

Robustness in essential biological circuits is an important feature of the living organism. A few pieces of evidence support the existence of robustness in vivo in the two-component system (TCS) with a bifunctional sensor kinase (SK). The assays were done under physiological conditions in which the SK was much lower than the response regulator (RR). Here, using a synthetic circuit, we varied the concentrations of the SK and RR of a representative TCS to monitor output robustness in vivo. In vitro assays were also performed under conditions where the concentration of the SK was greater than that of the RR. Our results demonstrate the extent of output robustness in the TCS signaling pathway with respect to the concentrations of the two components.
KEYWORDS: two-component system, TCS, bifunctional sensor kinase, transcription, gene regulation, synthetic circuit, response regulator
ABSTRACT
Variation in the concentration of biological components is inescapable for any cell. Robustness in any biological circuit acts as a cushion against such variation and enables the cells to produce homogeneous output despite the fluctuation. The two-component system (TCS) with a bifunctional sensor kinase (that possesses both kinase and phosphatase activities) is proposed to be a robust circuit. Few theoretical models explain the robustness of a TCS, although the criteria and extent of robustness by these models differ. Here, we provide experimental evidence to validate the extent of the robustness of a TCS signaling pathway. We have designed a synthetic circuit in Escherichia coli using a representative TCS of Mycobacterium tuberculosis, MprAB, and monitored the in vivo output signal by systematically varying the concentration of either of the components or both. We observed that the output of the TCS is robust if the concentration of MprA is above a threshold value. This observation is further substantiated by two in vitro assays, in which we estimated the phosphorylated MprA pool or MprA-dependent transcription yield by varying either of the components of the TCS. This synthetic circuit could be used as a model system to analyze the relationship among different components of gene regulatory networks.
IMPORTANCE Robustness in essential biological circuits is an important feature of the living organism. A few pieces of evidence support the existence of robustness in vivo in the two-component system (TCS) with a bifunctional sensor kinase (SK). The assays were done under physiological conditions in which the SK was much lower than the response regulator (RR). Here, using a synthetic circuit, we varied the concentrations of the SK and RR of a representative TCS to monitor output robustness in vivo. In vitro assays were also performed under conditions where the concentration of the SK was greater than that of the RR. Our results demonstrate the extent of output robustness in the TCS signaling pathway with respect to the concentrations of the two components.
INTRODUCTION
Gene regulation is central to the growth and survival of any living organism under different environmental conditions. Any organism that uses a certain gene regulatory network under its normal growth conditions makes appropriate changes in the network when conditions are not favorable. Thus, each of these organisms must employ a mechanism to sense changes in the micromilieu and accordingly induce a regulator(s) to the system for switching on the appropriate gene regulatory network.
One such example is the gene regulation by a two-component system (TCS), each comprising a paired sensor kinase with a response regulator (1, 2). In the presence of an external stimulus, the sensor kinase gets phosphorylated and concomitantly transfers the phosphoryl group to its cognate response regulator. The phosphorylated regulator subsequently controls one or more genes as an outcome of the external stimuli. Thus, an output of the signaling pathway is related to the input. In this event, one may ask what happens to the input-output relationship if the concentrations of the paired components or the reactant for phosphorylation (ATP) vary from cell to cell. In the case of a monofunctional sensor kinase, e.g., when the enzyme possesses only the kinase activity, the output of the TCS varies with the concentration of the system components (3–5). However, for a bifunctional sensor kinase, e.g., a sensor kinase that possesses both kinase and phosphatase activities (6) (removing the phosphate from the phosphorylated regulator), the output is robust and does not depend on the concentration of the system components (7). Using the EnvZ-OmpR TCS as an example, Batchelor and Goulian (8) have provided evidence that the output is robust when the concentration of the sensor kinase (EnvZ) is much lower than the regulator (OmpR). They have proposed a kinetic model which supports their experimental data. On the other hand, Uri Alon and colleagues have theoretically shown that if the phosphatase activity of a bifunctional sensor kinase is ATP dependent, the output of the TCS is robust only when the concentration of the response regulator is above a certain threshold (7, 9). Developing an in vivo phosphorylation estimation method, Gao and Stock have further studied the PhoB/PhoR TCS of E. coli under in vivo conditions where the level of PhoB is much higher than PhoR and showed that phosphatase activity is essential for robustness (10).
Here, we have designed a synthetic circuit in Escherichia coli using a representative two-component system, MprAB of Mycobacterium tuberculosis (11, 12), to monitor the in vivo output signal by systematically varying the concentration of either component or both. Using in vitro assays, we have further estimated the phosphorylated MprA (MprA∼P) pool or MprA∼P-dependent transcription yield by varying the concentration of each of the components of the TCS. We showed that the output of the MprAB signaling system remains constant with the variation of the concentration of components if the level of the response regulator is above a certain threshold limit. Thus, our results demonstrate the extent of dependence of input-output robustness on the concentration of the system components in the TCS signaling pathway.
RESULTS
In the MprAB TCS, the sensor kinase MprB gets activated by surface stress signals, undergoes autophosphorylation at a conserved histidine residue (His-249), and subsequently transfers the phosphate group to its cognate response regulator MprA (13). The aspartic acid residue at position 48 of MprA is known to receive the phosphate group and the phosphorylated MprA (MprA∼P) then acts as a transcription factor in downstream gene regulation. Thus, in MprAB TCS, MprA∼P is the output and MprA, and MprB are the components of the system. Here, we monitored how the population of MprA∼P changes with the system components, i.e., varying concentration of MprA and MprB. We performed three sets of experiments: (i) an in vivo assay to monitor MprA∼P-dependent gene expression in the presence of varying concentrations of either MprA or MprB or both (Fig. 1 and Fig. 2); (ii) an in vitro assay to monitor the changes in the MprA∼P pool with varying concentrations of MprA and MprB (Fig. 3); and (iii) an in vitro assay to see the effects of varying concentrations of MprA and MprB on the MprA∼P-dependent transcription yield (Fig. 4).
FIG 1.

