

 This work is licensed under a
This work is licensed under a Abstract
Polycystic ovary syndrome (PCOS) is a chronic endocrine and metabolic disease. Gut microbiota is closely related to many chronic diseases. In this study, we conducted a cross-sectional study and recruited 30 obese (OG) and 30 non-obese (NG) women with PCOS, 30 healthy women (NC) and 11 healthy but obese women (OC) as controls to investigate the characteristic gut microbiota and its metabolic functions in obese and non-obese patients with PCOS. The blood and non-menstrual faecal samples of all the participants were collected and analysed. As a result, the Hirsutism score, LH/FSH and serum T level in NG and OG both increased significantly compared with their controls (P < 0.05). High-throughput 16S rRNA gene sequencing revealed that the abundance and diversity of the gut microbiota changed in patients with PCOS. The linear discriminant analysis (LDA) indicated that Lactococcus was the characteristic gut microbiota in NG, while Coprococcus_2 in OG. Correlation heatmap analysis revealed that the sex hormones and insulin levels in human serum were closely related to the changes in the gut microbiota of NG and OG. Functional prediction analysis demonstrated that the citrate cycle pathway enriched both in NG and OG, and other 12 gut bacterial metabolic pathways enriched in NG. This study highlighted significant differences in the gut microbiota and predictive functions of obese and non-obese women with PCOS, thereby providing insights into the role and function of the gut microbiota that may contribute to the occurrence and development of PCOS in obese and non-obese women.
Keywords: polycystic ovary syndrome, gut microbiota, metabolic functions, clinical parameters
Introduction
Polycystic ovary syndrome (PCOS) is characterised by persistent anovulation, hyperandrogenism and insulin resistance (IR), involving a group of reproductive, endocrine and metabolic disorders. The pathophysiology of PCOS is centred on hyperandrogenism and IR and can cause many short- or long-term complications, such as spontaneous abortion, metabolic syndrome and endometrial cancer (1). The clinical manifestations of PCOS are highly heterogeneous, and obesity is a common clinical phenotype in patients with PCOS. Hyperinsulinemia and hyperandrogenism can mutually aggravate each other to form a ‘vicious circle’. Excessive fat storage in obese patients can increase insulin concentration in the blood and trigger and aggravate IR, promoting the development of PCOS (2).
The interrelation between gut microbiota and PCOS is attracting increased attention. Compared with healthy women, Lindheim et al. (3) found that the abundance of ML615J-28 and 124-7 from phylum Tenericutes was considerably low in the intestine of patients with PCOS. According to Liu et al. (4), the ratio of Escherichia/Shigella and the abundance of Streptococcus in intestine increase, whereas the abundance of Akkermansia and Ruminococcaceae decreases in patients with PCOS. Although Torres et al. (5) revealed that the gut microbiota diversity in patients with PCOS was closely related to hyperandrogenism, they did not conduct further studies on the correlation between obesity and gut microbiota in patients with PCOS, nor did they identify any specific genera closely related to the onset of PCOS. Based on the above research, Insenser et al. (6) determined the characteristics of gut microbiota from obese and non-obese patients with PCOS, non-hyperandrogenic women and healthy men, and found the specific correlations among gut microbiota, gender, sex hormone and obesity, which may promote the occurrence and development of PCOS. However, the study involved only 46 subjects divided into three groups. Additional interference factors were included when the researchers compared women with PCOS with healthy men. In addition, the altered metabolic functions of gut microbiota in patients with PCOS were not revealed. The purpose of our study was to investigate gut microbiota composition, reveal gut microbiota characteristics and predict metabolic functions and clinical manifestations in 101 obese and non-obese PCOS women and healthy women.
Materials and methods
Participants
A total of 60 women with PCOS aged 16–35 years who visited the Department of the Traditional Chinese Medicine Gynaecology of Changhai Hospital in Shanghai during June 2016 to June 2019 were recruited. These patients with PCOS consisted of 30 obese women (body mass indices, BMI ≥28 kg/m2) (OG) and 30 non-obese women (BMI 18.5–23.9 kg/m2) (NG). The PCOS diagnosis in the present study was based on the Rotterdam criteria revised on the 2003 Conference in the Netherlands. In addition, 30 healthy young women (BMI 18.5–23.9 kg/m2) from the Naval Medical University were selected as the non-obese control group (NC), and 11 healthy but obese women from Changhai Hospital Outpatient (BMI ≥28 kg/m2) as obese control group (OC). Individuals with one of the following conditions were excluded: pregnancy, smoking, tumour, chronic diarrhoea, autoimmune diseases, history of gastrointestinal surgery and other endocrine diseases. None of the individuals were treated with oral contraceptives, antiandrogens, insulin sensitizers, antibiotics and probiotics. Women in the control group showed no history of menstrual dysfunction, infertility, endocrine diseases or diagnosed PCOS by Rotterdam criteria. The study was approved by the China Registered Clinical Trial Ethics Review Committee (ethical review number: CHiECRT-20160050), and all of the participants were provided informed consent.
