

Abstract
Activation of the stimulator of interferon genes (STING) pathway by both exogenous and endogenous cytosolic DNA results in the production of interferon beta (IFN-β) and is required for the generation of cytotoxic T-cell priming against tumor antigens. In the clinical setting, pharmacological stimulation of the STING pathway has the potential to synergize with immunotherapy antibodies by boosting anti-tumor immune responses. We report the discovery of two highly potent cyclic dinucleotide STING agonists, IACS-8803 and IACS-8779, which show robust activation of the STING pathway in vitro and a superior systemic anti-tumor response in the B16 murine model of melanoma when compared to one of the clinical benchmark compounds.
Keywords: STING agonists, Cyclic dinucleotide, Immune response, T-cell priming, Phosphorothioate esters
Graphical Abstract
Stimulator of interferon genes (STING) is an innate pattern recognition receptor natively localized to the endoplasmic reticulum which plays a key role in host defense through innate detection of pathogenic or damage-associated nucleic acids.1–4 STING has recently been shown to be required for generation of optimal adaptive anti-tumor immune responses, thus deliberate pharmacological stimulation of the STING pathway is an exciting approach to boost anti-cancer immunity with the potential to synergize with FDA-approved immunotherapy antibodies.3,4 The STING pathway is activated upon direct binding to STING of cyclic dinucleotides (CDNs),5,6 second messenger molecules that are either of exogenous bacterial origin (such as 3’,3’-cyclic guanosine-adenosine monophosphate, cGAMP, Figure 1), or produced by a host cyclic GMP-AMP synthase (cGAS).7 cGAS acts as a sensor of double stranded cytosolic DNA of viral, malignant, or endogenous origin, generating the structurally distinct CDN ligand 2’,3’-cyclic guanosine-adenosine monophosphate (2’,3’-cGAMP, Figure 1), which features a non-canonical 2’,3’-phosphodiester linkage.8,9 Binding of CDN ligands to STING results in its activation and initiates a signaling cascade culminating in robust engagement of the interferon regulatory factor 3 (IFR3) and nuclear factor kappa-B (NF-kB) pathways, ultimately stimulating production of interferon beta (IFN-β) and other pro-inflammatory cytokines.10 Beyond its canonical role in immune defense against viral or bacterial pathogens, activation of the STING pathway and resulting IFN-β production have been recognized as critical components of the innate immune sensing of tumors, which is a critical upstream event required for optimal cytotoxic T-cell priming against tumor antigens.11 Recent reports demonstrated that pharmacological activation of the STING pathway via intratumoral administration of synthetic CDN agonists results in significant antitumor responses in a variety of preclinical models, achieves robust tumor regression at both injected and distal uninjected lesions, and is reported to facilitate generation of long-lived immunologic memory.3,4,12 Clinical evaluation of synthetic STING agonists is underway, with ADU-S100/MIW815,12 a 2’,3’-cyclic diadenosine monophosphate analogue featuring a thiophosphatediester bond (Figure 1) and MK-1454, a second agent of undisclosed structure, escalating in Phase I.13
Figure 1.

Cyclic dinucleotide STING agonists.
Given the key role proposed for the STING pathway in modulating the innate antitumor immune response, we set out to generate high-potency STING agonists and to investigate the role of key nucleobase and ribose modifications with the goal of identifying STING agonists with improved translational potential as cancer therapeutics and ultimately expand the number of available clinical tools. Herein we report the rational design and the discovery of IACS-8779 (1a) and IACS-8803 (3a, Figure 2), two highly potent 2’,3’- thiophosphate CDN analogs that show robust activation of the STING pathway in vitro and a superior systemic anti-tumor response when compared to the clinical benchmark ADU-S100 in the B16 melanoma murine model.
Figure 2.

The 2’,3’-linked phosphorothioate diadenosine monophosphate (2’,3’-S2-CDA) STING agonists herein described.
