

Abstract
Asthma is defined as chronic inflammation of the airways and is characterized by airway remodeling, hyperresponsiveness, and acute bronchoconstriction of airway smooth muscle (ASM) cells. Clinical findings suggest a higher incidence and severity of asthma in adult women, indicating a concrete role of sex steroids in modulating the airway tone. Estrogen, a major female sex steroid mediates its role through estrogen receptors (ER) ERα and ERβ, which are shown to be expressed in human ASM, and their expression is upregulated in lung inflammation and asthma. Previous studies suggested rapid, nongenomic signaling of estrogen via ERs reduces intracellular calcium ([Ca2+]i), thereby promoting relaxation of ASM. However, long-term ER activation on [Ca2+]i regulation in human ASM during inflammation or in asthma is still not known. In Fura-2-loaded nonasthmatic and asthmatic human ASM cells, we found that prolonged (24 h) exposure to ERα agonist (PPT) increased [Ca2+]i response to histamine, whereas ERβ activation (WAY) led to decreased [Ca2+] compared with vehicle. This was further confirmed by ER overexpression and knockdown studies using various bronchoconstrictor agents. Interestingly, ERβ activation was more effective than 17β-estradiol in reducing [Ca2+]i responses in the presence of TNF-α or IL-13, while no observable changes were noticed with PPT in the presence of either cytokine. The [Ca2+]i-reducing effects of ERβ were mediated partially via L-type calcium channel inhibition and increased Ca2+ sequestration by sarcoplasmic reticulum. Overall, these data highlight the differential signaling of ERα and ERβ in ASM during inflammation. Specific ERβ activation reduces [Ca2+]i in the inflamed ASM cells and is likely to play a crucial role in regulating ASM contractility, thereby relaxing airways.
Keywords: asthma, calcium, estrogen, inflammation, sex steroid, signaling
INTRODUCTION
Asthma is a chronic inflammatory respiratory disease characterized by airway hyperresponsiveness (AHR) and remodeling (18, 54). Epidemiological studies show sex differences in asthma incidence, prevalence, and severity (15, 19, 29, 65, 67, 70, 85). During childhood, boys have nearly twice the risk of developing asthma over girls. However, interestingly, during adulthood, there is a transition toward a female predominance, and asthma incidence is highest in women of reproductive age, suggesting a role for sex steroids in asthma genesis (13, 22). Although higher asthma prevalence in women hints toward the detrimental role of female sex hormone estrogen, a closer examination into the fluctuations of asthma exacerbations during different phases of menstrual cycle suggests symptom severity in asthma during lower plasmatic estrogen levels (21, 30, 71, 75). Overall, whether and how estrogens influence airway structure/function is still unclear. It should be noted here that, the issue of sex steroid effects in the airways is important beyond “sex differences” in asthma and is relevant to disease pathophysiology in both males and females and needs to be addressed.
Among various factors leading to airway obstruction, airway smooth muscle (ASM) cell hypertrophy, hyperplasia, and contraction are the prominent factors for determining the airway tone, and ultimately AHR (36). Maintenance of airway tone through ASM cells is dependent on the intracellular calcium ([Ca2+]i) levels, which regulates bronchoconstriction and bronchodilation (6, 8, 59). At baseline, [Ca2+]i is maintained at ~80 to ~120 nM, which is necessary to sustain the optimum airway tone (12, 27, 59, 72). Studies have demonstrated that increased airway tone in diseases such as asthma involves increased [Ca2+]i (34). Since [Ca2+]i regulation plays a determining role in airway smooth muscle contraction and the overall AHR in asthma, it is essential to explore the contribution of sex steroids in this direction.
Estrogen mediates its action prominently through estrogen receptors (ERs) ERα and ERβ (33, 37), as well as slightly through membrane-associated G protein-coupled estrogen receptor 30 (GPR30/GPER) (10, 58). All of these receptors are present and exert their actions in a wide variety of tissues/cells, such as vascular endothelial, cardiomyocytes, neuron, and smooth muscle (46, 48). ERs function through activation of target genes through a genomic pathway causing transcription of genes (33, 48, 49, 60), as well as by rapid activation of signaling pathways (2, 16, 41, 60, 83). These rapid effects of estrogen are produced within seconds to minutes and involves simple biological processes, such as activation of kinases and phosphatases, and increase or decrease in ion fluxes across membranes. Although ER signaling per se is established in other cell types and is highly cell and context-dependent, knowledge of their mechanisms, especially in disease states, such as asthma, seems limited (2, 16).
Previous studies have shown the constitutive expression of ERs in the lungs, and the expression levels seem to be comparable in both men and women (3, 38, 47). Studies from our own group suggested that physiologically relevant concentrations of estrogens significantly reduce [Ca2+]i responses to bronchoconstrictor agonists in human ASM cells and that the acute estrogen effect on the reductions in [Ca2+]i are largely mediated via ERα (81). Here, we further confirmed the full-length expression of ERα and ERβ in ASM with no notable expression of GPR30 in human ASM (81). Furthermore, ER isoform-specific agonists with short-term exposure resulted in increased cAMP, thereby, producing an overall relaxation of ASM (80). Interestingly, our recent studies have shown upregulated ER (both ERα and ERβ) expression in human ASM cells during inflammation and/or in asthma, with multifold increase observed with ERβ. In addition, recent studies suggested the noticeable difference in ER isoform-specific signaling in human ASM proliferation, especially during inflammation or in asthma (3, 4). However, it is not clear whether and how ERs specific signaling impacts the ASM [Ca2+]i and airway contractility in asthma. In this study, we hypothesized that the conflict regarding estrogen’s effects on the airway involves differential roles of ERs in ASM. Using a molecular approach and specific agonists/antagonists for ERα and ERβ, we examined [Ca2+]i regulation in ASM cells of nonasthmatic and asthmatic patients in the presence of proinflammatory cytokines.
MATERIALS AND METHODS
Chemicals and reagents.
Cell culture media and reagents, such as Dulbecco’s modified Eagle’s medium/F12 (DMEM/F12), trypsin and AbAm were purchased from Gibco (Carlsbad, CA). Charcoal-stripped fetal bovine serum (FBS) was used throughout the studies to avoid hormonal contamination in the media and was purchased from Sigma (St. Louis, MO). Proinflammatory cytokines tumor necrosis factor-α (TNF-α) and IL-13 were purchased from Invitrogen (Carlsbad, CA). Chemicals and supplies were purchased from Sigma, unless otherwise specified. Pharmacological modulators for ERs, 17-β estradiol (E2), ERα agonists, ERα-agonist (PPT, propylpyrazoletriol; THC, tetrahydro-2,8-chrysenediol) and ERβ agonists (WAY-200070; FERΒ-033; DPN, diarylpopionitrile), and ERα and ERβ antagonists (MPP and PHTPP, respectively) were obtained from Tocris (Minneapolis, MN). β-actin antibody was obtained from Applied Biological Sciences (cat. no. G043), SERCA2 antibody was obtained from Abcam (cat. no. ab2861). IRDye at goat anti-mouse secondary antibody was used followed by scanning in Li-Cor Odyssey imaging system (Li-Cor Systems, Omaha, NE).
ESR1 silencer select predesigned siRNA (Ambion, Austin, TX; cat. no. 4392420), ON-TARGETplus human ESR2 (2100) siRNA-Individual (Dharmacon, Lafayette, CO; cat. no. J-003402-13-0005) were used to knockdown ERα and ERβ, respectively. For negative controls, the silencer negative control 1 (cat. no. AM4611) from Ambion was used. For overexpression studies, ERα and ERβ plasmids tagged with GFP were created and gifted by Dr. Ratna Valdamudi (obtained via Addgene, cat. no. 65211 and 65212). Zymopure plasmid midiprep isolation kit (Zymoresearch, cat. no. D4200 and D4201) was used for isolating the plasmids. Imaging experiments were performed in HBSS obtained from Gibco (Carlsbad, CA). Calcium-sensitive dye, Fura-2 AM was obtained from Invitrogen, and calibration was done using a calibration kit from Invitrogen (cat. no. C3008MP).
Human ASM cells.
The technique for isolating human ASM cells has been previously described (1, 69). Briefly, third- to sixth-generation human bronchi were obtained from lung specimens from patients incidental to thoracic surgeries at Mayo Clinic for focal, noninfectious causes. The Mayo Clinic’s Institutional Review Board approved protocols allowed for initial review of patient histories, with complete deidentification of samples for storage and subsequent usage. This particular study was limited to samples from 25 male and female nonsmoker adults of ages 21 to 65 yr from both nonasthmatic and asthmatic (mild to moderate) patients. For each study, cells from five to seven nonasthmatic and asthmatic patients were chosen randomly and used independently. After removing the debris, airway samples were denuded of epithelium, and ASM tissue was enzymatically dissociated, according to previously described procedures (56) following the manufacturer’s instructions (Worthington Biochemical, Lakewood, NJ) to generate ASM cells. Cultures (<5th passage) were maintained under optimal conditions of 37°C (5% CO2, 95% air) in DMEM/F12 growth media. Periodically, ASM phenotype was verified by Western blot analysis for smooth muscle α-actin and Caldesmon (51). Cells were serum starved for 24 h before treatment with agonists or antagonists.
Cell culture and treatment.
