

Abstract
Stem cell factor (SCF) and its receptor c-kit have been implicated in inflammation, tissue remodeling, and fibrosis. Ingenuity Integrated Pathway Analysis of gene expression array data sets showed an upregulation of SCF transcripts in idiopathic pulmonary fibrosis (IPF) lung biopsies compared with tissue from nonfibrotic lungs that are further increased in rapid progressive disease. SCF248, a cleavable isoform of SCF, was abundantly and preferentially expressed in human lung fibroblasts and fibrotic mouse lungs relative to the SCF220 isoform. In fibroblast-mast cell coculture studies, blockade of SCF248 using a novel isoform-specific anti-SCF248 monoclonal antibody (anti-SCF248), attenuated the expression of COL1A1, COL3A1, and FN1 transcripts in cocultured IPF but not normal lung fibroblasts. Administration of anti-SCF248 on days 8 and 12 after bleomycin instillation in mice significantly reduced fibrotic lung remodeling and col1al, fn1, acta2, tgfb, and ccl2 transcript expression. In addition, bleomycin increased numbers of c-kit+ mast cells, eosinophils, and ILC2 in lungs of mice, whereas they were not significantly increased in anti-SCF248-treated animals. Finally, mesenchymal cell-specific deletion of SCF significantly attenuated bleomycin-mediated lung fibrosis and associated fibrotic gene expression. Collectively, these data demonstrate that SCF is upregulated in diseased IPF lungs and blocking SCF248 isoform significantly ameliorates fibrotic lung remodeling in vivo suggesting that it may be a therapeutic target for fibrotic lung diseases.
Keywords: cytokines, fibrosis, mast cells
INTRODUCTION
With a high global prevalence, chronic diseases often have severe complications, including end-stage tissue fibrosis that can ultimately culminate in organ/tissue dysfunction. Chronic fibrotic diseases, such as idiopathic pulmonary fibrosis (IPF), have been challenging to treat clinically, where many affected patients eventually require organ transplantation or succumb to the disease (9, 25, 43, 67). While many profibrotic mediators have been identified, therapeutically targeting many of these mediators have failed to modulate fibrotic progression and promote tissue regeneration (54, 57). Recently, nintedanib (OFEV) and pirfenidone (Esibret) have been Food and Drug Administration approved for the treatment of patients with IPF (23, 39, 46, 59, 60). These drugs have significant side effects and were only effective in slowing down, but not halting, disease progression. Thus, great effort is underway to develop next-generation therapeutics that are more effective at halting the fibrotic remodeling of the lung and promoting lung regeneration.
Fibrotic diseases have been often observed to have a strong inflammatory component, suggesting a role of inflammation in tissue fibrosis. Indeed, there is evidence for inflammatory processes and/or inflammatory cell infiltrates in fibrotic tissues, including the lung (30, 32, 50), kidney (48), and liver (61). While the role of inflammation in lung injury and disrepair in IPF remains controversial, clinical evidence suggests a correlation between inflammatory infiltrates and disease progression (4), and preclinical studies suggest that targeting inflammatory responses may ameliorate fibrotic lung remodeling (10, 29, 38, 56). However, immunosuppressive therapies have failed to modulate IPF disease progression clinically (33), suggesting that targeting profibrotic mechanisms may also be required to modulate clinical disease progression. Stem cell factor (SCF) is a cytokine involved in hematopoietic cell development of multiple lineages as well as mast cell differentiation and activation (24, 47). SCF binds to its surface receptor, c-Kit, which is a member of the receptor tyrosine kinase family (3, 24). Studies targeting c-kit activation using receptor tyrosine kinase inhibitors, imatinib, or nilotinib, have shown promising results in animal models but have not shown similar outcomes in an initial study in IPF using a nonselective patient population (2, 12, 58, 69). Since c-kit inhibitors have broad effects on homeostatic function (as well as the off-target RTKs inhibited), a specific and refined approach such as targeting the ligand may be more effective. Endogenous SCF occurs primarily in two forms, a 248 amino acid (AA) cleavable form (SCF248) and a 220 AA “noncleavable” form (SCF220) that differ by the presence or absence of exon 6 that encodes for protease cleavage site(s). Both isoforms of SCF are inserted into the plasma membrane, with the extracellular domain (ECD) of SCF248 more efficiently cleaved and shed from the surface of the cell during inflammation. While SCF-ECD is abundantly detected in circulation (~800 pg/mL in humans), circulating SCF-ECD is primarily monomeric that cannot cross-link and activate c-kit (31). Thus, c-kit activation is thought to primarily occur through the membrane-associated forms of SCF, which can efficiently cross-link the receptor leading to its phosphorylation and activation (13) (49).
The physiological roles of membrane-associated SCF were determined using Sl/Sld mice (lacking both forms of membrane-associated SCF) that are runted, anemic, and have altered myelopoiesis and inflammatory/immune responses. A fundamental study examined whether the two splice variants of SCF have differential biologic effects using Sl/Sld mutant mice embryonically transfected with either SCF248 or SCF220. The phenotype observed in these mice suggests an important and nonredundant role of SCF220 in development and erythropoiesis as its expression corrected the defects. However, SCF248 expression in these mice promoted normal myelopoiesis with little effect on runting and anemic phenotypes of SI/Sid mice, suggesting that the SCF248 isoform of SCF may be associated with myelopoiesis and the propagation of inflammation responses (36). Finally, the role of the SCF variant, SCF220, was confirmed in another study examining mice expressing SCF220 (but not SCF248) isoform. These mice were observed to develop normally (with no runting or anemia); however, there was a notable alteration in myelopoiesis and an absence of mast cells (65). Together, these studies suggest that SCF isoforms may play divergent roles in development, erythropoiesis, and myelopoiesis, where SCF220 may be required during development and erythropoiesis and SCF248 may be required for normal myelopoiesis and mast cell development and/or differentiation.
In the present study, we show evidence that KITLG expression (gene name for SCF) and protein are elevated in IPF. Further, SCF248 is preferentially and significantly elevated in human lung fibroblasts. Targeting this isoform using anti-SCF248–specific antibodies significantly reduced COL1A1, COL3A1, and FN1 transcript expression in IPF, but not normal lung fibroblasts, cocultured with mast cells. SCF248, but not SCF220, was markedly upregulated in fibrotic murine lungs and targeting SCF248 with specific antibodies significantly ameliorated bleomycin-induced lung fibrosis and profibrotic transcript expression. Finally, mesenchymal cell-specific deletion of SCF significantly ameliorated bleomycin-mediated lung remodeling. Collectively, our results suggest that the ability to target the SCF248 isoform, which is upregulated during fibrotic pulmonary diseases (including IPF), may be central to preserve important homeostatic functions of SCF (such as erythropoiesis) while blocking the detrimental pro-fibrotic effects of c-Kit+ cell activation.
MATERIALS AND METHODS
Study approval.
Institutional Review Boards at the University of Michigan approved all experiments with primary human cells and serum. All patients were consented before inclusion in the studies described herein, and all samples were deidentified before utilization.
Ingenuity pathway analysis.
Publicly available gene expression data sets (GSE24206) were mined from the National Center for Biotechnology Information (NCBI) geo data sets database. Groups were defined as follows: IPF lung biopsies (n = 8) versus normal lungs (n = 6). Gene expression values were extracted using NCBI’s Geo2R gene expression analysis tool and the expression data were uploaded onto Ingenuity Integrated Pathway Analysis (IPA) (QIAGEN Redwood City, https://www.qiagen.com/ingenuity). Ingenuity IPA was set to only consider changes in gene expression of 1.5-fold or greater and P ≤ 0.05. To generate KITLG interaction network, Ingenuity’s path-designer tool was utilized. Briefly, KITLG was added to the custom pathway designer and Ingenuity was set to grow the pathway using known direct downstream activation molecules (based on Ingenuity’s knowledge base). For transcription factor targets, Ingenuity was set to grow the transcription factor network by only considering molecules known to be direct downstream targets of the highlighted transcription factor (based on Ingenuity’s knowledge base). After the generation of a KITLG interaction network, gene expression data sets from IPF lung biopsies relative to normal lung explant were overlaid and exported.
