

Abstract
Prozymes are pseudoenzymes that stimulate the function of weakly active enzymes through complex formation. The major Trypanosoma brucei protein arginine methyltransferase, TbPRMT1 enzyme (ENZ), requires TbPRMT1 prozyme (PRO) to form an active heterotetrameric complex. Here we present the x-ray crystal structure of the TbPRMT1 ENZ-Δ52PRO tetrameric complex with the cofactor product S-adenosyl-L-homocysteine (AdoHcy) at 2.4 Å resolution. The individual ENZ and PRO units adopt the highly-conserved PRMT domain architecture and form an antiparallel heterodimer that corresponds to the canonical homodimer observed in all previously reported PRMTs. In turn, two such heterodimers assemble into a tetramer both in the crystal and in solution with twofold rotational symmetry. ENZ is unstable in absence of PRO and incapable of forming a homodimer due to a steric clash of an ENZ-specific tyrosine within the dimerization arm, rationalizing why PRO is required to complement ENZ to form a PRMT dimer that is necessary, but not sufficient for PRMT activity. The PRO structure deviates from other, active PRMTs in that it lacks the conserved η2 310-helix within the Rossmann fold, abolishing co-factor binding. In addition to its chaperone function for ENZ, PRO substantially contributes to substrate binding. Heterotetramerization is required for catalysis, since heterodimeric ENZ-PRO mutants lack binding affinity and methyltransferase activity towards the substrate protein TbRGG1. Together, we provide a structural basis for TbPRMT1 ENZ activation by PRO heterotetramer formation, which is conserved across all kinetoplastids, and describe a chaperone function of the TbPRMT1 prozyme, which represents a novel mode of PRMT regulation.
Keywords: PRMT1, pseudoenzyme, chaperone, Trypanosoma brucei, oligomerization
Graphical Abstarct
Introduction
Trypanosoma brucei is a protozoan parasite that can cause fatal sleeping sickness in humans and various animal diseases in Sub-Saharan Africa. An estimated 70 million people are at risk of infection [1]. African trypanosomes adopt two distinct replicative forms during their life cycle, the bloodstream form in the mammal and the procyclic form in the midgut of its insect vector, the tsetse fly [2]. Control of gene expression and life-cycle progression primarily takes place on the post-transcriptional level [3]. However, recent evidence points towards a role of chromatin proteins in this process as well [4]. Among the posttranslational modifications that impact gene expression and other cellular functions, arginine methylation is prevalent with at least 15% of the proteome being modified, including proteins involved in a wide spectrum of processes such as RNA processing, DNA repair, metabolism, and protein trafficking [5, 6]. Arginine methylation is catalyzed by S-adenosyl-L-methionine (AdoMet)-dependent protein arginine (R) methyltransferases (PRMTs) [7]. Type I PRMTs (PRMT1, 2, 3, 4, 6, and 8) generate monomethyl arginine (MMA) and asymmetric dimethyl arginine (ADMA); type II PRMTs (PRMT5 and 9) generate MMA and symmetric dimethyl arginine (SDMA); and type III PRMTs (PRMT7) only produce MMA [8, 9]. Their product specificities are restricted by the size and architecture of their active-site pockets [9–11].
TbPRMT1 enzyme (ENZ) is the predominant type I PRMT1 in T. brucei that produces MMA and ADMA [12–14]. It contributes to parasite virulence, metabolic regulation, and nutritional stress response [15]. RGG/RG motifs are often the target of asymmetric arginine dimethylation in substrates, including the TbPRMT1 substrates TbRBP16 and TbRGG1 [16]. The TbPRMT1 ENZ shares 51% identical residues with type I human and rat PRMT1 [13, 17], and 41% identical residues with type I rat PRMT3 [18]. The PRMT structures suggest that the PRMT fold and the catalytic mechanism are conserved and that at least dimerization of the PRMT cores is required for AdoMet binding and catalysis [17–19]. Human, rat, and yeast PRMT1s are homo-oligomeric complexes with molecular weights of 300–400 kDa in solution [19–21], while rat PRMT3 exists in a monomer-dimer equilibrium in the cell with an activity of 0.3% with respect to rat PRMT1 [18, 22]. By contrast, TbPRMT1 ENZ forms a stable heterotetrameric complex with a catalytically inert TbPRMT1 prozyme (PRO), which was previously termed TbPRMT3 [14]. The methyltransferase activity of TbPRMT1 ENZ by itself is not detectable, but catalytically inert TbPRMT1 PRO is necessary and sufficient to enable TbPRMT1 activity. The mRNA level and protein amount of TbPRMT1 ENZ are constant in both the procyclic and the bloodstream form [23–26]. The protein expression levels of ENZ and PRO are strongly synchronized and interdependent [27]: Repression of TbPRMT1 PRO mRNA reduces the TbPRMT1 ENZ protein level, although the TbPRMT1 ENZ mRNA level remained unchanged [13, 27]. We therefore proposed that the amount of catalytically active TbPRMT1 ENZ is regulated by the catalytically inert TbPRMT1 PRO [14].
In addition to TbPRMT1, three other enzymatic activities activated by prozymes have been discovered in T. brucei to date: AdoMet decarboxylase (TbAdoMetDC), TbRNase III endonucleases within the editosome, and deoxyhypusine synthase (TbDHS) in the polyamine synthesis pathway [14, 28–32]. Prozymes are a subgroup of pseudoenzymes, which are estimated to represent ~10% of the human proteome and are thought to serve as regulators of enzymes [33, 34]. So far, six modes of pseudoenzyme function have been proposed [34]. The TbAdoMetDC enzyme homodimer is inactive due to blockage of the active-site pocket by an N-terminal fragment [29]. The TbAdoMetDC prozyme allosterically unblocks the active site in the catalytically active enzyme-prozyme heterodimer, facilitating enzymatic activity [29]. As for TbRNase III endonucleases within the editosome, three prozymes adopt a chaperone function to form enzymatically competent heterodimeric complexes, but their detailed mechanism of action is presently unclear [32].
