

Abstract
Objective
Cytochrome b561 (CYB561) generates ascorbic acid, a cofactor in the enzymatic conversion of dopamine to norepinephrine by dopamine β-hydroxylase. We propose that the clinical relevance of this pathway can be revealed by characterizing the autonomic and biochemical characteristics of patients with CYB561 mutations.
Methods
We performed autonomic evaluations in 4 patients with lifelong orthostatic hypotension in whom CYB561 mutations were determined by genomic sequencing.
Results
Patients had disabling lifelong orthostatic hypotension (OH) and impaired blood pressure response to the Valsalva maneuver (VM), with exaggerated hypotension during phase 2 and lack of overshoot during phase 4. Heart rate ratios for sinus arrhythmia and the VM were normal. Plasma norepinephrine and metabolites were undetectable, and plasma dopamine and metabolites were normal. Droxidopa restored norepinephrine levels and improved OH. Patients 1 and 2 were sisters and homozygous for a nonsense mutation in exon 2, c.131G>A, p.Trp44 (Circ Res 2018). Their brother (patient 3) died at age 16 and his DNA was not available. Patient 4 was compound heterozygous; one allele had a missense mutation in exon 2, c157C>T, p.His.53Tyr, and the other had an exon 2 deletion.
Conclusion
CYB561 deficiency is characterized by selective sympathetic noradrenergic failure with lifelong, disabling OH but with normal sympathetic cholinergic (sweating) and parasympathetic (heart rate regulation) functions. We report a novel case of CYB561 deficiency due to an exon 2 deletion in one allele and a missense mutation in the other. These patients highlight the critical role CYB561 plays in sympathetic function and cardiovascular regulation.
Norepinephrine is a critical neurotransmitter in the CNS and in efferent sympathetic noradrenergic pathways that regulate cardiovascular function. We previously described patients lacking norepinephrine due to congenital deficiency of dopamine β-hydroxylase (DBH), the enzyme responsible for its synthesis.1–3 Recently we reported 2 families with congenital deficiency of cytochrome b561 (CYB561),4 the enzyme that generates ascorbic acid, a cofactor for DBH enzyme function. Here we report a novel genetic mutation causing CYB561 deficiency, and provide a detailed description of the clinical autonomic and biochemical characteristics of this disorder.
Methods
Autonomic evaluation
Patients were evaluated in Vanderbilt Autonomic Dysfunction Center (VADC), on a sodium-controlled diet and off medications. Autonomic evaluation included Valsalva maneuvers, sinus arrhythmia during deep breathing, orthostatic challenge, and determination of catecholamines and their metabolites using high-performance liquid chromatography with electrochemical detection.
Genetic testing
In patients 1 and 2, exonic and flanking intronic sequences of the CYB561 gene (NM_001915.3) were amplified by PCR and sequenced by the Sanger method as previously reported.4 In patient 4, genomic DNA was enriched for the coding exons using hybrid capture technology and prepared DNA libraries were sequenced using next-generation sequencing technology (Fulgent Diagnostics, Temple City, CA). Following alignment, variants were detected manually and interpreted using locus-specific databases. Variants were confirmed by Sanger sequencing.
Data availability
Data not provided in the article will be shared by request from any qualified investigator.
Standard protocol approvals, registrations, and patient consents
All data collections were approved by Vanderbilt's institutional review board and written informed consent was obtained from participants.
Results
We describe 4 patients with CYB561 deficiency. Two sisters (patients 1 and 2, Caucasian non-Hispanic) were initially evaluated by one of the authors (M.B.G.)5 and at VADC at ages 38 and 39. Their brother (patient 3) was evaluated by one of the authors (A.W.R.) at age 13; formal autonomic or genetic evaluations were not available, but his clinical picture is consistent with the diagnosis. Patient 4 is an individual of Indian origin initially evaluated by one of the authors (C.J.M.) and at VADC at age 31.
All 4 patients had normal birth and mental development. Patients 1, 2, and 3 had episodes of hypoglycemia severe enough that a partial pancreatectomy was performed in patient 2. All 4 patients had profound orthostatic hypotension. Ptosis was mild or questionable. Patient 4 had retrograde ejaculation. Patients 1, 2, and 3 were treated for seizures in early childhood, but it is not clear if these episodes corresponded to documented epilepsy, hypoglycemic seizures, or the tonic-clonic movements that occur during hypotensive syncope.
