

Abstract
The aging brain is associated with atrophy along with functional and metabolic changes. In this study, we examined age-related changes in resting brain functions and the vulnerability of brain physiology to hypoxic exposure in humans in vivo. Brain functions were examined in 81 healthy humans (aged 18–62 years) by acquisitions of gray and white matter volumes, cerebral blood flow, cerebral oxygen consumption, and concentrations of lactate, N-acetylaspartate, and glutamate+glutamine using magnetic resonance imaging and spectroscopy. We observed impaired cerebral blood flow reactivity in response to inhalation of hypoxic air (p = 0.029) with advancing age along with decreased cerebral oxygen consumption (p = 0.036), and increased lactate concentration (p = 0.009), indicating tissue hypoxia and impaired metabolism. Diminished resilience to hypoxia and consequently increased vulnerability to metabolic stress could be a key part of declining brain health with age. Furthermore, we observed increased resting cerebral lactate concentration with advancing age (p = 0.007), which might reflect inhibited brain clearance of waste products.
Keywords: Aging, lactate, cerebral blood flow, oxygen extraction fraction, energy metabolism
Introduction
Aging is associated with structural, functional, and metabolic changes in the brain and characterized by atrophy,1 reduced cerebral blood flow (CBF),2 and altered oxygen metabolism.3 Age is also the predominant risk factor for several brain diseases, particularly the neurodegenerative diseases such as mild cognitive impairment (MCI), Alzheimer's disease, vascular dementia, and other types of dementia.4 The precise causality between these diseases and age-related physiological changes in the brain is currently unknown; however, a hypoxic environment in brain cells likely plays a role in the development of these diseases.5 The disease trajectory, ultimately ending with dementia, starts decades before disease manifestation.6 As an example, subtle cognitive decline and abnormal accumulation of β-amyloid is observed years before overt disease manifestation appears in patients with Alzheimer's disease.7 Therefore, predictors of healthy versus unhealthy brain aging leading to neurodegenerative diseases are highly relevant. Much focus has been on changes in brain metabolism with advanced age. Particular attention has been paid to the tendency of oxidative metabolism to produce harmful reactive oxygen species (ROS), such as superoxide and hydrogen peroxide, which damage the vascular endothelium and trigger mitophagy and eventual apoptosis of the cell if encountered at high concentrations in mitochondria.8–11 The amelioration of ROS production is governed by the activity of the so-called uncoupling proteins (UCP) found in the inner mitochondrial membrane, which uncouple electron flow from the production of ATP in the electron transport chain. The ability of UCP to reduce ROS production is inhibited with age, rendering the brain more susceptible to oxidative stress, degeneration, and atrophy.12
Hypoxia also affects oxidative metabolism and ROS generation, with studies showing, paradoxically, that a hypoxic environment in the mitochondria accelerates ROS generation.13,14 In addition to inducing ROS production, hypoxia initiates other potentially harmful pathways that may lead to neuron loss and neurodegenerative disease. Hypoxia inhibits mitochondrial function by impairing calcium homeostasis, diminishing Na+/K+-ATPase activity, and declining membrane resting potential, leading to inhibited energy production.11,14 Hypoxia may also accelerate β-amyloid production11,15 and impair β-amyloid clearance due to dysfunction of the blood–brain barrier (BBB) from vascular damage.11
Hypoxic exposure also invokes adaptive mechanisms that affect metabolism, primarily increasing glycolysis by enhancing the catalytic effect of phosphofructokinase-1 and pyruvate kinase, thereby increasing the lactate concentration. Prolonged hypoxia stimulates hypoxia-inducible factors (HIF), which alters gene expression, inactivates anabolism, and inhibits mitochondrial aerobic metabolism by inhibiting pyruvate dehydrogenase, resulting in lactate production. Consequently, lactate accumulation is a manifestation of the loss of metabolic homeostasis during hypoxic exposure.
Using magnetic resonance spectroscopy (MRS) and magnetic resonance imaging (MRI), we previously demonstrated a large increase (∼50%–80%) in lactate concentration in the visual cortex during hypoxic conditions in young, healthy human subjects, indicating the upregulation of glycolysis.16,17 Lactate production also occurs in the normally functioning normoxic brain. Aerobic glycolysis (AG), roughly defined as the conversion of glucose to lactate, even in the presence of normal levels of oxygen (known as the Warburg effect in cancer cells), is constantly occurring in the normal functioning brain.18 AG is readily observed during neural activation related to the energy demand of the membrane pump Na+/K+-ATPase in astrocytes19 and seems also to be associated with biosynthesis of glycogen, protein, and nucleic acids.20 AG activity is highest in childhood and young adulthood and is most prevalent in brain areas with a high level of synaptic growth and remodeling.3 The actual brain lactate concentration also depends on the clearance of lactate. Surplus lactate is cleared from the brain through the blood stream and cerebrospinal fluid (CSF). Animal studies have demonstrated that during sleep, the brain tissue is cleared of metabolites (including lactate) by flow of CSF into the perivascular space, often called the glymphatic system, and that the suppression of this function by acetazolamide or aquaporin-4 deletions decrease lactate clearance during sleep.21 A study has also reported that the function of the glymphatic system declines with age in rats.22 In summary, aging seems to affect both the production and clearance of lactate in otherwise normal functioning brain.
The present study aimed to provide a thorough understanding of how normal aging from young adulthood to late mid-life affects cerebral metabolism with regard to resting and hypoxia-induced lactate concentration and its correlation with cerebral perfusion and oxygen consumption in vivo in healthy humans. Such information could enable the use of lactate as a biomarker of healthy versus unhealthy brain aging, paving the way for possible metabolic intervention and treatment. In 81 healthy subjects with a large age range (18–62 years), we measured the global CBF, global cerebral metabolic rate of oxygen (CMRO2) and cerebral lactate concentration using functional MRI and MRS techniques. In addition, we measured N-acetylaspartate (NAA) and glutamate + glutamine (glx) concentrations by MRS as markers of neuronal function. Total brain volume, gray matter volume, and white matter volume were acquired from anatomical MRI. By acquiring not only resting parameters but also parameters during hypoxic exposure, we examine resilience to metabolic stress, which will provide a much better understanding of possible malfunction of the cerebral metabolic system.