Fully confluent T-75 flasks of ASM cells were trypsinized mixed in 10% FBS (charcoal stripped) growth medium (DMEM/F-12 with 1% AbAm), counted, and seeded ~10,000 cells/well into Biotek eight-well chamber plates. Cells were allowed to adhere, grown up to 60–70% confluence, and incubated in serum-free medium (DMEM/F12 without FBS) for 24 h to mature. ASM cells were treated with proinflammatory cytokines in the presence and absence of ER-specific agonists, such as PPT and THC (10 nM), WAY, DPN, and FERB-033 (10 nM) and E2 (1 nM). Briefly, after 2 h of preincubation with respective agonist or antagonist treatment groups, cells were then exposed to TNF-α (20 ng/ml) or IL-13 (50 ng/ml) for 24 h. Serum-free media alone served as a vehicle. ASM cells were grown on 60-mm petri plates and treated similarly to collect lysates for mRNA used in quantitative PCR techniques.
Small interfering RNA transfection and overexpression.
Technique to knockdown the ERα and ERβ by interfering RNA (siRNA) has been recently described by our group (4). Briefly, human ASM cells were grown in eight-well plates to ~50% confluence. Transfection was achieved using 20 nM siRNA and Lipofectamine 3000 transfection reagent (Invitrogen) in DMEM/F-12 media without FBS and antibiotics. Six hours after transfection, growth medium was added and maintained for 24 h. Subsequently, cells were serum starved for another 24 h before being subjected to treatments followed by live-cell imaging. Efficiency of knockdown and specificity was verified by mRNA analysis by performing transfection of ASM in 60-mm plates.
Overexpression studies were performed with ER-specific plasmids tagged with GFP obtained in bacterial stabs from Addgene (Watertown, MA). The respective bacteria were streaked onto agar plate, and single colonies were obtained. Using the LB broth, we inoculated single colonies overnight, and plasmids were isolated using Zymopure plasmid Midiprep isolation kit and verified using gel electrophoresis. Before plasmid transfection, ASM cells were grown in eight-well plates to ~50% confluence. Transfection was achieved using Lipofectamine 3000 transfection reagent (Invitrogen) in reduced DMEM/F-12 (5% FBS and no antibiotics) with 0.5 μg of plasmids. Cells were incubated as such for 24 h in reduced media and then withdrawn to serum-free media for 24 h for further imaging studies. More than 70% transfection efficiency was achieved, and the efficiency of the transfection was confirmed using Olympus microscope with FITC filter. The effect of siRNA and overexpression on the [Ca2+]i regulation in the presence of E2, WAY, or PPT were analyzed by real-time [Ca2+]i imaging.
Western blot analysis.
Previously described standard techniques were used to perform Western blot analysis (4). Briefly, cells were washed, harvested in lysis buffer (Cell Signaling Technologies, Beverly, MA) containing protease inhibitors, and resultant supernatants were assayed for total protein content using the DC protein assay kit (Bio-Rad, Hercules, CA). Thirty micrograms equivalent protein from each treatment group lysate were loaded on 4–15% gradient gels (Criterion Gel System; Bio-Rad, Hercules, CA) and transferred into 0.22 µm PVDF membranes using a Bio-Rad Trans-Blot Turbo rapid transfer system (Bio-Rad, Hercules, CA). Nonspecific binding was blocked using 5.0% bovine serum albumin in Tris-buffered saline (TBS) for 1 h at room temperature, and the membranes were probed overnight at 4°C with specific primary antibodies of interest. Following three washes with TBS with 0.1% Tween (TBST), blots were then incubated with LiCOR near-infrared conjugated secondary antibody. Protein expression was detected by imaging the membrane on a Li-Cor Odyssey XL system. β-actin was used as a loading control. The normalized values were obtained by dividing the raw values of proteins of interest with the raw values of β-actin. The obtained values were then normalized to vehicle.
Quantitative PCR analysis.
Cells were washed with RNA-grade DPBS, trypsinized, and centrifuged. Total RNA was extracted using Quick-RNA MiniPrep kit (Zymo Research, Irvine, CA) following the manufacturer’s protocol, and cDNA was synthesized using OneScript cDNA synthesis kit (Applied Biological Materials, Richmond, BC, Canada) using 500 ng of quantified RNA (Take3 Micro volume plate, Synergy HTX, Biotek) for each sample. BrightGreen 2× qPCR Master Mix (Applied Biological Materials cat. no. MasterMix-S-XL) protocol was followed using QuantStudio 3 qPCR system as per the manufacturer’s instructions. The following primers were used for quantitative PCR analysis: ERα (forward 5′-GCC TGA ATG GCG AAT GGA-3′ and reverse 5′-GAA GGG AAG AAA GCG AAA GGA-3′); ERβ (forward 5′-GCT TAC TCC GAC CAT GAT TTC T-3′ and reverse 5′-GCC GAT GCT TGC AAT AGT TTA G-3′); SERCA 2 (forward 5′-ATT GGA CCC AGA GAA GTT GAC-3′ and reverse 5′-TCG GAC CAT TTA AGT CTT CAG AGA T-3′), s16 (forward 5′-CAA TGG TCT CAT CAA GGT GAA CGG-3′ and reverse 5′-CTG GAT AGC ATA AAT CTG GGC-3′). The fold changes in mRNA expression were calculated by normalization of cycle threshold (Ct) value of target genes to reference gene s16 using the ΔΔCt method, as previously described (3, 4, 43).
Real-time [Ca2+]i imaging.
The technique for [Ca2+]i imaging of human ASM cells using Fura-2 has been previously described (64, 65, 69, 82). After 24 h of incubation with the respective treatments, solutions containing various treatments were aspirated, and human ASM cells were washed with freshly prepared HBSS (1×) buffer solution having 2.5 mM calcium and 1 mM magnesium ions (from HBSS 10× Gibco). After the wash, ASM cells were incubated with 4 μM Fura-2 AM (prepared in HBSS solution) for 1 h at room temperature. Following 1 h, the dye was aspirated, and cells were washed three times with 1× HBSS buffer solution. The ASM cells were imaged at ×20 magnification with ratiometric Fura-2 filters (alternatively excited at 340 and 380 nm), and emissions at 510 nm were collected in the Olympus fluorescence microscope (Fluoview FV300) equipped with Hamamatsu camera using Cool-LED lamp. A custom-built fluid level controller allowed cell perfusion with a rapid exchange of perfusate with the help of Gilson minipump and vacuum. [Ca2+]i responses of at least 10 cells/chamber were obtained. Results were obtained in the ratio of 340/380-nm wavelengths, and quantification of [Ca2+]i was performed using previously described calibration procedures (31, 56). The change in fluorescence intensities in the individual software facilitated selected regions of interest, and the corresponding [Ca2+]i responses were recorded in response to 10 μM histamine, 1 μM ACh, and 1 μM bradykinin. The area under the curve (AUC) of the agonist-induced [Ca2+]i response curve was determined using GraphPad Prism software (50). For the overexpression studies, regions of interest were selected in those areas of the cells showing high levels of green fluorescence protein (GFP) with the help of FITC filter, and the then real-time live-cell imaging was performed with Fura-2 filter.
To isolate the cells from the calcium environment, following initial incubation with Fura-2 AM containing HBSS with calcium, cells were perfused in zero calcium containing HBSS, and the Ca2+ channels were nonspecifically blocked with 1 μM LaCl3 in HBSS for the “0” Ca2+ experiments. The rate of decline of the [Ca2+]i was calculated in terms of time to decay (ms). For the purpose of studying the L-type calcium channels (LTCC), the cells were perfused in calcium containing HBSS and before histamine, 1 μM nifedipine in HBSS was exposed to the cells for 15 min, resulting in blockage of LTCC. Further confirmatory studies on LTCC were performed using 100 mM KCl instead of histamine, which effectively depolarizes the membrane through voltage-gated channels.
Statistical analysis.
Up to 20 ASM cells from different nonasthmatic and asthmatic patients were used. Each set of [Ca2+]i experiments were performed in at least five randomly selected different patient cells, although not all protocols were performed in each sample. All the experiments were repeated at least three times for each patient sample. Statistical analysis was performed using GraphPad Prism version 8.0.0 for Windows (San Diego, CA). Statistical differences between the experimental groups were analyzed using Student’s t test or one-way ANOVA followed by Dunnett’s or Tukey’s post hoc test for multiple comparisons, where appropriate. Statistical significance was established at a minimum of P ≤ 0.05. All values are expressed as means ± SE.
RESULTS
Effect of ER-specific activation on [Ca2+]i response to agonist.
To determine the role of long-term ER signaling in [Ca2+]i regulation, ASM cells were exposed to ERα (PPT, 10 nM), ERβ (WAY, 10 nM) agonists, and E2 (1 nM) for 24 h, and the [Ca2+]i response was evaluated using Fura-2 AM [Ca2+]i response was measured and presented as AUC in both asthmatic and nonasthmatic ASM cells using three different agonists to induce elicitation: histamine (10 µM), ACh (1 µM), and bradykinin (10 nM). ERα agonist PPT-treated cells showed a significant increase in [Ca2+]i response to histamine compared with the vehicle in nonasthmatic ASM cells (P ≤ 0.05), and asthmatic ASM cells (P ≤ 0.01). Nonselective ER agonist E2 did not show any significant changes in the [Ca2+]i compared with vehicle (Fig. 1B); however, ERβ agonist significantly decreased the histamine induced [Ca2+]i response in asthmatic ASM cells (P ≤ 0.05). Representative traces of [Ca2+]i response to histamine in the presence of ER agonists in both nonasthmatic and asthmatic cells are shown in Fig. 1A. In response to bradykinin and ACh, the effect of PPT in increasing the [Ca2+]i response was similar for both asthmatic and nonasthmatic ASM cells (Fig. 1C, P ≤ 0.001). In ACh elicited [Ca2+]i response, E2 showed a significant increase in [Ca2+]i response in nonasthmatic ASM cells (P ≤ 0.05). Notably, there were no differences observed in baseline calcium levels.
