

One is not enough: Two PD-1 pTyr motifs bind to SHP2 simultaneously, inducing a conformational change and phosphatase activation.
Abstract
In cancer, the programmed death-1 (PD-1) pathway suppresses T cell stimulation and mediates immune escape. Upon stimulation, PD-1 becomes phosphorylated at its immune receptor tyrosine–based inhibitory motif (ITIM) and immune receptor tyrosine–based switch motif (ITSM), which then bind the Src homology 2 (SH2) domains of SH2-containing phosphatase 2 (SHP2), initiating T cell inactivation. The SHP2–PD-1 complex structure and the exact functions of the two SH2 domains and phosphorylated motifs remain unknown. Here, we explain the structural basis and provide functional evidence for the mechanism of PD-1-mediated SHP2 activation. We demonstrate that full activation is obtained only upon phosphorylation of both ITIM and ITSM: ITSM binds C-SH2 with strong affinity, recruiting SHP2 to PD-1, while ITIM binds N-SH2, displacing it from the catalytic pocket and activating SHP2. This binding event requires the formation of a new inter-domain interface, offering opportunities for the development of novel immunotherapeutic approaches.
INTRODUCTION
In signal transduction, cytoskeletal remodeling, cell survival, and cell proliferation, phosphorylation and dephosphorylation of tyrosine residues are major regulators of protein activity. The addition and removal of phosphate groups from the aromatic ring of tyrosine residues are catalyzed by protein tyrosine kinases and protein tyrosine phosphatases (PTPs), respectively. Among the PTPs, the cytoplasmic Src homology 2 (SH2) domain–containing phosphatase 2 (SHP2), encoded by the gene ptpn11, acts as a positive transducer of proliferative and antiapoptotic signals from receptor tyrosine kinases, in the context of the Ras/mitogen-activated protein kinase (MAPK) pathway (1).
SHP2 is composed of three folded domains and a C-terminal tail (Fig. 1A). The N-terminal SH2 (N-SH2) and C-terminal SH2 (C-SH2) domains bind phosphopeptides, thereby modulating the phosphatase activity (2). The PTP domain harbors the catalytic functionality in the conserved signature motif HCX5R, which contains the catalytic cysteine. The disordered C-terminal tail contains two potential phosphorylation sites (Y542 and Y584) and has a putative regulatory function (3).
Fig. 1. Interaction of the N-SH2 and C-SH2 domains with ITIM and ITSM.
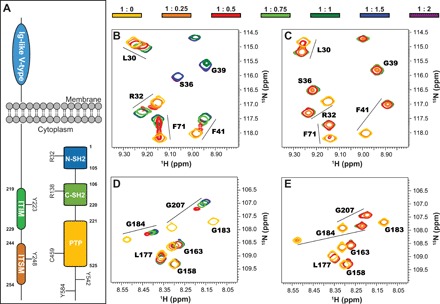
(A) Domain composition of SHP2 and PD-1. Highlighted are the conserved R32 and R138 required for binding phosphopeptides, the catalytic C459, and the putative phosphorylation sites Y542 and Y584 on SHP2, as well as the phosphorylation sites on the PD-1 cytoplasmic domain. (B and C) Excerpts from 1H-15N heteronuclear single-quantum coherence (HSQC) spectra of N-SH2 upon addition of ITIM (B) and ITSM (C). ppm, parts per million. (D and E) Excerpts from 1H-15N HSQC spectra of C-SH2 upon addition of ITIM (D) and ITSM (E). The protein concentration was 200 μM; the color code (top) indicates the protein:peptide molar ratios. All spectra were recorded at 298 K and 600 MHz.
SHP2 has been found to adopt an autoinhibited, catalytically inactive conformation, in which the DE loop of the N-SH2 domain occludes the active site of the PTP domain (fig. S1A) (2). Consequently, phosphotyrosine (pY)–bearing peptides binding to N-SH2 have been proposed to activate SHP2 by inducing a conformational change that disrupts the N-SH2–PTP interaction (2). Bidentate phosphopeptides, which contain two pY residues and can potentially bind to both N-SH2 and C-SH2 domains simultaneously, are stronger activators than phosphopeptides with a single pY site (4); however, mutational analysis has demonstrated that binding of phosphopeptides to N-SH2, but not to C-SH2, is essential for activation in vitro (3). Overall, the exact roles of the two SH2 domains in SHP2 activation remain unclear.
Ptpn11 has been reported to be both a proto-oncogene and a tumor suppressor in different cellular contexts (5). SHP2 mutations are associated with Noonan and LEOPARD syndromes as well as sporadic juvenile myelomonocytic leukemia (6–8) and have been implicated in breast and gastric cancers (9, 10). Consequently, SHP2 is considered a potential target for anticancer therapy (11, 12), with two phase 1 clinical trials currently underway (13). Besides its function as a positive regulator of Ras/MAPK signaling, SHP2 is a key downstream effector of the immune checkpoint receptor programmed death-1 (PD-1) in T cells (14). Once stimulated by its natural ligands PD-L1 or PD-L2, PD-1 is phosphorylated by Src kinases at two tyrosine-containing motifs, located in its cytoplasmic tail (15). The two motifs, termed immune receptor tyrosine–based inhibitory motif (ITIM) and immune receptor tyrosine–based switch motif (ITSM), recruit and activate SHP2, which then removes phosphate groups from nearby effector proteins (16). Ultimately, this leads to a reduction of both cytokine production and T cell proliferation, which results in the suppression of T cell–mediated immune response. The atomic details of the interaction between PD-1 and SHP2 are still unknown, with conflicting literature data indicating that ITIM is either involved (16–18) or not involved (15, 19) in SHP2 recruitment or activation.
The success of anticancer therapy based on monoclonal antibodies that disrupt PD-1 signaling (nivolumab and pembrolizumab), currently approved for the treatment of metastatic melanoma, non–small cell lung cancer, and renal cancer (20, 21), suggests that blocking the SHP2–PD-1 interaction by targeting SHP2 may result in an efficient and fairly inexpensive anticancer strategy. This hypothesis is supported by many reports demonstrating the role of SHP2 in T cell activation (14–16, 18, 19).
In this study, we elucidate the mechanism of PD-1–mediated SHP2 activation using a combination of biochemical, structural, and cellular biology approaches. We find that phosphorylated ITIM and ITSM bind the N-SH2 and C-SH2 domains with distinct affinities and rationalize the binding specificity by solving the structures of the C-SH2–ITSM, N-SH2–ITIM, and N-SH2–ITSM complexes. We demonstrate that a bidentate peptide, containing both ITIM and ITSM, activates SHP2 much more efficiently than either motif in isolation, implying simultaneous binding of ITIM and ITSM to the two SH2 domains. In agreement with this result, we find that, in cells, both motifs are necessary for full activation. Our structural data reveal that the concurrent binding of the two phosphorylated sites of the bidentate peptide to SHP21–220 (containing both SH2 domains) occurs at low concentrations in a specific register, with ITIM binding to N-SH2 and ITSM binding to C-SH2. The formation of the 1:1 complex between the bidentate peptide and SHP21–220 requires a conformational change of the protein, which alters the N-SH2:C-SH2 interface and may represent a way to regulate SHP2 activation.
RESULTS
PD-1 ITSM and ITIM interact with both N-SH2 and C-SH2 domains of SHP2
We used nuclear magnetic resonance (NMR) spectroscopy to test whether both pY motifs of PD-1 are able to bind the isolated N-SH2 (SHP21–105) and C-SH2 (SHP2106–220) domains of SHP2 in vitro (see Fig. 1A for domain numbering). We assigned the backbone resonances of the N-SH2 and C-SH2 domains and monitored the chemical shift perturbations (CSPs) observed in two-dimensional (2D) 1H-15N spectra upon addition of either ITIM (FSVDpYGELDFQ) or ITSM (EQTEpYATIVFP) peptides (Fig. 1 and fig. S2). In the following text, ITIM and ITSM always indicate the phosphorylated form of the motifs, unless stated otherwise. ITSM binds the N-SH2 and C-SH2 domains with dissociation constant (Kd) values in the high nanomolar range or lower, as indicated by the behavior of the NMR peaks during titration (slow-to-intermediate exchange regime; Fig. 1 and fig. S2). Accordingly, isothermal titration calorimetry (ITC) reported Kd values of 13 and 167 nM for ITSM in complex with C-SH2 and N-SH2, respectively. In contrast, upon addition of ITIM, the behavior of the 1H-15N peaks was consistent with Kd values in the micromolar range (intermediate-to-fast exchange regime). ITC data confirmed that ITIM binds N-SH2 and C-SH2 with Kd values of 1.9 and 26.4 μM, respectively. Thus, ITSM has the higher affinity for both SH2 domains with a 10-fold preference for C-SH2; conversely, ITIM displays a 10-fold preference for N-SH2. The CSPs induced by binding of ITIM and ITSM are in agreement with the canonical binding mode of phosphopeptides to SH2 domains (fig. S2) (22). The nonphosphorylated form of ITIM does not bind either of the domains, while the affinity of nonphosphorylated ITSM is greatly reduced with respect to the phosphorylated form (fig. S3).
Structures of N-SH2 and C-SH2 domains in complex with ITIM and ITSM
To understand the structural basis for the preferential binding of ITIM and ITSM to either N-SH2 or C-SH2, we determined the structure of the complexes. We used x-ray crystallography to solve the structures of the N-SH2–ITIM and N-SH2–ITSM complexes [Protein Data Bank (PDB) entries 6ROY and 6ROZ, respectively; Fig. 2 and table S1] and NMR spectroscopy for the C-SH2–ITSM complex (PDB entry 6R5G; Fig. 2, fig. S4, and table S2).
Fig. 2. Structure of SH2 domains in complex with ITIM and ITSM.
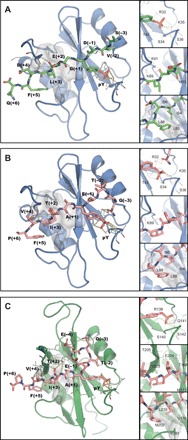
(A) Crystallographic structure of N-SH2–ITIM. N-SH2, blue; ITIM, green. (B) Crystallographic structure of N-SH2–ITSM. N-SH2, blue; ITSM, pink. (C) Lowest energy structure of the NMR ensemble of C-SH2–ITSM. C-SH2, green; ITSM, pink. In each panel, an overview of the complete structure is shown on the left (with protein hydrophobic patches interacting with the peptide C-terminal region shown as gray surfaces). Insets on the right show the interactions of the phosphate group (top), H bonds formed between the peptide and the BG loop of the protein (middle), and hydrophobic interactions of the peptide C-terminal region (bottom). The peptide residues are numbered starting from the pY residue (zero) as positive and negative numbers in the direction of the C and N terminus, respectively.
In all structures, both ITIM and ITSM adopt the typical extended conformation observed for other phosphopeptides bound to SH2 domains (22), with the peptide backbone arranged perpendicularly to the central β sheet bundle. The pY moiety is held in place by a network of hydrogen bonds to R32, S34, S36, and T42 of N-SH2 or R138, S140, and S142 of C-SH2. A patch of hydrophobic amino acids provides an interaction surface for the nonpolar C-terminal part of ITSM: In C-SH2–ITSM, residues V170, V181, M202, and L210 surround the ITSM A(+1)–F(+5) stretch; analogously, in N-SH2–ITSM, the peptide C-terminal stretch is in contact with I54, L65, and L88. The packing of the peptide is tighter in C-SH2–ITSM than in N-SH2–ITSM, and C-SH2–ITSM contains two hydrogen bonds from the backbone HN and the side-chain hydroxyl of ITSM-T(+2) to the side-chain carbonyl of C-SH2–E204, which are absent in N-SH2–ITSM (Fig. 2). These differences result in the higher affinity of ITSM for C-SH2 as compared to N-SH2. Conversely, comparison of the x-ray structures of N-SH2–ITSM and N-SH2–ITIM (Fig. 2) does not explain why N-SH2 binds ITSM more strongly than ITIM, suggesting that the difference between the free energies of the complexes might be due principally to entropic contributions.