Methods
Data reported in this study were obtained from healthy control subjects in multiple studies on the effect of inhalation of hypoxic air as follows: patients with obstructive sleep apnea (study A),23 patients with migraine (study B),24,25 healthy subjects treated with erythropoietin (study C),17,26 freedivers (study D),16 and a pilot study on healthy subjects (study E). By combining data from multiple studies, we obtained sufficient power of the statistics to examine the subtle effects from normal aging. Exclusion criteria for all participants were any severe somatic or psychiatric disease, any daily medication (apart from oral contraceptives), and regular or recent exposure to hypoxia, including mountaineering and freediving. All studies used similar project protocols, MR-scanner setup, and MR-sequences. The final cohort for the resting data included 81 participants (22 females) with a large age range (18–62 years). Among these, 68 went on to a hypoxic challenge after the acquisition of baseline data (subjects from pilot study E did not undergo a hypoxic challenge). Anatomical images, CBF, and MRS were acquired in all studies. In study C, MRS during hypoxic exposure was acquired during simultaneous visual stimulation; therefore, these measurements were not included in this analysis. Identical techniques were used to acquire CMRO2 data in studies A and D, and these measurements are included in this analysis. In study C, CMRO2 data were acquired using a different MRI sequence and post-processing and therefore are not included in this study. The resulting number of acquisitions used in the final analysis for each parameter is summarized in Figure 1. All participants provided written informed consents to participate in this study after detailed oral and written information before the study. The studies were approved by the Danish National Committee on Health Research Ethics (H-4-2012-167, H-15003589, H-4-2012-182, H-15008282) and were conducted according to the Declaration of Helsinki.
Figure 1.
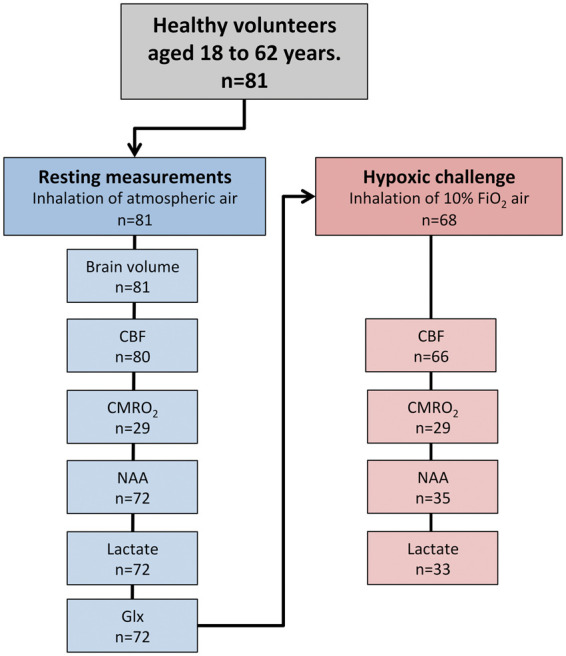
Flowchart of the acquired cerebral physiology parameters. Overall, 81 healthy subjects were included in the study and underwent the acquisition of resting values in a normoxic environment. In subsets of participants, CBF and CMRO2 were acquired using MRI. NAA, lactate, and glx concentrations were acquired using MRS. Hereafter, 68 of the 81 subjects underwent a hypoxic challenge by inhalation of air with approximately 10% O2. In subsets of participants, the measurements of CBF, CMRO2, NAA, lactate, and glx were reacquired during the hypoxic challenge.
CBF: cerebral blood flow; CMRO2: cerebral metabolic rate of oxygen; NAA: N-acetylaspartate; glx: glutamate + glutamine.
Procedure
The data were acquired during a single MRI session. Brain physiology was assessed through the measurements of global CBF and global CMRO2 using phase-contrast MRI techniques and cerebral NAA, glx, and lactate concentrations by MR spectroscopy. Brain volumes (gray matter, white matter, and CSF) were determined by anatomical MRI.
The participants were initially examined during normoxia by inhalation of air from the surrounding environment. Thereafter, participants were scanned while inhaling hypoxic air with approximately 10% O2. The participants inhaled hypoxic air for approximately 25 min to obtain stable oxygen saturation before the MRI parameters were reacquired. Fourteen participants (subjects in study B) inhaled hypoxic air for approximately 120 min before the parameters were reacquired.
All scans were performed on a Philips 3 Tesla Achieva MRI scanner (Philips Medical Systems, Best, The Netherlands) using a 32-channel phase array head coil. Hypoxic air was delivered to the participants while lying in the scanner through a mouthpiece or a face-mask connected to a 7-m tube and a one-way valve to prohibit rebreathing and dead space. The hypoxic air was delivered either by a gas-bottle or an AltiTrainer oxygen mixing system (SMTEC, Nyon, Switzerland). Mean arterial blood pressure (MAP) was acquired before scanning and during the hypoxic challenge in a subset of 53 subjects to assess the effect of hypoxia on MAP. Arterial oxygen saturation and heart rate were continually measured through the entire scan using a Veris Monitor system (MEDRAD, Pittsburgh, Pennsylvania, USA). In a subset of 23 subjects, a total of 92 arterial blood samples were drawn at normoxia and during hypoxia to calibrate the pulse oximetry equipment to ensure correct measurements during the hypoxic challenge. In the same subset of participants, arterial CO2 pressure was acquired from the blood samples; in the remaining subjects, end-tidal CO2 was continually acquired as a surrogate for arterial CO2 pressure using the Veris Monitor system.
All MRI data were processed using Matlab (Mathworks, Natick, MA, USA) scripts developed in house. Before each scan, a blood sample was drawn to determine the hematocrit and hemoglobin concentration. The arterial oxygen concentration (CaO2) was calculated as the hemoglobin concentration multiplied by the arterial saturation.
MRI
Structural imaging
Anatomical images were acquired by a 3D T1-weighted turbo field echo sequence (field of view (FOV), 241 × 180 × 165 mm3; voxel size, 1.1 × 1.1 × 1.1 mm3; echo time (TE), 2.78 ms; repetition time (TR), 6.9 ms; flip angle, 9°). Gray matter, white matter, and whole brain mask, including hemispheres, brainstem, and the cerebellum and excluding the CSF were segmented from the anatomical MRI scans using the FSL-functions BET and FAST (FMRIB Software Library, Oxford University, Oxford, UK).27 Examples of white and gray matter segmentation from anatomical images are displayed in Figure 2(a). Age-related atrophy of brain matter volume is demonstrated in Figure 3.
Figure 2.