In vivo recombinant reporter assay for assessing the MprAB output as a function of increasing concentration of both MprA and MprB. (A) Schematic diagram of tuning and functioning of the MprAB TCS circuit, in which the MprA and MprB proteins can be systematically controlled by the tetRO-inducible promoter. The MprB protein subsequently phosphorylates and dephosphorylates MprA to produce an MprA∼P pool, which in turn induces the mCherry gene through the ppk1 promoter. The output of the circuit (mCherry fluorescence) can be monitored as a function of the tetRO inducer anhydrotetracycline (ATC). Mtb, M. tuberculosis. (B) Design of the synthetic circuit using a three-plasmid reporter system in E. coli composed of pAcYc Duet-rpoA and sigE, and pCOLA Duet-rpoB-rpoC for the expression of the M. tuberculosis RNAP-σE holoenzyme, pFPV mCherry with the tetRO-inducible mprAB genes, and the mCherry reporter gene under the control of the MprA∼P-inducible ppk1promoter. Origins of replication and antibiotic resistances of each plasmid are highlighted. (C) Relative fluorescence intensities of mCherry expression with respect to control (no inducer) for OD 1 cells (average from 3 replicates with standard deviation) were plotted against various concentrations of MprA (average from 3 replicates) induced by 0, 10, 20, 40, 80, 160, 200, 240, and 320 ng/ml of anhydrotetracycline. (D) Representative Western blot analysis of OD 1 cells from each point of induction against respective antibodies of MprB and MprA and corresponding protein concentration are shown.
FIG 2.

In vivo recombinant reporter assay for assessing the MprAB output as a function of increasing concentration of MprA with constant MprB and vice versa. (A) Schematic diagram of tuning and functioning of the MprAB TCS circuit, which is the same as Fig. 1A except the concentration of MprB is controlled by the ara promoter. (B) Design of the synthetic circuit using a four-plasmid reporter system in E. coli, in which the first two plasmids were used for the expression of the Mtb RNAP-σE holoenzyme, a third plasmid pFPV mCherry with the tetRO-inducible mprA genes and the mCherry reporter gene placed under the control of an MprA∼P-inducible ppk1 promoter as in Fig. 1B, and then a fourth plasmid pCDF has the mprB gene inducible by l-arabinose. (C) (Top) Relative fluorescence intensities of mCherry expression with respect to control for 1 OD cells (average from 3 replicates with standard error) were plotted against various concentrations (0.10, 0.31, 0.60, 0.91, 1.05, 1.15, 1.29, 1.40, and 1.56 μg/OD/ml) of MprA (average from 3 replicates) while keeping MprB fixed at 0.14 μg/OD/ml (solid line). In the control, cells had no induction of MprA but had a fixed concentration of MprB (0.14 μg/OD/ml). The assay was further repeated with a higher value of MprB (0.26 μg/OD/ml) (dashed line) and with a lower value of MprB (0.12 μg/OD/ml) (dotted line) (Kruskal-Wallis test; df = 2, P > 0.05). (Bottom) Western blot analysis of the expression of MprA at each point of induction and corresponding protein concentrations. (D, top) Same as panel C, top, but with various concentrations (0.06, 0.12, 0.14, 0.18, 0.24, 0.26, 0.30, 0.33, and 0.38 μg/OD/ml) of MprB (average from 3 replicates), keeping MprA fixed at 0.96 μg/OD/ml (solid line). Control cells had no induction of MprB but had a fixed concentration of 0.96 μg/OD/ml MprA. The assay was further repeated with MprA fixed at a higher value of 1.15 μg/OD/ml (dashed line) and with a lower value of MprA (0.31 μg/OD/ml) (dotted line). Statistical analysis showed that the MprA~P pool remained unchanged (one-way analysis of variance [ANOVA]; Fcritical > F value) with various concentrations of MprB. (Bottom) Same as panel C, bottom, but for MprB.
FIG 3.
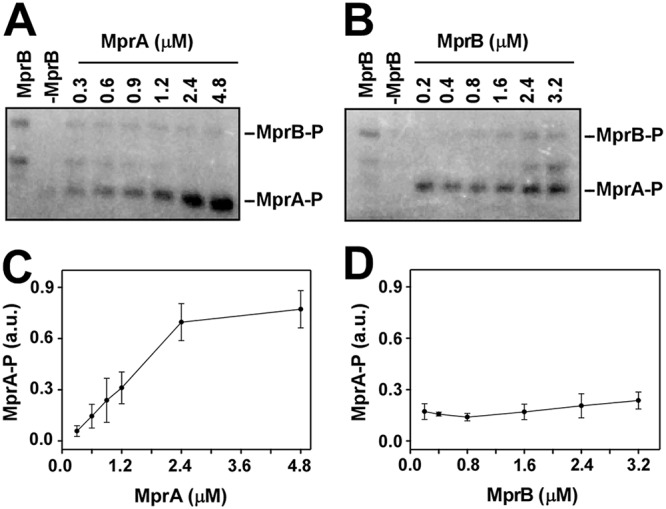
In vitro phosphorylation assay using radiolabeled [γ-32P]ATP. (A) Lanes 1 and 2, autophosphorylation of 0.8 μM purified MprB (truncated) only and 2 μM MprA only, respectively; lanes 3 to 8, transfer of phosphate from 0.8 μM MprB∼P to various concentrations of MprA (0.3, 0.6, 0.9, 1.2, 2.4, and 4.8 μM). (B) Lanes 1 and 2, autophosphorylation of MprB (truncated) only and MprA only, respectively; lanes 3 to 8 transfer of a phosphate group from various concentrations of MprB (0.2, 0.4, 0.8, 1.6, 2.4, and 3.2 μM) to 0.75 μM MprA. (C and D) Average pools of MprA∼P from three replicates were plotted against concentrations of MprA and MprB, respectively. Standard errors are shown.
FIG 4.