Comparison of the structures of the unbound (PDB entry 2SHP) (23) and peptide-bound N-SH2 domain shows that the BG and EF loops undergo large conformational changes that are necessary to accommodate the C-terminal part of the phosphopeptide (fig. S4). It has been previously reported that the conformational change of the EF loop induced by peptide binding is not compatible with the autoinhibited conformation of SHP2 (23), thus providing an explanation as to how singly phosphorylated peptides activate SHP2 by binding to the N-SH2 domain and preventing its interaction with the PTP active site (fig. S1, C and D).
Activation of SHP2 by a peptide containing both ITIM and ITSM
Next, we tested the activation of SHP2 in the presence of ITIM and ITSM, as well as with the bidentate peptide ITIM-[dPEG4]2-ITSM, which contains both ITIM and ITSM joined by a polyethylenglycol-based linker. The length of the linker was chosen to mimic that of an extended chain of the 14 amino acids that separate ITIM and ITSM in the PD-1 tail, following the strategy proposed by Chen et al. (24). We tested the effect of these peptides on the catalytic activity of SHP2 by an in vitro phosphatase assay, which monitors the conversion of the substrate 6,8-difluoro-4-methylumbelliferone phosphate (DiFMUP) into the fluorescent product 6,8-difluoro-4-methylumbelliferone.
We observed dose-dependent stimulation of SHP2-dependent DiFMUP dephosphorylation upon addition of increasing amounts of ITSM and ITIM peptides, starting at concentrations of 5 and 10 μM, respectively. Both ITSM and ITIM activated SHP2 with moderate potency, with ITSM producing a greater maximal response than ITIM (Fig. 3A). An equimolar mixture of ITIM and ITSM yielded a higher maximal velocity of DiFMUP dephosphorylation than either of the individual peptides, but also in this case, the activation of SHP2 was only moderate and did not reach saturation. In none of these assays could we determine the half-maximal stimulation concentration (EC50), as the response did not reach a plateau at the highest achievable concentrations of ITIM and ITSM.
Fig. 3. SHP2 activation in vitro by different PD-1–derived phosphopeptides.
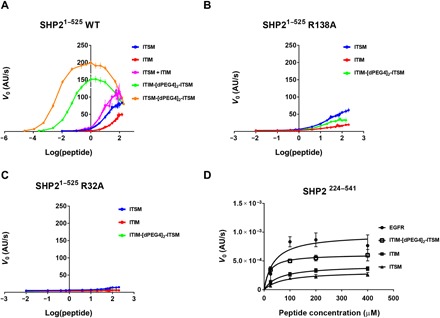
(A) DiFMUP-based activation assays of WT SHP2 with increasing concentrations of ITIM, ITSM, ITIM-[dPEG4]2-ITSM, and ITSM-[dPEG4]2-ITSM peptides. (B and C) DiFMUP-based activation assay of the SHP2-R32A (B) or the SHP2-R138A mutant (C) with increasing concentrations of ITIM, ITSM, and ITIM-[dPEG4]2-ITSM. In (A) to (C), the individual data points represent mean values with the error bars corresponding to the SEM of three independent experiments, each performed in triplicate. (D) EnzChek-based phosphatase activity assay of SHP2224–541 with peptides at concentrations of 25, 100, 200, and 400 μM. The slope values from the linear range of the kinetic curves are plotted as initial velocities. The individual data points represent mean values with error bars corresponding to the SEMs of three independent experiments (each performed in duplicate) for all cases except for ITIM-[dPEG4]2-ITSM, where only two independent experiments were performed. AU, arbitrary units; V0, initial velocity.
To determine the contribution of the individual SH2 domains to PD-1–mediated SHP2 activation, we generated SHP2 mutants, in which the ability of either N-SH2 (SHP2-R32A) or C-SH2 (SHP2-R138A) to bind phosphopeptides was disrupted. Neither the ITIM nor the ITSM peptide was able to activate the SHP2-R32A mutant (Fig. 3C), while the SHP2-R138A mutant responded to both peptides, although to a lesser extent than did wild-type (WT) SHP2 (Fig. 3B). The ability of the N-SH2 domain to bind phosphopeptides is therefore essential for activation, while binding of phosphopeptides by both N-SH2 and C-SH2 is required for full activation.
The bidentate phosphorylated peptide ITIM-[dPEG4]2-ITSM stimulated SHP2 catalytic activity with much higher efficiency than either of the monophosphorylated motifs alone or in combination and yielded an EC50 of 39.9 ± 1.6 nM. Another bidentate peptide, ITSM-[dPEG4]2-ITSM, with two ITSMs, was even more potent than ITIM-[dPEG4]2-ITSM, with an EC50 of 3.7 ± 0.4 nM (Fig. 3A). Both ITIM-[dPEG4]2-ITSM and ITSM-[dPEG4]2-ITSM stimulated SHP2 activity up to a concentration of ~1 μM, with higher peptide concentrations showing an inhibitory effect.
The ITIM-[dPEG4]2-ITSM bidentate peptide failed to activate SHP2-R32A (Fig. 3C) and displayed a weak activation profile toward SHP2-R138A (Fig. 3B). Overall, these data indicate that the efficacy of the ITIM-[dPEG4]2-ITSM peptide is strictly related to its bidentate functionality and requires simultaneous binding of ITIM and ITSM to both SH2 domains.
To rationalize the inhibition of SHP2 activity seen at high concentration of the bidentate peptides, we tested whether ITIM and ITSM are substrates of SHP2 themselves. We used a coupled enzymatic assay (EnzChek phosphate assay) to monitor the SHP2-dependent release of phosphate. As a positive control, we used a peptide derived from the epidermal growth factor receptor (EGFR), which is a known substrate of SHP2 (25). The catalytic domain of SHP2 (PTP, residues 224 to 541) dephosphorylated the ITIM, ITSM, and ITIM-[dPEG4]2-ITSM peptides (Fig. 3D) at high peptide concentrations (ranging from 25 to 400 μM) and with lower efficiency compared to the EGFR peptide. This result indicates that the inhibition of SHP2 activity observed in the DiFMUP activation assay at high ITIM-[dPEG4]2-ITSM concentrations is due to substrate competition and suggests that the dephosphorylation of ITIM and ITSM may represent another layer of regulation of SHP2 activation.
Simultaneous binding of SHP21–220 to ITIM and ITSM of ITIM-[dPEG4]2-ITSM requires formation of a new interdomain interface
Next, we used 2D 1H-15N NMR spectra to monitor the binding of ITIM, ITSM, and ITIM-[dPEG4]2-ITSM to SHP21–220, which contains both SH2 domains (tandem SH2). The peak positions of unbound SHP21–220 matched closely with those of isolated N-SH2 and C-SH2, demonstrating that the two domains do not share a large interaction surface or substantially perturb each other’s internal structure (fig. S5A). In addition, the CSPs induced by either ITIM or ITSM on SHP21–220 were very similar to those induced on isolated N-SH2 and C-SH2, confirming that the peptide binding sites on the two SH2 domains are the same in the SHP21–220 construct as in the isolated domains. The low affinity of ITIM for C-SH2 was apparent, as CSPs of C-SH2 peaks upon addition of ITIM occurred only after saturation of the N-SH2 binding site. Similarly, in the presence of ITSM, CSPs of N-SH2 peaks occurred at higher peptide concentrations than those producing CSPs of the C-SH2 peaks (fig. S5, B to G).
Because of the different CSPs induced by ITIM and ITSM on both N-SH2 and C-SH2, the binding of the two phosphorylated motifs of ITIM-[dPEG4]2-ITSM to each of the SH2 domains of SHP21–220 could be closely monitored during the titration (Fig. 4A). Using a protein concentration of 200 μM, we found that C-SH2 exclusively binds ITSM at all protein:peptide ratios, while a more complex picture emerged for N-SH2. At peptide concentrations <200 μM, N-SH2 was either unbound or bound to ITIM; at peptide concentrations >200 μM, the amount of ITIM-bound N-SH2 increased as expected, but evidence of N-SH2 binding to ITSM also started to appear. Raising the peptide concentration to 400 μM and higher resulted in an increase in the intensity of the peaks corresponding to N-SH2–ITSM at the expense of those corresponding to N-SH2–ITIM. The latter had disappeared with a fourfold excess of peptide, due to the complete displacement of ITIM by ITSM, resulting in the formation of a 1:2 protein:peptide complex.
Fig. 4. A new N-SH2:C-SH2 interface forms upon simultaneous binding of ITIM-[dPEG4]2-ITSM to both SH2 domains of SHP21–220.
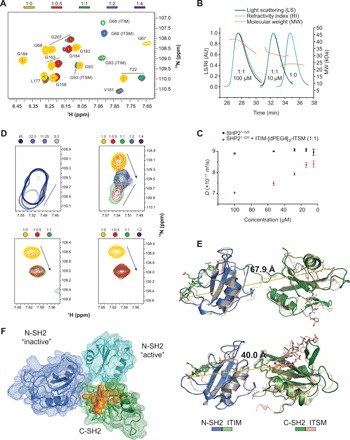
(A) Excerpt from the 1H-15N NMR spectrum of SHP21–220 in the presence of different stoichiometric ratios of ITIM-[dPEG4]2-ITSM shows two sets of peaks for amino acids 1 to 110, matching those of the N-SH2–ITIM and N-SH2–ITSM complexes. With excess peptide, the ITSM-bound peak becomes dominant. Colors represent protein:peptide molar equivalents. (B) Size-exclusion chromatography (SEC)–multiangle light scattering (MALS) profiles of equimolar SHP21–220:ITSM-[dPEG4]2-ITSM mixtures at either 10 or 100 μM, together with the profile of the unbound protein. (C) Translation diffusion coefficients measured by NMR DOSY (diffusion-ordered spectroscopy) for equimolar SHP21–220:ITIM-[dPEG4]2-ITSM mixtures at concentrations in the 5 to 100 μM range. (D) Top left: S189 CSPs upon dilution of a 1:1.2 SHP21–220:ITIM-[dPEG4]2-ITSM mixture. Shades of blue represent different concentrations. Top right: S189 CSPs upon titration of increasing molar ratios of ITIM-[dPEG4]2-ITSM ([SHP21–220] = 100 μM). The straight lines and arrows indicate concentration-dependent CSPs and CSPs from the unbound to the bound state, respectively. The peak indicative of the new interdomain interface reaches its maximum intensity at equimolar SHP21–220:ITIM-[dPEG4]2-ITSM and disappears with excess peptide. Bottom left and right: Titration of a mixture of isolated N-SH2 and C-SH2 at 200 μM each (left) or C-SH2 alone (right) with ITIM-[dPEG4]2-ITSM. The S189 peak corresponding to the new interdomain interface is not visible. (E) Homology models of SHP1–220 with ITIM and ITSM bound to the N-SH2 and C-SH2 domain, respectively, obtained by superposition of the N-SH2–ITIM and C-SH2–ITSM structures on the respective SH2 domains of PDB entry 2SHP (top, autoinhibited state of SHP2) and PDB entry 3PS5 (bottom, open state of SHP1). The dashed lines represent the distance between the ITIM C terminus and the ITSM N terminus. The length of the extended connecting linker in PD-1 is ~40 Å. (F) Superposition of the SHP21–220 models of (E) aligned on the C-SH2 domain (green). The N-SH2 domains from PDB entries 2SHP and 3PS5 are in blue and cyan, respectively. Residues in orange show CSPs upon dilution of the equimolar SHP21–220:ITIM-[dPEG4]2-ITSM mixture (D) and are located at the N-SH2:C-SH2 interface in either structure.