Sampling
All of the subjects enrolled in the current study were required to complete the following items: (1) measurement of anthropometric indicators, including height, weight, waistline and hipline, and acne, hairiness and black acanthosis scores; (2) detection of biochemical indicators, such as the sex hormone levels including luteinizing hormone (LH), follicle-stimulating hormone (FSH), testosterone, oestradiol (E2) and DHEA on the third day of the menstrual cycle and during menopause excluding pregnancy, and the glucose and insulin levels during an oral glucose tolerance test; and (3) the collection of faecal samples is described as follows. Approximately 3–5 g fresh faeces from each participant were collected during non-menstrual period and transferred to the laboratory in an ice box within 2 h. Each stool sample was then separately loaded into 4 EP tubes and frozen at −80°C until use.
DNA extraction and PCR amplification
The total DNA of the gut microbiota was obtained using the EZNA® soil kit (Omega Bio-tek, Norcross, GA, USA). The concentration and purity of the extracted DNA were analysed using NanoDrop2000. The DNA extraction quality was detected by 1% agarose gel electrophoresis. The V3–V4 variable region of the bacterial 16sRNA was amplified via PCR using the 338F (5′-ACTCCTACGGGAGGCAGCAG-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′) primers. Amplification was conducted by ABI GeneAmp® 9700, and the setting is as follows: pre-denaturation at 95°C for 3 min, 27 cycles (denaturation at 95°C for 30 s, annealing at 55°C for 30 s, extension at 72°C for 30 s) and extension at 72°C for 10 min. The amplification mixture volume was 20 µL, including 4 µL of 5*FastPfu buffer, 2 µL of 2.5 mM dNTPs, 0.8 µL of primer (5 µM), 0.4 µL of FastPfu polymerase and 10 ng of DNA template.
Illumina MiSeq sequencing
The PCR amplification products were collected using a 2% agarose gel and purified using AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA). The purified products were detected using 2% agarose electrophoresis after elution with Tris–HCl. The quantitative detection was then performed using QuantiFluorTM-ST (Promega). The purified amplified fragment was constructed into a library of PE 2*300 according to the standard operating protocol of the Illumina MiSeq platform (Illumina, San Diego, USA). The library was constructed as follows: (1) connecting the ‘Y’-shaped joint; (2) using magnetic beads to remove the self-ligating fragment; (3) enriching the library template by PCR amplification; and (4) producing ssDNA fragments after the denaturalization of sodium hydroxide. The sequencing processes were performed on Miseq PE300 platform of Illumina.
Sequencing process
The original sequence was quality controlled by Trimmomatic software and spliced by FLASH software. The detailed process was as follows: (1) bases below 20 at the end of reads were filtered and a window of 50 bp was set. If the average mass in the window was less than 20, we truncated the back-end bases from the window. Reads below 50 bp were filtered after quality control, and reads containing an N base were removed; (2) according to the overlapping relationship between sequencing data, paired reads were spliced into a sequence with a minimum 10 bp overlap length; (3) the maximum allowable mismatch ratio of the overlap region of stitching sequence was 0.2, and the mismatch sequence was screened out and (4) the barcode and primers at both ends of the sequence were used to distinguish the samples and adjust the sequence direction. The allowable and maximum mismatch number of barcodes and primers was 0 and 2, respectively. Usearch software of Qiime platform (version 7.0 http://drive5.com/uparse/) was then used to cluster the sequences according to 97% similarity. The representative sequence after clustering was annotated and clustered using the RDP classifier (http://rdp.cme.msu.edu/) and compared with Silva database (SSU128/16s bacteria) at 70% comparison threshold.
Bioinformatics analysis
Silva database was selected for comparison with threshold of 70% to obtain the community composition of each sample at different taxonomic levels. The OTU scale generated after flattened was used to analyse the diversity and abundance of gut microbiota among four groups. Alpha diversity indexes of gut microbiota were analysed by mothur software (version v.1.30.1). Sobs, chao and ace indexes reflected community richness. Simpson and Shannon indexes reflected community diversity. Coverage index reflected community coverage. Beta diversity was reflected by principal co-ordinates analysis (PCoA) charts conducted by PCoA statistical analysis method with R language. Kruskal–Wallis H test and multiple test correction were used to complete the analysis of species difference at the level of phylum and genus among four groups. Wilcoxon rank-sum test and GraphPad Prism 8.0 were used to analyse the difference between the two groups and map species diversity at phylum level. The gut microbiota with significant difference in abundance was detected by non-parametric factorial Kruskal–Wallis sum-rank test. Linear discriminant analysis (LDA) was used to estimate the effect of each species contributing to the difference and formed the LDA discriminant result table. R language (pheatmap package) was used to obtained the Spearman correlation coefficients between environmental factors and selected species, and heatmap diagram was used to display the obtained numerical matrix. The OTU abundance table was standardized by PICRUSt, and the Kyoto Encyclopedia of Genes and Genomes (KEGG) information corresponding to each OTU were obtained through Greengene ID according to the information of KEGG database. The abundance of each functional category was calculated according to OTU abundance. KEGG pathway at level 3 was used to analyse possible differential metabolic pathways among four groups with Kruskal–Wallis sum-rank test and then Wilcoxon rank-sum test.
Statistical analysis of clinical data
Quantitative demographic and clinical data of normal distribution were presented by mean ± s.d., and t-test was used to compare the two groups. Quantitative data of non-normal distribution were presented by median with interquartile range (IQR) and the two groups were compared by Wilcoxon rank-sum test. Qualitative demographic and clinical data were presented as percentages and compared by chi-square test. SPSS 21.0 was used for analysis. All statistical tests were selected as double-tailed. When P < 0.05, the difference was considered statistically significant.