In vitro transcription assays using radiolabeled [α-32P]CTP. (A) Schematic diagram of functioning of the MprAB TCS circuit in the transcription assay. Mtb, M. tuberculosis. (B) M. tuberculosis RNAP core (0.4 μM) and 0.8 μM σE were incubated with 0.1 μM ppkI promoter DNA and various concentrations of phosphorylated MprA as in the reaction in Fig. 3A (0.3, 0.6, 1.2, 2.4, and 4.8 μM while keeping MprB fixed at 1.0 μM). Transcription reactions were initiated by adding NTP mix (250 μM ATP, GTP, UTP, and 20 μM CTP containing 0.4 μCi [α-32P]CTP) and heparin (25 μg/ml) at 37°C for 30 min and terminated using formamide dye. Reaction products were kept for 2 min at 95°C, run on a 12.5% urea-PAGE gel, and scanned in a phosphorimage scanner. Transcript size was 71 nt. (C) Assay repeated as panel B but MprA∼P was added from the reaction as in panel B (keeping MprA fixed at 0.50 μM with 0.25, 0.5, 1.0, 2.0, and 4.0 μM MprB). (D and E) Average relative transcript yield (with respect to RNAP holosample in the presence of either MprA or MprB) from 3 replicates with standard error are plotted against concentrations of MprA and MprB, respectively.
In vivo assay suggests the MprA∼P pool is independent of the concentrations of MprB and MprA.
A synthetic circuit was designed using a three-plasmid expression system in E. coli: two plasmids for the expression of M. tuberculosis RNA polymerase (RNAP) (pAcYcDuet-rpoA-sigE and pCOLA Duet-rpoB-rpoC, which when induced with IPTG produces the M. tuberculosis RNA polymerase-SigE holoenzyme) (14, 15), along with a third plasmid for the differential expression of MprA and MprB, as well as MprA∼P, dependent upon mCherry expression (Fig. 1B). We used a ppk1 DNA fragment as an MprA∼P-inducible promoter for the mCherry gene (16). In the same plasmid (pFPV-mCherry), the mprAB gene was cloned under the control of the tetRO-inducible promoter. When E. coli BL21 cells containing all three plasmids were induced with IPTG, the assembly of the M. tuberculosis RNAP-SigE holoenzyme resulted in a basal level of mCherry expression. The fluorescence intensity of these cells was 7- to 8-fold higher than that of the control cells that produced only the M. tuberculosis RNAP core (see Materials and Methods). When the cells were induced with anhydrotetracycline to produce MprA and MprB, we observed a significant increase (∼2- to 3-fold) in the level of mCherry fluorescence until saturation (Fig. 1C). When the cells were not treated with IPTG before tetRO induction, we did not observe any change in the mCherry fluorescence intensity, suggesting that MprA∼P is not sufficient to induce protein expression from the ppk1 promoter in the absence of M. tuberculosis RNAP. The levels of the MprA and MprB at each point of induction by anhydrotetracycline were estimated by Western blotting assay using respective antibodies against MprA and MprB (Fig. 1D). The expression of both proteins increased with anhydrotetracycline induction. However, the level of MprA was almost 2-fold higher than the level of MprB at higher inducer concentrations. The corresponding MprA∼P-dependent mCherry expression also increased with the increase in protein levels. The mCherry expression saturated when levels of the proteins reached a certain threshold value. Thus, the mCherry expression data corroborate the variation in the levels of MprAB and indicates how the level of MprA∼P (output) varies with the changes in MprAB concentration. However, the dependence of MprA∼P (output) on the concentration of either MprA or MprB separately could not be monitored from this assay.
The level of MprA∼P-dependent gene expression in the presence of a fixed amount of MprA and varying concentrations of MprB, or vice versa, was tested by a similar in vivo reporter assay but with a four-plasmid synthetic circuit (Fig. 2A and B). While the first two plasmids for expression of M. tuberculosis RNAP remained the same, the third plasmid (pFPV-mCherry) contained the mprA gene under the control of the tetRO promoter, rather than mprAB, whereas the fourth plasmid was a pCDF Duet vector that contained a truncated (amino acids 196 to 504) mprB gene under the control of the ara promoter (Fig. 2B). Zahrt and colleagues (13) used the truncated derivative of MprB to increase the solubility of the recombinant protein and we have adopted the method in our assay. Thus, by varying the concentration of anhydrotetracycline (0, 10, 20, 40, 80, 160, 200, 240, and 320 ng/ml) while keeping the l-arabinose concentration fixed at 1.0 μg/ml, we were able to change the level of MprA (0.10, 0.31, 0.60, 0.91, 1.05, 1.15, 1.29, 1.40, and 1.56 μg/OD/ml) at a fixed level of MprB (0.14 μg/OD/ml). For each point of the data set, the fluorescence intensity of the cells was monitored. The result (Fig. 2C, top, solid line) showed that MprA∼P-dependent mCherry expression initially varied with the level of MprA until saturation was reached. Thus, the output varied linearly with the total concentration of MprA until it became higher than the level where the mCherry fluorescence was saturated. When a similar assay was performed as described above but with a higher level of MprB (0.26 μg/OD/ml) at 8 μg/ml of arabinose and with a lower level of MprB (0.12 μg/OD/ml) at 0.5 μg/ml of arabinose, the nature of the curve remained same within the error limit (Fig. 2C, top, dashed and dotted lines, respectively). The results indicate that the phosphorylated MprA saturates when the protein concentrations reach a threshold value.
In a complementary assay, the l-arabinose concentrations were varied (0, 0.5, 1.0, 2.0, 4.0, 8.0, 10.0, 12.0, and 16.0 μg/ml) while keeping the anhydrotetracycline concentration fixed (at 50 ng/ml). At each l-arabinose concentration, the level of MprB was estimated (0.06, 0.12, 0.14, 0.18, 0.24, 0.26, 0.30, 0.33, and 0.38 μg/OD/ml) at a fixed level of MprA (0.96 μg/OD/ml) and the fluorescence intensities of the cells were monitored. The assay was repeated thrice, and the results (Fig. 2D, top, solid line) show that the mCherry intensities of the cells did not change with various concentrations of MprB, suggesting that the MprA∼P pool remained unchanged under various concentrations of MprB. The results indicate that the output is independent of variations in the MprB level if the level of MprA is constant. Similar assays were performed keeping the concentrations of MprA at a constant higher level and a constant lower level than described above. At a higher concentration of MprA, the mCherry expression level remained almost constant with different levels of MprB (Fig. 2D, top, dashed line). At a lower concentration of MprA, the mCherry level also remains constant within the error limit, but at a lower value (Fig. 2D, top, dotted line).
