxia type 2: relationship to memory and motor performances. Sleep Med. 2019;60:188–96. doi: 10.1016/j.sleep.2019.04.005. [DOI] [PubMed] [Google Scholar]
- 91.Abele M, Burk K, Laccone F, Dichgans J, Klockgether T. Restless legs syndrome in spinocerebellar ataxia types 1, 2, and 3. J Neurol. 2001;248(4):311–4. doi: 10.1007/s004150170206. [DOI] [PubMed] [Google Scholar]
- 92.Boesch SM, Frauscher B, Brandauer E, Wenning GK, Hogl B, Poewe W. Disturbance of rapid eye movement sleep in spinocerebellar ataxia type 2. Mov Disord. 2006;21(10):1751–4. doi: 10.1002/mds.21036. [DOI] [PubMed] [Google Scholar]
- 93.Velazquez-Perez L, Voss U, Rodriguez-Labrada R, et al. Sleep disorders in spinocerebellar ataxia type 2 patients. Neurodegener Dis. 2011;8(6):447–54. doi: 10.1159/000324374. [DOI] [PubMed] [Google Scholar]
- 94.Velazquez-Perez L, Rodriguez-Labrada R, Alvarez-Gonzalez L, et al. Lisuride reduces involuntary periodic leg movements in spinocerebellar ataxia type 2 patients. Cerebellum. 2012;11(4):1051–6. doi: 10.1007/s12311-012-0382-6. [DOI] [PubMed] [Google Scholar]
- 95.Rodriguez-Labrada R, Velazquez-Perez L, Ortega-Sanchez R, et al. Insights into cognitive decline in spinocerebellar Ataxia type 2: a P300 event-related brain potential study. Cerebellum Ataxias. 2019;6:3. doi: 10.1186/s40673-019-0097-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Olivito G, Lupo M, Iacobacci C, et al. Structural cerebellar correlates of cognitive functions in spinocerebellar ataxia type 2. J Neurol. 2018;265(3):597–606. doi: 10.1007/s00415-018-8738-6. [DOI] [PubMed] [Google Scholar]
- 97.Olivito G, Cercignani M, Lupo M, et al. Neural substrates of motor and cognitive dysfunctions in SCA2 patients: a network based statistics analysis. Neuroimage Clin. 2017;14:719–25. doi: 10.1016/j.nicl.2017.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Luo L, Wang J, Lo RY, et al. The initial symptom and motor progression in spinocerebellar ataxias. Cerebellum. 2017;16(3):615–22. doi: 10.1007/s12311-016-0836-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gan SR, Wang J, Figueroa KP, et al. Postural tremor and ataxia progression in spinocerebellar ataxias. Tremor Other Hyperkinet Mov (N Y) 2017;7:492. doi: 10.7916/D8GM8KRH. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lai RY, Tomishon D, Figueroa KP, et al. Tremor in the degenerative cerebellum: towards the understanding of brain circuitry for tremor. Cerebellum. 2019. [DOI] [PMC free article] [PubMed]
- 101.Markovic V, Dragasevic-Miskovic NT, Stankovic I, Petrovic I, Svetel M, Kostic VS. Dystonia in patients with spinocerebellar ataxia type 2. Mov Disord Clin Pract. 2016;3(3):292–5. doi: 10.1002/mdc3.12274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Schmitz-Hubsch T, Coudert M, Bauer P, et al. Spinocerebellar ataxia types 1, 2, 3, and 6: disease severity and nonataxia symptoms. Neurology. 2008;71(13):982–9. doi: 10.1212/01.wnl.0000325057.33666.72. [DOI] [PubMed] [Google Scholar]
- 103.Kuo PH, Gan SR, Wang J, et al. Dystonia and ataxia progression in spinocerebellar ataxias. Parkinsonism Relat Disord. 2017;45:75–80. doi: 10.1016/j.parkreldis.2017.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Leadbetter R, Weatherall M, Pelosi L. Nerve ultrasound as a diagnostic tool for sensory neuronopathy in spinocerebellar ataxia syndrome. Clin Neurophysiol. 2019;130(4):568–72. doi: 10.1016/j.clinph.2018.12.010. [DOI] [PubMed] [Google Scholar]
- 105.Jensen K, Beylergil SB, Shaikh AG. Slow saccades in cerebellar disease. Cerebellum Ataxias. 2019;6:1. doi: 10.1186/s40673-018-0095-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Burk K, Fetter M, Abele M, et al. Autosomal dominant cerebellar ataxia type I: oculomotor abnormalities in families with SCA1, SCA2, and SCA3. J Neurol. 1999;246(9):789–97. doi: 10.1007/s004150050456. [DOI] [PubMed] [Google Scholar]
- 107.Luis L, Costa J, Munoz E, et al. Vestibulo-ocular reflex dynamics with head-impulses discriminates spinocerebellar ataxias types 1, 2 and 3 and Friedreich ataxia. J Vestib Res. 2016;26(3):327–34. doi: 10.3233/VES-160579. [DOI] [PubMed] [Google Scholar]
- 108.Seshagiri DV, Pal PK, Jain S, Yadav R. Optokinetic nystagmus in patients with SCA: a bedside test for oculomotor dysfunction grading. Neurology. 2018;91(13):e1255–e61. doi: 10.1212/WNL.0000000000006250. [DOI] [PubMed] [Google Scholar]