Mice.
Female C57BL6 mice (6–8 wk old), SCFfl/fl mice, and Col1-CreERT2 mice were purchased from Jackson Laboratory (Bar Harbor, ME). The SCFfl/fl mice were cross-ed with the Col1-CreERT2 mice to generate SCFfl/fl–Col1CreERT2 C57BL6 mice that can be treated with tamoxifen (1 mg/mouse intraperitoneally) to activate Cre in cells expressing Col1 and deleting SCF specifically from those cells only when treated with tamoxifen. All animal studies were reviewed and approved by the University Committee on Use and Care of Animals at the University of Michigan, an AAALAC-accredited institution.
Bleomycin-induced pulmonary fibrosis.
Mice were given bleomycin (Bleomycin, Hospira, Lake Forest, IL) at a dose of 2.5 U/kg body weight as previously described (15). Control mice received the same volume of sterile PBS only. Where indicated, mice were treated with monoclonal control or anti-SCF248 antibodies or given tamoxifen to activate Cre in the SCFfl/fl –Col1CreERT2 transgenic mice by intraperitoneal injection. After 16 days, the animals were euthanized and serum and lung tissue were harvested for histologic, mRNA, and protein analyses as described below.
Production and administration of anti-SCF248 monoclonal antibodies.
A peptide from exon 6 of SCF248 was generated and used as an immunogen in mice by a contract research organization (GenScript, Inc., Newark, NJ) and hybridomas were made after several rounds of boosting immune responses. Twelve different hybridoma clones were identified as producing SCF248 peptide specific antibody and further characterized for binding and function. A primary antibody with high affinity was identified, further expanded, and purified to generate endotoxin free reagent for use in our analyses. The monoclonal antibody (mAb) is of the IgG1 isotype class. Since exon 6 is completely conserved across mammalian species, the monoclonal antibody is fully cross-reactive and binds to mouse and human SCF248. Antibody suspended in PBS was administered into mice by intraperitoneal injection at a concentration of 20 mg/kg with a control isotype matched control monoclonal antibody given at the same concentration.
Flow cytometric analysis.
Differential binding of anti-SCF248 mAb to SCF isoforms was determined using American Type Culture Collection (ATCC) cell lines that specific expressed either human SCF220 or SCF248 and flow cytometry analysis using SCF248 mAb directly labeled with Alexis Cy5 fluorescent marker. Briefly, cells were stained with Alexis Cy5 conjugated anti-SCF248 mAb for 15 min and analyzed using a BD LSRII flow cytometer (BD Biosciences).
Lung flow cytometry.
The lungs were removed, and single cells were isolated by enzymatic digestion with 1 mg/mL collagenase A (Roche, Indianapolis, IN) and 20 U/mL DNaseI (Sigma, St. Louis, MO) in RPMI 1640 containing 10% FCS. Tissues were further dispersed through an 18-gauge needle (10-mL syringe), red blood cells were lysed, and samples were filtered through 100-μm nylon mesh twice. Cells were resuspended in PBS and live cells were identified using LIVE/DEAD Fixable Yellow Dead Cell Stain kit (Thermo Fisher Scientific, Waltham, MA), then washed and resuspended in PBS with 1% FCS and Fc receptors were blocked with purified anti-CD16/32 (clone 93; BioLegend, San Diego, CA). Surface markers were identified using Abs (clones) against the following antigens, all from BioLegend: anti-Gr-1 (RB6-8C5), B220 (RA3-6B2), CD3 (145-2C11), Ter119 (Ter-119), CD11b (M1/70), CD25 (PC61), CD45 (30-F11), CD127 (SB/199), ST2 (DIH9), c-KIT (2B8), and CD90 (30-H12). SiglecF (E50-2440) was purchased from BD Biosciences (San Jose, CA). For innate lymphoid cell staining, lineage markers were anti-CD3, CD11b, B220, Gr-1, and TER119. ILC2: Lin-CD45+CD25+CD90+ST2+ c-Kit +CD127+. Eosinophils: SSChCD45+11b+SinglecF+. Mast cells: CD45+CD11b+c-KIT+FcεR1+. Data were collected in NovoCyte flow cytometer (ACEA Bioscience, Inc. San Diego, California). Data analysis was performed using FlowJo software (Tree Star, Oregon).
Human lung fibroblast and mast cell cocultures.
IPF lung biopsies were obtained from patients at the University of Michigan Medical Center with written informed consent. Normal nonfibrotic explanted lungs were obtained from rejected donor lungs. Lung fibroblasts were generated and cultured as previously described (35). For in vitro studies, cells were transferred into 6-well plates and used with or without coculture with SCF-dependent LAD2 human mast cells, which were generously supplied by Dr. Dean Metcalfe (National Institute of Allergy and Infectious Diseases, NIH, Bethesda, MD) under an MTA agreement. In experiments where the role of SCF248 was examined in the Fibroblast:LAD2 cocultures, anti-SCF248 or control mAb (20 ug/mL) was preincubated with the fibroblasts for 15 min before adding the LAD2 mast cells, followed by an overnight (18 h) coculture period.
RNA isolation and quantitative PCR.
RNA were extracted with TRIzol (Invitrogen) following the manufacturer’s protocol, and total RNA were reverse transcribed to cDNA to determine gene expression using Taqman gene expression primer/probe sets and SYBR for SCF220 and SCF248 transcripts as described (31). Detection was performed in ABI 7500 Real-time PCR system. Gene expression was calculated using ΔΔCt method and normalized with 18 s control expression for all other genes.
Hydroxyproline assay.
Lung hydroxyproline content was measured in whole lung homogenates as previously described (27). The results are expressed as μg of hydroxyproline per left lobe of the lung.
Statistical analysis.
Data were analyzed by Prism6 (GraphPad). Data presented are mean values ± SE. Comparison of two groups was performed in unpaired, two-tailed Student’s t-test. Comparison of three or more groups was analyzed by ANOVA with Tukey’s posttests. Significance was indicated at the level of *:P < 0.05, unless otherwise indicated.
RESULTS
Expression of SCF in pulmonary disease.
Our previous studies using a bleomycin model of pulmonary fibrosis demonstrated that therapeutically blocking SCF in the remodeling phase attenuated the development of fibrosis (15). To assess potential role(s) of these proteins in clinical IPF, publicly available data sets (GSE10667) from IPF, exacerbated IPF, and nondiseased lungs were analyzed using Ingenuity IPA. As shown in Figs. 1. A and B, Ingenuity upstream regulatory analyses, which indicate the activation of genes (orange/red), predicted enhanced activation of KITLG (SCF) in IPF relative to normal (Fig. 1A), and SCF was further increased in exacerbated IPF relative to stable IPF lungs (Fig. 1B). Upregulated KITLG downstream mediators identified by this analysis included prosurvival and proinflammatory transcripts, several of which have been identified as potential therapeutic targets. Consistent with Ingenuity’s analyses, there was a significant increase in SCF protein in serum samples of patients with IPF relative to normal donor serum controls (Fig. 1C). These data support findings from previous reports (15, 72) and illustrate that SCF (KITLG) is upregulated in IPF patients and may play a role in IPF disease.
Fig. 1.

SCF is highly expressed in IPF lungs and blood. A: ingenuity IPA was utilized to generate KIT-KITLG interaction and transcriptional activation network. The resulting network was then overlaid with gene expression data sets from IPF lung biopsies relative to normal lung explants (GSE24206). KIT-activated kinases and transcription factors are shown in large font and direct downstream targets for the activated transcription factors are shown in small fonts. Solid arrowheads indicated activation (A) or expression (E). Significantly upregulated (≥ 1.5-fold change and P ≤ 0.05) and downregulated (≥ −1.5-fold change and P ≤ 0.05) are depicted in red and green color, respectively. B: serum from normal (n = 9) or patients newly diagnosed with IPF by high-resolution computed tomography assessment (n = 41) were measured for SCF levels using a specific ELISA (R&D Systems, Rochester, MN). Levels of SCF were measured in serum collected after diagnosis. IPA, Integrated Pathway Analysis; IPF, idiopathic pulmonary fibrosis; SCF, stem cell factor.