Results
Clinical and hormonal variables in obese and non-obese women with PCOS
We recruited 60 patients with PCOS and 41 women without PCOS as controls. No significant difference in age among four groups was presented. According to the willingness of volunteers, we obtained the data of sex hormone levels on the third day of menstruation from all participants, but only obtained the data of serum insulin levels from patients with PCOS, for the poor compliance of control women (Table 1). Compared with NC, Hirsutism score, Acne score, LH, LH/FSH, testosterone, E2 and DHEA in NG increased significantly (P < 0.05). Compared with OC, Hirsutism score, LH/FSH and testosterone in OG increased significantly (P < 0.05), while FSH in OG decreased significantly (P < 0.05).
Table 1.
Clinical and biochemical characteristics of participants (mean with s.d.).
| Parameters | NC (n = 30) | NG (n = 30) | OC (n = 11) | OG (n = 30) |
|---|---|---|---|---|
| Age (years) | 22.1 ± 1.64 | 25.1 ± 4.27 | 25.3 ± 1.61 | 26.9 ± 4.86 |
| Hirsutism score | 0.53 ± 2.06 | 7.73 ± 2.65b | 0.64 ± 0.67 | 6.13 ± 3.22d |
| Acne score | 0.47 ± 0.52 | 1.43 ± 1.47b | 0.45 ± 0.52 | 0.55 ± 0.95 |
| LH (IU/L) | 6.63 ± 1.08 | 11.3 ± 1.15a | 6.23 ± 0.75 | 5.80 ± 1.61 |
| FSH (IU/L) | 7.07 ± 0.67 | 6.62 ± 1.59 | 7.76 ± 1.76 | 5.97 ± 1.17c |
| LH/FSH | 0.54 ± 0.20 | 1.72 ± 0.50b | 0.83 ± 0.15 | 0.87 ± 0.49c |
| Testosterone (μg/L) | 0.48 ± 0.03 | 0.77 ± 0.06b | 0.52 ± 0.10 | 0.76 ± 0.16c |
| E2 (pg/mL) | 40.6 ± 2.13 | 50.8 ± 5.68b | 39.4 ± 2.36 | 43.2 ± 9.54 |
| DHEA (μg/dL) | 200 ± 66.2 | 276 ± 105b | 232 ± 37.1 | 290 ± 114.5 |
| FBG (µIU/mL) | – | 2.38 ± 3.10 | 6.32 ± 7.14 | |
| IAUC | – | 0.023 ± 0.017 | 0.012 ± 0.03 | |
| HOMA-IR | – | 2.36 ± 3.20 | 6.35 ± 0.17 | |
| ISI | – | 0.029 ± 0.01 | 0.014 ± 0.02 | |
| Insulin_0min (µIU/mL) | – | 11.9 ± 2.10 | 21.2 ± 4.03 | |
| Insulin_30min (µIU/mL) | – | 89.2 ± 2.93 | 104 ± 1.71 | |
| Insulin_60min (µIU/mL) | – | 85.3 ± 4.31 | 134 ± 3.17 | |
| Insulin_120min (µIU/mL) | – | 45.5 ± 2.57 | 74.6 ± 2.39 | |
| Insulin_180min (µIU/mL) | – | 24.9 ± 2.2 | 45.2 ± 2.55 |
aP < 0.05 compared with NC; bP < 0.01 compared with NC; cP < 0.05 compared with OC; dP < 0.01 compared with OC.
DHEA, dehydroepiandrosterone; E2, estradiol; FBG, fasting blood glucose; FSH, follicle-stimulating hormone; HOMA-IR, homeostatic model assessment for insulin resistance; IAUC, insulin area under the curve; Insulin_Xmin, serum insulin levels after a 75 g glucose load at X minutes; ISI, insulin sensitive index; LH, luteinizing hormone; NC, non-obese control group; NG, non-obese group with PCOS; OC, obese control group; OG, obese group with PCOS.
Gut bacterial diversity in obese and non-obese women with PCOS
When the sequence depth of random sampling was from 1000 to 15,000, the dilution curve tended to be flat, indicating that the sequence depth was sufficient to reflect the information of most species in the sample (Fig. 1A). FLASH and Trimmomatic software were used to obtain a total of 5,374,374 sequences. The number of optimised bases was 2,337,573,915 bp, and the average length of the optimised sequence was 434.948. Usearch software was used to cluster OTU at 97% similarity level and obtain 865 OTUs in total (Fig. 1B and C). No significant difference was found in Beta diversity among the four groups (Fig. 1D). The Sobs (Fig. 1E), Chao (Fig. 1F) and Ace (Supplementary Fig. 1A, see section on supplementary materials given at the end of this article) indexes of OTU throughout in NG were significantly lower than those in NC (P < 0.05), but there was no significant difference between OC and OG (P > 0.05). The Simpson index of OTU throughout in NG was significantly higher than that in NC (P < 0.05) (Fig. 1G), and no significant difference was found between OC and OG (P > 0.05). We have not found the significant differences of the Shannon and Coverage indexes of OTU throughout between NC and NG, or between OC and OG (P > 0.05) (Supplementary Fig. 1B and C).
Figure 1.

