

Summary
Acute myeloid leukemia (AML) is a severe, mostly fatal hematopoietic malignancy. We were interested in whether transcriptomic-based machine learning could predict AML status without requiring expert input. Using 12,029 samples from 105 different studies, we present a large-scale study of machine learning-based prediction of AML in which we address key questions relating to the combination of machine learning and transcriptomics and their practical use. We find data-driven, high-dimensional approaches—in which multivariate signatures are learned directly from genome-wide data with no prior knowledge—to be accurate and robust. Importantly, these approaches are highly scalable with low marginal cost, essentially matching human expert annotation in a near-automated workflow. Our results support the notion that transcriptomics combined with machine learning could be used as part of an integrated -omics approach wherein risk prediction, differential diagnosis, and subclassification of AML are achieved by genomics while diagnosis could be assisted by transcriptomic-based machine learning.
Subject Areas: Artificial Intelligence, Biological Sciences, Cancer, Computer Science, Omics, Transcriptomics
Graphical Abstract
Highlights
-
•
Study presents one of the largest transcriptomics datasets to date for AML prediction
-
•
Effective classifiers can be obtained by high-dimensional machine learning
-
•
Accuracy increases with dataset size
-
•
Includes challenging scenarios such as cross-study and cross-technology
Artificial Intelligence; Biological Sciences; Cancer; Computer Science; Omics; Transcriptomics
Introduction
Recommendations for the diagnosis and management of malignant diseases are organized by international expert panels. For example, the first edition of the European LeukemiaNet (ELN) recommendations for the diagnosis and management of acute myeloid leukemia (AML) in adults was published in 2010 (Döhner et al., 2010) and recently revised in 2017 (Döhner et al., 2017). Based on recent DNA sequencing results, such as those derived from The Cancer Genome Atlas, AML can be subdivided into multiple subclasses (Arber et al., 2016, Ding et al., 2012, Ley et al., 2008, Ley et al., 2010, Loriaux et al., 2008, Papaemmanuil et al., 2016, The Cancer Genome Atlas Research Network (TCGA) et al., 2013, Welch et al., 2012, Yan et al., 2011). Leukemias are characterized by strong transcriptomic signals, as seen in a pioneering study almost two decades ago by Golub et al. (Golub et al., 1999) and a rich body of subsequent work (Debernardi et al., 2003, Kohlmann et al., 2003, Ross et al., 2004, Schoch et al., 2002, Virtaneva et al., 2001). These findings led to the suggestion that gene expression profiling (GEP) could be utilized to define leukemia subtypes and derive useful predictive gene signatures (Andersson et al., 2007, Bullinger et al., 2004). Nevertheless, according to the ELN recommendations primary diagnosis still relies on classical approaches including assessment of morphology, immunophenotyping, cytochemistry, and cytogenetics (Döhner et al., 2017). Although undoubtedly effective in detecting disease, these existing diagnostic approaches rely on large investments in human expertise (training and employment of specialists) and physical infrastructure, whose costs scale with the number of samples. This has implications for accessibility (e.g., in rural areas or outside developed regions) and on cost and logistical grounds alone limits the scope to consider alternatives to the overall decision pipeline. In contrast to classical diagnostic pipelines that are centered on interpretation of results by human experts, artificial intelligence- (AI) and machine learning- (ML) based approaches have the potential for low marginal cost (i.e., cost per additional sample once the system is trained) (Esteva et al., 2017), and this key aspect of AI and ML is widely appreciated in the economics literature (see, e.g., Brynjolfsson and McAfee, 2014).
The potential of GEP for leukemia diagnosis has been recognized. A decade after the pioneering work of Golub et al., the International Microarray Innovations in Leukemia Study Group proposed GEP by microarray analysis to be a robust technology for the diagnosis of hematologic malignancies with high accuracy (Haferlach et al., 2010). The utility of GEP by RNA sequencing (RNA-seq) has been also demonstrated for other tumor entities, for example, breast cancer (Ciriello et al., 2015, Kristensen et al., 2012, Parker et al., 2009), bladder cancer, or lung cancer (Hoadley et al., 2014, Robertson et al., 2017). Furthermore, in AML research large RNA-seq datasets have been described in the meantime (Garzon et al., 2014, Lavallee et al., 2016, Lavallée et al., 2015, Macrae et al., 2013, Pabst et al., 2016).
In parallel, a series of advances in ML, AI, and computational statistics have transformed our understanding of prediction using high-dimensional data. A variety of approaches are now an established part of the toolkit, and for some models (including sparse linear and generalized linear models), there is a rich mathematical theory concerning their performance in the high-dimensional setting (Bühlmann and van de Geer, 2011). In a nutshell, the body of empirical and theoretical research has shown that learning predictive models over large numbers of variables is often feasible and remarkably effective. In applied ML, there has been a deepening understanding of practical issues, e.g., relating to the transferability of predictions across contexts (Quiñonero-Candela et al., 2009), that is very relevant to the clinical setting.
Based on these developments in the data sciences and the increasing availability of GEP data derived from peripheral blood including AML, we sought to develop near-automated approaches in which ML tools automatically learn suitable patterns directly from the global transcriptomic data without pre-selection of genes. To this end, we built the probably largest reference blood GEP dataset comprising 105 individual studies with, in total, more than 12,000 patient samples. We applied high-dimensional ML approaches to build genome-wide predictors in an unbiased, entirely data-driven manner and tested predictive accuracy in held-out data. We emphasize that our goal was not to outperform classical diagnostic methods, but to ask whether we could match human annotation in a near-automated and scalable manner. This aim is common to a number of recent efforts to use ML and AI advances in the diagnostic setting (see, e.g., Esteva et al., 2017) wherein human-derived labels are used to guide learning. We did not address the question of subclassification of leukemic disease, where the mutation status of the leukemic cells is currently the dominant approach (Arber et al., 2016, Heath et al., 2017, Papaemmanuil et al., 2016, The Cancer Genome Atlas Research Network (TCGA) et al., 2013), but rather focused on primary diagnosis, which continues to rely mostly on classical approaches (morphology, immunophenotyping, cytochemistry). We carried out extensive tests designed to address specific concerns relevant to practical use, including the case of transferring predictive models between entirely disjoint studies (that could be subject to batch effects or other unwanted variation) and even between transcriptomic platforms. Our results show that combining ML and blood transcriptomics can yield highly effective and robust classifiers. This supports the notion that transcriptomic-based ML could be used to assist AML diagnostics, particularly in settings wherein hematological expertise is not sufficiently available and/or costly.