SCF248 is upregulated in IPF patient-derived fibroblasts and in bleomycin-induced pulmonary fibrosis.
When the expression of both SCF isoforms was examined, SCF248 was present in lung fibroblasts from both normal patients and IPF patients at higher levels (10- to 20-fold) than SCF220 with increased SCF248 significantly higher (P = 0.02) in IPF patient-derived fibroblasts (Fig. 2A). We also examined the differential expression of SCF isoforms during bleomycin-induced lung fibrosis in mice and found that the primary SCF isoform-induced is SCF248, with little or no increase in SCF220 (Fig. 2B). Together these results suggest that SCF248 is highly expressed in fibroblasts from lungs of patients and during pulmonary fibrosis and that fibroblast-derived SCF248 is preferentially expressed in fibrotic mouse lungs. These results are consistent with our recent publication in chronic allergen-induced asthma indicating that SCF248 is highly upregulated in chronic remodeling disease (21) and supports the preferential expression of SCF248 over SCF220 in fibroblasts.
Fig. 2.

SCF248 is highly expressed by IPF lung fibroblasts and in fibrotic mouse lungs. A: RNA was extracted from normal and IPF lung fibroblasts and subjected to quantitative PCR analyses using SCF220- and SCF248-specific primer sets. Data are expressed as fold change of SCF248 over SCF220 to compare relative levels between the isoforms in patient-derived fibroblast cell lines. Shown is the mean ± SE of three cell lines. B: full-length SCF, SCF220, and SCF248 transcript expression levels in lungs of 16-day bleomycin-treated B6 mice expressed as fold increase over control untreated mice. Data represents mean ± SE from five mice/group. IPF, idiopathic pulmonary fibrosis; SCF, stem cell factor.
Characterization of mAb to SCF248.
Our results suggest that SCF248 is the predominant SCF isoform expressed in the fibrotic lungs of bleomycin-treated mice and in fibroblasts from IPF patients. Thus, a sophisticated targeting strategy was pursued to make mAb against the portion of SCF that differentiates SCF248 from SCF220 as complete loss of SCF/c-kit signaling has adverse effects on developmental pathways and erythropoiesis. MAb were generated against a peptide found in exon 6 of SCF248 that is not present in SCF220 to specifically target SCF248 but not SCF220. Due to the cleavage of exon 6 we chose a peptide on the membrane side of the cleavage domain; thus, this mAb did not bind to the portion of SCF248-extracellular domain that is shed into the circulation after cleavage (21). Flow cytometric analysis of ATCC cell lines that express no SCF, only SCF220, or only SCF248 isoforms demonstrated that the mAb against this peptide in exon 6 only binds to membrane-associated SCF248 and not to SCF220 (Fig. 3A). In addition, Biacore surface plasmon resonance analysis of anti-SCF248 showed that this mAb has an affinity to peptide of 4.5 × 10−9 M (4.5 nM; Fig. 3B). Thus, these results demonstrate that anti-SCF248 mAb is specific to SCF248 isoform, and it binds its ligand with high affinity.
Fig. 3.

Characterization of anti-SCF248 mAb. A: using ATCC cell lines CRL-2452, CRL-2453, and CRL-2454 that express no SCF, SCF220, or SCF248, respectively, the monoclonal antibody was used for flow cytometry binding assays to demonstrate specificity. B: monoclonal antibody generated to a peptide from exon 6 of SCF248 was subjected to Biacore surface plasmon resonance (SPR) analysis to the specific peptide. The data generated dose response curves that were assessed to have a fast on rate and a slow off rate with a 4.5 nM KD binding. C: image flow cytometry photos of ATCC SCF248-expressing cells incubated for 5 or 60 min with anti-SCF248 or control IgG1 mAb coupled with Phrodo-red pH-sensitive fluorescent dye. D: mean fluorescent intensity (MFI) of ATCC SCF248-expressing cells incubated with control or anti-SCF248 mAb coupled with phrodo-red dye over time to demonstrate internalization. E: IPF patient lung fibroblast cultures incubated for 15 min with control or Anti-SCF248 mAb coupled with phrodo-red dye showing internalization only in the anti-SCF48 mAb-incubated cells. F: naïve Balb/c/J mice were injected intravenously with either control IgG (250 mg/kg), polyclonal anti-SCF (250 mg/kg), or anti-SCF248 monoclonal ab (100 mg/kg) on day 0, 2, 4, and 6. The peripheral blood was assessed for reticulocyte numbers on day 8 as an indication of reduced erythropoiesis as a % of total cells. Data represent the mean ± SE from eight mice/group. ATCC, American Type Culture Collection; IPF, idiopathic pulmonary fibrosis; SCF, stem cell factor.
We hypothesized that antibody binding to surface SCF248 would promote the internalization, negating its ability to signal c-kit. To examine this, we used a pH-sensitive fluorescent tag (Phrodo-red) coupled to our mAb, where fluorescence is only observed when the pH drops to below six in endosomes. SCF248-expressing cells treated with the labeled anti-SCF248 antibody showed an increase in fluorescence starting at 5 min and continued to increase by 60 min (Fig. 3, C and D). In addition, cultured IPF patient-derived fibroblasts were incubated with anti-SCF248 or control antibody coupled with the pH-sensitive fluorescent tag and showed a similar internalization at 15 min (Fig. 3E). These results suggest that anti-SCF248 mAb induce internalization of cell surface SCF248 protein into endosomes and lysosomes allowing the clearance from the surface.
Previous studies that utilized specific expression of the different isoforms have demonstrated that the erythropoiesis function of SCF-c-kit is due to SCF220 and not SCF248 (36). To determine if anti-SCF248 mAb altered erythropoiesis, mice were intravenously administered a polyclonal rabbit anti-mouse antibody (250 mg/kg) that recognizes both SCF isoforms or anti-SCF248 mAb (100 mg/kg) every other day. At day 8, the peripheral blood was examined for levels of circulating hematopoietic cells, with a focus on reticulocytes that have recently entered circulation from bone marrow. The data in Fig. 3F illustrate that polyclonal anti-SCF, but not anti-SCF248 mAb, caused a significant decrease in reticulocytes in the peripheral blood. These results support previous studies and suggest that SCF220, but not SCF248, is required for normal erythropoiesis (36).
Mast cell-induced IPF patient-derived fibroblast activation is SCF248 dependent.
Previous studies from our laboratory have demonstrated that fibroblasts upregulate SCF and can activate mast cells (28), and studies examining tissue fibrosis suggest that mast cells play a role in the progression and severity of the disease (1, 8, 11, 41, 68, 72). We examined whether IPF lung fibroblasts activated mast cells and whether anti-SCF248 was effective at blocking activation. We utilized the LAD2 mast cell line, which is SCF dependent (40). SCF-dependent LAD2 cells were layered onto normal or IPF lung fibroblasts for 24 h in the presence of control or anti-SCF248 antibodies. Anti-SCF248 significantly reduced COL1A1, COL3A1, and FN1 transcript expression in cocultured IPF lung fibroblasts versus IgG-treated cells with no increase activation when layered onto normal fibroblasts (Fig. 4A). When we used the anti-SCF248 mAb for immunohistochemical analysis, abundant staining for SCF in IPF lung tissues especially in the fibrotic tissue (Fig. 4B) was observed compared with nonfibrotic lung samples from respiratory bronchiolitis-associated interstitial lung disease (RBILD) patients with staining only detected in macrophage populations. Thus, blocking SCF248 has a functional effect on fibrotic genes and it is highly expressed in areas of active remodeling in IPF disease.
Fig. 4.