- 109.Ronsin S, Hannoun S, Thobois S, et al. A new MRI marker of ataxia with oculomotor apraxia. Eur J Radiol. 2019;110:187–92. doi: 10.1016/j.ejrad.2018.11.035. [DOI] [PubMed] [Google Scholar]
- 110.Rosini F, Pretegiani E, Mignarri A, et al. The role of dentate nuclei in human oculomotor control: insights from cerebrotendinous xanthomatosis. J Physiol. 2017;595(11):3607–20. doi: 10.1113/JP273670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Liang L, Chen T, Wu Y. The electrophysiology of spinocerebellar ataxias. Neurophysiol Clin. 2016;46(1):27–34. doi: 10.1016/j.neucli.2015.12.006. [DOI] [PubMed] [Google Scholar]
- 112.Velazquez-Perez L, Seifried C, Santos-Falcon N, et al. Saccade velocity is controlled by polyglutamine size in spinocerebellar ataxia 2. Ann Neurol. 2004;56(3):444–7. doi: 10.1002/ana.20220. [DOI] [PubMed] [Google Scholar]
- 113.Velazquez-Perez L, Seifried C, Abele M, et al. Saccade velocity is reduced in presymptomatic spinocerebellar ataxia type 2. Clin Neurophysiol. 2009;120(3):632–5. doi: 10.1016/j.clinph.2008.12.040. [DOI] [PubMed] [Google Scholar]
- 114.Rodriguez-Labrada R, Velazquez-Perez L, Auburger G, et al. Spinocerebellar ataxia type 2: measures of saccade changes improve power for clinical trials. Mov Disord. 2016;31(4):570–8. doi: 10.1002/mds.26532. [DOI] [PubMed] [Google Scholar]
- 115.Rodriguez-Labrada R, Vazquez-Mojena Y, Canales-Ochoa N, Medrano-Montero J, Velazquez-Perez L. Heritability of saccadic eye movements in spinocerebellar ataxia type 2: insights into an endophenotype marker. Cerebellum Ataxias. 2017;4:19. doi: 10.1186/s40673-017-0078-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kim YE, Jeon B, Farrer MJ, et al. SCA2 family presenting as typical Parkinson’s disease: 34 year follow up. Parkinsonism Relat Disord. 2017;40:69–72. doi: 10.1016/j.parkreldis.2017.04.003. [DOI] [PubMed] [Google Scholar]
- 117.Woo KA, Lee JY, Jeon B. Familial spinocerebellar ataxia type 2 parkinsonism presenting as intractable oromandibular dystonia. Tremor Other Hyperkinet Mov (N Y) 2019;9:611. doi: 10.7916/D8087PB6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nkiliza A, Mutez E, Simonin C, et al. RNA-binding disturbances as a continuum from spinocerebellar ataxia type 2 to Parkinson disease. Neurobiol Dis. 2016;96:312–22. doi: 10.1016/j.nbd.2016.09.014. [DOI] [PubMed] [Google Scholar]
- 119.Kim YE, Oh KW, Noh MY, et al. Analysis of ATXN2 trinucleotide repeats in Korean patients with amyotrophic lateral sclerosis. Neurobiol Aging. 2018;67:201 e5–e8. doi: 10.1016/j.neurobiolaging.2018.03.019. [DOI] [PubMed] [Google Scholar]
- 120.de Silva R, Greenfield J, Cook A, et al. Guidelines on the diagnosis and management of the progressive ataxias. Orphanet J Rare Dis. 2019;14(1):51. doi: 10.1186/s13023-019-1013-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Rodriguez-Diaz JC, Velazquez-Perez L, Rodriguez Labrada R, et al. Neurorehabilitation therapy in spinocerebellar ataxia type 2: a 24-week, rater-blinded, randomized, controlled trial. Mov Disord. 2018;33(9):1481–7. doi: 10.1002/mds.27437. [DOI] [PubMed] [Google Scholar]
- 122.Zesiewicz TA, Wilmot G, Kuo SH, et al. Comprehensive systematic review summary: treatment of cerebellar motor dysfunction and ataxia: report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology. Neurology. 2018;90(10):464–71. doi: 10.1212/WNL.0000000000005055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Anderson CJ, Figueroa KP, Dorval AD, Pulst SM. Deep cerebellar stimulation reduces ataxic motor symptoms in the shaker rat. Ann Neurol. 2019. [DOI] [PMC free article] [PubMed]
- 124.Huynh DP, Figueroa K, Hoang N, Pulst SM. Nuclear localization or inclusion body formation of ataxin-2 are not necessary for SCA2 pathogenesis in mouse or human. Nat Genet. 2000;26(1):44–50. doi: 10.1038/79162. [DOI] [PubMed] [Google Scholar]
- 125.Kasumu AW, Hougaard C, Rode F, et al. Selective positive modulator of calcium-activated potassium channels exerts beneficial effects in a mouse model of spinocerebellar ataxia type 2. Chem Biol. 2012;19(10):1340–53. doi: 10.1016/j.chembiol.2012.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Egorova PA, Zakharova OA, Vlasova OL, Bezprozvanny IB. In vivo analysis of cerebellar Purkinje cell activity in SCA2 transgenic mouse model. J Neurophysiol. 2016;115(6):2840–51. doi: 10.1152/jn.00913.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Egorova PA, Gavrilova AV, Bezprozvanny IB. In vivo analysis of the climbing fiber-Purkinje cell circuit in SCA2-58Q transgenic mouse model. Cerebellum. 