Results
Establishment of a Unique GEP Dataset for Classifier Development
We hypothesized that the determination and comprehensive evaluation of GEP- and ML-based AML classifiers requires large datasets, should include samples from many sources to mimic the situation in real-world deployment, and should include several technical platforms to better understand their influence on classifier performance. To achieve these goals, we wanted to include the largest number of peripheral blood mononuclear cells (PBMC) or bone marrow samples possible and therefore systematically searched the National Center for Biotechnology Gene Expression Omnibus (GEO; Edgar, 2002) database for PBMC and bone marrow studies (Figure 1A). We identified 153,922 datasets, of which 111,632 contained human samples. To include only whole sample series and to avoid duplicate samples, we filtered for GEO series (GSE) and excluded the so-called super series, which resulted in 2,715 studies. We then focused the analysis on studies with samples analyzed on one of three platforms including the HG-U133A microarray, the HG-U133 2.0 microarray, and Illumina RNA-seq. Next, duplicated samples and studies working with pre-filtered cell subsets were excluded. This study search strategy resulted in 105 studies with a total of 12,029 samples (Figure 1) including 2,500 samples assessed by HG-U133A microarray (Dataset 1), 8,348 samples by HG-U133 2.0 microarray (Dataset 2), and 1,181 samples by RNA-seq (Dataset 3). In total, the dataset contained 4,145 AML samples of diverse disease subtypes and 7,884 other samples derived from healthy controls (n = 904), patients with acute lymphocytic leukemia (ALL, n = 3,466), chronic myeloid leukemia (CML, n = 162), chronic lymphocytic leukemia (CLL, n = 770), myelodysplastic syndrome (MDS, n = 267), and other non-leukemic diseases (n = 2,312) (Figures 1B, S1, and S2). Unless otherwise noted, all samples derived from patients with AML are referred to as cases and non-AML samples as controls. We additionally considered a differential diagnosis-like setting, in which case the controls comprised non-AML leukemias. According to the three platform types, the whole sample cohort was divided into three datasets referred to as datasets 1, 2, and 3 (Table S1, Figure S1).
Figure 1.

Establishing Datasets for the Largest AML Meta-study to Date
(A) Flowchart for the inclusion of studies. The gene expression omnibus (GEO) database was systematically searched for GEO Series of human PBMC and bone marrow samples processed with microarray platforms (Affymetrix HG-U133A and HG-U133 2.0) or next-generation RNA sequencing (RNA-seq) data. These data were filtered for inclusion of AML samples, samples of other leukemia, and healthy samples or other diseases. After manual revision and exclusion of duplicates and experiments using sorted cell populations (“expert filter”), the data were combined and normalized independently for each dataset.
(B) Detailed overview of the three datasets established in this study after filtering as given in (A).
See also Figure S1.
Effective AML Classification Using High-Dimensional Models
Here, we sought to assess classification of AML versus non-AML. Microarray data were RMA normalized using the R package affy (Gautier et al., 2004), whereas RNA-seq data were normalized as implemented in the R package DESeq2 (Love et al., 2014). For further analysis and better comparison between the different datasets, we trimmed the data to 12,708 genes, which were annotated within all datasets. No filtering of low expressed genes was performed (Figure 2A). The size of the test set was 20% of the total sample size, and random sampling of training and test sets was repeated 100 times. As main performance metrics, we considered (held-out) accuracy, sensitivity, and specificity. Classification was performed using l1-regularized logistic regression (the lasso; see also later).
Figure 2.

Prediction of AML in Random and Cross-study Sampling Scenarios
(A) Schema illustrating the approach to predict AML in random and cross-study sampling scenarios.
(B–D) AML classification accuracies based on the lasso model of AML versus all other samples and for both sampling strategies are shown for dataset 1 (B), dataset 2 (C), and dataset 3 (D).
(E and F) Classification accuracies for the differential diagnosis case (AML versus other leukemic samples, namely, AML, ALL, CML, CLL, and MDS) for both sampling strategies are shown for dataset 1 (E) and dataset 2 (F). Mean accuracies of the lasso models are shown as a function of the training sample size ntrain. Results are over 100 random training and test sets, with error bars indicating the standard deviation.
(G) Comparison of the performance of the LASSO models introduced in panels A to F with a neural network approach using either 5 or 10 layers. Error bars indicate the standard deviation.
See also Figures S3–S8 and S13, and Tables S2 and S4.
First, we included all non-AML samples, consisting of healthy controls and non-leukemic diseases, among the controls (Figures 2B, 2D, and 2F, light blue lines, Table S2). The goal was to classify unseen samples as AML or control. To understand how much data is needed in this setting, we plotted learning curves showing the test set accuracy as a function of training sample size ntrain. For each gene expression platform, this was done by randomly subsampling ntrain samples and testing on held-out test data with fixed sample size ntest (as shown). We see that prediction in this setting is already highly effective with a small number of training samples, although accuracy still increases with increasing ntrain (note that the total number of samples and hence range of ntrain differs by platform).
In many clinical settings, the control group does not contain healthy controls, but rather related diseases. To test effectiveness in a differential diagnosis setting, we repeated the experiments but with controls sampled only from other leukemic diseases, such as ALL, CLL, CML, and MDS (Figures 2C, 2E, S3–S5, and S13). We observed similar prediction results, which indicated that prediction accuracy is not only due to large differences between AML and non-leukemic conditions.
In additional experiments we considered performance of nine different classification methods (Figures S3–S5, Table S4). We could predict AML with good accuracy with all tested classification algorithms on microarray platforms (Figures S3 and S4). For RNA-seq data, the lasso, k nearest neighbors, linear support vector machines, linear discriminant analysis, and random forests were able to predict with high sensitivity and specificity (Figure S5, for details on used packages see Transparent Methods). Lasso-type methods have several advantages, including extensive theoretical support and interpretability, so we focused on these as our main predictive tool. Deep neural networks provided similar prediction performance to the lasso (Figure 2G) on dataset 2. We preferred the latter in this setting due to interpretability, because the lasso provides explicit variable selection, facilitating model interpretation.
Evaluation of Positive Predictive Value under Various Prevalence Scenarios
For diagnostic utility, the positive predictive value (PPV; the probability of disease given a positive test result) is an important quantity. The PPV depends not only on sensitivity and specificity but also on prevalence, as it is harder to achieve a high PPV for a condition that is rare in the population of interest. This has implications for any change to the effective threshold at which a potential case enters the diagnostic pipeline. As this threshold is relaxed, the prevalence (in the tested population) decreases, which in turn reduces the PPV. Thus, although we found high accuracy, sensitivity, and specificity already at moderate ntrain, depending on the use case, this could still imply that large training sample sizes would be useful to reach acceptable PPVs. For example, the predictive gains in increasing ntrain from the lowest to the highest values indicated in Figure 2C, which is for the dataset with largest total sample size, correspond to a doubling of PPV from ∼20% to ∼40% at an assumed prevalence of 1% (Figure 3). This illustrates the fact that although after a certain point increasing ntrain tends to increase accuracy only slowly, the gains, even if small in absolute terms, can be highly relevant with respect to PPV in low-prevalence settings.