Diversity analysis of gut microbiota among four groups. (A) Shannon–Wiener curves, showing that the amount of sequencing data is large enough to reflect the vast majority of microbial information in the samples. (B) Rank–Abundance curves. The X axis represents the OTU level, and the Y axis represents the relative percentage of the number of sequences in the OTU level. (C) Venn diagram, showing the number of common and unique OTUs, and the similarity and overlap of OTUs among groups. (D) Principal co-ordinates analysis (PCoA analysis). PC1 and PC2 represent the two suspected influencing factors of microbial composition migration. Percentage represents the contribution of principal coordinate components to sample composition differences. The closer the two sample points are, the more similar the species composition of the two samples is. (E) Sobs and (F) Chao indexes of OTU throughout in NC are significantly higher than that in NG (P < 0.05). (G) Simpson index of OTU throughout in NC is significantly lower than that in NG (P < 0.05). Sobs and Chao indexes of OTU throughout reflect the diversity of the community, while Simpson reflects the abundance. *P < 0.05; NC, non-obese control group; NG, non-obese group with PCOS; OC, obese control group; OG, obese group with PCOS.
Gut bacterial composition in obese and non-obese women with PCOS
At phylum level, significant differences in the abundance of gut microbiota among the four groups were revealed (Fig. 2A). The abundance of Tenericutes and Synergistets in NG was significantly lower than that in NC (P < 0.05), while the abundance of Fusobacteria was significantly higher than that in NC (P < 0.05) (Table 1). However, no differences of the abundance of bacteria at phylum were found between OC and OG (Table 1).
Figure 2.

Composition and Linear discriminant analysis of gut microbiota among four groups. (A) Composition of species at phylum level among four groups. (B) Composition of species at genus level among four groups. (C) Linear discriminant analysis (LDA) discriminant histogram of gut microbiota between NC and NG. (D) LDA discriminant histogram of gut microbiota between OC and OG. NC, non-obese control group; NG, non-obese group with PCOS; OC, obese control group; OG, obese group with PCOS.
At genus level, the abundance of 13 species in NG was significantly lower compared with NC (P < 0.05), such as Lactococcus and Alloprevotella. Compared with OC, the abundance of Coprococcus_2 in OG was significantly higher (P < 0.05), while the abundance of Coprococcus_3, Lactobacillus and Prevotella_7 were significantly lower (P < 0.05). (Fig. 2B and Table 2).
Table 2.
Relative abundances of bacteria at phylum and genus levels with significant difference between groups.
| Species at phylum level | % of total bacteria in NC (n = 30) | % of total bacteria in NG (n = 30) | P value | % of total bacteria in OC (n = 11) | % of total bacteria in OG (n = 30) | P value | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Median | IQR (1/4, 3/4) | Median | IQR (1/4, 3/4) | Median | IQR (1/4, 3/4) | Median | IQR (1/4, 3/4) | |||
| Firmicutes | 61.0 | 24.3 (44.5, 68.8) | 53.8 | 18.9 (45.5, 64.4) | NS | 50.6 | 31.2 (31.1, 62.3) | 50.4 | 29.8 (38.4, 68.2) | NS |
| Bacteroidetes | 32.9 | 28.0 (21.3, 49.3) | 37.2 | 23.0 (25.2, 48.2) | NS | 44.1 | 30.5 (30.9, 61.4) | 31.3 | 26.9 (22.8, 49.7) | NS |
| Proteobacteria | 2.2 | 2.0 (3.4, 1.4) | 2.0 | 4.3 (1.1, 5.4) | NS | 2.5 | 1.5 (1.4, 2.9) | 2.3 | 2.7 (1.3, 4.0) | NS |
| Actinobacteria | 1.6 | 3.9 (0.4, 4.3) | 0.75 | 3.8 (0.2, 4.0) | NS | 0.39 | 1.1 (0.1, 1.2) | 0.80 | 1.6 (0.3, 1.9) | NS |
| Tenericutes | 0 | 0.048 (0, 0.048) | 0 | 0 (0, 0) | <0.05 | 0 | 0 (0, 0) | 0 | 0.0019 (0, 0.0019) | NS |
| Fusobacteria | 0 | 0.0059 (0, 0.0059) | 0.0027 | 0.28 (0, 0.28) | <0.05 | 0 | 0.0068 (0, 0.0068) | 0.0025 | 0.059 (0, 0.059) | NS |
| Synergistets | 0 | 0.017 (0, 0.017) | 0 | 0.0019 (0, 0019) | <0.05 | 0 | 0.0080 (0, 0080) | 0 | 0 (0, 0) | NS |
| Species at genus level | Median | IQR (1/4, 3/4) | Median | IQR (1/4, 3/4) | P value | Median | IQR (1/4, 3/4) | Median | IQR (1/4, 3/4) | P value |
| aLachnospiraceae_ND3007_group | 0.15 | 0.18 (0.08, 0.26) | 0.041 | 0.18 (0, 0.18) | <0.01 | 0.096 | 0.18 (0.03, 0.21) | 0.087 | 0.16 (0.04, 0.20) | NS |
