

A new vaccine immunostimulatory (adjuvant) system was developed based on fully defined polymers built from native amino acids.
Abstract
To be optimally effective, peptide-based vaccines need to be administered with adjuvants. Many currently available adjuvants are toxic, not biodegradable; they invariably invoke adverse reactions, including allergic responses and excessive inflammation. A nontoxic, biodegradable, biocompatible, self-adjuvanting vaccine delivery system is urgently needed. Herein, we report a potent vaccine delivery system fulfilling the above requirements. A peptide antigen was coupled with poly-hydrophobic amino acid sequences serving as self-adjuvanting moieties using solid-phase synthesis, to produce fully defined single molecular entities. Under aqueous conditions, these molecules self-assembled into distinct nanoparticles and chain-like aggregates. Following subcutaneous immunization in mice, these particles successfully induced opsonic epitope-specific antibodies without the need of external adjuvant. Mice immunized with entities bearing 15 leucine residues were able to clear bacterial load from target organs without triggering the release of soluble inflammatory mediators. Thus, we have developed a well-defined and effective self-adjuvanting delivery system for peptide antigens.
INTRODUCTION
Vaccines are one of the most powerful tools to combat infectious diseases. Although whole-pathogen and protein-based vaccines are able to provide efficient protection against pathogens, they are not always entirely safe and may induce undesirable immune responses. An obvious alternative are peptide-based vaccines because they can be designed to induce specific immune responses against selected epitopes (1, 2). For instance, the majority of anticancer vaccines in the late-phase clinical trials are composed of peptide antigens (3). All recent vaccines against group A Streptococcus (GAS) that have entered clinical trials are based on peptides derived from a major GAS virulence factor, M-protein, but do not contain the whole protein itself (4). GAS is responsible for a variety of mild infections and life-threatening autoimmune diseases such as rheumatic fever and rheumatic heart disease (5), which are estimated to kill 1.4 million people worldwide per year (6). M-protein is the major antigen responsible for triggering these autoimmune responses (7). Nevertheless, it is possible to identify M-protein–based peptide antigens that do not stimulate an autoimmune response and are conserved among the vast number of GAS serotypes. These antigens need to be coadministered with an adjuvant and/or an appropriate delivery system to induce an effective immune response. However, there is limited choice of safe and efficient adjuvants that can stimulate the desired immune response against a peptide epitope (8). In general, alum is the only adjuvant widely used for the formulation of commercial vaccines. A few other adjuvants have been approved for specific vaccine formulations (9). Unfortunately, alum is too weak an adjuvant to stimulate a strong immune response against peptides. Powerful adjuvants, such as the “gold standard” complete Freund’s adjuvant (CFA), are effective in triggering recognition of the peptide antigen by the immune system. However, they are often toxic and/or are poorly chemically defined fragments of bacteria or toxins and vary from batch to batch. Adjuvant typically includes mixtures of lipids, polysaccharides, polymers, or various microbial components. Vaccines with added Toll-like receptor (TLR) stimulators or inflammasome activators necessarily induce inflammation to stimulate immunity. These vaccines, therefore, intrinsically carry the potential of unwanted inflammatory side effects mediated largely by soluble mediators such as interleukin-1 (IL-1) or tumor necrosis factor (TNF). These can also potentially limit their heterologous (nonspecific) beneficial effects (10) and be associated with the downstream induction of immunosuppressive control mechanisms (11). Thus, a chemically defined single compound/adjuvant that can help stimulate an immune response, without triggering the release of soluble mediators of inflammation, especially against weakly immunogenic antigens, is in high demand.
As nanoparticles have been shown to have promising self-adjuvanting properties (12, 13), amphiphiles that self-assemble into them are often used in peptide vaccine delivery (14–16). For example, hydrophobic dendritic poly(tert-butyl acrylate) has previously been conjugated to a variety of peptide epitopes, including GAS-derived peptides, and the conjugates have been self-assembled to form particles (17). These particles induced strong humoral and cellular immune responses against the incorporated peptide antigen/s without any indication of adverse effects (17–19). However, these polymers had serious limitations for commercial application because they are not biodegradable and have undefined stereochemistry and number of units in each polymer. Although this is typical for classical polymers, the batch-to-batch variability affects in vitro and in vivo results and makes them unsuitable for clinical trial.
Herein, we propose a self-adjuvanting peptide-based vaccine delivery system designed to overcome these disadvantages. This system is based on fully defined and biodegradable polymers built from natural hydrophobic amino acids (HAAs). In contrast to the often laborious methods required for polymer production, especially in dendritic forms, poly-HAAs (pHAAs) were synthesized using the classical solid-phase peptide synthesis (SPPS) method. This method could be fully automated and allowed incorporation of the desired peptide epitopes into the molecule in the same procedure. The properties of the pHAA unit (e.g., water solubility, conformational behavior) can easily be altered by changing its length and amino acid composition. We selected three typical HAAs (valine, phenylalanine, and leucine) to examine whenever their polymeric arrangement conjugated to a peptide epitope could self-assemble into nanoparticles. A conserved B cell epitope derived from GAS M-protein (J8, QAEDKVKQSREAKKQVEKALKQLEDKVQ), which recently reached phase 1 clinical trials, and a universal T helper epitope (PADRE, AKFVAAWTLKAAA) were incorporated into the vaccine design and modified with pHAA (Fig. 1). The pHAAs were built from valine, phenylalanine, and leucine: typical representatives of HAAs. The resulting amphiphilic compounds were self-assembled into particles that induced the maturation of antigen-presenting cells (APCs) in vitro and the production of high antibody titers. Mechanistically, they were able to induce dendritic cell (DC) maturation without induction of potentially damaging soluble inflammatory mediators in a mouse model. These antibodies opsonized clinical GAS isolates in vitro and greatly reduced the bacterial burden in mice challenged with the M1 GAS strain.
Fig. 1. Schematic structures of compounds 1 to 5.
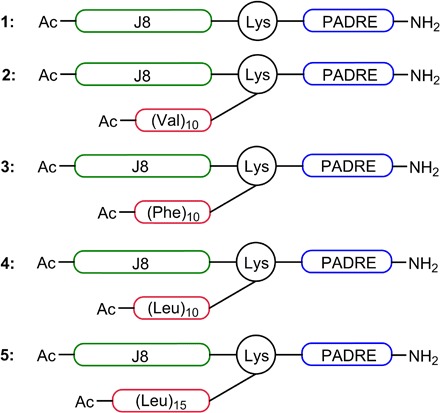
The vaccine candidates were constructed from three building blocks: the B cell epitope (J8), the T helper epitope (PADRE), and a poly-hydrophobic amino acid (pHAA) unit.
RESULTS
Synthesis of pHAA-peptide conjugates
The B cell epitope J8 and T helper PADRE were conjugated on resin using the standard Boc-SPPS method, with the lysine as a spacer and branching moiety for attachment of pHAA, to produce peptide 1 (Fig. 1). The peptide 1 was modified with 10 U of valine, 10 U of phenylalanine, 10 U of leucine, or 15 U of leucine to yield compounds 2, 3, 4, and 5, respectively. pHAAs of these lengths were expected to induce conjugate aggregation under aqueous conditions. All compounds were acetylated on their N terminus. An attempt to synthesize a compound that bore 15 valines was unsuccessful due to its high hydrophobicity, aggregative nature, and insolubility, consistent with the properties of previously reported poly-valine sequences (20). Similar problems were encountered during the synthesis and purification of a 15-phenylalanine derivative. Although the compound that bore 15 leucines precipitated when concentration was higher than 1.5 mg/ml, this was still practical for immunological evaluation.
Self-assembly and characterization of pHAA-peptide nanoparticles
Hydrophilic peptide epitope 1 upon conjugation with hydrophobic pHAA sequences forms conjugates (2 to 5). Therefore, 2 to 5 have amphiphilic properties and are able to self-assemble to form particles. Compounds (2 to 5) were self-assembled in phosphate-buffered saline (PBS) and examined by dynamic light scattering (DLS) and transmission electron microscopy (TEM). All compounds self-assembled into a mixture of small nanoparticles (10 to 30 nm) and larger aggregates with high polydispersity indexes according to DLS (table S1 and fig. S1) and TEM (Fig. 2, A to D). Distinct nanoparticles and chain-like aggregates of nanoparticles (CLAN) are visible in the TEM images (Fig. 2), especially for compound 5 (fig. S2).
