

Abstract
Background and Purpose
A critical role for sphingosine kinase/sphingosine‐1‐phosphate (S1P) pathway in the control of airway function has been demonstrated in respiratory diseases. Here, we address S1P contribution in a mouse model of mild chronic obstructive pulmonary disease (COPD).
Experimental Approach
C57BL/6J mice have been exposed to room air or cigarette smoke up to 11 months and killed at different time points. Functional and molecular studies have been performed.
Key Results
Cigarette smoke caused emphysematous changes throughout the lung parenchyma coupled to a progressive collagen deposition in both peribronchiolar and peribronchial areas. The high and low airways showed an increased reactivity to cholinergic stimulation and α‐smooth muscle actin overexpression. Similarly, an increase in airway reactivity and lung resistances following S1P challenge occurred in smoking mice. A high expression of S1P, Sph‐K2, and S1P receptors (S1P2 and S1P3) has been detected in the lung of smoking mice. Sphingosine kinases inhibition reversed the increased cholinergic response in airways of smoking mice.
Conclusions and Implications
S1P signalling up‐regulation follows the disease progression in smoking mice and is involved in the development of airway hyperresponsiveness. Our study defines a therapeutic potential for S1P inhibitors in management of airways hyperresponsiveness associated to emphysema in smokers with both asthma and COPD.
What is already known
An efficient therapeutic approach to control airway dysfunction associated to smoke‐induced emphysema is still lacking.
S1P signalling is involved in the control of airways function.
What this study add
S1P signalling contributes to cigarette smoke‐induced airway hyperresponsiveness (AHR) in a time‐dependent manner walking together with the disease progression.
What is the clinical significance
S1P pathway represents a promising novel therapeutic target for treatment of AHR associated to emphysema and asthma in COPD.
Abbreviations
- DAB
3,3′‐diaminobenzidine tetra hydrochloride
- AHR
airway hyperresponsiveness
- α‐SMA
α‐smooth muscle actin
- COPD
chronic obstructive pulmonary disease
- CS
cigarette smoke
- Sph‐K
sphingosine kinase
- S1P
sphingosine‐1‐phosphate
1. INTRODUCTION
Sphingosine‐1‐phosphate (https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=911) is a bioactive lysophospholipid generated from intracellular sphingosine, a product of the cell membrane component sphingomyelin. https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=773 isoenzymes catalyse the breakdown of sphingomyelin by cleavage of the phosphorycholine linkage releasing ceramide that in turn is converted to sphingosine (Pyne & Pyne, 2002). Sphingomyelinase is activated by several stress inductors such as shear stress, TNF, oxidative stress, and cigarette smoke (Levy, Khan, Careaga, & Goldkorn, 2009; Uhlig & Gulbins, 2008). The catabolically generated sphingosine is phosphorylated by sphingosine kinases, namely, Sph‐K1 and Sph‐K2. Once formed, S1P takes one of three pathways. In one, the sphingosine moiety of S1P is recycled to ceramide synthesis after dephosphorylation by specific phosphatases. In a second, S1P is irreversibly degraded by S1P lyase. This reaction facilitates transfer of substrate from the sphingolipid to the glycerolipid pathway. In the third pathway, intracellular S1P is released to the extracellular environment, a process that is highly efficient in red blood cells, platelets, and endothelial cells. One exported out of cells by cell‐specific transporters S1P interacts with five plasma membrane GPCRs (S1PRn), which promote intracellular signals to regulate cell behaviour (Proia & Hla, 2015).
In the last years, a critical role for the Sph‐K/S1P pathway in the control of airway function and pulmonary inflammation has been demonstrated (Price et al., 2013; Roviezzo et al., 2004; Roviezzo et al., 2007; Roviezzo et al., 2010; Roviezzo et al., 2015; Roviezzo et al., 2016). Compelling evidence from rodent models suggests that S1P signalling represents feasible therapeutic target(s) for lung diseases (Arish, Alaidarous, Ali, Akhter, & Rub, 2017; Ebenezer, Fu, & Natarajan, 2016). In particular, the inhibition of S1P biosynthesis reduces allergen‐induced asthma‐like features (Price et al., 2012; Roviezzo et al., 2007),while the administration of S1P worsens antigen‐induced airway inflammation (Chiba, Takeuchi, Sakai, & Misawa, 2010) and airway hyperresponsiveness (AHR). Furthermore, the systemic administration of S1P, without additional adjuvant factors, triggers in the mouse a disease closely mimicking several features of severe asthma such as AHR, pulmonary inflammation, high circulating levels of IgE, and a predominance of CD4+T cell‐derived IL‐4 (Roviezzo et al., 2015). This finds a match in the human disease, where S1P levels significantly increase in bronchoalveolar lavage of asthmatic patients following segmental allergen challenge and correlate to pulmonary inflammation (Ammit et al., 2009).
An involvement of S1P signalling in asthma pathogenesis finds further support in genome‐wild associated studies that have linked Orm/ORMDL family proteins, negative regulators of sphingolipid metabolism, to asthma onset in childhood (Breslow et al., 2010; Miller et al., 2017; Miller, Rosenthal, Beppu, Gordillo, & Broide, 2017; Moffatt et al., 2010). In addition, changes in sphingolipid metabolism have been related to chronic obstructive pulmonary disease (COPD) (Ahmed et al., 2014; Yang & Uhlig, 2011).
The most important risk factor for COPD is cigarette smoking (Mannino et al., 2006). COPD is characterized by progressive obstruction of airflow, not fully reversible, which is accompanied by a chronic inflammatory response, caused by deleterious particles or gases, in airways and lung parenchyma (Fabbri & Rabe, 2007). There are currently no specific COPD treatments, and smoking cessation remains the most effective therapeutic intervention (Scanlon et al., 2000). Several studies in humans and animals demonstrated that, once COPD is initiated, the pulmonary inflammatory response continues (De Cunto et al., 2018; Gamble et al., 2007; Lapperre et al., 2006; Scanlon et al., 2000; Willemse, ten Hacken, Rutgers, Postma, & Timens, 2005), leading to a progressive loss of alveolar wall that cannot be reversed. Abnormal amounts of sphingomyelinase have been found in smokers with emphysema and the increased ceramide levels in alveolar septal cells and macrophages well correlated to lung destruction and dysfunction (Petrache et al., 2005; Petrache et al., 2008; Telenga et al., 2014).
Thus, persistent lung inflammation, associated with a progressive deterioration of respiratory function and infections in these patients, is an increasingly important clinical problem. Most of the clinical features and physiological abnormalities of mild‐to‐moderate COPD may be linked to AHR state (Cockcroft & Wenzel, 2016; Hospers, Postma, Rijcken, Weiss, & Schouten, 2000). Smokers with both asthma and COPD (Asthma‐COPD Overlap Syndrome, ACOS) recapitulate the above described clinical features and are exposed to an increased risk of disease progression and mortality (Tashkin et al., 1996; Vestbo et al., 2013). Clinically, the asthma‐COPD phenotype is identified by methacholine provocation test in one out of four patients (Tkacova et al., 2016).
It is well established that the airway smooth muscle dysfunction plays a key role in COPD pathogenesis being involved in the size of the airways, alterations of contractility, inflammatory response, and the asthmatic phenotype. These changes are recognized as an important cause of airflow obstruction in smoker individuals (Barnes, 2017; Jones, Noble, Elliot, & James, 2016; Page, O'Shaughnessy, & Barnes, 2017) and are reproduced in animal models (Bartalesi et al., 2005;Cavarra et al., 2001 ; De Cunto et al., 2018). The aim of this study is to assess, by using a well‐characterised model of mild COPD, the role of S1P signalling in airway dysfunction associated to smoke‐induced emphysematous lesions.
2. METHODS
2.1. Animal model
C57BL/6J (MGI Cat# 5659366, RRID:5659366) mice were housed in a controlled temperature, humidity, and light/dark cycle environment and with food and water ad libitum (Mucedola Global Diet 2018; Harlan, Correzzana, Italy). All animal experiments were conducted under license number 186/2015_PR released by the Italian Ministry of Health and in agreement with the Guiding Principles for Research Involving Animals and Human Beings. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010) and with the recommendations made by the British Journal of Pharmacology. Mice were exposed to either room air (control mice) or to smoke from cigarettes (CS; Marlboro Red, 12 mg of tar and 0.9 mg of nicotine) in especially designed macrolon cages (Tecniplast, Buguggiate, Italy). The methodological approach for the exposure to cigarette smoke has been previously described in details (Cavarra et al., 2001; De Cunto et al., 2018). Briefly, mice were exposed to cigarette smoke from three cigarettes per day, 5 days a week, for an overall period of 11 months.
2.2. Bronchial reactivity
Mice were killed at different time points, and bronchial tissues were rapidly dissected and cleaned from fat and connective tissue. Rings of 1–2 mm length were cut and placed in organ baths filled with Krebs' solution (mM: 118 NaCl, 4.7 KCl, 1.2 MgCl2, 1.2 KH2PO4, 2.5 CaCl2, 25 NaHCO3, and 11 glucose) at 37°C and gassed with a mixture of O2/CO2 (95/5%). Rings were mounted to isometric force transducers (7006, Ugo Basile, Comerio, Italy) and connected to a Powerlab 800 (AD Instruments, Sidney, Australia). Rings were initially stretched until a resting tension of 0.5 g was reached and allowed to equilibrate for at least 30 min. In each experiment, bronchial rings were challenged with carbachol (10 μM) until the response was reproducible. Once a reproducible response was achieved, bronchial reactivity was assessed performing a cumulative concentration–response curve to carbachol (10 nM–30 μM) or S1P (10 nM–30 μM). In another set of experiments, bronchial tissue reactivity was assessed in presence of S1P2 antagonist (https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=10310, 10 μM, 15 min; Tocris Bioscience, UK), S1P3 antagonist (https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2917, 10 μM, 15 min), or Sph‐K inhibitor (https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6041, 10 μM, 30 min; Tocris, Bristol, UK).
