

Cardiovascular diseases are the leading cause of death worldwide. Approximately 85% of all deaths from cardiovascular disease are due to acute myocardial infarction (AMI) and stroke. Strokes are associated with high mortality, and 15% to 30% of patients have permanent disability.1 Although biomarkers like cardiac TnT (troponin T; cTnT) and creatine kinase (CK) are successfully used for diagnosis of AMI, there is an ongoing need for markers that, for example, help identify patients with vulnerable plaques. In addition, no biomarker exists for the diagnosis of stroke or the identification of patients at higher risk for stroke.1
Atherosclerosis is the underlying cause of most cardiovascular disease and involves plaque formation, intima thickening caused by lipid deposition, inflammation, and thickening of the arterial wall.2, 3 The plaque development starts with early atherosclerotic lesions and proceeds to advanced atherosclerotic lesions, which can evolve into complicated and unstable vulnerable plaques. Vulnerable plaques prone to rupture have intraplaque hemorrhage, apoptosis, and calcification. Rupture of unstable plaques causes atherothrombogenic events and leads to severe clinical symptoms.2
Oxidative modifications of LDL (low‐density lipoprotein) to oxidized LDL (oxLDL) play an important role in the initiation and progression of atherosclerosis. OxLDL promotes foam cell and fatty streak formation, induces proinflammatory pathways, triggers vascular smooth muscle cell migration and proliferation, and induces cell death and apoptosis. Thus, oxLDL contributes to plaque growth and instability.2 Several oxLDL receptors have been described. LOX‐1 (lectin‐like oxLDL receptor 1) was identified as the major oxLDL receptor in endothelial cells.4 In early atherosclerotic lesions, LOX‐1 is mainly expressed in endothelial cells but extends its expression to smooth muscle cells and macrophages in advanced lesions.5, 6 Under physiological conditions, LOX‐1 expression is very low, but its expression increases in response to oxLDL, proinflammatory cytokines, pro‐oxidative and biomechanical stimuli.6, 7, 8, 9, 10 LOX‐1 activation promotes secretion of proinflammatory cytokines and reactive oxygen species formation.6, 10 Furthermore, LOX‐1 deletion reduces MMP‐2 (matrix metalloproteinase 2) and MMP‐9 in atherosclerotic lesions, whereas activation of oxLDL–LOX‐1 pathways increases MMP activity.11, 12, 13 This connects LOX‐1 to advanced lesion formation and plaque vulnerability.6, 14, 15 LOX‐1 expression is increased in experimental and clinical studies of hypertension, myocardial infarction, carotid artery atherosclerosis, type 2 diabetes mellitus, and obesity.5, 16, 17, 18, 19
A soluble form of LOX‐1 (sLOX‐1) has been identified and is generated by ectodomain shedding. This process is triggered by oxLDL, CRP (C‐reactive protein), TNF‐α (tumor necrosis factor α), IL‐8 (interleukin‐8), and IL‐18 and mediated by the action of MMPs and ADAMs (a disintegrin and metalloproteinase; Figure 1).20, 21, 22 A large number of studies have demonstrated the diagnostic potential of sLOX‐1 regarding severity of stable coronary artery disease (CAD) and acute coronary syndrome (ACS), even allowing discrimination between different types of AMI.17, 23, 24, 25, 26 Furthermore, sLOX‐1 concentrations are increased in ischemic and hemorrhagic stroke, and patients with high sLOX‐1 levels have an increased risk of future clinical events.22, 27, 28 Quantification of sLOX‐1 might be an interesting novel biomarker to improve the diagnosis of patients at risk.
Figure 1.
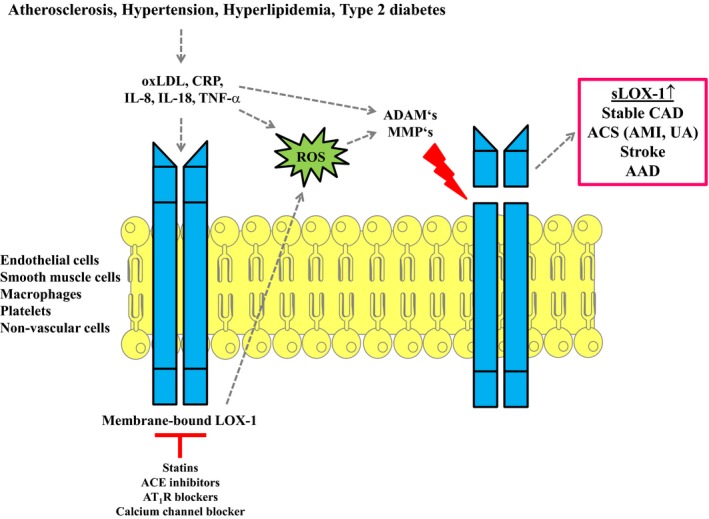
Generation of sLOX‐1 (soluble lectin‐like oxidized low‐density lipoprotein receptor 1) from the membrane‐bound form. LOX‐1 expression is found on endothelial cells, smooth muscle cells, macrophages, platelets, and nonvascular cells. Proinflammatory mediators oxidized LDL (oxLDL), CRP (C‐reactive protein), IL‐8 (interleukin 8), IL‐18, or TNF‐α (tumor necrosis factor α) induce the shedding of cell‐membrane‐bound LOX‐1. This process involves reactive oxygen species (ROS) and the activation of matrix‐degrading enzymes ADAMs (a disintegrin and metalloprotease) and MMPs (matrix metalloproteases). ROS are generated by different enzymatic sources in vascular cells. Their formation is increased on activation of LOX‐1 and direct effects of oxLDL or TNF‐α on ROS producing enzymes. The proteolytic cleavage in the extracellular neck domain of membrane‐bound LOX‐1 leads to the release of the shorter sLOX‐1. Elevated sLOX‐1 concentrations are shown in stable coronary artery disease (CAD), acute myocardial infarction (AMI), stroke, and acute aortic dissections (AAD). Proinflammatory mediators that stimulate sLOX‐1 release are increased in atherosclerosis, hypertension, hyperlipidemia or type 2 diabetes mellitus. In addition, sLOX‐1 may even present a pathophysiologic link among these diseases. Interpretation of results has to involve adjustment for other comorbidities and pharmacologic therapy with statins, angiotensin‐converting enzyme (ACE) inhibitors, angiotensin receptor II type 1 (ATR 1) blockers, or calcium channel blockers that might affect LOX‐1 expression and sLOX‐1 release. Figure adapted from SMART—Servier Medical Art by Servier (https://smart.servier.com). UA indicates unstable angina.
Acute aortic dissection (AAD) is a life‐threatening disease caused by a tear in the vessel wall. Risk factors are poorly controlled hypertension, age, and male sex.29, 30 LOX‐1 expression is increased in hypertension.6 Impaired aortic vascular function has been described in mouse models.31 Furthermore, effects on smooth muscle cell apoptosis were shown.32, 33 No experimental data on LOX‐1 and aortic disease are available, but LOX‐1 might be involved in the degeneration of the vessel wall. Therefore, sLOX‐1 could be a marker in AAD that helps to set the diagnosis and, furthermore, allows discrimination among patients with AMI.
This review provides a detailed overview on studies of sLOX‐1 in CAD, AMI, stroke, and AAD (Table). The findings are described and critically discussed. The potential of sLOX‐1 as a biomarker is evaluated by comparison to current clinical biomarkers and imaging techniques. The aim of this review is to evaluate the potential of sLOX‐1 for the diagnosis of CAD and AMI and its implications to predict the prognosis of these patients. In addition, the use of sLOX‐1 as a novel biomarker in patients with stroke and AAD will be discussed.
Table 1.