Example of magnetic resonance imaging and spectroscopy techniques used in the study. (a) Axial and sagittal slice of a high-resolution T1-weighted anatomical MRI-image with segmentation of white and gray matter visualized. The location of the MR spectroscopy voxel (30 × 35 × 30 mm3) covering the calcarine fissure in the occipital lobe is also demonstrated. (b) Example of an acquired spectrum to determine the concentrations of NAA, lactate, and glx. (c) Coronal and sagittal view of MRI angiogram highlighting the cerebral carotids and vertebral arteries with the imaging plane perpendicular to the arteries to acquire velocity maps by phase-contrast mapping. (d) Example of velocity maps acquired from phase-contrast mapping perpendicular to the carotid and vertebral arteries. (e) Sagittal and coronal view of MRI angiogram highlighting the sagittal sinus with the imaging plane for susceptibility-based oximetry. (f) Example of susceptibility-weighted MR-image used in susceptibility-based oximetry. The susceptibility difference between the venous blood in the sagittal sinus and surrounding tissue, which is related to oxygen saturation, is clearly visible.
MRS: magnetic resonance spectroscopy; NAA: N-acetylaspartate; glx: glutamate+glutamine.
Figure 3.

Age-related decline in total brain (a), gray matter (b), and white matter volume (c). The dark gray line demonstrates the correlation from a linear regression. The regression slopes (β), R2-coefficients, and p-values from the regressions are noted in each panel.
CBF
The mean global CBF was acquired by measuring the blood velocity in the cerebral feeding arteries using phase-contrast mapping (PCM) MRI.28 Blood velocity maps were acquired with a turbo field echo sequence (1 slice, FOV, 240 × 240 mm2; voxel size, 0.75 × 0.75 ×8 mm3; TE, 7.33 ms; TR, 27.63 ms; flip angle, 10°; velocity encoding, 100 cm/s, without cardiac gating). Examples of the imaging plane perpendicular to the carotid and vertebral arteries and the related velocity map are demonstrated in Figure 2(c) and (d). The total blood flow to the brain was calculated by multiplying the mean blood velocity by the cross-sectional area of the cerebral feeding arteries in the region of interests (ROIs) on the velocity maps. ROIs were drawn on the magnitude images and transferred to the velocity maps. CBF was calculated by normalizing the total blood flow to the brain weight as determined from the brain volume estimated from anatomical MRI images and assuming a brain density of 1.05 g/ml.29 The cerebral delivery of oxygen (CDO2) was calculated by multiplying the CBF by CaO2.
CMRO2
CMRO2 was calculated using the Fick principle (equation (1))
| (1) |
where BFss is the blood flow in the sagittal sinus in ml/min scaled to the global CBF, SaO2, and SvO2 are the arterial and venous oxygen saturation, respectively, and [Hgb] is the oxygen-carrying hemoglobin concentration (mmol/l) at full saturation. SaO2 was measured using pulse oximetry or arterial blood samples. SvO2 and blood flow in the sagittal sinus were measured simultaneously using an MRI sequence combining PCM for acquisition of blood flow and susceptibility-based oximetry (SBO) for acquisition of oxygen saturation. SBO uses the principle that difference in magnetic susceptibility between deoxyhemoglobin in venous blood and the surrounding tissue can be related to oxygen saturation.30,31 The susceptibility difference between venous blood and the surrounding tissue was obtained by acquiring susceptibility-weighted maps using a dual-echo gradient-echo sequence (1 slice, FOV, 220 × 190 mm2; voxel size, 0.5 × 0.5 × 8 mm3; echo time 1, 10.89 ms; echo time 2, 24.16 ms; flip angle, 30°; 5 repeated measures, total duration = 1 min 30 s; SENSE-factor, 2). Susceptibility-weighted maps were computed by subtracting phase-value maps from the two images generated with the short and long echo times. Examples of the imaging plane perpendicular to the sagittal sinus and the acquired susceptibility-weighted image are demonstrated in Figure 2(e) and (f). Any aliased phase values in the sagittal sinus and immediately surrounding tissue were corrected manually on each map. The difference in susceptibility between venous blood and tissue was calculated by drawing two ROI – one covering the sagittal sinus and one covering the immediately surrounding tissue.
Acquisition of each map was repeated with phase-contrast velocity encoding for simultaneous measurement of blood flow in the sagittal sinus. The blood flow in the sagittal sinus was calculated by multiplying the cross-sectional area and blood velocity similar to the processing of global CBF values described earlier. The acquired blood flow in the sagittal sinus was scaled to the global CBF measured at baseline in each participant to normalize CMRO2 to individual global brain values. The global arteriovenous oxygen saturation difference (A-V O2) was also calculated as SaO2−SvO2. A more in-depth discussion of the methods and data processing was reported previously.16,23,32
MRS
MRS was used to measure the NAA, lactate, and glx concentrations in the occipital lobe using a water-suppressed point-resolved spectroscopy pulse sequence (TR, 3000 or 5000 ms; TE, 36 ms; voxel size, 30 × 35 × 30 mm3). Example of placement of MRS voxel is demonstrated in Figure 2(a). Post-processing and quantification of the spectra were performed using LCModel (LCModel [Version 6.3-1F], Toronto, Canada). Example of an acquired spectrum is shown in Figure 2(b). A blinded investigator excluded spectra of poor quality, wherein metabolite quantification was erroneous. Because in vivo 3 Tesla MRS in humans cannot differentiate between glutamate and glutamine concentrations, only the combined concentration (glx) is reported. The water peak acquired in the spectrum was used to quantify the measured metabolites. The water concentration in the spectroscopy voxel was estimated from the content of gray matter, white matter, and CSF within the voxel from the segmentation of the high-resolution anatomical images. Age-related atrophy causes differences in the gray matter volume in the voxel; therefore, it is important to correct for these differences, as the water concentration directly affects the quantification. The metabolite concentrations are presented in Figure 4(a). As the measured metabolites are predominately present in gray matter, we also calculated the concentrations normalized to only gray matter volume (Figure 4(b)).
Figure 4.

Correlations between age, CBF, CMRO2, NAA concentration, glx concentration, lactate concentration, hemoglobin, pCO2, and brain volume in the resting brain. (a) The panels demonstrate scatterplots of the measured parameters. The dark gray lines demonstrate the linear correlations. The regression slopes (β), R2-coefficients, and p-values for the regression are noted in each panel. The histogram of the measured parameters is shown in the diagonal of the panel matrix. CBF, NAA concentration, and brain volume correlated negatively with age. CMRO2 was unaffected by age. NAA concentration correlated with glx concentration, lactate concentration, and brain volume. (b) The panels demonstrate the correlations between age and the concentration of NAA, glx, and lactate when the concentrations are normalized to only gray matter. (c) The panels demonstrate the correlations between pCO2 and hemoglobin and CBF, CMRO2, and age. Variations in hemoglobin and pCO2 partly explain the variation in CBF. CMRO2 and age were not affected by hemoglobin concentration.