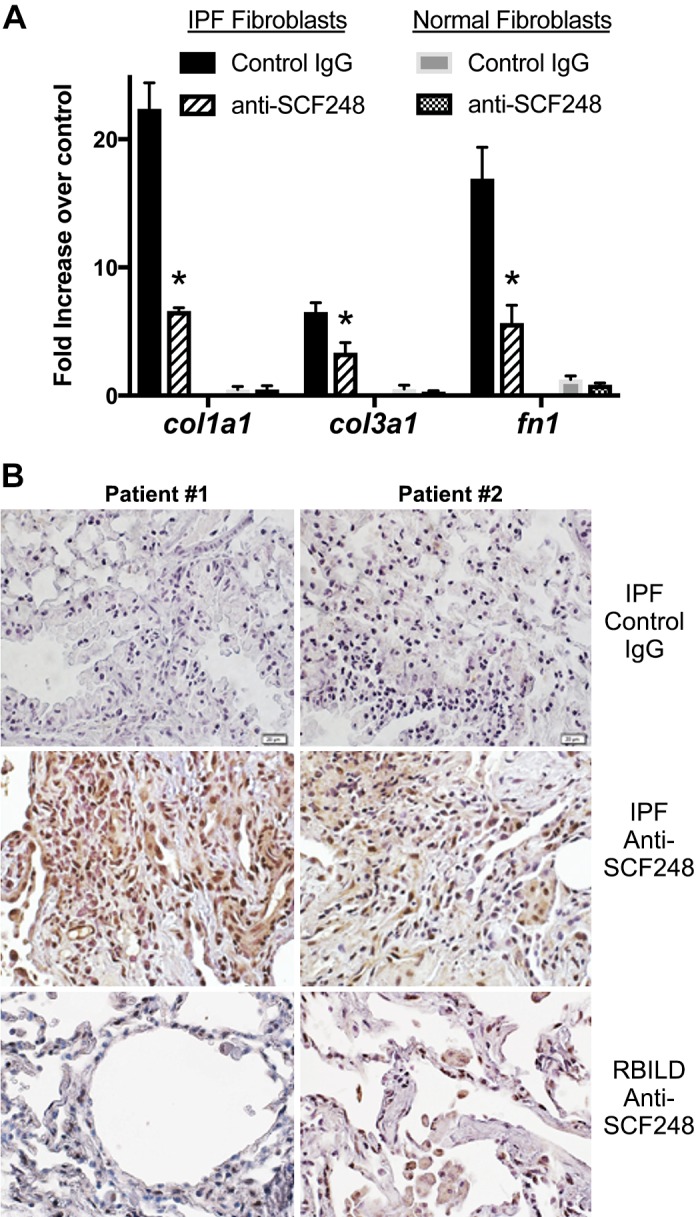
Monoclonal Ab to SCF248 blocks LAD2 mast cell-induced myofibroblast activation. A: fibroblast cell lines from either nonfibrotic (“normal”) or IPF fibrotic lung biopsies were plated in 48-well plates to confluent monolayers. Control or anti-SCF248 monoclonal antibodies (10 ug/mL) were added 30 min before layering of 2 × 105 LAD2 mast cells onto the monolayers for 24 h and assessed for increased matrix gene expression compared with control fibroblasts with no mast cells added to the culture. Data represent mean ± SE *P < 0.05 compared with the control ab-treated cells. B: immunohistochemistry using anti-SCF248 monoclonal antibody on tissue biopsy sections from two patients with IPF or RBILD. IgG control antibodies were used in IPF patient sections and did not show any nonspecific staining. A secondary alkaline phosphatase antibody was used to visualize the staining. IPF, idiopathic pulmonary fibrosis; RBILD, respiratory bronchiolitis-associated interstitial lung disease.
Specific inhibition of the SCF248 isoform inhibits lung remodeling responses.
Our previous data using polyclonal antibodies that recognize all isoforms of SCF demonstrated an attenuation of bleomycin-induced fibrosis (15); however, the differential roles of SCF220 and SCF248 are not known. To assess the role of SCF248 in lung fibrosis, anti-SCF248 or control ab (20 mg/kg) were given by intraperitoneal injection in bleomycin-exposed mice on day 8 and day 12 post-bleomycin treatment (fibrotic phase). Figure 5A illustrates that anti-SCF248 antibody treatment reduced the development of fibrosis and the consolidation of alveolar space. Biochemical quantification of hydroxyproline from lung tissue indicated a significant decrease in the anti-SCF248 versus control antibody-treated mice (Fig. 5B). Transcriptomic analysis by quantitative PCR demonstrated a significant decrease in several profibrotic transcripts, including tgfb, ccl2, col1a1, fn1, acta2, and SCF itself (Fig. 5C). Pulmonary function tests performed on day 16 demonstrate that FEV100 and FEF25 were significantly reduced in the control IgG bleomycin mice compared with naïve mice, whereas anti-SCF248-treated animals were not significantly different from naïve mice (Fig. 5D). Pulmonary pressure increases due to development of fibrosis and was significantly ameliorated in the anti-SCF248-treated mice versus control ab and further appeared to fully recover to normal levels (dPpl; Fig. 5D). Finally, c-kit+ mast cells, eosinophils, and ILC2 were significantly increased in the control IgG-treated bleomycin mice but not in the anti-SCF248-treated animals (Fig. 5E). Together, these data demonstrate that therapeutic targeting of SCF248 using a specific mAb attenuated the development and progression of severe lung fibrosis.
Fig. 5.

Therapeutically targeting of SCF248 using an isoform-specific mAb significantly ameliorated bleomycin-induced lung fibrosis. A: representative histology from 10-wk-old B6 mice 17 days after exposure to intratracheal bleomycin and treated with control or anti-SCF248 monoclonal antibody (20 mg/kg) on day 8 and 12 after the bleomycin challenge. B: the single left lobe was harvested from the normal and bleomycin-treated animals and assessed for hydroxyproline. C: the upper right lobe of the lung was used for isolation of mRNA and assessed for expression of the indicated genes by RT-PCR. D: mice were examined 17 days post-bleomycin treatment for pulmonary lung function using anesthetized and ventilated animals with bleomycin-treated animals given control or anti-SCF248 mAb on days 8 and 12 post-bleomycin instillation. E: mice were treated with IgG control or anti-SCF248 mAb on days 8 and 12 post-bleomycin instillation and examined for c-kit+ cell infiltration by flow cytometry on day 17. Data represent the mean ± SE from 6 to 8 mice/group. SCF, stem cell factor.
Selective deletion of SCF in col1a1 expressing cells attenuates Bleomycin-induced fibrosis.
Genetic proof-of-concept can be important to establish function of specific molecules during disease. However, complete deletion of SCF is lethal, having significant systemic consequences. Therefore, we generated a mouse that lacks SCF in Collagen 1-positive cells, SCFfl/fl-Col1-CreERT2 mouse, when induced by tamoxifen treatment. Col1-Cre- and Col1-Cre+ littermates were given bleomycin intratracheally and treated with tamoxifen daily from days 6–12 post-bleomycin to delete SCF from mesenchymal cells in the Cre+ mice. Histologic assessment demonstrated that the Col1-Cre+ mice showed markedly less remodeling and reduced consolidation of the lungs versus Col1-Cre- littermate mice (Fig. 6A). The bleomycin-challenged mice showed a significant decrease in ccl2, col1a1, and fn1 expression and a trending, but not significant, reduction in tgfb and acta2 expression in the Cre+ mice (Fig. 6B). Hydroxyproline quantification showed a significant decrease in tamoxifen-treated Col1-Cre+ mice versus Col1-Cre- littermate control mice (Fig. 6C). These results suggest that fibroblast-associated SCF promotes lung remodeling in vivo and further supports with genetic proof-of-concept that SCF248 may be an effective therapeutic target in lung fibrosis.
Fig. 6.