2018;17(5):590–600. doi: 10.1007/s12311-018-0951-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Aguiar J, Fernandez J, Aguilar A, et al. Ubiquitous expression of human SCA2 gene under the regulation of the SCA2 self promoter cause specific Purkinje cell degeneration in transgenic mice. Neurosci Lett. 2006;392(3):202–6. doi: 10.1016/j.neulet.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 129.Damrath E, Heck MV, Gispert S, et al. ATXN2-CAG42 sequesters PABPC1 into insolubility and induces FBXW8 in cerebellum of old ataxic knock-in mice. PLoS Genet. 2012;8(8):e1002920. doi: 10.1371/journal.pgen.1002920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hansen ST, Meera P, Otis TS, Pulst SM. Changes in Purkinje cell firing and gene expression precede behavioral pathology in a mouse model of SCA2. Hum Mol Genet. 2013;22(2):271–83. doi: 10.1093/hmg/dds427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Pflieger LT, Dansithong W, Paul S, et al. Gene co-expression network analysis for identifying modules and functionally enriched pathways in SCA2. Hum Mol Genet. 2017;26(16):3069–80. doi: 10.1093/hmg/ddx191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Dansithong W, Paul S, Figueroa KP, et al. Ataxin-2 regulates RGS8 translation in a new BAC-SCA2 transgenic mouse model. PLoS Genet. 2015;11(4):e1005182. doi: 10.1371/journal.pgen.1005182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chuang CY, Yang CC, Soong BW, et al. Modeling spinocerebellar ataxias 2 and 3 with iPSCs reveals a role for glutamate in disease pathology. Sci Rep. 2019;9(1):1166. doi: 10.1038/s41598-018-37774-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Marthaler AG, Schmid B, Tubsuwan A, et al. Generation of spinocerebellar ataxia type 2 patient-derived iPSC line H196. Stem Cell Res. 2016;16(1):199–201. doi: 10.1016/j.scr.2015.12.049. [DOI] [PubMed] [Google Scholar]
- 135.Maguire JA, Gagne AL, Gonzalez-Alegre P, et al. Generation of spinocerebellar ataxia type 2 induced pluripotent stem cell lines, CHOPi002-A and CHOPi003-A, from patients with abnormal CAG repeats in the coding region of the ATXN2 gene. Stem Cell Res. 2019;34:101361. doi: 10.1016/j.scr.2018.101361. [DOI] [PubMed] [Google Scholar]
- 136.Todd TW, Lim J. Aggregation formation in the polyglutamine diseases: protection at a cost? Mol Cells. 2013;36(3):185–94. doi: 10.1007/s10059-013-0167-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Koyano S, Yagishita S, Kuroiwa Y, Tanaka F, Uchihara T. Neuropathological staging of spinocerebellar ataxia type 2 by semiquantitative 1C2-positive neuron typing. Nuclear translocation of cytoplasmic 1C2 underlies disease progression of spinocerebellar ataxia type 2. Brain Pathol. 2014;24(6):599–606. doi: 10.1111/bpa.12146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ueda M, Li S, Itoh M, et al. Polyglutamine expansion disturbs the endoplasmic reticulum formation, leading to caspase-7 activation through Bax. Biochem Biophys Res Commun. 2014;443(4):1232–8. doi: 10.1016/j.bbrc.2013.12.114. [DOI] [PubMed] [Google Scholar]
- 139.Cornelius N, Wardman JH, Hargreaves IP, et al. Evidence of oxidative stress and mitochondrial dysfunction in spinocerebellar ataxia type 2 (SCA2) patient fibroblasts: effect of coenzyme Q10 supplementation on these parameters. Mitochondrion. 2017;34:103–14. doi: 10.1016/j.mito.2017.03.001. [DOI] [PubMed] [Google Scholar]
- 140.Lo RY, Figueroa KP, Pulst SM, et al. Coenzyme Q10 and spinocerebellar ataxias. Mov Disord. 2015;30(2):214–20. doi: 10.1002/mds.26088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Brown AS, Meera P, Altindag B, et al. MTSS1/Src family kinase dysregulation underlies multiple inherited ataxias. Proc Natl Acad Sci U S A. 2018;115(52):E12407–E16. doi: 10.1073/pnas.1816177115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Verbeek DS, Goedhart J, Bruinsma L, Sinke RJ, Reits EA. PKC gamma mutations in spinocerebellar ataxia type 14 affect C1 domain accessibility and kinase activity leading to aberrant MAPK signaling. J Cell Sci. 2008;121(Pt 14):2339–49. doi: 10.1242/jcs.027698. [DOI] [PubMed] [Google Scholar]