| aRuminococcaceae_UCG-003 | 0.11 | 0.31 (0.03, 0.34) | 0.052 | 0.13 (0.004, 0.13) | <0.05 | 0.024 | 0.13 (0.004, 0.13) | 0.0079 | 0.10 (0, 0.10) | NS |
| aCoprococcus_3 | 0.098 | 0.21 (0.04, 0.25) | 0.056 | 0.14 (0, 0.14) | NS | 0.17 | 0.21 (0.13, 0.34) | 0.038 | 0.18 (0.002, 0.18) | <0.05 |
| aRuminococcaceae_UCG-014 | 0.067 | 0.73 (0, 0.73) | 0 | 0.0020 (0, 0.0020) | <0.05 | 0 | 0.010 (0, 0.10) | 0 | 0.0086 (0, 0.0086) | NS |
| aCoprococcus_1 | 0.063 | 0.12 (0.005, 0.12) | 0.012 | 0.043 (0, 0.043) | <0.05 | 0.043 | 0.042 (0.016, 0.057) | 0.025 | 0.054 (0, 0.054) | NS |
| anorank_f_Ruminococcaceae | 0.043 | 0.16 (0.0006, 0.16) | 0 | 0.017 (0, 0.017) | <0.05 | 0.0028 | 0.043 (0, 0.043) | 0.012 | 0.058 (0, 0.058) | NS |
| aLachnospiraceae_FCS020_group | 0.037 | 0.057 (0.016, 0.073) | 0.012 | 0.027 (0.003, 0.03) | <0.05 | 0.037 | 0.025 (0.025, 0.050) | 0.022 | 0.045 (0.005, 0.05) | NS |
| aCoprococcus_2 | 0 | 0.18 (0, 0.18) | 0 | 0.0055 (0, 0.005) | NS | 0 | 0 (0, 0) | 0.0011 | 0.087 (0, 0.087) | <0.05 |
| aHoldemanella | 0 | 0.054 (0, 0.054) | 0 | 0 (0, 0) | <0.05 | 0 | 0 (0, 0) | 0 | 0.11 (0, 0.11) | NS |
| aLactobacillus | 0 | 0.0045 (0, 0.0045) | 0 | 0.0024 (0, 0.0024) | NS | 0.0063 | 0.0096 (0.0015, 0.011) | 0 | 0.0052 (0, 0.0052) | <0.05 |
| aLactococcus | 0 | 0.0066 (0, 0.0066) | 0 | 0 (0, 0) | <0.05 | 0 | 0 (0, 0) | 0.0010 | 0.0087 (0, 0.0087) | NS |
| aSellimonas | 0 | 0 (0, 0) | 0 | 0.0081 (0, 0.0081) | <0.05 | 0 | 0 (0, 0) | 0 | 0.0068 (0, 0.0068) | NS |
| anorank_f_Clostridiales_vadinBB60_group | 0 | 0.0075 (0, 0.0075) | 0 | 0 (0, 0) | <0.01 | 0 | 0 (0, 0) | 0 | 0 (0, 0) | NS |
| aParaprevotella | 0.011 | 0.34 (0, 0.34) | 0 | 0.0040 (0, 0.0040) | <0.05 | 0.11 | 1.7 (0, 1.7) | 0 | 0.27 (0, 0.27) | NS |
| bAlloprevotella | 0 | 0 (0, 0) | 0 | 0 (0, 0) | <0.05 | 0 | 0 (0, 0) | 0 | 0 (0, 0) | NS |
| bPrevotella_7 | 0 | 0 (0, 0) | 0 | 0 (0, 0) | NS | 0 | 0.0014 (0, 0.0014) | 0 | 0 (0, 0) | <0.05 |
| cnorank_o_Mollicutes_RF39 | 0 | 0.048 (0, 0.048) | 0 | 0 (0, 0) | <0.05 | 0 | 0 (0, 0) | 0 | 0.0019 (0, 0.0019) | NS |
P value was calculated by Wilcoxon rank-sum test with multiple test correction by fdr.
aFirmicutes; bBacteroidetes; cTenericutes.
IOR, interquartile range; NC, non-obese control group; NG, non-obese group with PCOS; NS, no significant difference; OC, obese control group; OG, obese group with PCOS .
Characteristic gut microbiota differences in obese and non-obese women with PCOS
The characteristic gut microbiota was identified using LEfSe multilevel species discrimination and LDA. LDA discriminant histogram was used to count the significant gut microbiota between two groups and interpret the degree of consistent difference of relative abundance between groups. The larger the LDA score, the more significant the difference between the two groups was. Only those genera that obtained a log LDA score >2 were ultimately considered. Compared with the 16 characteristic species found in NC, 1 characteristic bacteria (Lactococcus) were found in NG (Fig. 2C). The Coprococcus_3 and other two characteristic species were found in OC, and only one kind of species (Coprococcus_2) was found to be characteristic microbiota in OG (Fig. 2D).
Correlation between gut microbiota and serum hormone levels in patients with PCOS
According to the Spearman correlation Heatmap analysis at the genus level, specific linear correlations between gut microbiota and serum hormone levels in NG and OG were observed (Fig. 3). In NG, 26 species were associated with serum insulin levels after a 75 g glucose load, which was much more than 12 species in OG (Supplementary Table 1). In OG, 16 species were associated with serum sex hormones level, much more than those in NG group (Supplementary Table 2). Comparing the two groups, Lachnoclostridium was positively correlated with prolactin (PRL) in OG, but negatively correlated with PRL in NG group. Streptococcus was negatively correlated with insulin level in both groups. Lachnospiraceae_UCG-004 and insulin level were negatively correlated in OG, but positively correlated in NG. Klebsiella, Veillonella, Collinsella, Dialister, Ruminococcaceae_UCG-014, Ruminococcus_gnavus_group, Agathobacter, Megasphaera, Roseburia and Parasutterella were found in both groups to be closely correlated with serum hormones level.
Figure 3.