sLOX‐1 Concentrations in CAD, ACS, Stroke, and AAD
| Disease | Control | Disease | Sample (n) | Time Point of Blood Collection |
|---|---|---|---|---|
| CAD34 |
Control, 268 (111–767) pg/mL |
1–2 diseased vessels, 611 (346–1313) pg/mL; 3–4 diseased vessels, 2143 (824–3201) pg/mL |
Control: 29; CAD: 60 |
Before CAG |
| CAD26 |
Simple lesion, 0.426 (0.195–1.075) ng/mL |
Complex lesion, 0.914 (0.489–1.296) ng/mL |
Simple: 72; complex: 50 | Before CAG |
| CAD, ACS26 |
Stable CAD, 0.579 (0.256–1.172) ng/mL |
ACS, 1.610 (0.941–2.264) ng/mL |
CAD: 122; ACS: 58 |
Before CAG |
| ACS26 |
No complex lesion, 1.003 (0.783–1.668) ng/mL |
1 complex lesion, 1.456 (0.923–2.124) ng/mL; multiple complex lesions, 2.171 (1.067–3.247) ng/mL |
No complex lesion: 11; 1 complex lesion: 23;Multiple complex lesions: 24 | Before CAG |
| CAD35 |
Distal segment LAD lesion, 0.70±0.17 ng/mL |
Proximal/middle segment LAD lesion, 1.07±0.33 ng/mL | Distal: 51; proximal/middle: 64 | After CAG |
| ACS23 |
Intact coronary, <0.5 (<0.5–1.3) ng/mL Controlled CHD, <0.5 (<0.5–3.4) ng/mL; ischemic CHD, 0.73 (<0.5–14.0) ng/mL; acute noncardiac illness, <0.5 (<0.5–6.4) ng/mL; chronic illness, <0.5 (<0.5–3.3) ng/mL |
ACS, 2.91 (<0.5–170) ng/mL |
ACS: 80; intact coronary: 52; controlled CHD: 122; ischemic CHD: 173; acute cardiac illness: 34; chronic illness: 60 | At CAG or time of visit (acute and chronic illness) |
| ACS36 |
NSTEMI, 133.3 (106.6–238.5) ng/mL |
STEMI, 204.2 (135.7–456.0) ng/mL |
NSTEMI: 19; STEMI: 56 |
Before CAG |
| ACS37 |
Non‐ACS, 104.1 (67.9–128.6) ng/mL |
NSTEMI, 143.9 (96.6–255.1) ng/mL; STEMI, 259.0 (134.5–488.9) ng/mL |
Non‐ACS: 40; NSTEMI: 44; STEMI: 116 |
In ER |
| ACS25 |
Non‐ACS, 0.096 (0.0645–0.162) ng/mL |
ACS, 1.13 (0.168–3.46) ng/mL |
Non‐ACS: 89; ACS: 18 |
After CAG |
| PCI38 |
PCI without RPMI, 99±68 pg/mL |
PCI+RPMI, 167±89 pg/mL |
PCI without RPMI: 181; PCI+RPMI: 33 |
Every 6 h after PCI; in total, up to 24 h |
| ACS39 |
Non‐AMI, 64.3 (54.4–84.3) ng/mL |
STEMI, 241.0 (132.4–472.2) ng/mL; NSTEMI, 147.3 (92.9–262.4) ng/mL |
Non‐AMI: 125; NSTEMI: 44; STEMI: 125 |
Before CAG |
| ACS40 |
Event‐free survival, 2.54 ng/mL |
Recurrence‐ACS/and death, 6.60 ng/mL |
Event‐free: 81; recurrence: 13 | During acute stage |
| AAD41 | NSTEMI | AAD | NSTEMI: 39; AAD: 19 | In ER and before CAG |
| Stroke, ischemic stroke42 |
Mean: Q1, 558 ng/mL; Q2, 925 ng/mL; Q3, 1289 ng/mL; Q4, 2367 ng/mL |
Q1, n=21;Q2, n=20;Q3, n=25;Q4, n=25 | Median: 11 y | |
| Stroke27 |
Control (ischemic stroke), 486 (321–703) ng/L; control (ICH), 513 (307–770) ng/L; control (ABI), 496 (337–781) ng/L; control (cardioembolic stroke), 462 (333–652) ng/L; control (lacunar infarction), 558 (302–850) ng/L |
Ischemic stroke, 526 (330–883) ng/L; ICH, 720 (459–1125) ng/L; ABI, 641 (429–1302) ng/L; cardioembolic stroke, 442 (225–840) ng/L; lacunar infarction, 529 (341–743) ng/L |
Control: 250; ischemic stroke: 250—control: 127; ICH: 127—control: 43; ABI: 43—control: 59; cardioembolic stroke: 59—control: 56; lacunar infarction: 56 |
3 d after onset of stroke |
| Stroke28 |
Healthy control 0.67 ng/mL |
Carotid atherosclerosis, 0.99 ng/mL; TIA, 0.95 (0.23–7.31) ng/mL; ischemic stroke, 1.0 (0.11–2.63) ng/mL |
Healthy control: 81; carotid atherosclerosis: 232; TIA: 61; ischemic stroke: 104 | 48 h before operation |
| Stroke28 |
sLOX‐1: tertile 1, 3.48±0.279 (au); tertile 2, 4.04±0.134 (au); tertile 3, 4.79±0.480 (au) |
Tertile 1, 1567; tertile 2, 1568; tertile 3, 1568 | Baseline until first hospitalization for acute stroke (median: 16.5±3.6 y) |
Summary of the most important studies analyzing sLOX‐1 in patients with CAD, AAD, and stroke. AAD indicates acute aortic dissection; ABI, atherothrombogenic brain infarction; ACS, acute coronary syndrome; AMI, acute myocardial infarction; CAD, coronary artery disease; CAG, coronary angiography; CHD, coronary heart disease; ER, emergency room; ICH, intracerebral haemorrhage; LAD, left anterior descending; NSTEMI, non‐ST‐segment–elevation myocardial infarction; PCI, percutaneous coronary intervention; Q, quartile; RPMI, related periprocedural myocardial infarction; sLOX‐1, soluble lectin‐like oxidized low‐density lipoprotein receptor‐1; STEMI, ST‐segment–elevation myocardial infarction; TIA, transient ischemic attack.
Release of sLOX‐1 Involves Inflammatory Cytokines and Matrix‐Degenerating Enzymes
The polypeptide sLOX‐1 consists of 187 amino acids and derives from the proteolytic cleavage of LOX‐1 expressed on the cell surface.15 The molecular weight of sLOX‐1 is 35 kDa and reflects the shortening of the mature LOX‐1 protein with a size of 40 kDa.20 LOX‐1 structurally belongs to the C‐type lectin receptor family and consists of 4 functional domains: a short N‐terminal cytoplasmic domain, a connecting neck domain, a transmembrane domain, and the lectin‐like ligand binding domain, located extracellularly at the C‐terminus.16, 43 Detailed analysis showed that PMSF (phenylmethylsulfonyl fluoride)–sensitive serine proteases cleave at 2 possible sites in the LOX‐1 extracellularly located neck domain.20, 34, 44 In this study, PMSF was applied to cultured cells. Therefore, it is difficult to conclude that only PMSF‐sensitive proteases cleave LOX‐1 because most studies emphasize the importance of MMPs in this context. A PMSF‐sensitive process might be involved in sLOX‐1 generation but rather in an indirect way. A cleavage site between Arg88 and Gln89 has been identified in the human LOX‐1 protein. This cleavage site is located between 2 stable and highly conserved regions at the beginning of a flexible region. Increases in flexibility are mainly due to the replacement of hydrophobic amino acid residues in polar or charged amino acids, thereby mediating the proteolytic attack.44
It has been postulated that elevations in sLOX‐1 may reflect increased expression of the membrane‐bound form.20, 24 However, no clear correlation between plasma sLOX‐1 and plaque LOX‐1 mRNA expression was demonstrated in carotid atherosclerosis.28 One explanation could be that sLOX‐1 is generated not only from atherosclerotic carotid arteries but also from other atherosclerotic vessels45 and nonvascular cells.46, 47 Instead of correlations with tissue LOX‐1 expression, positive correlations between markers of macrophages and endothelial cells were found.28 Therefore, sLOX‐1 might be an inflammatory marker and a surrogate marker of tissue expression. In vulnerable atherosclerotic plaques, LOX‐1 is dominantly expressed in smooth muscle cells and macrophages and contributes to apoptosis and production of MMPs.12, 13, 23 MMPs are involved in the proteolytic cleavage of cell surface–expressed LOX‐1 and thus may represent a possible link between tissue LOX‐1 and sLOX‐1.21, 23
Ectodomain shedding is a proteolytic process at transmembrane proteins that is driven by catalytically active ADAMs. The function of ectodomain shedding is the modulation of signaling pathways between host and neighboring cells by downregulating the expression of cell surface receptors or by increasing their soluble ligands.48 For sLOX‐1, the proinflammatory cytokines TNF‐α, IL‐8,28 IL‐18,49 and CRP50 are known to stimulate its release. In macrophages, CRP stimulated sLOX‐1 release by a mechanism involving p47phox phosphorylation, production of reactive oxygen species, and activation of TACE (TNF‐α converting enzyme; also known as ADAM‐17).50 ADAM‐10 is also involved in the shedding of membrane‐bound LOX‐1.49 In endothelial cells, soluble sMMP‐1 and/or sMMP‐2 is secreted and may induce LOX‐1 ectodomain shedding.21 In addition, depletion of membrane cholesterol (eg, by statins) enhances the release of full‐length LOX‐1 and sLOX‐1 in exosomes that originated from endothelial cells.21 Although many of these studies revealed contributions of MMPs and ADAMs in the shedding of sLOX‐1, no direct evidence shows that these enzyme mediate the cleavage of LOX‐1. It is possible that they act as activators of the real and, to date, unknown proteases. A recent study provided evidence that oxLDL induces the release of sLOX‐1 from endothelial cells, supporting the fact that sLOX‐1 originates from cells that were pre‐exposed to oxLDL.22 Interestingly, the authors demonstrated that native LDL, VLDL (very low‐density lipoprotein), HDL (high‐density lipoprotein), TGF‐β (transforming growth factor β) and IL‐1β had no effect on sLOX‐1 in endothelial cells.22
Methods for Quantification of sLOX‐1 and Interpretation of Results
Most of the previous studies used a “self‐made” sandwich ELISA with 2 different anti–human LOX‐1 polyclonal antibodies. Antibodies were raised against the extracellular domain of LOX‐1. The capture antibody was coated on the plates, and the other was fragmented into Fab, with labeling of horseradish peroxidase for enzymatic quantification. In this assay, the lower limit of the detection was 0.5 ng/mL (500 pg/mL). A major disadvantage was the missing ability to detect sLOX‐1 in healthy patients. Therefore, Nakamura and colleagues developed a chemiluminescent enzyme immunoassay. This assay uses a combination of 2 different monoclonal antibodies and could detect sLOX‐1 at concentrations as low as 8 pg/mL.51 This assay was used by the majority of subsequent studies in patients with CAD, ACS, or AAD. Several commercially available ELISA kits exist. Their sensitivity is in the range of 1 to 5 pg/mL. Besides high sensitivity, a major advantage is the low interassay variation during analysis of a high numbers of patients.
Markstadt et al discussed LOX‐1 cleavage at the 187 residue in the neck domain. Elevated sLOX‐1 levels could be caused by cleavage of the cell‐bound LOX‐1 at the same site.22 Furthermore, mechanical cleavage during tissue disruption may contribute to sLOX‐1 in tissue homogenates.22 Potential cross‐reactivity of sLOX‐1 antibodies with oxLDL was analyzed. Zhao et al added high concentrations of oxLDL and found no interference.50 To exclude this possibility, oxLDL should be added to the analyzed matrix while using the ELISAs for the first time.