In all of the above-described assays, we observed a robust output when the level of MprA was above a specific value. However, whether this robustness was due to the constant level of MprA∼P or to the saturation of the promoter of the reporter gene remained unclear. To test the possibility of promoter saturation, we performed a similar assay in which the entire cassette of mprA and mCherry genes along with promoters (as from the pFPV plasmid) was cloned into a high copy number plasmid (pETDuet). In this assay, when the M. tuberculosis RNAP holoenzyme was expressed without MprA and MprB, the mCherry intensity with the high copy number plasmid was more than twice the signal seen with the low copy number plasmid, indicating an increase in the promoter concentration of mCherry in the assay system. However, when the M. tuberculosis RNAP holoenzyme was expressed along with MprA and MprB, the respective fold increase in the output signals with respect to controls (RNAP holoenzyme only) was similar with both the high and low copy number plasmids (see Fig. S1 in the supplemental material). As the mprA gene was expressed from high copy number plasmids, the levels of MprA were almost 2-fold higher than with low copy number plasmids at a higher inducer concentration. Even with a higher concentration of MprA, the robustness was observed. This is due to constant MprA∼P level in the assays and thus excludes the possibility of promoter saturation.
A nearly identical set of experiments were done by replacing the ppk1 promoter with the sigE promoter (17), and identical results were observed (Fig. S2). Interestingly, when the mprA gene was replaced by a mutant derivative (mprA-D48A) defective in phosphorylation (13), no MprA∼P-dependent mCherry expression was observed (Fig. S3). Thus, the mCherry induction was only observed when MprA∼P was present, and the gene expression was controlled by any MprA∼P-dependent promoter.
In vitro assays confirm the robustness of the MprA∼P pool.
(i) In vitro phosphorylation assay. To test the dependence of the generation of an MprA∼P pool on the concentration of MprA and MprB in vitro, we performed a phosphorylation assay. Here, we used a truncated MprB derivative (amino acids 196 to 504) (13) that lacks the N-terminal periplasmic domain and a transmembrane region. This protein derivative, when expressed in E. coli, appeared mostly in a soluble form. In the presence of ATP, MprB became autophosphorylated within 10 min, whereas autophosphorylation did not occur for MprA. MprA became phosphorylated only when the protein was incubated with autophosphorylated MprB. When various concentrations of MprA (0.3, 0.6, 0.9, 1.2, 2.4, and 4.8 μM) were incubated with a fixed amount of autophosphorylated MprB (0.8 μM), the transfer of phosphoryl groups between the proteins occurred initially in a concentration-dependent manner and then became saturated (Fig. 3A, lanes 3 to 8). Thus, the amount of MprA∼P was initially found to be linearly dependent on the total concentration of MprA, until total MprA reached a certain level (Fig. 3C).
To determine the dependence of MprA∼P on the system component MprB, various concentrations of autophosphorylated MprB (0.2, 0.4, 0.8, 1.6, 2.4, and 3.2 μM) were incubated with a fixed amount of MprA (0.75 μM) (Fig. 3B, lanes 3 to 8). In this case, the amount of MprA∼P did not change with the MprB∼P (Fig. 3D). Thus, at a constant level of MprA, the MprA∼P pool is independent of variation in the concentration of MprB∼P.
Similar assays were performed keeping the concentration of either MprA or MprB constant but at a higher level than described above (Fig. S4). When MprB was fixed at a higher level with various concentrations of MprA, the amount of MprA∼P increased with MprA as before, and the saturation of MprA∼P occurred at the same concentration (Fig. S4A). On the other hand, when MprA was fixed at a higher level, the level of MprA∼P (Fig. S4B) remained almost constant with different concentrations of MprB, but at a higher value as in Fig. 3.
(ii) In vitro transcription assay. To further study the effect of MprA∼P as a transcription factor, we performed an in vitro transcription assay with the promoter region of the ppk1 gene (16) and purified M. tuberculosis RNA polymerase and SigE. In the presence of the RNAP-SigE holoenzyme, a basal level of transcription occurred. When phosphorylation assay mixture containing MprA∼P was added to the transcription reaction, the yield of transcripts increased, suggesting a transcriptional activation of the ppk1 promoter by MprA∼P.
To check the effect of varying the concentrations of total MprA on MprA∼P pool-dependent gene regulation, an in vitro transcription assay was performed with 0.3, 0.6, 1.2, 2.4, and 4.8 μM MprA, keeping MprB fixed at 1.0 μM. The level of the transcript (71 nucleotides) increased with the increase in concentration of MprA up to 2.4 μM of MprA, and then saturated as the total concentration of MprA became much higher than that of MprB (Fig. 4B and D). In contrast, when a gradient of MprB (0.25, 0.5, 1.0, 2.0, and 4.0 μM) was used in the transcription assay while keeping MprA fixed at 0.50 μM, the transcription yield remained almost unchanged at each MprB concentration (Fig. 4C and E). However, the levels of transcripts were higher than in the control where the MprB concentration was zero. Similar assays were performed keeping the concentration of either MprA or MprB at a constant but higher level than described above. When MprB was fixed at a higher level, the nature of the curve (Fig. S5C, transcription yield versus protein concentration) was similar to that with lower MprB concentrations as in Fig. 4C. On the other hand, when MprA was fixed at higher level, the amount of transcript (Fig. S5D) remained almost constant with different concentrations of MprB, but at a higher level as in Fig. 4D. In a control experiment, addition of MprB∼P or bovine serum albumin (BSA) did not increase the yield of the transcript. Thus, these experiments demonstrate that the output is independent of variations in the concentration of the sensor kinase and also independent of the response regulator when the level of the phosphorylated response regulator is higher than a threshold value.