Next, we measured diffusion-ordered spectroscopy and 2D 1H-15N spectra of protein:peptide mixtures at 1:1.2 molar ratio over a dilution series corresponding to protein concentrations of 100, 53, 28, 14, and 5 μM. The diffusion coefficients (D) increased steadily upon dilution and remained stable after the second-to-last dilution point (Fig. 4C). At the concentration of 14 μM, the value of the diffusion coefficient was compatible with a 1:1 SHP21–220–ITIM-[dPEG4]2-ITSM complex, while at higher concentration, the D values suggested the formation of oligomers. In the absence of ITIM-[dPEG4]2-ITSM, SHP21–220 remained monomeric at all concentrations, indicating that these oligomers are held together by the two phosphorylated motifs of a single molecule of ITIM-[dPEG4]2-ITSM. The formation of multimers in the presence of ITIM-[dPEG4]2-ITSM was also observed for SHP21–525 (fig. S6).
The dependence of the stoichiometry of the complex on protein concentration was confirmed by size-exclusion chromatography (SEC) coupled with multiangle light scattering (MALS). We measured two equimolar mixtures of SHP21–220 and ITSM-[dPEG4]2-ITSM, prepared at concentrations of 10 and 100 μM. For this experiment, we used the bidentate ITSM-[dPEG4]2-ITSM peptide, instead of ITIM-[dPEG4]2-ITSM, to avoid the loss of binding between the protein and ITIM that could occur at low concentrations on the SEC column. The SEC-MALS profile of the mixture prepared at 100 μM displayed a significant degree of heterogeneity, as detected by the misalignment of the light scattering and refractive index curves, and a mass distribution ranging from 42 to 35 kDa (Fig. 4B, left peak). The solution prepared at 10 μM was far more homogeneous, with a molecular weight of 30 to 32 kDa (Fig. 4B, middle peak). These data confirm that the oligomerization of the protein-peptide complex is dependent on protein concentration with concentrations <<100 μM favoring the formation of 1:1 particles.
Next, we tested whether the transition from oligomeric protein-peptide complexes to a homogeneous 1:1 complex was visible in the 1H-15N spectra of SHP21–220 by monitoring SHP21–220 1H-15N peaks of a protein:peptide mixture at 1:1.2 molar ratio while decreasing the total concentration. Upon dilution, we observed several CSPs (Fig. 4D), all of which mapped to amino acids localized at the interface between the two SH2 domains rather than to the N-SH2 or C-SH2 peptide binding sites (Fig. 4F). This demonstrates that the formation of the 1:1 SHP21–220–ITIM-[dPEG4]2-ITSM complex, where both ITIM and ITSM of the bidentate peptide are bound to the N-SH2 and C-SH2 domains of one SHP21–220 molecule, generates a new interface between the SH2 domains. These concentration-dependent CSPs were not present when titrating a mixture of N-SH2 and C-SH2 with ITIM-[dPEG4]2-ITSM or C-SH2 with ITSM (Fig. 4D). In addition, the CSPs disappeared from the 1H-15N spectrum of SHP21–220 upon addition of a twofold molar excess of ITIM-[dPEG4]2-ITSM, as expected, due to the formation of the 1:2 protein:peptide complex, in which both SH2 domains are bound to ITSM.
The relative orientation of the SH2 domains in the autoinhibited SHP21–525 structure of PDB entry 2SHP is not compatible with the simultaneous binding of the two phosphorylated motifs of ITIM-[dPEG4]2-ITSM to both SH2 domains, due to the spatial constraint imposed by the length of the linker (Fig. 4E). In the structure of the constitutively active SHP2-E76K mutant solved recently (26, 27), the reorientation of both N-SH2 and C-SH2 domains relative to the PTP domain leaves the active site of the PTP domain fully accessible to the substrate (PDB entry 6CRF; fig. S7). However, this arrangement of the N-SH2 and C-SH2 domains is also incompatible with the simultaneous binding of the two phosphorylated motifs of ITIM-[dPEG4]2-ITSM, as the distance of 65 Å between the C-terminal end of ITIM and the N-terminal end of ITSM would be much longer than the linker (~40 Å). In contrast, the structure of full-length SHP1 seen in PDB entry 3PS5 (28) would allow binding of both ITIM and ITSM of one bidentate peptide molecule to its SH2 domains (Fig. 4E and fig. S7). In PDB entry 3PS5, the N-SH2 domain is moved away from the PTP domain and located behind the C-SH2 domain, which is rotated by 110° with respect to the structure of PDB entry 2SHP; in this conformation, the PTP catalytic pocket is wide open and accessible to the substrate.
Both ITIM and ITSM are necessary for full SHP2 activation
Our in vitro studies support the notion that both ITIM and ITSM are required for full activation of SHP2, which leads to suppression of T cell activation. To test this hypothesis in T cells, we used a functional readout of the consequences of ITIM and ITSM mutations on PD-1–mediated suppression of T cell receptor (TCR) signaling (Fig. 5A).
Fig. 5. Effect of PD-1 mutants on SHP2 binding and TCR-induced T cell activation.
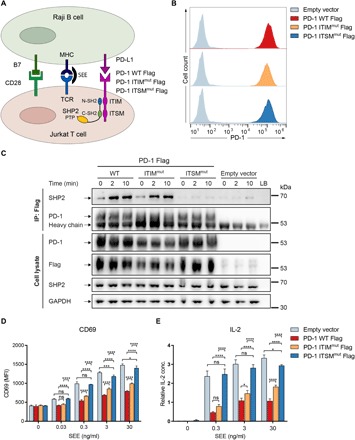
(A) Experimental setup for quantifying the inhibitory effect of PD-1 on TCR-mediated T cell activation. Raji B cells were lentivirally transduced with PD-L1 and used as antigen-presenting cells. Jurkat T cells were transduced with either WT PD-1, PD-1 ITIMmut, PD-1 ITSMmut, or an empty vector. For TCR stimulation, the B cells were incubated with superantigen [staphylococcal enterotoxin E (SEE)] and cocultured with the T cells. Upon activation of PD-1, ITIM and ITSM become phosphorylated and recruit SHP2. (B) Quantification of the surface expression of WT PD-1 (red), PD-1 ITIMmut (orange), and PD-1 ITSMmut (blue) in the different Jurkat cell lines. Cells containing an empty vector are used as negative control. (C) Coimmunoprecipitation (IP) experiment demonstrating SHP2 binding to the PD-1 variants after incubation of T cells with SEE-loaded B cells (30 ng/ml) for the indicated time points. PD-1, Flag, and SHP2 protein levels were analyzed; glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. (D) CD69 expression levels of the Jurkat T cell lines (WT PD-1, PD-1 ITIMmut, and PD-1 ITSMmut) were measured by flow cytometry after 5 hours of stimulation with B cells preincubated with different SEE concentrations. The mean fluorescence intensity (MFI) is shown. (E) Relative interleukin-2 (IL-2) levels in the supernatant of the Jurkat cell cultures (WT PD-1, PD-1 ITIMmut, and PD-1 ITSMmut) were measured by enzyme-linked immunosorbent assay (ELISA) after 24 hours of stimulation with SEE-loaded B cells. Statistics are based on two-way analysis of variance (ANOVA) and Bonferroni multiple comparison test. Data points represent one of three independent experiments, each performed in triplicate. ns, P > 0.05,*P < 0.05, ***P < 0.001, ****P < 0.0001; n = 3.
The Jurkat T cell line contained some cells with endogenous PD-1 expression in the unstimulated state, as detected by flow cytometry; and hence, cells were first fluorescence-activated cell sorting (FACS)–sorted to obtain only those that were PD-1 negative. These cells were then lentivirally transduced with Flag-tagged WT PD-1, PD-1–Y223F (called PD-1 ITIMmut), or PD-1–Y248F (called PD-1 ITSMmut) and sorted for green fluorescent protein (GFP) expression (the lentiviral plasmids also encoded for GFP in addition to the PD-1 variants). Similar PD-1 expression levels were confirmed by flow cytometry, using anti–PD-1 antibody staining (Fig. 5B). The Jurkat TCR can be stimulated with major histocompatibility complex (MHC) II molecules loaded with the superantigen staphylococcal enterotoxin E (SEE). We used human Raji B cells as MHC II–presenting cells (29); these lack endogenous PD-L1 expression and were therefore lentivirally transduced with PD-L1 and enriched by GFP sorting. Jurkat and Raji cells also express the CD28 receptor and its ligand B7.1, respectively. Thus, SEE-loaded MHC II stimulates the TCR and B7.1 stimulates CD28, while PD-L1 binds to PD-1 (or the PD-1 mutants) to suppress the TCR- and CD28-derived activation signals (Fig. 5A). This system was used in a previous study of suppression of TCR/CD28 signaling by PD-1 (16).
First, we tested how the PD-1 variants influenced the binding of endogenous SHP2 in the Jurkat T cells. The PD-L1–expressing Raji B cells were loaded with SEE and incubated with the PD-1 variant–expressing Jurkat T cells for 0, 2, or 10 min. The Flag-tagged PD-1 variants were immunoprecipitated using a Flag antibody, and coprecipitation of SHP2 was analyzed by SDS–polyacrylamide gel electrophoresis (PAGE) and immunoblotting (Fig. 5C). Upon coculture of the two cell lines, SHP2 was recruited to WT PD-1, as shown before (16). Similar levels of SHP2 recruitment were observed with PD-1 ITIMmut, while SHP2 could not be coprecipitated with PD-1 ITSMmut. As a control, immunoblotting with anti-PD-1 antibodies showed similar amounts of PD-1. This result indicates that ITSM is required for SHP2 recruitment by PD-1, while ITIM is dispensable.
Second, we interrogated the functional role of ITIM and ITSM in suppressing TCR/CD28 signaling. The PD-1 variant–expressing Jurkat cells were stimulated with the PD-L1–expressing Raji B cells using different concentrations of SEE for 5 hours. TCR-induced expression of the early activation marker CD69 was quantified by flow cytometry. In the absence of PD-1, stimulation with SEE resulted in a dose-dependent up-regulation of CD69 (Fig. 5D), while presence of WT PD-1 reduced the level of CD69 expression, most likely due to recruitment and activation of SHP2. Expression of PD-1 ITSMmut, which did not recruit SHP2, was unable to reduce CD69 levels compared to the empty vector control. In contrast, PD-1 ITIMmut, which recruited SHP2, was able to reduce CD69 expression, albeit to a lesser extent than WT PD-1. This result indicates that ITIM, despite being dispensable for SHP2 binding, does play a role in PD-1 function. As a control, the CD69 levels of the Raji B cells were not affected by the coculture with the T cells (fig. S8).
Third, we quantified the TCR-induced secretion of the late activation marker interleukin-2 (IL-2) by an enzyme-linked immunosorbent assay (ELISA). To this end, we cocultured the Jurkat and SEE-loaded Raji cells for 24 hours, to allow IL-2 to accumulate in the culture supernatant. The results recapitulated those obtained by CD69 quantification (Fig. 5, D and E). Briefly, WT PD-1 reduced TCR-induced IL-2 production, while the PD-1 ITSMmut did not reduce IL-2 levels. In contrast, PD-1 ITIMmut was able to inhibit TCR signaling but to a lesser extent than that by WT PD-1.