Finally, careful interpretation of findings is necessary with respect to comorbidities and medical treatments. Because sLOX‐1 is increased in obesity, type 2 diabetes mellitus, metabolic syndrome, and peripheral artery disease,17, 45, 52, 53, 54 adjustment of findings to these comorbidities is important. In addition, in vitro and in vivo studies revealed that LOX‐1 expression is affected by angiotensin‐converting enzyme inhibitors,55, 56 angiotensin II receptor type 1 blockers,57 and statins.57, 58, 59 These pharmacologic therapies may also have an impact on sLOX‐1 concentrations.
Is Soluble LOX‐1 a Diagnostic, Prognostic or Inflammatory Marker in Patients With CAD and MI?
The evaluation of patients with suspected CAD and its acute complications involves assessment of risk factors (age, sex, weight, blood pressure, plasma lipids, or smoking) and imaging tools in combination with biochemical laboratory markers.60 The most common strategy is coronary angiography (CAG), providing a high diagnostic specify. Nonetheless, this technique is invasive, expensive, might involve clinical complications and in rare cases, the death of patients. Measurement of cTnT or cardiac TnI (troponin I), FABP (fatty acid‐binding protein) or CK and CK‐muscle/brain (CK‐MB) are routinely used in combination with CAG.61, 62 These markers are released after cardiomyocyte injury and used for the diagnosis of AMI in patients with chest pain.60 The marker cTnT is widely used in diagnosing AMI and predicting death.61, 62 It is highly specific and sensitive for cardiac injury. Elevations in cTnT are associated with a nearly 90% frequency of CAD detected by CAG.61 Because cTnT or CK are released after ischemic injury, neither is useful in detecting plaque vulnerability or plaque rupture at a stage before myocardial damage. Developing markers that reflect plaque instability and rupture would allow diagnosis at early stages before ACS becomes clinically apparent.23
In recent years, novel risk markers have been developed in the cardiovascular field.60 IL‐6, CRP, monocyte subsets, and microRNAs, for example, are under intensive investigation. However, there are still controversies about confounding factors, methodological limitations, and statistics used for data analysis.60 In addition, several specific requirements are necessary to make tests reliable and useful in clinics, involving test sensitivity, specificity, potential to make a correct diagnosis, and “pretest probability” that match noninvasive findings with imaging or descriptions of clinical symptoms.63
Stable CAD encompasses patients who have stable angina pectoris, patients with CAD‐related symptoms who become asymptomatic because of medical treatments, and those who reported symptoms for the first time but are in a chronic stable condition.64 A typical symptom of stable CAD is chest discomfort during exercise, emotions, or other stressors. The symptoms are caused by a mismatch between myocardial oxygen demand and supply.64 Basic assessments involve fasting lipid profile, screening for type 2 diabetes mellitus, estimation of renal function, resting ECG, and transthoracic echocardiography.64 Low concentrations of TnT have good prognostic value in patients with stable CAD but require ultrasensitive laboratory assays.64 No additional prognostic markers are recommended to manage patients with stable CAD.64
Lubrano and colleagues analyzed 60 patients with CAD who underwent coronary catheterization. Their study showed elevated sLOX‐1, which tended to be increased with the severity of CAD, reflected by the number of affected vessels (control: 268 pg/mL, n=29; 1–2 diseased vessels: 611 pg/mL, n=30; 3–4 diseased vessels: 2143 pg/mL, n=30). Furthermore, sLOX‐1 positively correlated with TNF‐α, IL‐6, CRP, and age.34 A cross‐sectional study by Zhao et al measured ≈2.8‐fold higher sLOX‐1 levels in patients with ACS (1.610 ng/mL, n=58) compared with stable CAD (0.579 ng/mL, n=122).26 In ACS patients, sLOX‐1 positively correlated with the number of complex coronary lesions and was independently associated with the presence of multiple complex lesions (odds ratio: 1.967). It is known that the type of lesion predicts plaque vulnerability.24, 26 Interestingly, sLOX‐1 was 2‐fold higher in patients with complex lesions (n=50) compared with simple lesions (0.914 versus 0.426 pg/mL, n=72). In addition, sLOX‐1 was an independent predictor of complex lesions (odds ratio: 1.964), as demonstrated by multivariate analysis.26 Limitations were the exclusion of patients with type 2 diabetes mellitus and the rather small sample size.26
Patients with atherosclerosis in the proximal or middle segment of the left anterior descending artery (LAD) represent a high‐risk subgroup in CAD. Balin et al showed 1.5‐fold elevated sLOX‐1 (1.07±0.33 ng/mL) in patients with proximal‐ or middle‐segment atherosclerosis compared with distal lesions (0.70±0.17 ng/mL).35 Interestingly, sLOX‐1 allowed identification of plaques in the proximal segment and thus may represent a predictor of plaque vulnerability in patients with stable CAD.35 Lesions in the proximal segment become clinically more apparent than those in distal segments. Although the numbers of patients were limited (n=51–64), this finding underscores the importance of sLOX‐1 as a potential predictor of the complexity of coronary lesions. Another study enrolled 94 patients with suspected CAD who underwent CAG and demonstrated positive correlations between sLOX‐1 and markers of oxidative stress (extracellular superoxide dismutase and 8‐isoprostane).65 This study suggests, that sLOX‐1 levels may reflect increased vascular oxidative stress. However, no correlations were observed among sLOX‐1, high‐sensitivity CRP (hs‐CRP), or brain natriuretic peptide.65
ACS occurs by myocardial ischemia in patients with CAD. Unstable angina pectoris, non–ST‐segment–elevation MI (NSTEMI) and ST‐segment‐elevation MI (STEMI) are part of ACS.66 Although the clinical symptoms have the same underlying mechanism, a specific diagnosis is required to separate patients with the overall symptom of chest pain. MI is defined as myocardial cell death due to prolonged ischemia and diagnosed by ECG changes and monitored using the rise and/or fall of cardiac troponin levels.66 Cardiac troponins are the most commonly used biomarker and their high‐sensitivity assays are recommend for clinical use.66 Concentrations of TnT peak 4 hours after the onset of symptoms. Before this period, negative results may be obtained.39 In addition, TnT remains high for several days, making it difficult to detect recurrent MI. Because of the shorter half‐life, CK‐MB is often used for detection of reinfarction and periprocedural MI.67 An adequate diagnosis of MI requires a specific description of the patient's symptoms, detection of ischemic ECG changes, echocardiography for wall motion abnormalities, and identification of the thrombus by CAG.66 The most common treatments are revascularization by percutaneous coronary intervention (PCI) or coronary artery bypass grafting.68
A large‐scale study analyzing the relevance of sLOX‐1 in CAD patients was conducted by Hayashida and colleagues in 2005.23 This study demonstrated elevated sLOX‐1 in patients with ACS (n=80) compared with those with intact coronary arteries (n=52), controlled coronary heart disease (n=122), and stable angina pectoris (n=173). Serum sLOX‐1 distinguished patients with ACS from those with non‐ACS (odds ratio: 1.51) and identified patients before the typical TnT increase. In a second study, sLOX‐1 and TnT were serially analyzed in 40 patients with ACS. Blood was taken on admission to the emergency room (ER), after PCI, and at days 1, 3, 5, and 7. Levels of sLOX‐1 peaked on admission to the ER and after PCI, whereas TnT peaked on day 1. Interestingly, sLOX‐1 did not correlate with TnT or CK‐MB. The authors concluded that sLOX‐1 is not a marker for cardiomyocyte injury and may again reflect plaque stability before myocardial damage.23 This study clearly emphasized the potential of sLOX‐1 as an early marker for detection of plaque vulnerability before elevations of TnT.69
Significant differences in sLOX‐1 have been shown in ACS with (n=116) and without (n=44) ST‐segment elevation. The highest concentrations were found in those with STEMI (median: 259.0 pg/mL) compared with those with NSTEMI (median: 143.9 pg/mL) and without ACS (median: 104.1 pg/mL). Interestingly, hs‐TnT did not differ between STEMI and NSTEMI at this early stage of ACS,37 which supports the concept of sLOX‐1 as an early marker of ACS. Over time, sLOX‐1 declined after onset of pain and arrival at the ER, whereas hs‐TnT levels increased.37 The authors concluded that sLOX‐1 and hs‐TnT alone did not have the diagnostic accuracy to determine early stages of ACS, but a combination of both parameters may be useful.37 A study by Kume and colleagues showed a similar increase in sLOX‐1 in patients with ACS (n=18) versus without ACS (n=89).25 Compared with TnT or heart‐type FABP, sLOX‐1 had the highest sensitivity and specificity to diagnose ACS. It was detectable even without elevations of TnT. No correlations of sLOX‐1 with age, total cholesterol, triglycerides, and LDL cholesterol were found. In addition, sLOX‐1 did not correlate with TnT or heart‐type FABP.24, 25 A major limitation was the small number of patients, but this study highlights the role of LOX‐1 in diagnosis of early ACS without an TnT increase. In addition, these data suggest that sLOX‐1 is important in the late stages of CAD, where atherosclerotic plaques are prone to rupture, and not during the early stages of initiation and progression of atherosclerosis, for which risk factors play a central role.