DISCUSSION
Robustness in essential biological circuits is proposed to be an important feature of the living organism. In a robust circuit, the output varies only with the input and does not depend on the system components (18). This could be advantageous for the organism when the concentrations of the components varies from cell to cell. Thus all the cells would respond in the same way for a given input, and there would be no heterogeneity among the cells (19, 20). Using a model of phosphotransfer kinetics in a bifunctional two-component system, Batchelor and Goulian (8) and Shinar et al. (9) showed that the output of a TCS is directly proportional to the input signal. This robustness is valid for the TCS in which the sensor kinase possesses both kinase and phosphatase activities. However, there is a minor difference in the extent of robustness of the TCS between the two models. According to Batchelor and Goulian, the TCS output is robust if the concentration of the sensor kinase is lower than that of the response regulator. They used EnvZ-OmpR TCS as a model system where the concentration of EnvZ is much lower than the concentration of OmpR (21). Based on this observation and those of other TCS studies, Batchelor and Goulian argued that, in general, the concentration of the sensor kinase in the cell is much lower than that of the response regulator. Using kinetic rate parameters, they proposed a model for robustness of the TCS. In contrast, Shinar et al. have theoretically shown that the output of a TCS is robust at any concentration of the system components, provided the phosphatase activity of the kinase is ATP mediated, and the level of the response regulator is above a certain threshold. Below this threshold value, the output may vary linearly with the concentration of the response regulator. Using chemical reactions network theory, Shinar and Feinberg have proposed a more generalized “absolute concentration robustness” (ACR) for certain mass-reaction networks, including the TCS (22). This ACR model holds for a bifunctional kinase whose cofactor for phosphatase activity is either ATP or ADP (23). Gao and Stock developed a novel method to estimate the level of phosphorylated response regulator in vivo and showed that the phosphatase activity is essential for maintaining the robustness of a TCS. Using a PhoB/PhoR TCS of E. coli, they have shown that the level of phosphorylated response regulator has little effect on the total concentration of the protein if it remains within a saturating range. Here, we designed a synthetic circuit using a TCS of M. tuberculosis, MprAB, for direct observation in vivo of the dependence of the system output on the system components. We have systematically varied the concentration of either MprA or MprB or both and measured the output as the level of MprA∼P-dependent mCherry expression in E. coli. This recombinant assay has two advantages. First, the assay does not require the removal of indigenous TCS of E. coli that could affect the overall behavior of the bacteria. Second, there is no cross talk between MprAB and the other TCS in E. coli that could jeopardize the measurement of the actual output. Since the exogenous TCS components are more likely to have cross talk than the endogenous one (24), we tested it by expressing MprA in the absence of MprB and comparing the level of induction of mCherry with respect to the M. tuberculosis RNAP holoenzyme. The lack of mCherry induction confirmed the absence of cross talk among the exogenous E. coli TCS and MprA in vivo (see Fig. S6 in the supplemental material). We have previously shown that the endogenous transcription factor in E. coli does not interfere with the function of its counterpart in M. tuberculosis when expressed in the bacteria (14). We had developed a recombinant reporter assay by successfully expressing M. tuberculosis RNAP and its transcription factor in E. coli to monitor the mCherry expression from a promoter that is inducible in the presence of the transcription factor (15). This recombinant reporter assay was further used for two orthologous transcription factors (25–27). Here, four plasmids were used for simultaneous expression of the proteins (M. tuberculosis RNAP, MprA, and MprB) and MprA∼P-dependent mCherry expression in E. coli. It is important to note that we did not intend to monitor the input-output relation; rather, we monitored how the output of the TCS varied when the concentrations of the components were altered. The synthetic circuit allowed us to artificially vary the concentration of recombinant TCS without affecting the normal cell growth. Assuming that saturating levels of ATP and RNAP are present in the cell, we observed that the MprA∼P-dependent mCherry expression is independent of any change in MprB level for a fixed MprA level, whereas the mCherry expression level at a fixed MprB could vary with the MprA level until the level of the regulator attains a certain threshold. Of note, the in vivo assay involved an MprB level which is lower than the MprA level. In our assay, the recombinant proteins MprA-MprB were expressed either from the same promoter (tetRO) or from two different promoters (tetRO and araB) using the E. coli transcriptional and translational machinery. Even at a high inducer concentration, the level of soluble MprB is 2- to 5-fold lower than MprA. This observation corroborates the in vivo relative concentrations of MprA and MprB (28). In the in vivo experiments, we were not able to perform the assay at a fixed concentration of sensor kinase, which is higher than the response regulator. There is no such limitation in the in vitro assays. In these assays, when the sensor kinase level is fixed, the saturation signal is achieved when the concentration of the response regulator is greater than or comparable to that of the sensor kinase. The results correspond to the in vivo observation as in Fig. 1. In contrast, at a fixed level of response regulator, the output signal is constant even when the concentration of sensor kinase is higher than that of the response regulator. Thus, although the actual level of sensor kinase is much lower than the response regulator in vivo, the robustness of a TCS does not necessarily require the level of sensor kinase to be lower than response regulator. However, this robustness holds true only when the response regulator is above a certain threshold value. This observation is consistent with the prediction of the theoretical model proposed by Shinar et al. (9) and with the report of Gao and Stock (10), i.e., that the robustness of TCS holds when the concentration of the response regulator is within the saturating range. On the other hand, for a fixed concentration of response regulator, the output is constant irrespective of the concentration of the sensor kinase. If the concentration of response regulator is less than the threshold value, the output is still constant, but at a lower level. Overall, our observation is somewhat contrary to the proposition by Batchelor and Goulian that the robustness does not hold well when the concentrations of response regulator and sensor kinase are comparable, but does corroborate the model proposed by Shinar et al. that the output of a TCS does not depend on the concentration of the system components if the concentration of the response regulator is above a certain threshold value.
We demonstrate that MprA∼P acts as a transcription factor for the ppk1 promoter, which has not been reported to date. Previously, the binding sites of RegX3 and SigE on the ppk1 promoter have been reported (16), but the mechanism of activation of the promoter remained unclear. We have observed that MprA∼P binds to the promoter region of ppk1 gene and activates transcription from the promoter in the presence of the RNAP-σE holoenzyme.