At the beginning of our exploration we decided to focus our efforts on 2’,3’-linked phosphorothioate analogs bearing a diadenosine monophosphate backbone (2’,3’-S2-CDA), due to the excellent feature of this class of synthetic CDNs and the compelling results reported for ADU-S100. The 2’,3’-phosphodiester linkage offers improved affinity for STING compared to the canonical 3’,3’-form.14,15 The introduction of sulfur atoms within the two thiophophodiester bonds confers improved resistance to phophodiesterase mediated degradation, resulting in enhanced activation of the STING pathway in vitro and significantly more robust antitumor responses in vivo.12,16 These advantages proved especially significant for one of the possible stereoisomers of the bis-thiophosphate analogs, the Rp,Rp-series in which both the stereogenic thiophosphate groups are in the R configuration. The 2,’3’- Rp,Rp-S2 analogs also proved to be potent agonists against both mouse STING as well as a number of human STING allelic isoforms, which is significant because these common polymorphic STING alleles are present in a large portion of the human population yet may possess differential responsivity to CDN agonists.12 At the onset of this project, the 2,’3’-Rp,Rp-S2-CDA analog was reported to have the best overall features, and we decided therefore to start our exploration from this lead molecule, targeting nucleobase and ribose modifications specifically designed to have a high probability of success in improving its antitumor efficacy and overall profile.
Amongst the several potential modifications evaluated for the nucleobase portion, we selected to start our exploration by substituting either of the two adenines within the 2’,3’-CDA structure with the 7-deaza-adenine core (Figure 2, compounds 1a/b and 2a/b). Despite being a rather conservative modification, the 7-deaza substitution has been reported within the context of antiviral nucleotide projects to have significant effects on boosting intrinsic potency against nucleotide recognizing molecular targets, and to offer improved stability against metabolic degradation by adenosine deaminase and phosphorylase.17
As for modifications within the ribose portion, we targeted the specific replacement of the 2’-hydroxyl within the 2’,3’-CDA structure with either a fluorine or a chlorine atom (Figure 2, compounds 3a/b and 4a/b). Both these substitutions were selected in view of their successful replacements of the 2’-OH moiety reported in the context of antiviral nucleotide projects that targeted the inhibition of RNA-dependent-RNA-polymerases, where recognition through the ribose 2’-OH motif is known to play a crucial role.18,19
All the above 2,’3- S2-CDA analogs were evaluated in vitro for their ability to stimulate the human and mouse STING pathway head to head with two benchmark CDN agonists, 3’,3’-cGMP and 2’,3’-RR-S2-CDA (ADU-S100). The compounds were tested at 1µg/mL in human THP-1-Dual™ and mouse J774-Dual™ cells (Invivogen), featuring stable integration of two inducible reporter constructs enabling simultaneous study of the two main signaling pathways activated by STING, the NF-kB pathway, and the interferon regulatory factor 3 (IRF3) pathway. Data for the activation of the IRF3 pathway are reported in Figure 3a, as assessed by activity of the luciferase reporter gene. Several of these newly synthetized CDN analogues proved able to act as human and mouse STING agonists, with a selection of them showing equivalent or superior activity relative to the clinical benchmark 2’,3’-RR-S2-CDA. A noticeable difference was observed in the effect of 7-deaza-adenine substitution, with regioisomers 1a/b significantly more active than regiosiomers 2a/b, and 1a (IACS-8779) showing equivalent activity relative to the clinical benchmark 2’,3’-RR-S2-CDA. Within the ribose modifications evaluated in the 2’-position, substitution of the 2’-OH with a 2’-F in 3a/b proved to be superior to the 2’-Cl analogs 4a/b, and showed a significant advantage compared to the clinical benchmark 2’,3’-RR-S2-CDA. The most promising analogs were then tested in a dose response manner over a range of concentrations (0.5 – 50 ug/mL, Figure 3b), where ‘8779 was found to be comparable, and the two 2’-F analogs 8802/8803 showed superior activity to the 2’,3’-RR-S2-CDA benchmark. In light of these compelling data, compounds IACS-8779 (1a) and IACS-8803 (3a) were selected for evaluation of antitumor activity in mice bearing bilateral B16-OVA melanomas, in a head to head comparison with the 2’,3’-cGAMP and 2’,3’-RR-S2-CDA benchmarks (Figure 4a).
Figure 3.

Evaluation of CDN potency in THP-1 (human) and J774 (mouse) reporter cells; a) CDNs were added at 1 µg/mL to 1×105 THP-1 Dual or 5×104 J774-dual reporter cells for 20 hours and IF3 activity was measured by luciferase assay. b) CDNs were added at the indicated concentrations in the 0.5 – 50 µg/mL dose range to 1×105 THP-1 Dual reporter cells for 20 hours and IF3 activity was measured by luciferase assay.
Figure 4.