Further studies were conducted to evaluate the role of sLOX‐1 in early stages of AMI. Elevated sLOX‐1 levels have been demonstrated in patients with STEMI (241.0 pg/mL, n=125) compared with those with NSTEMI (147.3 pg/mL, n=44) and without AMI (64.3 pg/mL, n=125). In contrast to other studies of CAD,26 sLOX‐1 was not affected by the number of diseased vessels. The second part of this study analyzed time‐dependent changes in sLOX‐1. The highest concentrations were found between the onset of symptoms and arrival at the ER. TnT and CK‐MB increased 6 hours after arrival at the ER.39 At this early stage of STEMI, sLOX‐1 had high sensitivity for diagnosis of ACS.24, 39 The authors concluded that sLOX‐1 is not a marker of cardiomyocyte injury but rather reflects plaque instability. In summary, sLOX‐1 may help improve diagnosis in the early stage of ACS.39
Additional studies were performed to analyze a putative link between sLOX‐1 and plaque vulnerability. A study in patients with ACS (n=128) showed elevated sLOX‐1 in case of ruptured compared with nonruptured plaques and stable angina pectoris (n=20). Plaque morphology was examined in coronary vessels by optical coherence tomography, and patients were categorized according to ACS with and without plaque rupture. Interestingly, sLOX‐1 correlated with peak values for CK‐MB, but no correlation was found for hs‐CRP and hs‐TnT. Again, sLOX‐1 was able to distinguish between patients with STEMI and NSTEMI. Multivariate analysis revealed independent associations between sLOX‐1 and plaque rupture.36 In addition, an inverse correlation between sLOX‐1 and thickness of the fibrous cap was found. Overall, sLOX‐1 had the highest sensitivity and specificity to detect patients with thin‐cap fibroatheroma. The authors concluded that sLOX‐1 is a marker for evaluation of plaque vulnerability and for differentiation of patients with and without plaque rupture in the ER. The release of sLOX‐1 may precede plaque rupture and myocardial damage. Therefore, sLOX‐1 might be a marker to identify patients with an increased risk of plaque rupture.24, 36
PCI is one of the most common strategies for revascularization of coronary arteries in ACS and can be associated with periprocedural complications, like PCI‐related periprocedural MI (PCI‐RPMI).70 Patients who underwent single‐vessel PCI (n=214) were divided into groups with (n=33) and without (n=181) RPMI. Levels of sLOX‐1, TnT, CK, and CK‐MB were higher in PCI‐RPMI (167±89 versus 99±68 pg/mL), and sLOX‐1 correlated with CK‐MB, CK, and TnT.38 The authors conclude that sLOX‐1 might help identify patients at increased risk for RPMI before PCI. However, sLOX‐1 correlated with CK and CK‐MB levels38 that are used successfully in the clinics for identifying reinfarctions. In this context, sLOX‐1 alone might have no additional benefit.
Other studies considered sLOX‐1 as a potent predictor for recurrence or death of ACS.40 Kume et al analyzed patients who underwent PCI and were subsequently followed for a median of 896 days. They found that sLOX‐1 did not correlate with total cholesterol, triglycerides, HDL cholesterol, hs‐CRP, or TnT and classical risk factors, inflammatory biomarkers, and markers of cardiomyocyte injury. Their most important finding showed that patients with recurrent ACS or ACS‐related death had higher sLOX‐1 than event‐free patients. This result was not observed for TnT or hs‐CRP. Patients in the upper quartile or tertile of sLOX‐1 had greater prevalence and earlier incidence of recurrent ACS or death than in the event‐free survival group. In summary, the study by Kume et al showed the highest sensitivity and specificity for sLOX‐1 in the prediction of recurrence or death after acute‐stage ACS compared with TnT or hs‐CRP.24, 25 Unfortunately, limitations of this single‐center study were the small number of patients and the missing analysis of patients in the chronic stage after ACS (without death or recurrence) and with stable CAD.25
The majority of studies analyzed sLOX‐1 in stable CAD and ACS with different types of MI. Results showed that sLOX‐1 correlated with the complicity of coronary artery lesions, allowed identification of plaques at different segments of coronary arteries, was independently associated with plaque rupture, and predicted the risk of recurrent ACS or ACS‐related death. The majority of studies demonstrated that sLOX‐1 is not a marker for cardiomyocyte injury because it increases earlier than TnT. sLOX‐1 seems to be rather a marker for plaque stability that allows identifying patients with vulnerable plaques. Consequently, measuring sLOX‐1 would be suitable in patients with stable CAD to identify those with high risk for developing ACS (Figure 2). However, this recommendation needs larger studies and the use of imaging techniques, for example, to measure the thickness of the fibrous cap. The field of sLOX‐1 research in CAD started to grow in recent years. Whether sLOX‐1 will be used a useful diagnostic tool in the clinical setting will be decided in future studies involving more patients, novel imaging tools, and laboratory markers. However, it is not well understood whether sLOX‐1 originating from tissue LOX‐1 in atherosclerotic plaques provides the only explanation for systemic concentrations. Furthermore, the detailed mechanism of how sLOX‐1 increases in ACS is still missing. Addressing these open questions would help strengthen the clinical perspective of sLOX‐1.
Figure 2.
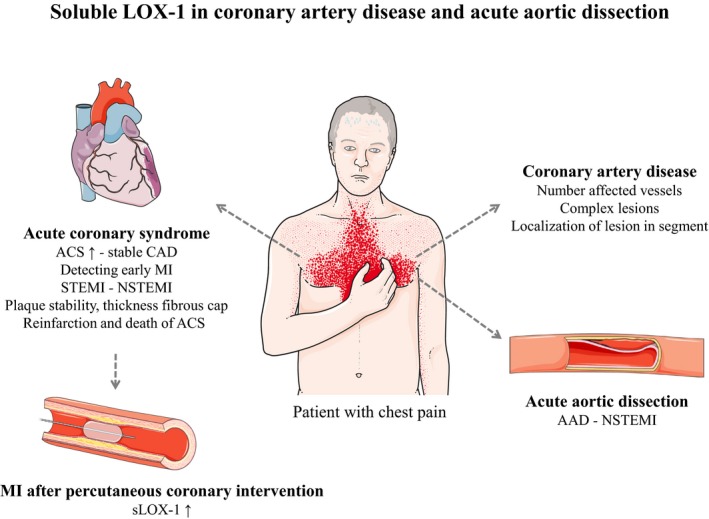
LOX‐1 (soluble lectin‐like oxidized LDL [low‐density lipoprotein] receptor 1; sLOX‐1) in coronary artery disease (CAD) and acute aortic dissection (AAD). sLOX‐1 was demonstrated to be elevated in acute coronary syndrome (ACS), especially in the early phase before elevations of cardiac troponin (TnT). Patients with ST‐segment–elevation myocardial infarction (STEMI) had higher sLOX‐1 than those with non‐STEMI (NSTEMI). sLOX‐1 was an indicator for plaque stability and thickness of the fibrous cap of coronary artery plaques. Concentrations of sLOX‐1 could predict reinfarction or death due to ACS. Patients with myocardial infarction (MI) after undergoing percutaneous coronary intervention (PCI) had higher sLOX‐1. In stable CAD, sLOX‐1 reflected the number of affected vessels and was an indicator for the complicity of lesions. sLOX‐1 was higher in patients with AAD compared with controls and with NSTEMI. sLOX‐1 had comparable specificity and sensitivity to TnT in discriminating between AAD and NSTEMI. Figure adapted from SMART—Servier Medical Art by Servier (https://smart.servier.com).
Is sLOX‐1 a Promising Biomarker in Patients With Stroke?
Stroke is the third leading cause of morbidity and mortality in Western industrialized countries. Patients who survive a stroke have an increased risk of having more strokes within the subsequent 5 years.1 Strokes are classified as ischemic or hemorrhagic. Most patients have ischemic strokes, which are caused by intracranial thrombosis or extracranial embolism. Intracranial thrombosis results from atherosclerosis in intracranial arteries (eg, middle meningeal artery), whereas extracranial embolism arises in extracranial arteries (eg, carotid arteries) or from the myocardium (cardioembolic).1 Intracerebral hemorrhage (ICH) is caused by weakened cerebral vessels that rupture and form hematomas.1 Diagnosis uses the National Institute of Health Stroke Scale (NIHSS), which quantifies clinical neurologic deficits and imaging techniques (computed tomography, magnetic resonance imaging) for detecting early infarction, localization, the degree of the infarct, and the vascular distribution.1 To date, no useful biomarker is available that provides additional information for the clinical diagnosis.1 An ideal biomarker has good diagnostic specify and sensitivity, is able to differentiate between hemorrhagic and ischemic stroke, increases early, is stable after the infarction, and has a known time frame for clearance. Furthermore, the biomarker has the potential to identify patients at high risk for further events and helps guide them to the right therapy. Finally, quantification should be fast, easy, and cost‐effective.1
A potential biomarker in this context could be a protein that is induced or regulated by oxidative stress. Oxidative stress is present within the first hours after stroke. Furthermore, LOX‐1 as a receptor of oxLDL is expressed in atherosclerotic carotid arteries.5, 71, 72 Therefore, sLOX‐1 might be generated from an increased membrane‐bound form of LOX‐1 and could be increased in stroke. A first cross‐sectional study by Yokota et al analyzed 377 patients of whom 250 had ischemic stroke and 127 had ICH. Control patients were age‐ and sex‐matched without a history of cardiovascular disease. Levels of sLOX‐1 were higher in those with ischemic stroke (526 versus 486 ng/L in controls) and ICH (720 versus 513 ng/L in controls). Differentiation of ischemic stroke revealed increased sLOX‐1 in atherothrombotic stroke (641 versus 496 ng/L controls). Cardioembolic stroke and lacunar infarction did not show any differences compared with controls. After adjusting for age, sex, and hypertension, ischemic stroke was independently associated with high levels of sLOX‐1.27 These findings support the hypothesis that increases in sLOX‐1 are caused by the number of atherosclerotic plaques and are not a consequence of ischemic stroke. However, the study by Yokota et al lacks statistical power (eg, for discrimination between different types of ischemic stroke) and might reflect variations in the small groups of patients.27 In addition, the time frame of blood sampling, which was 3 days after admission to the hospital, might be too late and underestimate sLOX‐1 peak levels.27 Huang and colleagues screened 148 patients with acute ischemic stroke with the subtype of large‐artery atherosclerosis. Concentrations of sLOX‐1 were elevated compared with corresponding controls (2.48±0.93 versus 2.22±0.79 ng/mL), and sLOX‐1 was an independent predictor of patients’ outcomes.73 Similar results were demonstrated by Li et al after screening 272 patients with ischemic stroke, in whom sLOX‐1 was elevated with severe stroke compared with mild stroke. The follow‐up revealed poor outcomes in patients with high sLOX‐1.74 However, this study has several limitations, such as small sample size, single‐center study design, and lack of data from different stages of ischemic stroke. In addition, the follow‐up did not account for rehabilitation, which might affect the results.74 In addition, the long sampling time (up to 72 hours) might have missed sLOX‐1 peak values.