TbPRMT1 ENZ and catalytically inactive TbPRMT1 PRO share several conserved features, including the Rossmann fold and motifs I and II (highlighted in bold in Fig. 1), albeit with a lower sequence identity (27%, 82 residues of 304) and similarity (44%, 136 residues of 304) than with human or rat PRMT1 [35]. A striking difference refers to conserved AdoMet binding residues [36, 37], many of which are lacking in TbPRMT1 PRO (Fig. 1). AdoMet crosslink experiments demonstrated that TbPRMT1 ENZ but not TbPRMT1 PRO binds AdoMet, consistent with the notion that PRO is a catalytically inactive enzyme [14].
Figure 1. Structure-based sequence alignment of TbPRMT1 enzyme and prozyme.

Structure-based alignment of TbPRMT1 enzyme (ENZ) and TbPRMT1 prozyme (PRO). α1-α7 refer to α-helices, η1-η8 to 310-helices, and β1-β15 to β-strands, indicating the secondary structure elements of TbPRMT1 ENZ and PRO. Residue numbering is shown above (ENZ) and below (PRO) the sequences. Red designates the N-terminal helical extension (ENZ residues 20–33, PRO residues 71–80), green the Rossmann fold (ENZ residues 34–157, and PRO residues 81–202), orange the β-barrel (ENZ residues 158–345, and PRO residues 203–389), and purple the dimerization arm (ENZ residues 168–199, and PRO residues 213–244). Similar and identical residues are marked as : and *, respectively. Residues highlighted in magenta are involved in tetramer formation, and residues highlighted in cyan are involved in ENZ-PRO dimer formation. Signature residues of the YFxxY motif, Motif-I, -II, the double E-loop, and the THW-loop are shown in bold. Key AdoMet-binding residues of ENZ are highlighted in yellow. Disordered regions are represented by dashed lines.
In order to elucidate the structural basis of enzyme activation by prozyme in TbPRMT1, we determined the crystal structure of the TbPRMT1 ENZ-PRO complex, analyzed its oligomeric state in solution, and performed substrate binding and methyltransferase assays of wild-type and mutant TbPRMT1 ENZ and PRO species. Our results reveal that TbPRMT1 PRO is required for TbPRMT1 ENZ stability, that the heterotetrameric architecture of the TbPRMT1 ENZ-PRO complex is necessary for substrate binding and catalytic activity, and that the features of the TbPRMT1 ENZ-PRO complex are conserved among kinetoplastids, implying a similar mode of PRMT1 regulation in these organisms.
Results
TbPRMT1 ENZ-PRO forms a heterotetrameric complex
TbPRMT1 ENZ-PRO from procyclic-form cells as well as the recombinantly expressed complex in Escherichia coli forms a heterotetramer as deduced from ultracentrifugation, size exclusion chromatography, and multiangle light scattering coupled to size exclusion chromatography (SEC-MALS) [14]. Because wild-type TbPRMT1 ENZ was insoluble or unstable [14] (Table 1), the wild-type TbPRMT1 ENZ (residues 1–345) and hexa-histidine-tagged wild-type TbPRMT1 PRO (residues 1–389) were co-expressed in E. coli using a pETDuet vector system (Novagen) and purified from the soluble fraction, followed by His-tag removal. According to SEC-MALS, the size of this complex was about 164 kDa, which corresponds to a heterotetrameric complex (theoretical molecular weight of 163.2 kDa), as previously described (Table 1) [14]. The heterotetrameric complex was further confirmed by small angle x-ray scattering coupled to SEC (SEC-SAXS) (Fig. 2a) and by negative-stain electron microscopy (EM) (Fig. 2b). In detail, the SEC-SAXS profile yielded a single peak with a constant radius of gyration across the peak (~43.0 Å) (Fig. 2a). Based on the volume of correlation (Vc) of this peak [38], a molecular weight of 187 kDa was estimated (Table 2). The Guinier and Kratky plots revealed that the complex was monodisperse and fully folded (Fig. 2a). The TbPRMT1 ENZ-PRO complex visualized by negative-stain EM showed four distinct globular masses symmetrically arranged at the vertices of a rhomboid structure in the most predominant 2D class average (Fig. 2b). The complex measures about 140 Å in its longest dimension, which fits the maximum distance of 143 Å obtained in a pair-distance distribution by SEC-SAXS (Fig. 2a). Finally, a stoichiometry of 1:1 was obtained for the TbPRMT1 heterotetramer with a Maltose Binding Protein (MBP)-fused TbRGG1 substrate by isothermal titration calorimetry (ITC) (Fig. 2c and Table 1) [14]. Collectively, these data demonstrate that the TbPRMT1 complex forms a rigid heterotetrameric unit in solution.
Table 1.
Methyltransferase activities, molecular weight, and binding affinities of TbPRMT1 ENZ-PRO and truncated TbPRMT1 PRO mutants
| ENZ (1–345) | PRO (1–389) | Expression | Solubility | MALS (kDa) | Methyltransferase activity | MBP-TbRGG1 binding (μM) |
|---|---|---|---|---|---|---|
| full-length | full-length | +++/+++ | +++/++++ | 164 | +++ | 9±1a |
| full-length | Δ11 | +++/+++ | +++/++++ | 162 | +++ | n.d.b |
| full-length | Δ21 | +++/+++ | +++/++++ | 165 | +++ | 36±8 |
| full-length | Δ31 | +++/+++ | +++/++++ | 171 | +++ | 86±10 |
| full-length | Δ41 | +++/+++ | +++/++++ | 160 | +++ | n.d.b |
| full-length | Δ45 | +++/+++ | +++/++++ | n.d.b | + | 160±20 |
| full-length | Δ52 | +++/+++ | +++/++++ | 157 | - | no binding |
| full-length | - | +++ | not soluble |
The standard deviations were calculated from two or three independent measurements
not determined
Figure 2. TbPRMT1 ENZ-PRO forms a heterotetrameric complex in solution.

(a) SEC–SAXS analysis of TbPRMT1 ENZ-PRO. Top left panel: SEC-SAXS integrated intensities (left y-axis) plotted against frame number (x-axis). The red dots indicate radius of gyration, Rg (on the right y-axis). Top right panel: Guinier plot calculated from averaging buffer-subtracted scattering intensities. The coefficient of determination, R2, is 0.9986. Bottom left panel: Pair-distance distribution function P(r), yielding a maximum molecular diameter of 143 Å. Bottom right panel: Normalized Kratky plot calculated from SEC–SAXS data.
(b) Negative-stain electron microsopy. EM micrograph with a 200 nm scale bar. Inset: Predominant 2D class average.