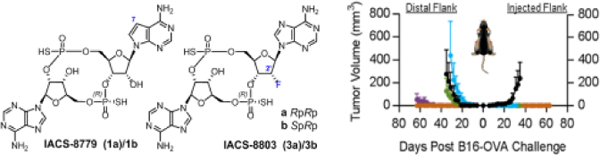
Antitumor activity of selected CDNs in the B16-Ova melanoma model. Mice (n=5 per group) were implanted bilaterally with 1×105 B16-Ova and received 10 ug of each CDN on day 6, 9, 12 post implantation. CDNs were administered by intratumoral injection on one flank only.
Mice were implanted bilaterally with 1×105 B16-Ova, and all the compounds were administered at the same dose (10 µg) with three intra-tumoral injections, on one flank only, on day 6, 9, 12 post implantation. While all the compounds showed comparable antitumor activity on the injected flank, IACS-8779 and IACS-8803 achieved superior regression on the untreated tumor in the contralateral flank, suggesting a more significant systemic immune response compared to the benchmark analogs. Treatment with ‘8779 and ‘8803 also resulted in a higher number of mice cured of both tumors compared to benchmarks (Figure 4b). Follow up studies in additional tumor models and dose down experiments are planned with IACS-8779 and IACS-8803, and will be reported in a forthcoming article.20
To enable the in vitro and in vivo studies above described, we required a robust synthetic approach that would allow a modular and efficient assembly of the targeted 2’,3’-S2-CDA analogs. We first secured multigram amounts of the necessary building blocks 3’-H-phosphonate intermediates A1–4 and the 2’-phosphoramidite intermediates B 1–2, depicted in Scheme 1.
Scheme 1.

Reaction conditions: a) Pyridine, TFA; b) DDTT, acetonitrile; c) aqueous Cl2HCCO2H; d) DMOC, pyridine; e) Beaucage reagent, pyridine; f) NH4OH, MeOH; g) NH4F, MeOH; h) Dowex®–50WX8 resin (Na+ form). Beaucage reagent = (3H-1,2-Benzodithiol-3-one 1,1-dioxide); Bz = benzoate; DDTT = 3-((dimethylaminomethylidene)amino)-3H-1,2,4-dithiazole-5-thione; DMOCP = 2-chloro-5,5-dimethyl-1,3,2-dioxaphosphorinane 2-oxide; TBS = tert-Butyldimethylsilyl.
We then employed the protocol developed by Gaffney et al. and its subsequent modifications to progress the above intermediates through an amidite-H-phophonate coupling and a first sulfurization step, employing 3-((dimethylaminomethylidene)amino)-3H-1,2,4-dithiazole-5-thione (DDTT).21–23,14 Intermediates 5–8 were often carried through to the next step without need of chromatographic purification, as a mixture of stereoisomers at the newly formed phopshorothioate stereogenic center.24 Cyclization was then achieved with 2-chloro-5,5-dimethyl-1,3,2-dioxaphosphorinane 2-oxide (DMOCP) in pyridine, and followed by a second sulfurization step with Beaucage reagent (3H-1,2-Benzodithiol-3-one 1,1-dioxide)25 in a two-step process reported to be selective for the Rp stereochemistry, 21,24 and hence resulting in the formation of two cyclized diatereosiomer products with RpRp and SpRp configuration respectively.¥
Removal of the cyanoethyl protecting group was achieved by treatment with base, and the two CDN disatereoisomers were typically separated at this stage by reverse phase HPLC. A desilylation step afforded the final 2’,3’-S2-CDN products as single stereoisomers, that were isolated as sodium salts after a second reverse phase HPLC followed by passage through ion exchange resin.§
The synthesis of the required building blocks intermediates was achieved either according to literature precedents (intermediates A2 and B1),22 or following the route described in Scheme 2 and Scheme 3 for intermediates A1-B2 and A3-A4 respectively. The 7-deaza-adenine intermediates A1 and B2 were both obtained from the same 4-chloro-7-deazaadenine ribose derivative 13. Following chlorine displacement with ammonia in MeOH, the resulting 4-amino group was selectively benzoylated via an in situ multi-step sequence, involving transient TMS-protection of the ribose hydroxyls.26 Selective tritylation of the 5’-hydroxyl, treament with TBSCl and chromatographic separation gave the regioisomeric mono-TBS protected alcohols 14 and 15, which were progressed to the 3’-H-phosphonate A2 and to the 2’-phosphoramidite B2 respectively.
Scheme 2.