Fig. 2. Physicochemical characterization of compounds 1 to 5.
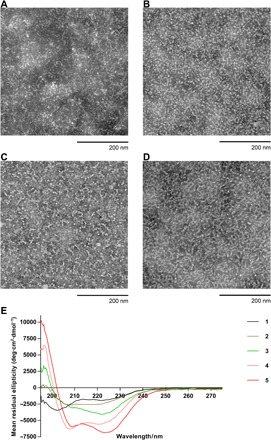
TEM photographs of the vaccine compounds (A) 2, (B) 3, (C) 4, and (D) 5 (scale bars, 200 nm). (E) Circular dichroism (CD) spectra of compounds 1 to 5.
Secondary structure analysis of the conjugates
The B cell epitope J8 was designed to adopt a helical conformation to trigger the production of antibodies that recognize native helical GAS M-protein. However, in aqueous solvents, without use of helicity inducing 2,2,2-trifluoroethanol, it adopted a random coil conformation rather than an α helix. Therefore, circular dichroism (CD) was used to test whether the J8-PADRE (1) epitope alone or after conjugation with pHAAs was able to adopt a helical conformation in PBS (Fig. 2E). As expected, the CD spectra of peptide 1 suggested the presence of a mixture of random coil (with typical minimum at 202 nm) and α helix (flat minimum at 222 nm). Only conjugate 5 adopted a typical helical conformation with a clear minimum at 223 nm and a lower-intensity minimum at 209 nm. Its analog bore a shorter leucine-repeat unit (4) and had a clear minimum at 207 nm and weaker at 221 nm. Compound 2 bore 10 valines and appeared as a mixture of an α helix and a β sheet with a flat minimum between 205 and 221 nm, which most likely have resulted from overlapping of α helix minimums at 208 and 222 nm, with a β sheet minimum at 215 nm. The tendency of valine-rich peptides to form β sheets is well documented (21). The CD spectrum of compound 3 displayed an unusual single minimum ellipticity at 222 nm, which was neither typical of an α helix nor a β sheet. However, this type of CD spectra has been observed for β sheet–rich proteins (22, 23).
Cytotoxicity of the conjugates 2 to 5
The vaccine delivery system/adjuvant was constructed entirely from natural amino acids; thus, the pHAA conjugates were not expected to be toxic. None of the compounds showed any cytotoxicity against SW620 (human colon carcinoma) and human embryonic kidney (HEK) 293 cell lines at the highest tested concentration (30 μM).
Ability of conjugates to stimulate the maturation of DCs, macrophages, and cellular responses
Maturation of APCs (CD11c + DCs and F4/80+ macrophages from murine splenocytes) was determined after their treatment with 2 to 5 by analyzing the expression of major histocompatibility complex II (MHC-II) and the CD40 costimulatory molecule in vitro (fig. S3). All conjugates induced higher expression of MHC-II compared with negative control, while only compound 5 induced significantly higher expression of CD40 in both DCs and macrophages (P = 0.011 and 0.0027, respectively). In the second experiment, long-term stimulation of macrophages and DCs was evaluated in vivo. Following two immunizations, spleens were collected 17 days after the final immunization. Maturation of APCs (CD11c + DCs and F4/80+ macrophages from murine splenocytes) was determined after their treatment with 5 and adjuvanted controls 1/AS04 [alum and monophosphoryl lipid A (MPLA) containing human-grade adjuvant]. Compound 5 stimulated significantly higher expression of CD40 and CD86 on DCs. 1/AS04 did not stimulate significant increases, albeit similar trends were observed. These makers were not significantly higher on macrophages compared with negative controls (PBS) by any adjuvant (fig. S4). Similar results were observed in inguinal lymph nodes, which were the lymph nodes draining the injection sites (data not shown). The endotoxin level in the vaccine formulation 5 was negligible (0.0044 EU/ml) and therefore could not influence the compound ability to stimulate DCs. In addition, to assess the immunogenicity of 5 in comparison to adjuvant control 1/AS04, we also measured cytokine-producing T cell responses from whole splenocytes by enzyme-linked immunosorbent spot (ELISpot; fig. S5). There were significant T helper 1 cell (TH1) responses observed in a number of animals after just two immunizations with the control adjuvant 1/AS04 recalled with 5 (none of six animals responding), 1 (one of six), or PADRE (three of six) with a similar pattern, albeit at lower overall levels, observed for compound 5 recalled with 5 (none of six), 1 (one of six), or PADRE (two of six). In terms of TH2 responses 1/AS04, there were only significant responses to 5 or 1 in one of six animals with a similar pattern for compound 5, with reactivity to 5 found in one of six animals, to 1 in one of six animals, and to PADRE in one of six animals. There was no TH17 reactivity induced by either 1/AS04 or compound 5 to any recall antigen in the culture. Together, these data show that compound 5 can induce early modest TH1 and TH2 reactivity comparable to 1/AS04.
Systemic and mucosal immune responses induced by compounds 2 to 5
C57BL/6 female mice received tail base subcutaneous immunization and three boosts with compounds 2 to 5 on days 0, 21, 28, and 35. All compounds elicited significant J8-specific immunoglobulin G (IgG) titers after the final immunization (Fig. 3A). Compound 5 induced significantly stronger responses than all other compounds, including 1 emulsified with CFA (P = 0.0219). While compound 5 induced significant antibody titers since the first immunization, one boost with compound 5 was not enough to induce production of antibody titters higher than that induced by oil-based adjuvants AS04 and CFA (fig. S6). However, two of the CFA-immunized mice died, illustrating the known concerns with toxicity associated with this gold standard adjuvant.
Fig. 3. J8-specific antibody titer and average opsonization percentage of different GAS strains.
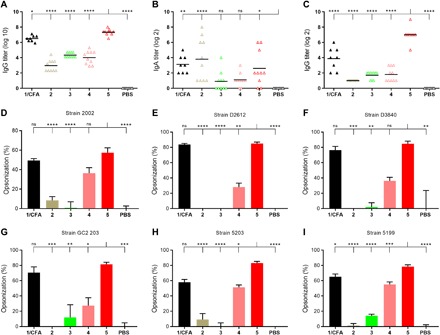
Experimental mice (C57BL/6, n = 10) were vaccinated subcutaneously (on days 0, 21, 28, and 35) with compounds 1 to 5. Each triangle in (A) to (C) represents an individual mouse; the mean J8-specific titers are presented as a bar. (A) Titers of J8-specific immunoglobulin G (IgG) in serum, (B) titers of J8-specific IgA in saliva, and (C) titers of J8-specific IgG in saliva, collected after final immunization. (D) Average percentage opsonization of GAS strain 2002, (E) D2612, (F) D3840, (G) GC2 203, (H) 5203, and (I) 5199. PBS: negative control group, mice immunized with PBS; 1/CFA: positive control group, compound 1 emulsified with CFA. Statistical analysis was performed using one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test compared with PBS indicated as ns, P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
The ability of compounds 1 to 5 to induce mucosal immune responses after subcutaneous immunization was also examined. Conjugates 2 and 5 and peptide 1 emulsified with CFA induced production of J8-specific IgA in mice (Fig. 3B). The IgA titers were relatively low, yet significantly higher than those observed in mice treated with PBS. Similarly, the salivary IgG titers induced by immunization with the conjugates (Fig. 3C) were very low (2), low (3 and 4), or moderate (1/CFA), with the exception of compound 5, which induced significantly higher (P < 0.0001) salivary IgG production than the positive control group (1/CFA).
The quality of these antibodies was tested in opsonization assays against a variety of GAS clinical isolates (Fig. 3, D to I). Sera collected from mice immunized with compounds that bore leucine-based pHAA units (4 and 5) and 1/CFA induced high levels of opsonization of all GAS strains when compared with sera derived from the mice treated with PBS. In particular, the sera collected from mice immunized with compound 5 demonstrated, in most cases, significantly better ability to kill bacteria than that from other groups (Fig. 3, D to I), including 1/CFA (Fig. 3I). Compound 4, which was less potent than 5, still generated better opsonic responses than 2 or 3.