(c) ITC thermogram (upper panel) and plot of corrected heat values (lower panel) for binding of the heterotetrameric TbPRMT1 ENZ-PRO complex to Maltose Binding Protein (MBP)-fused TbRGG1 protein.
Table 2.
SEC-SAXS analysis of ENZ-PRO wild-type, ENZ-Δ52PRO, and interface mutants
| ENZ | WT | WT | W46A/R47A/Y50A | WT | WT | WT | H270A |
|---|---|---|---|---|---|---|---|
| PRO | WT | Δ52 | WT | Y215A/M219A | Y215A/M219A/E223A | Y215A/T294A | Y215A |
| instrument | G1 beamline | ||||||
| wavelength (Å) | 1.25 | ||||||
| exposure time (s) | 1 | ||||||
| temperature (K) | 277 | ||||||
| protein concentration (mg/ml) | 3.8/8.0 | 1.5/4.0/8.0 | 2.3/3.2 | 3.0 | 3.0/16 | 6.0/12.0 | 6.0/16.0 |
| radius of gyration Rga,b (Å) | 42.9±0.2 | 40.9±0.6 | 32.1±0.7 | 33.4±0.1 | 32.6±0.2 | 32.0±0.1 | 32.1±0.3 |
| maximum
diameter, Dmaxb |
143±2 | 138±4 | 116±7 | 120 | 118±3 | 117±2 | 118±4 |
| M.W.b,c (kDa) | 187±2 | 166±9 | 84.7±0.7 | 88.3 | 83±3 | 84.1±0.8 | 82.6±0.8 |
| theoretical M.W.d (kDa) | 163.2 | 152.8 | 81.3 | 81.5 | 81.3 | 81.5 | 81.5 |
Rg determined from a Guinier plot.
The standard deviations were calculated from two or three independent measurements except for the ENZ(WT)/PRO(Y215A/M219A) mutant, where the error corresponds to the fitting error of the Guinier plot.
The M.W. was derived from the volume of correlation Vc [33].
The theoretical M.W. refers to a heterotetramer for the wild-type and the ENZ-Δ52PRO version and to a heterodimer for the mutants.
The TbPRMT1 PRO N-terminus contributes to substrate recognition
Previous studies have shown that all PRMTs contain a highly conserved core domain comprising ~310 residues [8] and that the N-terminal region of PRMTs is often flexible and involved in substrate recognition [39, 40]. Among kinetoplastids, the N-terminal regions of putative PRMT1 ENZ and PRO have highly diverged (Fig. S1 and S2). Therefore, we examined the role of the N-terminal residues of TbPRMT1 PRO and ENZ. Limited proteolysis on the full-length proteins identified a stable, N-terminally truncated TbPRMT1 PRO fragment spanning residues 53–389, referred to as TbPRMT1 Δ52PRO, while TbPRMT1 ENZ remained intact under the conditions tested. The TbPRMT1 Δ52PRO fragment was then co-expressed with full-length TbPRMT1 ENZ. SEC-MALS, SEC-SAXS, and SEC alone confirmed that the TbPRMT1 ENZ-Δ52PRO complex still formed a tetrameric complex (Fig. S3a and Tables 1 and 2). However, its binding affinity for MBP-TbRGG1 was abolished (Fig. S3b), consistent with methyltransferase inactivity, even in complex with full-length TbPRMT1 ENZ (Table 1). These data suggest that the N-terminus of TbPRMT1 PRO is essential for substrate binding, while the N-terminal region of TbPRMT1 ENZ is not sufficient for that. When we measured the methyltransferase activities and MBP-TbRGG1 protein binding affinities of a series of N-terminal TbPRMT1 PRO deletion mutants in complex with full-length wild-type TbPRMT1 ENZ, we found that the first ~40 N-terminal, non-conserved residues of TbPRMT1 PRO are dispensable for methyltransferase activity (Table 1 and Fig. S2). These data imply that the conserved N-terminal region between residues 41 and 52 of TbPRMT1 PRO is critical for substrate recognition, whereas the N-terminal residues 1–15 of ENZ are not sufficient for substrate binding, consistent with its non-conserved nature among kinetoplastids (Fig. S1).
TbPRMT1 ENZ and PRO form heterodimers that assemble into a heterotetramer
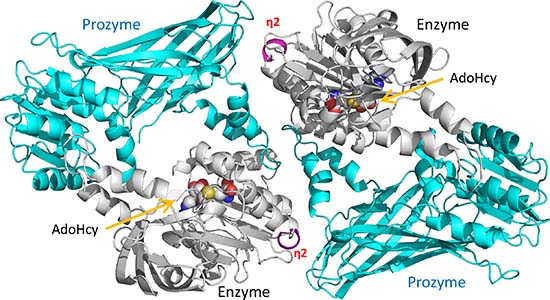
In order to obtain mechanistic insights into TbPRMT1 ENZ activation by TbPRMT1 PRO at atomic resolution, we crystallized the stable TbPRMT1 ENZ-Δ52PRO complex with the methylation cofactor product AdoHcy and solved the structure at 2.4 Å resolution from a seleno-methionine derivatized crystal using the single anomalous dispersion (SAD) phasing technique (Supplemental Table S1). Two TbPRMT1 ENZ (shown in gray) and two TbPRMT1 Δ52PRO (shown in cyan) molecules form the asymmetric unit (Fig. 3a). No electron density of the TbPRMT1 ENZ residues 1–20, the TbPRMT1 ENZ loop region between β9-β10 strands (residues 241–250), and TbPRMT1 Δ52PRO residues 53–70 was observed. Consistent with our previous biochemical finding [14], AdoHcy was only bound to TbPRMT1 ENZ (Fig. 3a). Notably, one TbPRMT1 ENZ and one TbPRMT1 Δ52PRO molecule form a canonical PRMT ring-like dimeric structure that has been observed in all other, homodimeric PRMTs thus far [17–19, 41] (Fig. 3b). In turn, two TbPRMT1 ENZ-Δ52PRO heterodimers touch each other side by side, exhibiting a 2-fold non-crystallographic symmetry (Fig. 3a). The two dimers are highly similar with a root-mean-square deviation (rmsd) of only 0.6 Å when comparing 637 pairs of Cα atoms. The dimensions of the TbPRMT1 ENZ-Δ52PRO heterotetrameric complex are 131 Å × 70 Å × 78 Å (Fig. 3a). Importantly, the molecular size and shape of the crystal structure are in good agreement with the results of SEC-MALS (Table 1), SEC-SAXS (Supplemental Fig. S3a, Fig. 2a, and Table 2), and electron microscopy (Fig. 2b), therefore corroborating the tetrameric form in solution, which is further confirmed by comparing the calculated radii of gyration of the TbPRMT1 ENZ-Δ52PRO heterotetramer and heterodimer (39 Å and 28 Å, respectively) with the experimental radius of gyration (41Å) [42].