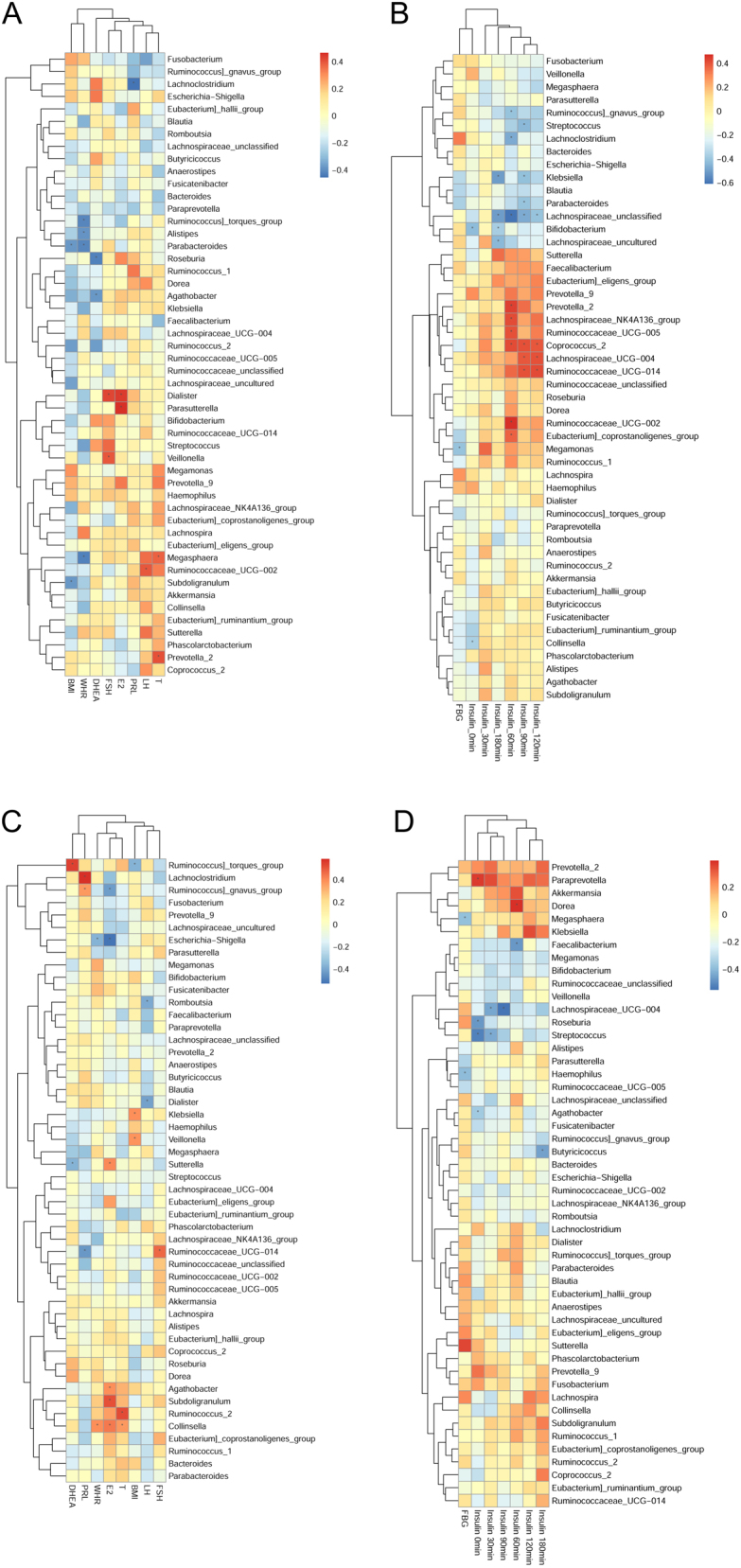
The Spearman Correlation Heat map between serum hormones and gut microbiota at genus level. The Spearman Correlation Heatmap between serum sex hormones (FSH, DHEA, PRL, TSH, LH, E2, testosterone) and gut microbiota in NG (A) and OG (C). The Spearman Correlation Heatmap between insulin release (FBG, insulin_0min, insulin_30min, insulin_60min, insulin_90min, insulin_120min, insulin_180min) and gut microbiota in NG (B) and OG (D). The r value is displayed in different colors in the graph. *P < 0.05; BMI, body mass index; WHR, waist-to-hip ratio; FSH, follicle-stimulating hormone; DHEA, dehydroepiandrosterone; PRL, prolactin; LH, luteinizing hormone; E2, estradiol; T, testosterone; PRL, prolactin; FBG, fasting blood glucose; Insulin_Xmin, serum insulin levels after a 75 g glucose load at X minutes.
Predicted metabolic functions of characteristic gut microbiota in obese and non-obese women with PCOS
In the present study, gut bacterial metabolic functions were enriched and predicted based on KEGG level-3. 82 KEGG pathways with significant differences were screened out in Table S3. Because of space limitation, 20 pathways were displayed in the Fig. 4. Compared with NC, there were 13 gut bacterial enrichment metabolic pathways in NG, mainly including Citrate cycle, Cell motility and secretion, Valine, leucine and isoleucine degradation, Glutathione metabolism, etc. (P < 0.05). Compared with OC, the only enrichment metabolic pathways found in OG was Citrate cycle (P < 0.05). Interestingly, the Citrate cycle pathway enriched both in NG and OG compared with their controls.