Postimmunization intranasal challenge with M1 GAS
Following intranasal bacterial challenge, the number of bacteria was measured using nasal shedding, throat swabs, and the colonization of nasal-associated lymphoid tissue (NALT; a murine functional homolog to human tonsils) and the spleen (Fig. 4). Both nasal shedding and throat swabs were analyzed at days 1 to 3. In both mucosal areas, compound 5 demonstrated the highest efficacy for clearing GAS bacteria, which, by day 3, were practically undetectable in mice immunized with this conjugate (Fig. 4, A and B). The NALT and spleen tissue gave similar results to the mucosal surfaces: by day 3, 5 induced the strongest reduction of bacterial burden of any tested compound, including CFA-adjuvanted peptide 1.
Fig. 4. Bacterial burden after intranasal challenge with M1 GAS strain in C57BL/6 mice (n = 10 per group).
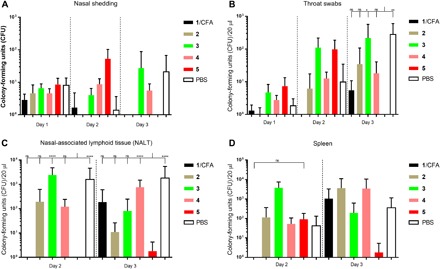
Bacterial burden results are represented as the mean CFU ± SEM for each group. (A) Nasal shedding, (B) throat swabs, (C) colonization of nasal-associated lymphoid tissue (NALT), and (D) colonization of spleen tissue. PBS: negative control group, mice immunized with PBS; 1/CFA: positive control group, compound 1 emulsified with CFA. On day 1, 10 mice were examined; on day 2, 5 mice were examined and euthanized for spleen and NALT organ harvesting; and the remaining 5 mice were processed on day 3. Statistical analysis was performed using a two-way ANOVA followed by Tukey’s post hoc test compared with PBS indicated as ns, P > 0.05; *P < 0.05; **P < 0.01; ****P < 0.0001.
Inflammatory responses
Activation of the inflammasome NLRP3 leads to autocatalytic caspase-1 activation, which, in turn, cleaves the pro-cytokines IL-1β and IL-18. Cleavage induces maturation of IL-1β and IL-18 into soluble and biologically active cytokines. Aside from lipopolysaccharide (LPS), silica are also well-known inflammasome activators (24). Previous studies have shown that prestimulation of LPS followed by silica triggered the maturation of NLRP3 inflammasome and, consequently, the cleavage of pro–IL-1β into active IL-1β in the immortalized bone marrow–derived macrophages (BMMs) (25). Prestimulating BMMs with LPS followed by silica stimulated the production of IL-1β (729 ± 509 pg/ml; Fig. 5A). However, prestimulation of LPS followed by compound 5 or 1 and either of the compounds alone did not trigger the release of IL-1β, further suggesting that compounds 5 and 1 do not induce the maturation and assembly of the inflammasome complex and are therefore noninflammatory.
Fig. 5. Cytokine production following stimulation with 5 and 1 with or without costimulation of LPS.
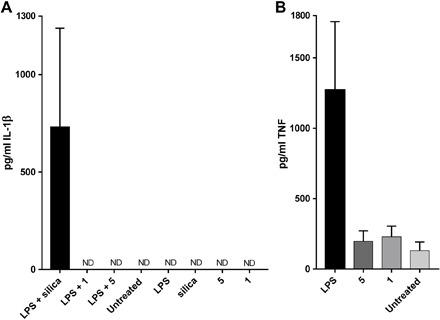
Cytokine production is expressed as pg/mL ± SD. (A) Production of IL-1β stimulated by LPS (0.1 μg/ml), silica (125 μg/ml), peptide 1 (0.1 mg/ml), and compound 5 (0.1 mg/ml). (B) Production of TNF stimulated by LPS (0.001 μg/ml), peptide 1 (0.1 mg/ml), and compound 5 (0.1 mg/ml). Positive control: cells stimulated with LPS alone or in combination with silica. Negative control: untreated cells. Cells were prestimulated with LPS for 3 hours followed by either 6 hours of stimulation with silica or 4 hours of stimulation with 5 and control peptide 1. ND, not detectable.
In addition, we analyzed the expression of TNF, a well-known inflammatory cytokine. When BMMs were stimulated with either LPS, 5, or control peptide 1, cells exhibited no differences in TNF production in comparison with untreated cells (Fig. 5B).
DISCUSSION
Adjuvants play a key role in modern vaccines (26). Despite some safety concerns, many currently available adjuvants are not fully defined and may include mixtures of lipids, polysaccharides, polymers, and various microbial components. Thus, a chemically defined single compound that can help stimulate an immune response against the antigen it carries would have clear benefits over many existing adjuvants. As nanoparticles have been shown to have promising self-adjuvanting properties (12), amphiphiles that self-assemble into nanoparticles are often used in peptide vaccine delivery (14, 15). Herein, we propose a universal peptide-based platform (pHAA), which upon conjugation to an antigen stimulates formation of nanoparticles. The properties of pHAA unit (e.g., water solubility and conformational) can easily be altered by changing its length and amino acid composition. We selected three HAAs (valine, phenylalanine, and leucine) to examine whether their polymeric arrangement conjugated to a peptide epitope could self-assemble into nanoparticles. GAS B cell epitope J8 conjugated to universal T helper epitope PADRE (1) was selected as a vaccine antigen and modified with 10 or 15 copies of HAA (Fig. 1). pHAAs of these lengths were expected to induce conjugate aggregation under aqueous conditions. Compounds that bore 15 repeats of phenylalanine and valine were poorly water soluble and were therefore discontinued from further studies. Although the compound that bore 15 leucines precipitated when concentration was higher than 1.5 mg/ml, this was still practical for immunological evaluation. Thus, the maximum length of pHAA sequence, which could be used to deliver peptide antigens, was essentially limited by the compounds’ aqueous solubility. Other compounds were easily water soluble; however, as they have amphiphilic properties due to the presence of highly hydrophobic pHAA units and hydrophilic peptide antigen (1), they promptly self-assembled under aqueous conditions. All of them (2 to 5) formed small nanoparticles and CLAN. Similar aggregates were previously reported for inorganic nanoparticles (27, 28). The aggregative potential of all compounds was generally similar with the exception of compound 5, which had a higher tendency to form CLAN (Fig. 2, A to D, table S1, and fig. S2). This can easily be explained by its more hydrophobic nature. The same compound adopted the most distinct secondary structure. A classical double-minimum α helix CD spectrum was observed for 5, while the spectra of its shorter analog 4 and peptide antigen 1 showed a shift of the helical curve toward random coil (Fig. 2E). In contrast, compounds 2 and 3 rather showed a tendency to form a β sheet. The antibodies induced against the J8 epitope need to recognize the parent helical M-protein; thus, the conformational properties of the vaccine have an important role in its antibacterial efficacy. It has been also recently reported that modification of p145 (LRRDLDASREAKKQVEKALE), the parent peptide of J8 that share same minimal B cell epitope (SREAKKQVEKAL), which stabilized helical folding, greatly induced its immunogenicity (29). Following immunization with 5, antibodies generated by mice showed the strongest ability to opsonize GAS clinical isolates in vitro (Fig. 4). At the same time, β sheet–rich compounds 2 and 3 appeared to induce production of nonprotective antibodies that were not able to kill GAS bacteria. Compound 5 also demonstrated excellent ability to protect mice in the M1 GAS challenge experiment (Fig. 5). Clearance of GAS bacteria was not observed in mice immunized with 4, which suggested that not only the opsonic ability but also the quantity of antibodies played an important role in protection. Compound 5 induced significantly higher J8-specific systemic and mucosal IgG titers than the other compounds. Relatively, low IgA titers were observed (as expected) after nonmucosal immunization, and therefore, the level of IgA production was not influential on the vaccine performance of the candidates. As antigen presentation and maturation of APCs are important for induction of effective humoral immunity, expression of MHC-II and CD40 was examined. MHC-II is an APC receptor required for peptide antigen presentation to T helper cells and, therefore, the induction of adaptive immunity, while the level of CD40 expression is typically used as a marker for DC maturation in mice. As expected, all compounds stimulated overexpression of both proteins. However, statistically significant overexpression of CD40 was observed only in mice treated with compound 5. When maturation was examined in vivo, a similar tendency was observed, even 17 days after stimulation of DCs, with compound 5 (fig. S4). Thus, compound 5 demonstrated the most promising properties among all examined pHAA-based immunogens. However, the mechanism of action (including DC maturation) for the adjuvanticity of nanoparticles, when these do not carry ligands for pattern recognition receptors, is still highly speculative (30, 31).