CBF: cerebral blood flow; CMRO2: cerebral metabolic rate of oxygen; NAA: N-acetylaspartate; glx: glutamate + glutamine.
Statistics
The post-processing of data was blinded regarding subject and measurement (hypoxic challenge vs. baseline measurement). Values are presented as the mean ± standard deviation. p-Values less than 0.05 were considered significant.
The significance of the correlations between the measured parameters was assessed by univariate linear regression analysis. The regression slopes (β), p-values, and R2 correlation coefficients from the linear regressions can be seen in Figure 4 in each panel. The histograms of the measured parameters are also presented in Figure 4. To address the likelihood of false positives in Figure 4(a) arising from multiple comparisons (21 comparisons), the false discovery rate method with a q-value of 0.05 was applied to calculate adjusted p-values (padj).
During the hypoxic challenge, the participants' ventilatory rates and consequently desaturation and pCO2 will vary. We therefore calculated the response to hypoxia as the change of the parameter normalized to the change in arterial saturation (equation (2)).
| (2) |
To examine the significance of age on the calculated response to hypoxia, we used linear regression analysis (equation (3)) with age and decrease in pCO2 as regressors. Arterial CO2 will vary between subjects during the hypoxic challenge due to differences in ventilation. CO2 has known effects on brain physiology, and thus we have to correct for this variation by including the change in pCO2 between baseline and hypoxia as a regressor in the linear model.
| (3) |
Scatterplots showing the responses to hypoxia with the correlations with age () with corresponding p-values and R2-coefficients are shown in Figure 5. A small number of the participants hyperventilated excessively during the hypoxic exposure, causing hypocapnia and only a slight desaturation. Due to hypocapnia-induced vasoconstriction, the CBF response would differ greatly from that of the other subjects; thus, the data collected during excessive hyperventilation were excluded from analysis. Saturation above 87% during hypoxic exposure was set as the threshold as hypoxia-induced increase in CBF starts at approximately this limit.33 The effects of hypoxia on heart rate and MAP were assessed by paired Student's t-test.
Figure 5.

Correlations between changes in CBF (a), CDO2 (b), A-V O2 (c), CMRO2 (d), lactate (e), and NAA (f) from desaturation during hypoxic challenge and age. The responses of the measured parameters to hypoxia are calculated as differences in value between baseline and hypoxia divided by the change in arterial oxygen saturation. The dark gray lines demonstrate the correlations from the linear regressions. The regression slopes, R2 coefficients, and p-values from the regressions are noted in each panel. The CBF reactivity and CMRO2 during the hypoxic challenge decline significantly with age. The lactate concentration increases during the hypoxic challenge for all ages and with a significantly higher increase with age. NAA concentration was unaffected by the hypoxic challenge and did not correlate with age.
CBF: cerebral blood flow; CMRO2: cerebral metabolic rate of oxygen; NAA: N-acetylaspartate; glx: glutamate + glutamine.
Results
Resting physiology
The total brain volume, gray matter volume, and white matter volume decreased (p < 10−5; p < 10−5; and p < 10−3, respectively) with age (Figure 3). Resting CBF decreased (p = 0.009; padj = 0.030) with age, but CMRO2 was unaffected (p = 0.89; padj = 0.94) (Figure 4(a)). CBF correlated with hemoglobin concentration (p < 10−4) and pCO2 (p = 0.003), but CMRO2 was unaffected by hemoglobin (p = 0.49) or pCO2 (p = 0.80) (Figure 4(c)).
NAA decreased with age (p = 0.016; padj = 0.047) and correlated with brain volume (p = 0.004; padj = 0.027). Lactate concentration increased with age (p = 0.007; padj = 0.029). Glx concentration correlated positively with CBF (p = 0.005; padj = 0.028) and NAA concentration (p < 10−4; padj < 10−3) and demonstrated a near-significant decrease with age (p = 0.077; padj = 0.19).
Normalization of the MRS metabolites to gray matter volume in the MRS-voxel eliminated the decrease in NAA with age (p = 0.24), but the increase in lactate with age was augmented (p < 10−3) (Figure 4(b)).
Hypoxic challenge
During hypoxic exposure, 11 CBF measurements, 2 CMRO2 measurements, and 1 MRS measurement were excluded from statistical analysis due to excessive hyperventilation (SaO2 > 87% O2). In the remaining subjects, the saturation decreased on average from 97.4 ± 1.0% at normoxia to 72.6 ± 7.3%, 72.8 ± 7.1%, and 73.1 ± 6.9% at the time of acquisition of CBF, CMRO2, and MRS, respectively. The pCO2 decreased on average from 5.1 ± 0.6 kPa at normoxia to 4.1 ± 0.8 kPa, 3.9 ± 0.8 kPa, and 4.1 ± 0.7 kPa at the time of acquisition of CBF, CMRO2, and MRS, respectively. Heart rate increased on average from 61.8 ± 9.5 bpm during normoxia to 75.4 ± 11.6 bpm during the hypoxic challenge (p < 10−7). MAP was unchanged from 90.6 ± 12.3 mmHg during normoxia to 91.4 ± 13.9 mmHg during hypoxia (p = 0.77).
The cerebrovascular reactivity (CVR) to inhalation of hypoxic air decreased with age (p = 0.029) (Figure 5(a)), as evidenced by a decrease in the magnitude of the hypoxia-stimulated increase in CBF. During the hypoxic challenge, the CMRO2 was maintained in younger subjects; however, CMRO2 decreased with age (Figure 5(d)). The effect of age was significant (p = 0.036). The A-V O2 during the hypoxic challenge demonstrated a tendency to decrease with age (p = 0.073) (Figure 5(c)). Lactate concentration increased during inhalation of hypoxic air for all ages, and this increase was significantly augmented with age (p = 0.009) (Figure 5(e)). NAA concentration was unaffected by the inhalation of hypoxic air, with no effect of age (Figure 5(f)).
