

Abstract
Fibroblast growth factor (FGF)13, a nonsecreted, X-linked, FGF homologous factor, is differentially expressed in adipocytes in response to diet, yet Fgf13’s role in metabolism has not been explored. Heterozygous Fgf13 knockouts fed normal chow and housed at 22°C showed hyperactivity accompanying reduced core temperature and obesity when housed at 30°C. Those heterozygous knockouts showed defects in thermogenesis even at 30°C and an inability to protect core temperature. Surprisingly, we detected trivial FGF13 in adipose of wild-type mice fed normal chow and no obesity in adipose-specific heterozygous knockouts housed at 30°C, and we detected an intact brown fat response through exogenous β3 agonist stimulation, suggesting a defect in sympathetic drive to brown adipose tissue. In contrast, hypothalamic-specific ablation of Fgf13 recapitulated weight gain at 30°C. Norepinephrine turnover in brown fat was reduced at both housing temperatures. Thus, our data suggest that impaired CNS regulation of sympathetic activation of brown fat underlies obesity and thermogenesis in Fgf13 heterozygous knockouts fed normal chow.—Sinden, D. S., Holman, C. D., Bare, C. J., Sun, X., Gade, A. R., Cohen, D. E., Pitt, G. S. Knockout of the X-linked Fgf13 in the hypothalamic paraventricular nucleus impairs sympathetic output to brown fat and causes obesity.
Keywords: brown adipose tissue, thermogenesis, fibroblast, growth factor homologous factor, FHF2
Alterations in body weight arise from disequilibrium between energy intake and energy expenditure (EE). Dysfunction of systems that regulate energy intake, satiety, and feeding behavior, such as leptin signaling, have dramatic consequences on animal body weight (1, 2). These systems involve complex crosstalk between the CNS and adipose tissues (3). Abnormal regulation of EE can have equally impressive effects on body weight (4–6). One significant source of EE above basal metabolic rate in rodent models is nonshivering thermogenesis, which occurs in brown adipose tissue (BAT) and is utilized to maintain body temperature in subthermoneutral environments (7).
Fibroblast growth factor (FGF) homologous factors (FHFs), also known as intracellular FGFs, are a subset of FGFs encoded by 4 genes (Fgf11–Fgf14). FHFs lack a recognizable secretory signal sequence, are not secreted from cells, and, unlike canonical FGFs, do not activate FGF receptors (8). Rather, their best characterized function is as modulators of voltage-gated Na+ channels (NaVs) (9), and mutations that affect their NaV modulatory ability have been identified in cardiac arrhythmias and neurologic diseases such as Brugada syndrome, autosomal dominant cerebral ataxia, and epilepsy (10–12). Beyond regulation of NaVs, FGF13, encoded by Fgf13 on the X chromosome, can regulate other voltage-gated ion channels (13, 14) and appears to affect a number of other cellular processes and contribute to physiology or disease states, including cancer (15), hypertrichosis (16), smooth and skeletal muscle cell development (17, 18), microtubule stabilization in developing neurons (19), and protection from mechanical stress in cardiomyocytes by regulation of caveolae (20). These observations suggest that the full complement of FGF13 functions is not fully defined. Moreover, multiple unbiased screens reported differential Fgf13 expression within adipose tissues in response to alterations in diet and in different obesogenic genetic backgrounds, as well as association of FGF13 with obesity in humans (21–26), but the roles of Fgf13 in metabolism and obesity have not been investigated directly.
We exploited a global Fgf13 knockout model to investigate the myriad roles attributed to Fgf13. As previously reported in Puranam et al. (12), male hemizygous Fgf13 knockout (Fgf13−/Y) mice on a C57BL/6J background were embryonically lethal—which we confirmed—so we focused on heterozygous Fgf13 knockout females and observed a striking metabolic phenotype, manifesting as a reduced ability to defend core body temperature and subsequent compensatory hyperactivity when housed at 22°C, but as obesity when mice were housed at 30°C and fed normal chow. Thermogenic activity of BAT in the heterozygous knockout animals was reduced, although adaptive thermogenesis could be directly activated in these animals through exogenous β3 adrenergic stimulation. Surprisingly, we detected minimal Fgf13 expression on a normal chow diet, and an adipocyte-specific Fgf13 heterozygous knockout failed to recapitulate the metabolic defects observed in the global Fgf13 heterozygous knockout. Instead, we found decreased hypothalamic regulation of sympathetic output to BAT and a consequent defect in thermogenesis, with no change in overall food intake. Because neurons in the hypothalamus are extensively interconnected and circuits controlling thermogenesis and feeding behavior are often linked, the isolated defect in thermogenesis disconnected from feeding behavior is unusual. Our findings establish a novel role for Fgf13 in the regulation of metabolism and thermogenesis.
MATERIALS AND METHODS
Animals
Animals were handled according to the Guide for the Care and Use of Laboratory Animals (National Institute of Health, Bethesda, MD, USA) (27). This study was approved by Weill Cornell Medical Center Institutional Animal Care and Use Committee (Protocol 2016-0042). Genetically modified mice were maintained on C57BL/6J (000664; The Jackson Laboratory, Bar Harbor, ME, USA) or 129S2/SvPasCrl (previously designated 129/SvPas) (Charles River Laboratories, Wilmington, MA, USA) genetic backgrounds or on a first filial (F1) generation intercross background derived by breeding C57BL/6J mice with 129S2/SvPasCrl mice. All mice were maintained on a standard rodent chow diet (PicoLab Rodent Diet 20; 5053; LabDiet, St. Louis, MO, USA) with a 12-h light/dark cycle (light cycle, 6:00 am to 6:00 pm; dark cycle, 6:00 pm to 6:00 am). Female mice with floxed Fgf13 alleles (Fgf13fl/fl) were generated as previously described in refs. 14, 20 by flanking exon 3 of the mouse Fgf13 gene with 2loxP (locus of X-over P1) sites. To generate global heterozygous knockout females, female Fgf13fl/fl mice were crossed with male DEAD-box polypeptide-4 promoter–driven Cre recombinant (Ddx4-Cre+; specifically, FVBTg(Ddx4-Cre)1Dcas/J) mice to produce an F1 generation with germline-specific deletion of the Fgf13 gene. In this F1 generation, incomplete recombination was observed in somatic cells, as was previously reported in Gallardo et al. (28). F1 generation Fgf13+/fl;Ddx4-Cre+ females were crossed with wild-type (WT) males to produce Fgf13+/− heterozygous females and Fgf13+/+ littermate controls. Although Fgf13−/Y males were genetically possible from this cross, we did not observe any viable hemizygous male offspring. For tissue-specific Cre studies, Fgf13+/fl females were crossed with male adiponectin promoter–driven Cre recombinant [Adipoq-Cre+; specifically, B6.FVB-Tg(Adipoq-Cre)1Evdr/J, 028020; The Jackson Laboratory] or nestin promoter–driven Cre recombinant [Nes-Cre+; specifically, B6.Cg-Tg(Nes-Cre)1Kln/J, 003771; The Jackson Laboratory] mice to produce Adipoq-Cre+;Fgf13+/fl or Nes-Cre+;Fgf13+/fl tissue-specific heterozygous female knockouts and Adipoq-Cre+;Fgf13+/+ or Nes-Cre+;Fgf13+/+ littermate controls. Mice were housed at either 22 or 30°C in an environmentally controlled cabinet.
Temperature recordings
Mouse core body temperatures were recorded by rectal thermometer at 6:00 pm, the beginning of the dark cycle. For cold challenge studies, mice raised for at least 8 wk at 30°C were transitioned to 22°C for 1 h, followed by transition to 4°C for 1 h, and had core body temperature recorded by rectal probe every 15 min until the end of the study. Mice were then returned to 30°C.
Metabolic measurement and data analysis
Metabolic studies were performed as previously described in refs. 29–31. Metabolic measurements were acquired through indirect calorimetry using the Promethion Metabolic Screening System (Sable Systems International, Las Vegas, NV, USA). Metabolic cages were contained within 2 temperature-controlled cabinets (DB034 Laboratory Incubator; Darwin Chambers, St. Louis, MO, USA) at stable ambient temperatures of either 22.0 ± 1.0°C or 30.0 ± 1.0°C with a 12-h light/dark cycle. Mice were single housed for the duration of the experiment and had ad libitum access to food and water. The mice were acclimated to the metabolic cages for 48 h, followed by 24 h of data recording. Food intake, water intake, and body mass were continuously monitored gravimetrically, and ambulatory activity was measured with an XY beam break array. Respirometry data, including Vo2 and Vco2, were acquired in 5 min intervals. EE was calculated using the Weir equation (32) as implemented in the Macro software provided with the Promethion system (ExpeData 1.9.14; UMC-10.1.8.1-mouse; Sable Systems International). Body composition including fat and lean mass was measured using NMR spectroscopy in an EchoMRI 3-in-1 Body Composition Analyzer (EchoMRI, Houston, TX, USA).
The raw Macro data were averaged hourly, with 12 data points per mouse comprising each hourly average. For EE, the total (6:00 am to 6:00 am) was summed and normalized via an ANCOVA using the VassarStats ANCOVA function (www.vassarstats.net) for differences in lean body mass (LBM) as previously described in Tschöp et al. (33). Food intake was converted from grams of pure food consumed into kcal of metabolizable energy (3.03 kcal/g) per the manufacturer’s information and also normalized for differences in LBM via ANCOVA. Outliers with P < 0.05 were removed via Grubbs’ test or removed if a known mechanical malfunction was present.
We observed significant weight loss in the Fgf13+/− mice during the metabolic cage experiment (see Supplemental Fig. 4), which necessitated an adjustment of the measured EE to account for this weight loss state. To accomplish this, we took the average of the fat and LBM change of both groups during the experiment. We converted this fat and lean mass change to kilocalories by multiplying by 9 and 4 kcal/g, respectively, and summed the resulting fat and lean caloric change to determine the total kilocalorie change over the course of the 4-d experiment. We then divided the total caloric change by 4 to get a daily change in kilocalories and added this to the ANCOVA corrected 24-h average EE, resulting in a significantly lower average EE adjusted for the metabolic cage weight loss in the Fgf13+/− mice. This weight loss adjustment was also performed on raw EE values and then normalized to LBM via ANCOVA after the adjustment had been incorporated, and this calculation maintained the significant reduction in 30°C Fgf13+/− mice EE (unpublished results).