The synthetic circuit used in this report involves four plasmids expressing seven recombinant proteins simultaneously in E. coli. We propose that a similar synthetic circuit could be used as a model system to identify/validate the relationship among different components of gene regulatory networks, protein-protein interaction networks, and protein-DNA interaction networks. Moreover, the synthetic circuit could be a better alternative approach for the in vivo reporter assays aiming toward characterization of interactions of biomolecules that are often masked by interference from cross talk and nonspecific interaction by other biomolecules.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
Bacterial strains used in this study are listed in Table S1 in the supplemental material. E. coli strains DH5α and BL21(DE3) were used for cloning and overexpression of recombinant proteins and the in vivo reporter assay, respectively. E. coli cells transformed with a plasmid(s) were plated in LB agar medium (10 g tryptone, 5 g yeast extract, and 10 g NaCl at pH 7.0) and further grown in either LB medium or in 2× YT medium (16 g tryptone, 8 g yeast extract, and 5 g NaCl at pH 7.0) supplemented with antibiotics (ampicillin [Amp] at 100 μg/ml, kanamycin [Kan] at 50 μg/ml, chloramphenicol [Chl] at 35 μg/ml, and streptomycin [Strep] at 50 μg/ml) as required (Table S2). Restriction endonucleases, DNA polymerases, and T4DNA ligase were supplied by New England BioLabs. All constructs reported in this study were sequenced to verify their integrity using an automated DNA Sequencer (Big dye terminator V3.1; Applied Biosystems).
Cloning strategies.
The plasmids used in this study and the construction of plasmids are described in Table S3.
Purification of recombinant MprA, MprB, and M. tuberculosis RNAP-SigEholo.
For purification of MprA and MprB, each of the genes was cloned into the pACYC Duet vector separately. For the mprB gene, only the coding sequence for the cytosolic part (amino acids 196 to 504) was cloned to increase the solubility of the protein (6, 13, 29). The genes were amplified by PCR from M. tuberculosis genomic DNA using the primers listed in Table S4. The mprA gene was cleaved and inserted into the vector with restriction enzymes BamHI and KpnI, while the mprB gene was inserted by blunt end ligation using BamHI and EcoRV.
The plasmids encoding MprA or truncated MprB were transformed into E. coli BL21(DE3) cells, and cells were grown in 2× YT medium with chloramphenicol (35 μg/ml) at 37°C until the optical density at 600 nm (OD600) reached 0.4, induced with 0.5 mM IPTG, and then further grown at 16°C for 14 to 16 h. Harvested cells were resuspended in buffer A (50 mM Tris-Cl [pH 7.5], 200 mM NaCl, 5% glycerol, and 5 mM β-mercaptoethanol) containing 2.5 mg/ml deoxycholic acid, 10 μg of DNase/ml, 10 μg of RNase/ml, and 1 mM phenylmethanesulfonyl fluoride, and were sonicated. Cell lysate was then centrifuged at 17,000 rpm for 30 min at 4°C. The pellet was discarded and the supernatant was passed through an Ni-nitrilotriacetic acid (Ni-NTA) column preequilibrated with buffer A. The column was washed several times with buffer A and buffer A plus 20 mM imidazole. Proteins were eluted with 200 mM immidazole in buffer A. In the case of MprB, an additional step of ammonium sulfate precipitation (0.25 g/ml) was carried out prior to the nickel affinity chromatography. The purity of eluted protein samples were judged by resolving them on 12.5% SDS-PAGE gel and staining with Coomassie brilliant blue.
M. tuberculosis RNAP core and M. tuberculosis RNAP-SigE holoenzyme were purified as described by Rudra et al. (27).
In vitro phosphorylation assay.
The specific amounts of sensor kinase MprB were autophosphorylated at 37°C in the presence of 5 mM (0.4 μCi, or 0.125 mCi/ml, or 37.5 Ci/mmol) [γ-32P]ATP for 10 min in 4 μl phosphorylation buffer (50 mM Tris-Cl [pH 7.6], 50 mM KCl, and 20 mM MgCl2). For transfer of the phosphoryl group to the cognate response regulator, specific amounts of MprA were incubated at 37°C for 30 min. The free ATP was removed by adding the reaction mixtures to 5 μl Ni-NTA beads (preequilibrated with phosphorylation buffer), and the mixtures were kept on ice for 5 min, followed by centrifugation at 10,600 × g, 4°C for 10 min. After removing the supernatant, the nickel beads were resuspended in 8 μl phosphorylation buffer, 800 mM imidazole, and 5 μl protein loading dye (5×). The radiolabeled proteins were resolved by 12.5% SDS-PAGE and viewed in a storage phosphorimager (Typhoon trio+; GE Healthcare).
In vitro transcription assay.
The ppk1 DNA fragment (from base −76 to +61) was amplified by PCR using primers listed in Table S2. Purified M. tuberculosis RNAP-SigE holoenzyme (200 nM) was incubated with 100 nM ppk1 promoter DNA for 15 min at 37°C to allow open complex formation in transcription buffer (50 mM Tris-Cl [pH 8], 100 mM KCl, 10 mM MgCl2, 1 mM dithiothreitol [DTT], and 5% glycerol). Various concentrations of phosphorylated MprA from the phosphotransfer assay were added at the open complex stage and further incubated for 5 min. RNA synthesis was initiated by addition of nucleoside triphosphate (NTP) mix (final concentration: 250 μM GTP, ATP, UTP and 50 μM CTP containing 0.4 μCi [α-32P]CTP) along with heparin (25 μg/ml) at 37°C for 30 min. Reactions were stopped by adding 2.5 μl of formamide dye (80% formamide, 10 mM EDTA, 0.01% bromophenol blue, 0.01% xylene cyanol), heating at 95°C for 5 min and chilling on ice. Samples were resolved in a 12% urea-PAGE gel (31) and were scanned by storage phosphor scanner (Typhoon trio+; GE Healthcare).
In vivo recombinant reporter assay in E. coli.