In summary, the functional experiments demonstrated that mutation of ITSM abolishes PD-1 signaling, while mutation of ITIM only reduces PD-1 signaling. These results correlate well with the lack of SHP2 coimmunoprecipitation with PD-1 ITSMmut, while PD-1 ITIMmut did bind SHP2. This correlation, together with our structural and biochemical data, indicates that the function of ITIM in supporting the PD-1–mediated suppression of the immune response is to enable full activation of SHP2 phosphatase activity.
DISCUSSION
Targeting of PD-1 by monoclonal antibodies that disrupt interaction with its ligands has proven a successful therapeutic strategy for several cancers (30). Nevertheless, the cost of such therapies imposes a burden on public health care, and there remains an unmet demand for affordable small-molecule drugs that are able to disrupt PD-1 signaling in T cells. Targeting SHP2, as the immediate downstream partner of PD-1, holds promise for the development of a new class of immune modulators. While this strategy has been recently questioned by Rota et al. (31), who used cells with chronically knocked-down SHP2 and found that the phosphatase is dispensable for PD-1 signaling in vivo, the concept is supported by many studies demonstrating that SHP2 plays an important role in T cell activation (14–16, 18, 19).
The design of small molecules targeting the SHP2–PD-1 interaction during immune response relies on the knowledge of the structure of the SHP2–PD-1 complex as well as on the discovery of the functional determinants of the signaling process. However, while the binding of SHP2 to PD-1 was shown to depend on its two SH2 domains recognizing the phosphorylated ITIM and ITSM of the cytoplasmic tail of PD-1, the exact roles of the C-SH2 domain of SHP2 and the ITIM of PD-1 could not be described unambiguously. In addition, the architecture of the SHP2–PD-1 complex, which contains two pY binding sites (the N-SH2 and C-SH2 domains of SHP-2) and two pY-peptide motifs (ITIM and ITSM of PD-1), remained obscure.
Here, we used a combination of structural and functional studies in vitro and in cells to determine the mechanism of activation of SHP2 by the cytoplasmic tail of PD-1. Using NMR spectroscopy and calorimetry, we find that ITSM has the highest affinity for both SH2 domains with a 10-fold preference for C-SH2. On the other hand, ITIM binds preferentially to N-SH2, although 10-fold weaker than ITSM. In agreement with these relative affinities, ITSM is a stronger activator of SHP2 activity in vitro and is indispensable for PD-1 signaling in cells. However, our structural and functional data demonstrate that full activation of the phosphatase is dependent on the simultaneous binding of both ITIM and ITSM, connected by a linker as parts of the same molecule, to SHP2, with each phosphorylated motif engaging one of the SH2 domains. ITSM binds to C-SH2 with strong affinity, recruiting PD-1 to SHP2, while ITIM binds to N-SH2, displacing it from the PTP catalytic pocket and activating the phosphatase (Fig. 6). In the absence of phosphorylated ITIM, ITSM binds to both C-SH2 and N-SH2, with a 10-fold preference for C-SH2. The N-SH2–ITSM binding event also activates the phosphatase, but this second activation mechanism (Fig. 6) is less efficient than the one involving the bidentate peptide, both in vitro and in cells. On the basis of our data, we can propose two reasons for this. First, activation of the phosphatase can be achieved by binding of phosphopeptides to only the N-SH2 domain, inducing the conformational change of the EF loop that releases N-SH2 from the PTP active site (2). However, this event has not been shown to yield a stable open conformation but likely generates a number of conformations, in some of which the N-SH2 domain would still partially occlude access to the PTP active site (Fig. 6). In contrast, simultaneous binding of ITIM and ITSM to both N-SH2 and C-SH2 yields a stable open conformation, with N-SH2 locked far away from the PTP active site. Second, in the absence of a second phosphorylated site, binding of ITSM to N-SH2 is outcompeted by the C-SH2 domain, which binds ITSM more strongly than does N-SH2. In vitro, the isolated ITIM can activate SHP2 through the same mechanism as ITSM. The failure to detect any SHP2-stimulating activity in the absence of phosphorylated ITSM in cells can be explained by the lower affinity of ITIM for N-SH2 as compared to ITSM, which is not sufficient to yield a measurable activation signal. Similarly, the low affinity of ITIM for both N-SH2 and C-SH2 (Kd > 1 μM) rationalizes the inability of PD-1 ITSMmut to pull down SHP2 in immunoprecipitation experiments.
Fig. 6. Two activation mechanisms of SHP2 by PD-1.
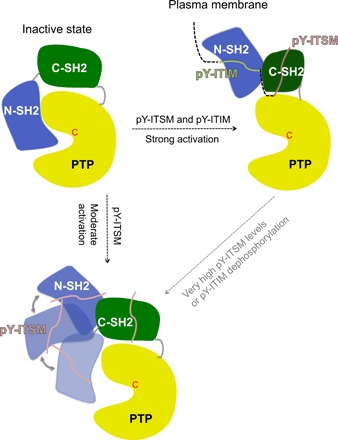
Binding of phosphoproteins activates the autoinhibited form of SHP2 (left) by acting to remove the N-SH2 domain from the PTP binding site. Activation can occur in two ways. Top right: When the doubly phosphorylated PD-1 tail binds simultaneously to both SH2 domains of SHP2, the phosphatase adopts a stable open conformation, where the N-SH2 is locked behind the C-SH2 due to the restraint imposed by the linker connecting ITIM and ITSM. Bottom left: When ITIM is dephosphorylated or ITSM is present in large excess, ITSM binds to the N-SH2 domain, thereby weakening its interaction with the PTP domain. However, in the absence of the divalent binding event, the N-SH2 domain is not locked behind the C-SH2 domain and may adopt many orientations (depicted schematically by three representative orientations connected by the gray double-headed arrows). In some of these orientations, the N-SH2 domain may occupy the space close to the PTP domain, partially occluding access to its active site. The switch between the two mechanisms may represent a way to regulate the efficiency of SHP2 activation depending on the concentration of the activating phosphorylated motifs.
The model of activation by the bidentate peptide shown in Fig. 6 is fully supported by the NMR data. Titration of the SHP21–220 construct, containing both SH2 domains, with the bidentate ITIM-[dPEG4]2-ITSM peptide shows that ITSM binds preferentially to C-SH2, while the N-SH2 domain is recruited by ITIM. The spectra show no indication of ITIM binding to the C-SH2 domain at any stoichiometric ratio or concentration, confirming the arrangement of the two binding motifs of the bidentate peptide as shown in Fig. 6. Overall, our data provide structural and biochemical support for the two-step model put forward by Peled et al. (17), who proposed that SHP2 is first recruited to PD-1 through the C-SH2–ITSM interaction and then activated by the N-SH2–ITIM interaction. However, there is no evidence of the temporal order of the two binding events, which could also occur concomitantly or involve additional intermediate steps.
Our biophysical data demonstrate that the SHP21–220–ITIM-[dPEG4]2-ITSM complex, with ITIM and ITSM recognized by N-SH2 and C-SH2, respectively, occurs only at concentrations below 15 μM. At higher concentrations, SHP21–220 forms multimers, held together by ITIM-[dPEG4]2-ITSM bridging two SHP21–220 molecules (Fig. 4, B and C). This fact implies that the formation of the stable 1:1 SHP21–220–ITIM-[dPEG4]2-ITSM complex, following the initial encounter of the protein with the peptide, occurs at a slower rate than an encounter with a second protein molecule at 15 μM concentration (diffusion-limited kon × protein concentration = 5 × 108 M−1 s−1 × 15 × 10−6 M = 7500 s−1). The relatively slow rate of the 1:1 complex formation suggests the involvement of significant conformational rearrangements in the protein. Accordingly, the NMR data reveal the formation of a new interaction surface between the N-SH2 and C-SH2 domains in the SHP21–220–ITIM-[dPEG4]2-ITSM complex. The change in the orientation of the SH2 domain is required because of the restrictions imposed by the length of the linker connecting ITIM and ITSM.
All the above data suggest that full activation of SHP2 by simultaneous binding of both ITIM and ITSM is regulated at several levels. First, the N-SH2 binding affinity of ITIM is less than that of ITSM, as a bidentate peptide containing two ITSMs (ITSM-[dPEG4]2-ITSM) activates SHP2 more efficiently than ITIM-[dPEG4]2-ITSM. The fact that such a combination of strong binding motifs does not exist in nature points to the necessity of tuning SHP2 activation to avoid hyperactivity. Accordingly, the SH2 domains are generally suboptimal with respect to phosphopeptide affinity, while they may be optimized for regulation (32). Second, SHP2 dephosphorylates ITIM more efficiently than ITSM, providing an additional way to tune down SHP2 activation, when required. Third, the full activation of SHP2 requires a slow conformational change, which is outcompeted by binding of ITIM to a second protein molecule at concentrations of SHP2 higher than 15 μM. While we cannot prove whether this mechanism is relevant in cells, it has been reported that the concentration of SHP2 in mammalian cells is in the micromolar range (33), where the regulation mechanism based on the oligomerization of the SHP-2–PD-1 complex would be applicable. Thus, it is conceivable that the formation of inactive or moderately active oligomers could contribute to the down-regulation of SHP2 activation in the presence of high amounts of either the phosphatase or PD-1. In support of this, Peled et al. (17) failed to find a correlation between PD-1 expression levels and TCR signaling in different T cell pools.
Previous reports have failed to reach consensus on the relevance of ITIM for SHP2 activation (15–19). Our results indicate that these discrepancies may be attributable to a fine regulation of the ITIM-dependent activation process, which is differently affected by the exact experimental setups. In addition, in the absence of phosphorylated ITIM, a second activation mechanism becomes relevant (Fig. 6), which, while less efficient, can have a compensatory effect within given cellular parameters, such as protein concentration or phosphorylation levels.
SHP2 is activated also by other bidentate phosphopeptides, such as that present in insulin receptor substrate-1 (IRS-1) in insulin signaling (34). It is conceivable that the mechanism of regulation of SHP2 activity described here applies generally to SHP2 activation by bidentate phosphopeptides in multiple contexts, albeit with different structural and biophysical parameters. In the case of the SHP2–PD-1 interaction, we believe that the interface between the N-SH2 and C-SH2 domains, which is formed upon full activation and holds SHP2 in the open active conformation, represents a potential target to affect SHP2 activation selectively in the context of PD-1 signaling (35). Preventing the formation of this interface would decrease, but not abolish, PD-1 signaling. This strategy could be exploited as part of a combination therapy to reduce cancer immune evasion, while sensitizing cancer cells to other drugs and preventing the side effects caused by full SHP2 inhibition.
MATERIALS AND METHODS
General experimental design
To elucidate the mechanism of SHP2 activation by PD1 and verify it in cells, we structured the work in three packages: structural, biochemical, and cellular experiments. The experimental design for each package is explained in the respective sections.
Structural studies
Experimental design
The protein-peptide complex structures were solved either by NMR or x-ray crystallography. The interaction of the N-SH2 and C-SH2 domains as well as of SHP21–220 with ITIM, ITSM, and ITIM-[dPEG4]2-ITSM was analyzed by NMR. The multimerization state of the complexes was studied by SEC-MALS. The binding affinities were determined by NMR and calorimetry.