All patients had profound orthostatic hypotension and impaired blood pressure response to the Valsalva maneuver, with exaggerated hypotension during phase 2 and absence of overshoot during phase 4 (table 1). On the other hand, they had intact heart rate responses to posture, sinus arrhythmia, and the hypotensive phase of the Valsalva maneuver. Cardiovagal responses to neck suction and phenylephrine were intact. Thermoregulatory testing showed normal sweating.
Table 1.
Autonomic evaluation

Plasma DBH activity was normal in patients 1 and 2, indicating intact noradrenergic fibers because plasma DBH originates from vesicular DBH released during sympathetic activation. Plasma levels of norepinephrine and its intraneuronal metabolic 3,4-dihydroxyphenylglycol (DHPG) were below limits of detection (figure). Plasma dopamine and its metabolites were comparable to normal participants, and much lower than in DBH deficiency.
Figure. Catecholamine biosynthesis and plasma levels in normal controls and patients with congenital deficiency of cytochrome b561 (CYB561) and dopamine β-hydroxylase (DBH) deficiencies.
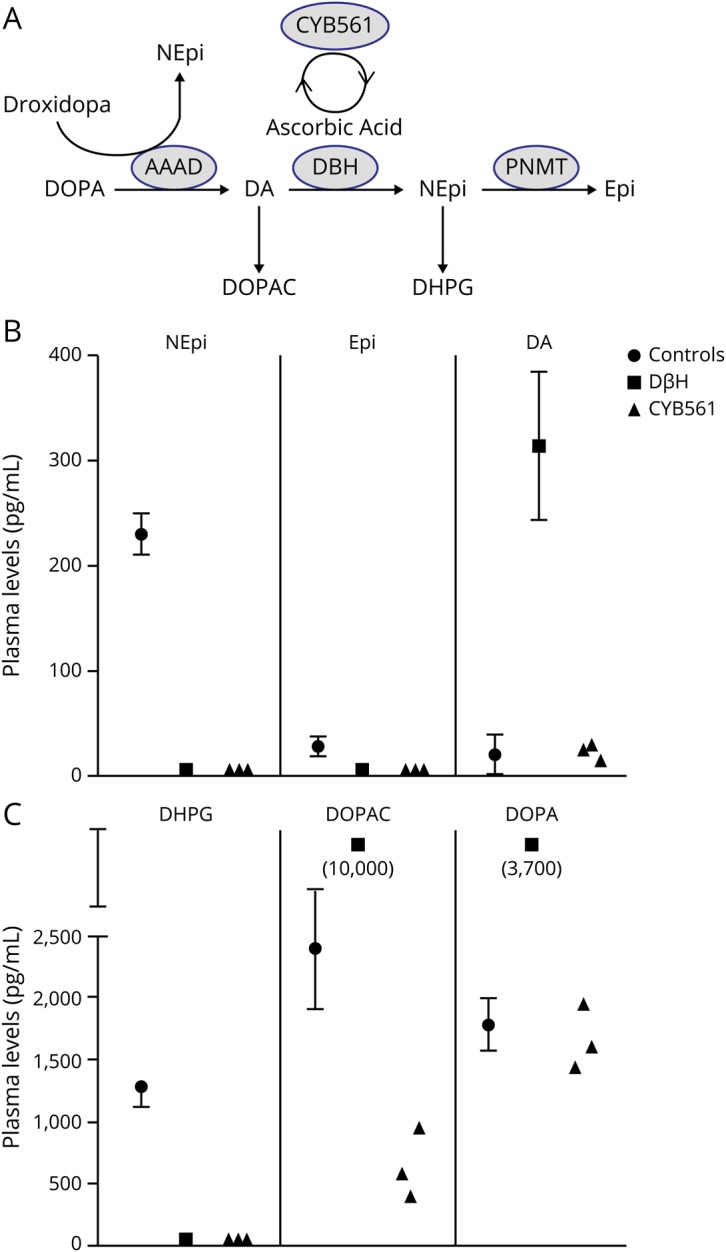
(A) Catecholamine biosynthesis with the conversion of DOPA to dopamine (DA) through l-aromatic amino acid decarboxylase (AAAD), to norepinephrine (NEpi) through DBH, to epinephrine (Epi) through phenylethanolamine N-methyltransferase (PNMT), and the intraneuronal metabolites of dopamine (3,4-dihydroxyphenylacetic acid [DOPAC]) and norepinephrine (3,4-dihydroxyphenylglycol [DHPG]). Cytochrome 561 generates ascorbic acid, a cofactor in the function of DBH. NEpi can also be produced therapeutically from droxidopa via AAAD, bypassing the enzymatic defect. (B) Plasma levels of NEpi, Epi, and DA in normal controls, patients with DBH deficiency, and patients 1, 2, and 4 with CYB561 deficiency. (C) Plasma levels of the intraneuronal metabolites DHPG and DOPAC and of DOPA.