- 143.Shimobayashi E, Kapfhammer JP. Calcium signaling, PKC gamma, IP3R1 and CAR8 link spinocerebellar ataxias and Purkinje cell dendritic development. Curr Neuropharmacol. 2018;16(2):151–9. doi: 10.2174/1570159X15666170529104000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Chopra R, Wasserman AH, Pulst SM, De Zeeuw CI, Shakkottai VG. Protein kinase C activity is a protective modifier of Purkinje neuron degeneration in cerebellar ataxia. Hum Mol Genet. 2018;27(8):1396–410. doi: 10.1093/hmg/ddy050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Meffert MK, Chang JM, Wiltgen BJ, Fanselow MS, Baltimore D. NF-kappa B functions in synaptic signaling and behavior. Nat Neurosci. 2003;6(10):1072–8. doi: 10.1038/nn1110. [DOI] [PubMed] [Google Scholar]
- 146.Ferro A, Qu W, Lukowicz A, Svedberg D, Johnson A, Cvetanovic M. Inhibition of NF-kappaB signaling in IKKbetaF/F;LysM Cre mice causes motor deficits but does not alter pathogenesis of spinocerebellar ataxia type 1. PLoS One. 2018;13(7):e0200013. doi: 10.1371/journal.pone.0200013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Li YX, Sibon OCM, Dijkers PF. Inhibition of NF-kappaB in astrocytes is sufficient to delay neurodegeneration induced by proteotoxicity in neurons. J Neuroinflammation. 2018;15(1):261. doi: 10.1186/s12974-018-1278-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Schwaller B, Meyer M, Schiffmann S. ‘New’ functions for ‘old’ proteins: the role of the calcium-binding proteins calbindin D-28k, calretinin and parvalbumin, in cerebellar physiology. Studies with knockout mice. Cerebellum. 2002;1(4):241–58. doi: 10.1080/147342202320883551. [DOI] [PubMed] [Google Scholar]
- 149.Kreiner L, Christel CJ, Benveniste M, Schwaller B, Lee A. Compensatory regulation of Cav2.1 Ca2+ channels in cerebellar Purkinje neurons lacking parvalbumin and calbindin D-28k. J Neurophysiol. 2010;103(1):371–81. doi: 10.1152/jn.00635.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kano M, Nakayama H, Hashimoto K, Kitamura K, Sakimura K, Watanabe M. Calcium-dependent regulation of climbing fibre synapse elimination during postnatal cerebellar development. J Physiol. 2013;591(Pt 13):3151–8. doi: 10.1113/jphysiol.2012.248252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Barnes JA, Ebner BA, Duvick LA, et al. Abnormalities in the climbing fiber-Purkinje cell circuitry contribute to neuronal dysfunction in ATXN1[82Q] mice. J Neurosci. 2011;31(36):12778–89. doi: 10.1523/JNEUROSCI.2579-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Long C, Grueter CE, Song K, et al. Ataxia and Purkinje cell degeneration in mice lacking the CAMTA1 transcription factor. Proc Natl Acad Sci U S A. 2014;111(31):11521–6. doi: 10.1073/pnas.1411251111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kawaguchi SY, Hirano T. Gating of long-term depression by Ca2+/calmodulin-dependent protein kinase II through enhanced cGMP signalling in cerebellar Purkinje cells. J Physiol. 2013;591(Pt 7):1707–30. doi: 10.1113/jphysiol.2012.245787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Fukumitsu K, Hatsukano T, Yoshimura A, Heuser J, Fujishima K, Kengaku M. Mitochondrial fission protein Drp1 regulates mitochondrial transport and dendritic arborization in cerebellar Purkinje cells. Mol Cell Neurosci. 2016;71:56–65. doi: 10.1016/j.mcn.2015.12.006. [DOI] [PubMed] [Google Scholar]
- 155.Di Bella D, Lazzaro F, Brusco A, et al. Mutations in the mitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28. Nat Genet. 2010;42(4):313–21. doi: 10.1038/ng.544. [DOI] [PubMed] [Google Scholar]
- 156.Alberts B. Molecular biology of the cell. 4th ed. New York: Garland Science; 2002. [Google Scholar]
- 157.Keebler MV, Taylor CW. Endogenous signalling pathways and caged IP3 evoke Ca(2+) puffs at the same abundant immobile intracellular sites. J Cell Sci. 2017;130(21):3728–39. doi: 10.1242/jcs.208520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hirai H, Kano M. Type 1 metabotropic glutamate receptor and its signaling molecules as therapeutic targets for the treatment of cerebellar disorders. Curr Opin Pharmacol. 2018;38:51–8. doi: 10.1016/j.coph.2018.02.002. [DOI] [PubMed] [Google Scholar]
- 159.Serra HG, Duvick L, Zu T, et al. RORalpha-mediated Purkinje cell development determines disease severity in adult SCA1 mice. Cell. 2006;127(4):697–708. doi: 10.1016/j.cell.2006.09.036. [DOI] [PubMed] [Google Scholar]