Recently, Skarpengland et al analyzed plasma sLOX‐1 in patients with moderate (50–69%) and severe (≥70%) carotid artery stenosis. Depending on preoperative symptoms, patients were classified into ischemic stroke or transient ischemic attack for <7 days (n=35), >7 days to ≤3 months (n=90), >3 months (n=40), or no reported symptoms (asymptomatic, n=67). The authors demonstrated that patients with carotid artery atherosclerosis had higher sLOX‐1 than age‐matched controls (0.67 versus 0.99 ng/mL). However, sLOX‐1 was not able to discriminate the different subgroups of patients and did not differ between transient ischemic attack and ischemic stroke. The authors concluded that the increase in sLOX‐1 might arise from the chronic atherosclerotic process in carotid artery plaques and is not related to the severity.28 Further limitations are the lack of longitudinal data and the missing association between sLOX‐1 and the prediction of future ischemic events. In addition, the time frame between the onset of symptoms and blood sampling is a critical issue28 because it is important to determine sLOX‐1 peak values, half‐life time, and clearance from the circulation. In 2010, Inoue et al published the first large cohort study in the Suita study group including the follow‐up of 2437 participants. In their study, sLOX‐1 and LAB (LOX‐1 ligand containing apolipoprotein B) were measured. By multiplying both parameters, a LOX‐1 index was calculated. The LOX‐1 index in the top quartile was associated with stroke (hazard ratio: 1.74) and coronary heart disease (hazard ratio: 2.09). This association was present after adjusting for sex, age, body mass index, drinking, smoking, hypertension, type 2 diabetes mellitus, non‐HDL cholesterol, and the use of lipid‐lowering drugs. The same interesting findings were seen for ischemic stroke. Compared with the bottom quartile of LOX index, the fully adjusted hazard ratios for ischemic stroke were constantly high from the second to the top quartile: 3.39 (95% CI 1.34–8.53), 3.15 (1.22–8.13) and 3.23 (1.24–8.37), respectively. This suggests a positive relationship between sLOX‐1 and LAB and ischemic stroke. Limitations of this study are the relatively small number of analyzed patients. The number of cardiovascular events did not allow a sex‐specific analysis.42
Markstad et al measured sLOX‐1 prospectively in a large cohort (n=4703) in the Malmö Diet Cancer study. Patients were analyzed for sLOX‐1 on the first hospitalization for acute ischemic stroke and followed‐up for 16.5±3.6 years. The authors showed an increase in sLOX‐1 with age and significant associations with CRP, hemoglobin A1c, total cholesterol, LDL and HDL cholesterol, and triglycerides.22 In particular, the correlation with CRP points toward a role for sLOX‐1 in proinflammatory states.22 This finding is interesting compared with ACS, in which sLOX‐1 was not always correlated with cardiovascular risk factors.25 A major finding of Markstad et al revealed that patients with higher baseline sLOX‐1 had higher incidence of ischemic stroke compared with those in the lowest tertile. This effect remained present after adjusting for age, sex, current smoking, diabetes mellitus, waist circumference, systolic blood pressure, LDL cholesterol, triglycerides, and CRP.22 In the second part of their study, a separate cohort that underwent carotid endarterectomy due to the presence of symptoms (stroke, amaurosis fugax, transient ischemic attack) with >70% stenosis of the internal carotid artery or in patients with >80% stenosis were analyzed. Patients in the highest sLOX‐1 tertile with additional carotid artery stenosis had a 2.4‐fold higher risk of further ischemic events. This effect remained significant after adjusting for risk factors.22 The authors concluded that sLOX‐1 might become clinically important in patients with established atherosclerosis in carotid arteries.22 Although this study is the first in a large population with long‐term follow‐up, no patients with established disease were monitored.22 This information would help establish sLOX‐1 as a prognostic marker for prediction of further ischemic events in the diseased population.
In summary, increases in sLOX‐1 have been shown in ischemic stroke and ICH. Overall, sLOX‐1 was not able to discriminate between the different types of ischemic stroke in any of these studies and did not reflect the duration of the symptoms. However, most of the studies in ischemic stroke due to atherosclerosis revealed an increase with the severity of disease. A recently published, promising study in patients with carotid artery atherosclerosis found an association of sLOX‐1 with higher incidence of ischemic stroke. Therefore, sLOX‐1 might be, as shown in CAD, a predictor of plaque stability and vulnerability and a useful parameter in patients with asymptomatic carotid artery stenosis or stroke to predict future events (Figure 3). It is most likely that sLOX‐1 arises from atherosclerosis plaques in stenotic arteries and is not a consequence of ischemic stroke. To further strengthen these relationships, blood collection at standardized time points in asymptomatic patients and in stroke patients are necessary.
Figure 3.
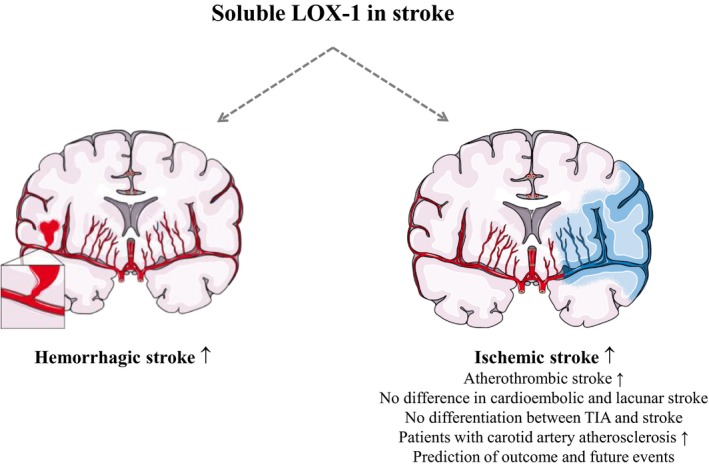
Role of sLOX‐1 (soluble lectin‐like oxidized low‐density lipoprotein receptor 1) in stroke. sLOX‐1 concentrations are elevated in hemorrhagic and ischemic stroke. Differentiation into different subtypes revealed elevations in atherothrombic stroke, but no differentiation between cardioembolic and lacunar stroke was possible. In ischemic stroke, sLOX‐1 was not able to discriminate between patients with transient ischemic attack (TIA) or carotid artery atherosclerosis. However, patients with higher sLOX‐1 levels have an increased risk of stroke. Figure adapted from SMART—Servier Medical Art by Servier (https://smart.servier.com).
Can sLOX‐1 Discriminate Between Patients With AAD or MI?
Acute aortic dissection (AAD) is defined by the disruption of the tunica intima of the vessel wall and provokes entry of blood, intramural bleeding, and the subsequent separation of aortic wall layers. In most cases, an intima tear is the underlying cause that attracts blood cells into the tunica media and leads to formation of a false lumen. Aortic rupture or reentry of blood into the aorta by a second tear are potential consequences. The false lumen thrombus attracts inflammatory cells that promote apoptosis and necrosis of vascular smooth muscle cells, which further worsens vessel wall degeneration.30 Several studies have demonstrated a role for LOX‐1 in induction of apoptosis in smooth muscle cells.16, 32, 33 In addition, ADAM‐17, MMP‐2, and MMP‐921, 49, 50 are involved in the shedding of sLOX‐1, and MMPs are key players in the degeneration of extracellular matrix in aortic diseases.75 Stanford type A aortic dissections affect the ascending thoracic aorta and/or aortic arch. Type B dissections are located within the descending thoracic aorta distal the subclavian artery and/or in the aortic abdominal part.75, 76 Observational studies revealed that hypertension is the most common comorbidity in AAD. Smoking, chronic renal insufficiency, chronic obstructive pulmonary disease, stroke, and transient ischemic attack were also observed.30 Diagnosing AAD includes computed tomography or magnetic resonance imaging and, in some cases, transesophageal echocardiography. Computed tomography and magnetic resonance imaging have the highest sensitivity and specificity.30 Currently, no biomarker is available that helps diagnose AAD. The most studied marker is D‐dimer with a low sensitivity and specificity to detect AAD.30
Myocardial ischemia is present in 10% to 15% of patients with AAD, and 10% show signs of myocardial ischemia on ECG. Besides ECG changes, 25% of patients with type A dissections have elevated TnT levels that may mislead the diagnosis toward MI and thereby delay the diagnosis and management of AAD.29 On this basis, Kobayashi and colleagues measured sLOX‐1 in patients with type A and B AAD (n=19) and with NSTEMI (n=39). Patients without significant coronary artery stenosis who underwent CAG served as controls (n=35). The authors found that sLOX‐1 was significantly elevated in AAD and NSTEMI, and levels in AAD were significantly higher than in NSTEMI. In contrast, TnT was elevated in NSTEMI only and did not show any differences between controls and AAD.41 Overall, sLOX‐1 clearly separated patients with AAD from controls. Effects on separation of AAD and NSTEMI were comparable to TnT.41 Limitations of this study were the rather low number of patients41 and the discussed “add‐on” to D‐dimer that was not analyzed in this study. In summary, sLOX‐1 could be an interesting biomarker to support the diagnosis of AAD; however, further studies including more patients and comparisons to imaging techniques are required.