Protein expression and purification
For full-length SHP2, the DNA sequence of SHP2, isoform 2 (protein sequence in UniProtKB accession code Q06124-2), was amplified and cloned into the pETM11-Sumo3 expression vector (European Molecular Biology Laboratory collection) using Bam HI and Xho I restriction sites. The DNA for the SHP21–525 mutant, with the deletion of the C-terminal tail, was generated using forward primer 5′-TCTTCTTCAATACGACGCTACAGGGTTTCGATATAATGC-3′ and reverse primer 5′-GCATTATATCGAAACCCTGTAGCGTCGTATTGAAGAAGA-3′. The SHP21–525 R32A and R138A mutants were generated with the QuikChange Lightning Site-Directed Mutagenesis protocol (Agilent Technologies). All constructs were confirmed by DNA sequencing.
Plasmid DNA encoding SHP21–525 was transformed into BL21 DE3 Escherichia coli, from which a single colony was selected to inoculate a 50-ml starter culture of LB with kanamycin (50 μg/ml), and allowed to grow overnight. The following morning, 10 ml of the saturated culture was used to inoculate 1 liter of LB with kanamycin (50 μg/ml). At an OD600 (optical density at 600 nm) of 0.5 to 0.6, expression of SHP21–525 was induced by the addition of 0.1 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG). Expression was continued overnight at 20°C. The cells were then pelleted at 4°C and 4000 rpm for 40 min; resuspended and incubated for 15 min in 30 ml of ice-cold 50 mM tris-HCl (pH 7.6), 1 M NaCl, 10% glycerol, 5 mM β-mercaptoethanol, 10 mM imidazole, and an EDTA-free protease inhibitor cocktail tablet (Roche); and then lysed via sonication. Total cell lysate was clarified at 18,000 rpm for 40 min at 4°C; the supernatant was filtered, then loaded onto a HisTrap FF (GE Healthcare) column washed with buffer A [50 mM tris-HCl (pH 7.6), 1 M NaCl, 2% glycerol, 5 mM β-mercaptoethanol, and 10 mM imidazole], and eluted with a gradient of buffer B [50 mM tris-HCl (pH 7.6), 1 M NaCl, 2% glycerol, 5 mM β-mercaptoethanol, and 500 mM imidazole]. The N-terminal histidine tag was then cleaved using sentrin-specific protease 2 protease (1:1000 protease/protein ratio) overnight in buffer C [20 mM tris-HCl (pH 8.0), 50 mM NaCl, and 5 mM β-mercaptoethanol]. The next day, the cleaved protein was loaded onto an ion-exchange column (HiTrap Q FF), washed with buffer C, and eluted with a gradient of 50 to 500 mM NaCl. Pure fractions were pooled, concentrated to 0.5 ml, and purified further on a HiLoad Superdex 200 16/600 size-exclusion column (GE Healthcare) equilibrated into 50 mM Hepes (pH 7.6), 150 mM NaCl, and 1 mM EDTA. Fractions were collected in 1.5-ml increments and assessed for purity via PAGE, and pure fractions were concentrated, dispensed into 10- to 50-μl single-use aliquots, and stored at −80°C with 20% glycerol and 1 mM dithiothreitol (DTT).
Human SHP2 domains N-SH2 (1 to 105, 11.9 kDa), C-SH2 (106 to 220, 13.3 kDa), and SHP21–220 were subcloned into the pETM22 plasmid, which bears a cleavable His6-tagged thioredoxin tag for improved solubility and folding. Expression was performed in BL21(DE3) E. coli strains, induced with 0.2 mM IPTG at 20°C for 18 hours. The cells were induced at an OD600 of 0.6 to 0.8. Preparation of uniformly 15N,13C-labeled protein for NMR studies was achieved by growing the bacteria in M9 minimal medium containing kanamycin (50 μg/ml), 15NH4Cl (1 g/liter), and 13C-d-glucose (4 g/liter). Isotopes were purchased from Cambridge Isotope Laboratories. After harvesting, cell pellets were stored at −20°C until purification.
Production of deuterated SHP21–220 was achieved with the same protocol, using BL21(DE3) Tuner E. coli strains. Sparse deuteration was accomplished by supplying the bacteria with a protonated carbon source and by growing them in 70% D2O (for a final deuteration level of 50%) or in 99% D2O (for a final deuteration level of >80%). D2O was purchased from Eurisotop. Perdeuterated isoleucine, leucine, and valine (ILV)–labeled SHP21–220 was produced growing the bacteria in fully deuterated M9 minimal medium and supplying them with d7-d-glucose (Cambridge Isotope Laboratories). At an OD600 of 0.5, methyl group precursors—α-ketobutyric acid, sodium salt (60 mg/liter) and α-ketoisovaleric acid, sodium salt (120 mg/liter)—were added, and expression was then induced at an OD600 of 0.6 (20°C for 18 hours). For side-chain assignment experiments, the following labeling scheme was adopted: α-ketobutyric acid, sodium salt (13C4, 98%; 3,3-D2, 98%) and α-ketoisovaleric acid, sodium salt (1,2,3,4-13C4, 99%; 3,4′,4′,4′-D4, 97 to 98%), whereas terminally labeled precursors were used for nuclear Overhauser effect spectroscopy (NOESY) experiments: α-ketobutyric acid, sodium salt (methyl-13C, 99%; 3,3-D2, 98%) and α-ketoisovaleric acid, sodium salt (3-methyl-13C, 99%; 3,4,4,4-D4, 98%). Cell pellets were resuspended in wash buffer [1 M NaCl, 50 mM tris, 2% glycerol, 10 mM imidazole, and 5 mM β-mercaptoethanol (pH 7.6)] with one tablet of EDTA-free protease inhibitor cocktail (Roche), 100 μg of lysozyme (Roth), and 50 μg of deoxyribonuclease (DNAse) [New England Biolabs (NEB)]. Lysis was performed on a high-pressure homogenizer (Avestin Emulsiflex C3). The lysate was spun at 19,000 rpm for 1 hour, and the filtered supernatant was loaded on a HisTrap HP column (GE Healthcare), equilibrated with wash buffer. Elution of the His6-tagged protein constructs was achieved with an imidazole gradient of 10 to 300 mM over a volume of 60 ml. Fractions containing the protein of interest were pooled, concentrated to approximately 15 ml, and loaded on a HiPrep 26/10 desalting column (GE Healthcare) to remove the excess of imidazole. The thioredoxin tag was cleaved by incubation with 3C protease (1:100 protease/protein ratio) at 4°C overnight. Purification continued with a second Ni2+-affinity chromatography step followed by SEC on a HiLoad 16/600 Superdex 75 pg (GE Healthcare) column, equilibrated with NMR buffer [150 mM NaCl and 100 mM MES (pH 6.8)]. Subsequently, the protein was concentrated to the desired concentration and either used directly for NMR experiments or flash-frozen with liquid nitrogen for long-term storage at −80°C.
The PTP domain of SHP2 (225 to 525) was expressed in BL21(DE3) Tuner E. coli cells and used in the activity assays. Bacteria were grown at 37°C until they reached an OD600 of 0.6 to 0.8. Protein expression was then induced with 0.1 mM IPTG and carried out at 16°C for 18 to 20 hours. After harvesting, cell pellets were resuspended in ice-cold wash buffer [50 mM tris-HCl, 1 M NaCl, 5% glycerol, 10 mM imidazole, and 5 mM β-mercaptoethanol (pH 7.6)], with one tablet of EDTA-free protease inhibitor cocktail (Roche), 100 μg of lysozime (Roth), and 50 μg of DNAse (NEB). Lysis was performed with a high-pressure homogenizer (Avestin Emulsiflex C3). The lysate was spun at 19,000 rpm for 1 hour, and the filtered supernatant was loaded on a HisTrap HP column (GE Healthcare), equilibrated with wash buffer. Elution was achieved with a step gradient of 100% elution buffer [50 mM tris-HCl, 1 M NaCl, 5% glycerol, 500 mM imidazole, and 5 mM β-mercaptoethanol (pH 7.6)]. Cleavage of the thioredoxin tag was performed overnight at 4°C with 3C protease, while the protein sample was being dialyzed against 2 liters of wash buffer. Purification continued with a second Ni2+-affinity chromatography step followed by SEC on a HiLoad 16/600 Superdex 200 pg column (GE Healthcare), equilibrated with 50 mM Hepes (pH 7.6), 150 mM NaCl, and 1 mM EDTA. Subsequently, the protein was concentrated to the desired concentration and either used directly for the activity assays or flash-frozen with liquid nitrogen for long-term storage at −80°C.
Peptides
The EGFR peptide (Ac-DADEpYLIPQQG-NH2) was synthesized in-house using standard Fmoc solid-phase peptide synthesis in a MultiPep RSi peptide synthesizer from Intavis Bioanalytical Instruments and purified by high-performance liquid chromatography (HPLC; Agilent Technologies, 1260 Infinity series). The other peptides were purchased from Caslo ApS (Lyngby, Denmark). The sequences of the peptides were as follows: ITIM, Ac-FSVD(pY)GELDFQ-NH2, ITSM, Ac-EQTE(pY)ATIVFP-NH2, diphosphorylated peptide ITIM-[dPEG4]2-ITSM, Ac-FSVD(pY)GELDFQ-(dPEG4)-(dPEG4)-EQTE(pY)ATIVFP-NH2, diphosphorylated peptide ITSM-[dPEG4]2-ITSM, Ac-EQTE(pY)ATIVFP-(dPEG4)-(dPEG4)-EQTE(pY)ATIVFP-NH2. The purity obtained in the synthesis was 95 to 98% and was determined by HPLC using a C18 column followed by mass spectrometry.
NMR spectroscopy
NMR spectra were recorded at a temperature of 298 K on Bruker Avance III HD 600- and 850-MHz spectrometers running TopSpin 3.2 software and equipped with N2-cooled and He-cooled inverse HCN triple-resonance cryogenic probe heads, respectively. NMR samples were prepared using protein concentrations of 0.3 to 0.8 mM in 150 mM NaCl and 100 mM MES (pH 6.8). Protein:peptide mixtures for protein-detected (isotope-edited) spectra contained peptide in a slight molar excess. Protein:peptide mixtures for peptide-detected (isotope-filtered) spectra contained protein and peptide at equimolar concentrations. All peptides were unlabeled. 2D 1H-15N correlation spectra for peptide titrations with the isolated C-SH2 and N-SH2 domains were recorded as 15N heteronuclear single-quantum coherence (HSQC) spectra (36) using uniformly 15N-labeled (U-15N) protein. 2D 1H-15N correlation spectra for titration of SHP21–220 with the ITIM-[dPEG4]2-ITSM peptide were recorded as 15N-TROSY (transverse relaxation-optimized spectroscopy)–HSQC spectra (37) using U-15N,50% 2H-labeled protein. Water suppression for amide-detected experiments was typically achieved using the WATERGATE sequence in combination with water flip-back to preserve the water magnetization. 2D 1H-13C correlation maps for selectively methyl-protonated (U-2H,ILV-{Me-1H,13C}–labeled) samples were recorded as methyl TROSY (13C heteronuclear multiple-quantum coherence) spectra (38).
Protein backbone and side-chain resonances of the isolated domains in the unbound and peptide-bound states, as well as the unbound SHP21–220, were assigned from standard triple-resonance backbone spectra (39) [2D 15N-HSQC, 3D HNCO, 3D HNCACB, and 3D HN(CO)CACB] and side-chain assignment spectra [3D HC(C)H–total correlation spectroscopy (TOCSY), 3D H(CCCO)NH-TOCSY, and 2D constant-time 13C-HSQC], respectively, recorded on samples containing U-{15N,13C}–labeled protein. In addition, 2D HBCB(CGCD)HD and HBCB(CGCDCE)HE spectra (40) were recorded for the C-SH2–ITSM complex to assist assignment of protein aromatic side-chain resonances. The 3D backbone assignment spectra for SHP21–220 were recorded with nonuniform sampling, using Poisson-gap sampling schedules generated with the web-based nus@HMS schedule generator from the group of Gerhard Wagner (Harvard Medical School).