2.3. Isolated perfused mouse lung preparation
Lung function was assessed using an isolated and perfused mouse lung model. Lungs were perfused in a non‐recirculating fashion through the pulmonary artery at constant flow of 1 ml·min−1 resulting in a pulmonary artery pressure of 2–3 cmH2O. The perfusion medium used was RPMI‐1640 lacking phenol red (37°C). The lungs were ventilated by negative pressure (−3 and −9 cmH2O) with 90 breaths per minute and a tidal volume of about 200 μl. A hyperinflation (−20 cmH2O) was performed every 5 min. Artificial thorax chamber pressure was measured with a differential pressure transducer (Validyne DP 45‐24, Instrumentation Devices, Como, Italy) and airflow velocity with pneumotachograph tube connected to a differential pressure transducer (Validyne DP 45‐15, Instrumentation Devices, Como, Italy). The lungs respired humidified air. The arterial pressure was continuously monitored by means of pressure transducer (Isotec Healthdyne, Irvine, USA) that was connected with the cannula ending in the pulmonary artery. All data were transmitted to a computer and analysed with Pulmodyn software (Hugo Sachs Elektronik, March Hugstetten, Germany). Data were analysed through the following formula: P = V·C−1 + RL·dV·dt−1, where P is chamber pressure, C pulmonary compliance, V tidal volume, and RL airway resistance. Successively, airway resistance value registered was corrected for the resistance of the pneumotacometer. Lungs were perfused and ventilated for 45 min without any treatment in order to obtain baseline state. Subsequently, lungs were challenged with carbachol. Dose–response curve of carbachol or S1P was administered as bolus. In another set of experiments, lungs were perfused with S1P inhibitors before carbachol challenge.
2.4. Morphology and morphometry
Lungs of six mice from each group were processed for histology, and lung slides were analysed for morphology and morphometry. The lungs from the different groups of mice were fixed intratracheally with buffered formalin (5%) at a constant pressure of 20 cmH2O at least for 24 hr. All lungs were then dehydrated, cleared in toluene, and embedded in paraffin. The lungs of mice were then processed for histological, morphometric, and immunohistochemical analyses. Lung slices were stained with haematoxylin/eosin. Morphometric assessment of emphysema included mean linear intercept (Lm) and internal surface area (ISA). Peribronchiolar fibrosis was evaluated in paraffin‐embedded lung slices after Masson's trichrome staining (Lucattelli et al., 2005).
2.5. Immunohistochemical analysis
The immuno‐related procedures used comply with the recommendations made by the British Journal of Pharmacology. Tissue sections from mice exposed to room air or CS for 9, 10, and 11 months were also stained for α‐smooth muscle actin (α‐SMA), sphingosine 1‐phosphate receptor 2 (S1P2), sphingosine 1‐phosphate receptor 3 (S1P3), sphingosine kinase 1 (Sph‐K1), and sphingosine kinase 2 (Sph‐K2). The sections were pretreated with 3% hydrogen peroxide for blocking the endogenous peroxidase. Antigen retrieval was performed by heating the sections in a microwave for 20 min in 0.01‐M pH 6.0 citrate buffer and allowing to cool slowly to room temperature. All the sections were incubated with 3% BSA for 30 min at room temperature to block non‐specific antibody binding. Sections were then incubated overnight at 4°C with the primary antibodies, rabbit Ab to mouse Sph‐K1 (Bioss Cat# bs‐2652R, RRID:AB_10856265) diluted 1:200; rabbit Ab to mouse Sph‐K2 (Abcam Cat# ab37977, RRID:AB_778046) diluted 1:70; rabbit Ab to mouse S1P3 (Bioss Antibodies, Woburn, US) diluted 1:100; rabbit Ab to mouse S1P2 (Biorbyt Cat# orb5558, RRID:AB_10938778) diluted 1:150. After rinsing with TBST, slides were incubated with sheep anti‐rabbit IgG (1:200) for 30 min at room temperature followed by incubation with peroxidase‐antiperoxidase complex, prepared from rabbit serum and diluted 1:200. Colour development was performed using 3,3′‐diaminobenzidine tetra hydrochloride (DAB; Vector Laboratories, Burlingame, CA) as a chromogen. Detection of α‐SMA protein was evaluated on paraffin sections by using mouse monoclonal α‐SMA Ab (Sigma, St. Louis, USA) diluted 1:400. Reaction was visualized using the MOM immunodetection kit (Vector Laboratories, Burlingame, USA) and DAB as substrate. The MOM immunodetection kit (Vector Laboratories) is designed specifically to localize mouse primary monoclonal and polyclonal antibodies on mouse tissues by using a novel blocking agent and reducing undesired background staining. As negative controls, all primary antibodies were replaced by non‐immunised‐specific serum.
2.6. S1P measurement
S1P quantification was performed by using a commercially available elisa kit (Echelon, Tebu‐bio, Magenta, Italy) on whole lung samples harvested from control and smoking mice at 11 months following CS exposure. Samples were mechanically homogenised in the following homogenization buffer: 20‐mM Tris–HCl, pH 7.4; 20% glycerol; 1 1. mM β‐mercaptoethanol; 1‐mM EDTA; 1‐mM Na‐orthovanadate; 15‐mM NaF; 1‐mM PMSF; protease inhibitor cocktail; 0.5‐mM deoxypyridoxine; and 40‐mM β‐glycerophosphate. Total protein concentration was measured, and homogenated samples were then frozen at −80°C until analysis. Samples were standardized according the kit protocol, and data were reported as picomoles of S1P per micrograms of total protein.
2.7. Randomization, blinding, and normalization
Experiments were performed by different operators, and animals were randomly used to set up control and CS groups. Drug treatments were randomized each day of experiment. Immunohistochemical experiments were performed by one operator, and analysis of images was carried out by a second one in a blind approach. Data were not normalized and were expressed as absolute values as they were obtained.
2.8. Unequal group sizes
The group size was not equal for all experiments, as detailed in figure legends. In some cases, n for control groups is higher than 5 since the controls have been repeated in all animal experiments. In addition, the procedure does not allow to run the whole set of treatments for each type of test. The number of animals has been determined by using G‐Power software with α = .05 and 1 − β = .85.
2.9. Data and statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2015). Results are presented as mean ± SEM or mean ± SD. Two‐way ANOVA followed by Bonferroni's post hoc test was performed throughout the data shown in Figures 2, 4, and 5. The Student's t test was run for data shown in Figure 8e (mean ± SEM) and Table 1 (mean ± SD). Bonferroni's post hoc test was run only when F was significant. Significance was determined as P < .05 (denoted by *, °, or # in figures). All data were plotted and analysed by using GraphPad Prism 5.
Figure 2.
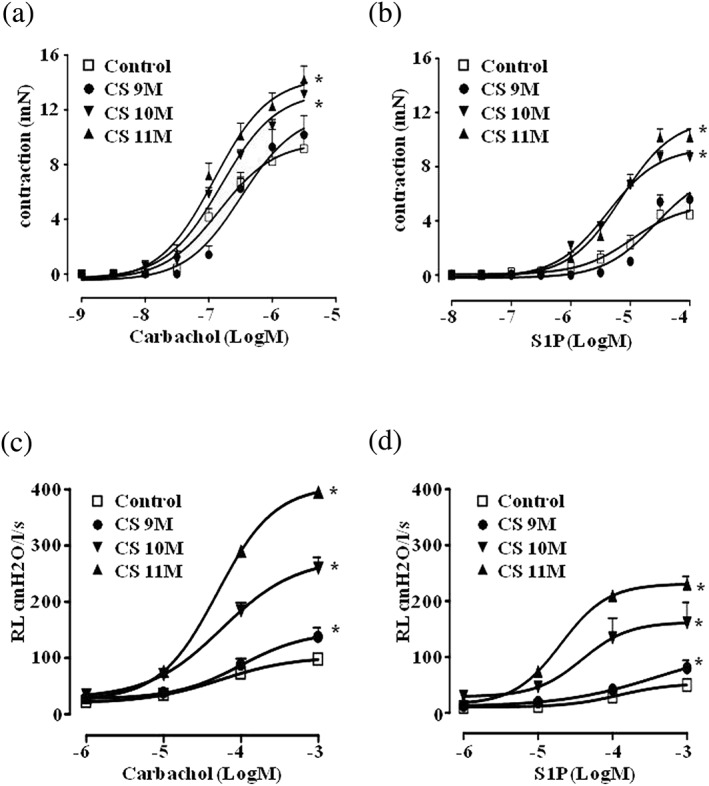
Cigarette smoke (CS) induces airway hyperresponsiveness. Mice were exposed to either room air (control) or to cigarette smoke for 9, 10, and 11 months, and bronchial reactivity to either (a) carbachol or (b) S1P was evaluated in vitro. Lung resistances (c) to carbachol or (d) to S1P were measured in an isolated and perfused mouse lung preparation. Data are expressed as mean ± SEM and have been analysed by using two‐way ANOVA followed by Bonferroni's post hoc analysis. n = 5 (control), 5 (CS 9 months), 5 (CS 10 months), and 8 (CS 11 months) for data showed in panel (a); n = 6 (control), 5 (CS 9 months), 5 (CS 10 months), and 6 (CS 11 months) for data showed in panel (b); n = 5 for all experimental groups showed in panel (c); n = 5 (control), 5 (CS 9 months), 5 (CS 10 months), and 6 (CS 11 months) for data showed in panel (d). *P < .05 versus control
Figure 4.