Conclusions
This review summarized findings on sLOX‐1 in CAD, MI, stroke, and AAD. The majority of studies analyzed patients with stable CAD and ACS in whom sLOX‐1 peaked earlier than markers of cardiomyocyte injury. Consequently, sLOX‐1 might be a marker for plaque stability and vulnerability. A combination of sLOX‐1 and novel imaging techniques for evaluating plaque morphology and fibrous cap thickness may provide novel opportunities for assessing plaque vulnerability in high‐risk patients with stable CAD or recurrent MI. Elevations in sLOX‐1 were found in ischemic stroke and ICH, but no discrimination between the different subtypes and the duration of stroke symptoms was possible. Yet missing time courses of sLOX‐1 levels in patients with previous stroke and their follow‐up for future events are necessary. In addition, time courses of sLOX‐1 release during onset of symptoms, half‐life time, and clearance must be evaluated. In aortic disease, sLOX‐1 may also support discrimination between patients with AAD and NSTEMI because its levels were higher in AAD and its sensitivity and specify to detect AAD were comparable to TnT. However, analyses in larger cohorts of patients are required, and comparisons to standard imaging techniques are necessary.
In summary, sLOX‐1 could be a useful marker for assessing plaque stability and vulnerability in CAD and carotid artery stenosis.
Sources of Funding
This study has been supported by the Else Kröner‐Fresenius‐Stiftung (210_A105), the Doktor Robert Pfleger Stiftung, Bamberg, Germany (adiposity and endothelial function; LOX‐1, aldosterone, mineralocorticoid receptors, and vascular function) to Dr Morawietz, the European Section of the Aldosterone Council Deutschland (aldosterone, LOX‐1, and adipose tissue to Dr Hofmann and Dr Morawietz), the Deutsche Forschungsgemeinschaft (MO 1695/5‐1), the Excellence Initiative by the German Federal State Governments (Institutional Strategy, measure “support the best,” grant 3‐2, F03661‐553‐41B‐1250000) to Dr Morawietz, the MeDDrive grant 2019 to Dr Hofmann, and Open Access Funding by the Publication Fund of the TU Dresden.
Disclosures
None.
J Am Heart Assoc. 2020;9:e013803 DOI: 10.1161/JAHA.119.013803.
References
- 1. Saenger AK, Christenson RH. Stroke biomarkers: progress and challenges for diagnosis, prognosis, differentiation, and treatment. Clin Chem. 2010;56:21–33. [DOI] [PubMed] [Google Scholar]
- 2. Negre‐Salvayre A, Auge N, Camare C, Bacchetti T, Ferretti G, Salvayre R. Dual signaling evoked by oxidized LDLs in vascular cells. Free Radic Biol Med. 2017;106:118–133. [DOI] [PubMed] [Google Scholar]
- 3. Heusch G, Libby P, Gersh B, Yellon D, Bohm M, Lopaschuk G, Opie L. Cardiovascular remodelling in coronary artery disease and heart failure. Lancet. 2014;383:1933–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sawamura T, Kume N, Aoyama T, Moriwaki H, Hoshikawa H, Aiba Y, Tanaka T, Miwa S, Katsura Y, Kita T, Masaki T. An endothelial receptor for oxidized low‐density lipoprotein. Nature. 1997;386:73–77. [DOI] [PubMed] [Google Scholar]
- 5. Kataoka H, Kume N, Miyamoto S, Minami M, Moriwaki H, Murase T, Sawamura T, Masaki T, Hashimoto N, Kita T. Expression of lectinlike oxidized low‐density lipoprotein receptor‐1 in human atherosclerotic lesions. Circulation. 1999;99:3110–3117. [DOI] [PubMed] [Google Scholar]
- 6. Hofmann A, Brunssen C, Morawietz H. Contribution of lectin‐like oxidized low‐density lipoprotein receptor‐1 and LOX‐1 modulating compounds to vascular diseases. Vascul Pharmacol. 2018;107:1–11. [DOI] [PubMed] [Google Scholar]
- 7. Morawietz H. LOX‐1 and atherosclerosis: proof of concept in LOX‐1‐knockout mice. Circ Res. 2007;100:1534–1536. [DOI] [PubMed] [Google Scholar]
- 8. Catar RA, Muller G, Heidler J, Schmitz G, Bornstein SR, Morawietz H. Low‐density lipoproteins induce the renin‐angiotensin system and their receptors in human endothelial cells. Horm Metab Res. 2007;39:801–805. [DOI] [PubMed] [Google Scholar]
- 9. Ogura S, Kakino A, Sato Y, Fujita Y, Iwamoto S, Otsui K, Yoshimoto R, Sawamura T. Lox‐1: the multifunctional receptor underlying cardiovascular dysfunction. Circ J. 2009;73:1993–1999. [DOI] [PubMed] [Google Scholar]
- 10. Pothineni NVK, Karathanasis SK, Ding Z, Arulandu A, Varughese KI, Mehta JL. LOX‐1 in atherosclerosis and myocardial ischemia: biology, genetics, and modulation. J Am Coll Cardiol. 2017;69:2759–2768. [DOI] [PubMed] [Google Scholar]
- 11. Sugimoto K, Ishibashi T, Sawamura T, Inoue N, Kamioka M, Uekita H, Ohkawara H, Sakamoto T, Sakamoto N, Okamoto Y, Takuwa Y, Kakino A, Fujita Y, Tanaka T, Teramoto T, Maruyama Y, Takeishi Y. LOX‐1‐MT1‐MMP axis is crucial for RhoA and Rac1 activation induced by oxidized low‐density lipoprotein in endothelial cells. Cardiovasc Res. 2009;84:127–136. [DOI] [PubMed] [Google Scholar]
- 12. Hu C, Dandapat A, Sun L, Chen J, Marwali MR, Romeo F, Sawamura T, Mehta JL. LOX‐1 deletion decreases collagen accumulation in atherosclerotic plaque in low‐density lipoprotein receptor knockout mice fed a high‐cholesterol diet. Cardiovasc Res. 2008;79:287–293. [DOI] [PubMed] [Google Scholar]
- 13. Li D, Liu L, Chen H, Sawamura T, Ranganathan S, Mehta JL. LOX‐1 mediates oxidized low‐density lipoprotein‐induced expression of matrix metalloproteinases in human coronary artery endothelial cells. Circulation. 2003;107:612–617. [DOI] [PubMed] [Google Scholar]
- 14. Li D, Mehta JL. Antisense to LOX‐1 inhibits oxidized LDL‐mediated upregulation of monocyte chemoattractant protein‐1 and monocyte adhesion to human coronary artery endothelial cells. Circulation. 2000;101:2889–2895. [DOI] [PubMed] [Google Scholar]
- 15. Xu S, Ogura S, Chen J, Little PJ, Moss J, Liu P. LOX‐1 in atherosclerosis: biological functions and pharmacological modifiers. Cell Mol Life Sci. 2013;70:2859–2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lu J, Mitra S, Wang X, Khaidakov M, Mehta JL. Oxidative stress and lectin‐like ox‐LDL‐receptor LOX‐1 in atherogenesis and tumorigenesis. Antioxid Redox Signal. 2011;15:2301–2333. [DOI] [PubMed] [Google Scholar]
- 17. Brinkley TE, Kume N, Mitsuoka H, Phares DA, Hagberg JM. Elevated soluble lectin‐like oxidized LDL receptor‐1 (sLOX‐1) levels in obese postmenopausal women. Obesity (Silver Spring). 2008;16:1454–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nomata Y, Kume N, Sasai H, Katayama Y, Nakata Y, Okura T, Tanaka K. Weight reduction can decrease circulating soluble lectin‐like oxidized low‐density lipoprotein receptor‐1 levels in overweight middle‐aged men. Metabolism. 2009;58:1209–1214. [DOI] [PubMed] [Google Scholar]
- 19. Wakabayashi I, Shimomura T, Nakanishi M, Uchida K. Elevation of circulating LOX‐1 ligand levels in Zucker obese and diabetic rats. Obes Res Clin Pract. 2015;9:26–30. [DOI] [PubMed] [Google Scholar]
- 20. Murase T, Kume N, Kataoka H, Minami M, Sawamura T, Masaki T, Kita T. Identification of soluble forms of lectin‐like oxidized LDL receptor‐1. Arterioscler Thromb Vasc Biol. 2000;20:715–720. [DOI] [PubMed] [Google Scholar]
- 21. Gioia M, Vindigni G, Testa B, Raniolo S, Fasciglione GF, Coletta M, Biocca S. Membrane cholesterol modulates LOX‐1 shedding in endothelial cells. PLoS One. 2015;10:e0141270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Markstad H, Edsfeldt A, Yao Mattison I, Bengtsson E, Singh P, Cavalera M, Asciutto G, Bjorkbacka H, Fredrikson GN, Dias N, Volkov P, Orho‐Melander M, Nilsson J, Engstrom G, Goncalves I. High levels of soluble lectinlike oxidized low‐density lipoprotein receptor‐1 are associated with carotid plaque inflammation and increased risk of ischemic stroke. J Am Heart Assoc. 2019;8:e009874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hayashida K, Kume N, Murase T, Minami M, Nakagawa D, Inada T, Tanaka M, Ueda A, Kominami G, Kambara H, Kimura T, Kita T. Serum soluble lectin‐like oxidized low‐density lipoprotein receptor‐1 levels are elevated in acute coronary syndrome: a novel marker for early diagnosis. Circulation. 2005;112:812–818. [DOI] [PubMed] [Google Scholar]