Figure 3. Crystal structure of the heterotetrameric TbPRMT1 ENZ-PRO complex.

(a) Tetrameric TbPRMT1 ENZ-PRO complex. One TbPRMT1 ENZ (gray) bound to AdoHcy shown in space-filling representation forms a heterodimer with TbPRMT1 PRO (cyan). The dimensions of the tetrameric complex are shown. The non-crystallographic twofold symmetry axis is shown as a black pointed oval. Key residues involving tetramerization (His270 of ENZ, and Tyr215 of PRO) are indicated.
(b) Anti-parallel TbPRMT1 ENZ-PRO heterodimer. The N-terminal helical extension (red), the Rossmann fold (green), the β-barrel (orange) and the dimerization arm (purple) are shown. AdoHcy, His270, and Tyr215 are highlighted.
(c) Monomeric TbPRMT1 ENZ structure.
(d) Monomeric TbPRMT1 PRO structure.
(e) Topology of TbPRMT1 ENZ.
(f) Topology of TbPRMT1 PRO.
(g) Superimposition of monomeric TbPRMT1 ENZ and PRO. Left panel: Overall structure. Right panel: Close-up view on Rossmann fold, illustrating rotated α-helices.
(h) Simulated-annealing omit electron density map for AdoHcy, contoured at 3.5σ above the mean.
TbPRMT1 ENZ adopts the canonical PRMT fold and features a type I active-site architecture
TbPRMT1 ENZ harbors seven α-helices, six 310-helices, and 15 β-strands (Figs. 1, 3c, and 3e). The two TbPRMT1 ENZ molecules within the asymmetric unit are highly similar (rmsd of 0.4 Å, comparing 318 pairs of Cα atoms). The overall monomeric structure of TbPRMT1 ENZ strongly resembles the monomeric class I PRMT core domain structure of rat PRMT1, rat PRMT3, mouse CARM1 (PRMT4), and yeast RMT1 (Supplemental Table S2), sharing an identical topology with the rat PRMT1 core domain (Fig. 3e and Supplemental Fig. S4) [17–19, 41]. Like other PRMTs, TbPRMT1 ENZ contains the four highly conserved modules of PRMTs: an N-terminal helical extension (residues 20–33 in red), a Rossmann fold (residues 34–157 in green), a dimerization arm (residues 168–199 in purple), and a β-barrel domain (residues 158–345 in orange) (Figs. 3b, 3c, and Supplemental Fig. S4). The electron density of the co-factor product AdoHcy is clearly observed in both TbPRMT1 ENZ molecules (Figs. 3a and 3h). AdoHcy interacts with residues that are highly conserved among human PRMT1, rat PRMT1, and TbPRMT1 ENZ for AdoMet binding [17, 18, 36]: Tyr22, His28, Arg37, Asp83, Cys84, Glu112, Glu126, Met137, Thr140, the main chain of Gly63 and Val111, and Asp59 via two water molecules (Fig. 3h). The TbPRMT1 ENZ active site possesses the previously described PRMT type I features [9–11]: An open subregion A is adjacent to the double E-loop, while subregion B towards the conserved THW loop is sterically more restricted, which enables conversion to mono- and asymmetric dimethyl arginine, but not to symmetric dimethylarginine (Supplemental Fig. S5). The distances between atoms of conserved TbPRMT1 ENZ residues and of the sulfur atom of AdoHcy recapitulate the type I enzyme active-site architecture and provide a structural basis for TbPRMT1 ENZ product specificity [14].
Lack of the η2 310-helix is a unique feature of the TbPRMT1 PRO core domain that twists its Rossmann fold
The two TbPRMT1 Δ52PRO molecules (cyan in Fig. 3a) within the asymmetric unit superimpose closely (rmsd of 0.4 Å, comparing 319 pairs of Cα atoms) and harbor the four canonical PRMT modules: an N-terminal helical extension (residues 71–80 in red), a Rossmann fold (residues 81–202 in green), a dimerization arm (residues 213–244 in purple), and a β-barrel domain (residues 203–389 in orange) (Figs. 3b and 3d). While the monomeric structures of TbPRMT1 ENZ and Δ52PRO are relatively similar (Fig. 3g, rmsd of 2.1 Å, comparing 293 pairs of Cα atoms), the topology of TbPRMT1 Δ52PRO bears a few marked differences with respect to TbPRMT1 ENZ and other PRMT1s (Figs. 3e and 3f) [17–19, 41]. Within the Rossmann fold, TbPRMT1 PRO lacks the η2 310-helix between the α2 helix and β1 strand, and has an extra η4 310-helix between the η3 310-helix and β4 strand (Figs. 1 and 3f). Importantly, the η2 310-helix is highly conserved among active enzymes (Supplemental Fig. S6) [8]. As a result of the lacking η2 310-helix, the α2, α3 and, α4 helices within the TbPRMT1 PRO Rossmann fold are tilted by 10–20°, whereas the β sheets of the TbPRMT1 ENZ and PRO Rossmann folds align well (Fig. 3g). In turn, these observed differences in the secondary structure elements of TbPRMT1 PRO twist and hence affect the dimerization interface, compromising cofactor AdoMet binding and providing a structural basis for TbPRMT1 PRO inactivity.