While the quality of the immune response elicited by compound 5 can be explained by its ability to adopt helical conformation due to the presence of a poly-leucine unit, the magnitude of response (e.g., high antibody titers) could not be explained by the conformation of the epitope and instead resulted from the higher hydrophobicity of the compound. When the same size and charge gold nanoparticles were examined, those modified with more hydrophobic groups induced stronger immune system activation (32). The hydrophobic properties and conformation can easily be modified by changing the type and number of amino acids in the pHAA unit, and therefore, the system can easily be customized for different epitopes. Moreover, in comparison to many other self-assembling systems, an external adjuvant was not needed to trigger a strong immune response (33).
Inflammation is one of the common side effects associated with the use of most vaccine adjuvants (30). For example, the commonly licensed adjuvant alum specifically activates the NLRP3 inflammasome, whereas other adjuvants in more restricted use, such as MPLA in Cervarix, activate inflammation via the TLR4 receptor. Undesirable side effects, such as toxicity, can be associated with the induction of excessive inflammatory responses (34). Moreover, inflammatory adjuvanted vaccines may lack the capacity to induce beneficial nonspecific systemic effects on immunity, which can be induced by some live vaccines (10, 35). Thus, to further analyze the safety of 5 and to gain mechanistic insight in the function of the nanoparticles, we examined the production of inflammatory cytokines associated with the presence of TLR or NLRP3 inflammasome activation. Inflammasomes are cytosolic molecular complexes involved in pathogen recognition and danger. The best characterized inflammasome is NLRP3, of which the assembly is associated with the production of IL-1β and IL-18 (36). Stimulation via TLR receptors, including the TLR4 receptor, activates the production of TNF (30), a well-known inflammatory cytokine. Under physiological conditions, during host invasion, the conserved structural elements of pathogens including LPS bind to TLRs and induce the secretion of TNF, which, in turn, exerts its antipathogen response by activating effector T cells (37, 38). Overall, TNF has a vast array of complex functions, of which some are not yet completely understood; recent studies have even showed that oligomerization of the NLRP3 inflammasome may be transcriptionally regulated by TNF (39).
When examined in vitro, no increased production of IL-1β and TNF was observed following BMM stimulation with compounds 5 and control 1 in comparison with untreated cells (Fig. 5). Cells exhibited a very high production of TNF when stimulated with LPS, indicating the readily available inflammatory responses of our model. These results suggest that the peptides are not triggering the NLRP3 complex assembly or the conventional TLR signaling pathways of these cells and, therefore, not inducing the production of inflammatory cytokines. These results support the contention that it is possible to induce high levels of immunity without the induction of the full spectrum of conventional inflammatory responses, initially described using model polystyrene nanoparticles (30), showing successful translation of this model concept into an approach with high translation potential.
The discovery of safe, biocompatible, and immunologically efficient adjuvants is a daunting challenge, with the vast majority of known adjuvants composed of microbiological components or their synthetic analogs. Here, we demonstrate that conjugating antigens to unnatural polymers constructed from natural HAAs leads to self-adjuvanting particles. This pHAA-based antigen delivery system can be fully customized, for example, to achieve desired aqueous solubility and particle formation characteristics of conjugates. This breakthrough opens the door for the discovery of safer, better, next-generation adjuvants that can lead to a revolution in the field of vaccine development.
MATERIALS AND METHODS
Reagents, cells, and equipment
All chemicals were used as received, without any purification. Butyloxycarbonyl (Boc)–protected l-amino acids were purchased from Novabiochem (Merck Chemicals, Darmstadt, Germany) and Mimotopes (Melbourne, Australia); methanol, trifluoroacetic acid (TFA), N,N′-diisopropylethylamine (DIPEA), N,N′-dimethylformamide, acetonitrile, and dichloromethane were purchased from Merck (Hohenbrunn, Germany); 4-methylbenzhydrylamine (pMBHA) resin was purchased from Peptides International (Kentucky, USA); 2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU) was purchased from Mimotopes (Melbourne, Australia); and p-cresol was obtained from Merck Millipore (Bayswater, Australia). AS04 adjuvant (MPLA-SM VacciGrade), complete Freund’s adjuvant, and goat anti-mouse IgG (H + L)–HRP (horseradish peroxidase) (IgG-HRP) conjugate were purchased from Millipore (Temecula, California, USA); goat anti-mouse IgA was obtained from InvivoGen (San Diego, USA), and analytical-grade Tween 20, (tris-hydroxymethyl)aminomethane, and glycine were acquired from VWR International (Queensland, Australia). Phenol-free Iscove’s Modified Dulbecco’s Medium (IMDM) Glutamax medium was purchased from Gibco (California, USA). Pierce Chromogenic Endotoxin Quant Kit, Streptex Latex Agglutination Test kit, and phenylmethylsulfonyl fluoride were purchased from Thermo Scientific (Victoria, Australia). Phycoerythrin (PE)/CY7 anti-mouse CD11C was purchased from eBioscience (California, USA), and BV421 anti-mouse MHC-II, fluorescein isothiocyanate (FITC) anti-mouse CD40, and BV605 anti-mouse F4/80 were purchased from BioLegend (California, USA). Fc-block was obtained from eBioscience (California, USA). Yeast extract was purchased from Merck Chemicals (Darmstadt, Germany). Todd-Hewitt broth (THB) was purchased from Oxoid (Thermo Fisher Scientific, South Australia, Australia), and horse blood was obtained from Serum Australis. C57BL/6 mice were purchased from the Animal Resources Centre (Western Australia, Australia). GAS strains 5448AP used in the GAS intranasal challenge were obtained from M. Walker (University Queensland). GAS strain ACM-2002 was obtained from Royal Brisbane and Women’s Hospital (human abscess–lymph gland), ACM-5199 [American Type Culture Collection (ATCC) 12344, National Center for Biotechnology Information (NCIB) 11841, scarlet fever], ACM-5203 (ATCC 19615, pharynx of child after a sore throat), GC2 203 (wound swab), D3840 (nasopharynx swabs), and D2612 (nasopharynx swabs). Enzyme-linked immunosorbent assay (ELISA) kits for TNF and IL-1b were purchased from Becton Dickinson and BioLegend, respectively. Silica (U.S. silica) and all other chemicals were purchased from Sigma-Aldrich (Victoria, Australia). Analytical reversed-phase high-performance liquid chromatography (RP-HPLC) was performed on a Shimadzu (Kyoto, Japan) instrument with 1 ml/min flow rate with compound detection at 214 nm. Preparative RP-HPLC was performed using Shimadzu (Kyoto, Japan) instrumentation (either LC-20AT, SIL-10A, CBM-20A, SPD-20AV, FRC-10A or LC-20AP ×2, CBM-20A, SPD-20A, FRC-10A) in linear gradient mode. The flow rate was 10 or 20 ml/min, and the compounds were detected at 230 nm. Separations were achieved with solvent A (100% H2O and 0.1% TFA) and solvent B (90% acetonitrile, 10% H2O, and 0.1% TFA) on a Vydac 214TP1022 preparative column (C4, 10 μm, 22 mm by 250 mm). A Perkin-Elmer-Sciex API3000 instrument with Analyst 1.4 software (Applied Biosystems/MDS Sciex, Toronto, Canada) was used for electrospray ionization mass spectrometry (ESI-MS). Particle size and morphology were analyzed by DLS using a Nanosizer (Zetasizer Nano Series ZS, Malvern Instruments, UK) with disposable capillary cuvettes using Dispersion Technology Software (Malvern Instruments, UK) and by TEM using a JEM-1010 microscope (JEOL Ltd., Japan). The secondary structure of compounds 1 to 5 was analyzed by a CD instrument (Jasco J710 spectropolarimeter, JASCO, Japan) with a 1-mm cell (Starna) at room temperature. The optical density (OD) of the endotoxin assay plate was measured using a Spectramax microplate reader (Molecular Devices, USA) at 405 nm. The absorbance of each well in cytotoxicity analysis was measured at 580 nm with a PowerWave XS Microplate Reader from Bio-Tek Instruments Inc. (Winooski, Vermont). An LSR II flow cytometry instrument (LSR II Flow cytometer, BD Biosciences, California, USA) was applied in the APC maturation study. An RS-VA 10 vortex mixer (Phoenix Instrument, Germany) was used to prepare vaccination solution. ELISA plates were analyzed in a Victor3 1420 multilabel counter (Perkin Elmer Life and Analytical Sciences, Shelton, USA) and a Multiskan Go (Thermo Scientific). ELISpot assay was performed with multiscreen 96-well plates (MSIPS4510, Millipore). Plates were coated with anti-mouse interferon-γ (IFN-γ; AN18, Mabtech), anti-mouse IL-4 (BVD4-1d11, BD Biosciences), or IL-17A (Mabtech), detected with anti-mouse IFN-γ–biotin (R4-6A2, Mabtech), anti-mouse IL-4–biotin (BVD6-24G2, Mabtech or BD Biosciences), or anti-mouse IL-17A–biotin (BD Biosciences) and streptavidin-alkaline phosphatase (ALP) or ExtrAvidin-ALP. Last, plates were developed with an AP colorimetric kit (Bio-Rad), and pots were counted using an AID ELISpot Reader system (AutoImmune Diagnostika, GmbH, Germany).