Because the weight loss observed during single housing warranted this unconventional correction, we further explored the validity of corrected average EE. We compared theFgf13+/− mice’s daily EE (previously corrected by ANCOVA for LBM, with that of WT mice and converted the deficit to grams by dividing by the caloric content of fat to obtain a predicted fat gain per day. We used the caloric content of fat because of the observations that weight gain of the Fgf13+/− mice was almost all fat mass and that there was no difference in the lean mass of the WT and Fgf13+/− mice and because it has been shown that from 13 to 17 wk, the majority of weight gain in female C57BL/6J mice is fat mass (34). Using these parameters, we extrapolated the change in weight to 16 wk of age by multiplying the daily fat gain by the number of days. Recognizing that weight gain is not a linear process, this model demonstrates validity of our calculation methods.
Morphology and histology
Excised intrascapular BAT (iBAT), gonadal white adipose tissue (gWAT), and inguinal white adipose tissue (iWAT) depots were washed with PBS and photographed immediately after dissection for gross morphologic comparison. Samples were then prepared for histology as previously described in Berry et al. (35). Briefly, a portion of each depot was fixed in 4% paraformaldehyde for 12 h at 4°C and dehydrated in a series of ethanol washes. Samples were then cleared in xylene and mounted in paraffin. Sections of 5 and 10 µm thickness were cut and stained with hematoxylin and eosin to analyze cellular morphology. Samples from different groups were processed in parallel.
Real-time quantitative PCR
After euthanasia, the brain and a sample of BAT were immersed in RNAlater (AM7020; Thermo Fisher Scientific, Waltham, MA, USA) and frozen at −80°C. Before isolating RNA, the hypothalamus was dissected from brain samples, and RNA extraction was performed on BAT and dissected hypothalamus using the RNeasy Plus Mini Kit (Qiagen, Germantown, MD, USA) according to the manufacturer’s instructions. Reverse transcription was performed using iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA). Real-time PCR was performed in triplicate for each sample with a QuantStudio 3 Machine (Thermo Fisher Scientific) using Sybr Green detection chemistry. Relative quantification was performed using the ΔΔCt method after determining the Ct values for the reference (glyceraldehyde 3-phosphate dehydrogenase for brain, actin for BAT) and target genes. See Supplemental Table S1 for a list of primers used.
Western blot
Tissues were immersed in ice cold RIPA buffer supplemented with Halt protease and phosphatase inhibitor cocktails (Thermo Fisher Scientific) and placed in a manual Dounce tissue homogenizer, in which they were homogenized for 1 min on ice. The homogenate was then centrifuged at 21,000 g at 4°C. Supernatant was collected, and protein concentration was determined by bicinchoninic acid assay. Equivalent amounts of protein from each tissue were subjected to SDS-PAGE and Western blot analysis. Rabbit anti-FGF13 antibody was generated as previously described in ref. 36. Mouse anti–glyceraldehyde 3-phosphate dehydrogenase was purchased from Thermo Fisher Scientific (MA5-15738) and used as a reference marker.
Intracranial injection of adeno-associated virus
The following viral vectors were used in this study: rAAV8/hsvn-EGFP (5.6 × 1012 vg/ml) and rAAV8/hsvn-GFP-Cre (6.5 × 1012 vg/ml). Both vectors were purchased from the University of North Carolina at Chapel Hill Vector Core (Chapel Hill, NC, USA). Female Fgf13fl/fl mice at least 8 wk of age were anesthetized with isoflurane and placed into a stereotaxic apparatus (David Kopf Instruments, Tujunga, CA, USA). The hair on the scalp was removed, and the skull was exposed via a small incision. Bregma was anatomically located, and 2 small holes were drilled into skull for virus injection. A 33-gauge Hamilton syringe loaded with virus was lowered to the following coordinates for bilateral injection in the paraventricular nucleus of the hypothalamus (PVH): bregma −0.86 mm; midline ±0.22 mm; dorsal surface −5.1 mm. Coronal sections used for imaging and verification of green fluorescent protein (GFP) expression were collected at bregma −0.86 mm. Seventy-five nanoliters of virus were injected at each injection site at a rate of 25 nl/min. The injection needle was left at site for 2 min before slowly retracting. Skull drill sites were sealed with Vetbond (3M, Maplewood, MN, USA), and the scalp was sutured closed. Metacam (Boehringer Ingelheim, Ingelheim am Rhein, Germany) was administered intraperitoneally for postoperative pain. Mice weight and food intake were monitored for 30 d after injection, after which mice were euthanized and perfusion fixed. Brains were removed and cryosectioned, and GFP expression was evaluated by fluorescence microscopy. Any animals that did not exhibit GFP expression within the region of the PVH of the hypothalamus were not used for analysis.
Norepinephrine turnover assay
Norepinephrine (NE) turnover (NETO) was measured using the α-methyl-p-tyrosine (AMPT) method (37, 38). AMPT inhibits tyrosine hydroxylase and prevents de novo synthesis of catecholamines, including NE. After catecholamine synthesis blockade, endogenous NE declines at a rate proportional to the rate of sympathetic nerve firing. One half of animals in each genotype and housing temperature condition were killed immediately to obtain basal NE levels. The other half were injected intraperitoneally with AMPT (300 mg/kg) initially and again 2 h later with an additional dose of AMPT (150 mg/kg) to ensure catecholamine synthesis inhibition. These mice were killed 4 h after the initial injection. BAT was rapidly dissected, weighed, snap frozen in liquid nitrogen, and stored at −80°C. NE content in harvested tissues was measured using reverse-phase HPLC with electrochemical detection. Tissue was homogenized in solution containing 0.2 M perchloric acid, 1 mg/ml ascorbic acid, and dihydroxybenzylamine as an internal standard. Catecholamines were then extracted using activated alumina and eluted into perchloric acid and ascorbic acid. NE was measured with an HPLC system with electrochemical detection (Coulochem II; Thermo Fisher Scientific). Concentrations were determined against a NE standard and normalized to dihydroxybenzylamine to control for extraction efficiency.
CL 316,243 injections
Mice acclimated to thermoneutrality, aged 12–16 wk, were weighed weekly for 2 wk. They were then given daily CL 316,243 (MilliporeSigma, Burlington, MA, USA) injections intraperitoneally at a dose of 0.2 mg/kg for 14 d and weighed daily. For Fig. 3D, E, mice acclimated to thermoneutrality, aged 12–16 wk were injected with saline on d 1 at 8:00 am, 1 h into an 8-h monitoring period (7:00 am to 3:00 pm) in Promethion metabolic cages. On d 2 they were injected with CL 316,243 at a dose of 0.2 mg/kg at 8:00 am. Respirometry data and EE were collected and calculated as described above. ANCOVA LBM normalization was performed on 8-h sum EE data.
Figure 3.

Metabolic cage data reveal a profound defect in thermogenesis in Fgf13+/− mice. A) Mass change during 4-d metabolic cage experiments, derived from body composition measurements made before and after mice were placed in metabolic cages; n = 7–8 for 22°C-raised animals and 11–13 for 30°C-raised animals. B) Core body temperature of mice raised at 30°C either group housed or single housed; n = 5 for both genotypes. Statistical significance was determined by 2-way repeated measures ANOVA followed by Holm-Sidak’s test for multiple comparisons. C) ANCOVA-normalized EE adjusted for weight change during metabolic measurements (see Materials and Methods section) n = 7–8 for 22°C-raised animals and 11–13 for 30°C-raised animals. Statistical significance was determined by 2-way ANOVA followed by Holm-Sidak’s test for multiple comparisons. D) Predicted weight over 3-wk period using weight change–adjusted EE; n = 7–8 for 22°C-raised animals and 11–13 for 30°C-raised animals. E) Core body temperature of mice housed at 22 or 30°C; n = 5–8/genotype at each temperature. Statistical significance was determined by 2-way ANOVA followed by Holm-Sidak’s test for multiple comparisons. F) Core body temperature of 30°C -housed mice in response to a thermal challenge of 1 h at 22°C followed by 1 h at 4°C; n = 5/genotype. Statistical significance was determined by 2-way repeated measures ANOVA followed by Holm-Sidak’s test for multiple comparisons. G) Weight of mice injected with CL 316,243 (CL; 0.2 mg/kg/d). Mice were injected daily starting on d 14 of a 28-d monitoring period; n = 5–7/genotype. Statistical significance was determined by 2-way repeated measures ANOVA followed by Holm-Sidak’s test for multiple comparisons. H) EE averaged from 15 min after a single injection of CL 316,243 (0.2 mg/kg) or saline (equivalent volume) to the end of an 8-h monitoring period. EE was normalized to LBM via ANCOVA; n = 4/genotype. Statistical significance was determined by 2-way ANOVA followed by Holm-Sidak’s test for multiple comparisons. I) EE as a function of time over 8-h monitoring period. Injections occurred at h 1; n = 4/genotype. Het, heterozygous; NS, not significant; TBM, total body mass. *P < 0.05, **P < 0.01 for comparisons between genotype at same temperature, time point, or treatment condition.
Multiplex fluorescent in situ hybridization
Brains of adult female WT mice were fixed with infusion of 4% paraformaldehyde, followed by incubation in fixing agent overnight at room temperature. Samples were then transferred to a PBS-sucrose buffer prior to being embedded in Optimal Cutting Temperature (OCT) compound and placed in cryomolds. Brains were sectioned coronally, in 10 µm sections, and stored at −80°C before further processing. RNAscope (Advanced Cell Diagnostics, Newark, CA, USA) dual multiplex fluorescence assay was used for in situ hybridization, following the manufacturer’s protocol for fixed frozen tissues. The following RNA probes were used for multiplex in situ hybridization: Mm-Fgf13-C3 (404021-C3) to detect Fgf13, Mm-slc17a6 (319171) to detect vesicular glutamate transporter-2, and Mm-Slc32a1 (319191) to detect vesicular GABA transporter. Nuclei were stained with DAPI. Representative images from multiple experiments are shown (N = 4 WT mice, with multiple sections collected, processed, and imaged from each animal).