E. coli BL21(DE3) cells were cotransformed with either three plasmids (pAcYc Duet-rpoA-sigE, pCOLA Duet-rpoB-rpoC, and pFPV-ppk1-mCherry-tetRO-MprAB) or four plasmids (pAcYc Duet-rpoA-sigE, pCOLA Duet-rpoB-rpoC, pCDF Duet-para-mprB, and pFPV-ppk1-mCherry-tetRO-MprA). The cells were grown in 50 ml LB broth at 37°C with appropriate antibiotics (Amp, Kan, and Chl for the three-plasmid expression system, or Amp, Kan, Chl, and Strep for the four-plasmid expression system) to an OD600 of 0.4, then induced by adding 0.5 mM IPTG and various concentrations of anhydrotetracycline (for the three-plasmid expression system) or 0.5 mM IPTG, 1.0 μg/ml l-arabinose, various concentrations of anhydrotetracycline or 0.5 mM IPTG, 50 ng/ml anhydrotetracycline, and various concentrations of l-arabinose (for the four-plasmid expression system). After induction, the cells were further grown at 16°C for 16 h and the fluorescence intensities of 1 ml of cells were monitored (excitation = 587 nm, emission = 610 nm; bandwidth, 5.5 nm) by a spectrofluorometer (PTI Inc.). As a background, the assay was performed with E. coli BL21(DE3) as above except pAcYc Duet rpoA-sigE was replaced by pAcYc Duet rpoA-rpoZ for RNAP core expression (15).
Each assay was repeated 3 times and the averages of the data were estimated after subtracting the background (i.e., samples with the RNAP core only). Fold increases in the fluorescence intensities of the samples with respect to the control containing the RNAP holoenzyme only (no inducer of MprA) were plotted as a function of protein concentration.
Identical assays were conducted as above replacing ppk1 with the promoter for sigE (p1p2) (30) for the MprA∼P-dependent expression of mCherry, and with a phosphorylation-deficient mutant of MprA (MprA[D48A]) (13) instead of active MprA.
Western blot analysis.
A part of the samples from the in vivo assay were harvested at 5,000 × g for 5 min and resuspended in 20 μl lysis buffer (Cell Signaling Technology) containing protease inhibitor cocktail (Roche Diagnostics) and incubated in ice for 20 min, followed by centrifugation at 13,000 rpm for 10 min at 4°C. Supernatants were collected and the total protein concentration of each sample was estimated using a BCA kit (Thermo Fisher Scientific). Equal amounts of protein were mixed with 5 μl protein loading dye, cooked for 10 min, and resolved in a 12.5% SDS-PAGE gel, followed by electrophoretic transfer of protein samples onto a polyvinylidene difluoride (PVDF) membrane (Millipore, USA) at a constant current of 0.8 mA/cm2. The membrane was then blocked by 5% skim milk in a 1× Tris-buffered saline with Tween 20 (TBST) buffer (50 mM Tris-Cl [pH 7.6], 150 mM NaCl, 0.1% [vol/vol] Tween 20) for 2 h, washed, and incubated overnight at 4°C with primary antibodies (BioBharti Lifesciences) raised against either MprA or MprB in 5% bovine serum albumin (BSA) dissolved in 1× TBST buffer, followed by washing and horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG secondary antibody incubation in 5% skim milk dissolved in 1× TBST buffer for 1 h. The membrane was then washed properly and immersed in the substrate solution (LumiGlo; Cell Signaling Technology), wrapped in a cellophane paper and exposed to X-ray film for development. The film was scanned in a Gel Documentation System (Bio-Rad Inc.) and the amounts of protein in the bands were quantified by comparing the intensities of the bands with the band of same protein of known quantity.
Statistical analysis.
The statistical analysis was performed using the R programming language (version 3.6.1), an open-source platform for statistical computing (R Core Team, 2013). Due to the low sampling size (n of <20), we computed a different nonparametric statistical test like the Kruskal-Wallis test (comparing three data sets). The results from these statistical tests have been incorporated accordingly in Results and Discussion.
Supplementary Material
ACKNOWLEDGMENTS
We thank Uri Alon (Weizmann Institute of Science) and Runa Sur (University of Calcutta) for critically reading the manuscript and giving comments and Arindam Roy (Bose Institute) for performing the statistical analysis.
This work is supported by a research grant from the Department of Biotechnology, India, number BT/PR5270/BRB/10/1066/2012.
We declare no conflicts of interest with the contents of this article.
J.M. and S.K.B. conceived and designed the experiments, A.D. and P.R. performed the experiments, A.D. and J.M. analyzed the data, and J.M. wrote the paper.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.West AH, Stock AM. 2001. Histidine kinases and response regulator proteins in two-component signaling systems. Trends Biochem Sci 26:369–376. doi: 10.1016/s0968-0004(01)01852-7. [DOI] [PubMed] [Google Scholar]
- 2.Laub MT, Goulian M. 2007. Specificity in two-component signal transduction pathways. Annu Rev Genet 41:121–145. doi: 10.1146/annurev.genet.41.042007.170548. [DOI] [PubMed] [Google Scholar]
- 3.Barkai N, Leibler S. 1997. Robustness in simple biochemical networks. Nature 387:913–917. doi: 10.1038/43199. [DOI] [PubMed] [Google Scholar]
- 4.Spudich JL, Koshland DE. Jr. 1976. Non-genetic individuality: chance in the single cell. Nature 262:467–471. doi: 10.1038/262467a0. [DOI] [PubMed] [Google Scholar]