Fig. 1.
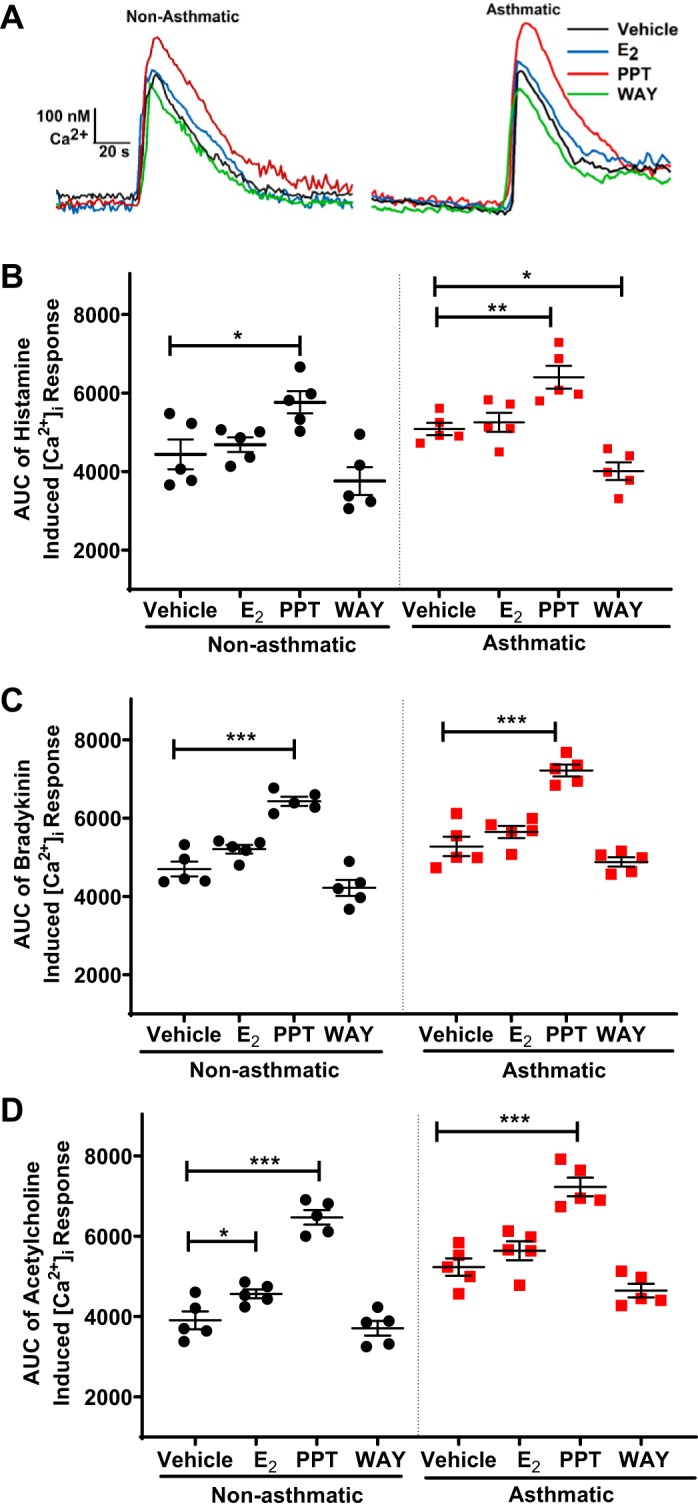
Effect of long-term exposure of estrogen receptor (ER)-specific agonists on intracellular Ca2+ ([Ca2+]i) response in nonasthmatic and asthmatic human airway smooth muscle (ASM) cells. A: representative traces showing transient [Ca2+]i responses in Fura-2-loaded cells with long-term (24 h) exposure of estrogen receptor α (ERα; PPT), ERβ (WAY) agonists, and nonselective 17-β estradiol (E2) in nonasthmatic and asthmatic ASM cells. In both nonasthmatic and asthmatic ASM cells treated with ER-specific agonists, the area under the curve (AUC) of the [Ca2+]i responses to 10 µM histamine (B), 1 µM bradykinin (C), and 1 µM ACh (D) were measured. *P < 0.05, **P < 0.01, ***P < 0.001 vs. vehicle. Data are presented as means ± SE; n = 5 patients each.
Effect of various ER selective agonists and antagonists on [Ca2+]i response.
Considering our previous studies, where acute ERα activation decreased ASM [Ca2+]i response (18), our results from Effect of ER-specific activation on [Ca2+]i response to agonist were distinct, and hence, additional validation of the long-term role of differential ER signaling in [Ca2+]i response in ASM was needed. Here, we used different ERα (PPT and THC) and ERβ (WAY, FERB-033, and DPN) agonists to study the histamine-elicited [Ca2+]i response in nonasthmatic human ASM (Fig. 2A). In addition, we used ER-specific antagonists, ERα (MPP) and ERβ (PHTPP) in the presence of E2 to study the specific ER effects on ASM [Ca2+]i response (Fig. 2B). 10 nM of ERα agonist THC, which has a dual role as an ERα agonist and an ERβ antagonist, significantly increased the [Ca2+]i response to histamine (P ≤ 0.001) compared with vehicle. Similarly, the effect of PPT on [Ca2+]i response was found to be significantly higher(P ≤ 0.01), albeit lower than that of THC. There were no significant changes observed in [Ca2+]i response in cells treated with ERβ agonists (10 nM: WAY, FERB-033, and DPN) compared with vehicle. In a separate set of experiments, cells pretreated with ERα antagonist MPP and followed by 24-h exposure to E2 showed no significant changes in the [Ca2+]i response to histamine, indirectly indicating the ERβ effect. Similarly, cells pretreated with ERβ antagonist before treatment with E2 for 24 h showed a significantly higher [Ca2+]i response, as compared with vehicle, pointing indirectly toward an ERα effect (P ≤ 0.001). All of these data indicate that ERα activation leads to increased [Ca2+]i response, whereas ERβ activation tends to decrease [Ca2+]i response in ASM cells but not significantly compared with vehicle.
Fig. 2.
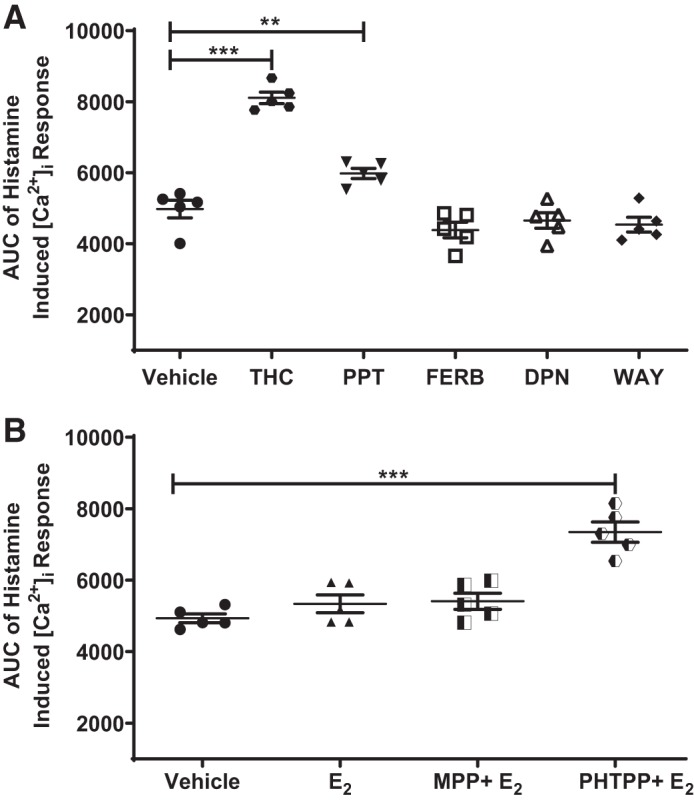
Effect of various estrogen receptor (ER) agonists and antagonists on intracellular Ca2+ ([Ca2+]i) response in nonasthmatic human airway smooth muscle (ASM) cells. A: long-term effects of various ERα agonists (THC and PPT), ERβ agonists (WAY, DPN, and FERB-033) on area under the curve (AUC) of the [Ca2+]i response to histamine were evaluated in nonasthmatic human ASM cells. B: in the presence of ERα antagonist (MPP) or ERβ antagonist (PHTPP), the effect of E2 on the AUC of the [Ca2+]i response to histamine was measured. **P < 0.01, ***P < 0.001 vs. vehicle. Data are presented as means ± SE; n = 5 patients each.
Effect of specific ER siRNA and overexpression on [Ca2+]i response.