TbPRMT1 PRO is required for dimerization and ENZ stability
The η1 310-helix and the α1 and α2 helices contribute to AdoMet binding (Fig. 3h) and dimerization of PRMTs (Fig. 3b) [8]. The total buried dimerization surface area between TbPRMT1 ENZ and PRO is ~1,600 Å2, which is similar to that of other, homodimeric PRMTs. Since dimerization arm mutants that lead to monomeric PRMTs do not have methyltransferase activity [10, 17, 19], dimerization of PRMTs is required for activity [9, 10, 17–19, 39, 41, 43]. As we previously reported, TbPRMT1 ENZ expressed by itself is an unstable protein that requires TbPRMT1 PRO to form a stable, catalytically active complex [14]. TbPRMT1 PRO, on the other hand, can be expressed by itself, albeit at a reduced amount, indicating decreased stability, and forms a homodimer [14]. Thus, the protein amount of folded TbPRMT1 ENZ is limited and hence may be regulated by TbPRMT1 PRO in vivo [14, 27]. As the molecular surface of the monomeric TbPRMT1 ENZ structure displays highly hydrophobic patches, covering these hydrophobic regions via dimerization with TbPRMT1 PRO is a likely mechanism to stabilize the TbPRMT1 ENZ protein. Particularly the dimerization arm of TbPRMT1 ENZ is highly hydrophobic and dominated by aromatic residues (Fig. 4a). In detail, the hydrophobic residues Ile179, Trp180, Val183, Ile186, Phe188, Tyr190, Phe191, and Leu194 of the ENZ dimerization arm contact the hydrophobic residues Ile72, Leu76, Ile79, Leu85, Met107, Leu109, Ile113, Ile130, Ala133, and Val137 on the η1 310-helix, α1, α2, and α3 helices of TbPRMT1 PRO (Fig. 4c). Similarly, the surface of the TbPRMT1 PRO dimerization arm is hydrophobic. The TbPRMT1 PRO residues Thr221, Phe224, Trp225, Val228, Tyr229, Phe231, Met233, Pro235, Met236, Leu239, and Val240 contact the hydrophobic TbPRMT1 ENZ residues Tyr25, Met29, Lys33, Cys35, Thr38, Thr39, Arg42, Trp46, Thr64, Ile66, Phe70, Val87, Gln90, Ile94 and Phe100 on the η1 310-helix, α1, α2, and α3 helices (Fig. 4b). Upon dimerization of TbPRMT1 ENZ and PRO, the hydrophobic patches of the η1 310-helix and α1 helix in TbPRMT1 ENZ become buried, leading to ENZ stabilization.
Figure 4. The dimerization arm of TbPRMT1 ENZ features non-conserved residues, preventing TbPRMT1 ENZ homodimerization.

(a) Sequence alignment of the dimerization arm regions of TbPRMT1 ENZ (TriTrypDB ID: Tb927.1.4690), rat PRMT1 (GenBank ID: NP_077339), rat PRMT3 (NP_446009), budding yeast RMT1 (NP_009590), fission yeast RMT1 (NP_594825), and TbPRMT1 PRO (TriTrypDB ID: Tb927.10.3560). Conserved interface residues are colored in green. Non-conserved interface residues are colored in yellow. The non-conserved TbPRMT1 ENZ Tyr190 is highlighted in magenta.
(b) Hydrophobic interaction of the TbPRMT1 PRO dimerization arm with the TbPRMT1 ENZ Rossmann fold.
(c) Hydrophobic interaction of the TbPRMT1 ENZ dimerization arm with the TbPRMT1 PRO Rossmann fold.
(d) Modelled TbPRMT1 PRO-PRO interface.
(e) Modelled TbPRMT1 ENZ-ENZ interface. The non-conserved Tyr190 causes a steric clash between two TbPRMT1 ENZ molecules highlighted by a red star, which interferes with dimerization.
The two dimerization arms of TbPRMT1 ENZ and PRO are structurally highly similar (rmsd of 0.7 Å, comparing 124 pairs of Cα atoms). When we generate homodimeric models of TbPRMT1 ENZ and PRO, respectively, we observe that TbPRMT1 PRO can indeed form homodimers without any severe steric clashes (Fig. 4d), while Tyr190 of TbPRMT1 ENZ, corresponding to Pro235 of TbPRMT1 PRO, clearly clashes with the side chains of Gln90, Glu93, and Ile94 on the α4 helix of TbPRMT1 ENZ (Fig. 4e). Consequently, TbPRMT1 ENZ is predicted to be unable to form a homodimer due to steric clashes and requires TbPRMT1 PRO to engage into a stable TbPRMT1 ENZ-PRO complex.
The TbPRMT1 heterotetrameric assembly is required for substrate binding and catalytic activity
Human and rat PRMT1 predominantly form oligomeric structures [17, 19–21], which are the active species in vivo [44]. PRMT3 exists in a monomer-dimer equilibrium in solution [18, 22]. However, PRMT3 crystal structures revealed the canonical dimeric arrangement, consistent with the notion that PRMT dimerization is necessary for catalytic activity [17–19]. TbPRMT1 ENZ and PRO form a stable heterotetramer (Figs. 2 and 3a) [14] that binds one substrate molecule (Fig. 2c). The interface between two heterodimers amounts to ~950 Å2 and is dominated by van der Waals’ contacts; Tyr50 of TbPRMT1 ENZ interacts with hydrophobic residues Val375 and Val376 of TbPRMT1 PRO, His270 of TbPRMT1 ENZ interacts with residues Asp292, Thr293, Thr294, Pro340, Leu341, and Val375 of PRO, Tyr215 of TbPRMT1 PRO interact with residues Asp43, Trp46, and Arg47 of TbPRMT1 ENZ, and Met219 of TbPRMT1 PRO interacts with Trp46 of TbPRMT1 ENZ (Fig. 5). We mutated various key dimer-dimer surface residues to alanine in order to test whether they break down the tetramer into dimers, and evaluated the size of the resulting mutants using SEC, SEC-MALS, SEC-SAXS, and EM (Fig. 6, Tables 2 and 3). Indeed, the TbPRMT1 ENZ triple mutant W46A/R47A/Y50A, the TbPRMT1 ENZ double mutant Y50A/H270A, the TbPRMT1 PRO double mutants Y215A/M219A, Y215A/E223A, Y215A/K232A, Y215A/T294A, as well as the TbPRMT1 PRO triple mutant Y215A/M219A/E223A all disrupt the tetrameric complex and form a stable heterodimer. Moreover, the combination of a single mutation in each protein, H270A in TbPRMT1 ENZ and Y215A in TbPRMT1 PRO, disrupted the tetramer as well (Tables 2 and 3). In contrast to the rhomboid shape of the wild-type, negative-stain EM analysis of a mutant revealed a square-shaped structure, consistent in shape and size with an TbPRMT1 ENZ-PRO heterodimeric unit (Figs. 3b and 6b). Extensive SEC-SAXS data obtained from these mutants confirm the substantially smaller size of the heterodimer with respect to the heterotetrameric wild-type structure (Fig. 6a, Table 2). These solution studies verify the dimer-dimer interface observed in the crystal structure. Importantly, ITC binding and methyltransferase assays showed that none of the dimeric mutants bound MBP-TbRGG1 and did not possess any detectable methyltransferase activity (Fig. 6c and Table 3). On the other hand, the single mutations K73A, W64A, R47A, Y50A, and H270A in TbPRMT1 ENZ, the double alanine mutant H270A/E271A in TbPRMT1 ENZ and single mutations Y215A and K232A in TbPRMT1 PRO retained the tetrameric assembly and methylate the substrate as the wild-type complex (Table 3). We conclude that the determined TbPRMT1 ENZ-PRO heterotetramer structure is the biological functional unit both in vitro and in vivo [14], and that heterotetramer assembly is required for substrate binding and methyltransferase activity.