Synthesis and purification of pHAA-peptide conjugates
Peptides 1 to 5 (Fig. 1) were synthesized by microwave-assisted standard Boc-SPPS method. Briefly, deprotection of the Boc group was performed twice (1× 2 min and 1× 3 min) with neat TFA, and double coupling of amino acids was applied (1× 5 min and 1× 10 min, 20 W, 70°C). Amino acids were activated by DIPEA and HATU. Peptides were cleaved by anhydrous hydrogen fluoride with addition of p-cresol as a scavenger. Compounds 1 to 5 were purified by RP-HPLC.
Compound 1. Yield: 34%. Molecular weight: 4823.63. ESI-MS [M + 3H]3+ m/z 1608.6 (calc. 1608.9), [M + 4H]4+ m/z 1206.8 (calc. 1206.9), [M + 5H]5+ m/z 965.8 (calc. 965.7), [M + 6H]6+ m/z 804.9 (calc. 804.9), [M + 7H]7+ m/z 690.1 (calc. 690.1), [M + 8H]8+ m/z 603.9 (calc. 604.0), [M + 9H]9+ m/z 537.0 (calc. 537.0). tR = 19.1 min (0 to 100% solvent B; C4 column); purity ≥ 99%.
Compound 2. Yield: 23%. Molecular weight: 5814.96. ESI-MS [M + 3H]3+ m/z 1938.6 (calc. 1939.3), [M + 4H]4+ m/z 1455.0 (calc. 1454.7), [M + 5H]5+ m/z 1164.0 (calc. 1164.0), [M + 6H]6+ m/z 969.9 (calc. 970.1), [M + 7H]7+ m/z 831.5 (calc. 831.7), [M + 8H]8+ m/z 728.0 (calc. 727.9). tR = 24.1 min (0 to 100% solvent B; C4 column); purity ≥ 99%.
Compound 3. Yield: 28%. Molecular weight: 6295.40. ESI-MS [M + 4H]4+ m/z 1574.7 (calc. 1574.9), [M + 5H]5+ m/z 1260.0 (calc. 1260.0), [M + 6H]6+ m/z 1050.3 (calc. 1050.2), [M + 7H]7+ m/z 900.4 (calc. 900.3), [M + 8H]8+ m/z 788.0 (calc. 787.9), [M + 9H]9+ m/z 700.7 (calc. 700.5). tR = 24.5 min (0 to 100% solvent B; C4 column); purity ≥ 99%.
Compound 4. Yield: 28%. Molecular weight: 5955.23. ESI-MS [M + 3H]3+ m/z 1985.2 (calc. 1986.1), [M + 4H]4+ m/z 1489.6 (calc. 1489.8), [M + 5H]5+ m/z 1192.0 (calc. 1192.0), [M + 6H]6+ m/z 993.5 (calc. 993.5), [M + 7H]7+ m/z 851.8 (calc. 851.7), [M + 8H]8+ m/z 745.4 (calc. 745.4). tR = 25.6 min (0 to 100% solvent B; C4 column); purity ≥ 99%.
Compound 5. Yield: 26%. Molecular weight: 6521.03. ESI-MS [M + 4H]4+ m/z 1631.8 (calc. 1631.3), [M + 5H]5+ m/z 1305.6 (calc. 1305.2), [M + 6H]6+ m/z 1088.0 (calc. 1087.8), [M + 7H]7+ m/z 932.9 (calc. 932.6), [M + 8H]8+ m/z 816.3 (calc. 816.1), [M + 9H]9+ m/z 725.6 (calc. 725.6). tR = 30.9 min (0 to 100% solvent B; C4 column); purity ≥ 99%.
Particle size measurements
Compounds 2 to 5 were self-assembled in PBS to prepare solutions (0.5 mg/ml). Then, the average particle sizes (nanometer) of the nanostructures were measured by DLS at 25°C. Sizes were analyzed using a noninvasive backscatter method, and measurements were taken with a 173° scattering angle. Correlation times were based on 10 s per run, and at least 10 consecutive runs were made per measurement. Each measurement was repeated five times. The same compound solutions were also analyzed by TEM.
Secondary structure analyses
The secondary structure of compounds 1 to 5 (0.1 mg/ml in PBS) was analyzed by CD. Spectra were tested with the following parameters: 5-nm bandwidth; scan rate, 50 nm/min; response time, 2 s; and 1-nm intervals over the wavelength range of 195 to 260 nm in a nitrogen atmosphere. Reported data are the mean of six accumulations. Mean residue molar ellipticity (deg∙cm2∙dmol−1) was calculated using the formula [θ] = mdeg/(l × c × n) * 1000, where l is the path length (0.1 cm), c is the peptide concentration (millimolar), and n is the number of residues in the peptide.
Endotoxin assay
The assay was performed as per the manufacturer’s instruction. Low concentration of lyophilized endotoxin (0.01 to 0.1 EU/ml) was used for the preparation of the standard curve (average blank-corrected OD at 405 nm versus endotoxin concentration) to determine the endotoxin concentration in compound 5 (1 mg/ml in PBS).
Ethics statement
All animal protocols were approved by the Griffith University Animal Ethics Committee, GU ref. no. GLY/07/14 and The University of Queensland Animal Ethics Committee (AEC), AEC approval no. SCMB/AIBN/069/17. This study was carried out in accordance with the National Health and Medical Research Council (NHMRC) of Australia guidelines for generating, breeding, caring for, and using genetically modified and cloned animals for scientific purposes (2007). Methods were chosen to minimize pain and distress to the mice, which were observed daily by trained animal care staff. The mice were terminated using a CO2 inhalation chamber.
Cytotoxicity results study
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cytotoxicity assay was performed using SW620 and HEK293 adherent cell lines. SW620 and HEK293 were cultured in RPMI 1640 medium and Dulbecco’s modified Eagle’s medium (DMEM), respectively, as adherent monolayers in flasks supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, penicillin (100 U/ml), and streptomycin (100 μg/ml) in a humidified 37°C incubator supplied with 5% CO2. Cells were then harvested with trypsin and dispensed into 96-well microtiter assay plates at 2000 cells per well for the two cell lines and incubated for 18 hours at 37°C with 5% CO2 (to allow cells to attach). Compounds 2 to 5 were dissolved in 5% dimethyl sulfoxide (DMSO) in PBS (v/v), and aliquots (10 μl) were tested over a series of final concentrations ranging from 10 nM to 30 μM. Control wells were treated with 5% aqueous DMSO. After a 68-hour incubation at 37°C with 5% CO2, an aliquot (10 μl) of MTT in PBS (5 mg/ml) was added to each well (final concentration of 0.5 mg/ml), and the microtiter plates were incubated for a further 4 hours at 37°C with 5% CO2. After this final incubation, the medium was aspirated and precipitated formazan crystals were dissolved in DMSO (100 μl per well). The absorbance of each well was then measured. Vinblastine was used as a positive control (20 mg/ml in H2O). All experiments were performed in duplicate.