Figure 3.
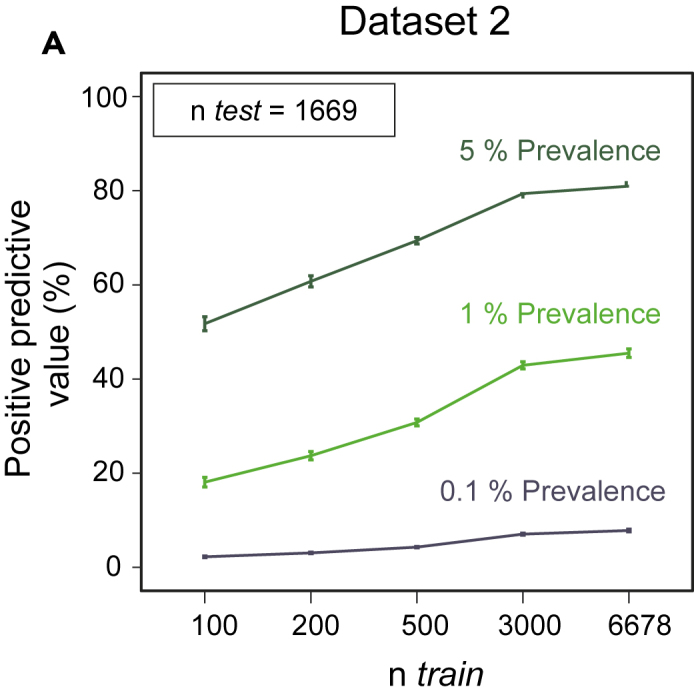
Positive Predictive Value
Positive predictive value as a function of ntrain corresponding to the setting as in Figure 2C and assumed prevalence of 0.1%, 1%, or 5% is shown (see text). Error bars depict the standard deviation.
Assessing the Effect of Cross-Study Variation on Predictive Performance
Microarray data and data generated by high-throughput sequencing are both known to be susceptible to batch effects (Leek et al., 2010). More generally, diverse study-specific effects and sources of study-to-study variation can pose problems in the context of predictive tests for clinical applications. Predictors that perform well within one study may perform worse when applied to data from new studies (Hornung et al., 2017) with implications for practical generalizability.
The aforementioned results spanned data from multiple heterogeneous studies. Provided training and test data are sampled in the same way, such heterogeneity does not necessarily pose problems for classification, as evidenced earlier. However, if the training and test data are from entirely different sites/studies (rather than randomly sampled from a shared pool), then the impact of batch/study effects may be more serious. We took advantage of the large number of studies in our dataset to sample training and testing sets in such a way that they were mutually disjoint with respect to studies. That is, any individual study from which any sample was included into the training dataset was entirely absent from the test set, and vice versa, and we use the term cross-study to refer to this strictly disjoint case. Results are shown in Figures 2B, 2D, and 2F (dark blue lines). As expected, performance was worse in the cross-study setting than under entirely random sampling (light blue lines). However, in the dataset with the largest sample size (dataset 2, platform HG-U133 2.0; Figure 2D) we see that the performance in the cross-study case gradually catches up to the random sampling case with only a small gap at the largest ntrain. The other two datasets have smaller total sample sizes, so they never reach comparable training sample sizes. Note that we did not carry out any batch effect removal using tools such as combat (Johnson et al., 2007), SVA (Leek et al., 2012), or RUV (Jacob et al., 2016), and in that sense our results are conservative. Despite the availability of these and other tools for batch effect correction, it is difficult to be fully assured of the removal of unwanted variation in practice. Our intention here was not to remove between-study variation but rather to (conservatively) quantify its effects on accuracy.
Owing to the large number of studies included in our analysis, we were able to carry out an entirely disjoint cross-study analysis also for the differential diagnosis case. These results are shown in Figures 2C and 2E (dark blue lines; cross-study sampling for differential diagnosis was not possible using dataset 3 due to lack of samples, see Figure S1) and are broadly similar, also across different classification algorithms (Figures S6–S8).
However, even in this strict cross-study sampling scenario, where samples from studies of the training and testing sets are entirely disjoint, the predictor matrices are still normalized together, meaning that the prediction rule still depends to some extent on features (not labels) in the test set. To address this issue, we performed addon RMA normalization (Hornung et al., 2017) as implemented in the R package bapred (Hornung et al., 2016). We split dataset 1 in training and testing data in a strict cross-study setting as in Figure 2A, performed RMA normalization on the training data, and then performed addon normalization of the test data onto the training data, meaning that the normalization of the training data does not in any sense depend on the testing data (Figure S9A). Accuracy, sensitivity, and specificity of this setting compare well to the “classical” cross-study setting described earlier (Figures 2 and S6A).
Classification Accuracy and AML Subtypes
Next, we sought to understand whether the accuracy of the classifiers depended on specific AML subtypes. As only a limited number of samples in our data were already annotated according to the new World Health Organization (WHO) classification, we utilized the French-American-British (FAB) classification of AML. The FAB classification was available for a total of 616 samples of dataset 1 and 1,269 samples in dataset 2. We utilized results from train/test splits of datasets 1 and 2 to quantify accuracy for each individual sample (Figures 4A and 4D). No particular AML subtype dominated classification accuracy in either dataset 1 or 2 (Figures 4C and 4F). Prediction accuracy was also consistent when broken down by non-AML disease category (Figures 4B and 4E). In dataset 1, 8 MDS samples and 10 samples from patients with Down syndrome transient myeloproliferative disorder were misclassified. However, both are diseases closely related to AML and represented by a very limited sample size in dataset 1. For dataset 2, correct classification of MDS appeared to depend on the individual sample, potentially reflecting disease heterogeneity.
Figure 4.

Accuracy of AML Classification in Different Leukemia Types and AML Subclasses
(A) Schema for determining accuracy for leukemia types and AML subclasses in dataset 1.
(B–D) Normalized dataset 1 was randomly split into training and test sets 100 times (same permutations as in Figure 2B), and prediction accuracy is reported for each individual sample. The bars in the figure correspond to individual samples broken down by leukemia type (B) and AML subtype (C). (D) Schema for determining accuracy for leukemia types and AML subclasses in dataset 2.