- 160.Gold DA, Baek SH, Schork NJ, et al. RORalpha coordinates reciprocal signaling in cerebellar development through sonic hedgehog and calcium-dependent pathways. Neuron. 2003;40(6):1119–31. doi: 10.1016/s0896-6273(03)00769-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Orr HT. SCA1-phosphorylation, a regulator of Ataxin-1 function and pathogenesis. Prog Neurobiol. 2012;99(3):179–85. doi: 10.1016/j.pneurobio.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Meera P, Pulst S, Otis T. A positive feedback loop linking enhanced mGluR function and basal calcium in spinocerebellar ataxia type 2. eLife. 2017;6. [DOI] [PMC free article] [PubMed]
- 163.Collins AJ, Foley RN, Herzog C, et al. US Renal Data System 2012 annual data report. Am J Kidney Dis. 2013;61(1 Suppl 1):A7. doi: 10.1053/j.ajkd.2012.11.031. [DOI] [PubMed] [Google Scholar]
- 164.Meera P, Pulst SM, Otis TS. Cellular and circuit mechanisms underlying spinocerebellar ataxias. J Physiol. 2016;594(16):4653–60. doi: 10.1113/JP271897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Puorro G, Marsili A, Sapone F, et al. Peripheral markers of autophagy in polyglutamine diseases. Neurol Sci. 2018;39(1):149–52. doi: 10.1007/s10072-017-3156-6. [DOI] [PubMed] [Google Scholar]
- 166.Paul S, Dansithong W, Figueroa KP, Scoles DR, Pulst SM. Staufen1 links RNA stress granules and autophagy in a model of neurodegeneration. Nat Commun. 2018;9(1):3648. doi: 10.1038/s41467-018-06041-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ferro A, Sheeler C, Rosa JG, Cvetanovic M. Role of microglia in ataxias. J Mol Biol. 2019. [DOI] [PMC free article] [PubMed]
- 168.Thion MS, Low D, Silvin A, et al. Microbiome influences prenatal and adult microglia in a sex-specific manner. Cell. 2018;172(3):500–16. doi: 10.1016/j.cell.2017.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Nakayama H, Abe M, Morimoto C, et al. Microglia permit climbing fiber elimination by promoting GABAergic inhibition in the developing cerebellum. Nat Commun. 2018;9(1):2830. doi: 10.1038/s41467-018-05100-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Ebner BA, Ingram MA, Barnes JA, et al. Purkinje cell ataxin-1 modulates climbing fiber synaptic input in developing and adult mouse cerebellum. J Neurosci. 2013;33(13):5806–20. doi: 10.1523/JNEUROSCI.6311-11.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Cvetanovic M, Ingram M, Orr H, Opal P. Early activation of microglia and astrocytes in mouse models of spinocerebellar ataxia type 1. Neuroscience. 2015;289:289–99. doi: 10.1016/j.neuroscience.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Qu W, Johnson A, Kim JH, Lukowicz A, Svedberg D, Cvetanovic M. Inhibition of colony-stimulating factor 1 receptor early in disease ameliorates motor deficits in SCA1 mice. J Neuroinflammation. 2017;14(1):107. doi: 10.1186/s12974-017-0880-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Llinas R, Sugimori M. Electrophysiological properties of in vitro Purkinje cell dendrites in mammalian cerebellar slices. J Physiol. 1980;305:197–213. doi: 10.1113/jphysiol.1980.sp013358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Llinas R, Sugimori M. Electrophysiological properties of in vitro Purkinje cell somata in mammalian cerebellar slices. J Physiol. 1980;305:171–95. doi: 10.1113/jphysiol.1980.sp013357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Raman IM, Bean BP. Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J Neurosci. 1997;17(12):4517–26. doi: 10.1523/JNEUROSCI.17-12-04517.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Raman IM, Bean BP. Ionic currents underlying spontaneous action potentials in isolated cerebellar Purkinje neurons. J Neurosci. 1999;19(5):1663–74. doi: 10.1523/JNEUROSCI.19-05-01663.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Nam SC, Hockberger PE. Analysis of spontaneous electrical activity in cerebellar Purkinje cells acutely isolated from postnatal rats. J Neurobiol. 1997;33(1):18–32. doi: 10.1002/(sici)1097-4695(199707)33:1<18::aid-neu3>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 178.Womack M, Khodakhah K. Active contribution of dendrites to the tonic and trimodal patterns of activity in cerebellar Purkinje neurons. J Neurosci. 2002;22(24):10603–12. doi: 10.1523/JNEUROSCI.22-24-10603.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Smith SL, Otis TS. Persistent changes in spontaneous firing of Purkinje neurons triggered by the nitric oxide signaling cascade. J Neurosci. 2003;23(2):367–72. doi: 10.1523/JNEUROSCI.23-02-00367.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.De Zeeuw CI, Hoebeek FE, Bosman LW, Schonewille M, Witter L, Koekkoek SK. Spatiotemporal firing patterns in the cerebellum. Nat Rev Neurosci. 2011;12(6):327–44. doi: 10.1038/nrn3011. [DOI] [PubMed] [Google Scholar]