Figure 4.

Comparison of predicted microbial function among groups based on KEGG level-3. Kruskal–Wallis H test was used to analyze the significant differences of species among four groups. KEGG, Kyoto Encyclopedia of Genes and Genomes; NC, non-obese control group; NG, non-obese group with PCOS; OC, obese control group; OG, obese group with PCOS.
Discussion
In the present study, we uncovered clinical manifestation differences, serum hormone levels and characteristic gut microbiota in Chinese obese and non-obese women with PCOS. The Hirsutism score, LH/FSH and serum testosterone level in obese and non-obese PCOS patients were significantly higher than those in their control groups, and the diversity of gut microbiota was lower than control groups. The characteristic species of gut microbiota in obese and non-obese women with PCOS were revealed. The close relationships between several types of gut microbiota at genus level and serum hormones level were presented. The metabolic functions of differential gut microbiota were enriched and predicted in obese and non-obese women with PCOS.
Alpha diversity analysis showed that the gut microbiota abundance and diversity in women with PCOS changed. At the phylum level, Lim et al. (7) found that the abundance and diversity of Tenericutes in the intestine of healthy people are higher than those of patients with metabolic syndrome. Lindheim et al. (3) compared the abundance of Tenericutes in the intestine of patients with PCOS with healthy women decreased significantly and was negatively correlated with the total blood lymphocyte count. Our study further uncovered that the abundance of Tenericutes in non-obese women with PCOS was significantly lower than that in healthy women, but no difference was found between obese women with PCOS and healthy but obese women. Synergistets plays an important pathogenic role in oral infections (8, 9). Synergistets can promote the production of natural Immunoglobulin M and exhibits vital function in inhibiting autoimmune diseases (10). Synergistetes exists in the human oral cavity, intestine, vagina and skin, but this type of bacteria is still difficult to isolate and culture (11). Our study showed that the abundance of intestinal Synergistets in non-obese patients with PCOS was significantly lower than that in healthy women. At the genus level, we identified the specific bacteria in PCOS patients, such as Coprococcus_2, Lactobacillus and Prevotella_7 in the intestine of obese women with PCOS, Lactococcus, Paraprevotella and Alloprevotella in the intestine of non-obese women with PCOS.
According to the genus-level correlation Heatmap analysis, environmental factors exhibited different correlation strengths with gut microbiota in obese and non-obese women with PCOS. Interestingly, much more species closely related to the serum insulin level and less number of species closely related to serum sex hormone level was detected in non-obese PCOS patients than in obese PCOS patients. In this study, we found Lachnoclostridium was closely related to the serum PRL level both in obese and non-obese women with PCOS, which never reported. We found that the dysbacteriosis in the intestine of PCOS patients had a correlation with the serum sex hormones level, which suggested that we may regulate the status of gut microbial community to adjust the patients’ serum sex hormones level.
The serum insulin level in patients with PCOS is highly correlated with several intestinal bacteria such as Collinsella, suggesting that gut microbiota can significantly affect glucose metabolic function. Before our study, Chen et al. (12) demonstrated that oral administration of Collinsella can reduce the expression of tight junction protein in the intestinal cells and increase the permeability of the intestinal wall. Dietary adjustment can reduce the abundance of Collinsella and improves intestinal function (13). We speculated that patients with PCOS can adjust their diet structure to optimise the structure of gut microbiota and improve the quality of physical condition.
To identify the key bacterial genus in the intestinal tract of patients with PCOS, we used LEfSe method. The results showed that Lactococcus played vital roles in the intestine of non-obese patients with PCOS, and Coprococcus_2 played a role in the intestine of obese patients with PCOS. We hope the characteristic genus we found could be the targeted clinical diagnostic objectives, although there is still a lot work need to do, such as validation of clinical diagnosis.
To uncover the enriched metabolic functions of gut microbiota, we used predictive microbial function analysis. The results indicated that the altered gut microbiota in non-obese patients with PCOS was closely related to the excessive production of lipopolysaccharides and the abnormal functions of endocrine system in the host. Actually, hyperandrogenism and dysfunction of glucose and insulin metabolism are the common symptoms in patients with PCOS and may be promoted by the abnormal gut microbiota. And the patients in this group, their microbiota had more significant effects on the digestion and absorption of nutrients and glucose metabolism, which might cause the excessive accumulation of energy in host. In addition, the gut microbiota of non-obese patients with PCOS was abnormal active in amino acid, pyrimidine and purine metabolism compared with healthy women. These metabolic functions are closely related to the transport and absorption of nutrients and critically affected the levels of serum hormones, cytokines and other substances in the human body. Notably, citrate cycle pathway, the key link of carbohydrate, lipid and amino acid metabolism (14), enriched both in obese and non-obese patients with PCOS. Based on the above evidence, the altered gut microbiota had a tightly relationship with the activity of these metabolic pathways and may affect human endocrine function and cause the occurrence and development of the diseases.
The gut microbiota establishes a complex interdependent relationship with the host. Different regions, races and habits will make the gut microbiota behave increasingly heterogeneous among individuals. Therefore, the results from different countries may be variable. Originally, we expected that nearly 100 samples in each group were used, so the conclusion would be accurate, and the common characteristic gut microbiota of obese and non-obese patients with PCOS from different countries would be determined. However, collecting samples, especially faeces, blood samples and clinical data under specified conditions was very difficult. Even with total 101 participants, we made tremendous efforts to obtain the samples. Anyway, we found the characteristic gut microbiota and the enriched metabolic functions of obese and non-obese patients with PCOS to provide assistance for the diagnosis and prognosis of PCOS.