Figure 5. The interface between two TbPRMT1 ENZ-PRO heterodimers is dominated by van der Waals’ contacts.

Residues forming key contacts between two TbPRMT1 ENZ-PRO heterodimers are displayed in dotted-sphere representation, indicating their van der Waals’ radii. Mutation of the underlined residues breaks the heterotetrameric assembly into TbPRMT1 ENZ-PRO heterodimers.
Figure 6. Structure-based mutagenesis of the tetrameric interface yields stable TbPRMT1 ENZ-PRO heterodimers.

(a) SEC–SAXS analysis of a representative heterodimeric TbPRMT1 ENZ-PRO mutant (the ENZ W46A/R47A/Y50A triple mutant). Top left panel: SEC-SAXS integrated intensities (left y-axis) plotted against frame number (x-axis). The red dots indicate radius of gyration, Rg (on the right y-axis). Top right panel: Guinier plot calculated from averaging buffer-subtracted scattering intensities. The coefficient of determination, R2, is 0.9989. Bottom left panel: Pair-distance distribution function P(r), yielding a maximum molecular diameter of 116 Å. Bottom right panel: Normalized Kratky plot calculated from SEC–SAXS data.
(b) Negative-stain electron microscopy analysis of a representative TbPRMT1 ENZ-PRO mutant (the PRO Y215A/M219A/E223A triple mutant). EM micrograph with a 200 nm scale bar. Inset: Predominant 2D class average.
(c) ITC thermogram (upper panel) and plot of corrected heat values (lower panel) for binding of the TbPRMT1 ENZ Y215A/M219A/E223A triple mutant to Maltose Binding Protein (MBP)-fused TbRGG1 protein.
Table 3.
Methyltransferase activity, molecular weight, and MBP-TbRGG1 binding affinities of ENZ-PRO and tetrameric interface mutants
| ENZ (1–345) | PRO (1–389) | Expression | Solubility | MALS (kDa) | SEC elution volume (mL) | Methyl-transferase activity | MBP-TbRGG1 Binding (μM) |
|---|---|---|---|---|---|---|---|
| full-length | full-length | +++/+++ | +++/+++ | 164 | 68 | +++ | 9±1a |
| K73A | full-length | +++/+++ | +++/+++ | 164 | 68 | n.d. | n.d.b |
| H270A, E271A | full-length | +++/+++ | +++/+++ | 142 | 71 | +++ | n.d. |
| W46A | full-length | +++/+++ | +++/+++ | 167 | 67 | n.d. | n.d. |
| R47A | full-length | +++/+++ | +++/+++ | 162 | 69 | n.d. | n.d. |
| Y50A | full-length | +++/+++ | +++/+++ | 152 | 69 | n.d. | n.d. |
| H270A | full-length | +++/+++ | +++/+++ | 149 | 69 | n.d. | n.d. |
| W46A, R47A, | |||||||
| Y50A | full-length | +++/+++ | +++/+++ | 80 | 80 | - | no binding |
| Y50A, H270A | full-length | +++/+++ | +++/+++ | 89 | 74 | - | n.d. |
| full-length | Y215A | +++/+++ | +++/+++ | 107 | 70 | + | n.d. |
| full-length | K232A | +++/+++ | +++/+++ | 165 | 68 | n.d. | n.d. |
| full-length | Y215A, M219A | +++/+++ | +++/+++ | 84 | 76 | - | no binding |
| full-length | Y215A, M219A, E223A | +++/+++ | +++/+++ | 81 | 79 | - | no binding |
| full-length | Y215A, E223A | +++/+++ | +++/+++ | 84 | 77 | - | no binding |
| full-length | Y215A, K232A | +++/+++ | +++/+++ | 90 | 75 | - | n.d. |
| full-length | Y215A, T294A | +++/+++ | +++/+++ | 81 | 79 | - | no binding |
| H270A | Y215A | +++/+++ | +++/+++ | 80 | 79 | - | no binding |
The standard deviations were calculated from two or three independent measurements
not determined
Discussion
Catalytically inert pseudoenzymes are abundant in the proteome and perform diverse functions, serving as scaffold proteins [45], modulators of enzyme activity and signaling pathway components [30], or competitors of active paralogs for substrate(s) [34]. Prozymes are a subgroup of pseudoenzymes that specifically stimulate an otherwise inactive enzyme by complex formation. Here, we describe a chaperone function of the TbPRMT1 prozyme (PRO) in complex with the TbPRMT1 enzyme (ENZ) in T. brucei. Structural and functional analyses reveal distinct structural features that set TbPRMT1 PRO apart from its active counterpart TbPRMT1 ENZ and elucidate the mechanism of TbPRMT1 ENZ activation by TbPRMT1 PRO through oligomerization.