Protein backbone resonances of SHP21–220 in complex with the ITIM-[dPEG4]2-ITSM peptide were assigned from TROSY-based triple-resonance backbone spectra [2D 15N-TROSY-HSQC, 3D TROSY-HNCO, 3D TROSY-HNCACB, and 3D TROSY-HN(CO)CACB] (41) recorded with U-{15N,13C},80% 2H-labeled protein. ILV methyl resonances of the SHP21–220–ITIM-[dPEG4]2-ITSM complex were assigned with the assistance of a 3D HMCM(CG)CBCA spectrum (39), recorded with U-{13C,15N,2H},ILV-{Me-1H} protein. Proton peptide resonances of the C-SH2–ITSM and SHP21–220–ITIM-[dPEG4]2-ITSM complexes were assigned from 2D 13C,15N-filtered TOCSY spectra (42) recorded on samples containing U-{15N,13C}–labeled C-SH2 and U-{12C,14N,2H},ILV-{Me-1H,13C}–labeled SHP21–220 proteins, respectively.
Distance restraints for structure calculation of the C-SH2–ITSM complex were derived from 3D NOESY–13C-HSQC, 3D 13C,15N-filtered NOESY–13C-edited HSQC, and 2D 13C,15N-filtered NOESY spectra (yielding protein intramolecular and protein-peptide intermolecular NOEs, protein-peptide intermolecular NOEs, and peptide intramolecular NOEs, respectively), recorded on C-SH2–ITSM samples containing U-{15N,13C}–labeled C-SH2 protein.
NMR data analysis
Uniformly sampled NMR spectra were processed with TopSpin 3.2 (Bruker) and nmrPipe (43). Nonuniformly sampled time-domain data were first reconstructed using iterative soft thresholding as implemented in the hmsIST program (44) with the assistance of the Linux command line tool “parallel.” Peak-picking, resonance assignment, and NOESY peak assignment were done using CcpNmr Analysis (45).
Structure calculation and analysis
Structure calculations were performed using ARIA v2.3/CNS v1.21 via the standard ARIA iterative assignment protocol (46). Distance restraints were generated from NOESY peak intensities with application of spin diffusion correction for the calibration factors. Dihedral angle restraints for backbone ϕ and ψ angles were generated from backbone chemical shifts using TALOS-N. Restraints were applied for residues where the TALOS-N classifications were “strong” or “generous,” using respective error ranges of twice and three times the uncertainty reported by TALOS-N. The numbers of steps for the hot, cool1, and cool2 stages of the molecular dynamics simulated annealing protocol were 20,000, 20,000, and 16,000, respectively. All distance restraints were used in every iteration. One hundred fifty structures were calculated in the final iteration, of which the 10 lowest-energy structures were refined in explicit water to generate the final structural ensemble (PDB entry 6R5G).
X-ray data collection, structure determination, and refinement
X-ray diffraction experiments were carried out on the beamline X06DA, operated by the Paul Scherrer Institute at the Swiss Light Source (Villigen, Switzerland), and the diffraction images were recorded with a Pilatus 2M-F pixel detector (DECTRIS Ltd., Switzerland). Before data collection, the crystals were flash-cooled to 100 K and kept at this temperature during x-ray exposure. The x-ray wavelength used was 100 pm. To process data, the recorded Bragg reflections were indexed and integrated with the x-ray detection software (XDS) (47) followed by scaling and merging in Aimless and Ctruncate, both part of the CCP4 software suite (48). In both datasets, data processing revealed the presence of four- and twofold rotational symmetry elements that were arranged in a way compatible with a tetragonal space group of the form P4x2y2 (x: ∅, 1, 2, 3; y: ∅, 1). An analysis of the data with phenix.xtriage (49) revealed an issue with unusual intensity statistics for both datasets, which could not be explained by twinning as there are no twin operators in P4x2y2. After reprocessing in lower-symmetry space groups, the unusual intensity statistics remained in place and still could not be attributed to twinning according to phenix.xtriage. They were most likely caused by a poor spot separation, which resulted from a combination of the large lattice parameter for axis c and the width of the Bragg peaks and which could not be overcome by adjustments to the experimental setup. The phase problem could be solved by molecular replacement with Phaser (50) in space group P43212. However, subsequent refinement of the structure in P43212 resulted in too high values for the residuals Rwork and Rfree (both >0.35), indicating the wrong choice of space group. To restore the correct space group, data were reprocessed with intentional low symmetry and analyzed with Zanuda (48). However, Zanuda analysis was inconclusive, probably due to the fact that the parameters it used for refinement with Refmac5 (51) did not fit the quality of the datasets. As a consequence, data were reprocessed in all subgroups of space group P43212 and subjected to refinement with Refmac5 through the PDB_REDO web server. This allowed us to obtain the most suitable refinement parameters, because PDB_REDO optimizes refinement settings such as geometric and B-factor restraint weights, the B-factor model, translation-libration-screw groups, and noncrystallographic symmetry restraints. The best, though still unusually high, values for the residuals were achieved in space group P43 with two copies of the N-SH2–peptide complexes in the asymmetric unit. The twofold symmetry axis that relates the two copies to each other was arranged perpendicular to the fourfold crystallographic screw axis and thus gave rise to the ambiguity of the space group. Throughout all the data processing and reprocessing, the upper resolution limits were assessed through careful observation of mean I/σI and correlation coefficient (CC) ½ values. The structures were finalized in repeated cycles of manual building in Coot (52) and refinement in Refmac5 using the optimized settings found with PDB_REDO. Refinement was stopped when Rwork and Rfree converged. The final models showed reasonable Ramachandran statistics with percent favored/allowed/disallowed of 94/6/0 for N-SH2–ITSM and of 97/3/0 for N-SH2–ITIM. The structures of N-SH2–ITSM and N-SH2–ITIM were deposited with the PDB (www.rcsb.org) with accession numbers 6ROZ and 6ROY, respectively. The complete data processing and refinement statistics are shown in table S1.
SEC-MALS experiments
The measurements were performed using a Superdex 200 10/300 (GE Healthcare) column in line with Optilab T-rEX and miniDAWN TREOS (Wyatt). All samples were prepared in 100 mM MES, 150 mM NaCl, and 5 mM DTT (pH 6.8).
Isothermal titration calorimetry
ITC measurements were performed on a Nano ITC (TA Instruments) in 100 mM MES and 150 mM NaCl (pH 6.8) at 25°C using an equilibration time of 3000 s and a stir rate of 200 rpm. The concentration of the SH2 domains in the cell was 0.1 mM, and the concentration of the peptides in the syringe was 1 mM. Analysis of the data was carried out with NanoAnalyze (v3.6.0, TA Instruments). The Kd was calculated from a single replicate, using a 1:1 binding model, based on the results of the NMR titrations.
Biochemical studies
Experimental design
To characterize the enzymatic activity of SHP2 in vitro in the presence of PD-1 ITIM and ITSM, we followed two lines of experiments. First, we measured the activation of SHP2 (WT and mutant forms) by ITIM and ITSM either in isolation (singly phosphorylated peptides) or combined in one molecule (doubly phosphorylated peptides). Different concentrations of peptides were incubated with a given concentration of SHP2; the enzymatic activity of SHP2 was measured using DiFMUP as a substrate. Second, we measured the ability of the catalytic domain of SHP2 (SHP2225–525 lacking the SH2 domains) to dephosphorylate the PD-1–derived peptides. In addition, in these assays, a given concentration of protein was incubated with different concentrations of peptides. To measure dephosphorylation, we used a commercial coupled enzymatic assay, in which the phosphate released from the peptide is used as a cofactor by a second enzyme present in the reaction mixture. All enzymatic measurements were carried out in three technical replicates and three independent experiments.
SHP2 phosphatase activity tests in the presence of peptide activators
SHP2 was diluted to 0.5 nM and treated with varying concentrations of either monophosphorylated ITIM peptide (10 nM to 150 μM), ITSM (10 nM to 150 μM), diphosphorylated peptide ITIM-[dPEG4]2-ITSM (240 pM to 200 μM), or ITSM-[dPEG4]2-ITSM (25 pM to 200 μM), in the following buffer conditions: 50 mM Hepes (pH 7.6), 150 mM NaCl, 1 mM EDTA, 5 mM DTT, and 0.05% Tween. The protein was incubated with the peptide at 25°C for 20 min, and then phosphatase activity was measured monitoring the dephosphorylation of DiFMUP (200 μM, excitation at 358 nm and emission at 452 nm) on a Tecan Infinite M1000 PRO plate reader. For each peptide concentration, a control reaction without enzyme was measured and subtracted as blank. The initial velocity data were fit to a sigmoidal dose-response curve (variable slope) by means of the GraphPad software
| (1) |
where V represents initial enzyme velocity, Vbasal is the basal velocity, Vmax is the maximal velocity, [Pep] is the concentration of the peptide activator, EC50 is the peptide concentration that yields a response equal to 50% of the difference between Vbasal and Vmax, and the HillSlope describes the steepness of the curve.
EnzCheck assay
The commercially available EnzCheck phosphate assay kit (Thermo Fisher Scientific) was used to measure the release of phosphate from the peptides under investigation. The kit was used according to the manufacturer’s instructions. The PTP domain of SHP2 (SHP2224–541) was incubated at 50 nM in the reaction mixture in 96-well plates with different concentrations of phosphopeptides for 30 min. The assay was conducted at 25°C in a BioTek Synergy H1 plate reader. The release of phosphate from the phosphopeptides was monitored measuring absorbance at 360 nm over time.
Cellular experiments
Experimental design
The aim of the cellular studies was to investigate whether both ITIM and ITSM are required for full activation of SHP2. Suppression of T cell activation was analyzed in lentivirally transduced stable PD-1 WT, PD-1 ITIMmut, and PD-1 ITSMmut T cell lines after stimulation with superantigen-loaded Raji cells. Expression of the T cell activation marker CD69 was analyzed via flow cytometry, IL-2 secretion was measured via ELISA, and SHP2 recruitment to PD-1 was analyzed using immunoprecipitation and Western blot assays.
Lentiviral expression constructs
Human WT PD-1 (protein sequence in UniProt Q15116) and PD-1 mutants with Y233F substitution in the ITIM (ITIMmut) and Y248F substitution in the ITSM (ITSMmut) were fused at the C terminus with the Flag tag peptide sequence DYKDDDDK. DNA sequences of the Flag-tagged PD-1 mutants and WT human PD-L1 (protein sequence in UniProt Q9NZQ7) were codon-optimized for expression in humans, assembled as gBlocks (Integrated DNA Technologies), and cloned into the pCDH-EF1-MCS-T2A-copGFP expression plasmid (System Biosciences) using the Xba I and Eco RI restriction sites. PD-1 and PD-L1 expression was coupled with a GFP reporter gene (copGFP) via a 2A-like sequence (T2A).
Lentiviral vector production
Human embryonic kidney–293T cells were transfected with the respective constructs cloned into pCDH-EF1-MCS-T2A-copGFP (System Biosciences) and the packaging plasmids pMD2.G and pCMVR8.74 (Addgene) using the transfection reagent polyethylenimine. The virus-containing supernatant was used for transduction of the immune cell lines.