In conclusion, this study highlighted the significant differences of gut microbiota among Chinese obese and non-obese women with PCOS. The abundance and diversity of gut microbiota in PCOS patients were lower than that in healthy people. Gut microbiota was closely related to clinical manifestations and serum hormone levels of patients with PCOS. The changed gut microbiota may affect the intestinal environment of the host by enriching different metabolic functions to contribute to the occurrence and development of PCOS in obese and non-obese women.
Supplementary Material
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This work was supported by the National Natural Science Foundation of China (grant numbers 81603646, 81202963 and 81973896).
References
- 1.Escobar-Morreale HF. Polycystic ovary syndrome: definition, aetiology, diagnosis and treatment. Nature Reviews Endocrinology 2018. 270–284. ( 10.1038/nrendo.2018.24) [DOI] [PubMed] [Google Scholar]
- 2.Chen L, Xu WM, Zhang D. Association of abdominal obesity, insulin resistance, and oxidative stress in adipose tissue in women with polycystic ovary syndrome. Fertility and Sterility 2014. 1167–1174. ( 10.1016/j.fertnstert.2014.06.027) [DOI] [PubMed] [Google Scholar]
- 3.Lindheim L, Bashir M, Münzker J, Trummer C, Zachhuber V, Leber B, Horvath A, Pieber TR, Gorkiewicz G, et al Alterations in gut microbiota composition and barrier function are associated with reproductive and metabolic defects in women with polycystic ovary syndrome (PCOS): a pilot study. PLoS ONE 2017. e0168390 ( 10.1371/journal.pone.0168390) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu R, Zhang C, Shi Y, Zhang F, Li L, Wang X, Ling Y, Fu H, Dong W, Shen J, et al Dysbiosis of gut microbiota associated with clinical parameters in polycystic ovary syndrome. Frontiers in Microbiology 2017. 324 ( 10.3389/fmicb.2017.00324) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Torres PJ, Siakowska M, Banaszewska B, Pawelczyk L, Duleba AJ, Kelley ST, Thackray VG. Gut microbial diversity in women with polycystic ovary syndrome correlates with hyperandrogenism. Journal of Clinical Endocrinology and Metabolism 2018. 1502–1511. ( 10.1210/jc.2017-02153) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Insenser M, Murri M, Del Campo R, Martínez-García MÁ, Fernández-Durán E, Escobar-Morreale HF. Gut microbiota and the polycystic ovary syndrome: influence of sex, sex hormones, and obesity. Journal of Clinical Endocrinology and Metabolism 2018. 2552–2562. ( 10.1210/jc.2017-02799) [DOI] [PubMed] [Google Scholar]
- 7.Lim MY, You HJ, Yoon HS, Kwon B, Lee JY, Lee S, Song YM, Lee K, Sung J, Ko G. The effect of heritability and host genetics on the gut microbiota and metabolic syndrome. Gut 2017. 1031–1038. ( 10.1136/gutjnl-2015-311326) [DOI] [PubMed] [Google Scholar]
- 8.Yu XL, Chan Y, Zhuang LF, Lai HC, Lang NP, Lacap-Bugler DC, Leung WK, Watt RM. Distributions of synergistetes in clinically-healthy and diseased periodontal and peri-implant niches. Microbial Pathogenesis 2016. 90–103. ( 10.1016/j.micpath.2015.11.029) [DOI] [PubMed] [Google Scholar]
- 9.Fernandes Cdo C, Rechenberg DK, Zehnder M, Belibasakis GN. Identification of synergistetes in endodontic infections. Microbial Pathogenesis 2014. 1–6. ( 10.1016/j.micpath.2014.05.001) [DOI] [PubMed] [Google Scholar]
- 10.López P, de Paz B, Rodríguez-Carrio J, Hevia A, Sánchez B, Margolles A, Suárez A. Th17 responses and natural IgM antibodies are related to gut microbiota composition in systemic lupus erythematosus patients. Scientific Reports 2016. 24072 ( 10.1038/srep24072) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marchandin H, Damay A, Roudière L, Teyssier C, Zorgniotti I, Dechaud H, Jean-Pierre H, Jumas-Bilak E. Phylogeny, diversity and host specialization in the phylum synergistetes with emphasis on strains and clones of human origin. Research in Microbiology 2010. 91–100. ( 10.1016/j.resmic.2009.12.008) [DOI] [PubMed] [Google Scholar]
- 12.Chen J, Wright K, Davis JM, Jeraldo P, Marietta EV, Murray J, Nelson H, Matteson EL, Taneja V. An expansion of rare lineage intestinal microbes characterizes rheumatoid arthritis. Genome Medicine 2016. 43 ( 10.1186/s13073-016-0299-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walker AW, Ince J, Duncan SH, Webster LM, Holtrop G, Ze X, Brown D, Stares MD, Scott P, Bergerat A, et al Dominant and diet-responsive groups of bacteria within the human colonic microbiota. ISME Journal 2011. 220–230. ( 10.1038/ismej.2010.118) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akram M. Citric acid cycle and role of its intermediates in metabolism. Cell Biochemistry and Biophysics 2014. 475–478. ( 10.1007/s12013-013-9750-1) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.