- 24. Pirillo A, Catapano AL. Soluble lectin‐like oxidized low density lipoprotein receptor‐1 as a biochemical marker for atherosclerosis‐related diseases. Dis Markers. 2013;35:413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kume N, Mitsuoka H, Hayashida K, Tanaka M, Kominami G, Kita T. Soluble lectin‐like oxidized LDL receptor‐1 (sLOX‐1) as a sensitive and specific biomarker for acute coronary syndrome–comparison with other biomarkers. J Cardiol. 2010;56:159–165. [DOI] [PubMed] [Google Scholar]
- 26. Zhao ZW, Zhu XL, Luo YK, Lin CG, Chen LL. Circulating soluble lectin‐like oxidized low‐density lipoprotein receptor‐1 levels are associated with angiographic coronary lesion complexity in patients with coronary artery disease. Clin Cardiol. 2011;34:172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yokota C, Sawamura T, Watanabe M, Kokubo Y, Fujita Y, Kakino A, Nakai M, Toyoda K, Miyamoto Y, Minematsu K. High levels of soluble lectinlike oxidized low‐density lipoprotein receptor‐1 in acute stroke: an age‐ and sex‐matched cross‐sectional study. J Atheroscler Thromb. 2016;23:1222–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Skarpengland T, Skjelland M, Kong XY, Skagen K, Holm S, Otterdal K, Dahl CP, Krohg‐Sorensen K, Sagen EL, Bjerkeli V, Aamodt AH, Abbas A, Gregersen I, Aukrust P, Halvorsen B, Dahl TB. Increased levels of lectin‐like oxidized low‐density lipoprotein receptor‐1 in ischemic stroke and transient ischemic attack. J Am Heart Assoc. 2018;7:e006479 DOI: 10.1161/JAHA.117.006479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Erbel R, Aboyans V, Boileau C, Bossone E, Bartolomeo RD, Eggebrecht H, Evangelista A, Falk V, Frank H, Gaemperli O, Grabenwoger M, Haverich A, Iung B, Manolis AJ, Meijboom F, Nienaber CA, Roffi M, Rousseau H, Sechtem U, Sirnes PA, Allmen RS, Vrints CJ; ESC Committee for Practice Guidelines . 2014 ESC guidelines on the diagnosis and treatment of aortic diseases: document covering acute and chronic aortic diseases of the thoracic and abdominal aorta of the adult. The Task Force for the Diagnosis and Treatment of Aortic Diseases of the European Society of Cardiology (ESC). Eur Heart J. 2014;35:2873–2926. [DOI] [PubMed] [Google Scholar]
- 30. Mussa FF, Horton JD, Moridzadeh R, Nicholson J, Trimarchi S, Eagle KA. Acute aortic dissection and intramural hematoma: a systematic review. JAMA. 2016;316:754–763. [DOI] [PubMed] [Google Scholar]
- 31. Hofmann A, Brunssen C, Poitz DM, Langbein H, Strasser RH, Henle T, Ravens U, Morawietz H. Lectin‐like oxidized low‐density lipoprotein receptor‐1 promotes endothelial dysfunction in LDL receptor knockout background. Atheroscler Suppl. 2017;30:294–302. [DOI] [PubMed] [Google Scholar]
- 32. Ding Z, Wang X, Khaidakov M, Liu S, Mehta JL. MicroRNA hsa‐let‐7g targets lectin‐like oxidized low‐density lipoprotein receptor‐1 expression and inhibits apoptosis in human smooth muscle cells. Exp Biol Med (Maywood). 2012;237:1093–1100. [DOI] [PubMed] [Google Scholar]
- 33. Ding Z, Wang X, Schnackenberg L, Khaidakov M, Liu S, Singla S, Dai Y, Mehta JL. Regulation of autophagy and apoptosis in response to ox‐LDL in vascular smooth muscle cells, and the modulatory effects of the microRNA hsa‐let‐7g. Int J Cardiol. 2013;168:1378–1385. [DOI] [PubMed] [Google Scholar]
- 34. Lubrano V, Del Turco S, Nicolini G, Di Cecco P, Basta G. Circulating levels of lectin‐like oxidized low‐density lipoprotein receptor‐1 are associated with inflammatory markers. Lipids. 2008;43:945–950. [DOI] [PubMed] [Google Scholar]
- 35. Balin M, Celik A, Kobat MA. Circulating soluble lectin‐like oxidized low‐density lipoprotein receptor‐1 levels are associated with proximal/middle segment of the LAD lesions in patients with stable coronary artery disease. Clin Res Cardiol. 2012;101:247–253. [DOI] [PubMed] [Google Scholar]
- 36. Kobayashi N, Takano M, Hata N, Kume N, Yamamoto M, Yokoyama S, Shinada T, Tomita K, Shirakabe A, Otsuka T, Seino Y, Mizuno K. Soluble lectin‐like oxidized LDL receptor‐1 (sLOX‐1) as a valuable diagnostic marker for rupture of thin‐cap fibroatheroma: verification by optical coherence tomography. Int J Cardiol. 2013;168:3217–3223. [DOI] [PubMed] [Google Scholar]
- 37. Kobayashi N, Hata N, Kume N, Shinada T, Tomita K, Shirakabe A, Kitamura M, Nozaki A, Inami T, Seino Y, Mizuno K. Soluble lectin‐like oxidized LDL receptor‐1 and high‐sensitivity troponin T as diagnostic biomarkers for acute coronary syndrome. Improved values with combination usage in emergency rooms. Circ J. 2011;75:2862–2871. [DOI] [PubMed] [Google Scholar]
- 38. Balin M, Celik A, Kobat MA, Baydas A. Circulating soluble lectin‐like oxidized low‐density lipoprotein receptor‐1 levels predict percutaneous coronary intervention‐related periprocedural myocardial infarction in stable patients undergoing elective native single‐vessel PCI. J Thromb Thrombolysis. 2012;34:483–490. [DOI] [PubMed] [Google Scholar]
- 39. Kobayashi N, Hata N, Kume N, Seino Y, Inami T, Yokoyama S, Shinada T, Tomita K, Kaneshige T, Mizuno K. Soluble lectin‐like oxidized low‐density lipoprotein receptor‐1 as an early biomarker for ST elevation myocardial infarction: time‐dependent comparison with other biomarkers: time‐dependent comparison with other biomarkers. Circ J. 2011;75:1433–1439. [DOI] [PubMed] [Google Scholar]
- 40. Kume N, Mitsuoka H, Hayashida K, Tanaka M, Kita T. Soluble lectin‐like oxidized low‐density lipoprotein receptor‐1 predicts prognosis after acute coronary syndrome—a pilot study. Circ J. 2010;74:1399–1404. [DOI] [PubMed] [Google Scholar]
- 41. Kobayashi N, Hata N, Kume N, Yokoyama S, Takano M, Shinada T, Tomita K, Shirakabe A, Inami T, Seino Y, Mizuno K. Detection of acute aortic dissection by extremely high soluble lectin‐like oxidized LDL receptor‐1 (sLOX‐1) and low troponin T levels in blood. Int J Cardiol. 2013;165:557–559. [DOI] [PubMed] [Google Scholar]
- 42. Inoue N, Okamura T, Kokubo Y, Fujita Y, Sato Y, Nakanishi M, Yanagida K, Kakino A, Iwamoto S, Watanabe M, Ogura S, Otsui K, Matsuda H, Uchida K, Yoshimoto R, Sawamura T. LOX index, a novel predictive biochemical marker for coronary heart disease and stroke. Clin Chem. 2010;56:550–558. [DOI] [PubMed] [Google Scholar]
- 43. Aoyama T, Sawamura T, Furutani Y, Matsuoka R, Yoshida MC, Fujiwara H, Masaki T. Structure and chromosomal assignment of the human lectin‐like oxidized low‐density‐lipoprotein receptor‐1 (LOX‐1) gene. Biochem J. 1999;339(Pt 1):177–184. [PMC free article] [PubMed] [Google Scholar]
- 44. Biocca S, Arcangeli T, Tagliaferri E, Testa B, Vindigni G, Oteri F, Giorgi A, Iacovelli F, Novelli G, Desideri A, Falconi M. Simulative and experimental investigation on the cleavage site that generates the soluble human LOX‐1. Arch Biochem Biophys. 2013;540:9–18. [DOI] [PubMed] [Google Scholar]
- 45. Fukui M, Tanaka M, Senmaru T, Nakanishi M, Mukai J, Ohki M, Asano M, Yamazaki M, Hasegawa G, Nakamura N. LOX‐1 is a novel marker for peripheral artery disease in patients with type 2 diabetes. Metabolism. 2013;62:935–938. [DOI] [PubMed] [Google Scholar]
- 46. Rasouli N, Yao‐Borengasser A, Varma V, Spencer HJ, McGehee RE Jr, Peterson CA, Mehta JL, Kern PA. Association of scavenger receptors in adipose tissue with insulin resistance in nondiabetic humans. Arterioscler Thromb Vasc Biol. 2009;29:1328–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Takanabe‐Mori R, Ono K, Sowa N, Wada H, Takaya T, Horie T, Satoh‐Asahara N, Shimatsu A, Fujita M, Sawamura T, Hasegawa K. Lectin‐like oxidized low‐density lipoprotein receptor‐1 is required for the adipose tissue expression of proinflammatory cytokines in high‐fat diet‐induced obese mice. Biochem Biophys Res Commun. 2010;398:576–580. [DOI] [PubMed] [Google Scholar]
- 48. Weber S, Saftig P. Ectodomain shedding and ADAMs in development. Development. 2012;139:3693–3709. [DOI] [PubMed] [Google Scholar]
- 49. Mitsuoka H, Kume N, Hayashida K, Inui‐Hayashiada A, Aramaki Y, Toyohara M, Jinnai T, Nishi E, Kita T. Interleukin 18 stimulates release of soluble lectin‐like oxidized LDL receptor‐1 (sLOX‐1). Atherosclerosis. 2009;202:176–182. [DOI] [PubMed] [Google Scholar]