APC maturation evaluation
Single-cell spleen suspensions were harvested from naive C57BL/6 mice by physical disruption of their spleens and passing the disrupted spleens through stainless steel mesh. Red blood cell lysis buffer was used to lyse the erythrocytes. Then, a 96-well plate was filled with 2 × 105 cells per well in phenol-free IMDM Glutamax medium supplemented with 10% FBS, 50 mM 2-mercaptoethanol, penicillin (100 U/ml), and streptomycin (100 mg/ml). For each well of the plates, 10 μM of compounds 2 to 5 was added, and the plates were incubated at 37°C for 6 hours. Cells that adhered to the wells were scraped first, and then Fc-block was added. Following this, the plates were put in an incubator for 30 min at 4°C. The cells were centrifuged and resuspended in a buffer containing CD11c, F4/80, CD40, CD80, and MHC-II antibodies for 30 min at 4°C. After the incubation, the plates were centrifuged and washed again. The cells were then resuspended in 0.5 ml of fluorescence-activated cell sorting (FACS) buffer (PBS, 0.02% sodium azide, 0.5% bovine serum albumin). The mean fluorescence intensity for CD11C or F4/80 cells and activation markers CD40 and MHC-II was used to identify the maturation of DCs or macrophages.
Vaccination and bacterial infection challenge
C57BL/6 female mice received a primary immunization and three boosts with compounds 2 to 5 on days 0, 21, 28, and 35. The compounds 2 to 5/PBS solutions and compound 1/CFA emulsion (1:1 volume ratio) were prepared fresh before each immunization. Compounds 2 to 5 were dissolved in PBS directly and then vortexed for 2 min. Mice in each group (10 mice per group) were immunized subcutaneously at the base of their tails with 150 μg of compound in 50 to 100 μl of PBS.
All immunized and control mice were challenged intranasally with the GAS strain M1, with a predetermined dose on the 61st day after the primary immunization. The throat swab was obtained on days 1 to 3 after the mice were challenged with the GAS bacteria. All throat swabs were streaked on Columbia base agar plates containing 2% defibrinated horse blood incubated at 37°C overnight. The plates were stored in a 4°C room for later determination of GAS colonization. Nasal shedding was determined on days 1 to 3 after challenge by pressing the nares of each mouse onto the surface of the prepared Columbia blood agar (CBA) plates 10 times (triplicate CBA plates per mouse per day), and exhaled particles were streaked out. On days 2 and 3 after challenge, five mice from each group were euthanized to harvest the NALT and spleen. The NALT and spleen samples were homogenized in PBS first and then plated in serial dilutions on Columbia base agar plates containing 2% defibrinated horse blood (37°C, overnight) to assess bacterial burden.
In the second vaccination experiment, C57BL/6 female mice received a primary immunization and one boost with compound 5 and the controls on days 0 and 14. The compound 5 solutions (150 μg of compound 5 in 100 μl of PBS) and compound 1/AS04 emulsion (5 μg of MPLA-SM/5 μl of DMSO mixed with 150 μg of compound 1/45 μl of PBS according to the manufacturer MPLA-SM VacciGrade’s instruction) were prepared freshly before each immunization. Compound 5 was dissolved in PBS directly and then vortexed for 2 min. Mice in each group (six mice per group) were immunized subcutaneously at the base of their tails with the formulation bearing150 μg of appropriate antigen.
In vivo assessment of DC maturation by flow cytometry
Spleens were collected and placed in complete RPMI media [RPMI, Gibco, 10% FBS, penicillin/streptomycin (100 U/ml), 0.4 mM 2-mercaptoethanol, 20 mM Hepes, 4 mM l-glutamine]. Single-cell suspension from spleens was obtained by passing the tissue through a 70-μm nylon strainer (Corning), and erythrocytes were eliminated with ACK Lysis buffer (0.15 M NH4Cl, 1 mM KHCO3, 0.1 mM EDTA). Single cells were washed and resuspend in 5 ml of complete RPMI (Gibco). Two-fifth of the spleen sample was used for the staining. Prior antibody staining mouse FC receptors were blocked using TruStain FCX (BioLegend, San Diego, California) at 1:200 dilution in FACS buffer (PBS, 2% FBS) for 15 min on ice. Surface markers were stained for 40 min using combinations of labeled monoclonal antibodies diluted in PBS: CD45.2-PE-dazzle (clone 104), CD11c-PE-Cy7 (clone N418), MHC-II-BV421 (clone M5/114.15.2), F4/80-APC-Cy7 (clone CI-A3–1), CD40-FITC (3/23), CD80-PE (clone 16-10A1), rat IgG2a, κ isotype Ctrl-FITC (clone RTK2758), Armenian hamster IgG isotype Ctrl-PE (clone HTK888), rat IgG2b, and κ isotype Ctrl-APC (RTK4530) (BioLegend). For Foxp3 intracellular staining, the Foxp3 staining buffer set kit (eBioscience) was used following the manufacturer’s recommendations. Before analysis, samples were washed twice with permeabilization buffer. All samples were resuspended in 200 μl of PBS before analysis, and 10-μl Beckman Coulter count beads were added for absolute number quantification. Stained samples were analyzed on Flow Cytometry BD LSRFortessa. Measurements of median were used to calculate the mean fluorescence intensity of cell populations using FlowJo software.
ELISpot assay
Splenocytes from immunized animals were isolated at 14 days following the last immunization and assessed by ELISpot for IFN-γ, IL-4, and IL-17A production. Multiscreen plates, 96 wells (MAHA, MAIP, or MSIP, Millipore), were coated overnight at 4°C with anti-mouse IFN-γ (5 μg/ml; AN18, Mabtech), anti-mouse IL-4 (5 μg/ml; BVD4-1d11, BD Biosciences), or IL-17A (5 μg/ml; Mabtech). Plates were washed five times with PBS and blocked with RPMI 1640 (Gibco, Life technologies) containing 10% FBS [supplemented with penicillin (100 U/ml), streptomycin (100 μg/ml), 2 mM l-glutamine, 1 M Hepes, and 0.1 mM 2-mercaptoethanol] for 2 hours at 37°C. Splenocytes were added at a final concentration of 0.5 × 106 per well and coincubated with recall antigens (1, 5, and PADRE conjugated to an unrelated peptide at a final concentration of 25 μg/ml) in complete media for 12 to 16 hours at 37°C (IFN-γ), 20 to 24 hours (IL-4), and 40 to 48 hours (IL-17A). Media alone and positive control concanavalin A (1 μg/ml final) wells were also added. Following incubation, plates were washed with PBS, and anti-mouse IFN-γ–biotin (1 μg/ml; R4-6A2, Mabtech), anti-mouse IL-4–biotin (BVD6-24G2, Mabtech or BD Biosciences), or anti-mouse IL-17A–biotin (BD Biosciences) in 0.5% FBS/PBS was added for 2 hours at room temperature (IFN-γ and IL-17A) or overnight at 4°C for IL-4. Plates were washed, and streptavidin-ALP (1 μg/ml) or ExtrAvidin-ALP (1 μg/ml) in 0.5% FBS/PBS was added for a further 1.5 hours at room temperature. Plates were given a final wash with PBS followed by reverse osmosis water, and spots were developed using an AP colorimetric kit (Bio-Rad). Once plates were dry, spots were counted using an AID ELISpot Reader system.
Antibody titer evaluation by ELISA
The J8-specific IgG in murine serum/saliva and J8-specific IgA in murine saliva collected from the C57BL/6 mice after each immunization were measured by ELISA. Polycarbonate plates were coated with J8 peptide (pH 9.6, 0.5 mg/ml in a carbonate coating buffer) with amount of 100 μl per well, and incubated at 4°C overnight. Then, after washing the plates five times with PBS–Tween 20 buffer, 150 μl of 5% skim milk PBS–Tween 20 was added. After incubation at 37°C for 2 hours, plates were washed again. Sera samples (200 μl of 1:100 dilution) or saliva samples (100 μl of 1:2 dilution) were added to the plate followed by serial dilution down the plate in 0.5% skim milk PBS–Tween 20 buffer. All plates were incubated for 1.5 hours in a 37°C incubator. The plates were washed five times, and 100 μl per well of 1:3000 diluted peroxidase-conjugated goat anti-mouse IgG (in 0.5% skim milk PBS–Tween 20) or 50 μl per well of 1:1000 diluted goat anti-mouse IgA (in 0.5% skim milk PBS–Tween 20) was added; the plates were incubated for 1.5 hours at 37°C. After incubation, plates were washed again, and 100 μl per well of o-phenylenediamine dihydrochloride (OPD) substrate was added. The plates were incubated for 30 min in a dark environment at room temperature, and then the absorbance of stained J8-specific IgG or IgA was measured at λ 450 nm.