Statistical analyses
Data are presented as means ± sem. Statistical significance between 2 groups was assessed using a 2-tailed unpaired Student’s t test. For experiments with 2 factors, such as genotype and housing temperature, 2-way ANOVA was performed, followed by Holm-Sidak’s test for multiple comparison to assess main effects, interaction effects, and direct comparisons between groups. For experiments with repeated measures over time, 2-way repeated measures ANOVA was performed to test for main effects and interaction effects, followed by Tukey’s test for multiple comparisons to compare differences between genotypes at each time point. For all tests, statistical significance was set at P < 0.05. Analyses were conducted with Prism 7 (GraphPad Software, La Jolla, CA, USA).
RESULTS
Fgf13+/− mice exhibit hyperactivity when housed at 22°C and develop obesity when housed at thermoneutrality temperatures
We and others have characterized Fgf13 effects on cardiac function (13, 14, 20, 39–41), neuronal migration during development (19), epileptogenesis (12), and ion channel trafficking and targeting in the axon initiation segment of cultured hippocampal neurons (42), yet gene ablation studies have not been employed to investigate roles for Fgf13 in metabolism, despite multiple studies reporting differential expression in adipose tissue in response to alterations in diet and in different obesogenic genetic backgrounds (21–25). To begin to investigate metabolic functions we generated a global Fgf13 knockout mouse model on a C57BL/6J background. We crossed female Fgf13fl/fl mice (14) with male Ddx4-Cre+ mice to generate germ cell–specific deletion of Fgf13 (Supplemental Fig. S1A, B). Ddx4-Cre+;Fgf13+/fl female mice were then crossed with male WT mice to generate global germline Fgf13+/− heterozygous female offspring in the C57BL/6 background. No viable Fgf13−/Y males were produced from this cross (n >210 offspring; 50% of males would be expected to be Fgf13−/Y), preventing the generation of homozygous Fgf13−/− females. The lack of viability of Fgf13−/Y on a C57BL/6 background was previously reported in ref. 12, but homozygous Fgf13−/− females and hemizygous Fgf13−/Y males were reported viable on a 129/Sv background (40). In an attempt to obtain Fgf13−/Y males and homozygous Fgf13−/− females, we performed an intercross between our C57BL/6 Fgf13−/− females and 129/Sv males. However, we only obtained 3 hemizygous males in the F1 intercross with the 129/Sv strain (of a total of 42 male offspring, 50% of which should have been hemizygous). These males were placed in breeding, but did not produce any offspring, so we were also unable to study complete knockouts in this mixed background. Nevertheless, while attempting to generate those complete knockouts, we placed the intercrossed Fgf13+/− females and WT littermate controls in metabolic cages at standard mouse housing temperature (22° in our facility). After a 48-h acclimation period, we observed a marked increase in physical activity for the intercrossed Fgf13+/− females compared with WT littermate control females, most prominently during the dark cycle, despite no difference in EE or food intake (Supplemental Fig. S2A–D). We repeated the metabolic cage analyses on the C57BL/6J background and again observed that the Fgf13+/− females showed increased activity during the dark cycle compared with WT littermate control females. As with the intercross mice, the C57BL/6J mice showed no genotype-dependent change in food intake or EE (Supplemental Fig. S2E–H).
We hypothesized that the increased activity in the Fgf13+/− females could result from thermal stress caused by impaired nonshivering thermogenesis, so we then housed the C57BL/6J mice at 30°C (thermoneutrality) starting at 5 wk of age and followed them until they were 16 wk old. Thermoneutral housing reduces thermal stress and minimizes the metabolic demand required to defend core body temperature (4). With 30°C housing, the increased physical activity in the Fgf13+/− mice observed at 22°C was no longer evident, and the level of activity in both genotypes was comparable to that of WT mice housed at 22°C (Fig. 1A, B). During the 11 wk of 30°C housing, the Fgf13+/− mice gained significant weight compared with WT mice (Fig. 1C, D). In contrast, Fgf13+/− mice housed for 11 wk at 22°C housing did not show an increase in weight compared with WT controls. Body composition data showed that weight gain in Fgf13+/− mice housed at 30°C was entirely due to an increase in fat mass (Fig. 1E). This increase in fat mass developed despite no change in food intake (WT, 37.1 ± 1.2 g per mouse measured over a 2-wk period in 7 mice averaged over 2 feeding cages; Fgf13+/−, 38.1 ± 0.6 g in 9 mice averaged over 2 feeding cages; Student’s t test, P = 0.44). Because the Fgf13+/− mice gained weight at 30°C without increasing food intake, their calculated metabolic efficiency (conversion of consumed energy into increased body mass, as opposed to expending energy on other body functions) was significantly higher (WT, 5.9 ± 0.1 mg weight gain/kcal consumed in 7 mice averaged over 2 feeding cages. Fgf13+/−; 33.7 ± 1.7 mg weight gain/kcal consumed in 9 mice averaged over 2 feeding cages; Student’s t test, P = 0.003). The development of obesity at thermoneutrality was not strain specific, because Fgf13+/− females from the intercross between C57BLl/6 Fgf13+/− females and 129/Sv WT males also developed an increase in body mass at 30°C housing that was absent when the mice were housed at 22°C (Supplemental Fig. S3). That the Fgf13+/− mice from both backgrounds gained significantly more weight when housed at 30°C than when housed at 22°C supported our hypothesis that the Fgf13+/− mice were deficient in thermogenesis and thus exerted additional energy at 22°C to maintain core body temperature and that the increased activity observed in the Fgf13+/− at 22°C mice reflected an alternative means to expend energy and defend core body temperature. We focused only on the C57BL/6J mice for the remainder of these studies.
Figure 1.

Fgf13+/− mice develop obesity at thermoneutrality. A, B) Cumulative hourly physical activity for 24-h measurement period (A) and 12-h average physical activity during the dark cycle (B) in mice raised at 30°C. Physical activity data at 22°C shown in Supplemental Fig. S2 are displayed for comparison. n = 5–8 per genotype for 22°C measurements; 11–13/genotype for 30°C measurements. Statistical comparison was made using Student’s t test. C) Growth curves of Fgf13+/− mice and WT littermate controls raised at 22 and 30°C. Mice were placed in environmentally controlled chambers starting at 5 wk of age and monitored weekly for 11 wk; n = 5–8/genotype for 22°C measurements and 11–13/genotype for 30°C measurements. 2-way repeated measures ANOVA was used to assess main effects over time, as well as interaction effects. Comparisons between genotypes at each time point were made using Tukey’s test for multiple comparisons. Indicated significance is for comparison between heterozygous (Het) and WT mice raised at 30°C. D) Representative image of Fgf13+/− mouse and WT littermate control raised at 30°C at 16 wk of age. E) Body composition by MRI measured at 12 wk of age; n = 5–8/genotype for 22°C measurements and 11–13 per genotype for 30°C measurements. Two-way ANOVA was performed on each mass type. Comparisons between genotypes at each temperature were made using Holm-Sidak’s test for multiple comparisons. Indicated significance is for comparison with WT at the same housing temperature. *P < 0.05, **P < 0.001.
We extended our metabolic cage analyses in Fig. 2. The Fgf13+/− mice housed at 22°C showed no statistical difference in 24-h EE or food intake compared with WT littermate controls when normalized to LBM via ANCOVA (Fig. 2A–F), despite the previously noted hyperactivity during the dark cycle (Supplemental Fig. S2). Body composition analysis performed at the end of the metabolic cage studies revealed no differences in lean or total body mass, whereas fat mass trended higher in Fgf13+/− mice (Fig. 2G). Interestingly, the observed hyperactivity in the Fgf13+/− mice housed at 22°C (Supplemental Fig. S2) was not mirrored by an increase in the respiratory exchange ratio (RER) (Fig. 2H, I). A diurnal cycle with a rising RER during the dark phase was observed in both genotypes, indicating that both WT and Fgf13+/− mice utilize dietary carbohydrates in oxidative organs during active periods.
Figure 2.

Metabolic testing of Fgf13+/− mice and littermate controls housed at 22 or 30°C. Mice were placed in metabolic cages singly and allowed to acclimate, after which EE, food intake, and RER were measured. A–C) EE measured over 24 h. EE was compared and normalized to LBM via ANCOVA by first plotting those values against MRI-measured LBM (A). Data are plotted over time, with the dark cycle indicated by shading, (B) and as 24-h means with 2-way ANOVA used on 24-h sum data (C). D–F) Food intake measured over 24 h. Food intake was compared and normalized to LBM via ANCOVA by first plotting those values against MRI-measured LBM (D). Data are plotted over time, with the dark cycle indicated by shading, (E) and as 24-h means with 2-way ANOVA used on 24-h sum data (F). G) Body composition by MRI was measured at 13 wk of age, immediately after the end of the 24-h monitoring period. H, I) RER measured over 24 h. Data are plotted over time, with the dark cycle indicated by shading, (H) and as 24-h means with 2-way ANOVA used on 24-h sum data and on data for each individual mass type to assess effects of genotype, temperature, and interaction effects (I); n = 7–8/genotype for 22°C measurements and 11–13/genotype for 30°C measurements. NS, not significant. Comparisons between genotypes at each temperature were made using Holm-Sidak’s test for multiple comparisons. *P < 0.05.
Additional analyses metabolic chamber data obtained from 13-wk-old mice housed at 30°C for the 8 previous wk further supported our hypothesis that the Fgf13+/− mice had a deficit in nonshivering thermogenesis. During 30°C housing, both WT and Fgf13+/− mice showed the expected reduction in total EE compared with mice that had been housed at 22°C (Fig. 2A–C). Both genotypes also showed the expected decrease in food intake (Fig. 2D–F) at 30°C housing compared with 22°C housing. For the Fgf13+/− mice, however, there was marked variability in food intake during 30°C housing (Supplemental Fig. S4), which we again attributed to variable levels of Fgf13 expression because of random X-inactivation of the WT allele. Therefore, the regression slopes of the fitted relationships between food consumed and LBM, as shown in Fig. 2D, were statistically different (P = 0.03) between WT and Fgf13+/− mice at 30°C. Because the P value was marginal and the regression slopes EE were similar (Fig. 2A, P > 0.05) we elected to proceed first with analysis by ANCOVA (Fig. 2F) (33) and then consider appropriate adjustments (see Fig. 3).