(E–H) Normalized dataset 2 was randomly split into training and test sets 100 times (D) and prediction accuracy is reported for each individual sample, listed by leukemia type (E) and AML subtype (F). Workflow for M3 subtype prediction using dataset 2 (G) Boxplots of prediction accuracy, sensitivity, and specificity over 100 train/test splits (H). Error bars depict the standard deviation.
While our main focus is on diagnosis, we asked whether the transcriptomic data could contribute to classifying AML subtypes. To exemplify this aspect, we focused on AML subtype M3, also named acute promyelocytic leukemia, as this is the only genetically defined subtype of the FAB classification that is also part of the WHO classification. Using dataset 2, we used a train/test approach, drawing subsets of dataset 2 with approximately the same class balance as in main results (here, one-third AML-M3 cases in every subset) (Figure 4G). M3 was distinguished from non-M3 AML with high accuracy, sensitivity, and specificity (Figure 4H). Although the data here do not allow rigorous testing of transcriptomics combined with genomics in an integrated fashion for subtype classification, and we would not recommend at this stage the use of a purely transcriptomic classifier for subtyping, these initial results suggest that it may be useful to further study the potential value of testing scalable ML- and GEP-based methodology in the area of subclassification as well.
Translation of Classifiers across Technical Platforms
Over the long term, clinical pipelines must cope with changes in technological platforms. It is therefore relevant to understand to what extent predictors can generalize not only between studies but also between different platforms. In other words, is it possible to take a model learned on data from platform A and deploy it using unseen data from platform B? To address this question, we constructed AML versus non-AML training and test sets in a cross-platform manner, i.e., training on one platform and testing on another (Figure 5A). That is, a model was learned using independently normalized data from one platform and then this model, used “as is,” with no further fine-tuning, was used to make predictions using expression data from a different platform. We see that classification accuracy varies greatly. Classifiers that were trained on HG-U133 A (dataset 1) work well when tested using data generated with the more advanced microarray HG-U133 2.0 (dataset 2) (Figures 5B and S10) and models trained on HG-U133 2.0 data can predict well using RNA-seq data (dataset 3) (Figures 5D and S11). However, models trained naively on HG-U133 A data cannot predict using RNA-seq data (Figures 5F, S12, and S13, Table S4).
Figure 5.

Translating Predictive Signatures across Technological Platforms
(A) Schema of signature translation across platforms. Datasets were normalized individually and trimmed to 12,708 common genes. The classifiers were trained on subsamples of different sizes on one platform and tested on all samples of another platform.
(B–G) Classification accuracies are shown as a function of training sample size (ntrain) without rank transformation (B, D, and F) and with rank transformation (C, E, and G). For the latter case, the training and test datasets (from different platforms) were separately rank transformed (see text for details). Error bars depict the standard deviation.
See also Figures S10–S13.
To explore the utility of simple transformations in this context, we then performed a rank transformation to normality on all datasets (see Transparent Methods). This is among the simplest and best known data transformations, has previously been shown to increase the performance of prognostic gene expression signatures, and can even outperform more complex variance-stabilizing approaches (Zwiener et al., 2014). With this approach, we reached very good overall performance across all platforms under study (Figures 5C, 5E, and 5G). This is particularly interesting for the prediction of dataset 3, which fails when the model is trained on the untransformed dataset 1 (Figures 5F, 5G, S11, and S12) and performs worse (on dataset 3) as ntrain increases. This is because as ntrain increases, the models learn a pattern that is increasingly fine-tuned to the data type in the training set. However, because the test set is from a different platform, test performance suffers. This is most likely not classical “overfitting,” because as shown in previous figures test error is well-behaved within dataset 1, but rather an example of a transfer learning/distribution-shift type problem, which in this case is solved simply by rank transformation. Note that the transformation is simply applied to each dataset independently and could be easily deployed in any practical use case without any need for prior input into, e.g., cross-platform designs such as inclusion of control samples.
Furthermore, we used the rich resource of the present dataset to explore whether prediction across leukemic diseases would be possible as well. For this, we trained a multilabel-classifier on dataset 2 using both datasets 1 and 3 as independent validation sets (Figure S14A). We found good prediction accuracy, sensitivity, and specificity over most tested diseases (Figure S14B); however, a rigorous study over all leukemic conditions would clearly require the inclusion of more training samples for CLL and CML.
Predictive Signatures and AML Biology
The predictive models derived from the lasso and used earlier are sparse in the sense that they automatically select a small number of genes to drive the prediction. The genes are selected in a unified global analysis, rather than by differential expression (DE) on a gene-by-gene basis. From a statistical point of view, global sparsity patterns for prediction and gene-by-gene DE are different criteria. Differentially expressed genes are those that individually have different levels between the groups, whereas genes selected for prediction are those that together perform well in a predictive sense. For the lasso, the selected set of genes also typically includes false-positives with respect to the truly relevant predictors. Furthermore, a good set of genes for prediction need not be mechanistic (in the sense of constituting causal drivers of the disease state). We therefore sought to understand the relationship between DE, known mechanisms, and predictive gene signatures.
Using dataset 2 (the largest dataset) we compared DE and the sparse predictive models (Figure S15). We performed DE analysis using the whole dataset and compared the results with the set of genes in the lasso model (“lasso genes”) based on the same data (Figure 6A). A total of 506 genes was differentially expressed (“DE genes”), of which 26 were associated with the disease ontology term or Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway for AML (“AML-related genes”). Of the 141 lasso genes, 7 genes were leukemia related and 46 were DE genes, meaning that many of the lasso genes were not differentially expressed, as clearly seen when overlaying the lasso gene selection on a volcano plot (Figure 6A). This underlines the fact that DE and predictive value in a signature sense are different criteria.
Figure 6.

Predictive Signatures and AML Biology
(A) Volcano plot of global differentially expressed (DE) genes and genes of the lasso model (“lasso genes”) in dataset 2, and Venn diagram indicating the overlap of both gene sets and the genes included in the KEGG pathway or the disease ontology term “AML.”
(B) Inclusion plot of DE genes and lasso genes in 100 random permutations of dataset 2. The plot is sorted according to DE gene rank, and a Venn diagram shows the overlap between genes with a minimum of 50% inclusion.
(C) Boxplot of accuracy, sensitivity, and specificity of the predictive model trained and tested on random subsets of dataset 2 with inclusion of all genes of the dataset and without 155 genes known to be relevant for AML biology. Error bars depict the standard deviation.
(D) Heatmap and hierarchical clustering of z-scaled expression values of 35 genes with >50% inclusion both in lasso and DE genes, as shown in (B). Genes with known associations with AML are marked red; genes associated with other types of leukemia are labeled in orange.