Cell lines
To generate Jurkat cell lines expressing the Flag-tagged PD-1 mutants, we sorted Jurkat cells to enrich for cells, which were both negative for PD-1 and positive for CD3 by costaining with anti–PD-1 and anti-CD3ε antibodies. PD-1−/CD3+ cells were transduced with lentivirus encoding the Flag-tagged PD-1 mutants. To obtain similar PD-1 expression levels, we sorted the transduced cells to enrich for cells with similar GFP expression. To generate Raji cells expressing PD-L1, cells were transduced with lentivirus encoding WT human PD-L1 followed by selection of the highest GFP-expressing cells. Jurkat cells were purchased from the American Type Culture Collection, and Raji cell lines were purchased from the German Collection of Microorganisms and Cell Cultures GmbH (DSMZ, Braunschweig, Germany) and maintained in culture as recommended by the manufacturer.
PD-1, PD-L1, and CD3 surface expression
PD-1 and PD-L1 expression was detected with biotin-coupled anti–PD-1 (clone EH12.2H7, BioLegend) and anti-PD-L1 (clone 29E.2A3, BioLegend) antibody staining followed by streptavidin-allophycocyanin (APC) (BioLegend) staining. CD3 surface expression was detected by APC-conjugated anti-CD3ε antibody (clone OKT3, BioLegend). Cells were analyzed by flow cytometry using a Gallios flow cytometer (Beckman Coulter). Cell sorting was performed using a BD FACS Aria III cell sorter (Becton Dickinson). Data were analyzed using the software FlowJo v10 (TreeStar Inc.).
Recruitment of SHP2 in an intact-cell costimulation experiment
PD-L1–expressing Raji cells were preincubated with superantigen SEE (30 ng/ml) (Toxin Technology) for 30 min at 37°C in serum-free media in culture dishes coated with bovine serum albumin (BSA). Jurkat cells were preincubated in serum-free media for 3 hours at 37°C in BSA-coated culture dishes. Jurkat and Raji cells were resuspended in complete growth media supplemented with 1% fetal bovine serum, transferred to 1.5-ml microfuge tubes in a 1:1 ratio, mixed, and immediately centrifuged for 10 s at room temperature to initiate cell-cell contact followed by incubation at 37°C. At the indicated time points, the cell pellets were snap-frozen in liquid nitrogen after the supernatant was aspirated. The frozen cell pellets were stored at −20°C. To obtain unstimulated samples for the immunopurification experiments, Jurkat and Raji cells were treated in the same way as the stimulated samples but were not mixed in media and combined later during cell lysis. The frozen cell mixtures consisting of 2 × 107 Jurkat and 2 × 107 Raji cells were lysed in 500 μl of lysis buffer containing 20 mM tris-HCl (pH 8.0), 137 mM NaCl, 2 mM EDTA, 10% glycerol, 1× protease inhibitor cocktail, 1 mM phenylmethylsulfonyl fluoride, 5 mM iodoacetamide, 0.5 mM sodium orthovanadate, 1 mM NaF, and 0.5% Brij 96 for 30 min at 4°C, followed by 15-min centrifugation to pellet the nuclei and insoluble material. For immunoprecipitation of the Flag-tagged PD-1 mutants, 450 μl of cleared cell lysate was incubated with 5 μl of protein G Sepharose slurry in phosphate-buffered saline (PBS) and 1 μg of anti-Flag tag antibody overnight at 4°C. After four washes, the immunoprecipitated material and aliquots of the cell lysate were separated by 10% SDS-PAGE. The separated proteins were transferred to polyvinylidene difluoride membranes by semidry transfer. After blocking with 5% milk in PBS containing 0.1% Tween 20, the membranes were incubated with antibodies against SHP2 (1:1000), PD-1 (1 μg/ml), Flag-tag (5 μg/ml), and glyceraldehyde-3-phosphate dehydrogenase (1:2500), followed by incubation with horseradish peroxidase (HRP)–conjugated secondary antibodies (1:2500). Western blot signals were recorded using an ImageQuant LAS 4000 Mini (GE Healthcare).
The following antibodies and reagents were used for biochemical analysis: mouse anti-Flag tag (clone M2, Sigma-Aldrich), rabbit anti–SHP-2 (#3752, Cell Signaling Technology), rabbit anti–PD-1 (#PA5-20350, Thermo Fisher Scientific) and HRP-coupled goat anti-mouse immunoglobulin G (IgG) (#A0168, Sigma-Aldrich), HRP-coupled donkey anti-rabbit IgG (#NA934, GE Healthcare), protein G Sepharose 4 fast flow (GE Healthcare), and the protease inhibitor cocktail from Sigma-Aldrich.
Expression of CD69
Raji cells (1 × 105) transduced with human PD-L1 were seeded per well in a V-shaped 96-well plate and preincubated in 1% fetal bovine serum starvation media with different amounts of superantigen SEE (30, 3, 0.3, 0.03, and 0 ng/ml). Jurkat cells transduced with the PD-1 mutants were stained with the cell dye eFluor670 (eBioscience) to allow for discrimination of B and T cells in flow cytometry analysis. Stained Jurkat cells (1 × 105) were mixed with SEE-loaded B cells and centrifuged to enable cell-cell contact followed by incubation at 37°C. After 5 hours, the cells were stained with pacific blue–conjugated anti-human CD69 (clone FN5O, BioLegend) and phycoerythrin-conjugated anti-human CD19 (clone HIB19, BioLegend) antibodies and analyzed by flow cytometry to test for up-regulation of CD69 expression in the B and T cells. Data were analyzed by the software FlowJo (TreeStar Inc.).
Secretion of IL-2
Raji cells (1 × 105) transduced with human PD-L1 were preincubated in complete growth media supplemented with 1% fetal calf serum and different amounts of superantigen SEE (30, 3, 0.3, 0.03, and 0 ng/ml) for 24 hours in V-shaped 96-wells. Relative IL-2 protein levels were analyzed by ELISA using the Human IL-2 Ready-SET-Go Kit (#88-7025-88, eBioscience) as described by the manufacturer. The absorbance at 450 nm was measured with a plate reader (Synergy BioTek). As reference wavelength, the absorbance at 570 nm was also measured to detect nonspecific absorbance of the 96-well plate material. To obtain the relative IL-2 concentrations, absorbance values at 570 nm were subtracted from those at 450 nm. Data were analyzed using the software Prism.
Statistical analysis
Results were analyzed with GraphPad Prism 6. Statistics are based on two-way analysis of variance (ANOVA) and Bonferroni multiple comparison test. Each data point represents one of three independent experiments, each performed in triplicate. The designations ns, *, ***, and **** denote P values for the measured differences of >0.05, ≤0.05, ≤0.001, and ≤0.0001, respectively (n = 3).
Supplementary Material
Acknowledgments
Funding: M.M. was supported by a fellowship from the Hannover School for Biomolecular Drug Research and was a member of the Hannover Biomedical Research School (HBRS) and the MD/PhD program “Molecular Medicine”. A.B. thanks the EMBL and Marie Curie actions EMBL interdisciplinary postdoctoral program (EIPOD) for a fellowship. This work was funded by the German Science Foundation DFG (grant CA 294/20-1, BIOSS EXC 294, and CIBSS EXC-2189—project ID 390939984). Author contributions: M.M. planned and carried out NMR experiments, structure calculations, and modeling. A.B. designed and set up the biophysical and biochemical studies and measured the activation of SHP2 by the peptides. J.W. and N.T. carried out the cell biology experiments. J.S. performed ITC experiments. J.S. and J.Kr. crystallized the complexes and solved the crystal structures. J.Ki. designed and supervised the NMR experiments. P.R. designed the biochemical experiments, supervised the laboratory work of H.J.B., carried out SHP2 activation assays, and analyzed the biochemical data. H.J.B. carried out the EnzChek assays. M.M., J.Ki., J.Kr., P.R., and J.W. wrote respective parts of the manuscript. W.W.S. designed the cell biology experiments and supervised the experiments. M.K. and T.C. designed and cosupervised the entire study. T.C. wrote the manuscript. M.K., W.W.S., and all other authors edited the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The crystallographic structures of N-SH2–ITSM and N-SH2–ITIM were deposited with the PDB with accession numbers 6ROZ and 6ROY, respectively. The NMR structure of C-SH2–ITSM was deposited with the PDB with accession number 6R5G. All data needed to evaluate the conclusions are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/5/eaay4458/DC1
Fig. S1. Autoinhibited conformation of SHP2.
Fig. S2. Interaction of the N-SH2 and C-SH2 domains with ITIM and ITSM.
Fig. S3. Lack of phosphorylation on ITIM and ITSM severely impairs their ability to bind both N-SH2 and C-SH2.
Fig. S4. NMR structure of the C-SH2–ITSM complex and overlay of unbound and phosphopeptide-bound structures of the SH2 domains.
Fig. S5. Overlay of the 1H-15N spectra of SHP21–220, SHP21–105, and SHP2110–220 in the bound and unbound forms.
Fig. S6. Small-angle scattering analysis of SHP21–220 and SHP21–525-C459S in the presence of the bidentate peptide.
Fig. S7. Superposition of the structures of N-SH2–ITIM and C-SH2–ITSM on the corresponding domains of SHP1 in the open state (PDB entry 3PS5) and SHP2-E76K in the open state (PDB entry 6CRF).
Fig. S8. CD69 expression levels of Raji B cells are not affected by coculture with Jurkat T cells.
Table S1. Crystallographic data collection and refinement statistics.
Table S2. NMR statistics for the structure of the C-SH2–ITSM complex.