- 181.Hoebeek FE, Stahl JS, van Alphen AM, et al. Increased noise level of purkinje cell activities minimizes impact of their modulation during sensorimotor control. Neuron. 2005;45(6):953–65. doi: 10.1016/j.neuron.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 182.Alvina K, Khodakhah K. The therapeutic mode of action of 4-aminopyridine in cerebellar ataxia. J Neurosci. 2010;30(21):7258–68. doi: 10.1523/JNEUROSCI.3582-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Dell’Orco JM, Wasserman AH, Chopra R, et al. Neuronal atrophy early in degenerative ataxia is a compensatory mechanism to regulate membrane excitability. J Neurosci. 2015;35(32):11292–307. doi: 10.1523/JNEUROSCI.1357-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Mark MD, Krause M, Boele HJ, et al. Spinocerebellar ataxia type 6 protein aggregates cause deficits in motor learning and cerebellar plasticity. J Neurosci. 2015;35(23):8882–95. doi: 10.1523/JNEUROSCI.0891-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Shakkottai VG, do Carmo Costa M, Dell’Orco JM, Sankaranarayanan A, Wulff H, Paulson HL. Early changes in cerebellar physiology accompany motor dysfunction in the polyglutamine disease spinocerebellar ataxia type 3. J Neurosci. 2011;31(36):13002–14. doi: 10.1523/JNEUROSCI.2789-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Walter JT, Alvina K, Womack MD, Chevez C, Khodakhah K. Decreases in the precision of Purkinje cell pacemaking cause cerebellar dysfunction and ataxia. Nat Neurosci. 2006;9(3):389–97. doi: 10.1038/nn1648. [DOI] [PubMed] [Google Scholar]
- 187.Chopra R, Shakkottai VG. Translating cerebellar Purkinje neuron physiology to progress in dominantly inherited ataxia. Future Neurol. 2014;9(2):187–96. doi: 10.2217/fnl.14.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Dell’Orco JM, Pulst SM, Shakkottai VG. Potassium channel dysfunction underlies Purkinje neuron spiking abnormalities in spinocerebellar ataxia type 2. Hum Mol Genet. 2017;26(20):3935–45. doi: 10.1093/hmg/ddx281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Bushart DD, Shakkottai VG. Ion channel dysfunction in cerebellar ataxia. Neurosci Lett. 2019;688:41–8. doi: 10.1016/j.neulet.2018.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Coutelier M, Coarelli G, Monin ML, et al. A panel study on patients with dominant cerebellar ataxia highlights the frequency of channelopathies. Brain. 2017;140(6):1579–94. doi: 10.1093/brain/awx081. [DOI] [PubMed] [Google Scholar]
- 191.Jones JM, Dionne L, Dell’Orco J, et al. Single amino acid deletion in transmembrane segment D4S6 of sodium channel Scn8a (Nav1.6) in a mouse mutant with a chronic movement disorder. Neurobiol Dis. 2016;89:36–45. doi: 10.1016/j.nbd.2016.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Lee KH, Mathews PJ, Reeves AM, et al. Circuit mechanisms underlying motor memory formation in the cerebellum. Neuron. 2015;86(2):529–40. doi: 10.1016/j.neuron.2015.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Lang EJ, Apps R, Bengtsson F, et al. The roles of the olivocerebellar pathway in motor learning and motor control. A consensus paper. Cerebellum. 2017;16(1):230–52. doi: 10.1007/s12311-016-0787-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Kuo SH, Lin CY, Wang J, et al. Climbing fiber-Purkinje cell synaptic pathology in tremor and cerebellar degenerative diseases. Acta Neuropathol. 2017;133(1):121–38. doi: 10.1007/s00401-016-1626-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Burroughs A, Wise AK, Xiao J, et al. The dynamic relationship between cerebellar Purkinje cell simple spikes and the spikelet number of complex spikes. J Physiol. 2017;595(1):283–99. doi: 10.1113/JP272259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Davie JT, Clark BA, Hausser M. The origin of the complex spike in cerebellar Purkinje cells. J Neurosci. 2008;28(30):7599–609. doi: 10.1523/JNEUROSCI.0559-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.van Roon-Mom WMC, Roos RAC, de Bot ST. Dose-dependent lowering of mutant Huntingtin using antisense oligonucleotides in Huntington disease patients. Nucleic Acid Ther. 2018;28(2):59–62. doi: 10.1089/nat.2018.0720. [DOI] [PubMed] [Google Scholar]