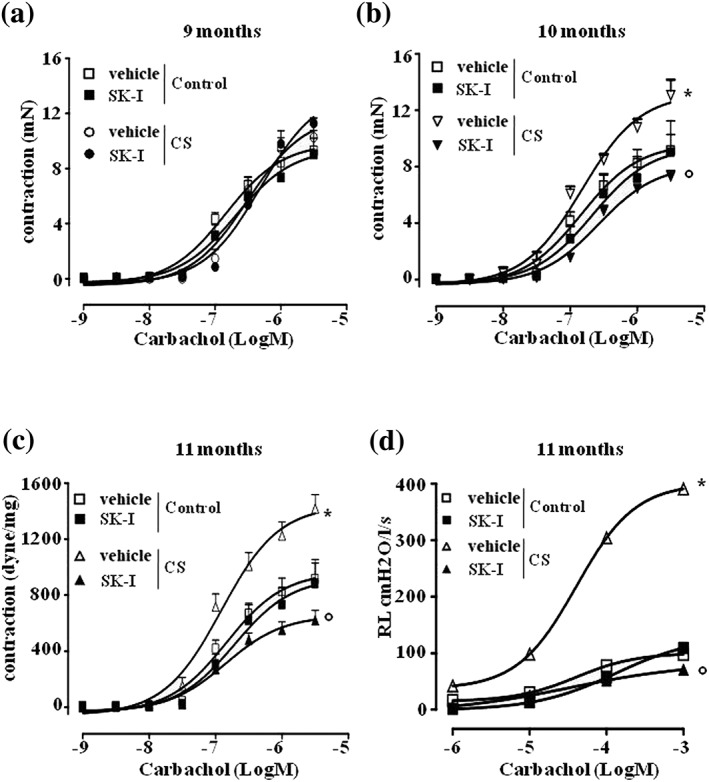
Inhibition of sphingosine kinases reverts airway hyperresponsiveness associated to CS. Carbachol‐induced contraction was assessed in bronchi harvested from air exposed mice (control) and (a) 9‐, (b) 10‐, and (c) 11‐month CS‐exposed mice, following pre‐incubation with sphingosine kinases inhibitor (SKI‐I; 30 min; 10 μM). In isolated and perfused lung protocol, SKI‐I was injected within the pulmonary artery, and lung resistances to carbachol (d) were measured in control or 11‐month CS‐exposed mice. Data are expressed as mean ± SEM and have been analysed by using two‐way ANOVA followed by Bonferroni's post hoc analysis. n = 7 mice (control), 5 mice (control + SKI‐I), 8 mice (CS 9 months), and 5 mice (CS 9 months + SKI‐I) for data showed in panel (a); n = 7 mice (control), 5 mice (control + SKI‐I), 8 mice (CS 10 months), and 5 mice (CS 10 months + SKI‐I) for data showed in panel (b); n = 8 mice (control), 5 mice (control + SKI‐I), 8 mice (CS 11 months), and 5 mice (CS 11 months + SKI‐I) for data showed in panel (c); n = 5 mice for all experimental groups showed in panel (d). *P < .05 versus control vehicle; °P < .05 versus CS vehicle
Figure 5.
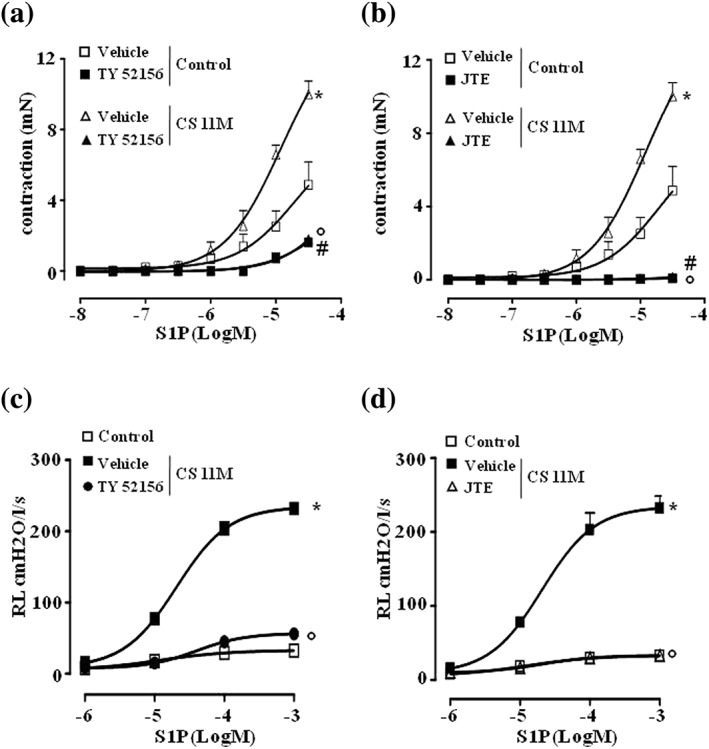
S1P effects on airways are mediated by S1P2 and S1P3 receptors. Bronchi harvested from control and smoking mice were incubated with (a) S1P3 antagonist TY‐52156 (10 μM) or (b) S1P2 antagonist JTE‐013 (10 μM) prior to S1P challenge. In another set of experiments, lung resistances to S1P were measured following administration of (c) TY‐52156 or (d) JTE‐013. Data are expressed as mean ± SEM and have been analysed by using two‐way ANOVA followed by Bonferroni's post hoc analysis. n = 5 mice for all experimental groups showed in panels (a), (b), (c), and (d). *P < .05 versus control vehicle; °P < .05 versus CS vehicle; #P < .05 versus (a) control + TY52156 or (b) control + JTE‐013
Figure 8.
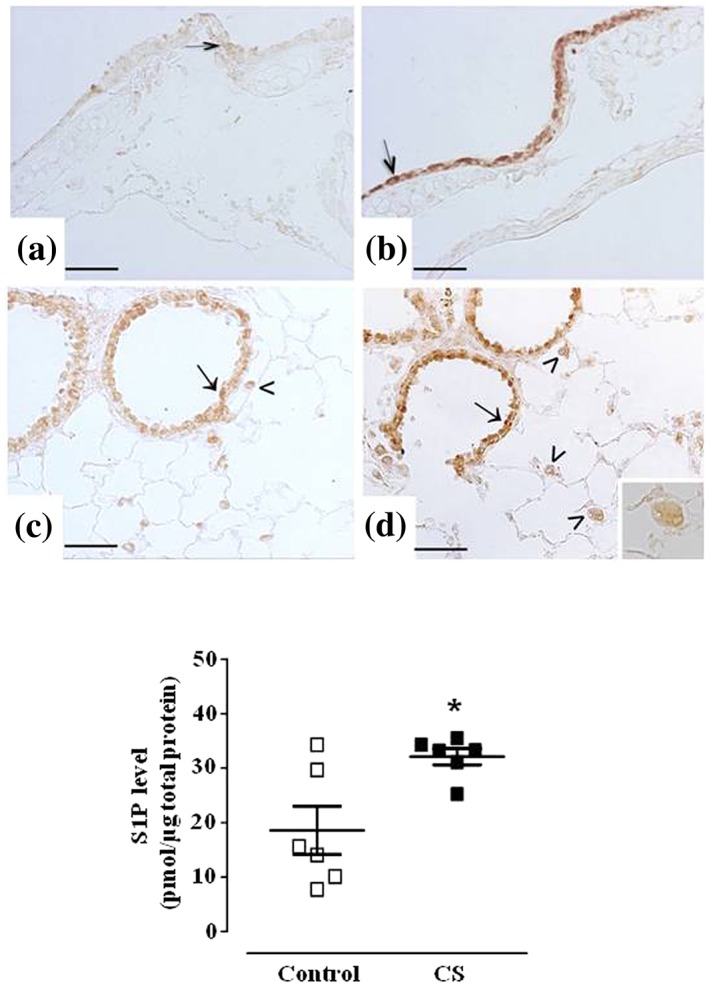
Cigarette smoke induces S1P release in the lung and a marked increase of sphingosine kinase 2. Immunostaining for Sph‐K2 in airways epithelium of main bronchi (arrow) is reported for control mice (panel a) or mice exposed to CS for 11 months (panel b). Bronchiolar epithelium (arrow) and macrophages (arrowhead) staining for Sph‐K2 is seen as a mild or strong signalling in control mice (panel c) or mice exposed to CS for 11 months (panel d), respectively. High magnification of a macrophage is shown in the inset. Scale bars = 50 μm. Six mice for each experimental group (control and CS mice) were used. S1P levels have been quantified in control and mice exposed to CS for 11 months (panel e). Data are expressed as the mean ± SEM. Seven mice for both experimental groups (control and CS mice). *P < .05 versus control
Table 1.