Sequence analysis alone had already indicated that TbPRMT1 PRO was lacking conserved residues that are critical for catalysis and methyl donor (AdoMet) binding and hence provided evidence that TbPRMT1 PRO would catalytically be inactive [14]. Indeed, the crystal structure of the TbPRMT1 ENZ-PRO complex showed that the cofactor product was only bound to TbPRMT1 ENZ, in agreement with an AdoMet crosslink experiment (Fig. 3h) [14]. However, our structural analysis revealed further marked differences on the secondary-structure level of TbPRMT1 PRO with respect to TbPRMT1 ENZ (Fig. 1), most notably the lack of the η2 310-helix in the Rossmann fold of PRO, which affect its tertiary structure (Fig. 3g) and which are ultimately critical for its catalytic inactivity. The lack of the η2 310-helix results in a tilt of several adjacent α-helices, which in turn affect dimerization and abolish cofactor binding in the prozyme (Fig. 3g).
While TbPRMT1 ENZ possesses all essential residues and secondary structure elements for cofactor AdoMet binding and catalysis (Figs. 1 and 3h), TbPRMT1 ENZ itself is unstable in solution in the absence of TbPRMT1 PRO and hence incapable of catalysis [27]. TbPRMT1 ENZ features highly hydrophobic patches in the dimerization arm that are thermodynamically unfavorable if exposed to solvent [46]. While hydrophobic patches are also found in other PRMTs, homodimerization covers these hydrophobic patches to stabilize the active enzymes [8, 17–19]. By contrast, TbPRMT1 ENZ homodimerization is sterically prevented, specifically by Tyr190 within the dimerization arm of TbPRMT1 ENZ (Fig. 4e). In mammalian and yeast class I PRMTs, as well as in TbPRMT1 PRO, the corresponding residue is mostly a cysteine or a proline, thus facilitating formation of a stable homodimer [14, 17–19] (Fig. 4a). However, we note that the Tyr190 mutation to Cys or Pro was not sufficient to form a homodimeric enzyme complex (data not shown), suggesting that homodimerization does not depend on a single residue.
Higher-order oligomerization beyond the homodimer is well established in PRMTs. For example, PRMT1s from various organisms form a hexamer in solution [17, 19–21]. While the oligomerization of mammalian and yeast PRMT1s is dependent on PRMT concentration [17], we did not detect such a dependence for the TbPRMT1 complex, as it always remained tetrameric under different protein concentrations (Supplemental Fig. S7). Dimerization is required for AdoMet binding in PRMTs [17–19], and even higher oligomerization of human PRMT1 is required for its catalysis in vivo [44]. As we demonstrated here, substrate binding and methyltransferase activities are essentially abolished in the stable TbPRMT1 ENZ-PRO dimeric complex mutants with respect to the wild-type tetrameric complex (Table 2). We conclude that oligomers beyond the dimer may generally constitute the active species of PRMT1s, with dimerization not being sufficient for its activities in vitro and/or in vivo.
Although the TbPRMT1 ENZ-PRO heterotetramer possesses two active sites, only one MBP-TbRGG1 substrate molecule is bound to the tetramer (Fig. 2c). In principle, an allosteric mechanism between the two heterodimers could explain this finding, whereby substrate binding to one heterodimer would induce changes in the other dimer that would prevent further substrate binding. Alternatively, a tetramer could provide a unique composite interaction surface not present in the heterodimer. Our ITC experiments show that substrate binding is abolished in heterodimer mutants (Table 2), supporting the latter mechanism. We cannot exclude though that both TbPRMT1 ENZ-PRO heterodimers of a tetramer may independently and in parallel be engaged in the methylation of other substrate proteins or even TbRGG1 without the bulky MBP fusion partner [13]. However, attempts to remove the MBP fusion partner from TbRGG1 have not yielded stable TbRGG1.
The concept that TbPRMT1 PRO is necessary to form a stable, catalytically active PRMT heterodimer with TbPRMT1 ENZ has important implications for regulation. In essence, the activity of TbPRMT1 ENZ would be regulated by the protein amount of TbPRMT1 PRO. As the knockdown of TbPRMT1 PRO has shown, its mRNA reduction did not affect the mRNA amount of TbPRMT1 ENZ, but affected the protein amount of TbPRMT1 ENZ [27]. Unlike all previously reported PRMTs, TbPRMT1 ENZ is unstable on its own because it cannot form a homodimer, in part due to Tyr190 of TbPRMT1 ENZ by steric clashes (Fig. 4e). TbPRMT1 PRO, but not ENZ, was found in stress granules, where it would not be accessible for TbPRMT1 ENZ translated in the cytosol, implying that sequestration of TbPRMT1 PRO may provide a means of controlling TbPRMT1 ENZ activity. We propose that TbPRMT1 PRO serves as a folding chaperone for its catalytic partner, providing a new paradigm for prozyme function. Thus, the expression and localization of TbPRMT1 PRO are ultimately determinants of TbPRMT1 ENZ activity.
Analogous folding chaperones have recently been described for TbRNase III enzymes within the RNA editing machinery of T. brucei [32], supporting the paradigm for prozyme function in this organism. However, a more detailed comparison between these systems must further await biochemical and structural studies of the editosome complexes. With respect to the other two well-characterized prozymes in T. brucei, the folding chaperone function of TbPRMT1 PRO is distinct from AdoMet decarboxylase (TbAdoMetDC) PRO, which serves as an allosteric activator for TbAdoMetDC ENZ [29], and from deoxyhypusine synthase (TbDHS) PRO, which activates TbDHS ENZ by direct active-site complementation [31]. While the former ENZ-PRO system also utilizes AdoMet as a substrate, the fold as well as the AdoMet binding mode of AdoMetDCs are unrelated to those of PRMTs [47, 48].
PRMT1 prozyme function has thus far not been well studied in other related protozoan parasites. Sequence alignments of ENZ and PRO with homologs from related protozoan kinetoplastids (Supplemental Figs. S1 and S2) including the human parasites T. cruzi causing Chagas disease and Leishmania spp. causing various forms of leishmaniasis suggest that PRMT1 ENZ-PRO complexes also exist in many other parasites and corroborate numerous features and conclusions that we present here for TbPRMT1. Among the putative TbPRMT1 PRO homologs, AdoMet binding residues are not conserved, and the residues forming the η2 310-helix are missing, consistent with catalytically inactive TbPRMT1 PRO. Furthermore, dimerization residues of TbPRMT1 ENZ and PRO are vastly conserved, indicating the same heterodimer formation in other kinetoplastids. Specifically, Tyr190 of TbPRMT1 ENZ is invariant, arguing that the PRMT1 ENZ proteins of other species are similarly incapable of homodimerization because of steric clashes and that they therefore necessitate a prozyme for stability. Intriguingly, tetrameric interface residues are largely conserved as well, which provides further evidence that heterotetramers are the active species and a prerequisite for methylation. Finally, the first 40 N-terminal TbPRMT1 PRO residues are not conserved, which coincides with our finding that this region is dispensable for substrate binding, while the adjacent residues 41–52 are conserved and critical for substrate recognition. In TbPRMT1 ENZ, the N-terminal residues 1–15 are not conserved and not sufficient for substrate binding. Among the kinetoplastid PRMTs, the enzyme-prozyme paradigm only exists for PRMT1, while TbPRMT5, TbPRMT6 [49], and TbPRMT7 [9, 10] do not have such prozymes for regulation.