SCF248 deletion in collagen 1-expressing cells significantly reduced bleomycin-induced lung fibrosis. A: representative histology from lung bleomycin-treated 10–12-wk-old B6 SCFfl/fl Col1a-Cre-ERT mice exposed to tamoxifen (days 6–12 after bleomycin) to delete SCF expression in mesenchymal cells (myofibroblasts). Animals were harvested on day 17 after bleomycin instillation. B: the upper right lobe of the lungs from day 17 bleomycin-treated mice was processed for mRNA analysis by RT-PCR and genes expressed as fold increased over control non-bleomycin-exposed mice. C: hydroxyproline analysis was performed on the single left lobe of the animals and expressed as total hydroxyproline in the left lobe. Data represent mean ± SE from 6 to 7 mice/group. SCF, stem cell factor.
DISCUSSION
The identification of novel therapeutic targets induced during tissue fibrosis will be central to development of new reagents to block the progression of end-stage fibrotic diseases. Previous studies have implicated SCF as a viable target during pulmonary fibrosis, primarily using animal models (5, 15, 17, 18, 45, 72). However, the present studies link the overexpression of SCF in patients with IPF, both by the examination of lung tissue microarray data and in serum from peripheral blood of IPF patients. We have also been able to identify that a primary isoform of SCF (SCF248, also known as sSCF) is preferentially expressed in fibroblasts and during induction of bleomycin-induced fibrosis. The overexpression of SCF248, compared with SCF220, in both normal and IPF fibroblast is consistent with earlier results that reported fibroblasts nearly exclusively express the longer isoform SCF248 containing exon 6 (3). By specifically blocking the SCF248 isoform with a mAb that does not recognize SCF220 or cleaved SCF-ECD, profibrotic responses were significantly reduced in both in vitro coculture studies of IPF fibroblast with mast cells and in vivo after bleomycin-induced lung remodeling. Thus, during a fibroproliferative response, there would be a predominance of the longer SCF248 isoform that could be central to the progression of disease due to the expansion of myofibroblasts. Experimentally, this is supported in our studies using fibroblast- and myofibroblast-specific genetic deletion and antibody neutralization (anti-SCF248) experiments that suggest that SCF248 is a promising target during the progression of a pulmonary fibrotic response.
A primary role for SCF in fibrosis has been suggested to involve mast cell activation and accumulation. Mast cells have been implicated in a number of fibrotic diseases including IPF, kidney fibrosis, scleroderma, and others, where they have often been linked to the overexpression of SCF in areas of active fibrosis (6, 19, 20, 34, 44, 63, 66). This link between SCF expression and mast cells in fibrotic tissue is logical, since SCF has long been shown to be involved in mast cell differentiation, survival, and activation. In particular, mast cells release a number of preformed mediators, such as histamine, chymase, and vasoactive amines, and produce a number of cytokines, chemokines, and growth factors known to initiate and contribute to chronic inflammation and fibrosis (7, 37, 42, 51, 52, 62). In present studies, transcriptomic analysis showed that SCF248 was ~20 times higher in the IPF patient lung fibroblast compared with SCF220, while normal fibroblasts showed a similar predominance of SCF248 expression. Indeed, blockade of fibroblast-associated SCF248 reduced mast cell-mediated expression of matrix transcripts by SCF248-rich IPF, but not normal, lung fibroblasts in mast cell fibroblast coculture studies. These results suggest that SCF248 is upregulated and preferentially expressed in fibroblasts from IPF patients, where it may mediate fibroblast activation through interaction with c-kit on cells in the microenvironment. In addition, immunohistochemical staining of lung tissue demonstrated that nonfibrotic RBILD tissue had some SCF248 staining in macrophage populations and no detectable structural cell staining compared with extensive staining of remodeled areas of the lung of IPF patient tissue.
In addition to mast cells, other immune cell populations also express the SCF receptor, c-kit, and may be activated by SCF and contribute to the progression of fibrotic disease. Eosinophils have also been shown to express c-kit and, when activated by SCF, produce profibrotic cytokines including TGFβ and FGF as well as a number of other lipid mediators, proteases, and chemokines (53). In vitro coculture studies have suggested that SCF/c-Kit-mediated eosinophil activation can promote fibroblast activation and matrix production (16). Further, our own data have previously implicated SCF in eosinophil activation and airway remodeling in allergic asthma models (5), and other studies have linked eosinophils to severe fibrotic disease progression (55, 64). More recently, another c-kit+ cell population, type two innate lymphoid cells (ILC2), have been associated with disease progression in IPF, potentially through the production of IL-5 and IL-13 (26, 70, 71). The infiltration of the c-kit+ cells were impaired when SCF248 was neutralized, including mast cells, eosinophils, and ILC2. While it is not clear to what extent each of these populations contributes to the fibrotic responses in different end-stage fibrotic diseases, the potential role for SCF in their activation and accumulation suggests a strong correlation to disease severity.
The biology of SCF and c-kit activation is complex and not fully understood. It is known that membrane-associated SCF isoforms (both 220 and 248), as compared with cleaved, soluble SCF-ECD, induce the most efficient activation of c-kit by allowing cross-linking and strong signaling (3). Furthermore, studies have shown that the majority of cleaved soluble SCF is monomeric in biologic solutions and therefore would not be able to activate c-kit efficiently (31). Seminal studies found that SCF220 and SCF248 have biologically different functions, with SCF220 providing the homeostatic functions of erythropoiesis and overall growth and health in mice, whereas SCF248 did not alter those same functions but did affect mast cells and myeloid cell populations (36). These differences provide fidelity in the system, where based upon regulated expression in specific cell populations or tissue, these isoforms can provide targeted function(s). The regulation of posttranscriptional modification has not been defined under homeostatic or disease conditions; however, it is speculated that these modifications must be tightly regulated. It is possible that fibroblasts preferentially produce the SCF248 isoform to modulate immune cell activation during normal growth and repair processes. The presence of exon 6 in the SCF248 isoform (that appears to be most closely associated with inflammatory and fibrotic responses) would allow more efficient cleavage of SCF from the surface of the expressing cells by the immune cell-derived proteases, such as mast cell chymase (14). Thus, the removal of SCF248 by this process would limit immune cell activation at the site of the response (unless continuously expressed on the surface such as in fibrosis) and therefore function to limit the ongoing inflammatory responses. Our previous data have demonstrated that SCF248, but not SCF220, is upregulated on fibroblasts by inflammatory and profibrotic cytokines, providing a “feed forward” loop in tissue fibrosis (21). Likewise, SCF220, which appears to provide a more membrane-stable isoform (without exon 6 cleavage sites), might be more functional during critical homeostatic processes, such as erythropoiesis (36).
Our understanding of the function of the different SCF isoforms continues to be incomplete due to a lack of appropriate reagents, especially those specific for SCF220, as well as the ability to properly detect each isoform's biologic function under homeostasis and disease conditions. Using novel genetic and antibody tools that have been generated to examine the function of SCF248, we have identified a specific, nonredundant role for the SCF248 isoform in lung remodeling using the bleomycin model. Together, the data provided in these studies demonstrate differential expression and disease relevance of the SCF248 isoform and suggest that SCF248 may be an important therapeutic target in fibrotic lung diseases.
GRANTS
These studies were funded in part by NIH National Heart, Lung, and Blood Institute Grants HL059178 and HL138013 (N. W. Lukacs) and by funds provided by Opsidio, LLC.
DISCLOSURES
The commercialization of an anti-SCF248 antibody is being pursued by Opsidio, LLC. M. Phillips is the CEO of the company and its founders are C. Hogaboam, S. L. Kunkel, and N. W. Lukacs. None of the other authors has any conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
M.P., C.H., and N.W.L. conceived and designed research; A.J.R., D.M.H., S.M., M.S., and N.W.L. performed experiments; A.J.R., D.M.H., S.M., M.S., B.B.M., C.H., and N.W.L. analyzed data; D.M.H., M.S., B.B.M., C.H., and N.W.L. interpreted results of experiments; D.M.H. and N.W.L. prepared figures; D.M.H., M.P., and N.W.L. drafted manuscript; A.J.R., D.M.H., M.S., B.B.M., S.H.P., S.L.K., M.P., C.H., and N.W.L. edited and revised manuscript; A.J.R., D.M.H., S.M., M.S., B.B.M., S.H.P., S.L.K., M.P., C.H., and N.W.L. approved final version of manuscript.