Lung morphometry
| Experimental groups | Time points since the onset of CS exposure | |||||
|---|---|---|---|---|---|---|
| 9 months | 10 months | 11 months | ||||
| Lm (μM) | ISA (cm2) | Lm (μM) | ISA (cm2) | Lm (μM) | ISA (cm2) | |
| Control | 41.6 ± 1.4 | 1,190 ± 11 | 41.7 ± 1.1 | 1,106 ± 9 | 42.0 ± 1.3 | 1,117 ± 7 |
| CS | 46.9 ± 1.3* | 1,091 ± 41* | 46.3 ± 1.4* | 1,015 ± 9* | 46.7 ± 1.7* | 962 ± 8* |
Note. Data are shown as mean ± SD, n = 6 for both experimental groups.
Abbreviations: CS, cigarette smoke; ISA, internal surface area of the lungs; Lm, mean linear intercept.
P < .05 versus control mice for each time point.
2.10. Drugs, chemicals, and reagents
The compounds used here were provided by the following suppliers: S1P by Enzo Life Science, Rome, Italy, and S1P2 antagonist (TY52156), S1P3 antagonist (JTE‐013), and nonselective Sph‐K inhibitor (SK‐I) by Tocris Bioscience United Kingdom. All other reagents were purchased from Sigma Aldrich, Milan, Italy.
2.11. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander, Christopoulos, et al., 2017; Alexander, Fabbro, et al., 2017).
3. RESULTS
3.1. Progressive emphysema development in cigarette smoking mice
As reported in our previous papers (Bartalesi et al., 2005; Cardini et al., 2012) exposure of mice to cigarette smoke resulted already at 3 months onwards in emphysematous changes disseminated throughout the lung parenchyma, additionally at 7 months only a small amount of collagen was found around bronchioles, distal and main bronchi of either control or smoking mice (De Cunto et al., 2016).
In our series, CS‐exposed mice developed more evident pulmonary changes at 11 months (Figure 1d). After 9 months of treatment, smoking mice showed a mild increase in collagen deposition (sea‐green stained areas) in peribronchiolar and peribronchial regions (Figure 1b) that progressively increased during the following 2 months of treatment (Figure 1c,d). As shown in the inset in Figure 1c, inflammatory infiltrate is evidently widespread in peribronchiolar/bronchial areas of the lung. No changes in morphology were seen after trichrome staining among air‐control mice (Figure 1a). At light microscopy examination, the lungs harvested from mice exposed to cigarette smoke for 9, 10, and 11 months showed significant higher values of mean linear intercepts (Lm) values (P < .05) and significant lower values of internal surface area of lungs (ISA) values (P < .05) when compared to mice exposed to room air at the same time points (Table 1).
Figure 1.
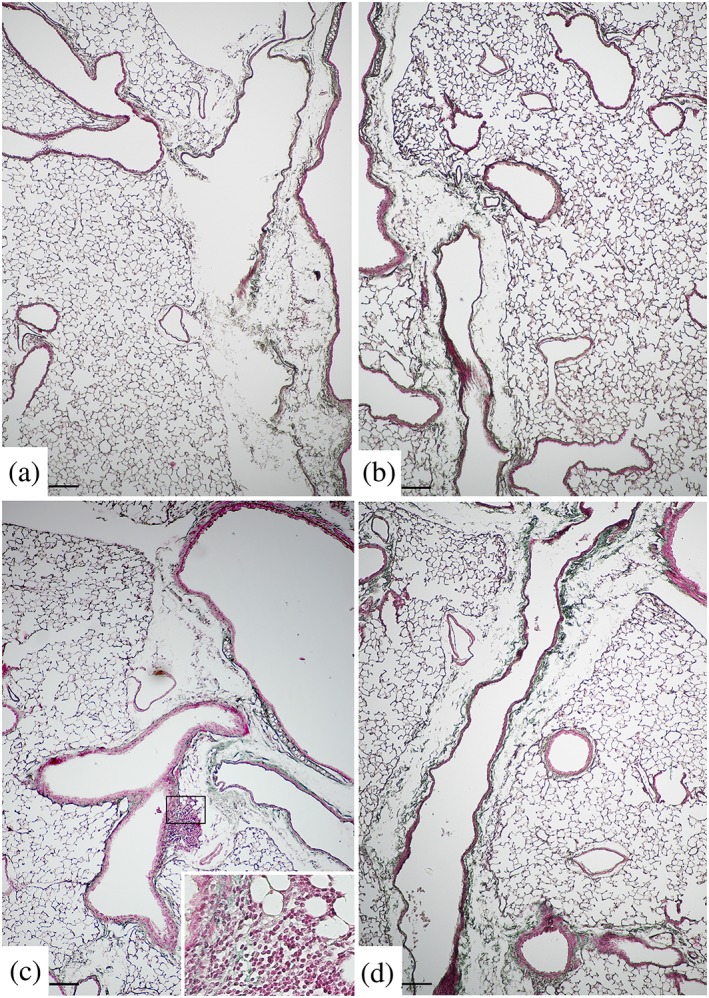
Representative images after Masson's trichrome stain. Chronic cigarette smoke (CS) exposure results in collagen accumulation in intrapulmonary and extrapulmonary airways. (a) Intrapulmonary and extrapulmonary airways from air exposed mice showed normal architecture, (b) while an increased collagen deposition is evident in mice after 9 months of CS exposure (see the sea‐green stained areas) around bronchioles and intraparenchymal bronchi. Collagen deposition around bronchioles and distal and main bronchi markedly increased at (c) 10 and (d) 11 months after CS exposure. In Figure 1c, inflammatory infiltrate is seen within a newly deposited matrix (inset). Scale bars = 150 μm; six mice for each experimental group (control and CS mice) were used
3.2. Smoking mice airways develops a progressive increased response to cholinergic stimulation
Airway function was tested evaluating both bronchial responsiveness in organ baths (upper airway) and lung resistances (RL; lower airways) measured in anaesthetized, tracheotomised, and ventilated mice using a whole‐body plethysmography. Airway function was found unaltered in mice exposed to cigarette smoke up to 7 months (Figure S1). First significant changes in the response to carbachol challenge became visible after 9 months of cigarette smoke exposure as compared to control mice (Figure 2). This effect was related to cigarette exposure time, reaching the plateau at 11 months (Figure 2a,c). Interestingly, the increased response to carbachol of the lower airways (Figure 2c), already visible at 9 months, preceded the hyperresponsiveness of the upper airways, which occurred at 10 months (Figure 2a).
Similarly, cigarette smoke induced an increased response of airways when challenged in vitro with S1P. In control mice, S1P induced a significant contraction of isolated bronchi (Figure 2b) only at very high concentrations (>10 μM). Conversely, when mice were exposed to cigarette smoke, S1P‐induced bronchial contractions were already present at concentrations as low as 1 μM. In addition, bronchi harvested from mice exposed to cigarette smoke for 10 and 11 months also showed a significant increase in the maximal contractile effect of S1P (9.99 mN vs. 4.78 mN; Figure 2b). Similarly, RL resulted significantly increased in smoking mice also following S1P challenge (Figure 2c,d). Conversely, in control mice, carbachol or S1P‐induced contractions did not change at all time points considered. In order to assess whether cigarette smoke‐induced hyperresponsiveness was sustained by airway remodelling, α‐SMA expression was evaluated by immunohistochemical analysis of pulmonary sections. Control mice displayed only a few and dispersed staining in bronchi or bronchioles of control mice (Figure 3a,c). Conversely, mice chronically exposed to cigarette smoke displayed a thick layer of α‐SMA‐positive staining that circled main and distal bronchi of mice (Figure 3b). An increased number of such cells was also found in peribronchiolar areas in smoking mice (Figure 3d).
Figure 3.
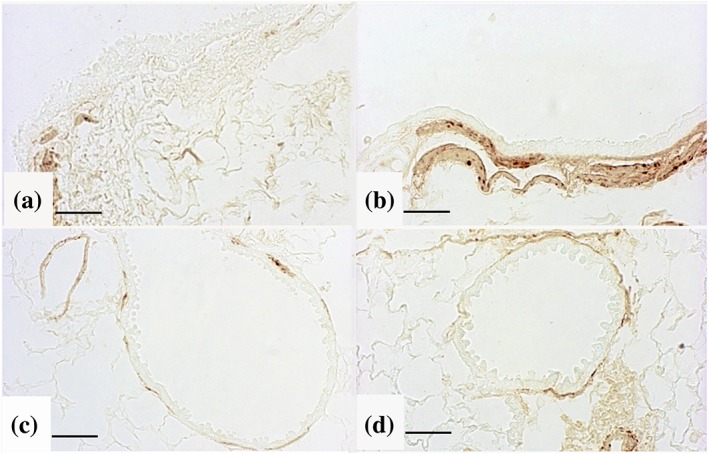
Cigarette smoke (CS) induces pulmonary α‐SMA up‐regulation. Immunohistochemical staining for α‐SMA showed reactivity in peribronchial region of (a) control and (b) CS‐exposed mice following 11‐month treatment. Peribronchiolar regions staining for α‐SMA are also shown in (c) control and (d) CS‐exposed mice following 11‐month treatment. Scale bars = 40 μm. Six mice for each experimental group (control and CS mice) were used
3.3. S1P signalling is involved in the cholinergic control of airway function of smoking mice
Cholinergic innervation is the predominant neural bronchoconstrictor pathway in airways; thus, we tested the effect of a pan sphingosine kinases inhibitor, namely, SK‐I, on carbachol‐induced contractions in bronchi from control or smoking mice. Pretreatment of bronchial rings harvested from control and smoking mice up to 9 months with SK‐I did not affect carbachol‐induced contractions of bronchi (Figure 4a). However, SK‐I reversed the increased reactivity to carbachol in bronchi of mice exposed to cigarette smoke for 10 or 11 months (Figure 4b,c). These in vitro findings were also confirmed by using whole body plethysmography experiments. Indeed, bolus administration of SK‐I in pulmonary artery, prior to carbachol challenge, abrogated the increased lung resistances observed in mice following 11 months of cigarette smoke exposure (Figure 4d).