To further confirm the observed pharmacological results on the ER-specific [Ca2+]i response, molecular level experiments were performed using ER overexpression or ER-specific siRNA on human ASM cells (Fig. 3). The ER-specific siRNA transfection was performed using standard protocols in nonasthmatic human ASM cells, and the efficiency of ER-specific siRNA (~70%) was evaluated using mRNA analysis (Fig. 3B). In ERα siRNA-transfected ASM cells, treatment with E2 (P ≤ 0.001), as well as with ERα agonist PPT (P ≤ 0.01), for 24 h showed a significantly reduced [Ca2+]i response to histamine as compared with the negative siRNA-treated cells (Fig. 3A). E2 and WAY exposure on ERβ siRNA transfected did not show any significantly changes in the [Ca2+]i response compared with negative siRNA (P ≤ 0.05). For overexpression studies, GFP-tagged ER-specific plasmids were used, and their transfection efficiency was confirmed through fluorescence imaging (Fig. 3E). ASM cells overexpressed with ERα-GFP plasmid, and when subsequently treated with E2 (24 h), showed a higher [Ca2+]i response (P ≤ 0.001) as compared with lipofectamine, whereas ERβ-GFP transfected cells treated with E2 showed a reduced [Ca2+]i response (P ≤ 0.001) compared with lipofectamine-treated cells (Fig. 3C). Representative traces of [Ca2+]i response to histamine regulated through specific overexpressed ERs in the presence of E2 are shown in the Fig. 3D.
Fig. 3.

Effect of specific estrogen receptor (ER) small interference RNA (siRNA) and overexpression on intracellular Ca2+ ([Ca2+]i) response in nonasthmatic human airway smooth muscle (ASM) cells. A: effects of E2, PPT, and WAY in ER-specific siRNA-transfected cells on the AUC of the [Ca2+]i response to histamine were evaluated in nonasthmatic human ASM cells. B: transfection efficiency of siRNA was evaluated using mRNA expression studies. C: ERα and ERβ overexpressed ASM cells were treated with nonselective agonist E2 and their AUC of the [Ca2+]i response to histamine were evaluated. D: representative traces showing effect of overexpression of specific ERs on the [Ca2+]i. E: representative images of ERα-GFP and ERβ-GFP transfections on nonasthmatic ASM cells. **P < 0.01, ***P < 0.001 vs. negative siRNA or lipofectamine (Lipofect). Data are presented as means ± SE; n = 5–7 patients.
Effect of specific ER signaling on TNF-α mediated [Ca2+]i response.
Nonasthmatic and asthmatic ASM cells were treated with E2, PPT, or WAY for 2 h and then treated with TNF-α (20 ng/ml) for 24 h in these experimental conditions. TNF-α alone produced a marked increase in [Ca2+]i responses to ACh, bradykinin, and histamine, as compared with the vehicle (P ≤ 0.001) in both asthmatic and nonasthmatic ASM cells (Fig. 4), which is consistent with our previous results (63, 69). In nonasthmatic and asthmatic ASM cells, ERβ agonist WAY (P ≤ 0.001), but not PPT, showed a reduction in the enhancing effect of TNF-α on [Ca2+]i response to all the three agonists (P ≤ 0.001, Fig. 4, A–F). Here, the effect of E2 in reducing the increased [Ca2+]i response varied depending on the conditions and showed a significant reduction in TNF-α-induced effect on [Ca2+]i responses to histamine (P ≤ 0.05, Fig. 4B), bradykinin, and ACh in asthmatic cells (P ≤ 0.01, Fig. 4, D and F).
Fig. 4.

Effect of differential estrogen receptor (ER) signaling on the TNF-α enhancement of intracellular Ca2+ ([Ca2+]i) response in human airway smooth muscle (ASM) cells. Human nonasthmatic and asthmatic cells were treated with E2, PPT, or WAY for 2 h before treatment with TNF-α (20 ng/ml) for 24 h and their effects on the AUC of the [Ca2+]i response to histamine (A and B), bradykinin (C and D), and ACh (E and F) were evaluated. ***P < 0.001 vs. vehicle. #P ≤ 0.05, ##P < 0.01, ###P < 0.001 vs. TNF-α. Data are presented as means ± SE; n = 5 patients each.
Effect of specific ER signaling on IL-13-mediated [Ca2+]i response.
Similar to the effect of TNF-α above, exposure of IL-13 (50 ng/ml) for 24 h also produced a significant increase in [Ca2+]i response to ACh, bradykinin, and histamine, as compared with the vehicle (P ≤ 0.001), in both nonasthmatic and asthmatic cells (Fig. 5), demonstrating Th2 effect in increasing [Ca2+]i in human ASM cells, which is comparable to our previous findings (63, 69). In nonasthmatic and asthmatic ASM cells, significant reduction in the [Ca2+]i response to histamine was observed in IL-13-exposed cells treated with WAY (P ≤ 0.001, Fig. 5, A and B). In asthmatic ASM cells (Fig. 5B), E2 showed a reduction in IL-13-induced [Ca2+]i response to histamine (P ≤ 0.01). No significant effects were observed in cells treated with PPT in the presence of IL-13. With respect to bradykinin and ACh-elicited [Ca2+]i response, WAY in the presence of IL-13 significantly reduced the [Ca2+]i response in both nonasthmatic cells (P ≤ 0.01, Fig. 5, C and E) and asthmatic ASM cells (P ≤ 0.001, Fig. 5, D and F). In asthmatic ASM cells, E2 significantly decreased the IL-13-induced increase in [Ca2+]i response to bradykinin (P ≤ 0.01, Fig. 5D) and to a lesser extent (but significant) with ACh (P ≤ 0.05, Fig. 5F). Overall, WAY invariably showed a decreased [Ca2+]i response in IL-13-treated cells in both nonasthmatic and asthmatic cells (P ≤ 0.001), whereas PPT did not show any significant changes in [Ca2+]i response.
Fig. 5.

Effect of differential estrogen receptor (ER) signaling on the IL-13 enhancement of intracellular Ca2+ ([Ca2+]i) response in airway smooth muscle (ASM) cells. Human nonasthmatic and asthmatic cells were treated with E2, PPT, or WAY for 2 h before treatment with IL-13 (50 ng/ml) for 24 h, and their effects on the area under the curve (AUC) of the [Ca2+]i response to histamine (A and B), bradykinin (C and D), and ACh (E and F) were evaluated. ***P < 0.001 vs. vehicle, #P ≤ 0.05, ##P < 0.01, ###P < 0.001 vs. IL-13. Data are presented as means ± SE; n = 5 patients each.
Effect of differential ER signaling on [Ca2+]i reuptake.
To confirm the [Ca2+]i regulation of ER through intracellular stores, cells were isolated from the calcium-containing environment (“0” Ca2+ HBSS), and their [Ca2+]i response to histamine was studied in nonasthmatic human ASM cells. Cells were treated with ER agonists for 2 h and then exposed to TNF-α or IL-13 for 24 h, and the [Ca2+]i response was evaluated in the absence of extracellular Ca2+ and reduced plasma membrane Ca2+ fluxes. Proinflammatory cytokines, TNF-α, or IL-13 both showed a significant increase in the [Ca2+]i response to histamine, as compared with vehicle (P ≤ 0.001, Fig. 6, A and B). As expected, the [Ca2+]i responses in the environment devoid of Ca2+ was lower, indicating the effect only on the intracellular stores. In the presence of either of these proinflammatory cytokines, ERβ agonist WAY-treated ASM cells significantly reduced the [Ca2+]i response (P ≤ 0.001), whereas PPT or E2-treated cells showed negligible effects on the [Ca2+]i response in the “0” Ca2+ HBSS environment. In these experiments, the effects of ERs on sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA2) reuptake were evaluated by calculating time to decay (Fig. 6, C and D). Time to decay was significantly higher in TNF-α or IL-13 alone-treated groups (P ≤ 0.001) and was significantly lower in cells treated with WAY (P ≤ 0.001). E2 and PPT did not effectively change the TNF-α- or IL-13-induced increase in time to decay. Furthermore, SERCA2 protein expressions were significantly reduced in TNF-α- or IL-13-treated cells compared with vehicle (Fig. 6, E and F; P ≤ 0.01). ERβ agonist WAY treatment reversed this trend in TNF-α (P ≤ 0.01) or IL-13 (P ≤ 0.001)-exposed ASM cells and showed increased SERCA2 expression, but there were no differences observed in PPT or E2-treated cells. This protein expression results were further confirmed by SERCA2 mRNA expression (not shown here). Overall, these support the concept that ERβ activation can blunt [Ca2+]i responses by increasing Ca2+ sequestration at intracellular level.
Fig. 6.

Effect of differential estrogen receptor (ER) signaling on sarcoplasmic reticulum (SR) intracellular Ca2+ ([Ca2+]i) reuptake. In zero calcium environment, the effect on the area under the curve (AUC) of the [Ca2+]i response due to proinflammatory cytokines TNF-α (A) and IL-13 (B) in the presence of specific ER agonists were evaluated in nonasthmatic human airway smooth muscle (ASM) cells. The rate of fall of [Ca2+]i response representing [Ca2+]i reuptake by SR was measured in TNF-α- (C) or IL-13- (D) exposed ASM cells. SERCA2 protein expression analysis of ER agonists treated TNF-α or IL-13-exposed nonasthmatic human ASM cells (E and F). ***P < 0.001, **P < 0.01 vs. vehicle; ##P < 0.01, ###P < 0.001 vs. cytokines. Data are presented as means ± SE; n = 5 or 6 patients.
Effect of ERβ signaling on L-type calcium channel influx.