Reaction conditions: a) NH3, MeOH, 110 °C; b) i. TMSCl, pyridine, 0 °C; ii. BzCl, 0 °C to RT; iii. H2O, then aq. NH3 0 °C; c) DMTrCl, pyridine; d) TBSCl, pyridine, AgNO3; e) PivCl, pyridine, H3PO3; f) Cl2CHCO2H, H2O, DCM; g) 2-cyanoethyl diisopropylchlorophosphoramidite, DCI, DCM. Bz = benzoate; DMTr = 4,4-Dimethoxytrityl; TBS = tert-Butyldimethylsilyl.
Scheme 3.

Reaction conditions: a) LiCl, DMF, 50 °C; b) BzCl, pyridine, 0–25 °C; c) TBAF, THF; d) aq. NH3, THF; e) DMTrCl, pyridine; f) PivCl, H3PO3, pyridine, 0–30 °C; g) Cl2HCCO2H, H2O, DCM. Bz = benzoate; DMTr = 4,4- Dimethoxytrityl; Tf = trifluoromethansulfonate; TBS = tert-Butyldimethylsilyl.
The synthesis of intermediate A3 started with chloride displacement of triflate 16,27 followed by protecting group manipulations to obtain the 2’-deoxy-2’-chlorine-adenosine derivative 17a. The 5’hydroxyl was temporary protected with dimethoxytrityl to install the 3’-H-phosphonate moiety, and then liberated to yield intermediate A3. The same synthetic sequence was employed to convert 17b in the 2’-deoxy-2’-fluorine building block A4.
In summary, we set out to generate high-potency CDN STING agonists by introducing a focused set of rationally selected modifications within the nucleobase and ribose portions of the 2’,3’-CDA structure. This focused approach led to the discovery of IACS-8779 and IACS-8803, two 2’,3’-phophothioate-CDA analogs that show robust activation of the STING pathway in vitro and a superior systemic anti-tumor response when compared to the clinical benchmark ADU-S100 in the B16 murine model of melanoma. Additional studies are ongoing towards positioning these molecules for clinical translation.
Supplementary Material
Acknowledgements
We would like to acknowledge the TL1 Fellowship (NIH NCATS TL1TR000369) for generous funding to CRA during this study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
In the case of the chlorine analogue 4, two additional stereoisomers were isolated as final products, suggesting that the cyclization-sulfurization process might have resulted in both Rs and Rp stereochemistry. These additional analogs were found inactive, data not presented.
Stereochemical assignent of the final products as depicted in Scheme 1 was based on comparison of 1H and 31P-NMR data with authentic samples of 2’,3’-RpRp-S2-CDA and 2’,3’-SpRp-S2-CDA prepared according to published procedures.12
Supplementary material
Experimental procedures and characterization data for synthetic intermediates A1, A3, A4, B1-B2, and final compounds IACS-8779 (1a) / 1b, and IACS-8803 (3a) / 3b are available at the link https “add link”. Procedure and data for the additional final compounds are available through reference n. 28.
References and notes
- 1.Ishikawa H; Barber GN; STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling; Nature, 2008, 455 (7213), 674–678; doi: 10.1038/nature07317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ishikawa H; Ma Z; Barber GN; STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity; Nature, 2009, 461(7265), 788–792; doi: 10.1038/nature08476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Chandra D; Quispe-Tintaya W; Jahangir A; Asafu-Adjei D; Ramos I; Sintim HO; Zhou J; Hayakawa Y; Karaolis DK, Gravekamp C; STING Ligand c-di-GMP Improves Cancer Vaccination against Metastatic Breast Cancer; Cancer Immunol. Res, 2014, September;2(9):901–10; doi: 10.1158/2326-6066.CIR-13-0123; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fu J; Kanne DB; Leong M; Hix Glickman L; McWhirter SM; Lemmens E; Mechette K; Leong JJ; Lauer P; Liu W; Sivick KE; Zeng Q; Soares KC; Zheng L; Portnoy DA; Woodward JJ; Pardoll DM; Dubensky TW Jr.; Young K; STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade; Science Translational Medicine, 2015, 7(283), 283ra52; doi: 10.1126/scitranslmed.aaa4306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ager CR; Reilley MJ; Nicholas C; Bartkowiak T; Jaiswal