Droxidopa 100 mg three times a day improved orthostatic hypotension (table 1) and increased plasma norepinephrine from undetectable to 65 and 139 pg/mL in patients 1 and 2. Plasma DHPG, the intraneuronal metabolite of norepinephrine, also increased from undetectable to 771 and 1,220 pg/mL, indicating restoration of catecholamine synthesis in noradrenergic fibers. Doses >100 mg TID were not tolerated because of excessive pressor responses (patients 1 and 2) or nausea (patient 4).
CYB561 genetic mutations of these patients are summarized in table 2. Variants of patients 1 and 2 were reported recently.4 Patient 4, not reported previously, had compound heterozygous mutations of the CYB561 gene. One allele had a deletion of exon 2; the other allele had a c.157C>T variant not previously reported in the Human Gene Mutation Database. This is a missense mutation (p.His53Tyr) in a highly conserved site, predictive of impaired protein function. Variants were identified on separate chromosomes (in trans), indicating biparental inheritance.
Table 2.
Known CYB561 mutations

Patients 1 and 2 were lost to follow-up and died at ages 59 and 48 and patient 3 died at age 16. The causes of death were not ascertained. Patient 4 remains stable and recently fathered a healthy child. His wife has a normal CYB561 gene sequence.
Discussion
We report a novel genetic defect causing CYB561 deficiency characterized by lifelong selective sympathetic failure with disabling orthostatic hypotension, but intact sympathetic cholinergic (sweating) and parasympathetic (normal autonomic control of heart rate) functions. Hypoglycemia in infancy was prominent. This is also seen in DBH deficiency and thought to be explained by the underlying autonomic abnormalities, because of the loss of α2-adrenergic receptor-mediated inhibition, and conservation of parasympathetic-mediated stimulation of insulin secretion.6 These episodes did not persist through childhood, perhaps due to adaptation of other counterregulatory mechanisms or the development of insulin resistance we have documented in DBH deficiency.6
Both DBH and CYB561 deficiency have undetectable plasma norepinephrine and DHPG (figure). However, dopamine does not accumulate in CYB561 deficiency as it does in DBH deficiency. We speculate that the presence of ascorbic acid within noradrenergic vesicles provides antioxidant capacity, and its absence in CYB561 deficiency leads to degradation of DOPA and dopamine, preventing their accumulation.4 The clinical and autonomic characteristics are otherwise similar in both conditions. Renal impairment is seen in DBH deficiency and it has been proposed to be due at least partially to dopamine excess. However, patients with CYB561 deficiency also appear to develop impaired renal function, even though they do not have dopamine accumulation.4
These cases of congenital CYB561 deficiency reveal the critical importance of this protein in norepinephrine synthesis and autonomic function. Previous studies have suggested associations between CYB561 polymorphisms and cardiovascular regulation. The rs2058203 variant is associated with lower resting plasma norepinephrine,7 and the rs3087776 variant is associated with lower heart rate responses to the cold pressor test.8
Droxidopa is converted to norepinephrine by DOPA-decarboxylase (figure), bypassing the enzymatic defect.9,10 Accordingly, droxidopa restored norepinephrine levels and improved orthostatic hypotension in these patients. Restoration of ascorbic acid with oral or parenteral administration is not feasible because ascorbic acid is not taken up by catecholaminergic vesicles.