Discussion
The main finding of this study is that the human brain demonstrates increased metabolic vulnerability to hypoxic exposure with advancing age from young adulthood to early 60s, as evidenced by decreased CVR and CMRO2 and increased lactate concentration as indicators of tissue hypoxia. We further observed an age-related increase in resting cerebral lactate concentration along with the expected atrophy and declining CBF.
During a hypoxic challenge in young healthy humans, the CVR is activated and CBF increases to compensate for arterial desaturation, and the CMRO2 is maintained or increased slightly.16,17,23,24,34,35 Interestingly, acute short-term hypoxic exposure also causes a large increase in cerebral lactate concentration (but without significant increase in blood lactate if the subjects remain inactive), despite maintained oxygen consumption.16,17 In the present analysis, we found that advancing age caused significant alterations in the response to hypoxia. With advancing age, the CVR decreased, oxygen consumption decreased, and lactate concentration increased during the hypoxic challenge, demonstrating increased vulnerability of the brain to metabolic stress and consequent tissue hypoxia. Tissue hypoxia can have a number of harmful consequences, including increased ROS production; vascular damage causing impaired BBB and mitochondrial dysfunction, leading to mitophagy, neurodegeneration, and accelerated aging of the brain, and may be a precursor for the development of Alzheimer's disease.11,13–15
Impaired CVR (from hypercapnic or acetazolamide challenge) has been observed in a number of age-related diseases associated with stroke and cognitive decline; conversely, a high CVR is positively associated with cognitive abilities in elderly adults.36 Patients with metabolic syndrome and related diseases, such as type 2 diabetes and obstructive sleep apnea (OSA), have reduced baseline CBF and CVR,37,38 and decreased CVR may correlate with the development of Alzheimer's disease.39 Our hypoxic challenge data demonstrate a possible causality, as the age-related decrease in CVR is associated with decreased oxygen consumption and increased lactate production, indicating tissue hypoxia and neurometabolic uncoupling.
Concurrent with the decreased CVR, we observed a tendency of the A-V O2 during the hypoxic challenge to decrease with age; however, the difference was only near-significant. Uneven distribution of capillary perfusion due to vascular insults and disease in the small vessels has been proposed to inhibit the extraction of oxygen with increased blood flow.40,41 This phenomenon could cause age-related impairment of oxygen extraction during hyperperfusion from the hypoxic challenge, manifesting as decreased A-V O2 and decreased CMRO2. This observation further suggests that normal CVR in older subjects does not necessarily result in normal oxygen usage, as the increased flow causes further uneven perfusion distribution.41 Interestingly, patients with Alzheimer's disease, despite having impaired CVR, demonstrate intact cerebral autoregulation in response to change in blood pressure from orthostatic challenge, which similarly suggest deficits in the small vessels.42 In summary, the age-related decrease in CVR and oxygen consumption, increase in lactate concentration, and tendency toward decreased oxygen extraction demonstrated increased frailty and vulnerability of the brain to metabolic stress and consequent tissue hypoxia.
Our data further demonstrated an age-related decrease in resting CBF, as noted in previous studies.2,43 Decreased CBF may be a sign of impaired cerebrovascular function and correlates with overall mortality in aging subjects that may be related to insufficient maintenance of homeostasis.44 Interestingly, cerebral oxygen consumption, when correcting for atrophy, is maintained or even slightly increased with age, suggesting that the remaining neurons rely more on oxygen with age.45
We also observed an age-related increase in resting cerebral lactate concentration that was further augmented by normalization to gray matter volume. The interpretation of resting lactate concentration is not clear and should be approached carefully. The measurement of each individual lactate flux in vivo in humans is not possible; only the net resulting lactate concentration is measureable. Lactate is continuously produced, cleared, and used by brain cells.46 During a resting state, lactate is produced primarily in astrocytes, and neurons may be prone to use lactate in oxidative phosphorylation. The production of lactate is further amplified during activation.47
With advancing age, cerebral metabolism shifts toward more oxidative phosphorylation in proportion to glycolysis, causing the ratio of oxygen to glucose consumption (OGI-ratio) to increase with age toward the theoretical upper limit of six.3 In children and young adults, the proportionally greater glucose consumption results in lactate production. The larger net lactate concentration we observed with advanced age is therefore unlikely to have resulted from higher production. Instead, this increase could be the buildup of lactate resulting from decreased clearance or less usage. Surplus lactate is cleared from the brain through the bloodstream and CSF. Recent studies on rats show that during sleep, brain tissue is drained of metabolites (including lactate) by the flow of CSF in the perivascular space,21 known as the glymphatic system, and that glymphatic system function declines with age.22 This observed increase in lactate could therefore result from the diminished function of the glymphatic system or from age-related decreases in perfusion. In addition, the lactate concentration did not correlate with CMRO2, glx, or NAA concentrations, further indicating that the increase in lactate does not arise from a change in metabolism but rather from decreased clearance or usage.
Inhibited clearance of waste products in the brain has been discussed as a major contributor to declining brain health and the development of neurodegenerative diseases.22,48 Thus, cerebral lactate concentration could be used as a marker of brain clearance function. Studies of resting lactate in humans are very limited. To our knowledge, no studies have examined the effect of age on resting lactate in humans. The one study conducted on rats reports an increase in resting cerebral lactate, as observed in our study, and attributes this increase to a shift in lactate dehydrogenase equilibrium toward lactate.49 We cannot exclude that the increase in lactate we observed in humans is from altered lactate dehydrogenase equilibrium; however, this hypothesis cannot be examined in a non-invasive manner in the human brain.
We further found that the NAA concentration decreased with age and correlated with a decrease in brain volume, as found in previous studies. NAA is abundant in the CNS and is predominantly synthesized in neurons.50 NAA is typically associated with neuronal integrity and mitochondrial function, and decreased NAA concentration correlates with the clinical severity of atrophic brain diseases.51 The NAA concentration declines in brain areas affected by stroke and correlates with neuron loss.50 Decreased NAA concentration has also been shown to correlate with decreased axonal density in patients with multiple sclerosis (MS).52 This observation has led to the use of NAA concentration as a marker of neuronal integrity. However, the decrease in NAA concentration has also been shown to be reversible in patients with traumatic brain injury and to closely follow reversal of impaired metabolism.53 Furthermore, NAA increases in patients with MS after treatment,54 indicating that the NAA concentration more likely reflects mitochondrial activity and only correlates secondarily with neuron loss.50 The age-related decrease in NAA observed here could therefore indicate declining metabolic function in the brain. However, with normalization to gray matter volume, the NAA concentration remains unchanged with age, indicating that the metabolic function is intact in the neurons remaining after correction for atrophy. Similarly, we observed a slight and near-significant decrease in glx with age that did not remain after normalization to gray matter volume.