Body composition analysis post metabolic measurement (Fig. 2G) showed a large increase in fat mass and total body mass for Fgf13+/− animals raised at 30°C, whereas LBM was decreased, which was unexpected when compared with data obtained in group-housed mice that had not been placed in metabolic cages (see Fig. 1E). Fgf13+/− mice also displayed a consistently lower RER than WT when studied at 30°C (Fig. 2J, K), indicating increased catabolism of endogenous fat stores. This lower RER was surprising given the larger fat mass of Fgf13+/− mice. Additionally, there was reduced diurnal rhythmicity in the RER of both WT and Fgf13+/− animals housed at 30°C compared with 22°C housing, which has been previously observed in ref. 43. We hypothesized that the increased utilization of fat as a fuel source was due to altered behavior of the Fgf13+/− mice during the measurement period in metabolic cages compared with standard thermoneutral housing, possibly because of the necessity of housing the animals singly in the metabolic cages.
Moreover, the striking weight gain of Fgf13+/− mice at 30°C over a 16-wk period (Fig. 1A) in the context of the metabolic cage data showing similar EE and food intake between the Fgf13+/− mice and their WT controls (Fig. 2) prompted us to reexamine the metabolic cage data. During the 4 d of acclimation and measurements, we noted that the Fgf13+/− mice raised at 30°C exhibited a significant weight loss (1.82 ± 0.55 g) when compared with WT littermates, whose body weight remained relatively constant (a gain of 0.17 ± 0.13 g) over the 4-d experiment (Fig. 3A). We hypothesized that the weight loss resulted from a both deficit in thermogenesis and the inability for the Fgf13+/− mice to huddle when singly housed. To test this directly, we measured core body temperature in single and group housed WT and Fgf13+/− mice at 30°C (Fig. 3B). Indeed, Fgf13+/− mice demonstrated a reduced core body temperature when singly housed at 30°C but not when group housed (5 mice per cage). Because the drop in core body temperature and the accompanying weight loss during single housing indicated nonhomeostatic conditions for the Fgf13+/− mice in the metabolic cages, we performed an additional calculation of EE to capture the negative energy balance. Specifically, we adjusted EE in all groups by the daily caloric equivalence of the observed weight change by using the differences in body composition data between group housing (Fig. 1C) and when housed in the metabolic cages (Fig. 2I) (see Materials and Methods for details). This adjustment yields a steady state EE calculation independent of the weight loss observed from single housing and estimates what would be observed in normal group housing conditions. This calculation showed that the weight loss in the Fgf13+/− mice at 30°C equaled an expenditure of 3.64 ± 0.85 kcal/d caused by single housing. Subtracting the respective values for each group from the EE values obtained in the metabolic cages suggests that, when Fgf13+/− mice were group housed at 30°C (as in Fig. 1) and did not lose weight, the Fgf13+/− mice expended significantly reduced energy compared with WT controls. To test the validity of these calculations, we used these adjusted EE data to calculate the predicted weight change in group-housed mice from 13 wk of age (when the metabolic cage dates were obtained) to 16 wk of age (the end of the group housing data not perturbed by metabolic cage analyses, as in Fig. 1A). Remarkably, the predicted weights at 16 wk (Fig. 3D) are in excellent agreement with observed weights (see Fig. 1A).
Fgf13+/− mice exhibit impaired ability to defend body temperature espite β3 adrenergic–responsive BAT
Motivated by altered metabolic efficiency and reduced core body temperature in singly housed 30°C Fgf13+/− mice, we further tested our hypothesis that Fgf13+/− mice showed defects in thermogenesis by examining core body temperature during 22°C group housing in mice never subjected to single housing (Fig. 3E). Indeed, Fgf13+/− mice displayed a reduced core body temperature compared with WT. In contrast, no genotype difference was observed in perpetually group-housed animals living at 30°C, recapitulating the observation made in the cohort of animals shown in Fig. 3B. We also tested response to an acute cold challenge (Fig. 3F) by placing mice acclimated to 30°C housing into a 22°C environment. Within 60 min, core body temperature dropped in Fgf13+/− mice, but not in WT mice. When both genotypes were subsequently exposed to an ambient temperature of 4°C, core body temperature fell more rapidly in Fgf13+/− mice than in WT. Together, these data support our hypothesis that thermogenesis was defective in Fgf13+/− mice.
Impaired thermogenesis could result from decreased sympathetic drive to activate BAT or from decreased BAT function. To test whether BAT function was intact, we activated BAT in mice acclimated to thermoneutrality by the daily administration of CL 316,243, a specific β3 adrenoreceptor agonist, for 14 d. CL 316,243 elicits weight loss in obese mice (but not in nonobese models), because it induces an increase in EE that exceeds any increase in food intake (44). In response to administration, we noted an abrupt arrest and reversal of weight gain exhibited by Fgf13+/− mice, with no weight changes observed in WT littermate injected mice (Fig. 3G). Additionally, Fgf13+/− mice acutely injected with CL 316,243 exhibited the same increase in EE as WT littermates (Fig. 3H, I). Together, these data suggest that the defect in thermogenic response in Fgf13+/− mice resulted from decreased sympathetic activation of BAT rather than a defect in BAT itself.
To test our hypothesis further, we examined BAT directly. Consistent with the observed weight gain at 30°C in group-housed Fgf13+/− mice, dissected iBAT and gWAT depot weights were also increased compared with WT controls (Fig. 4A). On gross examination, isolated Fgf13+/− iBAT pads were larger and lacked the characteristic brown color of typical BAT, with a paler appearance than WT pads at both housing temperatures. gWAT and iWAT depots were notably larger in 30°C raised Fgf13+/− animals (Fig. 4B) but otherwise appeared morphologically similar to WT white adipose tissue depots. Consistent with gross appearance, upon histologic analysis, BAT adipocytes from Fgf13+/− animals were larger and displayed a more unilocular appearance, regardless of housing temperature, indicating lipid accumulation and whitening of BAT, which may have been attributable to impaired cell function, inflammation, and adipocyte death (45) (Fig. 4C). gWAT and iWAT adipocytes appeared similar between genotypes at 22°C, whereas Fgf13+/− adipocytes exhibited hypertrophy at 30°C. Furthermore, although thermoneutral group housing inhibits expression of the thermogenic gene uncoupling protein 1 (Ucp1), responsible for nonshivering thermogenesis in BAT, Fgf13+/− animals exhibited impaired Ucp1 expression also at 22°C (Fig. 4D), a temperature at which nonshivering thermogenesis is activated to maintain body temperature. The gross and histologic appearance of adipose depots and the impaired Ucp1 expression in the Fgf13+/− mice at 22°C housing are all consistent with the reduced thermogenesis observed in the Fgf13+/− mice.
Figure 4.

Fgf13+/− mice exhibit increased adiposity at thermoneutrality and abnormal BAT in both 22 and 30°C housing. A) Dissected iBAT and unilateral gWAT average weight; n = 7–11/genotype at each housing temperature. Two-way ANOVA was followed by Holm-Sidak’s test for multiple comparisons. B) Representative images of dissected iBAT, gWAT, and iWAT adipose depots. C) Representative images of hematoxylin and eosin–stained adipose tissues. Scale bar, 100 µm. D) Real-time quantitative PCR gene expression of Ucp1; n = 4 /genotype at each temperature. Two-way ANOVA was followed by Holm-Sidak’s test for multiple comparisons. Het, heterozygous; NS, not significant; WAT, white adipose tissue. **P < 0.01 for comparisons between genotype at same temperature.
Loss of Fgf13 does not cause obesity through effects on adipose tissue
That the β3 adrenoreceptor agonist stimulation revealed no intrinsic defect in BAT in Fgf13+/− mice was unexpected in the context of multiple studies that reported differential expression of Fgf13 in adipose in response to high-fat feeding or starvation (21–25). We therefore investigated specifically whether Fgf13 in adipose contributed to the observed metabolic phenotypes. First, we examined Fgf13 expression. Using a validated antibody against FGF13 (20, 36), we observed abundant Fgf13 expression in the heart and the brain (Fig. 5A), where we and others (12, 19, 20, 40, 42) previously documented Fgf13 expression and function. Moreover, we saw a relative reduction in FGF13 protein in Fgf13+/− mice compared with WT in both heart and brain, confirming efficacy of the heterozygous knockout. We detected only weak signals in BAT and white adipose tissue (which were reduced to undetectable levels in the Fgf13+/− mice). The strong expression in heart and brain is consistent with several previous studies that identified heart and the nervous system as major sites of Fgf13 expression. The limited expression in adipose on standard chow is also consistent with global surveys of Fgf13 expression (46, 47). Nevertheless, because of the multiple studies detecting diet-induced differential Fgf13 expression in adipose tissue, we generated an adipose-specific heterozygous knockout using Adipoq-Cre to test directly for possible contributions of Fgf13 in adipose to the observed obesity phenotype. Because neither male Fgf13 knockouts nor female homozygous Fgf13 global knockouts were viable, we restricted our analyses here to tissue-specific Fgf13+/− knockouts for a direct comparison. When housed at thermoneutrality, Adipoq-Cre+;Fgf13+/fl mice did not show any weight gain compared with Adipoq-Cre+ mice (Fig. 5B). Thus, although Fgf13 is differentially regulated by diet in adipose tissue, we conclude that it is expressed at low levels when mice are fed normal chow, and Fgf13 in adipose does not contribute to the metabolic phenotypes under the conditions studied here. In conjunction with the data showing that BAT in Fgf13+/− mice could be exogenously activated, we instead suspected a defect in sympathetic stimulation of BAT.
Figure 5.
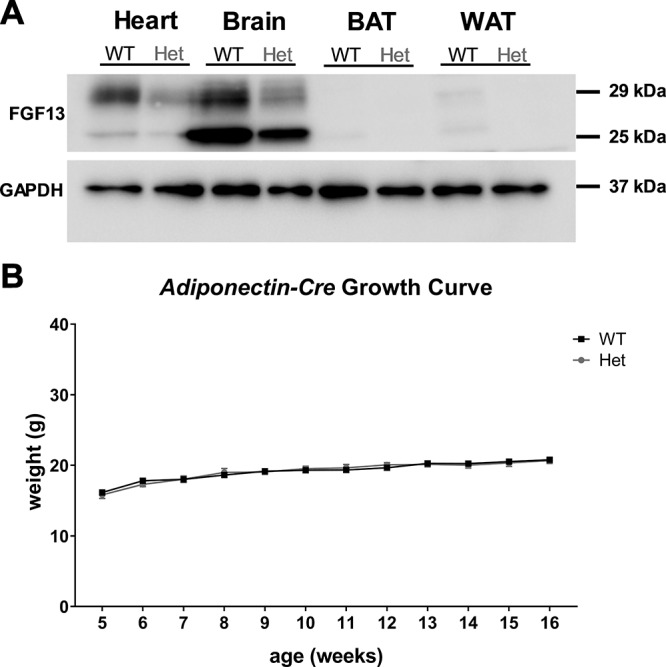
FGF13 is not abundant in adipose tissues, and adipose-specific Fgf13 heterozygous (Het) knockout does not recapitulate weight gain. A) Western blot demonstrating FGF13 expression in various tissues. This is a representative image of 3 successful experiments. B) Growth chart of Adipoq-Cre+;Fgf13+/fl mice and Adipoq-Cre+ littermate controls housed at 30°C; n = 5–6/genotype. GAPDH, glyceraldehyde 3-phosphate dehydrogenase. Two-way repeated measures ANOVA was used to assess main effects of genotype and time, as well as interaction effects. Comparisons between genotypes at each time point were made using Tukey’s test for multiple comparisons.