AR; Curran MA; Intratumoral STING Activation with T-cell Checkpoint Modulation Generates Systemic Antitumor Immunity; Cancer Immunol Res, 2017, 5(8), 676–684; doi: 10.1158/2326-6066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burdette DL; Monroe KM; Sotelo-Troha K; Iwig JS; Eckert B; Hyodo M; Hayakawa Y; Vance RE; STING is a direct innate immune sensor of cyclic di-GMP; Nature, 2011, 478(7370), 515–518; doi: 10.1038/nature10429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burdette DL; Vance RE; STING and the innate immune response to nucleic acids in the cytosol; Nat. Immunol, 2013, 14, 19–26; doi: 10.1038/ni.2491 [DOI] [PubMed] [Google Scholar]
- 7.Sun L; Wu J; Du F; Chen X; Chen ZJ; Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway; Science, 2013, 339, 786–791; doi: 10.1126/science.1232458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ablasser A; Goldeck M; Cavlar T; Deimling T; Witte G; Rohl I; Hopfner KP; Ludwig J; Hornung V; cGAS produces a 2,5-linked cyclic dinucleotide second messenger that activates STING; Nature, 2013, 498, 380–384; doi: 10.1038/nature12306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diner EJ; Burdette DL; Wilson SC; Monroe KM; Kellenberger CA; Hyodo M; Hayakawa Y; Hammond MC; Vance RE; The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING; Cell Rep, 2013, 3, 1355–1361; doi: 10.1016/j.celrep.2013.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Q; Sun L; Chen ZJ; Regulation and function of the cGAS–STING pathway of cytosolic DNA sensing; Nature Immunology, 2016, 17(10), 1142–1149; doi: 10.1038/ni.3558 [DOI] [PubMed] [Google Scholar]
- 11.a) Woo SR; Fuertes MB; Corrales L; Spranger S; Furdyna MJ; Leung MY; Duggan R; Wang Y; Barber GN; Fitzgerald KA; Alegre ML; Gajewski TF; STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors; Immunity, 2014, 41(5), 830–842; doi: 10.1016/j.immuni.2014.10.017; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Klarquist J; Hennies CM; Lehn MA; Reboulet RA; Feau S; Janssen EM; STING-mediated DNA sensing promotes antitumor and autoimmune responses to dying cells; J. Immunol, 2014, 193(12), 6124–6134; doi: 10.4049/jimmunol.1401869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corrales L; Glickman LH; McWhirter SM; Kanne DB; Sivick KE; Katibah GE; Woo SR; Lemmens E; Banda T; Leong JJ; Metchette K; Dubensky TW Jr; Gajewski TF; Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity; Cell Rep, 2015, 11(7), 1018–1030; doi: 10.1016/j.celrep.2015.04.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harrington K et al. ; Preliminary results of the first-in-human (FIH) study of MK-1454, an agonist of stimulator of interferon genes (STING), as monotherapy or in combination with pembrolizumab (pembro) in patients with advanced solid tumors or lymphomas; Abstract of Papers, European Society for Medical Oncology (ESMO), Munich, Germany, 2018; Abstract LBA15. [Google Scholar]
- 14.Zhang X; Shi H; Wu J; Zhang X; Sun L; Chen C; Chen ZJ; Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING; Molecular Cell, 2013, 51(2), 226–235; doi: 10.1016/j.molcel.2013.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao P; Ascano M; Zillinger T; Wang W; Dai P; Serganov AA; Gaffney BL; Shuman S; Jones RA; Deng L; Hartmann G; Barchet W; Tuschl T; Patel DJ; Structure-function analysis of STING activation by c[G(2’,5’)pA(3’,5’)p] and targeting by antiviral DMXAA; Cell, 2013, 154, 748–762; doi: 10.1016/j.cell.2013.07.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li L; Yin Q; Kuss P; Maliga Z; Millán JL; Wu H; Mitchison TJ; Hydrolysis of 2’3’-cGAMP by ENPP1 and design of nonhydrolyzable analogs; Nat. Chem. Biol, 2014, 10(12), 1043–1048; doi: 10.1038/nchembio.1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Olsen DB; Eldrup AB; Bartholomew L; Bhat B; Bosserman MR; Ceccacci A; Colwell LF; Fay JF; Flores OA; Getty KL; Grobler JA; LaFemina RL; Markel EJ; Migliaccio G; Prhavc M; Stahlhut MW; Tomassini JE; MacCoss M; Hazuda DJ; Carroll SS; A 7-deaza-adenosine analog is a potent and, selective inhibitor of hepatitis C virus replication with excellent pharmacokinetic properties; Antimicrob Agents Chemother, 2004, 48(10), 3944–3953; doi: 10.1128/AAC.48.10.3944-3953.