Alzheimer's disease and other geriatric neurodegenerative diseases often appear later in life than the oldest subjects included in this study. However, the track to develop Alzheimer's disease starts years or even decades before manifestation of clinical symptoms,6 and therefore the deficits observed in this study could be part of the prior non-symptomatic declining brain health. Patients with Alzheimer's disease also have alterations of resting cerebral metabolism such as decreased glucose consumption,6 deficits in several steps in the glycolytic pathway, tricarboxylic acid cycle and oxidative phosphorylation,55 as well as alteration of the phosphocreatine–creatine kinase system.56 How these metabolic deficits and the development of neurodegenerative diseases relate to the impairment we demonstrated in this study would be highly relevant to examine in future longitudinal studies.
The main strength of this study is that we acquired in vivo MRI data during acute hypoxic exposure in a relatively large human cohort. A further strength is that we measured multiple parameters in the same subjects in the same session (approximately 45 min for all parameters) and can correlate these parameters. We used non-invasive, rapid techniques, which enabled us to measure all parameters in the same experiment session during both resting and the hypoxic challenge. Furthermore, we take into account confounding factors such as hemoglobin and blood gases, which is missing in many studies. To our knowledge, we are the first to examine the effect of aging on the resilience of the human brain against cerebral hypoxia with respect to oxygen metabolism and lactate production.
This study also has some limitations. We only acquired global values for CBF and CMRO2, making it impossible to examine potential regional variations in the brain. Furthermore, the MRS was only acquired in the occipital lope, and we did not investigate the levels of NAA, glx, and lactate in other areas of the brain.
We used SBO MRI technique to measure the venous saturation in the sagittal sinus to calculate CMRO2. Direct measurements from blood samples from the jugular veins would have been preferred; however, this is an invasive maneuver that is difficult to carry out in the MRI scanner setting, and using the SBO MRI technique was therefore opted as the best solution. Also, as we applied the same methods in all subjects, possible biases in the MRI techniques would not affect the conclusions on the effects from aging.
The quantitative values of CBF and CMRO2 have large normal variations, both in this study and in previous published results.57 The variation in CBF is explained by many factors where hemoglobin, blood pCO2, and innate intra-subject differences are likely the most important factors, demonstrating the importance of obtaining hemoglobin and pCO2 when studying brain perfusion to draw the correct conclusions.57 We found that hemoglobin and pCO2 partly explained the variation in CBF; however, hemoglobin and pCO2 were unaffected by age, precluding that the age-related decline in CBF is caused by increased hemoglobin concentration or differences in pCO2. Importantly, we also found that hemoglobin and pCO2 did not affect CMRO2; possible alterations in CMRO2 are therefore likely caused by actual brain energy demand and consumption and not by changes in the perfusion state.
We also observed some confounding factors that may influence the age-related effects we observed. Body mass index (BMI) and MAP increased with age (Figure 6(a) and (b)); such changes are known to contribute to declining health with advanced age and might also affect cerebral physiology. The observed increase in BMI and MAP reflects that of the general Danish population58 from which our subjects were recruited; therefore, we chose not to correct for these parameters in the study.
Figure 6.

Demonstration of possible confounding factors to age. The panels demonstrate the associations between age and BMI (a), MAP (b), and scan time (c) for the study participant.
MAP: mean arterial blood pressure; BMI: body mass index.
A further confounding factor is that cerebral metabolism and perfusion may undergo a circadian rhythm and variation throughout the day, although the exact effect is not well described. Cerebral lactate decreases during sleep and may steadily accumulate throughout the day. In general, the older volunteers in this study were scanned in the morning (Figure 6(c)), when lactate should be lower according to circadian rhythms. Thus, circadian rhythms are not the cause of the observed age-related increase in lactate.
Lastly, 14 subjects (those who participated in study C) inhaled hypoxic air for a longer time than the other subjects (120 min vs. 25 min, respectively). This difference might introduce errors if the response to hypoxia is time dependent in this time range. However, a comparison of the two groups revealed no significant difference in saturation or measured MRI parameters; thus, any time-dependent effect is likely minimal and without significance. These subjects were therefore included in the study to increase the power of the statistics. With exclusion of these subjects, we still observed a significant decrease in CMRO2 and significant increase in lactate with advanced age during hypoxic exposure; however, the reduced CVR became insignificant (p = 0.12).
In conclusion, this study demonstrated an increase in the metabolic vulnerability of the human brain to hypoxic exposure with advancing age from young adulthood to early 60s. During the inhalation of hypoxic air, we found an age-related decrease in CVR and CMRO2 and increase in cerebral lactate concentration, indicating tissue hypoxia. These results provide a possible causality between declining brain health and aging. With advanced age, the brain cannot withstand metabolic stress in the same way as young, healthy individuals, which causes episodes of tissue hypoxia that can initiate a number of harmful pathways, e.g. higher ROS production, vascular insults, mitochondrial damage, and mitophagy.
This study also demonstrated the importance of maintaining a well-functioning cerebrovascular mechanism that can oppose metabolic stress and counteract declining brain health caused by aging. We further observed a higher resting cerebral lactate concentration with advanced age. Increased lactate concentrations might result from diminished clearance of waste products from the brain and is a potential biomarker for evaluating brain clearance function.
Acknowledgments
We would like to thank Poul Jennum for providing control data from study A on patients with obstructive sleep apnea and Messoud Ashina for providing control data from study B on patients with migraine. We would also like to thank Niels Vidiendal Olsen, Peter Rasmussen, Ian Law and Otto Mølby Henriksen for providing control data from study C on the effect of EPO on brain physiology, and Kristian Lisbjerg, Søren Just Christensen and Niels Jacob Aachmann-Andersen for helping with recruitment and data collection for that study. Lastly, we thank laboratory technician Helle Juhl Simonsen for assistance with blood sample analysis.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The University of Copenhagen, Capital Region of Denmark Foundation for Health Research (A4620), Danish Council for Independent Research (DFF-4004-00169B and 10-094110), Lundbeck Foundation through the Center for Neurovascular Signaling (LUCENS), A.P. Møller Fonden, Læge Sofus Carl Emil Friis og Hustru Olga Doris Friis Legat, the Augustinus Foundation (13-3794), Simon Fougner Hartmanns Familiefond, and the European Union’s Seventh Framework programme (2007–2003) under grant agreement no. 602633.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors' contributions
MBV and HBWL initiated and formulated the study, developed the study protocol, performed the data acquisition, data processing, analysis, statistical analysis, and interpretation, and the drafting and revision of the article. MLFJ contributed to data acquisition, data processing and analysis, and revision of the paper. NA contributed to data acquisition, data processing and analysis, and revision of the paper. UL contributed to data acquisition, data processing, and analysis and revision of the paper. All authors have given final approval of the article.