Fgf13 heterozygous knockout decreases sympathetic drive to BAT
To directly assay sympathetic drive to BAT, we performed an NETO assay. We measured catecholamine levels in BAT by HPLC after injecting mice with AMPT to inhibit de novo catecholamine synthesis. The rate of disappearance of NE from BAT after AMPT treatment is proportional to the rate of sympathetic nerve firing on the tissue (37, 48, 49). We found that, although there was no difference in the basal levels of NE between genotypes at either temperature (Fig. 6A), NE declined less in the Fgf13+/− mice than in the WT mice after 4 h of AMPT inhibition. NE levels in WT mice declined by 76% at both housing temperatures, whereas NE levels in Fgf13+/− mice declined by 44 and 42% at 22 and 30°C, respectively (Fig. 6B). This demonstrates decreased NETO and a clear defect in sympathetic drive to BAT in Fgf13+/− mice. We also measured expression of β3 adrenergic receptor (Adrb3) in BAT, through which catecholamines activate the thermogenic program. In previous BAT surgical denervation models, Adrb3 was up-regulated (50) as a presumed sensitizing response. Here, we observed that Adrb3 was up-regulated in Fgf13+/− mice compared with WT mice in animals housed at both 22 and 30°C, consistent with reduced sympathetic drive to BAT in the Fgf13+/− mice (Fig. 6C).
Figure 6.

Fgf13 loss causes decreased sympathetic drive to BAT. A) Basal levels of NE in BAT of Fgf13+/− mice and littermate controls at 22 and 30°C. B) NE percentage decline after 4 h of inhibition of tyrosine hydroxylase with AMPT. NE percentage decline is proportional to rate of sympathetic nerve firing. C) Real-time quantitative PCR gene expression of Adrb3. For all experiments; n = 6/genotype at each temperature, half of which were injected with AMPT; data are displayed for mice not injected with AMPT. Het, heterozygous; NS, not significant. Two-way ANOVA was followed by Holm-Sidak’s test for multiple comparisons indicated significance. *P < 0.05, **P < 0.001.
Hypothalamic-specific deletion of Fgf13 causes weight gain with no changes in food intake, and hypothalamic expression of Fgf13 is inversely correlated with severity of obesity in Fgf13+/− mice
Decreased sympathetic drive to BAT in Fgf13+/− mice could result from defects in sympathetic ganglia or CNS-mediated regulation of the sympathetic ganglia, where Fgf13 is prominently expressed (46). We therefore investigated whether expression of Fgf13 in neurons contributed to our observed phenotypes by generating Fgf13 heterozygous knockouts using Nes-Cre. When housed at thermoneutrality, Nes-Cre+;Fgf13+/fl mice recapitulated the weight gain observed in global Fgf13+/− mice. Nes-Cre+ controls, like WT controls, exhibited no weight gain (Fig. 7A). This weight gain developed despite known metabolic effects of Nes-Cre, which causes reduced body size (51). Thus, we conclude that Fgf13 in neurons contributes to the regulation of obesity. We then focused on hypothalamus, which exerts multiple effects on metabolism, such as regulation of the autonomic nervous system, feeding behavior, energy balance, and thermogenesis (5, 52–55). We started by asking whether the variable obesity in the global Fgf13+/− mice correlated with Fgf13 expression levels within the hypothalamus. We measured Fgf13 expression by real-time quantitative PCR in hypothalami dissected from Fgf13+/− mice housed at 30°C and found an inverse linear correlation between the amount of Fgf13 expression and weight (Fig. 7B). The hypothalamus is highly heterogeneous, and examination of the Allen Brain Atlas (56) does not demonstrate marked concentration of Fgf13 within a particular nucleus or nuclei. Moreover, our analysis of recent single-cell RNA sequencing data shows that Fgf13 is expressed within multiple neuronal types within the hypothalamus, including both glutamatergic and GABA-ergic neurons (57), which we confirmed by RNAScope in situ hybridization (Supplemental Fig. S5). Because the best characterized role for FGF13 is as a regulator of NaVs, we queried the single-cell RNA sequencing data and compared the expression profiles of the various NaVs expressed within the hypothalamus (NaV1.1, NaV1.2, NaV1.3, NaV1.6, and NaV1.7) and found that the Fgf13 expression pattern most closely matched that of NaV1.1 [Na+ voltage-gated channel α subunit 1 (Scn1a)] and NaV1.7 (Scn9a) (Supplemental Table S2). We were particularly intrigued by the similar expression pattern with NaV1.7, because elimination of NaV1.7 within the PVH led to rapid obesity (58). Moreover, exclusion of FGF13 from heterologous expression systems reduces NaV1.7-dependent Na+ currents, and deletion of Fgf13 in neurons leads to a marked reduction in Na+ currents (59). We therefore hypothesized that reduced Fgf13 expression in the PVH of Fgf13+/− mice phenocopied the NaV1.7 ablation from the hypothalamus, and so we chose to test whether Fgf13 deletion in the PVH recapitulated the global heterozygous knockout phenotype. We therefore targeted the PVH with viral Cre-GFP (or GFP as a control) by intracranial injection into Fgf13fl/fl mice. We consistently achieved unilateral targeting of the PVH (Fig. 7C and Supplemental Fig. S6), as indicated by GFP immunofluorescence analyzed in brain histologic slices at the completion of the experiment. We monitored the animals’ weight for 30 d after injection while animals were housed at thermoneutrality. By d 9, Cre-GFP injected mice gained significantly more weight than GFP-injected mice, and the Cre-GFP injected mice continued to gain weight throughout the 30-d monitoring period (Fig. 7D) despite no increase in food intake (117.8 ± 2.8 g 30-d food intake for Cre-GFP injected animals; 123.4 ± 1.6 g for GFP injected animals; P > 0.05) Together, these data indicate that loss of Fgf13 in the hypothalamus decreased sympathetic output to BAT and led to obesity at thermoneutrality.
Figure 7.

CNS- and hypothalamic-specific Fgf13 ablation recapitulates weight gain phenotype. A) Growth chart of Nes-Cre+;Fgf13+/fl mice and Nes-Cre+ littermate controls housed at 30°C. Two-way repeated measures ANOVA was followed by Tukey’s test for multiple comparisons. B) Correlation of mouse weight and hypothalamic Fgf13 expression in Fgf13+/− mice raised at thermoneutrality. C) Image of Cre-GFP expression in hypothalamus of Fgf13fl/fl mouse. Scale bar, 500 µm. D) Weight gain of Cre-GFP and GFP injected Fgf13fl/fl mice over 30 d. Two-way repeated measures ANOVA was followed by Tukey’s test for multiple comparisons; n = 5–6/genotype or injection condition. GAPDH, glyceraldehyde 3-phosphate dehydrogenase; Het, heterozygous indicated significance. *P < 0.05, **P < 0.001.
DISCUSSION
Here, we demonstrated that Fgf13 regulates nonshivering thermogenesis and metabolism. We found that heterozygous knockout of Fgf13 resulted in hyperactivity and impaired body temperature defense in mice housed at 22°C and caused obesity in mice housed at thermoneutrality, when thermal stress was minimized. Interestingly, the Fgf13+/− mice demonstrated a residual thermogenesis deficit even at 30°C, as revealed by weight loss during single-housing conditions compared with group housing, during which mice can exploit huddling as means to preserve core body temperature core (60). Consistent with those physiologic consequences, we observed that BAT from Fgf13+/− mice showed an abnormal histologic appearance and reduced expression of Ucp1. Although thermogenesis in the mutants was abnormal, we determined that BAT in Fgf13+/− mice remained responsive to β3 stimulation, suggesting that the deficit in the heterozygous knockout animals resulted from reduced sympathetic drive to BAT. Indeed, we found reduced sympathetic activity in BAT by measuring NETO. In agreement with that result, CNS- or PVH-specific Fgf13 knockout recapitulated the weight gain observed in the global Fgf13 heterozygous knockout mice. The established reduction of neuronal Na+ currents after Fgf13 elimination (59) provides a suggestion for the underlying mechanism. Because of the limited tools to identify specific hypothalamic neurons expressing Fgf13 in slices, confirmation of reduced NaV currents in the Fgf13+/− mice is not yet possible. Nevertheless, these data advance our understanding of FGF13 and establish novel roles for this protein.
Although our identification of FGF13 as a regulator of obesity fits with multiple previous reports correlating Fgf13 transcription with changes in diet (2, 21–24), we were unable to detect significant FGF13 in adipose depots in mice fed normal chow. Instead, we implicated a CNS role for Fgf13. Moreover, we observed an inverse correlation with Fgf13 transcript levels in the hypothalamus and weight, in contrast to previous studies that found a direct correlation between Fgf13 expression levels and obesogenic diets. Although we did not evaluate the effects of diet on Fgf13 expression or function in the CNS, our study in combination with those previous studies suggests that FGF13 has multiple roles in metabolism.
Even though we did not detect significant Fgf13 expression in adipose, it is interesting to compare the consequences in our model with consequences of Ucp1 knockout. Like Fgf13+/− mice, Ucp1 knockout animals only develop significant obesity when housed at thermoneutrality (4). At standard mouse housing temperatures (∼22°C), mice generally utilize nonshivering thermogenesis mediated by BAT to maintain core body temperature. To compensate for decreased BAT activity, alternative mechanisms of thermogenesis, such as increased motor activity (as seen in our model) or shivering, are recruited to respond to the thermal challenge present at 22°C. When acutely challenged with an even colder environment, in which increased activity or shivering could not compensate for a deficit in BAT activity, Fgf13+/− mice were unable to defend core body temperature, similar to Ucp1 knockouts (61). On the other hand, when the thermogenic drive was reduced (by housing at 30°C), the excess energy used at 22°C to maintain core body temperature was stored as adipose, as indicated by the significant weight gain in both Fgf13+/− mice and Ucp1 knockouts. That our model recapitulates salient features of Ucp1 knockouts, yet direct activation of β3 receptors in the Fgf13+/− confirms intact BAT activity, adding further support to our hypothesis that obesity in our model results from decreased activation of BAT because of reduced sympathetic drive.