3.4. S1P effects on airways are mediated by S1P2 and S1P3 receptors
To further assess the role of S1P pathway in cigarette smoke‐induced AHR, bronchi were pretreated with S1P2 (JTE‐013) or S1P3 (TY52156) receptor antagonists and then exposed to S1P. TY52156 (S1P3) and JTE‐013 (S1P2) abrogated S1P‐induced contractions of bronchi harvested by smoking mice (Figure 5a,c). Accordingly, bolus administration of TY52156 or JTE‐013 in pulmonary artery abrogated the increased lung resistances induced by S1P challenge when mice were exposed to cigarette smoke (Figure 5b,d).
3.5. Cigarette smoke induces pulmonary S1P pathway up‐regulation
In air‐exposed mice, a weak signal for S1P3 was found in airway epithelium (Figure 6a–c) and pulmonary macrophages (Figure 6c). The reaction for S1P3 was more marked in cigarette smoke‐exposed mice at 11 months. An evident increase in expression of S1P3 was observed in bronchial (Figure 6d), bronchiolar epithelium (Figure 6e,f), and in macrophages (inset in Figure 6f). On the other hand, S1P2 up‐regulation was evident in pulmonary epithelium of intra‐parenchymal airways (Figure 7b), as well as in airway smooth muscle cells and vessels of CS‐exposed mice (Figure 7c). An up‐regulation of S1P2 was observed in smooth muscle cells of bronchioles and main bronchi of smoking mice at the different time points (Figure 7d). A trivial reaction for this receptor was seen in control mice (Figure 7a).
Figure 6.
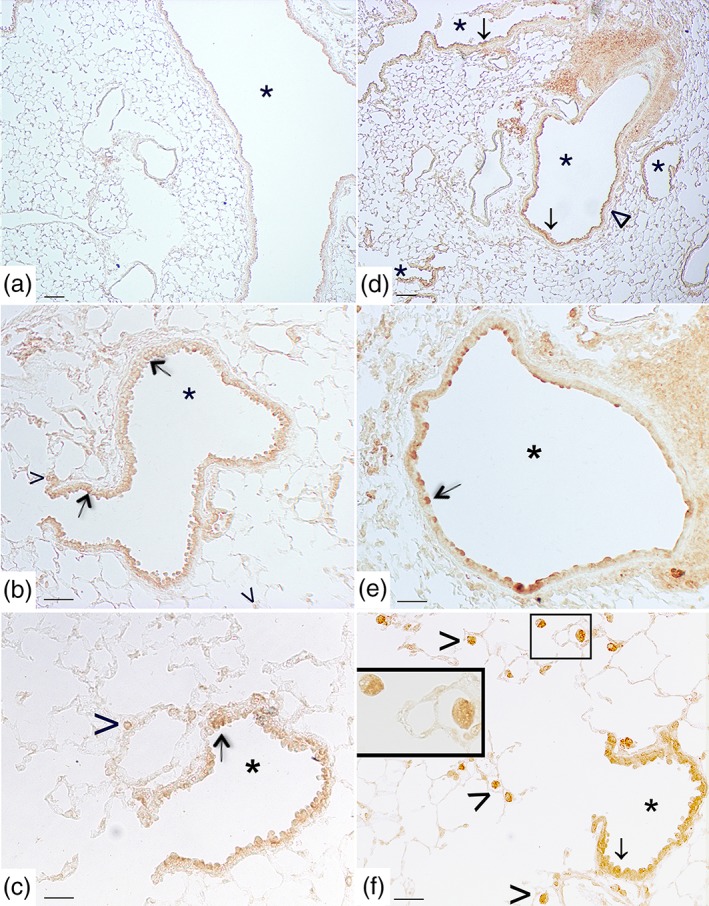
Cigarette smoke induces pulmonary S1P3 up‐regulation. Panels (a), (b), and (c) representative immunohistochemical reaction for S1P3 in lung tissue from air‐exposed mice at 11 months. In panel (a), a mild positivity for S1P3 in epithelial cells of main bronchi as well as of bronchioles from air‐exposed mice is evident. In panel (b), a faint S1P3 staining is detected in epithelial cells (arrow) of small airways and in a limited number of macrophages (arrowhead). (c) A faint reaction on epithelial cells of bronchiole (arrow) and on macrophages (arrowhead) is present in air control mice. (d–f) Micrographs of lung tissue from smoking mice at 11 months. (d) The staining for S1P3 is evident and diffuse on airways epithelium (arrow) and on the smooth muscle cell layer (wedge). (e) S1P3 staining is evident on bronchial epithelial cells (arrow). (f) Epithelial cells from small airways (arrow) and macrophages (arrowhead) show a strong positivity for S1P3 in smoking mice. High magnification of two macrophages is shown in the inset. Scale bars = 100 μm (panels a and d), 60 μm (panels b and e), and 30 μm (panels c and f). Six mice for each experimental group (control and CS mice) were used
Figure 7.
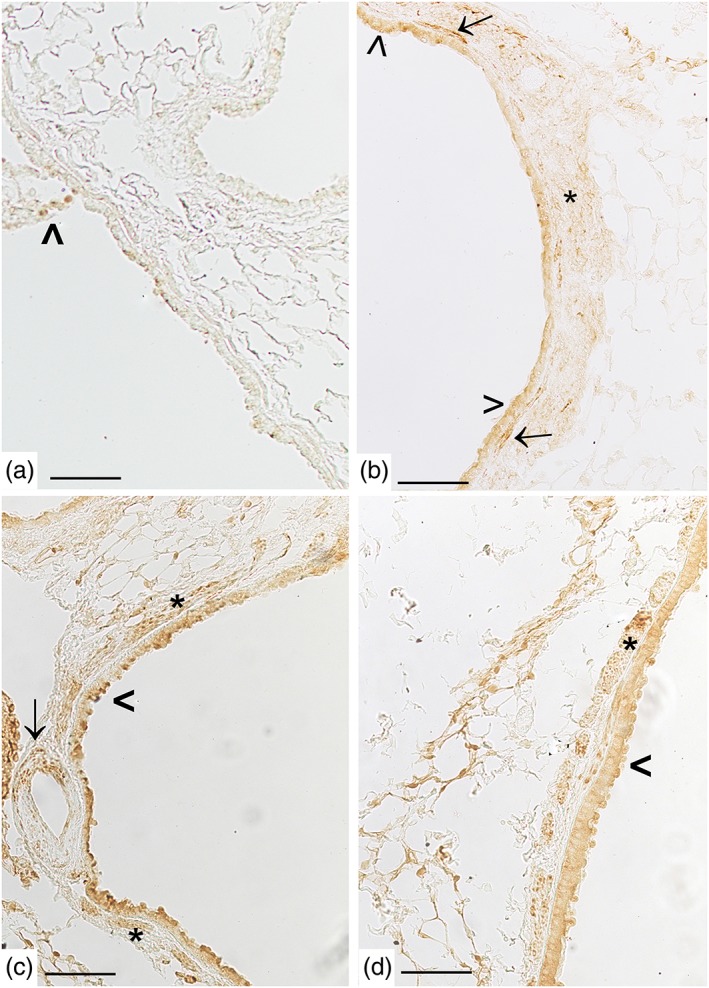
Cigarette smoke induces pulmonary S1P2 up‐regulation. A mild positivity for S1P2 is present in some cells from airways epithelium from control mice (panel a, arrowhead). In panel (b), S1P2 staining is evident on epithelial cells of intraparenchymal bronchiole (arrowhead), on spindle‐shaped cells (arrow), and smooth muscle cell layer (asterisk) from mice exposed to CS for 11 months. In panel (c), S1P2 staining is evident in smooth muscle cells (asterisk), on epithelial cells of the bronchus (arrowhead), and blood vessel (arrow) from mice exposed to CS for 11 months. In panel (d), a representative lung section from a mouse exposed to CS for 9 months shows an S1P2 positivity in smooth muscle cells (asterisk). Scale bar = 100 μm. Six mice for each experimental group (control and CS mice) were used
Since the SK‐I is a pan inhibitor, we performed an immunohistochemistry study with selective antibodies to identify the Sph‐K isoforms involved. With respect to Sph‐K1 expression, no significant changes in signal were observed between smoking and air control mice (data not shown). On the other hand, Sph‐K2 staining was mild in air exposed mice (Figure 8a,c), while resulted markedly increased in airways epithelium (Figure 8b,d) and pulmonary macrophages (insert in Figure 8d) from mice exposed to cigarette smoke for 11 months. Finally, in perfect tune with the molecular and functional studies, we found a significant increase in S1P levels in the lungs harvested from CS mice at 11 months as compared to control mice (Figure 8e).