- 50. Zhao XQ, Zhang MW, Wang F, Zhao YX, Li JJ, Wang XP, Bu PL, Yang JM, Liu XL, Zhang MX, Gao F, Zhang C, Zhang Y. CRP enhances soluble LOX‐1 release from macrophages by activating TNF‐alpha converting enzyme. J Lipid Res. 2011;52:923–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nakamura M, Ohta H, Kume N, Hayashida K, Tanaka M, Mitsuoka H, Kaneshige T, Misaki S, Imagawa K, Shimosako K, Ogawa N, Kita T, Kominami G. Generation of monoclonal antibodies against a soluble form of lectin‐like oxidized low‐density lipoprotein receptor‐1 and development of a sensitive chemiluminescent enzyme immunoassay. J Pharm Biomed Anal. 2010;51:158–163. [DOI] [PubMed] [Google Scholar]
- 52. Brinkley TE, Wang X, Kume N, Mitsuoka H, Nicklas BJ. Caloric restriction, aerobic exercise training and soluble lectin‐like oxidized LDL receptor‐1 levels in overweight and obese post‐menopausal women. Int J Obes (Lond). 2011;35:793–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Civelek S, Kutnu M, Uzun H, Erdenen F, Altunoglu E, Andican G, Seven A, Sahin AO, Burcak G. Soluble lectin‐like oxidized LDL receptor 1 as a possible mediator of endothelial dysfunction in patients with metabolic syndrome. J Clin Lab Anal. 2015;29:184–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tan KC, Shiu SW, Wong Y, Leng L, Bucala R. Soluble lectin‐like oxidized low density lipoprotein receptor‐1 in type 2 diabetes mellitus. J Lipid Res. 2008;49:1438–1444. [DOI] [PubMed] [Google Scholar]
- 55. Morawietz H, Erbs S, Holtz J, Schubert A, Krekler M, Goettsch W, Kuss O, Adams V, Lenk K, Mohr FW, Schuler G, Hambrecht R. Endothelial protection, AT1 blockade and cholesterol‐dependent oxidative stress: the EPAS trial. Circulation. 2006;114:I296–I301. [DOI] [PubMed] [Google Scholar]
- 56. Morawietz H, Rueckschloss U, Niemann B, Duerrschmidt N, Galle J, Hakim K, Zerkowski HR, Sawamura T, Holtz J. Angiotensin II induces LOX‐1, the human endothelial receptor for oxidized low‐density lipoprotein. Circulation. 1999;100:899–902. [DOI] [PubMed] [Google Scholar]
- 57. Chen J, Li D, Schaefer R, Mehta JL. Cross‐talk between dyslipidemia and renin‐angiotensin system and the role of LOX‐1 and MAPK in atherogenesis studies with the combined use of rosuvastatin and candesartan. Atherosclerosis. 2006;184:295–301. [DOI] [PubMed] [Google Scholar]
- 58. Kang BY, Wang W, Palade P, Sharma SG, Mehta JL. Cardiac hypertrophy during hypercholesterolemia and its amelioration with rosuvastatin and amlodipine. J Cardiovasc Pharmacol. 2009;54:327–334. [DOI] [PubMed] [Google Scholar]
- 59. Hofnagel O, Luechtenborg B, Eschert H, Weissen‐Plenz G, Severs NJ, Robenek H. Pravastatin inhibits expression of lectin‐like oxidized low‐density lipoprotein receptor‐1 (LOX‐1) in Watanabe heritable hyperlipidemic rabbits: a new pleiotropic effect of statins. Arterioscler Thromb Vasc Biol. 2006;26:604–610. [DOI] [PubMed] [Google Scholar]
- 60. Thomas MR, Lip GY. Novel risk markers and risk assessments for cardiovascular disease. Circ Res. 2017;120:133–149. [DOI] [PubMed] [Google Scholar]
- 61. Jaffe AS, Babuin L, Apple FS. Biomarkers in acute cardiac disease: the present and the future. J Am Coll Cardiol. 2006;48:1–11. [DOI] [PubMed] [Google Scholar]
- 62. van der Linden N, Wildi K, Twerenbold R, Pickering JW, Than M, Cullen L, Greenslade J, Parsonage W, Nestelberger T, Boeddinghaus J, Badertscher P, Rubini Gimenez M, Klinkenberg LJJ, Bekers O, Schoni A, Keller DI, Sabti Z, Puelacher C, Cupa J, Schumacher L, Kozhuharov N, Grimm K, Shrestha S, Flores D, Freese M, Stelzig C, Strebel I, Miro O, Rentsch K, Morawiec B, Kawecki D, Kloos W, Lohrmann J, Richards AM, Troughton R, Pemberton C, Osswald S, van Dieijen‐Visser MP, Mingels AM, Reichlin T, Meex SJR, Mueller C. Combining high‐sensitivity cardiac troponin I and cardiac troponin T in the early diagnosis of acute myocardial infarction. Circulation. 2018;138:989–999. [DOI] [PubMed] [Google Scholar]
- 63. Balfour PC Jr, Gonzalez JA, Kramer CM. Non‐invasive assessment of low‐ and intermediate‐risk patients with chest pain. Trends Cardiovasc Med. 2017;27:182–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Task Force M , Montalescot G, Sechtem U, Achenbach S, Andreotti F, Arden C, Budaj A, Bugiardini R, Crea F, Cuisset T, Di Mario C, Ferreira JR, Gersh BJ, Gitt AK, Hulot JS, Marx N, Opie LH, Pfisterer M, Prescott E, Ruschitzka F, Sabate M, Senior R, Taggart DP, van der Wall EE, Vrints CJ, Guidelines ESCCfP , Zamorano JL, Achenbach S, Baumgartner H, Bax JJ, Bueno H, Dean V, Deaton C, Erol C, Fagard R, Ferrari R, Hasdai D, Hoes AW, Kirchhof P, Knuuti J, Kolh P, Lancellotti P, Linhart A, Nihoyannopoulos P, Piepoli MF, Ponikowski P, Sirnes PA, Tamargo JL, Tendera M, Torbicki A, Wijns W, Windecker S; Document R , Knuuti J, Valgimigli M, Bueno H, Claeys MJ, Donner‐Banzhoff N, Erol C, Frank H, Funck‐Brentano C, Gaemperli O, Gonzalez‐Juanatey JR, Hamilos M, Hasdai D, Husted S, James SK, Kervinen K, Kolh P, Kristensen SD, Lancellotti P, Maggioni AP, Piepoli MF, Pries AR, Romeo F, Ryden L, Simoons ML, Sirnes PA, Steg PG, Timmis A, Wijns W, Windecker S, Yildirir A, Zamorano JL. 2013 ESC guidelines on the management of stable coronary artery disease: the Task Force on the management of stable coronary artery disease of the European Society of Cardiology. Eur Heart J. 2013;34:2949–3003. [DOI] [PubMed] [Google Scholar]
- 65. Kamezaki F, Yamashita K, Tasaki H, Kume N, Mitsuoka H, Kita T, Adachi T, Otsuji Y. Serum soluble lectin‐like oxidized low‐density lipoprotein receptor‐1 correlates with oxidative stress markers in stable coronary artery disease. Int J Cardiol. 2009;134:285–287. [DOI] [PubMed] [Google Scholar]
- 66. Thygesen K, Alpert JS, Jaffe AS, Chaitman BR, Bax JJ, Morrow DA, White HD; Executive Group on behalf of the Joint European Society of Cardiology/American College of Cardiology/American Heart Association/World Heart Federation Task Force for the Universal Definition of Myocardial Infarction I . Fourth universal definition of myocardial infarction (2018). Circulation. 2018;138:e618–e651. [DOI] [PubMed] [Google Scholar]
- 67. Kumar A, Cannon CP. Acute coronary syndromes: diagnosis and management, part I. Mayo Clin Proc. 2009;84:917–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Stone GW. Revascularization decisions in coronary artery disease: hitting a moving target. JACC Cardiovasc Interv. 2014;7:507–509. [DOI] [PubMed] [Google Scholar]
- 69. Keaney JF Jr. Circulating biomarkers in acute coronary syndromes: something different or more of the same? Circulation. 2005;112:778–780. [DOI] [PubMed] [Google Scholar]
- 70. Park DW, Kim YH, Yun SC, Ahn JM, Lee JY, Kim WJ, Kang SJ, Lee SW, Lee CW, Park SW, Park SJ. Frequency, causes, predictors, and clinical significance of peri‐procedural myocardial infarction following percutaneous coronary intervention. Eur Heart J. 2013;34:1662–1669. [DOI] [PubMed] [Google Scholar]
- 71. Akhmedov A, Camici GG, Reiner MF, Bonetti NR, Costantino S, Holy EW, Spescha RD, Stivala S, Schaub Clerigue A, Speer T, Breitenstein A, Manz J, Lohmann C, Paneni F, Beer JH, Luscher TF. Endothelial LOX‐1 activation differentially regulates arterial thrombus formation depending on oxLDL levels: role of the Oct‐1/SIRT1 and ERK1/2 pathways. Cardiovasc Res. 2017;113:498–507. [DOI] [PubMed] [Google Scholar]
- 72. Li DY, Chen HJ, Staples ED, Ozaki K, Annex B, Singh BK, Vermani R, Mehta JL. Oxidized low‐density lipoprotein receptor LOX‐1 and apoptosis in human atherosclerotic lesions. J Cardiovasc Pharmacol Ther. 2002;7:147–153. [DOI] [PubMed] [Google Scholar]
- 73. Huang W, Li Q, Chen X, Lin Y, Xue J, Cai Z, Zhang W, Wang H, Jin K, Shao B. Soluble lectin‐like oxidized low‐density lipoprotein receptor‐1 as a novel biomarker for large‐artery atherosclerotic stroke. Int J Neurosci. 2017;127:881–886. [DOI] [PubMed] [Google Scholar]
- 74. Li XM, Jin PP, Xue J, Chen J, Chen QF, Luan XQ, Zhang ZR, Yu TE, Cai ZY, Zhao K, Shao B. Role of sLOX‐1 in intracranial artery stenosis and in predicting long‐term prognosis of acute ischemic stroke. Brain Behav. 2018;8:e00879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Cifani N, Proietta M, Tritapepe L, Di Gioia C, Ferri L, Taurino M, Del Porto F. Stanford‐A acute aortic dissection, inflammation, and metalloproteinases: a review. Ann Med. 2015;47:441–446. [DOI] [PubMed] [Google Scholar]
- 76. Gawinecka J, Schonrath F, von Eckardstein A. Acute aortic dissection: pathogenesis, risk factors and diagnosis. Swiss Med Wkly. 2017;147:w14489. [DOI] [PubMed] [Google Scholar]