Previous studies showed decreased Ca2+ influx in human ASM due to acute action of estrogen. To identify specific contribution of ERβ signaling in ASM [Ca2+]i regulation, an additional possible mechanism through LTCC was also tested. After the 24-h treatment (with WAY and cytokines), ASM cells were exposed to LTCC inhibitor nifedipine (1 µM) for 20 min before being subjected to histamine in calcium-containing HBSS. Acute nifedipine exposure significantly decreased the [Ca2+]i response (P ≤ 0.001) compared with vehicle (Table 1). WAY-treated ASM cells in the presence of nifedipine showed similar effects to that of nifedipine-exposed cells in decreasing the [Ca2+]i response, indicating the role of WAY effect partially through LTCC. TNF-α- or IL-13-induced increases in the [Ca2+]i response was significantly reduced by acute nifedipine exposure. Furthermore, WAY treatment substantially and significantly reduced [Ca2+]i response in TNF-α- or IL-13-exposed ASM cells in the presence of nifedipine (P ≤ 0.001), and this was comparable to nifedipine or WAY-alone-treated groups. To further confirm the involvement of ERβ in Ca2+ influx mechanisms, a subsequent set of studies was performed in similar experimental conditions, where KCl (100 mM), instead of histamine, was used as a depolarizing agent. It was found that [Ca2+]i response to KCl was drastically reduced (P ≤ 0.001) in the presence of nifedipine, as compared with vehicle (Table 2), suggesting that KCl depolarizes the ASM cell membrane primarily through LTCC. WAY-treated cells showed a [Ca2+]i response significantly lower than vehicle (P ≤ 0.001), indicating an inhibitory effect primarily via LTCC. Furthermore, TNF-α or IL-13 increased the KCl-induced [Ca2+]i response in ASM cells (P ≤ 0.001), and WAY-treated cells showed decreased [Ca2+]i response (P ≤ 0.001) in the presence of either of the cytokines.
Table 1.
Effect of ERβ signaling on histamine-induced [Ca2+]i response through L-type calcium channel inhibition
| Treatment Groups | AUC ± SE (−Nifedipine) | AUC ± SE (+Nifedipine) |
|---|---|---|
| Vehicle | 5,150.18 ± 127.83 | 3,268.98 ± 175.17* |
| WAY | 4,798.59 ± 225.98 | 2,804.56 ± 340.72* |
| TNF-α | 9,684.00 ± 315.96 | 7,780.38 ± 37.45# |
| WAY + TNF-α | 5,039.17 ± 210.63 | 2,522.55 ± 211.08†§ |
| IL-13 | 9,131.13 ± 250.73 | 7,392.80 ± 213.06# |
| WAY + IL-13 | 5303.31 ± 163.01 | 2734.72 ± 190.93†§ |
Data are presented as means ± SE; n = 7 patients. Human nonasthmatic airway smooth muscle (ASM) cells exposed to TNF-α or IL-13 in the presence of WAY, were subjected to intracellular Ca2+ ([Ca2+]i) response to histamine, and comparisons of their area under the curve (AUC) were obtained with blunting effect of L-type calcium channel (LTCC) inhibitor nifedipine (1 µM, 20 min).
P ≤ 0.05, vs. vehicle or WAY (nifedipine effect),
P < 0.001 vs. respective cytokines (nifedipine effect).
P < 0.001 vs. WAY + cytokines (nifedipine effect).
P < 0.001 vs. cytokines + nifedipine (WAY effect).
Table 2.
Effect of ERβ signaling on KCl-induced [Ca2+]i response
| Treatment Groups | AUC ± SE |
|---|---|
| Vehicle | 3,217.07 ± 225.25 |
| Nifedipine | 899.83 ± 109.22* |
| WAY | 2,002.80 ± 116.53* |
| TNFα | 4,232.01 ± 148.54* |
| WAY + TNF-α | 2,094.94 ± 137.31# |
| IL-13 | 4,528.09 ± 72.89* |
| WAY + IL-13 | 1,995.08 ± 97.52# |
Data are presented as means ± SE; n = 5 patients. Human nonasthmatic airway smooth muscle (ASM) cells exposed to TNF-α or IL-13 in the presence of WAY, were subjected to [Ca2+]i response to KCl. The intracellular Ca2+ ([Ca2+]i) response to KCl-induced depolarization was evaluated and compared with nifedipine.
P < 0.001 vs. vehicle.
P < 0.01 vs. cytokines.
DISCUSSION
In asthma, various inflammatory pathways increase AHR and airway obstruction, leading to recurrent episodes of coughing and wheezing (18, 54). These hallmark features of asthma are mainly mediated through ASM layer, which balances between bronchodilation and bronchoconstriction to maintain the tone of airways (6, 35, 55). [Ca2+]i in ASM cells is one of the major factors for the overall constriction of airways (8, 34, 53, 59, 78). A bronchoconstrictor stimulation (muscarinic or histaminergic) causes transient increase in [Ca2+]i, due to influx of extracellular calcium and release of calcium from the intracellular stores (7). As such, there are several channels and pumps, which affect the [Ca2+]i. Therefore, it is imperative to study various signaling mechanisms, which affect the [Ca2+]i regulation in ASM cells.
Several clinical findings have stressed the sex disparity existing in asthma prevalence and severity (15, 42, 79, 85). Because of increased evidence of asthma prevalence in women, focus has been diverted toward the role of estrogen on ASM cells (29, 67, 85). Estrogen mediates its action via ERα and ERβ, which are ligand-activated transcription factors that are mostly associated with genomic signaling pathways that result in target gene expression (10, 58, 61). Another membrane-associated GPR30 can mediate the acute nongenomic action of estrogens in different cell contexts (2, 10, 16, 83). In the present study, we attempt to establish the link between long-term activation of ERs on the [Ca2+]i regulation in both nonasthmatic and asthmatic ASM cells in the presence of inflammation.
The first study on the modulatory role of estrogen in the airway dates back to 1983 when it was found that these sex steroids cause potentiation of isoproterenol-mediated relaxation (28). More recent studies, including our own, have shown the expression of both ERα and ERβ in human ASM cells (81). Here, acute exposure of physiological concentration of E2, via ERα, inhibits plasma membrane influx, thereby reducing ASM [Ca2+]i (81). Furthermore, cAMP and protein kinase A were found to be increased as a result of acute exposure of E2 (80). In our previous studies, the long-term effect of ERs on the [Ca2+]i levels in healthy and inflamed ASM cells was not addressed owing to the complexities of signaling mechanisms of ER isoforms.
In our recent studies, it was found that during asthma, expression of ERs is found to be increased, which laid the foundation for examining the specific role of ERs in asthmatics (9). Interestingly, ERβ shows a marked increase in expression, as compared with ERα. Furthermore, we have recently demonstrated the differential role of ERs in PDGF-induced ASM cell proliferation (4), as well as in the mixed allergen-induced AHR in the murine models of asthma (3). However, the significance of a transmembrane estrogen receptor GPR30 remains unclear, owing to its lack of expression observed using Western blot analysis (9) and its ineffective G1 (GPR30 agonist) role in ASM proliferation. The results of the present study will establish the differential contribution of ER isoforms in the maintenance of [Ca2+]i and overall airway tone in normal and inflamed conditions.
Our first set of studies demonstrated that the long-term ERα activation led to increased [Ca2+]i response in both asthmatic and nonasthmatic ASM cells. Here, a physiological concentration of E2 (1 nM) has no significant effect on the [Ca2+]i response to histamine, bradykinin, or ACh with 24-h treatment. These results were interesting, owing our previous studies, which showed acute exposure of E2 decreased the [Ca2+]i response (81), indicating the differences in the ER signaling depend on the duration of action. In our studies, we used three different contractile agonists to confirm our results, due to Gq-coupled receptor activation and involvement of voltage-gated, receptor-operated, store-operated [Ca2+]i changes in varying degrees (14, 24). Notably, ERβ activation showed a significant reduction in [Ca2+]i in the asthmatic cells under normal conditions with histamine exposure. It is important to note that the baseline [Ca2+]i levels were observed to be higher in all the asthmatic cells compared with the nonasthmatic cells (data not shown) and not changed with ER-specific agonist treatment.
Modulation of receptors pharmacologically may not be precise, owing to cross-reactivity of the chemical compounds with a variety of other receptor types. Therefore, to confirm our results, various ER-specific agonists and antagonists were randomly screened, and an ERα activation-mediated increase in ASM [Ca2+]i was validated. Particularly, ERα agonist THC, which also shows antagonism toward ERβ receptor (77) exhibited highest [Ca2+]i response, suggesting divergent [Ca2+]i regulation by ERα versus ERβ. The selection of estrogen receptor-selective agonists/antagonists and concentrations used were based on our own and others’ previous studies. For example, WAY200070 has an IC50 of 2.3 nM for ERβ and 155 nM for ERα with 410 times more selectivity to ERβ over ERα (32, 45). Similarly, PPT acts as an ERα-selective agonist, as it is inactive through ERβ (76). All other pharmacological ligands were also selected on the basis of their IC50 and binding affinity reported, as well as from our own studies (4, 20, 26, 45, 52, 77, 86). Hence, on the basis of their IC50 values, it is safe to consider that the agonist concentrations used in our studies (10 nM) have high selectivity toward specific ER activation with minimal crossover.