Knockout models of Fgf13 have previously been reported in refs. 12, 19, 40, but obesity was not noted. The lack of observed obesity in these models may be explained by strain differences, variable X-chromosome inactivation, and—most likely—the absence of thermoneutral housing, which we found necessary to reveal the obesity phenotype. It is interesting that Wu et al. (19) observed hyperactivity, as we also observed in Fgf13+/− mice housed at 22°C. Because we did not observe the increased activity in Fgf13+/− mice when they were housed at 30°C, our data suggest that the hyperactivity at 22°C served as a compensatory thermogenic mechanism. Moreover, the hyperactivity reported by Wu et al. (19), in which Emx1-Cre (rather than the Ddx4-Cre that we used here) suggests that the phenotypes we observed are not due to off-target effects of the recently described ∼1 Mb deletion on chromosome 18 in the Ddx4-Cre+ mice (62). The ∼1 Mb deletion affects neuropilin and tolloid like 1 and cerebellin 2 precursor, neither of which have been associated with a metabolic phenotype. That our Nes-Cre knockouts and our PVH-specific knockouts generated by Cre injection displayed obesity phenotypes similar to the Ddx4-Cre–generated Fgf13+/− mice provides further reassurance that the phenotypes we studied are not related to the heterozygous loss of neuropilin and tolloid like 1 or cerebellin 2 precursor.
Further, X inactivation, which occurs randomly in each cell of the developing embryo (63), may have clouded the obesity phenotype in previous studies. Indeed, we observed variable differences in weight and fat mass distribution in mice housed at 22°C. After elimination of metabolic demand to meet a thermal challenge (by thermoneutral housing) we observed a larger effect on obesity, yet under these conditions we still observed significant interanimal variation that inversely correlated with the level of Fgf13 expression in the hypothalamus (Fig. 7E). Interestingly, the degree of random X inactivation has been shown to affect phenotype severity of X-linked disorder knockout models such as Rett syndrome, suggesting that modulation of X inactivation and subsequent mutant and WT allele expression may offer a novel therapeutic avenue for these disorders (64, 65).
Although our injections of Cre into the hypothalamus do not exclude contributions of other areas of the CNS to the observed phenotype, they do identify a major area within which Fgf13 affects metabolism. Moreover, the effects after targeting Fgf13 in the hypothalamus are consistent with the well-characterized role of the hypothalamus as a critical regulator of energy balance, thermogenesis, and metabolism (2, 6, 66, 67). Here, we observed that Fgf13 knockout by Cre injection targeting the PVH in the hypothalamus recapitulated the global Fgf13+/− mice and the Nes-Cre nervous system–specific Fgf13+/−. There are, however, several nuclei and neuronal subtypes within the hypothalamus, with overlapping roles in regulation of food intake and thermogenesis (5, 52, 53, 68, 69).
It will be of interest to define the complete complement of nuclei and neuronal subtypes within the hypothalamus in which Fgf13 is expressed to define further the mechanisms by which Fgf13 exerts its effects, especially in the context of the observed thermogenic phenotype with no effects on feeding behavior. Although our antibody against FGF13 has been validated and used successfully for protein detection by Western blot, optimizing conditions for immunohistochemical detection in the CNS has proven challenging. Once we surmount this challenge, we will be able to dissect the contribution of Fgf13 to complex circuitry within the PVH that regulates BAT. Indeed, whether PVH output to BAT (via the raphe pallidus) is excitatory or inhibitory is unclear, as there are both excitatory and inhibitory influences on a tonic inhibitory PVH to BAT regulation (70). The ability to identify the specific neuronal subpopulations within the hypothalamus in which FGF13 is expressed will allow us to directly measure FGF13’s contribution to NaV1.7-mediated Na+ current in those neurons. Because some NaVs such as NaV1.7 are expressed in both excitatory and inhibitory neurons, in conjunction with evidence from us and others that FGF13, which regulates NaVs (and thus neuronal action potential output), is expressed in both excitatory and inhibitory neurons (12, 19, 42), the effect of Fgf13 knockout in the PVH could result in decreased action potential output from either excitatory or inhibitory neurons within the PVH, thus producing the observed effect of reduced sympathetic drive to BAT.
Our hypothesis that haploinsufficiency of FGF13 leads to reduced NaV1.7 Na+ currents fits with the best-characterized role for FHFs as modulators of NaVs (9, 19, 71) to which FHFs directly bind (39, 72, 73), and our focus on FGF13 regulation of NaVs in the PVH is consistent with recent reports identifying NaV1.7 as a regulator of body weight, specifically within the hypothalamus (58). Virally targeted deletion of Scn9a, which encodes NaV1.7, within the PVH resulted in a doubling in body weight over 30 d. Thus, we speculate that depletion of Fgf13 in the hypothalamus decreases NaV1.7-dependent Na+ currents and therefore reduces the ability of the affected neurons to fire action potentials. As a consequence, sympathetic output to BAT is reduced, and thermogenesis is impaired.
Our data establish that FGF13 regulates thermogenesis and energy balance. FGF13 supports effective sympathetic output to metabolically active BAT and promotes EE through nonshivering thermogenesis. This function resides, at least in part, within the hypothalamus. These findings contribute to a more complete understanding of the multifunctional FHFs, and this model provides a platform to investigate further how FGF13 regulates metabolism within the CNS.
Supplementary Material
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
ACKNOWLEDGMENTS
The authors thank Dr. Roberto Levi and Dr. Alice Marino (Weill Cornell Medical College, New York, NY, USA) for valuable assistance with norepinephrine turnover assay sample preparation and data acquisition; Dr. Conor Liston and Dr. Thu Huynh (Weill Cornell, Medical College, New York, NY, USA) for valuable technical assistance with intracranial injections; Dr. Jeffrey Friedman and Dr. Luca Parolari (The Rockefeller University, New York, NY, USA) for assistance with RNAscope in situ hybridization protocols and imaging; and Dr. James Lo (Weill Cornell, Medical College, New York, NY, USA) and Dr. Paul Cohen (Rockefeller University, NY, NY, USA) for insightful discussion and comments on the manuscript. This work was supported by U.S. National Institutes of Health (NIH) National Heart, Lung, and Blood Institute Grants F30 HL131217 to D.S.S., R01 HL112918, R01 HL071165, and R01 HL122967 to G.S.P., U.S. National Institutes of Health, National Institute of General Medical Sciences Grant T32 GM007171 to D.S.S., and NIH National Institute of Diabetes and Digestive and Kidney Diseases Grants R01 DK103046, R01 DK056626, and R37 DK048873 to D.E.C. The authors declare no conflicts of interest.
Glossary
- Adipoq-Cre+
adiponectin promoter–driven Cre recombinant
- Adrb3
β3 adrenergic receptor
- AMPT
α-methyl-p-tyrosine
- BAT
brown adipose tissue
- Ddx4-Cre+
DEAD-box polypeptide-4 promoter–driven Cre recombinant
- EE
energy expenditure
- F1
first filial
- FGF
fibroblast growth factor
- Fgf13−/Y
male hemizygous Fgf13 knockout
- Fgf13fl/fl
floxed Fgf13 alleles
- FHF
FGF homologous factor
- GFP
green fluorescent protein
- gWAT
gonadal white adipose tissue
- iBAT
inguinal BAT
- iWAT
inguinal white adipose tissue
- LBM
lean body mass
- NaV
voltage-gated Na+ channel
- NE
norepinephrine
- Nes-Cre+
nestin promoter–driven Cre recombinant
- NETO
NE turnover
- PVH
paraventricular nucleus of the hypothalamus
- RER
respiratory exchange ratio
- Scn1a
Na+ voltage-gated channel α subunit 1
- Ucp1
uncoupling protein 1
- WT
wild type
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
AUTHOR CONTRIBUTIONS
D. S. Sinden and G. S. Pitt designed experiments and wrote the manuscript; D. S. Sinden, C. D. Holman, C. J. Bare, X. Sun, and A. R. Gade conducted experiments and analyzed data; D. E. Cohen designed experiments and contributed essential equipment and analytic tools; and D. S. Sinden, C. D. Holman, C. J. Bare, D. E. Cohen, and G. S. Pitt edited the manuscript.