Bactericidal assessment
An opsonization assay was performed with serum collected from immunized mice to evaluate the bactericidal efficacy. Clinical isolates were provided from Princess Alexandra Hospital that includes (i) ACM-2002 (Royal Brisbane Hospital, human abscess–lymph gland), (ii) ACM-5199 (ATCC 12344, NCIB 11841, scarlet fever), (iii) ACM-5203 (ATCC 19615, pharynx of child followed by episode of sore throat), (iv) GC2 203 (wound swab), (v) D3840 (nasopharynx swabs), (vi) D2612 (nasopharynx swabs). The bacteria were prepared by streaking on THB agar supplemented with 5% yeast extract and incubated (37°C, 24 hours). A single colony from the bacterium was transferred to THB (5 ml) supplemented with 5% yeast extract and incubated for 24 hours at 37°C to give approximately 4.6 × 106 colony-forming units (CFU)/ml. The culture was serially diluted to 10-2 in PBS, and an aliquot (10 μl) was mixed with heat-inactivated sera (10 μl) and horse blood (80 μl). The inactivated sera were prepared by heating in a 50°C water bath for 30 min. The bacteria incubation was conducted in the presence of sera in a 96-well plate at 37°C for 3 hours. Ten microliters of this suspension was plated on Todd-Hewitt agar plates supplemented with 5% yeast extract and 5% horse blood. The plates were incubated at 37°C for 24 hours. The bacteria survival rate was analyzed on the basis of the CFU enumerated from the incubated Todd-Hewitt agar plates. The assay was performed in triplicate from three independent cultures.
Cell lines and ELISA inflammatory cytokine detection
Immortalized wild-type C57BL/6 macrophages were grown in DMEM (Gibco) supplemented with 10% heat-inactivated FBS and 2 mM glutamine. 1 × 106 cells/ml were seeded in six-well plates, left to settle for an hour, and subsequently stimulated with LPS (100, 50, and 1 ng/ml), 1 (0.1 mg/ml), control (0.1 mg/ml), silica (250 μg/ml; U.S. Silica), or left untreated. Cells were stimulated for either 4 or 6 hours and maintained at 37°C in a 5% CO2 incubator. For the detection of IL-1b and TNFα, culture supernatants were harvested by washing with PBS and detached with Glutamax. Debris were pelleted at 201,000g, 5 min, 4°C, and either 50 or 100 μl of undiluted sample supernatant was used in a mouse IL-1b or TNFα ELISA according to the manufacturer’s specifications.
Statistical analysis
GraphPad Prism 7 software (GraphPad Software Inc., California, USA) was used for all statistical analysis. For the APC maturation results, mean fluorescence intensity was recorded. Two-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test was applied during the statistical analysis, with P < 0.05 considered statistically significant. The titer of the J8-specific IgG or IgA was described as the lowest dilution that offered an absorbance of greater than 3 SD above the mean absorbance of the negative control wells (wells coated with serum from mice immunized with PBS). A one-way ANOVA followed by Tukey’s post hoc test was applied to statistical analysis of the antibody titers and ELISpot, with P < 0.05 considered statistically significant. The opsonic activity of the antibody (anti-peptide) sera (% reduction in mean CFU) was calculated as [1 − (CFU in the presence of anti-peptide sera)/(mean CFU in the presence of PBS)] × 100. One-way ANOVA followed by Tukey’s post hoc test was applied for the opsonization statistical analysis, with P < 0.05 considered statistically significant. The bacteria infection challenge evaluation of CFU was recorded and analyzed by one-way ANOVA followed by Tukey’s post hoc test, with P < 0.05 considered statistically significant.
Supplementary Material
Acknowledgments
We thank the facilities and the scientific and technical assistance of the Australian Microscopy and Microanalysis Research Facility at the Centre for Microscopy and Microanalysis, The University of Queensland. We also acknowledge A. D. Paterson and P. Harris (The University of Queensland Centre for Clinical Research) for providing Streptococcus isolates, GC2 203, D3840, and D2612. We thank T. Guerin for the critical editing of the manuscript. Funding: This work was supported by the National Health and Medical Research Council (program grant APP1132975 to M.F.G. and I.T., and project grant APP1099999 to J.W.W.). Author contributions: M.S. and I.T. designed the compounds and experiments and directed the study. M.P., M.R.B., and M.F.G. designed the immunization and challenge experiments. R.J.C. and Z.G.K. designed the opsonization experiment. J.C.B. and M.P. designed the cytokine study. G.Z., J.C.B., V.O., A.A., J.G.C., A.K.G., Z.G.K., M.A.S., W.M.H., R.J.N., and J.W.W. performed the experiments. M.S., G.Z., J.C.B., J.G.C., M.P., A.K.G., W.M.H., J.W.W., and V.O. analyzed the data. M.S. and G.Z. wrote the manuscript. M.P. and I.T. edited the paper. Competing interests: M.S., G.Z., and I.T. are co-inventors in a patent application entitled “Self assembling, self adjuvanting system for delivery of vaccines” filed by The University of Queensland (application number AU 2019900328). The remaining authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/5/eaax2285/DC1
Fig. S1. Particle size distribution for compounds 2 to 5.
Fig. S2. TEM photographs of compound 5.
Fig. S3. Expression of APC maturation markers MHC-II and CD40 on CD11c+ DCs and F4/80+ macrophages in response to stimulation with vaccine candidates.
Fig. S4. Expression of APC maturation markers CD40, CD80, CD86, and MHC-II on CD11c+ DCs and F4/80+ macrophages in response to stimulation with vaccine candidates in vivo.
Fig. S5. ELISpot analysis of IFN-γ– and IL-4–producing T cells.
Fig. S6. Antibody titers measured in two distinct in vivo experiments.
Fig. S7. Photos of mouse tails 55 days after primary immunization.
Fig. S8. MS and HPLC spectra of compounds 1 to 5.
Fig. S9. Latex agglutination test results of bacteria collected from the challenge experiment.
Table S1. Particle size distribution for compounds 2 to 5 analyzed by intensity.