Furthermore, the differential role of ERα and ERβ activation in the regulation of [Ca2+]i, was confirmed by molecular biology techniques, where effects of ERs were prominently observed as a result of their specific overexpression, using plasmids or knockdown through siRNA. Here, an interesting point to note was that the effectiveness of ERβ in reducing the [Ca2+]i was much more pronounced in ERβ-overexpressed cells, suggesting a regulatory role of ERβ isoform under higher expression.
The important role of TNF-α and IL-13 in airway inflammation and hyperreactivity is well established (5, 7, 23, 25). These cytokines affect several regulatory pathways through Th1 and Th2 responses, thereby increasing the overall [Ca2+]i levels. Previous studies have demonstrated an increased expression of both ERα and ERβ in the TNF-α- or IL-13-treated human ASM cells, hinting toward possible modulation of inflammation due to differential signaling mechanisms of ER isoforms (9). In our present functional studies, in the context of inflammation, intriguingly, downregulation of [Ca2+]i response in the presence of cytokines was particularly highly pronounced with ERβ activation than that with ERα activation, indicating the possible bronchodilatory effects the ERβ isoform could exert on the inflamed or asthmatic airways.
Our work presented here is the first to demonstrate the role of long-term ER signaling on the mechanisms of [Ca2+]i regulation. Notably, the regulatory role of ERβ in reducing [Ca2+]i during inflammation was more prominent in the zero Ca2+ environment. We have previously shown that the human ASM does not express the regulatory phospholamban (66, 68) and that inflammation or asthma leads to decreased SERCA expression (57, 68). Interestingly, in our previous acute exposure studies, we did not find any observable effect of estrogen on the SR Ca2+ reuptake (81). Whereas long-term ERβ activation reverses the trend of cytokine effect with increased SERCA2 expression and function, leading to increased Ca2+ sequestration. Additional possible mechanisms of ERβ also appear to be inhibition of LTCC. Here, KCl was used as a depolarizing agent to elicit the transient [Ca2+]i response because it effectively causes depolarization of the membrane primarily through voltage-gated channels (73). These results corroborate other reported studies on LTCC inhibitory effect of E2 (39, 40, 81). The [Ca2+]i homeostasis in ASM cells is due to the interplay between the vast majority of protein channels and pumps (11, 34, 59, 62), among which LTCC and SERCA have a major contribution (17, 57, 68). If ER signaling affects the SR function (as evidenced by the LaCl3 experiments), it may also modulate regulatory proteins, such as STIM1 and Orai1 that are critical to store-operated calcium entry (SOCE) (17, 27, 44, 74). Furthermore, as transient receptor potential channels and sodium-calcium exchange pumps modulate the SOCE (64, 72, 82, 84), involvement of these mechanisms of long-term ER signaling remain to be examined.
Overall, our present study demonstrates the differential role of ERs in regulating [Ca2+]i in baseline and inflamed conditions. ERα activation at baseline shows increased [Ca2+]i response and is not effective in regulating the cytokine-induced increase in [Ca2+]i responses. Whereas ERβ activation tends to decrease [Ca2+]i response slightly at baseline and significantly in the inflamed conditions. ERβ contributes to decreased [Ca2+]i response through increased SERCA2 function, as evident by the increased rate of sequestration of Ca2+. Furthermore, ERβ also exerts its signaling through inhibition of pathways involved in activating the voltage-gated LTCC. Overall, these results suggest that estrogen signaling is maintained in the presence of inflammation, and indeed, more enhanced via ERβ activation, providing a potential avenue to blunt effects of inflammation on [Ca2+]i in ASM.
GRANTS
This study was supported by National Heart, Lung, and Blood Institute Grants R01-HL-123494 and R01-HL-123494-02S1 (Venkatachalem), R01-HL-088029 (Prakash), and R01-HL-142061 (Pabelick, Prakash).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.B., C.M.P., Y.S.P., and V.S. conceived and designed research; S.B., J.C., and V.S. performed experiments; S.B., J.C., and V.S. analyzed data; S.B., C.M.P., Y.S.P., and V.S. interpreted results of experiments; S.B. and V.S. prepared figures; S.B., J.C., C.M.P., Y.S.P., and V.S. drafted manuscript; S.B., J.C., C.M.P., Y.S.P., and V.S. edited and revised manuscript; S.B., J.C., C.M.P., Y.S.P., and V.S. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Dr. Nilesh Ambhore, Department of Pharmaceutical Sciences, North Dakota State University, for providing technical support in completion of this study.
REFERENCES
- 1.Abcejo AJ, Sathish V, Smelter DF, Aravamudan B, Thompson MA, Hartman WR, Pabelick CM, Prakash YS. Brain-derived neurotrophic factor enhances calcium regulatory mechanisms in human airway smooth muscle. PLoS One 7: e44343, 2012. doi: 10.1371/journal.pone.0044343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Acconcia F, Kumar R. Signaling regulation of genomic and nongenomic functions of estrogen receptors. Cancer Lett 238: 1–14, 2006. doi: 10.1016/j.canlet.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 3.Ambhore NS, Kalidhindi RSR, Loganathan J, Sathish V. Role of differential estrogen teceptor activation in airway hyperreactivity and remodeling in a murine model of asthma. Am J Respir Cell Mol Biol 61: 469–480, 2019. doi: 10.1165/rcmb.2018-0321OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ambhore NS, Katragadda R, Raju Kalidhindi RS, Thompson MA, Pabelick CM, Prakash YS, Sathish V. Estrogen receptor beta signaling inhibits PDGF induced human airway smooth muscle proliferation. Mol Cell Endocrinol 476: 37–47, 2018. doi: 10.1016/j.mce.2018.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amrani Y, Chen H, Panettieri RA Jr. Activation of tumor necrosis factor receptor 1 in airway smooth muscle: a potential pathway that modulates bronchial hyper-responsiveness in asthma? Respir Res 1: 49–53, 2000. doi: 10.1186/rr12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amrani Y, Panettieri RA. Airway smooth muscle: contraction and beyond. Int J Biochem Cell Biol 35: 272–276, 2003. doi: 10.1016/S1357-2725(02)00259-5. [DOI] [PubMed] [Google Scholar]
- 7.Amrani Y, Panettieri RA Jr. Cytokines induce airway smooth muscle cell hyperresponsiveness to contractile agonists. Thorax 53: 713–716, 1998. doi: 10.1136/thx.53.8.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amrani Y, Panettieri RA Jr. Modulation of calcium homeostasis as a mechanism for altering smooth muscle responsiveness in asthma. Curr Opin Allergy Clin Immunol 2: 39–45, 2002. doi: 10.1097/00130832-200202000-00007. [DOI] [PubMed] [Google Scholar]
- 9.Aravamudan B, Goorhouse KJ, Unnikrishnan G, Thompson MA, Pabelick CM, Hawse JR, Prakash YS, Sathish V. Differential expression of estrogen receptor variants in response to inflammation signals in human airway smooth muscle. J Cell Physiol 232: 1754–1760, 2017. doi: 10.1002/jcp.25674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barton M, Filardo EJ, Lolait SJ, Thomas P, Maggiolini M, Prossnitz ER. Twenty years of the G protein-coupled estrogen receptor GPER: historical and personal perspectives. J Steroid Biochem Mol Biol 176: 4–15, 2018. doi: 10.1016/j.jsbmb.2017.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bartoszewski R, Matalon S, Collawn JF. Ion channels of the lung and their role in disease pathogenesis. Am J Physiol Lung Cell Mol Physiol 313: L859–L872, 2017. doi: 10.1152/ajplung.00285.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bazán-Perkins B, Flores-Soto E, Barajas-López C, Montaño LM. Role of sarcoplasmic reticulum Ca2+ content in Ca2+ entry of bovine airway smooth muscle cells. Naunyn Schmiedebergs Arch Pharmacol 368: 277–283, 2003. doi: 10.1007/s00210-003-0806-4. [DOI] [PubMed] [Google Scholar]
- 13.Bellia V, Augugliaro G. Asthma and menopause. Monaldi Arch Chest Dis 67: 125–127, 2007. [DOI] [PubMed] [Google Scholar]
- 14.Billington CK, Penn RB. Signaling and regulation of G protein-coupled receptors in airway smooth muscle. Respir Res 4: 2, 2003. doi: 10.1186/rr195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bjornson CL, Mitchell I. Gender differences in asthma in childhood and adolescence. J Gend Specif Med 3: 57–61, 2000. doi: 10.4081/monaldi.2007.482. [DOI] [PubMed] [Google Scholar]
- 16.Björnström L, Sjöberg M. Mechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genes. Mol Endocrinol 19: 833–842, 2005. doi: 10.1210/me.2004-0486. [DOI] [PubMed] [Google Scholar]