See also Figure S15.
Next, we extended this analysis to focus on DE and lasso genes whose selection was robust to data subsampling. This was done by subsampling half the dataset randomly 100 times and in each such subsample carrying out the full DE and lasso analyses. For the lasso this type of approach has been studied under the name stability selection (Meinshausen and Bühlmann, 2010). DE and lasso genes were then scored according to the frequency with which they appeared among the 100 rounds of selection (Figure 6B). Thus, an inclusion score of 100% for a DE gene means that the gene is selected as differentially expressed in all 100 iterations, and similarly for the lasso genes. In total, 669 genes passed the DE cutoffs in at least 50% of the iterations, whereas 80 genes were called in at least 50% of the iterations by the lasso model (Figure 6B). Of these genes, 35 were called according to both criteria. The above-mentioned results show that even among the genes that are included in the lasso models with high frequency (i.e., those genes that are robustly selected for prediction), many are not differentially expressed.
Next, we excluded the 155 known AML genes that are associated with the disease ontology term or KEGG pathway for AML from the prediction, which did not affect disease prediction at all (Figure 6C), highlighting the strong robustness of the classifier. To better understand the potential biological relevance for AML of the 35 genes that were robustly called under both DE and lasso criteria (Figure 6B), we visualized the top-ranked genes over all 8,348 samples within the dataset by hierarchical clustering of z-transformed expression values (Figure 6D). We identified one distinct cluster of genes with the majority of genes being elevated in AML compared with other leukemias and non-leukemic samples (cluster 1, n = 29). Although we identified several well-known AML-related genes (gene name in red color) such as the KIT Proto-Oncogene Receptor Tyrosine Kinase (KIT) (Gao et al., 2015, Heo et al., 2017, Ikeda et al., 1991), RUNX2 (Kuo et al., 2009), and FLT3 (Bullinger et al., 2008, Carow et al., 1996) in this cluster, many genes have not yet been linked to AML biology, and, although not the focus of the present article, further study of these genes may be interesting from a mechanistic point of view. Within the other cluster (cluster 2, n = 6 genes), genes had reduced expression values in AML compared with other leukemias and two of these genes have been linked to other types of leukemias (gene names in orange color).
Discussion
Despite the pioneering studies by Golub and others (Debernardi et al., 2003, Kohlmann et al., 2003, Ross et al., 2004, Schoch et al., 2002, Virtaneva et al., 2001) suggesting high potential value of GEP for primary AML diagnosis and differential diagnosis, current recommendations for diagnosing this disease currently center on classical approaches including assessment of morphology, immunophenotyping, cytochemistry, and cytogenetics (Döhner et al., 2017). Analyzing more than 12,000 samples from more than 100 individual studies, we provide evidence that combining large transcriptomic data with ML allows for the development of robust disease classifiers. Such classifiers could, in the future, potentially assist in primary diagnosis of this deadly disease particularly in settings where hematological expertise is not sufficiently available and/or costly. Considering the increased utilization of whole-genome and whole-transcriptome sequencing in the management of patients with cancer, we propose that application of GEP- and ML-based classifiers for diagnosis needs to be re-evaluated. This is in line with previous suggestions by the International Microarray Innovations in Leukemia Study Group (Haferlach et al., 2010). Furthermore, we suggest that similar analyses may be useful for other diseases when analyzing whole blood or PBMC-derived gene expression profiles, or for multiple conditions in parallel (see later).
We sought to understand and address some of the bottlenecks in the way of clinical deployment of transcriptomic-based ML tools for diagnosis. To this end, we considered a range of practical scenarios, including cross-study issues and prediction across different technological platforms. We found that accurate prediction is possible across a range of scenarios and, in many cases, with relatively few training samples. However, we also showed that depending on the use case and the associated prevalence, large training sets may be required to reach accuracies high enough to yield acceptable PPVs.
Our results show that with existing technologies it is potentially possible to achieve good performance in a near-automated fashion. An ML-plus-genomics approach can be run at very low marginal cost: the RNA assays can already be done at <$100 (and this continues to fall), and in the long-term these costs will drop still further. To our knowledge, this is already in a cost range that is lower than the combined use of morphology, immunophenotyping, and cytochemistry for primary AML diagnosis. Furthermore, the sparse models we considered, once trained, require only a small subset of the genome, hence custom sequencing pipelines could be used. Marginal cost is important precisely because it opens up the possibility of a truly scalable detection/diagnosis strategy. One example of a recently developed, very-low-cost whole-transcriptomics protocol is BRB-seq which allows generating genome-wide transcriptomic data at a similar cost as profiling four genes using RT-qPCR (Alpern et al., 2019), which could be a candidate for future clinical development. Furthermore, recent developments in nanopore sequencing (Byrne et al., 2017) suggest that in the future, delivery of transcriptomic assays could be greatly simplified, and this, combined with cloud- or local-device-based ML prediction, would represent a paradigm shift in terms of scalability and accessibility. Such transcriptome-based ML might therefore also be utilized at an earlier time point in the disease course, when patients present with non-specific symptoms to their primary care physician. Here, ML-based diagnostics might assist a faster transfer of the patient to specialized hematology centers for complete diagnostics and therapeutic management.
The next steps toward better understanding ML-based diagnosis for AML would include prospective studies specifically aimed at assessing diagnostic utility. Before any development in the future pivotal clinical trials for approval with the respective regulatory bodies would be required. Naturally, any such development would require additional, independent studies with the development of deployment-ready pipelines, which by itself is a nontrivial undertaking (as discussed in Keane and Topol, 2018). However, initial prospective studies have already been started, such as the 5000 genomes project (https://www.mll.com/en/science/5000-genome-project.html), which also performs RNA-seq to develop such a classifier for the clinics. It is also important to emphasize that just as regulatory standards have evolved for classical diagnostics, so too will new regulatory frameworks be needed for ML-assisted diagnostics in the future (Keane and Topol, 2018).
An additional point concerns explicit and implicit thresholds at which a suspected case is entered into the pipeline in the first place. A lower threshold for entry could lower false-negatives and reduce the risk of delayed treatment (which has been associated with worse outcomes, notably in younger patients; Sekeres et al., 2008). Using current diagnostic systems any such change would dramatically increase the overall costs; in contrast, more efficient solutions would allow thresholds to be optimized for patient benefit while keeping the overall costs controlled. Naturally any modification to the overall diagnostic strategy would need a full health economic and decision analysis (accounting in particular for a necessarily higher false-positive rate) and case-by-case assessment. For some diseases it may be the case that earlier entry into a diagnostic pipeline would overall not be beneficial, a point that is widely appreciated in the context of population-level screening (see, e.g., Jacobs et al., 2016). Nevertheless, the point is that scalable diagnostic strategies increase the scope for optimization of decision making for patient benefit.