We report a novel case of CYB561 deficiency due to compound heterozygosity, with an exon deletion in one allele, and a missense mutation in the other. The specific genetic defect is diverse among cases (table 2), but the clinical phenotype is similar, and characterized by selective sympathetic noradrenergic failure causing lifelong disabling orthostatic hypotension, intact parasympathetic function, low to undetectable plasma norepinephrine, and normal plasma dopamine. These patients highlight the critical role CYB561 plays in norepinephrine synthesis, sympathetic function, and cardiovascular regulation, previously unrecognized.
Glossary
- CYB561
cytochrome b561
- DBH
dopamine β-hydroxylase
- DHPG
3,4-dihydroxyphenylglycol
- VADC
Vanderbilt Autonomic Dysfunction Center
Appendix. Authors
Study funding
The study was supported by Vanderbilt CTSA grant 1UL1 RR000445 from NCATS/NIH and NIH U54 NS065736.
Disclosure
C. Shibao received grant support from the Office of Orphan Products Development, Food and Drug Administration, grant FD-R-04778-01-A3. C.A.S. received speaker honorarium from Lundbeck Pharmaceuticals. E. Garland, B. Black, C. Mathias, M. Grant, A. Root, and D. Robertson report no disclosures relevant to the manuscript. I. Biaggioni received consulting honoraria from Lundbeck and Theravance Biopharma. I.B. received grant support from Office of Orphan Products Development, Food and Drug Administration, grant FD-R-04778-01-A3. Go to Neurology.org/N for full disclosures.
References
- 1.Robertson D, Goldberg MR, Onrot J, et al. Isolated failure of autonomic noradrenergic neurotransmission: evidence for impaired beta-hydroxylation of dopamine. N Engl J Med 1986;314:1494–1497. [DOI] [PubMed] [Google Scholar]
- 2.Man in't Veld AJ, Boomsma F, Moleman P, Schalekamp MADH. Congenital dopamine-beta-hydroxylase deficiency: a novel orthostatic syndrome. Lancet 1987;1:183–187. [DOI] [PubMed] [Google Scholar]
- 3.Mathias CJ, Bannister RB, Cortelli P, et al. Clinical, autonomic and therapeutic observations in two siblings with postural hypotension and sympathetic failure due to an inability to synthesize noradrenaline from dopamine because of a deficiency of dopamine beta hydroxylase. QJM 1990;75:617–633. [PubMed] [Google Scholar]
- 4.van den Berg MP, Almomani R, Biaggioni I, et al. Mutations in CYB561 causing a novel orthostatic hypotension syndrome. Circ Res 2018;122:846–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Misbin RI, Grant MB, Pecker MS, Atlas SA. Elevated levels of plasma prorenin (inactive renin) in diabetic and nondiabetic patients with autonomic dysfunction. J Clin Endocrinol Metab 1987;64:964–968. [DOI] [PubMed] [Google Scholar]
- 6.Arnold AC, Garland EM, Celedonio JE, et al. Hyperinsulinemia and insulin resistance in dopamine beta-hydroxylase deficiency. J Clin Endocrinol Metab 2017;102:10–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ghimire LV, Kohli U, Li C, et al. Catecholamine pathway gene variation is associated with norepinephrine and epinephrine concentrations at rest and after exercise. Pharmacogenet Genomics 2012;22:254–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang K, Deacon DC, Rao F, et al. Human heart rate: heritability of resting and stress values in twin pairs, and influence of genetic variation in the adrenergic pathway at a microribonucleic acid (microRNA) motif in the 3'-UTR of cytochrome b561 [corrected]. J Am Coll Cardiol 2014;63:358–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Biaggioni I, Robertson D. Endogenous restoration of noradrenaline by precursor therapy in dopamine-beta-hydroxylase deficiency. Lancet 1987;2:1170–1172. [DOI] [PubMed] [Google Scholar]
- 10.Man in 't Veld AJ, Boomsma F, van den Meiracker AH, Schalekamp MA. Effect of unnatural noradrenaline precursor on sympathetic control and orthostatic hypotension in dopamine-beta-hydroxylase deficiency. Lancet 1987;2:1172–1175. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data not provided in the article will be shared by request from any qualified investigator.