REFERENCES
- 1.Andersson CK, Andersson-Sjöland A, Mori M, Hallgren O, Pardo A, Eriksson L, Bjermer L, Löfdahl CG, Selman M, Westergren-Thorsson G, Erjefält JS. Activated MCTC mast cells infiltrate diseased lung areas in cystic fibrosis and idiopathic pulmonary fibrosis. Respir Res 12: 139, 2011. doi: 10.1186/1465-9921-12-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aono Y, Nishioka Y, Inayama M, Ugai M, Kishi J, Uehara H, Izumi K, Sone S. Imatinib as a novel antifibrotic agent in bleomycin-induced pulmonary fibrosis in mice. Am J Respir Crit Care Med 171: 1279–1285, 2005. doi: 10.1164/rccm.200404-531OC. [DOI] [PubMed] [Google Scholar]
- 3.Ashman LK. The biology of stem cell factor and its receptor C-kit. Int J Biochem Cell Biol 31: 1037–1051, 1999. doi: 10.1016/S1357-2725(99)00076-X. [DOI] [PubMed] [Google Scholar]
- 4.Balestro E, Calabrese F, Turato G, Lunardi F, Bazzan E, Marulli G, Biondini D, Rossi E, Sanduzzi A, Rea F, Rigobello C, Gregori D, Baraldo S, Spagnolo P, Cosio MG, Saetta M. Immune inflammation and disease progression in idiopathic pulmonary fibrosis. PLoS One 11: e0154516, 2016. doi: 10.1371/journal.pone.0154516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berlin AA, Hogaboam CM, Lukacs NW. Inhibition of SCF attenuates peribronchial remodeling in chronic cockroach allergen-induced asthma. Lab Invest 86: 557–565, 2006. doi: 10.1038/labinvest.3700419. [DOI] [PubMed] [Google Scholar]
- 6.Brito JM, Borojevic R. Liver granulomas in schistosomiasis: mast cell-dependent induction of SCF expression in hepatic stellate cells is mediated by TNF-alpha. J Leukoc Biol 62: 389–396, 1997. doi: 10.1002/jlb.62.3.389. [DOI] [PubMed] [Google Scholar]
- 7.Caughey GH. Mast cell proteases as protective and inflammatory mediators. Adv Exp Med Biol 716: 212–234, 2011. doi: 10.1007/978-1-4419-9533-9_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cha SI, Chang CS, Kim EK, Lee JW, Matthay MA, Golden JA, Elicker BM, Jones K, Collard HR, Wolters PJ. Lung mast cell density defines a subpopulation of patients with idiopathic pulmonary fibrosis. Histopathology 61: 98–106, 2012. doi: 10.1111/j.1365-2559.2012.04197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chauhan D, Karanam AB, Merlo A, Tom Bozzay PA, Zucker MJ, Seethamraju H, Shariati N, Russo MJ. Post-transplant survival in idiopathic pulmonary fibrosis patients concurrently listed for single and double lung transplantation. J Heart Lung Transplant 35: 657–660, 2016. doi: 10.1016/j.healun.2015.12.030. [DOI] [PubMed] [Google Scholar]
- 10.Chen ES, Greenlee BM, Wills-Karp M, Moller DR. Attenuation of lung inflammation and fibrosis in interferon-gamma-deficient mice after intratracheal bleomycin. Am J Respir Cell Mol Biol 24: 545–555, 2001. doi: 10.1165/ajrcmb.24.5.4064. [DOI] [PubMed] [Google Scholar]
- 11.Chyczewski L, Debek W, Chyczewska E, Debek K, Bankowski E. Morphology of lung mast cells in rats treated with bleomycin. Exp Toxicol Pathol 48: 515–517, 1996. doi: 10.1016/S0940-2993(96)80070-4. [DOI] [PubMed] [Google Scholar]
- 12.Daniels CE, Lasky JA, Limper AH, Mieras K, Gabor E, Schroeder DR; Imatinib-IPF Study Investigators . Imatinib treatment for idiopathic pulmonary fibrosis: randomized placebo-controlled trial results. Am J Respir Crit Care Med 181: 604–610, 2010. doi: 10.1164/rccm.200906-0964OC. [DOI] [PubMed] [Google Scholar]
- 13.Dastych J, Metcalfe DD. Stem cell factor induces mast cell adhesion to fibronectin. J Immunol 152: 213–219, 1994. [PubMed] [Google Scholar]
- 14.de Paulis A, Minopoli G, Dal Piaz F, Pucci P, Russo T, Marone G. Novel autocrine and paracrine loops of the stem cell factor/chymase network. Int Arch Allergy Immunol 118: 422–425, 1999. doi: 10.1159/000024153. [DOI] [PubMed] [Google Scholar]
- 15.Ding L, Dolgachev V, Wu Z, Liu T, Nakashima T, Wu Z, Ullenbruch M, Lukacs NW, Chen Z, Phan SH. Essential role of stem cell factor-c-Kit signalling pathway in bleomycin-induced pulmonary fibrosis. J Pathol 230: 205–214, 2013. doi: 10.1002/path.4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dolgachev V, Berlin AA, Lukacs NW. Eosinophil activation of fibroblasts from chronic allergen-induced disease utilizes stem cell factor for phenotypic changes. Am J Pathol 172: 68–76, 2008. doi: 10.2353/ajpath.2008.070082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dolgachev VA, Ullenbruch MR, Lukacs NW, Phan SH. Role of stem cell factor and bone marrow-derived fibroblasts in airway remodeling. Am J Pathol 174: 390–400, 2009. doi: 10.2353/ajpath.2009.080513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.El Kossi MM, El Nahas AM. Stem cell factor and crescentic glomerulonephritis. Am J Kidney Dis 41: 785–795, 2003. doi: 10.1016/S0272-6386(03)00026-X. [DOI] [PubMed] [Google Scholar]
- 19.El-Koraie AF, Baddour NM, Adam AG, El Kashef EH, El Nahas AM. Role of stem cell factor and mast cells in the progression of chronic glomerulonephritides. Kidney Int 60: 167–172, 2001. doi: 10.1046/j.1523-1755.2001.00783.x. [DOI] [PubMed] [Google Scholar]
- 20.Fireman E, Kivity S, Shahar I, Reshef T, Mekori YA. Secretion of stem cell factor by alveolar fibroblasts in interstitial lung diseases. Immunol Lett 67: 229–236, 1999. doi: 10.1016/S0165-2478(99)00020-6. [DOI] [PubMed] [Google Scholar]
- 21.Fonseca W, Rasky AJ, Ptaschinski C, Morris SH, Best SKK, Phillips M, Malinczak CA, Lukacs NW. Group 2 innate lymphoid cells (ILC2) are regulated by stem cell factor during chronic asthmatic disease. Mucosal Immunol 12: 445–456, 2019. doi: 10.1038/s41385-018-0117-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fukihara J, Kondoh Y. Nintedanib (OFEV) in the treatment of idiopathic pulmonary fibrosis. Expert Rev Respir Med 10: 1247–1254, 2016. doi: 10.1080/17476348.2016.1249854. [DOI] [PubMed] [Google Scholar]
- 24.Galli SJ, Tsai M, Wershil BK. The c-kit receptor, stem cell factor, and mast cells. What each is teaching us about the others. Am J Pathol 142: 965–974, 1993. [PMC free article] [PubMed] [Google Scholar]
- 25.George TJ, Arnaoutakis GJ, Shah AS. Lung transplant in idiopathic pulmonary fibrosis. Arch Surg 146: 1204–1209, 2011. doi: 10.1001/archsurg.2011.239. [DOI] [PubMed] [Google Scholar]