We saw also encouraging results across other conditions. Although the data used in the present study do not allow rigorous study of diagnosis across multiple conditions, we conjecture that diagnosis of multiple conditions from blood transcriptomes may be possible, opening up the possibility of training multi-class classifiers on blood transcriptomic data. Note that this would allow diagnosis of several conditions at essentially the same marginal cost per additional sample, bolstering the economic case outlined earlier. Rigorous study would require new pan-disease study designs, but we think that such approaches could lead to large efficiency gains in the future.
All our models were learned in an unbiased manner, directly from the full transcriptome data with no prior biological knowledge or any pre-selection of genes. We showed that genes relevant for prediction were often not differentially expressed and that prediction was robust to removal of known AML-related genes. These observations illustrate two points of relevance to clinical applications. First, for prediction it can be more fruitful to consider signatures derived in data-driven, genome-wide fashion than to think in terms of single genes or DE. Second, high-dimensional analyses, although complex relative to more classical methods, can be highly predictive as well as robust to the presence or absence of specific genes. Taken together, our results underline the immense value of making GEP data publicly available, allowing for new and large-scale multi-study analyses. Furthermore, we support the notion that the application of ML approaches based on sequencing data to identify gene signatures for certain diseases such as AML will become part of recommendations for diagnosis and management of AML. We envision that combining whole-genome and whole-transcriptome analysis based on ML algorithms will ultimately allow early detection, diagnosis, differential diagnosis, subclassification, and outcome prediction in an integrated fashion.
Limitations of the Study
It is important to note that the data used here were pooled from multiple studies with different designs and goals. Further work, including suitably designed prospective studies, would be needed to better understand the diagnostic utility of an ML-plus-transcriptomics approach. Site- and study-specific effects may be relevant for clinical applications. This is because a classifier once learned might be deployed in a range of new settings (sites, regions) that could lead in a number of ways to unwanted variation. If training and test sets are very different, this can impact performance. In clinical applications of predictive models it will be important to continually track performance even after deployment and the possibility of distributional shifts that require more complex analyses cannot be ruled out.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy—EXC2151—390873048. This project has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No 733100. J.L.S is member of the Helmholtz network Sparse2Big.
Author Contributions
Conceptualization, J.L.S. and S.M.; Methodology, S.M., J.L.S., and S.W.-H.; Software, S.W.-H., K.P., and B.T.; Validation, B.T.; Formal Analysis, S.W.-H., T.U., K.P., J.S.-S., K.K., M.B., L.S.; Investigation, S.W.-H., T.U., P.G., K.B.; Resources, T.H., S.W.-H.; Data Curation, S.W.-H.; Writing – Original Draft, J.L.S., S.M., S.W.-H.; Visualization, S.W.-H., Supervision, J.L.S., S.M., M.B.
Declaration of Interests
There are no competing interests.
Published: January 24, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.100780.
Contributor Information
Sach Mukherjee, Email: sach.mukherjee@dzne.de.
Joachim L. Schultze, Email: j.schultze@uni-bonn.de.
Data and Code Availability
Processed data can be accessed via the SuperSeries GSE122517 or via the individual SubSeries GSE122505 (dataset 1), GSE122511 (dataset 2), and GSE122515 (dataset 3). The code for preprocessing and for predictions can be found at GitHub (https://github.com/schultzelab/aml_classifer). In addition, all data and package versions are stored in a docker container on Docker Hub (https://hub.docker.com/r/schultzelab/aml_classifier, Table S3).
Supplemental Information
References
- Alpern D., Gardeux V., Russeil J., Mangeat B., Meireles-Filho A.C.A., Breysse R., Hacker D., Deplancke B. BRB-seq: ultra-affordable high-throughput transcriptomics enabled by bulk RNA barcoding and sequencing. Genome Biol. 2019;20:71. doi: 10.1186/s13059-019-1671-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson A., Ritz C., Lindgren D., Edén P., Lassen C., Heldrup J., Olofsson T., Råde J., Fontes M., Porwit-MacDonald A. Microarray-based classification of a consecutive series of 121 childhood acute leukemias: prediction of leukemic and genetic subtype as well as of minimal residual disease status. Leukemia. 2007;21:1198–1203. doi: 10.1038/sj.leu.2404688. [DOI] [PubMed] [Google Scholar]
- Arber D.A., Orazi A., Hasserjian R., Thiele J., Borowitz M.J., Le Beau M.M., Bloomfield C.D., Cazzola M., Vardiman J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–2405. doi: 10.1182/blood-2016-03-643544. [DOI] [PubMed] [Google Scholar]
- Brynjolfsson E., McAfee A. W W Norton & Co; 2014. The Second Machine Age: Work, Progress, and Prosperity in a Time of Brilliant Technologies. [Google Scholar]
- Bühlmann P., van de Geer S. Springer; 2011. Statistics for High-Dimensional Data: Methods, Theory and Applications. [Google Scholar]
- Bullinger L., Döhner K., Bair E., Fröhling S., Schlenk R.F., Tibshirani R., Döhner H., Pollack J.R. Use of gene-expression profiling to identify prognostic subclasses in adult acute myeloid leukemia. N. Engl. J. Med. 2004;350:1605–1616. doi: 10.1056/NEJMoa031046. [DOI] [PubMed] [Google Scholar]
- Bullinger L., Döhner K., Kranz R., Stirner C., Fröhling S., Scholl C., Kim Y.H., Schlenk R.F., Tibshirani R., Döhner H. An FLT3 gene-expression signature predicts clinical outcome in normal karyotype AML. Blood. 2008;111:4490–4495. doi: 10.1182/blood-2007-09-115055. [DOI] [PubMed] [Google Scholar]
- Byrne A., Beaudin A.E., Olsen H.E., Jain M., Cole C., Palmer T., DuBois R.M., Forsberg E.C., Akeson M., Vollmers C. Nanopore long-read RNAseq reveals widespread transcriptional variation among the surface receptors of individual B cells. Nat. Commun. 2017;8:16027. doi: 10.1038/ncomms16027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carow C.E., Levenstein M., Kaufmann S.H., Chen J., Amin S., Rockwell P., Witte L., Borowitz M.J., Civin C.I., Small D. Expression of the hematopoietic growth factor receptor FLT3 (STK-UFIk2) in human leukemias. Blood. 1996;87:1089–1096. [PubMed] [Google Scholar]