- 17.Boie S, Chen J, Sanderson MJ, Sneyd J. The relative contributions of store-operated and voltage-gated Ca2+ channels to the control of Ca2+ oscillations in airway smooth muscle. J Physiol 595: 3129–3141, 2017. doi: 10.1113/JP272996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boulet LP, Chakir J, Dubé J, Laprise C, Boutet M, Laviolette M. Airway inflammation and structural changes in airway hyper-responsiveness and asthma: an overview. Can Respir J 5: 16–21, 1998. doi: 10.1155/1998/926439. [DOI] [PubMed] [Google Scholar]
- 19.Carey MA, Card JW, Voltz JW, Germolec DR, Korach KS, Zeldin DC. The impact of sex and sex hormones on lung physiology and disease: lessons from animal studies. Am J Physiol Lung Cell Mol Physiol 293: L272–L278, 2007. doi: 10.1152/ajplung.00174.2007. [DOI] [PubMed] [Google Scholar]
- 20.Chamkasem A, Toniti W. Sequence to structure approach of estrogen receptor α and ligand interactions. Asian Pac J Cancer Prev 16: 2161–2166, 2015. doi: 10.7314/APJCP.2015.16.6.2161. [DOI] [PubMed] [Google Scholar]
- 21.Chhabra SK. Premenstrual asthma. Indian J Chest Dis Allied Sci 47: 109–116, 2005. [PubMed] [Google Scholar]
- 22.de Marco R, Locatelli F, Sunyer J, Burney P. Differences in incidence of reported asthma related to age in men and women. A retrospective analysis of the data of the European Respiratory Health Survey. Am J Respir Crit Care Med 162: 68–74, 2000. doi: 10.1164/ajrccm.162.1.9907008. [DOI] [PubMed] [Google Scholar]
- 23.Delmotte P, Yang B, Thompson MA, Pabelick CM, Prakash YS, Sieck GC. Inflammation alters regional mitochondrial Ca2+ in human airway smooth muscle cells. Am J Physiol Cell Physiol 303: C244–C256, 2012. doi: 10.1152/ajpcell.00414.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deshpande DA, Penn RB. Targeting G protein-coupled receptor signaling in asthma. Cell Signal 18: 2105–2120, 2006. doi: 10.1016/j.cellsig.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 25.Dogan M, Han YS, Delmotte P, Sieck GC. TNFα enhances force generation in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 312: L994–L1002, 2017. doi: 10.1152/ajplung.00550.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Farzaneh S, Zarghi A. Estrogen receptor ligands: a review (2013–2015). Sci Pharm 84: 409–427, 2016. doi: 10.3390/scipharm84030409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flores-Soto E, Reyes-García J, Sommer B, Montaño LM. Sarcoplasmic reticulum Ca(2+) refilling is determined by L-type Ca2+ and store operated Ca2+ channels in guinea pig airway smooth muscle. Eur J Pharmacol 721: 21–28, 2013. doi: 10.1016/j.ejphar.2013.09.060. [DOI] [PubMed] [Google Scholar]
- 28.Foster PS, Goldie RG, Paterson JW. Effect of steroids on β-adrenoceptor-mediated relaxation of pig bronchus. Br J Pharmacol 78: 441–445, 1983. doi: 10.1111/j.1476-5381.1983.tb09409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fuseini H, Newcomb DC. Mechanisms driving gender differences in asthma. Curr Allergy Asthma Rep 17: 19, 2017. doi: 10.1007/s11882-017-0686-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gibbs CJ, Coutts II, Lock R, Finnegan OC, White RJ. Premenstrual exacerbation of asthma. Thorax 39: 833–836, 1984. doi: 10.1136/thx.39.11.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 260: 3440–3450, 1985. [PubMed] [Google Scholar]
- 32.Harris HA. Estrogen receptor-β: recent lessons from in vivo studies. Mol Endocrinol 21: 1–13, 2007. doi: 10.1210/me.2005-0459. [DOI] [PubMed] [Google Scholar]
- 33.Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, Tujague M, Ström A, Treuter E, Warner M, Gustafsson JA. Estrogen receptors: how do they signal and what are their targets. Physiol Rev 87: 905–931, 2007. doi: 10.1152/physrev.00026.2006. [DOI] [PubMed] [Google Scholar]
- 34.Hirota S, Helli P, Janssen LJ. Ionic mechanisms and Ca2+ handling in airway smooth muscle. Eur Respir J 30: 114–133, 2007. doi: 10.1183/09031936.00147706. [DOI] [PubMed] [Google Scholar]
- 35.Hirst SJ. Airway smooth muscle as a target in asthma. Clin Exp Allergy 30, Suppl 1: 54–59, 2000. doi: 10.1046/j.1365-2222.2000.00099.x. [DOI] [PubMed] [Google Scholar]
- 36.James AL, Noble PB, Drew SA, Mauad T, Bai TR, Abramson MJ, McKay KO, Green FHY, Elliot JG. Airway smooth muscle proliferation and inflammation in asthma. J Appl Physiol (1985) 125: 1090–1096, 2018. doi: 10.1152/japplphysiol.00342.2018. [DOI] [PubMed] [Google Scholar]
- 37.Jia M, Dahlman-Wright K, Gustafsson JA. Estrogen receptor α and β in health and disease. Best Pract Res Clin Endocrinol Metab 29: 557–568, 2015. doi: 10.1016/j.beem.2015.04.008. [DOI] [PubMed] [Google Scholar]
- 38.Jia S, Zhang X, He DZ, Segal M, Berro A, Gerson T, Wang Z, Casale TB. Expression and function of a novel variant of estrogen receptor-α36 in murine airways. Am J Respir Cell Mol Biol 45: 1084–1089, 2011. doi: 10.1165/rcmb.2010-0268OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kitazawa T, Hamada E, Kitazawa K, Gaznabi AK. Non-genomic mechanism of 17 β-oestradiol-induced inhibition of contraction in mammalian vascular smooth muscle. J Physiol 499: 497–511, 1997. doi: 10.1113/jphysiol.1997.sp021944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kurata K, Takebayashi M, Kagaya A, Morinobu S, Yamawaki S. Effect of β-estradiol on voltage-gated Ca2+ channels in rat hippocampal neurons: a comparison with dehydroepiandrosterone. Eur J Pharmacol 416: 203–212, 2001. doi: 10.1016/S0014-2999(01)00880-9. [DOI] [PubMed] [Google Scholar]
- 41.Lai YJ, Yu D, Zhang JH, Chen GJ. Cooperation of genomic and rapid nongenomic actions of estrogens in synaptic plasticity. Mol Neurobiol 54: 4113–4126, 2017. doi: 10.1007/s12035-016-9979-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liptzin DR, Landau LI, Taussig LM. Sex and the lung: observations, hypotheses, and future directions. Pediatr Pulmonol 50: 1159–1169, 2015. doi: 10.1002/ppul.23178. [DOI] [PubMed] [Google Scholar]
- 43.Loganathan J, Pandey R, Ambhore NS, Borowicz P, Sathish V. Laser-capture microdissection of murine lung for differential cellular RNA analysis. Cell Tissue Res 376: 425–432, 2019. doi: 10.1007/s00441-019-02995-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mahn K, Ojo OO, Chadwick G, Aaronson PI, Ward JP, Lee TH. Ca2+ homeostasis and structural and functional remodelling of airway smooth muscle in asthma. Thorax 65: 547–552, 2010. doi: 10.1136/thx.2009.129296. [DOI] [PubMed] [Google Scholar]
- 45.Malamas MS, Manas ES, McDevitt RE, Gunawan I, Xu ZB, Collini MD, Miller CP, Dinh T, Henderson RA, Keith JC Jr, Harris HA. Design and synthesis of aryl diphenolic azoles as potent and selective estrogen receptor-β ligands. J Med Chem 47: 5021–5040, 2004. doi: 10.1021/jm049719y. [DOI] [PubMed] [Google Scholar]
- 46.Miller VM, Duckles SP. Vascular actions of estrogens: functional implications. Pharmacol Rev 60: 210–241, 2008. doi: 10.1124/pr.107.08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mollerup S, Jørgensen K, Berge G, Haugen A. Expression of estrogen receptors α and β in human lung tissue and cell lines. Lung Cancer 37: 153–159, 2002. doi: 10.1016/S0169-5002(02)00039-9. [DOI] [PubMed] [Google Scholar]
- 48.Montgomery S, Shaw L, Pantelides N, Taggart M, Austin C. Acute effects of oestrogen receptor subtype-specific agonists on vascular contractility. Br J Pharmacol 139: 1249–1253, 2003. doi: 10.1038/sj.bjp.0705368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nilsson S, Mäkelä S, Treuter E, Tujague M, Thomsen J, Andersson G, Enmark E, Pettersson K, Warner M, Gustafsson JA. Mechanisms of estrogen action. Physiol Rev 81: 1535–1565, 2001. doi: 10.1152/physrev.2001.81.4.1535. [DOI] [PubMed] [Google Scholar]
- 50.Orfanos S, Jude J, Deeney BT, Cao G, Rastogi D, van Zee M, Pushkarsky I, Munoz HE, Damoiseaux R, Di Carlo D, Panettieri RA Jr. Obesity increases airway smooth muscle responses to contractile agonists. Am J Physiol Lung Cell Mol Physiol 315: L673–L681, 2018. doi: 10.1152/ajplung.00459.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Panettieri RA., Jr Isolation and culture of human airway smooth muscle cells. Methods Mol Med 56: 155–160, 2001. doi: 10.1385/1-59259-151-5:155. [DOI] [PubMed] [Google Scholar]