- 26.Hams E, Armstrong ME, Barlow JL, Saunders SP, Schwartz C, Cooke G, Fahy RJ, Crotty TB, Hirani N, Flynn RJ, Voehringer D, McKenzie AN, Donnelly SC, Fallon PG. IL-25 and type 2 innate lymphoid cells induce pulmonary fibrosis. Proc Natl Acad Sci USA 111: 367–372, 2014. doi: 10.1073/pnas.1315854111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hashimoto N, Jin H, Liu T, Chensue SW, Phan SH. Bone marrow-derived progenitor cells in pulmonary fibrosis. J Clin Invest 113: 243–252, 2004. doi: 10.1172/JCI200418847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hogaboam C, Kunkel SL, Strieter RM, Taub DD, Lincoln P, Standiford TJ, Lukacs NW. Novel role of transmembrane SCF for mast cell activation and eotaxin production in mast cell-fibroblast interactions. J Immunol 160: 6166–6171, 1998. [PubMed] [Google Scholar]
- 29.Howell DC, Johns RH, Lasky JA, Shan B, Scotton CJ, Laurent GJ, Chambers RC. Absence of proteinase-activated receptor-1 signaling affords protection from bleomycin-induced lung inflammation and fibrosis. Am J Pathol 166: 1353–1365, 2005. doi: 10.1016/S0002-9440(10)62354-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoyne GF, Elliott H, Mutsaers SE, Prêle CM. Idiopathic pulmonary fibrosis and a role for autoimmunity. Immunol Cell Biol 95: 577–583, 2017. doi: 10.1038/icb.2017.22. [DOI] [PubMed] [Google Scholar]
- 31.Hsu YR, Wu GM, Mendiaz EA, Syed R, Wypych J, Toso R, Mann MB, Boone TC, Narhi LO, Lu HS, Langley KE. The majority of stem cell factor exists as monomer under physiological conditions. Implications for dimerization mediating biological activity. J Biol Chem 272: 6406–6415, 1997. doi: 10.1074/jbc.272.10.6406. [DOI] [PubMed] [Google Scholar]
- 32.Huang Y, Ma SF, Espindola MS, Vij R, Oldham JM, Huffnagle GB, Erb-Downward JR, Flaherty KR, Moore BB, White ES, Zhou T, Li J, Lussier YA, Han MK, Kaminski N, Garcia JGN, Hogaboam CM, Martinez FJ, Noth I; COMET-IPF Investigators . Microbes are associated with host innate immune response in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 196: 208–219, 2017. doi: 10.1164/rccm.201607-1525OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 366: 1968–1977, 2012. doi: 10.1056/NEJMoa1113354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ishii M, Iwai M, Harada Y, Morikawa T, Okanoue T, Kishikawa T, Tsuchihashi Y, Hanai K, Arizono N. A role of mast cells for hepatic fibrosis in primary sclerosing cholangitis. Hepatol Res 31: 127–131, 2005. doi: 10.1016/j.hepres.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 35.Jakubzick C, Choi ES, Carpenter KJ, Kunkel SL, Evanoff H, Martinez FJ, Flaherty KR, Toews GB, Colby TV, Travis WD, Joshi BH, Puri RK, Hogaboam CM. Human pulmonary fibroblasts exhibit altered interleukin-4 and interleukin-13 receptor subunit expression in idiopathic interstitial pneumonia. Am J Pathol 164: 1989–2001, 2004. doi: 10.1016/S0002-9440(10)63759-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kapur R, Majumdar M, Xiao X, McAndrews-Hill M, Schindler K, Williams DA. Signaling through the interaction of membrane-restricted stem cell factor and c-kit receptor tyrosine kinase: genetic evidence for a differential role in erythropoiesis. Blood 91: 879–889, 1998. doi: 10.1182/blood.V91.3.879. [DOI] [PubMed] [Google Scholar]
- 37.Katsanos GS, Anogeianaki A, Orso C, Tetè S, Salini V, Antinolfi PL, Sabatino G. Mast cells and chemokines. J Biol Regul Homeost Agents 22: 145–151, 2008. [PubMed] [Google Scholar]
- 38.Keane MP, Belperio JA, Moore TA, Moore BB, Arenberg DA, Smith RE, Burdick MD, Kunkel SL, Strieter RM. Neutralization of the CXC chemokine, macrophage inflammatory protein-2, attenuates bleomycin-induced pulmonary fibrosis. J Immunol 162: 5511–5518, 1999. [PubMed] [Google Scholar]
- 39.King TE Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, Gorina E, Hopkins PM, Kardatzke D, Lancaster L, Lederer DJ, Nathan SD, Pereira CA, Sahn SA, Sussman R, Swigris JJ, Noble PW; ASCEND Study Group . A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 370: 2083–2092, 2014. doi: 10.1056/NEJMoa1402582. [DOI] [PubMed] [Google Scholar]
- 40.Kirshenbaum AS, Akin C, Wu Y, Rottem M, Goff JP, Beaven MA, Rao VK, Metcalfe DD. Characterization of novel stem cell factor responsive human mast cell lines LAD 1 and 2 established from a patient with mast cell sarcoma/leukemia; activation following aggregation of FcepsilonRI or FcgammaRI. Leuk Res 27: 677–682, 2003. doi: 10.1016/S0145-2126(02)00343-0. [DOI] [PubMed] [Google Scholar]
- 41.Kondo S, Kagami S, Kido H, Strutz F, Müller GA, Kuroda Y. Role of mast cell tryptase in renal interstitial fibrosis. J Am Soc Nephrol 12: 1668–1676, 2001. [DOI] [PubMed] [Google Scholar]
- 42.Krishnaswamy G, Kelley J, Johnson D, Youngberg G, Stone W, Huang SK, Bieber J, Chi DS. The human mast cell: functions in physiology and disease. Front Biosci 6: d1109–d1127, 2001. doi: 10.2741/krishnas. [DOI] [PubMed] [Google Scholar]
- 43.Lehmann S, Uhlemann M, Leontyev S, Seeburger J, Garbade J, Merk DR, Bittner HB, Mohr FW. Bilateral versus single lung transplant for idiopathic pulmonary fibrosis. Exp Clin Transplant 12: 443–447, 2014. doi: 10.6002/ect.2014.0060. [DOI] [PubMed] [Google Scholar]
- 44.Li Y, Zhou L, Liu F, Peng Y, Li J, Sun L, Duan S, Ling G, Chen X, Jiang W, Xia Y. Mast cell infiltration is involved in renal interstitial fibrosis in a rat model of protein-overload nephropathy. Kidney Blood Press Res 33: 240–248, 2010. doi: 10.1159/000317102. [DOI] [PubMed] [Google Scholar]
- 45.Liu H, Liu F, Peng Y, Liu Y, Li L, Tu X, Cheng M, Xu X, Chen X, Ling G, Sun L. Role of mast cells, stem cell factor and protease-activated receptor-2 in tubulointerstitial lesions in IgA nephropathy. Inflamm Res 59: 551–559, 2010. doi: 10.1007/s00011-010-0159-7. [DOI] [PubMed] [Google Scholar]
- 46.Mazzei ME, Richeldi L, Collard HR. Nintedanib in the treatment of idiopathic pulmonary fibrosis. Ther Adv Respir Dis 9: 121–129, 2015. doi: 10.1177/1753465815579365. [DOI] [PubMed] [Google Scholar]
- 47.McNiece IK, Briddell RA. Stem cell factor. J Leukoc Biol 58: 14–22, 1995. doi: 10.1002/jlb.58.1.14. [DOI] [PubMed] [Google Scholar]
- 48.Meng XM, Nikolic-Paterson DJ, Lan HY. Inflammatory processes in renal fibrosis. Nat Rev Nephrol 10: 493–503, 2014. doi: 10.1038/nrneph.2014.114. [DOI] [PubMed] [Google Scholar]
- 49.Miyazawa K, Williams DA, Gotoh A, Nishimaki J, Broxmeyer HE, Toyama K. Membrane-bound Steel factor induces more persistent tyrosine kinase activation and longer life span of c-kit gene-encoded protein than its soluble form. Blood 85: 641–649, 1995. doi: 10.1182/blood.V85.3.641.bloodjournal853641. [DOI] [PubMed] [Google Scholar]