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.a) Liu P; Sharon A; Chu CK; Fluorinated Nucleosides: Synthesis and Biological Implication; J. Fluor. Chem, 2008, 129(9), 743–766; doi: 10.1016/j.jfluchem.2008.06.007; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sofia MJ; Bao D; Chang W; Du J; Nagarathnam D; Rachakonda S; Reddy PG; Ross BS; Wang P; Zhang HR; Bansal S; Espiritu C; Keilman M; Lam AM; Steuer HM; Niu C; Otto MJ; Furman PA; Discovery of a β-d-2’-deoxy-2’-α-fluoro-2’-β-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus; J. Med. Chem, 2010, 53(19), 7202–7218; doi: 10.1021/jm100863x [DOI] [PubMed] [Google Scholar]
- 19.Alexandre FR; Badaroux E; Bilello JP; Bot S; Bouisset T; Brandt G; Cappelle S; Chapron C; Chaves D; Convard T; Counor C; Da Costa D; Dukhan D; Gay M; Gosselin G; Griffon JF; Gupta K; Hernandez-Santiago B; La Colla M; Lioure MP; Milhau J; Paparin JL; Peyronnet J; Parsy C; Pierra Rouvière C; Rahali H; Rahali R; Salanson A; Seifer M; Serra I; Standring D; Surleraux D; Dousson CB; The discovery of IDX21437: Design, synthesis and antiviral evaluation of 2’-α-chloro-2’-β-C-methyl branched uridine pronucleotides as potent liver-targeted HCV polymerase inhibitors; Bioorg. Med. Chem. Lett, 2017, 27(18), 4323–4330; doi: 10.1016/j.bmcl.2017.08.029 [DOI] [PubMed] [Google Scholar]
- 20.Ager CA; Boda A; Castro-Pando S; Whitfield B; Molldrem J; AlAtrash G; Di Francesco ME; Jones P; Curran MA; Identification of Nonfunctional Alternatively Spliced Isoforms of STING in Human AML; 2019, submitted to Cancer Immunology Research. [DOI] [PMC free article] [PubMed]
- 21.Gaffney BL; Veliath E; Zhao J; Jones RA; One-flask syntheses of c-di-GMP and the [Rp,Rp] and [Rp,Sp] thiophosphate analogues; Org Lett, 2010, 12(14), 3269–3271; doi: 10.1021/ol101236b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao P; Ascano M; Wu Y; Barchet W; Gaffney BL; Zillinger T; Serganov AA; Liu Y; Jones RA; Hartmann G, Tuschl T, Patel DJ; Cyclic [G(2’,5’)pA(3’,5’)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase; Cell, 2013, 153(5), 1094–1107; doi: 10.1016/j.cell.2013.04.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lioux T; Mauny MA; Lamoureux A; Bascoul N; Hays M; Vernejoul F; Baudru AS; Boularan C; Lopes-Vicente J; Qushair G; Tiraby G; Design, Synthesis, and Biological Evaluation of Novel Cyclic Adenosine-Inosine Monophosphate (cAIMP) Analogs That Activate Stimulator of Interferon Genes (STING); J. Med. Chem 2016, 59(22), 10253–10267; [DOI] [PubMed] [Google Scholar]
- 24.Battistini C; Fustinoni S; Brasca MG; Borghi D; Stereoselective synthesis of cyclic dinucloetide phosphorothioates; Tetrahedron, 1993, 49 (5), 1115–1132. [Google Scholar]
- 25.Iyer RP; Egan W; Regan JB; Beaucage SL; 3H-1,2-Benzodithiole-3-one 1,1-dioxide as an improved sulfurizing reagent in the solid-phase synthesis of oligodeoxyribonucleoside phosphorothioates; J. Am. Chem. Soc, 1990, 112 (3), 1253–1254. [Google Scholar]
- 26.Zhu XF; Williams HJ; Ian SA; An Improved Transient Method for the Synthesis of N-Benzoylated Nucleosides; Synthetic Communications, 2003, 33(7), 1233–1243 [Google Scholar]
- 27.Robins MJ; Nowak I; Wnuk SF; Hansske F; Madej D; Deoxygenative [1,2]-hydride shift rearrangements in nucleoside and sugar chemistry: analogy with the [1,2]-electron shift in the deoxygenation of ribonucleotides by ribonucleotide reductases; J. Org. Chem, 2007, 72(22), 8216–8221. [DOI] [PubMed] [Google Scholar]
- 28.Curran MA; Di Francesco ME; Jones P; Preparation of cyclic dinucleotides as agonists of stimulator of interferon gene dependent signaling; PCT Int. Appl (2018), WO 2018156625.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.