- Ciriello G., Gatza M.L., Beck A.H., Wilkerson M.D., Rhie S.K., Pastore A., Zhang H., McLellan M., Yau C., Kandoth C. Comprehensive molecular portraits of invasive lobular breast cancer. Cell. 2015;163:506–519. doi: 10.1016/j.cell.2015.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debernardi S., Lillington D.M., Chaplin T., Tomlinson S., Amess J., Rohatiner A., Lister T.A., Young B.D. Genome-wide analysis of acute myeloid leukemia with normal karyotype reveals a unique pattern of homeobox gene expression distinct from those with translocation-mediated fusion events. Genes Chromosomes Cancer. 2003;37:149–158. doi: 10.1002/gcc.10198. [DOI] [PubMed] [Google Scholar]
- Ding L., Ley T.J., Larson D.E., Miller C.A., Koboldt D.C., Welch J.S., Ritchey J.K., Young M.A., Lamprecht T., McLellan M.D. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481:506–510. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Döhner H., Estey E.H., Amadori S., Appelbaum F.R., Buchner T., Burnett A.K., Dombret H., Fenaux P., Grimwade D., Larson R.A. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115:453–474. doi: 10.1182/blood-2009-07-235358. [DOI] [PubMed] [Google Scholar]
- Döhner H., Estey E., Grimwade D., Amadori S., Appelbaum F.R., Büchner T., Dombret H., Ebert B.L., Fenaux P., Larson R.A. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129:424–447. doi: 10.1182/blood-2016-08-733196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteva A., Kuprel B., Novoa R.A., Ko J., Swetter S.M., Blau H.M., Thrun S. Dermatologist-level classification of skin cancer with deep neural networks. Nature. 2017;542:115–118. doi: 10.1038/nature21056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X., Lin J., Gao L., Deng A., Lu X., Li Y., Wang L., Yu L. High expression of c-kit mRNA predicts unfavorable outcome in adult patients with t(8;21) acute myeloid leukemia. PLoS One. 2015;10:e0124241. doi: 10.1371/journal.pone.0124241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garzon R., Volinia S., Papaioannou D., Nicolet D., Kohlschmidt J., Yan P.S., Mrózek K., Bucci D., Carroll A.J., Baer M.R. Expression and prognostic impact of lncRNAs in acute myeloid leukemia. Proc. Natl. Acad. Sci. U S A. 2014;111:18679–18684. doi: 10.1073/pnas.1422050112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier L., Cope L., Bolstad B.M., Irizarry R.A. affy–analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20:307–315. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- Golub T.R., Slonim D.K., Tamayo P., Huard C., Gaasenbeek M., Mesirov J.P., Coller H., Loh M.L., Downing J.R., Caligiuri M.A. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- Haferlach T., Kohlmann A., Wieczorek L., Basso G., Te Kronnie G., Béné M.-C., De Vos J., Hernández J.M., Hofmann W.-K., Mills K.I. Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: report from the International Microarray Innovations in Leukemia Study Group. J. Clin. Oncol. 2010;28:2529–2537. doi: 10.1200/JCO.2009.23.4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath E.M., Chan S.M., Minden M.D., Murphy T., Shlush L.I., Schimmer A.D. Biological and clinical consequences of NPM1 mutations in AML. Leukemia. 2017;31:798–807. doi: 10.1038/leu.2017.30. [DOI] [PubMed] [Google Scholar]
- Heo S.-K., Noh E.-K., Kim J.Y., Jeong Y.K., Jo J.-C., Choi Y., Koh S., Baek J.H., Min Y.J., Kim H. Targeting c-KIT (CD117) by dasatinib and radotinib promotes acute myeloid leukemia cell death. Sci. Rep. 2017;7:15278. doi: 10.1038/s41598-017-15492-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoadley K.A., Yau C., Wolf D.M., Cherniack A.D., Tamborero D., Ng S., Leiserson M.D.M., Niu B., McLellan M.D., Uzunangelov V. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell. 2014;158:929–944. doi: 10.1016/j.cell.2014.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung R., Boulesteix A.-L., Causeur D. Combining location-and-scale batch effect adjustment with data cleaning by latent factor adjustment. BMC Bioinformatics. 2016;17:27. doi: 10.1186/s12859-015-0870-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung R., Causeur D., Bernau C., Boulesteix A.-L. Improving cross-study prediction through addon batch effect adjustment or addon normalization. Bioinformatics. 2017;33:397–404. doi: 10.1093/bioinformatics/btw650. [DOI] [PubMed] [Google Scholar]
- Ikeda H., Kanakura Y., Tamaki T., Kuriu A., Kitayama H., Ishikawa J., Kanayama Y., Yonezawa T., Tarui S., Griffin J. Expression and functional role of the proto-oncogene c-kit in acute myeloblastic leukemia cells. Blood. 1991;78:2962–2968. [PubMed] [Google Scholar]
- Jacob L., Gagnon-Bartsch J.A., Speed T.P. Correcting gene expression data when neither the unwanted variation nor the factor of interest are observed. Biostatistics. 2016;17:16–28. doi: 10.1093/biostatistics/kxv026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs I.J., Menon U., Ryan A., Gentry-Maharaj A., Burnell M., Kalsi J.K., Amso N.N., Apostolidou S., Benjamin E., Cruickshank D. Ovarian cancer screening and mortality in the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS): a randomised controlled trial. Lancet. 2016;387:945–956. doi: 10.1016/S0140-6736(15)01224-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson W.E., Li C., Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- Keane P.A., Topol E.J. With an eye to AI and autonomous diagnosis. NPJ Digit. Med. 2018;1:40. doi: 10.1038/s41746-018-0048-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlmann A., Schoch C., Schnittger S., Dugas M., Hiddemann W., Kern W., Haferlach T. Molecular characterization of acute leukemias by use of microarray technology. Genes Chromosomes Cancer. 2003;37:396–405. doi: 10.1002/gcc.10225. [DOI] [PubMed] [Google Scholar]