4. DISCUSSION
Multiple dysfunctions of the airway smooth muscle contribute to alter both the airway response to stimuli and the compliance of lungs. In COPD patients, the pathological abnormalities occur both in the small and large airways and are responsible of the air flow limitation and lung inflammation. Here, we demonstrate the involvement of S1P signalling in the progression of airway dysfunction associated to emphysematous lesions following chronic exposure to cigarette smoke.
Exposure of mice to the smoke of three cigarettes per day, 5 days per week up to 11 months caused evident pulmonary changes disseminated throughout the lung. Emphysema per se affects two key elements: (a) the airway structure through a reduction of lung elastic recoil, which decreases the driving force of air from the lung and (b) the attachments between airways and the surrounding parenchyma, which are lost causing a dynamic airway collapse during expiration. In our experimental conditions, C57BL/6J mice developed significant emphysema when exposed to cigarette smoke for 6–7 months (Rahman, Cunto, Sundar, & Lungarella, 2017). From 9 months onwards, the emphysematous changes were accompanied by an increased collagen deposition in peribronchiolar and peribronchial regions. All these smoke‐associated features lead to significant structural changes of the airways. The histological study showed that there was a time‐dependent evolution of the lung damage that in turn affected the airway function. At early time (up to 9 months), morphological pulmonary changes were not accompanied by significant differences in bronchial reactivity to carbachol in vitro. At this time point, there were only slight but significant changes in lung resistances. This latter finding indicated that, with the progression of the lung damage, the lower airways were the first to be interested. Indeed, a significant increase in the contractility to carbachol of the main bronchi (upper airways), harvested from smoking mice, becomes significantly marked at 10 months, reaching a plateau at 11 months of smoke exposure. Accordingly, lung resistance measurements demonstrated an even more evident and time‐related increase in cholinergic activation, with a threefold and fivefold increase in maximal response at 10 and 11 months, respectively. The immunohistochemical study showed the presence of a thick layer of α‐SMA‐positive cells in the lung of cigarette smoke exposed mice. In particular, α‐SMA‐positive reaction is substantial around the main and distal bronchi, and it is increased in peribronchiolar areas of smoking mice at 11 months implying the activation of remodelling process within the peripheral airways. This is in line with clinical data showing that patients with COPD show evident signs of remodelling in the large airways, measured as an increased expression of α‐SMA‐positive cells (Hogg et al., 2004; Jones et al., 2016; Skold, 2010). However, at the present stage, only few studies have investigated the presence of histopathological changes due to remodelling in the large airways of COPD patients. These processes might be responsible of the structural changes in the airway wall leading to lung function impairment in COPD.
In our experimental setting, cigarette smoke triggers a lung inflammation, which slowly develops and promotes the airway structural changes responsible for the cholinergic hyperresponsiveness of both lower (early response) and upper (late response) airways from 9 months onwards. Cholinergic innervation of airways is the predominant neural bronchoconstrictor pathway. However, chronic irritants such as cigarette smoke and air pollution or mediators released from inflammatory cells further stimulate airway sensory nerve, which in turn release ACh causing a more sustained stimulation of muscarinic receptors. In addition, more recent findings suggest that ACh regulates additional functions in the respiratory tract, including inflammation and remodelling in airway diseases. Indeed, a recent clinical trial demonstrates that repeated methacholine inhalations cause airway remodelling, characterised by collagen deposition and increased TGF‐ß expression (Grainge et al., 2011). Muscarinic receptors on airway smooth muscle cells also play an important role in regulating immunomodulatory function of airway smooth muscle (Zuyderduyn, Sukkar, Fust, Dhaliwal, & Burgess, 2008). Indeed, inhaled bronchodilator treatment with a long acting muscarinic antagonist reduces symptoms and the risk of exacerbations of COPD as well as of asthma (Decramer et al., 2009; Tashkin et al., 2008). Measurement of S1P total levels in pulmonary homogenates shows that in smoking mice, there is a significant increase that is almost doubled as compared to control mice. This led us to hypothesize that the higher S1P level might be associated with the hyperresponsiveness to cholinergic stimuli. To address this issue, we evaluated the effect of a pan sphingosine kinases inhibitor on carbachol‐induced contractions in vitro. The incubation of bronchial rings with the nonselective inhibitor of Sph‐Ks (SK‐I) did not affect carbachol‐induced contractions of bronchi harvested from either control (air challenged) or smoking mice in the early phase. Conversely, SK‐I abrogated the increased reactivity to carbachol following 10 and 11 months of cigarette smoke exposure reporting the response to carbachol back to the control values. Similarly, injection of SK‐I in pulmonary artery before carbachol challenge abrogated the increased lung resistances in smoking mice. Thus, in mice that have been exposed to cigarette smoke from 9 months onwards, the increased response to cholinergic stimulation involves the Sph‐K/S1P pathway signalling.
Immunohistochemistry study further confirmed the role of Sph‐Ks in the airway dysfunction associated to cigarette smoke exposure. Indeed, in the lungs of mice exposed for 11 months at CS exposure, there was an increased expression of Sph‐K2, mainly localised in the airway epithelium and pulmonary macrophages. The strong positivity found in the airway epithelium well fits with previous evidence showing epithelium as the major source for S1P in the airways (Tran et al., 2017). Recent clinical data also suggest a potential link between the S1P pathway and the defective macrophage phagocytic function in COPD patients (Barnawi et al., 2015). Furthermore, it has been demonstrated that Sph‐K2 contribute is more relevant and directed towards intracellular targets thereby affecting transcription and mitochondrial integrity leading to apoptosis, distinct from inside–out signalling via S1P receptors. Indeed, Sph‐K2 instead of Sph‐K1 enhances ceramide biosynthesis and promotes caspase‐mediated apoptosis (Kharel et al., 2012; Santos & Lynch, 2015). Animal models have confirmed the importance of apoptosis in the pathogenesis of COPD, since these mechanisms are induced in cigarette smoke‐exposed mice that develop emphysema and induction of either endothelial or epithelial cell apoptosis is sufficient to cause murine emphysema (Sauler, Bazan, & Lee, 2019). The increased expression of Sph‐K2 was coupled also to up‐regulation of both S1P2 and S1P3 receptors. In particular, while in air‐exposed mice there was a low signal for S1P3 in airway epithelium and pulmonary macrophages, its expression increased in bronchial and bronchiolar epithelium after 11 months of cigarette smoke exposure. Similarly, the signal for S1P2 is greatly enhanced in pulmonary epithelium of intraparenchymal as well as in main bronchi and in smooth muscle cells of airways and vessels harvested from smoking mice.
In order to further corroborate our hypothesis, we evaluated if the response to exogenous S1P was altered in the same experimental conditions. Indeed, the data obtained so far indicated that (a) there is an increased production of S1P and (b) the carbachol response is reduced by blocking sphingosine kinases. S1P binds to specific GPCRs and generates downstream signals that play a crucial role in the development of a number of lung pathologies (Yang & Uhlig, 2011). S1P receptors such as S1P2 and S1P3 are present on airways, and they regulate many of their actions, such as airway contraction and remodelling. In physiological conditions, that is, in control air‐exposed mice, S1P induced a significant contraction of bronchi in vitro at very high concentrations within a millimolar range, which is well over the physiological S1P concentration. Conversely, in smoking mice, bronchial response to S1P in vitro occurred within a molar range. An analogous effect was observed when lung resistances were measured following S1P injection in pulmonary artery. These increased responses in both upper and lower airways of cigarette exposed mice were markedly evident since the S1P effect was almost doubled. The role played by S1P2 and S1P3was confirmed by the inhibition obtained by pretreatment with the selective receptor antagonists. S1P2 and S1P3 are the receptor subtypes involved in the pro‐remodelling response in airways where they promote activation of both epithelial and smooth muscle cells. Indeed, S1P can induce smooth muscle contraction by interacting with S1P2, while it may indirectly affect airways function upon binding to S1P3. All together, the data obtained support our hypothesis of an active role of Sph‐K/S1P axis in the altered cholinergic control of airway function triggered by cigarette smoke. In addition, our data imply a possible complementary role of S1P intracellular (e.g., via Sph‐K) and extracellular (e.g., S1Pn) pathways that promotes both the lung inflammatory response and the airway remodelling processes by converging on different downstream signalling pathways.
In conclusion, the up‐regulation of Sph‐K/S1P pathway develops progressively in smoking mice and follows the disease development leading to an increased response to carbachol. This hyperresponsiveness to cholinergic stimulation involves Sph‐K2, S1P2, and S1P3 receptors. Thus, S1P pathway represents a feasible therapeutic target in AHR associated to emphysema and particularly in smokers with both asthma and COPD (ACOS patients), a patient category more exposed to the disease progression and mortality.
AUTHOR CONTRIBUTIONS
G.D.C. and V.B. designed, performed, and analysed the data and contribute to write the manuscript; E.C. and B.B. performed lung tissue histology and immunohistochemistry; M.A.R., A.B., and I.C. performed in vitro study evaluating bronchial reactivity; G.S. and B.D. performed animal studies evaluating lung resistance. V.V. prepared lung tissue for histology; G.L. analysed experiments; F.R., M.L., and G.C. interpreted the data and wrote the manuscript.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14207, https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14208, and https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14206, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Cigarette smoke (CS) effect on bronchial reactivity. Mice were exposed to cigarette smoke for 1,2, 3 and 7 months and bronchial reactivity to carbachol was evaluated in vitro.