Data availability
The data derived from the MRI images and all other data supporting the findings of this study are available from the corresponding author upon request.
References
- 1.Tabatabaei-Jafari H, Shaw ME, Cherbuin N. Cerebral atrophy in mild cognitive impairment: a systematic review with meta-analysis. Alzheimer's Dement Diagnosis Assess Dis Monit 2015; 1: 487–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vernooij MW, van der Lugt A, Ikram MA, et al. Total cerebral blood flow and total brain perfusion in the general population: the Rotterdam Scan Study. J Cereb Blood Flow Metab 2008; 28: 412–419. [DOI] [PubMed] [Google Scholar]
- 3.Goyal MS, Hawrylycz M, Miller JA, et al. Aerobic glycolysis in the human brain is associated with development and neotenous gene expression. Cell Metab 2014; 19: 49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.George SM, Stephen WB, Paul IG, et al. Age, neuropathology, and dementia. N Engl J Med 2009; 360: 2302–2309. [DOI] [PubMed] [Google Scholar]
- 5.Peers C, Pearson HA, Boyle JP. Hypoxia and Alzheimer's disease. Essays Biochem 2007; 43: 153–164. [DOI] [PubMed] [Google Scholar]
- 6.Masters CL, Bateman R, Blennow K, et al. Alzheimer's disease. Nat Rev Dis Prim 2015; 1: 1–18. [DOI] [PubMed] [Google Scholar]
- 7.Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimer's Dement 2011; 7: 280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brand MD. The efficiency and plasticity of mitochondrial energy transduction. Biochem Soc Trans 2005; 33: 897–904. [DOI] [PubMed] [Google Scholar]
- 9.Ozben T. Oxidative stress and apoptosis: impact on cancer therapy. J Pharm Sci 2007; 96: 2181–2196. [DOI] [PubMed] [Google Scholar]
- 10.Wright AF, Jacobson SG, Cideciyan AV, et al. Lifespan and mitochondrial control of neurodegeneration. Nat Genet 2004; 36: 1153–1158. [DOI] [PubMed] [Google Scholar]
- 11.Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders Berislav. Nat Rev Neurosci 2011; 12: 723–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carney JM, Starke-Reed PE, Oliver CN, et al. Reversal of age-related increase in brain protein oxidation, decrease in enzyme activity, and loss in temporal and spatial memory by chronic administration of the spin-trapping compound N-tert-butyl-alpha-phenylnitrone. Proc Natl Acad Sci U S A 1991; 88: 3633–3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chandel NS, Maltepe E, Goldwasser E, et al. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A 1998; 95: 11715–11720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Solaini G, Baracca A, Lenaz G, et al. Hypoxia and mitochondrial oxidative metabolism. Biochim Biophys Acta 2010; 1797: 1171–1177. [DOI] [PubMed] [Google Scholar]
- 15.Sun X, He G, Qing H, et al. Hypoxia facilitates Alzheimer's disease pathogenesis by up-regulating BACE1 gene expression. Proc Natl Acad Sci 2006; 103: 18727–18732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vestergaard MB, Larsson HB. Cerebral metabolism and vascular reactivity during breath-hold and hypoxic challenge in freedivers and healthy controls. J Cereb Blood Flow Metab. Epub ahead of print 3 November 2017. DOI: 10.1177/0271678X17737909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vestergaard MB, Lindberg U, Aachmann-Andersen NJ, et al. Acute hypoxia increases the cerebral metabolic rate – a magnetic resonance imaging study. J Cereb Blood Flow Metab 2016; 36: 1046–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vaishnavi SN, Vlassenko AG, Rundle MM, et al. Regional aerobic glycolysis in the human brain. Proc Natl Acad Sci 2010; 107: 17757–17762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bélanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab 2011; 14: 724–738. [DOI] [PubMed] [Google Scholar]
- 20.Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol 2011; 27: 441–464. [DOI] [PubMed] [Google Scholar]
- 21.Lundgaard I, Lu ML, Yang E, et al. Glymphatic clearance controls state-dependent changes in brain lactate concentration. J Cereb Blood Flow Metab 2017; 37: 2112–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kress BT, Iliff JJ, Xia M, et al. Impairment of paravascular clearance pathways in the aging brain. Ann Neurol 2015; 76: 845–861. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jensen MLF, Vestergaard MB, Tønnesen P, et al. Cerebral blood flow, oxygen metabolism and lactate during hypoxia in patients with obstructive sleep apnea contributing. Sleep 2018; 41: 1–10. [DOI] [PubMed] [Google Scholar]
- 24.Arngrim N, Schytz HW, Britze J, et al. Migraine induced by hypoxia: a MRI spectrocopy and angiography study. Brain 2015; 139: 723–737. [DOI] [PubMed] [Google Scholar]
- 25.Arngrim N, Hougaard A, Schytz HW, et al. Effect of hypoxia on BOLD fMRI response and total cerebral blood flow in migraine with aura patients. J Cereb Blood Flow Metab. Epub ahead of print 7 July 2017. DOI: 10.1177/0271678X17719430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vestergaard MB, Henriksen OM, Lindberg U, et al. No evidence for direct effects of recombinant human erythropoietin on cerebral blood flow and metabolism in healthy humans. J Appl Physiol 2018; 124: 1107–1116. [DOI] [PubMed] [Google Scholar]
- 27.Jenkinson M, Beckmann CF, Behrens TEJ, et al. Fsl. Neuroimage 2012; 62: 782–790. [DOI] [PubMed] [Google Scholar]
- 28.Vestergaard MB, Lindberg U, Aachmann-Andersen NJ, et al. Comparison of global cerebral blood flow measured by phase-contrast mapping MRI with 15O-H2O positron emission tomography. J Magn Reson Imaging 2017; 45: 692–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Torack RM, Alcala H, Gado M, et al. Correlative assay of computerized cranial tomography (CCT), water content and specific gravity in normal and pathological postmortem brain. J Neuropathol Exp Neurol 1976; 35: 385–392. [DOI] [PubMed] [Google Scholar]