REFERENCES
- 1.Leibel R. L. (2008) Molecular physiology of weight regulation in mice and humans. Int. J. Obes. 32 (Suppl 7), S98–S108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morton G. J., Cummings D. E., Baskin D. G., Barsh G. S., Schwartz M. W. (2006) Central nervous system control of food intake and body weight. Nature 443, 289–295 [DOI] [PubMed] [Google Scholar]
- 3.Bartness T. J., Song C. K. (2007) Brain-adipose tissue neural crosstalk. Physiol. Behav. 91, 343–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feldmann H. M., Golozoubova V., Cannon B., Nedergaard J. (2009) UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metab. 9, 203–209 [DOI] [PubMed] [Google Scholar]
- 5.An J. J., Liao G. Y., Kinney C. E., Sahibzada N., Xu B. (2015) Discrete BDNF neurons in the paraventricular hypothalamus control feeding and energy expenditure. Cell Metab. 22, 175–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang X., Ruan H. B. (2015) Neuronal control of adaptive thermogenesis. Front. Endocrinol. (Lausanne) 6, 149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garland T., Jr., Schutz H., Chappell M. A., Keeney B. K., Meek T. H., Copes L. E., Acosta W., Drenowatz C., Maciel R. C., van Dijk G., Kotz C. M., Eisenmann J. C. (2011) The biological control of voluntary exercise, spontaneous physical activity and daily energy expenditure in relation to obesity: human and rodent perspectives. J. Exp. Biol. 214, 206–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olsen S. K., Garbi M., Zampieri N., Eliseenkova A. V., Ornitz D. M., Goldfarb M., Mohammadi M. (2003) Fibroblast growth factor (FGF) homologous factors share structural but not functional homology with FGFs. J. Biol. Chem. 278, 34226–34236 [DOI] [PubMed] [Google Scholar]
- 9.Goldfarb M. (2005) Fibroblast growth factor homologous factors: evolution, structure, and function. Cytokine Growth Factor Rev. 16, 215–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Swieten J. C., Brusse E., de Graaf B. M., Krieger E., van de Graaf R., de Koning I., Maat-Kievit A., Leegwater P., Dooijes D., Oostra B. A., Heutink P. (2003) A mutation in the fibroblast growth factor 14 gene is associated with autosomal dominant cerebellar ataxia [corrected]. Am. J. Hum. Genet. 72, 191–199; erratum: 1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hennessey J. A., Marcou C. A., Wang C., Wei E. Q., Wang C., Tester D. J., Torchio M., Dagradi F., Crotti L., Schwartz P. J., Ackerman M. J., Pitt G. S. (2013) FGF12 is a candidate Brugada syndrome locus. Heart Rhythm 10, 1886–1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Puranam R. S., He X. P., Yao L., Le T., Jang W., Rehder C. W., Lewis D. V., McNamara J. O. (2015) Disruption of Fgf13 causes synaptic excitatory-inhibitory imbalance and genetic epilepsy and febrile seizures plus. J. Neurosci. 35, 8866–8881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hennessey J. A., Wei E. Q., Pitt G. S. (2013) Fibroblast growth factor homologous factors modulate cardiac calcium channels. Circ. Res. 113, 381–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang X., Tang H., Wei E. Q., Wang Z., Yang J., Yang R., Wang S., Zhang Y., Pitt G. S., Zhang H., Wang C. (2017) Conditional knockout of Fgf13 in murine hearts increases arrhythmia susceptibility and reveals novel ion channel modulatory roles. J. Mol. Cell. Cardiol. 104, 63–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bublik D. R., Bursać S., Sheffer M., Oršolić I., Shalit T., Tarcic O., Kotler E., Mouhadeb O., Hoffman Y., Fuchs G., Levin Y., Volarević S., Oren M. (2017) Regulatory module involving FGF13, miR-504, and p53 regulates ribosomal biogenesis and supports cancer cell survival. Proc. Natl. Acad. Sci. USA 114, E496–E505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DeStefano G. M., Fantauzzo K. A., Petukhova L., Kurban M., Tadin-Strapps M., Levy B., Warburton D., Cirulli E. T., Han Y., Sun X., Shen Y., Shirazi M., Jobanputra V., Cepeda-Valdes R., Cesar Salas-Alanis J., Christiano A. M. (2013) Position effect on FGF13 associated with X-linked congenital generalized hypertrichosis. Proc. Natl. Acad. Sci. USA 110, 7790–7795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu H., Shi X., Wu G., Zhu J., Song C., Zhang Q., Yang G. (2015) FGF13 regulates proliferation and differentiation of skeletal muscle by down-regulating Spry1. Cell Prolif. 48, 550–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reddy M. A., Das S., Zhuo C., Jin W., Wang M., Lanting L., Natarajan R. (2016) Regulation of vascular smooth muscle cell dysfunction under diabetic conditions by miR-504. Arterioscler. Thromb. Vasc. Biol. 36, 864–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu Q. F., Yang L., Li S., Wang Q., Yuan X. B., Gao X., Bao L., Zhang X. (2012) Fibroblast growth factor 13 is a microtubule-stabilizing protein regulating neuronal polarization and migration. Cell 149, 1549–1564 [DOI] [PubMed] [Google Scholar]
- 20.Wei E. Q., Sinden D. S., Mao L., Zhang H., Wang C., Pitt G. S. (2017) Inducible Fgf13 ablation enhances caveolae-mediated cardioprotection during cardiac pressure overload. Proc. Natl. Acad. Sci. USA 114, E4010–E4019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hageman R. S., Wagener A., Hantschel C., Svenson K. L., Churchill G. A., Brockmann G. A. (2010) High-fat diet leads to tissue-specific changes reflecting risk factors for diseases in DBA/2J mice. Physiol. Genomics 42, 55–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim S. S., Choi K. M., Kim S., Park T., Cho I. C., Lee J. W., Lee C. K. (2016) Whole-transcriptome analysis of mouse adipose tissue in response to short-term caloric restriction. Mol. Genet. Genomics 291, 831–847 [DOI] [PubMed] [Google Scholar]
- 23.Macotela Y., Emanuelli B., Mori M. A., Gesta S., Schulz T. J., Tseng Y. H., Kahn C. R. (2012) Intrinsic differences in adipocyte precursor cells from different white fat depots. Diabetes 61, 1691–1699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller R. S., Becker K. G., Prabhu V., Cooke D. W. (2008) Adipocyte gene expression is altered in formerly obese mice and as a function of diet composition. J. Nutr. 138, 1033–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morton N. M., Nelson Y. B., Michailidou Z., Di Rollo E. M., Ramage L., Hadoke P. W., Seckl J. R., Bunger L., Horvat S., Kenyon C. J., Dunbar D. R. (2011) A stratified transcriptomics analysis of polygenic fat and lean mouse adipose tissues identifies novel candidate obesity genes. PLoS One 6, e23944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Modesto A., Jacas C. A., Kim S. M., Desman A., West I., Lebow M., Littlejohn C., Deeley K., Studen-Pavlovich D., Vieira A. R. (2019) Childhood obesity, genetic variation, and dental age. Pediatr. Dent. 41, 132–135 [PubMed] [Google Scholar]
- 27.Council N. R. (2011) Guide for the Care and Use of Laboratory Animals, 8th ed., The National Academies Press, Washington, DC [Google Scholar]
- 28.Gallardo T., Shirley L., John G. B., Castrillon D. H. (2007) Generation of a germ cell-specific mouse transgenic Cre line, Vasa-Cre. Genesis 45, 413–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Desai A., Alves-Bezerra M., Li Y., Ozdemir C., Bare C. J., Li Y., Hagen S. J., Cohen D. E. (2018) Regulation of fatty acid trafficking in liver by thioesterase superfamily member 1. J. Lipid Res. 59, 368–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Staffas A., Burgos da Silva M., Slingerland A. E., Lazrak A., Bare C. J., Holman C. D., Docampo M. D., Shono Y., Durham B., Pickard A. J., Cross J. R., Stein-Thoeringer C., Velardi E., Tsai J. J., Jahn L., Jay H., Lieberman S., Smith O. M., Pamer E. G., Peled J. U., Cohen D. E., Jenq R. R., van den Brink M. R. M. (2018) Nutritional support from the intestinal microbiota improves hematopoietic reconstitution after bone marrow transplantation in mice. Cell Host Microbe 23, 447–457.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu X., Krumm C., So J. S., Bare C. J., Holman C., Gromada J., Cohen D. E., Lee A. H. (2018) Preemptive activation of the integrated stress response protects mice from diet‐induced obesity and insulin resistance by fibroblast growth factor 21 induction. Hepatology 68, 2167–2181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weir J. B. (1949) New methods for calculating metabolic rate with special reference to protein metabolism. J. Physiol. 109, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tschöp M. H., Speakman J. R., Arch J. R., Auwerx J., Brüning J. C., Chan L., Eckel R. H., Farese R. V., Jr., Galgani J. E., Hambly C., Herman M. A., Horvath T. L., Kahn B. B., Kozma S. C., Maratos-Flier E., Müller T. D., Münzberg H., Pfluger P. T., Plum L., Reitman M. L., Rahmouni K., Shulman G. I., Thomas G., Kahn C. R., Ravussin E. (2011) A guide to analysis of mouse energy metabolism. Nat. Methods 9, 57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gargiulo S., Gramanzini M., Megna R., Greco A., Albanese S., Manfredi C., Brunetti A. (2014) Evaluation of growth patterns and body composition in C57Bl/6J mice using dual energy X-ray absorptiometry. Biomed Res. Int. 2014, 253067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berry R., Church C. D., Gericke M. T., Jeffery E., Colman L., Rodeheffer M. S. (2014) Imaging of adipose tissue. Methods Enzymol. 537, 47–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang C., Hennessey J. A., Kirkton R. D., Wang C., Graham V., Puranam R. S., Rosenberg P. B., Bursac N., Pitt G. S. (2011) Fibroblast growth factor homologous factor 13 regulates Na+ channels and conduction velocity in murine hearts. Circ. Res. 109, 775–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vaughan C. H., Zarebidaki E., Ehlen J. C., Bartness T. J. (2014) Analysis and measurement of the sympathetic and sensory innervation of white and brown adipose tissue. Methods Enzymol. 537, 199–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peaston R. T., Weinkove C. (2004) Measurement of catecholamines and their metabolites. Ann. Clin. Biochem. 41, 17–38 [DOI] [PubMed] [Google Scholar]