- 52.Paterni I, Granchi C, Katzenellenbogen JA, Minutolo F. Estrogen receptors alpha (ERα) and beta (ERβ): subtype-selective ligands and clinical potential. Steroids 90: 13–29, 2014. doi: 10.1016/j.steroids.2014.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pelaia G, Renda T, Gallelli L, Vatrella A, Busceti MT, Agati S, Caputi M, Cazzola M, Maselli R, Marsico SA. Molecular mechanisms underlying airway smooth muscle contraction and proliferation: implications for asthma. Respir Med 102: 1173–1181, 2008. doi: 10.1016/j.rmed.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 54.Prakash YS. Airway smooth muscle in airway reactivity and remodeling: what have we learned? Am J Physiol Lung Cell Mol Physiol 305: L912–L933, 2013. doi: 10.1152/ajplung.00259.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Prakash YS. Emerging concepts in smooth muscle contributions to airway structure and function: implications for health and disease. Am J Physiol Lung Cell Mol Physiol 311: L1113–L1140, 2016. doi: 10.1152/ajplung.00370.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Prakash YS, Kannan MS, Sieck GC. Regulation of intracellular calcium oscillations in porcine tracheal smooth muscle cells. Am J Physiol Cell Physiol 272: C966–C975, 1997. doi: 10.1152/ajpcell.1997.272.3.C966. [DOI] [PubMed] [Google Scholar]
- 57.Prakash YS, Sathish V, Thompson MA, Pabelick CM, Sieck GC. Asthma and sarcoplasmic reticulum Ca2+ reuptake in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 297: L794, 2009. doi: 10.1152/ajplung.00237.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pupo M, Maggiolini M, Musti AM. GPER mediates non-genomic effects of estrogen. Methods Mol Biol 1366: 471–488, 2016. doi: 10.1007/978-1-4939-3127-9_37. [DOI] [PubMed] [Google Scholar]
- 59.Reyes-García J, Flores-Soto E, Carbajal-García A, Sommer B, Montaño LM. Maintenance of intracellular Ca2+ basal concentration in airway smooth muscle. Int J Mol Med 42: 2998–3008, 2018. doi: 10.3892/ijmm.2018.3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Saczko J, Michel O, Chwiłkowska A, Sawicka E, Mączyńska J, Kulbacka J. Estrogen receptors in cell membranes: regulation and signaling. Adv Anat Embryol Cell Biol 227: 93–105, 2017. doi: 10.1007/978-3-319-56895-9_6. [DOI] [PubMed] [Google Scholar]
- 61.Sánchez DS, Fischer Sigel LK, Azurmendi PJ, Vlachovsky SG, Oddo EM, Armando I, Ibarra FR, Silberstein C. Estradiol stimulates cell proliferation via classic estrogen receptor-α and G protein-coupled estrogen receptor-1 in human renal tubular epithelial cell primary cultures. Biochem Biophys Res Commun 512: 170–175, 2019. doi: 10.1016/j.bbrc.2019.03.056. [DOI] [PubMed] [Google Scholar]
- 62.Sanders KM. Invited review: mechanisms of calcium handling in smooth muscles. J Appl Physiol (1985) 91: 1438–1449, 2001. doi: 10.1152/jappl.2001.91.3.1438. [DOI] [PubMed] [Google Scholar]
- 63.Sathish V, Abcejo AJ, VanOosten SK, Thompson MA, Prakash YS, Pabelick CM. Caveolin-1 in cytokine-induced enhancement of intracellular Ca(2+) in human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 301: L607–L614, 2011. doi: 10.1152/ajplung.00019.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sathish V, Delmotte PF, Thompson MA, Pabelick CM, Sieck GC, Prakash YS. Sodium-calcium exchange in intracellular calcium handling of human airway smooth muscle. PLoS One 6: e23662, 2011. doi: 10.1371/journal.pone.0023662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sathish V, Freeman MR, Long E, Thompson MA, Pabelick CM, Prakash YS. Cigarette smoke and estrogen signaling in human airway smooth muscle. Cell Physiol Biochem 36: 1101–1115, 2015. doi: 10.1159/000430282. [DOI] [PubMed] [Google Scholar]
- 66.Sathish V, Leblebici F, Kip SN, Thompson MA, Pabelick CM, Prakash YS, Sieck GC. Regulation of sarcoplasmic reticulum Ca2+ reuptake in porcine airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 294: L787–L796, 2008. doi: 10.1152/ajplung.00461.2007. [DOI] [PubMed] [Google Scholar]
- 67.Sathish V, Martin YN, Prakash YS. Sex steroid signaling: implications for lung diseases. Pharmacol Ther 150: 94–108, 2015. doi: 10.1016/j.pharmthera.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sathish V, Thompson MA, Bailey JP, Pabelick CM, Prakash YS, Sieck GC. Effect of proinflammatory cytokines on regulation of sarcoplasmic reticulum Ca2+ reuptake in human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 297: L26–L34, 2009. doi: 10.1152/ajplung.00026.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sathish V, Thompson MA, Sinha S, Sieck GC, Prakash YS, Pabelick CM. Inflammation, caveolae and CD38-mediated calcium regulation in human airway smooth muscle. Biochim Biophys Acta 1843: 346–351, 2014. doi: 10.1016/j.bbamcr.2013.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shah R, Newcomb DC. Sex bias in asthma prevalence and pathogenesis. Front Immunol 9: 2997, 2018. doi: 10.3389/fimmu.2018.02997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shames RS, Heilbron DC, Janson SL, Kishiyama JL, Au DS, Adelman DC. Clinical differences among women with and without self-reported perimenstrual asthma. Ann Allergy Asthma Immunol 81: 65–72, 1998. doi: 10.1016/S1081-1206(10)63111-0. [DOI] [PubMed] [Google Scholar]
- 72.Sommer B, Flores-Soto E, Gonzalez-Avila G. Cellular Na+ handling mechanisms involved in airway smooth muscle contraction. Int J Mol Med 40: 3–9, 2017. doi: 10.3892/ijmm.2017.2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sommer B, Montano LM, Chávez J, Carbajal V, García-Hernandez LM, Irles C, Jiménez-Garduno AM, Ortega A. ROCK1 translocates from non-caveolar to caveolar regions upon KCl stimulation in airway smooth muscle. Physiol Res 63: 179–187, 2014. [DOI] [PubMed] [Google Scholar]
- 74.Spinelli AM, Trebak M. Orai channel-mediated Ca2+ signals in vascular and airway smooth muscle. Am J Physiol Cell Physiol 310: C402–C413, 2016. doi: 10.1152/ajpcell.00355.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stanford KI, Mickleborough TD, Ray S, Lindley MR, Koceja DM, Stager JM. Influence of menstrual cycle phase on pulmonary function in asthmatic athletes. Eur J Appl Physiol 96: 703–710, 2006. doi: 10.1007/s00421-005-0067-7. [DOI] [PubMed] [Google Scholar]
- 76.Stauffer SR, Coletta CJ, Tedesco R, Nishiguchi G, Carlson K, Sun J, Katzenellenbogen BS, Katzenellenbogen JA. Pyrazole ligands: structure-affinity/activity relationships and estrogen receptor-α-selective agonists. J Med Chem 43: 4934–4947, 2000. doi: 10.1021/jm000170m. [DOI] [PubMed] [Google Scholar]
- 77.Sun J, Meyers MJ, Fink BE, Rajendran R, Katzenellenbogen JA, Katzenellenbogen BS. Novel ligands that function as selective estrogens or antiestrogens for estrogen receptor-α or estrogen receptor-β. Endocrinology 140: 800–804, 1999. doi: 10.1210/endo.140.2.6480. [DOI] [PubMed] [Google Scholar]
- 78.Tliba O, Panettieri RA Jr. Regulation of inflammation by airway smooth muscle. Curr Allergy Asthma Rep 8: 262–268, 2008. doi: 10.1007/s11882-008-0043-5. [DOI] [PubMed] [Google Scholar]
- 79.Townsend EA, Miller VM, Prakash YS. Sex differences and sex steroids in lung health and disease. Endocr Rev 33: 1–47, 2012. doi: 10.1210/er.2010-0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Townsend EA, Sathish V, Thompson MA, Pabelick CM, Prakash YS. Estrogen effects on human airway smooth muscle involve cAMP and protein kinase A. Am J Physiol Lung Cell Mol Physiol 303: L923–L928, 2012. doi: 10.1152/ajplung.00023.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Townsend EA, Thompson MA, Pabelick CM, Prakash YS. Rapid effects of estrogen on intracellular Ca2+ regulation in human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 298: L521–L530, 2010. doi: 10.1152/ajplung.00287.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vohra PK, Thompson MA, Sathish V, Kiel A, Jerde C, Pabelick CM, Singh BB, Prakash YS. TRPC3 regulates release of brain-derived neurotrophic factor from human airway smooth muscle. Biochim Biophys Acta 1833: 2953–2960, 2013. doi: 10.1016/j.bbamcr.2013.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vrtačnik P, Ostanek B, Mencej-Bedrač S, Marc J. The many faces of estrogen signaling. Biochem Med (Zagreb) 24: 329–342, 2014. doi: 10.11613/BM.2014.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xia Y, Xia L, Lou L, Jin R, Shen H, Li W. Transient receptor potential channels and chronic Airway inflammatory diseases: a comprehensive review. Hai 196: 505–516, 2018. doi: 10.1007/s00408-018-0145-3. [DOI] [PubMed] [Google Scholar]
- 85.Yung JA, Fuseini H, Newcomb DC. Hormones, sex, and asthma. Ann Allergy Asthma Immunol 120: 488–494, 2018. doi: 10.1016/j.anai.2018.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhu BT, Han GZ, Shim JY, Wen Y, Jiang XR. Quantitative structure-activity relationship of various endogenous estrogen metabolites for human estrogen receptor α and β subtypes: Insights into the structural determinants favoring a differential subtype binding. Endocrinology 147: 4132–4150, 2006. doi: 10.1210/en.2006-0113. [DOI] [PubMed] [Google Scholar]