ACKNOWLEDGEMENT
This work was supported in part by research grant Fondo 000005_FFABR_2017_A_Roviezzo_001_001.
De Cunto G, Brancaleone V, Riemma MA, et al. Functional contribution of sphingosine‐1‐phosphate to airway pathology in cigarette smoke‐exposed mice. Br J Pharmacol. 2020;177:267–281. 10.1111/bph.14861
Giovanna De Cunto and Vincenzo Brancaleone equally contributed to this work.
Monica Lucattelli and Fiorentina Roviezzo equally contributed to this work
REFERENCES
- Ahmed, F. S. , Jiang, X. C. , Schwartz, J. E. , Hoffman, E. A. , Yeboah, J. , Shea, S. , et al. (2014). Plasma sphingomyelin and longitudinal change in percent emphysema on CT. The MESA lung study. Biomarkers, 19(3), 207–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Marrion, N. V. , Peters, J. A. , et al. (2017). The concise guide to pharmacology 2017/18: G protein‐coupled receptors. British Journal of Pharmacology, 174(Suppl 1), S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , et al. (2017). The concise guide to pharmacology 2017/18: Enzymes. British Journal of Pharmacology, 174(Suppl 1), S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammit, A. J. , Burgess, J. K. , Hirst, S. J. , Hughes, J. M. , Kaur, M. , Lau, J. Y. , et al. (2009). The effect of asthma therapeutics on signalling and transcriptional regulation of airway smooth muscle function. Pulmonary Pharmacology & Therapeutics, 22(5), 446–454. [DOI] [PubMed] [Google Scholar]
- Arish, M. , Alaidarous, M. , Ali, R. , Akhter, Y. , & Rub, A. (2017). Implication of sphingosine‐1‐phosphate signaling in diseases: Molecular mechanism and therapeutic strategies. Journal of Receptor and Signal Transduction Research, 37(5), 437–446. [DOI] [PubMed] [Google Scholar]
- Barnawi, J. , Tran, H. , Jersmann, H. , Pitson, S. , Roscioli, E. , Hodge, G. , et al. (2015). Potential link between the sphingosine‐1‐phosphate (S1P) system and defective alveolar macrophage phagocytic function in chronic obstructive pulmonary disease (COPD). PLoS ONE, 10(10), e0122771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes, P. J. (2017). Cellular and molecular mechanisms of asthma and COPD. Clinical Science (London, England), 131(13), 1541–1558. [DOI] [PubMed] [Google Scholar]
- Bartalesi, B. , Cavarra, E. , Fineschi, S. , Lucattelli, M. , Lunghi, B. , Martorana, P. A. , et al. (2005). Different lung responses to cigarette smoke in two strains of mice sensitive to oxidants. The European Respiratory Journal, 25(1), 15–22. [DOI] [PubMed] [Google Scholar]
- Breslow, D. K. , Collins, S. R. , Bodenmiller, B. , Aebersold, R. , Simons, K. , Shevchenko, A. , et al. (2010). Orm family proteins mediate sphingolipid homeostasis. Nature, 463(7284), 1048–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardini, S. , Dalli, J. , Fineschi, S. , Perretti, M. , Lungarella, G. , & Lucattelli, M. (2012). Genetic ablation of the fpr1 gene confers protection from smoking‐induced lung emphysema in mice. American Journal of Respiratory Cell and Molecular Biology, 47(3), 332–339. [DOI] [PubMed] [Google Scholar]
- Cavarra, E. , Bartalesi, B. , Lucattelli, M. , Fineschi, S. , Lunghi, B. , Gambelli, F. , et al. (2001). Effects of cigarette smoke in mice with different levels of α1‐proteinase inhibitor and sensitivity to oxidants. American Journal of Respiratory and Critical Care Medicine, 164(5), 886–890. [DOI] [PubMed] [Google Scholar]
- Chiba, Y. , Takeuchi, H. , Sakai, H. , & Misawa, M. (2010). SKI‐II, an inhibitor of sphingosine kinase, ameliorates antigen‐induced bronchial smooth muscle hyperresponsiveness, but not airway inflammation, in mice. Journal of Pharmacological Sciences, 114(3), 304–310. [DOI] [PubMed] [Google Scholar]
- Cockcroft, D. W. , & Wenzel, S. (2016). Airway hyperresponsiveness and chronic obstructive pulmonary disease outcomes. The Journal of Allergy and Clinical Immunology, 138(6), 1580–1581. [DOI] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , et al. (2015). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175(7), 987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cunto, G. , Bartalesi, B. , Cavarra, E. , Balzano, E. , Lungarella, G. , & Lucattelli, M. (2018). Ongoing lung inflammation and disease progression in mice after smoking cessation: Beneficial effects of formyl‐peptide receptor blockade. The American Journal of Pathology, 188(10), 2195–2206. [DOI] [PubMed] [Google Scholar]
- De Cunto, G. , Lunghi, B. , Bartalesi, B. , Cavarra, E. , Fineschi, S. , Ulivieri, C. , et al. (2016). Severe reduction in number and function of peripheral T cells does not afford protection toward emphysema and bronchial remodeling induced in mice by cigarette smoke. The American Journal of Pathology, 186(7), 1814–1824. [DOI] [PubMed] [Google Scholar]
- Decramer, M. , Celli, B. , Kesten, S. , Lystig, T. , Mehra, S. , & Tashkin, D. P. (2009). Effect of tiotropium on outcomes in patients with moderate chronic obstructive pulmonary disease (UPLIFT): A prespecified subgroup analysis of a randomised controlled trial. Lancet, 374(9696), 1171–1178. [DOI] [PubMed] [Google Scholar]
- Ebenezer, D. L. , Fu, P. , & Natarajan, V. (2016). Targeting sphingosine‐1‐phosphate signaling in lung diseases. Pharmacology & Therapeutics, 168, 143–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbri, L. M. , & Rabe, K. F. (2007). From COPD to chronic systemic inflammatory syndrome? Lancet, 370(9589), 797–799. [DOI] [PubMed] [Google Scholar]
- Gamble, E. , Grootendorst, D. C. , Hattotuwa, K. , O'Shaughnessy, T. , Ram, F. S. , Qiu, Y. , et al. (2007). Airway mucosal inflammation in COPD is similar in smokers and ex‐smokers: A pooled analysis. The European Respiratory Journal, 30(3), 467–471. [DOI] [PubMed] [Google Scholar]
- Grainge, C. L. , Lau, L. C. , Ward, J. A. , Dulay, V. , Lahiff, G. , Wilson, S. , et al. (2011). Effect of bronchoconstriction on airway remodeling in asthma. The New England Journal of Medicine, 364(21), 2006–2015. [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , et al. (2018). The IUPHAR/BPS guide to pharmacology in 2018: Updates and expansion to encompass the new guide to immunopharmacology. Nucleic Acids Research, 46(D1), D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg, J. C. , Chu, F. , Utokaparch, S. , Woods, R. , Elliott, W. M. , Buzatu, L. , et al. (2004). The nature of small‐airway obstruction in chronic obstructive pulmonary disease. The New England Journal of Medicine, 350(26), 2645–2653. [DOI] [PubMed] [Google Scholar]
- Hospers, J. J. , Postma, D. S. , Rijcken, B. , Weiss, S. T. , & Schouten, J. P. (2000). Histamine airway hyper‐responsiveness and mortality from chronic obstructive pulmonary disease: A cohort study. Lancet, 356(9238), 1313–1317. [DOI] [PubMed] [Google Scholar]
- Jones, R. L. , Noble, P. B. , Elliot, J. G. , & James, A. L. (2016). Airway remodelling in COPD: It's not asthma! Respirology, 21(8), 1347–1356. [DOI] [PubMed] [Google Scholar]
- Kharel, Y. , Raje, M. , Gao, M. , Gellett, A. M. , Tomsig, J. L. , Lynch, K. R. , et al. (2012). Sphingosine kinase type 2 inhibition elevates circulating sphingosine 1‐phosphate. The Biochemical Journal, 447(1), 149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160(7), 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapperre, T. S. , Postma, D. S. , Gosman, M. M. , Snoeck‐Stroband, J. B. , ten Hacken, N. H. , Hiemstra, P. S. , et al. (2006). Relation between duration of smoking cessation and bronchial inflammation in COPD. Thorax, 61(2), 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy, M. , Khan, E. , Careaga, M. , & Goldkorn, T. (2009). Neutral sphingomyelinase 2 is activated by cigarette smoke to augment ceramide‐induced apoptosis in lung cell death. American Journal of Physiology. Lung Cellular and Molecular Physiology, 297(1), L125–L133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucattelli, M. , Bartalesi, B. , Cavarra, E. , Fineschi, S. , Lunghi, B. , Martorana, P. A. , et al. (2005). Is neutrophil elastase the missing link between emphysema and fibrosis? Evidence from two mouse models. Respiratory Research, 6, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannino, D. M. , Watt, G. , Hole, D. , Gillis, C. , Hart, C. , McConnachie, A. , et al. (2006). The natural history of chronic obstructive pulmonary disease. The European Respiratory Journal, 27(3), 627–643. [DOI] [PubMed] [Google Scholar]