- 39.Wang C., Wang C., Hoch E. G., Pitt G. S. (2011) Identification of novel interaction sites that determine specificity between fibroblast growth factor homologous factors and voltage-gated sodium channels. J. Biol. Chem. 286, 24253–24263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park D. S., Shekhar A., Marra C., Lin X., Vasquez C., Solinas S., Kelley K., Morley G., Goldfarb M., Fishman G. I. (2016) Fhf2 gene deletion causes temperature-sensitive cardiac conduction failure. Nat. Commun. 7, 12966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang J., Wang Z., Sinden D. S., Wang X., Shan B., Yu X., Zhang H., Pitt G. S., Wang C. (2016) FGF13 modulates the gating properties of the cardiac sodium channel Nav1.5 in an isoform-specific manner. Channels (Austin) 10, 410–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pablo J. L., Wang C., Presby M. M., Pitt G. S. (2016) Polarized localization of voltage-gated Na+ channels is regulated by concerted FGF13 and FGF14 action. Proc. Natl. Acad. Sci. USA 113, E2665–E2674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fischer A. W., Cannon B., Nedergaard J. (2018) Optimal housing temperatures for mice to mimic the thermal environment of humans: an experimental study. Mol. Metab. 7, 161–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xiao C., Goldgof M., Gavrilova O., Reitman M. L. (2015) Anti-obesity and metabolic efficacy of the β3-adrenergic agonist, CL316243, in mice at thermoneutrality compared to 22°C. Obesity (Silver Spring) 23, 1450–1459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kotzbeck P., Giordano A., Mondini E., Murano I., Severi I., Venema W., Cecchini M. P., Kershaw E. E., Barbatelli G., Haemmerle G., Zechner R., Cinti S. (2018) Brown adipose tissue whitening leads to brown adipocyte death and adipose tissue inflammation. J. Lipid Res. 59, 784–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smallwood P. M., Munoz-Sanjuan I., Tong P., Macke J. P., Hendry S. H., Gilbert D. J., Copeland N. G., Jenkins N. A., Nathans J. (1996) Fibroblast growth factor (FGF) homologous factors: new members of the FGF family implicated in nervous system development. Proc. Natl. Acad. Sci. USA 93, 9850–9857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fon Tacer K., Bookout A. L., Ding X., Kurosu H., John G. B., Wang L., Goetz R., Mohammadi M., Kuro-o M., Mangelsdorf D. J., Kliewer S. A. (2010) Research resource: comprehensive expression atlas of the fibroblast growth factor system in adult mouse. Mol. Endocrinol. 24, 2050–2064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Young J. B., Saville E., Rothwell N. J., Stock M. J., Landsberg L. (1982) Effect of diet and cold exposure on norepinephrine turnover in brown adipose tissue of the rat. J. Clin. Invest. 69, 1061–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Virtue S., Vidal-Puig A. (2013) Assessment of brown adipose tissue function. Front. Physiol. 4, 128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Granneman J. G., Lahners K. N. (1992) Differential adrenergic regulation of beta 1- and beta 3-adrenoreceptor messenger ribonucleic acids in adipose tissues. Endocrinology 130, 109–114 [DOI] [PubMed] [Google Scholar]
- 51.Declercq J., Brouwers B., Pruniau V. P., Stijnen P., de Faudeur G., Tuand K., Meulemans S., Serneels L., Schraenen A., Schuit F., Creemers J. W. (2015) Metabolic and behavioural phenotypes in nestin-cre mice are caused by hypothalamic expression of human growth hormone. PLoS One 10, e0135502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tolson K. P., Gemelli T., Gautron L., Elmquist J. K., Zinn A. R., Kublaoui B. M. (2010) Postnatal Sim1 deficiency causes hyperphagic obesity and reduced Mc4r and oxytocin expression. J. Neurosci. 30, 3803–3812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.De Luca B., Monda M., Amaro S., Pellicano M. P., Cioffi L. A. (1989) Lack of diet-induced thermogenesis following lesions of paraventricular nucleus in rats. Physiol. Behav. 46, 685–691 [DOI] [PubMed] [Google Scholar]
- 54.Kasahara Y., Takayanagi Y., Kawada T., Itoi K., Nishimori K. (2007) Impaired thermoregulatory ability of oxytocin-deficient mice during cold-exposure. Biosci. Biotechnol. Biochem. 71, 3122–3126 [DOI] [PubMed] [Google Scholar]
- 55.Ferguson A. V., Latchford K. J., Samson W. K. (2008) The paraventricular nucleus of the hypothalamus - a potential target for integrative treatment of autonomic dysfunction. Expert Opin. Ther. Targets 12, 717–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lein E. S., Hawrylycz M. J., Ao N., Ayres M., Bensinger A., Bernard A., Boe A. F., Boguski M. S., Brockway K. S., Byrnes E. J., Chen L., Chen L., Chen T. M., Chin M. C., Chong J., Crook B. E., Czaplinska A., Dang C. N., Datta S., Dee N. R., Desaki A. L., Desta T., Diep E., Dolbeare T. A., Donelan M. J., Dong H. W., Dougherty J. G., Duncan B. J., Ebbert A. J., Eichele G., Estin L. K., Faber C., Facer B. A., Fields R., Fischer S. R., Fliss T. P., Frensley C., Gates S. N., Glattfelder K. J., Halverson K. R., Hart M. R., Hohmann J. G., Howell M. P., Jeung D. P., Johnson R. A., Karr P. T., Kawal R., Kidney J. M., Knapik R. H., Kuan C. L., Lake J. H., Laramee A. R., Larsen K. D., Lau C., Lemon T. A., Liang A. J., Liu Y., Luong L. T., Michaels J., Morgan J. J., Morgan R. J., Mortrud M. T., Mosqueda N. F., Ng L. L., Ng R., Orta G. J., Overly C. C., Pak T. H., Parry S. E., Pathak S. D., Pearson O. C., Puchalski R. B., Riley Z. L., Rockett H. R., Rowland S. A., Royall J. J., Ruiz M. J., Sarno N. R., Schaffnit K., Shapovalova N. V., Sivisay T., Slaughterbeck C. R., Smith S. C., Smith K. A., Smith B. I., Sodt A. J., Stewart N. N., Stumpf K. R., Sunkin S. M., Sutram M., Tam A., Teemer C. D., Thaller C., Thompson C. L., Varnam L. R., Visel A., Whitlock R. M., Wohnoutka P. E., Wolkey C. K., Wong V. Y., Wood M., Yaylaoglu M. B., Young R. C., Youngstrom B. L., Yuan X. F., Zhang B., Zwingman T. A., Jones A. R. (2007) Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176 [DOI] [PubMed] [Google Scholar]
- 57.Romanov R. A., Zeisel A., Bakker J., Girach F., Hellysaz A., Tomer R., Alpár A., Mulder J., Clotman F., Keimpema E., Hsueh B., Crow A. K., Martens H., Schwindling C., Calvigioni D., Bains J. S., Máté Z., Szabó G., Yanagawa Y., Zhang M. D., Rendeiro A., Farlik M., Uhlén M., Wulff P., Bock C., Broberger C., Deisseroth K., Hökfelt T., Linnarsson S., Horvath T. L., Harkany T. (2017) Molecular interrogation of hypothalamic organization reveals distinct dopamine neuronal subtypes. Nat. Neurosci. 20, 176–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Branco T., Tozer A., Magnus C. J., Sugino K., Tanaka S., Lee A. K., Wood J. N., Sternson S. M. (2016) Near-perfect synaptic integration by Nav1.7 in hypothalamic neurons regulates body weight. Cell 165, 1749–1761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang L., Dong F., Yang Q., Yang P. F., Wu R., Wu Q. F., Wu D., Li C. L., Zhong Y. Q., Lu Y. J., Cheng X., Xu F. Q., Chen L., Bao L., Zhang X. (2017) FGF13 selectively regulates heat nociception by interacting with Nav1.7. Neuron 93, 806–821.e9 [DOI] [PubMed] [Google Scholar]
- 60.Glancy J., Groß R., Stone J. V., Wilson S. P. (2015) A self-organising model of thermoregulatory huddling. PLOS Comput. Biol. 11, e1004283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rowland L. A., Bal N. C., Kozak L. P., Periasamy M. (2015) Uncoupling protein 1 and sarcolipin are required to maintain optimal thermogenesis, and loss of both systems compromises survival of mice under cold stress. J. Biol. Chem. 290, 12282–12289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goodwin L. O., Splinter E., Davis T. L., Urban R., He H., Braun R. E., Chesler E. J., Kumar V., van Min M., Ndukum J., Philip V. M., Reinholdt L. G., Svenson K., White J. K., Sasner M., Lutz C., Murray S. A. (2019) Large-scale discovery of mouse transgenic integration sites reveals frequent structural variation and insertional mutagenesis. [E-pub ahead of print] Genome Res. 10.1101/gr.233866.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Augui S., Nora E. P., Heard E. (2011) Regulation of X-chromosome inactivation by the X-inactivation centre. Nat. Rev. Genet. 12, 429–442 [DOI] [PubMed] [Google Scholar]
- 64.Sripathy S., Leko V., Adrianse R. L., Loe T., Foss E. J., Dalrymple E., Lao U., Gatbonton-Schwager T., Carter K. T., Payer B., Paddison P. J., Grady W. M., Lee J. T., Bartolomei M. S., Bedalov A. (2017) Screen for reactivation of MeCP2 on the inactive X chromosome identifies the BMP/TGF-β superfamily as a regulator of XIST expression. Proc. Natl. Acad. Sci. USA 114, 1619–1624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Carrette L. L. G., Wang C. Y., Wei C., Press W., Ma W., Kelleher R. J., III, Lee J. T. (2018) A mixed modality approach towards Xi reactivation for Rett syndrome and other X-linked disorders. Proc. Natl. Acad. Sci. USA 115, E668–E675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Morrison S. F. (2016) Central neural control of thermoregulation and brown adipose tissue. Auton. Neurosci. 196, 14–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Labbé S. M., Caron A., Lanfray D., Monge-Rofarello B., Bartness T. J., Richard D. (2015) Hypothalamic control of brown adipose tissue thermogenesis. Front. Syst. Neurosci. 9, 150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Butler A. A., Marks D. L., Fan W., Kuhn C. M., Bartolome M., Cone R. D. (2001) Melanocortin-4 receptor is required for acute homeostatic responses to increased dietary fat. Nat. Neurosci. 4, 605–611 [DOI] [PubMed] [Google Scholar]
- 69.Morris M. J., Tortelli C. F., Filippis A., Proietto J. (1998) Reduced BAT function as a mechanism for obesity in the hypophagic, neuropeptide Y deficient monosodium glutamate-treated rat. Regul. Pept. 75–76, 441–447 [DOI] [PubMed] [Google Scholar]
- 70.Madden C. J., Morrison S. F. (2009) Neurons in the paraventricular nucleus of the hypothalamus inhibit sympathetic outflow to brown adipose tissue. Am. J. Physiol. Regul. Integr. Comp. Physiol. 296, R831–R843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wittmack E. K., Rush A. M., Craner M. J., Goldfarb M., Waxman S. G., Dib-Hajj S. D. (2004) Fibroblast growth factor homologous factor 2B: association with Nav1.6 and selective colocalization at nodes of Ranvier of dorsal root axons. J. Neurosci. 24, 6765–6775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu C. j., Dib-Hajj S. D., Waxman S. G. (2001) Fibroblast growth factor homologous factor 1B binds to the C terminus of the tetrodotoxin-resistant sodium channel rNav1.9a (NaN). J. Biol. Chem. 276, 18925–18933 [DOI] [PubMed] [Google Scholar]
- 73.Wang C., Chung B. C., Yan H., Lee S. Y., Pitt G. S. (2012) Crystal structure of the ternary complex of a NaV C-terminal domain, a fibroblast growth factor homologous factor, and calmodulin. Structure 20, 1167–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.