- 198.Becker LA, Huang B, Bieri G, et al. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature. 2017;544(7650):367–71. doi: 10.1038/nature22038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Ashizawa AT, Holt J, Faust K, et al. Intravenously administered novel liposomes, DCL64, deliver oligonucleotides to cerebellar purkinje cells. Cerebellum. 2019;18(1):99–108. doi: 10.1007/s12311-018-0961-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Chang YK, Chen MH, Chiang YH, et al. Mesenchymal stem cell transplantation ameliorates motor function deterioration of spinocerebellar ataxia by rescuing cerebellar Purkinje cells. J Biomed Sci. 2011;18:54. doi: 10.1186/1423-0127-18-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Vogel MW, Caston J, Yuzaki M, Mariani J. The Lurcher mouse: fresh insights from an old mutant. Brain Res. 2007;1140:4–18. doi: 10.1016/j.brainres.2005.11.086. [DOI] [PubMed] [Google Scholar]
- 202.Jones J, Jaramillo-Merchan J, Bueno C, Pastor D, Viso-Leon M, Martinez S. Mesenchymal stem cells rescue Purkinje cells and improve motor functions in a mouse model of cerebellar ataxia. Neurobiol Dis. 2010;40(2):415–23. doi: 10.1016/j.nbd.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 203.Bushart DD, Murphy GG, Shakkottai VG. Precision medicine in spinocerebellar ataxias: treatment based on common mechanisms of disease. Ann Transl Med. 2016;4(2):25. doi: 10.3978/j.issn.2305-5839.2016.01.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Womack MD, Khodakhah K. Somatic and dendritic small-conductance calcium-activated potassium channels regulate the output of cerebellar Purkinje neurons. J Neurosci. 2003;23(7):2600–7. doi: 10.1523/JNEUROSCI.23-07-02600.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Alvina K, Khodakhah K. KCa channels as therapeutic targets in episodic ataxia type-2. J Neurosci. 2010;30(21):7249–57. doi: 10.1523/JNEUROSCI.6341-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Cho LT, Alexandrou AJ, Torella R, et al. An intracellular allosteric modulator binding pocket in SK2 ion channels is shared by multiple chemotypes. Structure. 2018;26(4):533–44. doi: 10.1016/j.str.2018.02.017. [DOI] [PubMed] [Google Scholar]
- 207.Nagai Y, Tucker T, Ren H, et al. Inhibition of polyglutamine protein aggregation and cell death by novel peptides identified by phage display screening. J Biol Chem. 2000;275(14):10437–42. doi: 10.1074/jbc.275.14.10437. [DOI] [PubMed] [Google Scholar]
- 208.Nagai Y, Fujikake N, Ohno K, et al. Prevention of polyglutamine oligomerization and neurodegeneration by the peptide inhibitor QBP1 in Drosophila. Hum Mol Genet. 2003;12(11):1253–9. doi: 10.1093/hmg/ddg144. [DOI] [PubMed] [Google Scholar]
- 209.Popiel HA, Nagai Y, Fujikake N, Toda T. Delivery of the aggregate inhibitor peptide QBP1 into the mouse brain using PTDs and its therapeutic effect on polyglutamine disease mice. Neurosci Lett. 2009;449(2):87–92. doi: 10.1016/j.neulet.2008.06.015. [DOI] [PubMed] [Google Scholar]
- 210.Lecerf JM, Shirley TL, Zhu Q, et al. Human single-chain Fv intrabodies counteract in situ huntingtin aggregation in cellular models of Huntington’s disease. Proc Natl Acad Sci U S A. 2001;98(8):4764–9. doi: 10.1073/pnas.071058398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Wolfgang WJ, Miller TW, Webster JM, et al. Suppression of Huntington’s disease pathology in Drosophila by human single-chain Fv antibodies. Proc Natl Acad Sci U S A. 2005;102(32):11563–8. doi: 10.1073/pnas.0505321102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Wang CE, Zhou H, McGuire JR, et al. Suppression of neuropil aggregates and neurological symptoms by an intracellular antibody implicates the cytoplasmic toxicity of mutant huntingtin. J Cell Biol. 2008;181(5):803–16. doi: 10.1083/jcb.200710158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Chen X, Wu J, Luo Y, et al. Expanded polyglutamine-binding peptoid as a novel therapeutic agent for treatment of Huntington’s disease. Chem Biol. 2011;18(9):1113–25. doi: 10.1016/j.chembiol.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Reis SD, Pinho BR, Oliveira JMA. Modulation of molecular chaperones in Huntington’s disease and other polyglutamine disorders. Mol Neurobiol. 2017;54(8):5829–54. doi: 10.1007/s12035-016-0120-z. [DOI] [PubMed] [Google Scholar]
- 215.Kampinga HH, Bergink S. Heat shock proteins as potential targets for protective strategies in neurodegeneration. Lancet Neurol. 2016;15(7):748–59. doi: 10.1016/S1474-4422(16)00099-5. [DOI] [PubMed] [Google Scholar]