- 30.Jain V, Langham MC, Wehrli FW. MRI estimation of global brain oxygen consumption rate. J Cereb blood flow Metab 2010; 30: 1598–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jain V, Langham MC, Floyd TF, et al. Rapid magnetic resonance measurement of global cerebral metabolic rate of oxygen consumption in humans during rest and hypercapnia. J Cereb Blood Flow Metab 2011; 31: 1504–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodgers ZB, Leinwand SE, Keenan BT, et al. Cerebral metabolic rate of oxygen in obstructive sleep apnea at rest and in response to breath-hold challenge. J Cereb Blood Flow Metab 2016; 36: 755–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Willie CK, Macleod DB, Shaw AD, et al. Regional brain blood flow in man during acute changes in arterial blood gases. J Physiol 2012; 590: 3261–3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kety SS, Schmidt CF. The effects of altered arterial tensions of carbon dioxide and oxygen on cerebral blood flow and cerebral oxygen consumption of normal young men. J Clin Invest 1948; 27: 484–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu F, Liu P, Pascual JM, et al. Effect of hypoxia and hyperoxia on cerebral blood flow, blood oxygenation, and oxidative metabolism. J Cereb blood flow Metab 2012; 32: 1909–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eskes GA, Longman S, Brown AD, et al. Contribution of physical fitness, cerebrovascular reserve and cognitive stimulation to cognitive function in post-menopausal women. Front Aging Neurosci 2010; 2: 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tyndall AV, Argourd L, Sajobi TT, et al. Cardiometabolic risk factors predict cerebrovascular health in older adults: results from the Brain in Motion study. Physiol Rep 2016; 4: e12733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fülesdi B, Limburg M, Bereczki D, et al. Cerebrovascular reactivity and reserve capacity in type II diabetes mellitus. J Diabetes Complications 1999; 13: 191–199. [DOI] [PubMed] [Google Scholar]
- 39.Girouard H, Iadecola C. Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer disease. J Appl Physiol 2006; 100: 328–335. [DOI] [PubMed] [Google Scholar]
- 40.Kuschinsky W, Paulson OB. Capillary circulation in the brain. Cerebrovasc Brain Metab Rev 1992; 4: 261–286. [PubMed] [Google Scholar]
- 41.Jespersen SN, Østergaard L. The roles of cerebral blood flow, capillary transit time heterogeneity, and oxygen tension in brain oxygenation and metabolism. J Cereb Blood Flow Metab 2012; 32: 264–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Heus RAA, de Jong DLK, Sanders ML, et al. Dynamic regulation of cerebral blood flow in patients with Alzheimer disease novelty and significance. Hypertension 2018; 72: 139–150. [DOI] [PubMed] [Google Scholar]
- 43.Peng S-L, Dumas JA, Park DC, et al. Age-related increase of resting metabolic rate in the human brain. Neuroimage 2014; 98: 176–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sabayan B, Grond J Van Der, Westendorp RG, et al. Total cerebral blood flow and mortality in old age: a 12-year follow-up study. Neurology 2013; 81: 1922–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lu H, Xu F, Rodrigue KM, et al. Alterations in cerebral metabolic rate and blood supply across the adult lifespan. Cereb Cortex 2011; 21: 1426–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dienel GA. Brain lactate metabolism: the discoveries and the controversies. J Cereb Blood Flow Metab 2012; 32: 1107–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bednařík P, Tkáč I, Giove F, et al. Neurochemical and BOLD responses during neuronal activation measured in the human visual cortex at 7 Tesla. J Cereb Blood Flow Metab 2015; 35: 601–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tarasoff-Conway JM, Carare RO, Osorio RS, et al. Clearance systems in the brain – implications for Alzheimer disease. Nat Rev Neurol 2015; 11: 457–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ross JM, Öberg J, Brené S, et al. High brain lactate is a hallmark of aging and caused by a shift in the lactate dehydrogenase A/B ratio. Proc Natl Acad Sci U S A 2010; 107: 20087–20092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moffett RJ, Ross B, Arun P, et al. N-Acetylaspartate in the CNS: from neurodiagnostics to neurobiology. Prog Neurobiol 2007; 81: 89–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Block W, Jessen F, Träber F, et al. Regional N-acetylaspartate reduction in the hippocampus detected with fast proton magnetic resonance spectroscopic imaging in patients with Alzheimer disease. Arch Neurol 2002; 59: 828–834. [DOI] [PubMed] [Google Scholar]
- 52.Bitsch A, Bruhn H, Vougioukas V, et al. Inflammatory CNS demyelination: histopathologic correlation with in vivo quantitative proton MR spectroscopy. Am J Neuroradiol 1999; 20: 1619–1627. [PMC free article] [PubMed] [Google Scholar]
- 53.Signoretti S, Marmarou A, Tavazzi B, et al. N-acetylaspartate reduction as a measure of injury severity and mitochondrial dysfunction following diffuse traumatic brain injury. J Neurotrauma 2001; 18: 977–991. [DOI] [PubMed] [Google Scholar]
- 54.Narayanan S, De Stefano N, Francis GS, et al. Axonal metabolic recovery in multiple sclerosis patients treated with interferon beta-1b. J Neurol 2001; 248: 979–86. [DOI] [PubMed] [Google Scholar]
- 55.Murray IVJ, Proza JF, Sohrabji F, et al. Vascular and metabolic dysfunction in Alzheimer's disease: a review. Exp Biol Med 2011; 236: 772–782. [DOI] [PubMed] [Google Scholar]
- 56.Rijpma A, van der Graaf M, Meulenbroek O, et al. Altered brain high-energy phosphate metabolism in mild Alzheimer's disease: a 3-dimensional31P MR spectroscopic imaging study. NeuroImage Clin 2018; 18: 254–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Henriksen OM, Kruuse C, Olesen J, et al. Sources of variability of resting cerebral blood flow in healthy subjects: a study using (133)Xe SPECT measurements. J Cereb Blood Flow Metab 2013; 33: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hoffmann-Petersen N, Lauritzen T, Bech JN, et al. High prevalence of hypertension in a Danish population telemedical home measurement of blood pressure in citizens aged 55-64 years in Holstebro county. Am J Hypertens 2016; 29: 439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data derived from the MRI images and all other data supporting the findings of this study are available from the corresponding author upon request.