REFERENCES AND NOTES
- 1.Skwarczynski M., Toth I., Peptide-based synthetic vaccines. Chem. Sci. 7, 842–854 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Purcell A. W., McCluskey J., Rossjohn J., More than one reason to rethink the use of peptides in vaccine design. Nat. Rev. Drug Discov. 6, 404–414 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Melero I., Gaudernack G., Gerritsen W., Huber C., Parmiani G., Scholl S., Thatcher N., Wagstaff J., Zielinski C., Faulkner I., Mellstedt H., Therapeutic vaccines for cancer: An overview of clinical trials. Nat. Rev. Clin. Oncol. 11, 509–524 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Steer A. C., Carapetis J. R., Dale J. B., Fraser J. D., Good M. F., Guilherme L., Moreland N. J., Mulholland E. K., Schodel F., Smeesters P. R., Status of research and development of vaccines for Streptococcus pyogenes. Vaccine 34, 2953–2958 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Carapetis J. R., Steer A. C., Mulholland E. K., Weber M., The global burden of group A streptococcal diseases. Lancet Infect. Dis. 5, 685–694 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Paar J. A., Berrios N. M., Rose J. D., Cáceres M., Peña R., Pérez W., Chen-Mok M., Jolles E., Dale J. B., Prevalence of rheumatic heart disease in children and young adults in Nicaragua. Am. J. Cardiol. 105, 1809–1814 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gorton D., Govan B., Olive C., Ketheesan N., B- and T-Cell responses in group a streptococcus M-protein- or peptide-induced experimental carditis. Infect. Immun. 77, 2177–2183 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Azmi F., Ahmad Fuaad A. A., Skwarczynski M., Toth I., Recent progress in adjuvant discovery for peptide-based subunit vaccines. Hum. Vaccin. Immunother. 10, 778–796 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.R. J. Nevagi, I. Toth, M. Skwarczynski, Peptide-based vaccines, in Peptide Applications in Biomedicine, Biotechnology and Bioengineering, S. Koutsopoulos, Ed. (Woodhead Publishing, 2018), pp. 327–358. [Google Scholar]
- 10.Flanagan K. L., Plebanski M., Sex-differential heterologous (non-specific) effects of vaccines: An emerging public health issue that needs to be understood and exploited. Expert Rev. Vaccines 16, 5–13 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Wilson K. L., Xiang S. D., Plebanski M., Montanide, Poly I:C and nanoparticle based vaccines promote differential suppressor and effector cell expansion: A study of induction of CD8 T cells to a minimal Plasmodium berghei epitope. Front. Microbiol. 6, 29 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Skwarczynski M., Toth I., Recent advances in peptide-based subunit nanovaccines. Nanomedicine 9, 2657–2669 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Lebre F., Hearnden C. H., Lavelle E. C., Modulation of immune responses by particulate materials. Adv. Mater. 28, 5525–5541 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Zhao G., Chandrudu S., Skwarczynski M., Toth I., The application of self-assembled nanostructures in peptide-based subunit vaccine development. Eur. Polym. J. 93, 670–681 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Negahdaripour M., Golkar N., Hajighahramani N., Kianpour S., Nezafat N., Ghasemi Y., Harnessing self-assembled peptide nanoparticles in epitope vaccine design. Biotechnol. Adv. 35, 575–596 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barrett J. C., Ulery B. D., Trent A., Liang S., David N. A., Tirrell M. V., Modular peptide amphiphile micelles improving an antibody-mediated immune response to Group A Streptococcus. ACS Biomater. Sci. Eng. 3, 144–152 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Skwarczynski M., Zaman M., Urbani C. N., Lin I. C., Jia Z., Batzloff M. R., Good M. F., Monteiro M. J., Toth I., Polyacrylate dendrimer nanoparticles: A self-adjuvanting vaccine delivery system. Angew. Chem. Int. Ed. Engl. 49, 5742–5745 (2010). [DOI] [PubMed] [Google Scholar]
- 18.Chandrudu S., Bartlett S., Khalil Z. G., Jia Z., Hussein W. M., Capon R. J., Batzloff M. R., Good M. F., Monteiro M. J., Skwarczynski M., Toth I., Linear and branched polyacrylates as a delivery platform for peptide-based vaccines. Ther. Deliv. 7, 601–609 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Liu T. Y., Hussein W. M., Giddam A. K., Jia Z., Reiman J. M., Zaman M., McMillan N. A., Good M. F., Monteiro M. J., Toth I., Skwarczynski M., Polyacrylate-based delivery system for self-adjuvanting anticancer peptide vaccine. J. Med. Chem. 58, 888–896 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Sohma Y., Hayashi Y., Skwarczynski M., Hamada Y., Sasaki M., Kimura T., Kiso Y., O-N intramolecular acyl migration reaction in the development of prodrugs and the synthesis of difficult sequence-containing bioactive peptides. Biopolymers 76, 344–356 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Malkov S. N., Zivković M. V., Beljanski M. V., Hall M. B., Zarić S. D., A reexamination of the propensities of amino acids towards a particular secondary structure: Classification of amino acids based on their chemical structure. J. Mol. Model. 14, 769–775 (2008). [DOI] [PubMed] [Google Scholar]
- 22.Bouchard M., Zurdo J., Nettleton E. J., Dobson C. M., Robinson C. V., Formation of insulin amyloid fibrils followed by FTIR simultaneously with CD and electron microscopy. Protein Sci. 9, 1960–1967 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang C.-A., Yung-Han C., Shyue-Chu K., Yi-Ru C., Hsien-Bin H., Ta-Hsien L., Yi-Cheng C., Correlation of copper interaction, copper-driven aggregation, and copper-driven H2O2 formation with Aβ40 conformation. Intl. J. Alzheimer’s Dis. 2011, 607861 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hornung V., Bauernfeind F., Halle A., Samstad E. O., Kono H., Rock K. L., Fitzgerald K. A., Latz E., Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 9, 847–856 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li M., Chen L.-X., Chen S.-R., Deng Y., Zhao J., Wang Y., Li S.-P., Non-starch polysaccharide from Chinese yam activated RAW 264.7 macrophages through the Toll-like receptor 4 (TLR4)-NF-κB signaling pathway. J. Funct. Foods 37, 491–500 (2017). [Google Scholar]
- 26.Reed S. G., Orr M. T., Fox C. B., Key roles of adjuvants in modern vaccines. Nat. Med. 19, 1597–1608 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Liao J., Zhang Y., Yu W., Xu L., Ge C., Liu J., Gu N., Linear aggregation of gold nanoparticles in ethanol. Colloids Surf. A Physicochem. Eng. Asp. 223, 177–183 (2003). [Google Scholar]
- 28.Friedlander S. K., Ogawa K., Ullmann M., Elasticity of nanoparticle chain aggregates: Implications for polymer fillers and surface coatings. Powder Technol. 118, 90–96 (2001). [Google Scholar]
- 29.Nordström T., Pandey M., Calcutt A., Powell J., Phillips Z. N., Yeung G., Giddam A. K., Shi Y., Haselhorst T., von Itzstein M., Batzloff M. R., Good M. F., Enhancing vaccine efficacy by engineering a complex synthetic peptide to become a super immunogen. J. Immunol. 199, 2794–2802 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Powles L., Xiang S. D., Selomulya C., Plebanski M., The use of synthetic carriers in malaria vaccine design. Vaccines 3, 894–929 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reddy S. T., van der Vlies A. J., Simeoni E., Angeli V., Randolph G. J., O‘Neil C. P., Lee L. K., Swartz M. A., Hubbell J. A., Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat. Biotechnol. 25, 1159–1164 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Moyano D. F., Goldsmith M., Solfiell D. J., Landesman-Milo D., Miranda O. R., Peer D., Rotello V. M., Nanoparticle hydrophobicity dictates immune response. J. Am. Chem. Soc. 134, 3965–3967 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rad-Malekshahi M., Fransen M. F., Krawczyk M., Mansourian M., Bourajjaj M., Chen J., Ossendorp F., Hennink W. E., Mastrobattista E., Amidi M., Self-assembling peptide epitopes as novel platform for anticancer vaccination. Mol. Pharm. 14, 1482–1493 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hamesch K., Borkham-Kamphorst E., Strnad P., Weiskirchen R., Lipopolysaccharide-induced inflammatory liver injury in mice. Lab. Anim 49, 37–46 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Fish E. N., Flanagan K. L., Furman D., Klein S. L., Kollmann T. R., Jeppesen D. L., Levy O., Marchant A., Namachivayam S., Netea M. G., Plebanski M., Rowland-Jones S. L., Selin L. K., Shann F., Whittle H. C., Changing oral vaccine to inactivated polio vaccine might increase mortality. Lancet 387, 1054–1055 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abderrazak A., Syrovets T., Couchie D., El Hadri K., Friguet B., Simmet T., Rouis M., NLRP3 inflammasome: From a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol. 4, 296–307 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chattopadhyay K., Lazar-Molnar E., Yan Q., Rubinstein R., Zhan C., Vigdorovich V., Ramagopal U. A., Bonanno J., Nathenson S. G., Almo S. C., Sequence, structure, function, immunity: Structural genomics of costimulation. Immunol. Rev. 229, 356–386 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.I. Chatzidakis, C. Mamalaki, in Tnf Pathophysiology: Molecular and Cellular Mechanisms, G. Kollias, P. P. Sfikakis, Eds. (2010), vol. 11, pp. 105–118.
- 39.McGeough M. D., Wree A., Inzaugarat M. E., Haimovich A., Johnson C. D., Peña C. A., Goldbach-Mansky R., Broderick L., Feldstein A. E., Hoffman H. M., TNF regulates transcription of NLRP3 inflammasome components and inflammatory molecules in cryopyrinopathies. J. Clin. Invest. 127, 4488–4497 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/5/eaax2285/DC1
Fig. S1. Particle size distribution for compounds 2 to 5.
Fig. S2. TEM photographs of compound 5.
Fig. S3. Expression of APC maturation markers MHC-II and CD40 on CD11c+ DCs and F4/80+ macrophages in response to stimulation with vaccine candidates.
Fig. S4. Expression of APC maturation markers CD40, CD80, CD86, and MHC-II on CD11c+ DCs and F4/80+ macrophages in response to stimulation with vaccine candidates in vivo.
Fig. S5. ELISpot analysis of IFN-γ– and IL-4–producing T cells.
Fig. S6. Antibody titers measured in two distinct in vivo experiments.
Fig. S7. Photos of mouse tails 55 days after primary immunization.
Fig. S8. MS and HPLC spectra of compounds 1 to 5.
Fig. S9. Latex agglutination test results of bacteria collected from the challenge experiment.
Table S1. Particle size distribution for compounds 2 to 5 analyzed by intensity.