- Miller, M. , Rosenthal, P. , Beppu, A. , Gordillo, R. , & Broide, D. H. (2017). Oroscomucoid like protein 3 (ORMDL3) transgenic mice have reduced levels of sphingolipids including sphingosine‐1‐phosphate and ceramide. The Journal of Allergy and Clinical Immunology, 139(4), 1373–1376. e1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, M. , Tam, A. B. , Mueller, J. L. , Rosenthal, P. , Beppu, A. , Gordillo, R. , et al. (2017). Cutting edge: Targeting epithelial ORMDL3 increases, rather than reduces, airway responsiveness and is associated with increased sphingosine‐1‐phosphate. Journal of Immunology, 198(8), 3017–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffatt, M. F. , Gut, I. G. , Demenais, F. , Strachan, D. P. , Bouzigon, E. , Heath, S. , et al. (2010). A large‐scale, consortium‐based genomewide association study of asthma. The New England Journal of Medicine, 363(13), 1211–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page, C. , O'Shaughnessy, B. , & Barnes, P. (2017). Pathogenesis of COPD and asthma. Handbook of Experimental Pharmacology, 237, 1–21. [DOI] [PubMed] [Google Scholar]
- Petrache, I. , Medler, T. R. , Richter, A. T. , Kamocki, K. , Chukwueke, U. , Zhen, L. , et al. (2008). Superoxide dismutase protects against apoptosis and alveolar enlargement induced by ceramide. American Journal of Physiology. Lung Cellular and Molecular Physiology, 295(1), L44–L53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrache, I. , Natarajan, V. , Zhen, L. , Medler, T. R. , Richter, A. T. , Cho, C. , et al. (2005). Ceramide upregulation causes pulmonary cell apoptosis and emphysema‐like disease in mice. Nature Medicine, 11(5), 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MM, Oskeritzian CA, Falanga YT, Harikumar KB, Allegood JC, Alvarez SE , et al (2013). A specific sphingosine kinase 1 inhibitor attenuates airway hyperresponsiveness and inflammation in a mast cell‐dependent murine model of allergic asthma. The Journal of Allergy and Clinical Immunology 131(2): 501–511 e501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price, M. M. , Oskeritzian, C. A. , Falanga, Y. T. , Harikumar, K. B. , Allegood, J. C. , Alvarez, S. E. , et al. (2012). A specific sphingosine kinase 1 inhibitor attenuates airway hyperresponsiveness and inflammation in a mast cell‐dependent murine model of allergic asthma. The Journal of Allergy and Clinical Immunology, 131(2), 501–511. e501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proia, R. L. , & Hla, T. (2015). Emerging biology of sphingosine‐1‐phosphate: Its role in pathogenesis and therapy. Journal of Clinical Investigation, 125(4), 1379–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyne, S. , & Pyne, N. J. (2002). Sphingosine 1‐phosphate signalling and termination at lipid phosphate receptors. Biochimica et Biophysica Acta, 1582(1–3), 121–131. [DOI] [PubMed] [Google Scholar]
- Rahman I, De Cunto G, Sundar IK, Lungarella G (2017). Vulnerability and genetic susceptibility to cigarette smoke‐induced emphysema in mice. American Journal of Respiratory Cell and Molecular Biology 57(3): 270–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roviezzo, F. , D'Agostino, B. , Brancaleone, V. , De Gruttola, L. , Bucci, M. , De Dominicis, G. , et al. (2010). Systemic administration of sphingosine‐1‐phosphate increases bronchial hyperresponsiveness in the mouse. American Journal of Respiratory Cell and Molecular Biology, 42(5), 572–577. [DOI] [PubMed] [Google Scholar]
- Roviezzo, F. , Del Galdo, F. , Abbate, G. , Bucci, M. , D'Agostino, B. , Antunes, E. , et al. (2004). Human eosinophil chemotaxis and selective in vivo recruitment by sphingosine 1‐phosphate. Proceedings of the National Academy of Sciences of the United States of America, 101(30), 11170–11175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roviezzo, F. , Di Lorenzo, A. , Bucci, M. , Brancaleone, V. , Vellecco, V. , De Nardo, M. , et al. (2007). Sphingosine‐1‐phosphate/sphingosine kinase pathway is involved in mouse airway hyperresponsiveness. American Journal of Respiratory Cell and Molecular Biology, 36(6), 757–762. [DOI] [PubMed] [Google Scholar]
- Roviezzo, F. , Sorrentino, R. , Bertolino, A. , De Gruttola, L. , Terlizzi, M. , Pinto, A. , et al. (2015). S1P‐induced airway smooth muscle hyperresponsiveness and lung inflammation in vivo: Molecular and cellular mechanisms. British Journal of Pharmacology, 172(7), 1882–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roviezzo, F. , Sorrentino, R. , Iacono, V. M. , Brancaleone, V. , Terlizzi, M. , Riemma, M. A. , et al. (2016). Disodium cromoglycate inhibits asthma‐like features induced by sphingosine‐1‐phosphate. Pharmacological Research, 113(Pt A), 626–635. [DOI] [PubMed] [Google Scholar]
- Santos, W. L. , & Lynch, K. R. (2015). Drugging sphingosine kinases. ACS Chemical Biology, 10(1), 225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauler, M. , Bazan, I. S. , & Lee, P. J. (2019). Cell death in the lung: The apoptosis‐necroptosis axis. Annual Review of Physiology, 81, 375–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanlon, P. D. , Connett, J. E. , Waller, L. A. , Altose, M. D. , Bailey, W. C. , Buist, A. S. , et al. (2000). Smoking cessation and lung function in mild‐to‐moderate chronic obstructive pulmonary disease. The Lung Health Study. American Journal of Respiratory and Critical Care Medicine, 161(2 Pt 1), 381–390. [DOI] [PubMed] [Google Scholar]
- Skold, C. M. (2010). Remodeling in asthma and COPD—Differences and similarities. The Clinical Respiratory Journal, 4(Suppl 1), 20–27. [DOI] [PubMed] [Google Scholar]
- Tashkin, D. P. , Altose, M. D. , Connett, J. E. , Kanner, R. E. , Lee, W. W. , & Wise, R. A. (1996). Methacholine reactivity predicts changes in lung function over time in smokers with early chronic obstructive pulmonary disease. The Lung Health Study Research Group. American Journal of Respiratory and Critical Care Medicine, 153(6 Pt 1), 1802–1811. [DOI] [PubMed] [Google Scholar]
- Tashkin, D. P. , Celli, B. , Senn, S. , Burkhart, D. , Kesten, S. , Menjoge, S. , et al. (2008). A 4‐year trial of tiotropium in chronic obstructive pulmonary disease. The New England Journal of Medicine, 359(15), 1543–1554. [DOI] [PubMed] [Google Scholar]
- Telenga, E. D. , Hoffmann, R. F. , Ruben, t. K. , Hoonhorst, S. J. , Willemse, B. W. , van Oosterhout, A. J. , et al. (2014). Untargeted lipidomic analysis in chronic obstructive pulmonary disease. Uncovering sphingolipids. American Journal of Respiratory and Critical Care Medicine, 190(2), 155–164. [DOI] [PubMed] [Google Scholar]
- Tkacova, R. , Dai, D. L. Y. , Vonk, J. M. , Leung, J. M. , Hiemstra, P. S. , van den Berge, M. , et al. (2016). Airway hyperresponsiveness in chronic obstructive pulmonary disease: A marker of asthma‐chronic obstructive pulmonary disease overlap syndrome? The Journal of Allergy and Clinical Immunology, 138(6), 1571–1579. e1510 [DOI] [PubMed] [Google Scholar]
- Tran, H. B. , Jersmann, H. , Truong, T. T. , Hamon, R. , Roscioli, E. , Ween, M. , et al. (2017). Disrupted epithelial/macrophage crosstalk via Spinster homologue 2‐mediated S1P signaling may drive defective macrophage phagocytic function in COPD. PLoS ONE, 12(11), e0179577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlig, S. , & Gulbins, E. (2008). Sphingolipids in the lungs. American Journal of Respiratory and Critical Care Medicine, 178(11), 1100–1114. [DOI] [PubMed] [Google Scholar]
- Vestbo, J. , Hurd, S. S. , Agusti, A. G. , Jones, P. W. , Vogelmeier, C. , Anzueto, A. , et al. (2013). Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. American Journal of Respiratory and Critical Care Medicine, 187(4), 347–365. [DOI] [PubMed] [Google Scholar]
- Willemse, B. W. , ten Hacken, N. H. , Rutgers, B. , Postma, D. S. , & Timens, W. (2005). Association of current smoking with airway inflammation in chronic obstructive pulmonary disease and asymptomatic smokers. Respiratory Research, 6, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y. , & Uhlig, S. (2011). The role of sphingolipids in respiratory disease. Therapeutic Advances in Respiratory Disease, 5(5), 325–344. [DOI] [PubMed] [Google Scholar]
- Zuyderduyn, S. , Sukkar, M. B. , Fust, A. , Dhaliwal, S. , & Burgess, J. K. (2008). Treating asthma means treating airway smooth muscle cells. The European Respiratory Journal, 32(2), 265–274. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Cigarette smoke (CS) effect on bronchial reactivity. Mice were exposed to cigarette smoke for 1,2, 3 and 7 months and bronchial reactivity to carbachol was evaluated in vitro.