- 50.Moore BB, Fry C, Zhou Y, Murray S, Han MK, Martinez FJ, Flaherty KR; The COMET Investigators . Inflammatory leukocyte phenotypes correlate with disease progression in idiopathic pulmonary fibrosis. Front Med 1: 00056, 2014. doi: 10.3389/fmed.2014.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oliveira SH, Lukacs NW. Stem cell factor and igE-stimulated murine mast cells produce chemokines (CCL2, CCL17, CCL22) and express chemokine receptors. Inflamm Res 50: 168–174, 2001. doi: 10.1007/s000110050741. [DOI] [PubMed] [Google Scholar]
- 52.Oliveira SH, Lukacs NW. Stem cell factor: a hemopoietic cytokine with important targets in asthma. Curr Drug Targets Inflamm Allergy 2: 313–318, 2003. doi: 10.2174/1568010033483990. [DOI] [PubMed] [Google Scholar]
- 53.Oliveira SH, Taub DD, Nagel J, Smith R, Hogaboam CM, Berlin A, Lukacs NW. Stem cell factor induces eosinophil activation and degranulation: mediator release and gene array analysis. Blood 100: 4291–4297, 2002. doi: 10.1182/blood.V100.13.4291. [DOI] [PubMed] [Google Scholar]
- 54.Parker JM, Glaspole IN, Lancaster LH, Haddad TJ, She D, Roseti SL, Fiening JP, Grant EP, Kell CM, Flaherty KR. A phase 2 randomized controlled study of tralokinumab in subjects with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 197: 94–103, 2018. doi: 10.1164/rccm.201704-0784OC. [DOI] [PubMed] [Google Scholar]
- 55.Peterson MW, Monick M, Hunninghake GW. Prognostic role of eosinophils in pulmonary fibrosis. Chest 92: 51–56, 1987. doi: 10.1378/chest.92.1.51. [DOI] [PubMed] [Google Scholar]
- 56.Piñeiro-Hermida S, López IP, Alfaro-Arnedo E, Torrens R, Iñiguez M, Alvarez-Erviti L, Ruíz-Martínez C, Pichel JG. IGF1R deficiency attenuates acute inflammatory response in a bleomycin-induced lung injury mouse model. Sci Rep 7: 4290, 2017. doi: 10.1038/s41598-017-04561-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Raghu G, Brown KK, Collard HR, Cottin V, Gibson KF, Kaner RJ, Lederer DJ, Martinez FJ, Noble PW, Song JW, Wells AU, Whelan TP, Wuyts W, Moreau E, Patterson SD, Smith V, Bayly S, Chien JW, Gong Q, Zhang JJ, O’Riordan TG. Efficacy of simtuzumab versus placebo in patients with idiopathic pulmonary fibrosis: a randomised, double-blind, controlled, phase 2 trial. Lancet Respir Med 5: 22–32, 2017. doi: 10.1016/S2213-2600(16)30421-0. [DOI] [PubMed] [Google Scholar]
- 58.Rhee CK, Lee SH, Yoon HK, Kim SC, Lee SY, Kwon SS, Kim YK, Kim KH, Kim TJ, Kim JW. Effect of nilotinib on bleomycin-induced acute lung injury and pulmonary fibrosis in mice. Respiration 82: 273–287, 2011. doi: 10.1159/000327719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, Cottin V, Flaherty KR, Hansell DM, Inoue Y, Kim DS, Kolb M, Nicholson AG, Noble PW, Selman M, Taniguchi H, Brun M, Le Maulf F, Girard M, Stowasser S, Schlenker-Herceg R, Disse B, Collard HR; INPULSIS Trial Investigators . Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 370: 2071–2082, 2014. doi: 10.1056/NEJMoa1402584. [DOI] [PubMed] [Google Scholar]
- 60.Rodriguez-Portal JA. Efficacy and safety of nintedanib for the treatment of idiopathic pulmonary fibrosis: an update. Drugs R D 18: 19–25, 2018. doi: 10.1007/s40268-017-0221-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Seki E, Schwabe RF. Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology 61: 1066–1079, 2015. doi: 10.1002/hep.27332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shakoory B, Fitzgerald SM, Lee SA, Chi DS, Krishnaswamy G. The role of human mast cell-derived cytokines in eosinophil biology. J Interferon Cytokine Res 24: 271–281, 2004. doi: 10.1089/107999004323065057. [DOI] [PubMed] [Google Scholar]
- 63.Shen DZ. A target role for mast cell in the prevention and therapy of hepatic fibrosis. Med Hypotheses 70: 760–764, 2008. doi: 10.1016/j.mehy.2007.07.042. [DOI] [PubMed] [Google Scholar]
- 64.Shock A, Rabe KF, Dent G, Chambers RC, Gray AJ, Chung KF, Barnes PJ, Laurent GJ. Eosinophils adhere to and stimulate replication of lung fibroblasts ‘in vitro’. Clin Exp Immunol 86: 185–190, 1991. doi: 10.1111/j.1365-2249.1991.tb05793.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tajima Y, Moore MA, Soares V, Ono M, Kissel H, Besmer P. Consequences of exclusive expression in vivo of Kit-ligand lacking the major proteolytic cleavage site. Proc Natl Acad Sci USA 95: 11903–11908, 1998. doi: 10.1073/pnas.95.20.11903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tsuneyama K, Kono N, Yamashiro M, Kouda W, Sabit A, Sasaki M, Nakanuma Y. Aberrant expression of stem cell factor on biliary epithelial cells and peribiliary infiltration of c-kit-expressing mast cells in hepatolithiasis and primary sclerosing cholangitis: a possible contribution to bile duct fibrosis. J Pathol 189: 609–614, 1999. doi:. [DOI] [PubMed] [Google Scholar]
- 67.Tzouvelekis A, Bonella F, Spagnolo P. Update on therapeutic management of idiopathic pulmonary fibrosis. Ther Clin Risk Manag 11: 359–370, 2015. doi: 10.2147/TCRM.S69716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Veerappan A, O’Connor NJ, Brazin J, Reid AC, Jung A, McGee D, Summers B, Branch-Elliman D, Stiles B, Worgall S, Kaner RJ, Silver RB. Mast cells: a pivotal role in pulmonary fibrosis. DNA Cell Biol 32: 206–218, 2013. doi: 10.1089/dna.2013.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vittal R, Zhang H, Han MK, Moore BB, Horowitz JC, Thannickal VJ. Effects of the protein kinase inhibitor, imatinib mesylate, on epithelial/mesenchymal phenotypes: implications for treatment of fibrotic diseases. J Pharmacol Exp Ther 321: 35–44, 2007. doi: 10.1124/jpet.106.113407. [DOI] [PubMed] [Google Scholar]
- 70.Weiskirchen R, Tacke F. Interleukin-33 in the pathogenesis of liver fibrosis: alarming ILC2 and hepatic stellate cells. Cell Mol Immunol 14: 143–145, 2017. doi: 10.1038/cmi.2016.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wohlfahrt T, Usherenko S, Englbrecht M, Dees C, Weber S, Beyer C, Gelse K, Distler O, Schett G, Distler JH, Ramming A. Type 2 innate lymphoid cell counts are increased in patients with systemic sclerosis and correlate with the extent of fibrosis. Ann Rheum Dis 75: 623–626, 2016. doi: 10.1136/annrheumdis-2015-207388. [DOI] [PubMed] [Google Scholar]
- 72.Wygrecka M, Dahal BK, Kosanovic D, Petersen F, Taborski B, von Gerlach S, Didiasova M, Zakrzewicz D, Preissner KT, Schermuly RT, Markart P. Mast cells and fibroblasts work in concert to aggravate pulmonary fibrosis: role of transmembrane SCF and the PAR-2/PKC-α/Raf-1/p44/42 signaling pathway. Am J Pathol 182: 2094–2108, 2013. doi: 10.1016/j.ajpath.2013.02.013. [DOI] [PubMed] [Google Scholar]