- 5.Levin MD, Morton-Firth CJ, Abouhamad WN, Bourret RB, Bray D. 1998. Origins of individual swimming behavior in bacteria. Biophys J 74:175–181. doi: 10.1016/S0006-3495(98)77777-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hsing W, Russo FD, Bernd KK, Silhavy TJ. 1998. Mutations that alter the kinase and phosphatase activities of the two-component sensor EnvZ. J Bacteriol 180:4538–4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hart Y, Alon U. 2013. The utility of paradoxical components in biological circuits. Mol Cell 49:213–221. doi: 10.1016/j.molcel.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 8.Batchelor E, Goulian M. 2003. Robustness and the cycle of phosphorylation and dephosphorylation in a two-component regulatory system. Proc Natl Acad Sci U S A 100:691–696. doi: 10.1073/pnas.0234782100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shinar G, Milo R, Martinez MR, Alon U. 2007. Input output robustness in simple bacterial signaling systems. Proc Natl Acad Sci U S A 104:19931–19935. doi: 10.1073/pnas.0706792104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao R, Stock AM. 2013. Probing kinase and phosphatase activities of two-component systems in vivo with concentration-dependent phosphorylation profiling. Proc Natl Acad Sci U S A 110:672–677. doi: 10.1073/pnas.1214587110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bretl DJ, Demetriadou C, Zahrt TC. 2011. Adaptation to environmental stimuli within the host: two-component signal transduction systems of Mycobacterium tuberculosis. Microbiol Mol Biol Rev 75:566–582. doi: 10.1128/MMBR.05004-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kundu M. 2018. The role of two-component systems in the physiology of Mycobacterium tuberculosis. IUBMB Life 9. doi: 10.1002/iub.1872. [DOI] [PubMed] [Google Scholar]
- 13.Zahrt TC, Wozniak C, Jones D, Trevett A. 2003. Functional analysis of the Mycobacterium tuberculosis MprAB two-component signal transduction system. Infect Immun 71:6962–6970. doi: 10.1128/iai.71.12.6962-6970.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Banerjee R, Rudra P, Prajapati RK, Sengupta S, Mukhopadhyay J. 2014. Optimization of recombinant Mycobacterium tuberculosis RNA polymerase expression and purification. Tuberculosis (Edinb) 94:397–404. doi: 10.1016/j.tube.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 15.Banerjee R, Rudra P, Saha A, Mukhopadhyay J. 2015. Recombinant reporter assay using transcriptional machinery of Mycobacterium tuberculosis. J Bacteriol 197:646–653. doi: 10.1128/JB.02445-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanyal S, Banerjee SK, Banerjee R, Mukhopadhyay J, Kundu M. 2013. Polyphosphate kinase 1, a central node in the stress response network of Mycobacterium tuberculosis, connects the two-component systems MprAB and SenX3-RegX3 and the extracytoplasmic function sigma factor, sigma E. Microbiology 159:2074–2086. doi: 10.1099/mic.0.068452-0. [DOI] [PubMed] [Google Scholar]
- 17.He H, Hovey R, Kane J, Singh V, Zahrt TC. 2006. MprAB is a stress-responsive two-component system that directly regulates expression of sigma factors SigB and SigE in Mycobacterium tuberculosis. J Bacteriol 188:2134–2143. doi: 10.1128/JB.188.6.2134-2143.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hart Y, Madar D, Yuan J, Bren A, Mayo AE, Rabinowitz JD, Alon U. 2011. Robust control of nitrogen assimilation by a bifunctional enzyme in E. coli. Mol Cell 41:117–127. doi: 10.1016/j.molcel.2010.12.023. [DOI] [PubMed] [Google Scholar]
- 19.Maity AK, Bandyopadhyay A, Chaudhury P, Banik SK. 2014. Role of functionality in two-component signal transduction: a stochastic study. Phys Rev E Stat Nonlin Soft Matter Phys 89:032713. doi: 10.1103/PhysRevE.89.032713. [DOI] [PubMed] [Google Scholar]
- 20.Wei K, Moinat M, Maarleveld TR, Bruggeman FJ. 2014. Stochastic simulation of prokaryotic two-component signalling indicates stochasticity-induced active-state locking and growth-rate dependent bistability. Mol Biosyst 10:2338–2346. doi: 10.1039/c4mb00264d. [DOI] [PubMed] [Google Scholar]
- 21.Cai SJ, Inouye M. 2002. EnvZ-OmpR interaction and osmoregulation in Escherichia coli. J Biol Chem 277:24155–24161. doi: 10.1074/jbc.M110715200. [DOI] [PubMed] [Google Scholar]
- 22.Shinar G, Feinberg M. 2010. Structural sources of robustness in biochemical reaction networks. Science 327:1389–1391. doi: 10.1126/science.1183372. [DOI] [PubMed] [Google Scholar]
- 23.Zhu Y, Qin L, Yoshida T, Inouye M. 2000. Phosphatase activity of histidine kinase EnvZ without kinase catalytic domain. Proc Natl Acad Sci U S A 97:7808–7813. doi: 10.1073/pnas.97.14.7808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Capra EJ, Laub MT. 2012. Evolution of two-component signal transduction systems. Annu Rev Microbiol 66:325–347. doi: 10.1146/annurev-micro-092611-150039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prajapati RK, Sengupta S, Rudra P, Mukhopadhyay J. 2016. Bacillus subtilis delta factor functions as a transcriptional regulator by facilitating the open complex formation. J Biol Chem 291:1064–1075. doi: 10.1074/jbc.M115.686170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prajapati RK, Sur R, Mukhopadhyay J. 2016. A novel function of delta factor from Bacillus subtilis as a transcriptional repressor. J Biol Chem 291:24029–24035. doi: 10.1074/jbc.M116.746065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rudra P, Prajapati RK, Banerjee R, Sengupta S, Mukhopadhyay J. 2015. Novel mechanism of gene regulation: the protein Rv1222 of Mycobacterium tuberculosis inhibits transcription by anchoring the RNA polymerase onto DNA. Nucleic Acids Res 43:5855–5867. doi: 10.1093/nar/gkv516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schubert OT, Ludwig C, Kogadeeva M, Zimmermann M, Rosenberger G, Gengenbacher M, Gillet LC, Collins BC, Rost HL, Kaufmann SH, Sauer U, Aebersold R. 2015. Absolute proteome composition and dynamics during dormancy and resuscitation of Mycobacterium tuberculosis. Cell Host Microbe 18:96–108. doi: 10.1016/j.chom.2015.06.001. [DOI] [PubMed] [Google Scholar]
- 29.Bretl DJ, Bigley TM, Terhune SS, Zahrt TC. 2014. The MprB extracytoplasmic domain negatively regulates activation of the Mycobacterium tuberculosis MprAB two-component system. J Bacteriol 196:391–406. doi: 10.1128/JB.01064-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dona V, Rodrigue S, Dainese E, Palu G, Gaudreau L, Manganelli R, Provvedi R. 2008. Evidence of complex transcriptional, translational, and posttranslational regulation of the extracytoplasmic function sigma factor sigmaE in Mycobacterium tuberculosis. J Bacteriol 190:5963–5971. doi: 10.1128/JB.00622-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.