- Kristensen V.N., Vaske C.J., Ursini-Siegel J., Van Loo P., Nordgard S.H., Sachidanandam R., Sorlie T., Warnberg F., Haakensen V.D., Helland A. Integrated molecular profiles of invasive breast tumors and ductal carcinoma in situ (DCIS) reveal differential vascular and interleukin signaling. Proc. Natl. Acad. Sci. U S A. 2012;109:2802–2807. doi: 10.1073/pnas.1108781108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo Y.-H., Zaidi S.K., Gornostaeva S., Komori T., Stein G.S., Castilla L.H. Runx2 induces acute myeloid leukemia in cooperation with Cbfbeta-SMMHC in mice. Blood. 2009;113:3323–3332. doi: 10.1182/blood-2008-06-162248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavallee V.-P., Lemieux S., Boucher G., Gendron P., Boivin I., Armstrong R.N., Sauvageau G., Hebert J. RNA-sequencing analysis of core binding factor AML identifies recurrent ZBTB7A mutations and defines RUNX1-CBFA2T3 fusion signature. Blood. 2016;127:2498–2501. doi: 10.1182/blood-2016-03-703868. [DOI] [PubMed] [Google Scholar]
- Lavallée V.-P., Baccelli I., Krosl J., Wilhelm B., Barabé F., Gendron P., Boucher G., Lemieux S., Marinier A., Meloche S. The transcriptomic landscape and directed chemical interrogation of MLL-rearranged acute myeloid leukemias. Nat. Genet. 2015;47:1030–1037. doi: 10.1038/ng.3371. [DOI] [PubMed] [Google Scholar]
- Leek J.T., Scharpf R.B., Bravo H.C., Simcha D., Langmead B., Johnson W.E., Geman D., Baggerly K., Irizarry R.A. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet. 2010;11:733–739. doi: 10.1038/nrg2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek J.T., Johnson W.E., Parker H.S., Jaffe A.E., Storey J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28:882–883. doi: 10.1093/bioinformatics/bts034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley T.J., Mardis E.R., Ding L., Fulton B., McLellan M.D., Chen K., Dooling D., Dunford-Shore B.H., McGrath S., Hickenbotham M. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456:66–72. doi: 10.1038/nature07485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley T.J., Ding L., Walter M.J., McLellan M.D., Lamprecht T., Larson D.E., Kandoth C., Payton J.E., Baty J., Welch J. DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med. 2010;363:2424–2433. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loriaux M.M., Levine R.L., Tyner J.W., Fröhling S., Scholl C., Stoffregen E.P., Wernig G., Erickson H., Eide C.A., Berger R. High-throughput sequence analysis of the tyrosine kinome in acute myeloid leukemia. Blood. 2008;111:4788–4796. doi: 10.1182/blood-2007-07-101394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macrae T., Sargeant T., Lemieux S., Hébert J., Deneault E., Sauvageau G. RNA-Seq reveals spliceosome and proteasome genes as most consistent transcripts in human cancer cells. PLoS One. 2013;8:e72884. doi: 10.1371/journal.pone.0072884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinshausen N., Bühlmann P. Stability selection. J. R. Stat. Soc. 2010;72:417–473. [Google Scholar]
- Pabst C., Bergeron A., Lavallee V.-P., Yeh J., Gendron P., Norddahl G.L., Krosl J., Boivin I., Deneault E., Simard J. GPR56 identifies primary human acute myeloid leukemia cells with high repopulating potential in vivo. Blood. 2016;127:2018–2027. doi: 10.1182/blood-2015-11-683649. [DOI] [PubMed] [Google Scholar]
- Papaemmanuil E., Gerstung M., Bullinger L., Gaidzik V.I., Paschka P., Roberts N.D., Potter N.E., Heuser M., Thol F., Bolli N. Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 2016;374:2209–2221. doi: 10.1056/NEJMoa1516192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker J.S., Mullins M., Cheang M.C.U., Leung S., Voduc D., Vickery T., Davies S., Fauron C., He X., Hu Z. Supervised Risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009;27:1160–1167. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiñonero-Candela J., Sugiyama M., Schwaighofer A., Lawrence N.D. MIT Press; 2009. Dataset Shift in Machine Learning. [Google Scholar]
- Robertson A.G., Kim J., Al-Ahmadie H., Bellmunt J., Guo G., Cherniack A.D., Hinoue T., Laird P.W., Hoadley K.A., Akbani R. Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell. 2017;171:540–556.e25. doi: 10.1016/j.cell.2017.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross M.E., Mahfouz R., Onciu M., Liu H.-C., Zhou X., Song G., Shurtleff S.A., Pounds S., Cheng C., Ma J. Gene expression profiling of pediatric acute myelogenous leukemia. Blood. 2004;104:3679–3687. doi: 10.1182/blood-2004-03-1154. [DOI] [PubMed] [Google Scholar]
- Schoch C., Kohlmann A., Schnittger S., Brors B., Dugas M., Mergenthaler S., Kern W., Hiddemann W., Eils R., Haferlach T. Acute myeloid leukemias with reciprocal rearrangements can be distinguished by specific gene expression profiles. Proc. Natl. Acad. Sci. U S A. 2002;99:10008–10013. doi: 10.1073/pnas.142103599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekeres M.A., Elson P., Kalaycio M.E., Advani A.S., Copelan E.A., Faderl S., Kantarjian H.M., Estey E. Time from diagnosis to treatment initiation predicts survival in younger, but not older, acute myeloid leukemia patients. Blood. 2008;113:28–36. doi: 10.1182/blood-2008-05-157065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network (TCGA) Ley T.J., Miller C., Ding L., Raphael B.J., Mungall A.J., Robertson A.G., Hoadley K., Triche T.J., Laird P.W. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virtaneva K., Wright F.A., Tanner S.M., Yuan B., Lemon W.J., Caligiuri M.A., Bloomfield C.D., de La Chapelle A., Krahe R. Expression profiling reveals fundamental biological differences in acute myeloid leukemia with isolated trisomy 8 and normal cytogenetics. Proc. Natl. Acad. Sci. U S A. 2001;98:1124–1129. doi: 10.1073/pnas.98.3.1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch J.S., Ley T.J., Link D.C., Miller C.A., Larson D.E., Koboldt D.C., Wartman L.D., Lamprecht T.L., Liu F., Xia J. The origin and evolution of mutations in Acute Myeloid Leukemia. Cell. 2012;150:264–278. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X.-J., Xu J., Gu Z.-H., Pan C.-M., Lu G., Shen Y., Shi J.-Y., Zhu Y.-M., Tang L., Zhang X.-W. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat. Genet. 2011;43:309–315. doi: 10.1038/ng.788. [DOI] [PubMed] [Google Scholar]
- Zwiener I., Frisch B., Binder H. Transforming RNA-Seq data to improve the performance of prognostic gene signatures. PLoS One. 2014;9:e85150. doi: 10.1371/journal.pone.0085150. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Processed data can be accessed via the SuperSeries GSE122517 or via the individual SubSeries GSE122505 (dataset 1), GSE122511 (dataset 2), and GSE122515 (dataset 3). The code for preprocessing and for predictions can be found at GitHub (https://github.com/schultzelab/aml_classifer). In addition, all data and package versions are stored in a docker container on Docker Hub (https://hub.docker.com/r/schultzelab/aml_classifier, Table S3).