REFERENCES AND NOTES
- 1.Dance M., Montagner A., Salles J.-P., Yart A., Raynal P., The molecular functions of Shp2 in the Ras/mitogen-activated protein kinase (ERK1/2) pathway. Cell. Signal. 20, 453–459 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Hof P., Pluskey S., Dhe-Paganon S., Eck M. J., Shoelson S. E., Crystal structure of the tyrosine phosphatase SHP-2. Cell 92, 441–450 (1998). [DOI] [PubMed] [Google Scholar]
- 3.Neel B. G., Gu H., Pao L., The 'Shp'ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem. Sci. 28, 284–293 (2003). [DOI] [PubMed] [Google Scholar]
- 4.Pluskey S., Wandless T. J., Walsch C. T., Schoelson S. E., Potent stimulation of SH-PTP2 phosphatase activity by simultaneous occupancy of both SH2 domains. J. Biol. Chem. 270, 2897–2900 (1995). [DOI] [PubMed] [Google Scholar]
- 5.Li S., Hsu D. D., Wang H., Feng G.-S., Dual faces of SH2-containing protein-tyrosine phosphatase Shp2/PTPN11 in tumorigenesis. Front. Med. 6, 275–279 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Kontaridis M. I., Swanson K. D., David F. S., Barford D., Neel B. G., PTPN11 (Shp2) mutations in LEOPARD syndrome have dominant negative, not activating, effects. J. Biol. Chem. 281, 6785–6792 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Tartaglia M., Mehler E. L., Goldberg R., Zampino G., Brunner H. G., Kremer H., van der Burgt I., Crosby A. H., Ion A., Jeffery S., Kalidas K., Patton M. A., Kucherlapati R. S., Gelb B. D., Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat. Genet. 29, 465–468 (2001). [DOI] [PubMed] [Google Scholar]
- 8.Tartaglia M., Niemeyer C. M., Fragale A., Song X., Buechner J., Jung A., Hählen K., Hasle H., Licht J. D., Gelb B. D., Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat. Genet. 34, 148–150 (2003). [DOI] [PubMed] [Google Scholar]
- 9.Aceto N., Sausgruber N., Brinkhaus H., Gaidatzis D., Martiny-Baron G., Mazzarol G., Confalonieri S., Quarto M., Hu G., Balwierz P. J., Pachkov M., Elledge S. J., van Nimwegen E., Stadler M. B., Bentires-Alj M., Tyrosine phosphatase SHP2 promotes breast cancer progression and maintains tumor-initiating cells via activation of key transcription factors and a positive feedback signaling loop. Nat. Med. 18, 529–537 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Hatakeyama M., Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat. Rev. Cancer 4, 688–694 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Butterworth S., Overduin M., Barr A. J., Targeting protein tyrosine phosphatase SHP2 for therapeutic intervention. Future Med. Chem. 6, 1423–1437 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Scott L. M., Lawrence H. R., Sebti S. M., Lawrence N. J., Wu J., Targeting protein tyrosine phosphatases for anticancer drug discovery. Curr. Pharm. Des. 16, 1843–1862 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mullard A., Phosphatases start shedding their stigma of undruggability. Nat. Rev. Drug Discov. 17, 847–849 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Vang T., Miletic A. V., Arimura Y., Tautz L., Rickert R. C., Mustelin T., Protein tyrosine phosphatases in autoimmunity. Annu. Rev. Immunol. 26, 29–55 (2008). [DOI] [PubMed] [Google Scholar]
- 15.Okazaki T., Maeda A., Nishimura H., Kurosaki T., Honjo T., PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc. Natl. Acad. Sci. U.S.A. 98, 13866–13871 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hui E., Cheung J., Zhu J., Su X., Taylor M. J., Wallweber H. A., Sasmal D. K., Huang J., Kim J. M., Mellman I., Vale R. D., T cell costimulatory receptor CD28 is a primary target for PD-1–mediated inhibition. Science 355, 1428–1433 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peled M., Tocheva A. S., Sandigursky S., Nayak S., Philips E. A., Nichols K. E., Strazza M., Azoulay-Alfaguter I., Askenazi M., Neel B. G., Pelzek A. J., Ueberheide B., Mor A., Affinity purification mass spectrometry analysis of PD-1 uncovers SAP as a new checkpoint inhibitor. Proc. Natl. Acad. Sci. U.S.A. 115, E468–E477 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yokosuka T., Takamatsu M., Kobayashi-Imanishi W., Hashimoto-Tane A., Azuma M., Saito T., Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med. 209, 1201–1217 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chemnitz J. M., Parry R. V., Nichols K. E., June C. H., Riley J. L., SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J. Immunol. 173, 945–954 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Dömling A., Holak T. A., Programmed death-1: Therapeutic success after more than 100 years of cancer immunotherapy. Angew. Chem. Int. Ed. 53, 2286–2288 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brahmer J. R., Hammers H., Lipson E. J., Nivolumab: Targeting PD-1 to bolster antitumor immunity. Future Oncol. 11, 1307–1326 (2015). [DOI] [PubMed] [Google Scholar]
- 22.P. Gopalasingam, L. Quill, M. Jeeves, M. Overduin, in SH Domains: Structure, Mechanisms and Applications, N. Kurochkina, Ed. (Springer International Publishing, 2015), chap. 8, pp. 159–185. [Google Scholar]
- 23.Barford D., Neel B. G., Revealing mechanisms for SH2 domain mediated regulation of the protein tyrosine phosphatase SHP-2. Structure 6, 249–254 (1998). [DOI] [PubMed] [Google Scholar]
- 24.Chen Y.-N. P., LaMarche M. J., Chan H. M., Fekkes P., Garcia-Fortanet J., Acker M. G., Antonakos B., Chen C. H.-T., Chen Z., Cooke V. G., Dobson J. R., Deng Z., Fei F., Firestone B., Fodor M., Fridrich C., Gao H., Grunenfelder D., Hao H. X., Jacob J., Ho S., Hsiao K., Kang Z. B., Karki R., Kato M., Larrow J., La Bonte L. R., Lenoir F., Liu G., Liu S., Majumdar D., Meyer M. J., Palermo M., Perez L., Pu M., Price E., Quinn C., Shakya S., Shultz M. D., Slisz J., Venkatesan K., Wang P., Warmuth M., Williams S., Yang G., Yuan J., Zhang J.-H., Zhu P., Ramsey T., Keen N. J., Sellers W. R., Stams T., Fortin P. D., Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 535, 148–152 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Agazie Y. M., Hayman M. J., Molecular mechanism for a role of SHP2 in epidermal growth factor receptor signaling. Mol. Cell. Biol. 23, 7875–7886 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.LaRochelle J. R., Fodor M., Vemulapalli V., Mohseni M., Wang P., Stams T., LaMarche M. J., Chopra R., Acker M. G., Blacklow S. C., Structural reorganization of SHP2 by oncogenic mutations and implications for oncoprotein resistance to allosteric inhibition. Nat. Commun. 9, 4508 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pádua R. A. P., Sun Y., Marko I., Pitsawong W., Stiller J. B., Otten R., Kern D., Mechanism of activating mutations and allosteric drug inhibition of the phosphatase SHP2. Nat. Commun. 9, 4507 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang W., Liu L., Song X., Mo Y., Komma C., Bellamy H. D., Zhao Z. J., Zhou G. W., Crystal structure of human protein tyrosine phosphatase SHP-1 in the open conformation. J. Cell. Biochem. 112, 2062–2071 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gil D., Schamel W. W. A., Montoya M. I. A., Sánchez-Madrid F., Alarcón B., Recruitment of Nck by CD3ϵ reveals a ligand-induced conformational change essential for T cell receptor signaling and synapse formation. Cell 109, 901–912 (2002). [DOI] [PubMed] [Google Scholar]
- 30.Shen X., Zhao B., Efficacy of PD-1 or PD-L1 inhibitors and PD-L1 expression status in cancer: Meta-analysis. BMJ 362, k3529 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rota G., Niogret C., Dang A. T., Barros C. R., Fonta N. P., Alfei F., Morgado L., Zehn D., Birchmeier W., Vivier E., Guarda G., Shp-2 is dispensable for establishing T cell exhaustion and for PD-1 signaling in vivo. Cell Rep. 23, 39–49 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Kaneko T., Huang H., Cao X., Li X., Li C., Voss C., Sidhu S. S., Li S. S. C., Superbinder SH2 domains act as antagonists of cell signaling. Sci. Signal. 5, ra68 (2012). [DOI] [PubMed] [Google Scholar]
- 33.Sun J., Lu S., Ouyang M., Lin L.-J., Zhuo Y., Liu B., Chien S., Neel B. G., Wang Y., Antagonism between binding site affinity and conformational dynamics tunes alternative cis-interactions within Shp2. Nat. Commun. 4, 2037 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sugimoto S., Wandless T. J., Shoelson S. E., Neel B. G., Walsh C. T., Activation of the SH2-containing protein tyrosine phosphatase, SH-PTP2, by phosphotyrosine-containing peptides derived from insulin receptor substrate-1. J. Biol. Chem. 269, 13614–13622 (1994). [PubMed] [Google Scholar]
- 35.Zhao M., Guo W., Wu Y., Yang C., Zhong L., Deng G., Zhu Y., Liu W., Gu Y., Lu Y., Kong L., Meng X., Xu Q., Sun Y., SHP2 inhibition triggers anti-tumor immunity and synergizes with PD-1 blockade. Acta Pharm. Sin. B 9, 304–315 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sklenar V., Piotto M., Leppik R., Saudek V., Gradient-tailored water suppression for 1H-15N HSQC experiments optimized to retain full sensitivity. J. Magn. Reson. A 102, 241–245 (1993). [Google Scholar]
- 37.Pervushin K., Riek R., Wider G., Wüthrich K., Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc. Natl. Acad. Sci. U.S.A. 94, 12366–12371 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tugarinov V., Hwang P. M., Ollerenshaw J. E., Kay L. E., Cross-correlated relaxation enhanced 1H−13C NMR spectroscopy of methyl groups in very high molecular weight proteins and protein complexes. J. Am. Chem. Soc. 125, 10420–10424 (2003). [DOI] [PubMed] [Google Scholar]
- 39.Frueh D. P., Practical aspects of NMR signal assignment in larger and challenging proteins. Prog. Nucl. Magn. Reson. Spectrosc. 78, 47–75 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yamazaki T., Forman-Kay J. D., Kay L. E., Two-dimensional NMR experiments for correlating carbon-13.beta. and proton.delta./.epsilon. chemical shifts of aromatic residues in 13C-labeled proteins via scalar couplings. J. Am. Chem. Soc. 115, 11054–11055 (1993). [Google Scholar]
- 41.Salzmann M., Pervushin K., Wider G., Senn H., Wüthrich K., TROSY in triple-resonance experiments: New perspectives for sequential NMR assignment of large proteins. Proc. Natl. Acad. Sci. U.S.A. 95, 13585–13590 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zwahlen C., Legault P., Vincent S. J. F., Greenblatt J., Konrat R., Kay L. E., Methods for measurement of intermolecular NOEs by multinuclear NMR spectroscopy: Application to a bacteriophage λ N-peptide/boxB RNA complex. J. Am. Chem. Soc. 119, 6711–6721 (1997). [Google Scholar]
- 43.Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A., NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 (1995). [DOI] [PubMed] [Google Scholar]
- 44.Hyberts S. G., Milbradt A. G., Wagner A. B., Arthanari H., Wagner G., Application of iterative soft thresholding for fast reconstruction of NMR data non-uniformly sampled with multidimensional Poisson Gap scheduling. J. Biomol. NMR 52, 315–327 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vranken W. F., Boucher W., Stevens T. J., Fogh R. H., Pajon A., Llinas M., Ulrich E. L., Markley J. L., Ionides J., Laue E. D., The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins 59, 687–696 (2005). [DOI] [PubMed] [Google Scholar]
- 46.Rieping W., Habeck M., Bardiaux B., Bernard A., Malliavin T. E., Nilges M., ARIA2: Automated NOE assignment and data integration in NMR structure calculation. Bioinformatics 23, 381–382 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Kabsch W., XDS. Acta Crystallogr. 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S., Overview of the CCP4 suite and current developments. Acta Crystallogr. D67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A., Moriarty N., Oeffner R., Read R., Richardson D., Richardson J., Terwilliger T. C., Zwart P. H., PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. D66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J., Phaser crystallographic software. J. Appl. Cryst. 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A., REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D67, 355–367 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Emsley P., Lohkamp B., Scott W. G., Cowtan K., Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/5/eaay4458/DC1
Fig. S1. Autoinhibited conformation of SHP2.
Fig. S2. Interaction of the N-SH2 and C-SH2 domains with ITIM and ITSM.
Fig. S3. Lack of phosphorylation on ITIM and ITSM severely impairs their ability to bind both N-SH2 and C-SH2.
Fig. S4. NMR structure of the C-SH2–ITSM complex and overlay of unbound and phosphopeptide-bound structures of the SH2 domains.
Fig. S5. Overlay of the 1H-15N spectra of SHP21–220, SHP21–105, and SHP2110–220 in the bound and unbound forms.
Fig. S6. Small-angle scattering analysis of SHP21–220 and SHP21–525-C459S in the presence of the bidentate peptide.
Fig. S7. Superposition of the structures of N-SH2–ITIM and C-SH2–ITSM on the corresponding domains of SHP1 in the open state (PDB entry 3PS5) and SHP2-E76K in the open state (PDB entry 6CRF).
Fig. S8. CD69 expression levels of Raji B cells are not affected by coculture with Jurkat T cells.
Table S1. Crystallographic data collection and refinement statistics.
Table S2. NMR statistics for the structure of the C-SH2–ITSM complex.