- 216.Al-Ramahi I, Lam YC, Chen HK, et al. CHIP protects from the neurotoxicity of expanded and wild-type ataxin-1 and promotes their ubiquitination and degradation. J Biol Chem. 2006;281(36):26714–24. doi: 10.1074/jbc.M601603200. [DOI] [PubMed] [Google Scholar]
- 217.Williams AJ, Knutson TM, Colomer Gould VF, Paulson HL. In vivo suppression of polyglutamine neurotoxicity by C-terminus of Hsp70-interacting protein (CHIP) supports an aggregation model of pathogenesis. Neurobiol Dis. 2009;33(3):342–53. doi: 10.1016/j.nbd.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Cummings CJ, Sun Y, Opal P, et al. Over-expression of inducible HSP70 chaperone suppresses neuropathology and improves motor function in SCA1 mice. Hum Mol Genet. 2001;10(14):1511–8. doi: 10.1093/hmg/10.14.1511. [DOI] [PubMed] [Google Scholar]
- 219.Fujimoto M, Takaki E, Hayashi T, et al. Active HSF1 significantly suppresses polyglutamine aggregate formation in cellular and mouse models. J Biol Chem. 2005;280(41):34908–16. doi: 10.1074/jbc.M506288200. [DOI] [PubMed] [Google Scholar]
- 220.Helmlinger D, Bonnet J, Mandel JL, Trottier Y, Devys D. Hsp70 and Hsp40 chaperones do not modulate retinal phenotype in SCA7 mice. J Biol Chem. 2004;279(53):55969–77. doi: 10.1074/jbc.M409062200. [DOI] [PubMed] [Google Scholar]
- 221.Neef DW, Turski ML, Thiele DJ. Modulation of heat shock transcription factor 1 as a therapeutic target for small molecule intervention in neurodegenerative disease. PLoS Biol. 2010;8(1):e1000291. doi: 10.1371/journal.pbio.1000291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Nelson VK, Ali A, Dutta N, et al. Azadiradione ameliorates polyglutamine expansion disease in Drosophila by potentiating DNA binding activity of heat shock factor 1. Oncotarget. 2016;7(48):78281–96. doi: 10.18632/oncotarget.12930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.van Waarde-Verhagen M, Kampinga HH. Measurement of chaperone-mediated effects on polyglutamine protein aggregation by the filter trap assay. Methods Mol Biol. 2018;1709:59–74. doi: 10.1007/978-1-4939-7477-1_5. [DOI] [PubMed] [Google Scholar]
- 224.Pulsinelli WA. Selective neuronal vulnerability: morphological and molecular characteristics. Prog Brain Res. 1985;63:29–37. doi: 10.1016/S0079-6123(08)61973-1. [DOI] [PubMed] [Google Scholar]
- 225.Jaspers NG, Gatti RA, Baan C, Linssen PC, Bootsma D. Genetic complementation analysis of ataxia telangiectasia and Nijmegen breakage syndrome: a survey of 50 patients. Cytogenet Cell Genet. 1988;49(4):259–63. doi: 10.1159/000132673. [DOI] [PubMed] [Google Scholar]
- 226.Jung J, Bonini N. CREB-binding protein modulates repeat instability in a Drosophila model for polyQ disease. Science. 2007;315(5820):1857–9. doi: 10.1126/science.1139517. [DOI] [PubMed] [Google Scholar]
- 227.Sinha S, Goyal S, Somvanshi P, Grover A. Mechanistic insights into the binding of class IIa HDAC inhibitors toward spinocerebellar ataxia type-2: a 3D-QSAR and pharmacophore modeling approach. Front Neurosci. 2016;10:606. doi: 10.3389/fnins.2016.00606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Hubert L, Jr, Lin Y, Dion V, Wilson JH. Xpa deficiency reduces CAG trinucleotide repeat instability in neuronal tissues in a mouse model of SCA1. Hum Mol Genet. 2011;20(24):4822–30. doi: 10.1093/hmg/ddr421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Gao R, Liu Y, Silva-Fernandes A, et al. Inactivation of PNKP by mutant ATXN3 triggers apoptosis by activating the DNA damage-response pathway in SCA3. PLoS Genet. 2015;11(1):e1004834. doi: 10.1371/journal.pgen.1004834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Wan L, Xu K, Chen Z, Tang B, Jiang H. Roles of post-translational modifications in spinocerebellar ataxias. Front Cell Neurosci. 2018;12:290. doi: 10.3389/fncel.2018.00290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Asada A, Yamazaki R, Kino Y, et al. Cyclin-dependent kinase 5 phosphorylates and induces the degradation of ataxin-2. Neurosci Lett. 2014;563:112–7. doi: 10.1016/j.neulet.2014.01.046. [DOI] [PubMed] [Google Scholar]
- 232.Tsoi H, Lau TC, Tsang SY, Lau KF, Chan HY. CAG expansion induces nucleolar stress in polyglutamine diseases. Proc Natl Acad Sci U S A. 2012;109(33):13428–33. doi: 10.1073/pnas.1204089109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Zhang Q, Chen ZS, An Y, et al. A peptidylic inhibitor for neutralizing expanded CAG RNA-induced nucleolar stress in polyglutamine diseases. RNA. 2018;24(4):486–98. doi: 10.1261/rna.062703.117. [DOI] [PMC free article] [PubMed] [Google Scholar]