From an evolutionary standpoint, we speculate that PRMT1 ENZ and PRO have co-evolved to furnish a functional PRMT1 enzyme, as a mutation in PRMT1 ENZ such as Cys190-to-Tyr would render PRMT1 ENZ unstable, unless PRMT1 PRO concomitantly emerged to function as a folding chaperone for PRMT1 ENZ. Initially, PRMT1 PRO may have been catalytically active, but over time, mutations of the PRMT1 PRO AdoMet binding site may have transformed PRMT1 PRO into a catalytically dead enzyme, focusing on its primary role as a regulator of PRMT1 ENZ. Even if the AdoMet binding residues were not mutated, lack of η2 310-helix alone may have compromised AdoMet binding within the Rossmann fold. The fact that PRMT1 ENZ-PRO complexes are conserved throughout kinetoplastids suggests that this regulatory mechanism proved to be valuable to these organisms and may constitute a general mechanism of PRMT regulation beyond kinetoplastids.
Materials and Methods
Protein expression and purification
DNA fragments of TbPRMT1 PRO (TriTrypDB: Tb927.10.3560) and TbPRMT1 ENZ (TriTrypDB: Tb927.1.4690) were amplified by PCR from genomic DNA and cloned into the multiple coning sites (MCS) 1 and 2 using the NcoI/NotI and NdeI/XhoI restriction sites of a modified pETDuet-1 vector (Novagen) containing an N-terminal PreScission protease (GE Healthcare) cleavable His6-tag prior to MCS 1. The constructs were overexpressed in E. coli BL21-CodonPlus(DE3)-RIL cells (Stratagene) and grown in LB media containing appropriate antibiotics. Mutations in ENZ and PRO were introduced by overlap extension PCR mutagenesis. Protein expression was induced at OD600 of ≈0.4 with 0.1 mM isopropyl-β-D-thiogalactoside at 18 °C for 16 h. The cells were harvested by centrifugation at 7,500 × g and 4°C and lysed with a cell disrupter (Avestin) in a buffer containing 20 mM Tris, pH 8.0, 300 mM NaCl, 5 mM β-mercaptoethanol, 0.5 mM 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride (Sigma), 2 mM bovine lung aprotinin (Sigma), and complete EDTA-free protease inhibitor cocktail (Roche). After centrifugation at 35,000 × g for 45 min, the cleared lysate was loaded onto a Ni-NTA column (Qiagen) and eluted with an imidazole gradient. Protein-containing fractions were pooled, dialyzed against a buffer containing 20 mM Tris, pH 8.0, 100 mM NaCl, and 5 mM dithiothreitol (DTT), and subjected to cleavage with PreScission protease (GE Healthcare) for 5 h at 4°C. Following His6-tag removal, the cleaved protein was bound to a heparin column (GE Healthcare) and eluted with a NaCl gradient. Protein-containing fractions were pooled, concentrated, and purified on a HiLoad Superdex 200 16/60 gel filtration column (GE Healthcare) in a buffer containing 20 mM HEPES, pH 7.5, 150 mM NaCl, and 0.5 mM Tris(2-carboxyethyl)phosphine hydrochloride (TCEP).
MBP-tagged TbRGG1 was cloned into a pMAL-c2X vector (NEB) using BamHI and SalI restriction sites. TbRGG1 was overexpressed in E. coli BL21-CodonPlus(DE3)-RIL cells (Stratagene) and grown in LB media containing appropriate antibiotics. Protein expression was induced at OD600 of ≈0.4 with 0.1 mM isopropyl-β-D-thiogalactoside at 18 °C for 16 h. The cells were harvested by centrifugation at 7,500 × g and 4°C and lysed with a cell disrupter (Avestin) in a buffer containing 20 mM Tris, pH 8.0, 300 mM NaCl, 5 mM β-mercaptoethanol, 0.5 mM 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride (Sigma), 2 mM bovine lung aprotinin (Sigma), and complete EDTA-free protease inhibitor cocktail (Roche). After centrifugation at 35,000 × g for 45 min, the cleared lysate was loaded onto an amylose resin (NEB) and eluted with a maltose gradient. Protein-containing fractions were pooled, dialyzed against a buffer containing 20 mM Tris, pH 7.5, 20 mM NaCl, and 5 mM dithiothreitol (DTT). The protein was bound to a SP column (GE Healthcare) and eluted with a NaCl gradient. Protein-containing fractions were pooled, concentrated, and purified on a HiLoad Superdex 200 16/60 gel filtration column (GE Healthcare) in a buffer containing 20 mM HEPES, pH 7.5, 150 mM NaCl, and 0.5 mM TCEP.
Crystallization, data collection, structure determination and refinement
For formation of the complex with the AdoHcy, 4.5 mg/ml of purified TbPRMT1 ENZ-Δ52PRO was mixed in a 1:8 molar ratio with AdoHcy and incubated for 4 h on ice. The crystallization solution consisted of 7% PEG 4000 and 0.1 M Tris, pH 7.4. Crystals grew in space group C2 at room temperature within a week. X-ray diffraction data were collected at the 24ID-C beamline at the NE-CAT at the Advanced Photon Source (APS) of Argonne National Laboratory (ANL). Diffraction data were processed in HKL2000 [50]. The structure was solved by the single anomalous dispersion (SAD) phasing technique in the program AutoSol of the PHENIX package [51], using data obtained from seleno-L-methionine-labeled crystals. The asymmetric unit contained one tetramer. Model building was performed in O [52] and Coot [53]. The final model spanning residues 21–240 and 251–345 of ENZ and residues 71–389 of PRO was refined in Phenix [51] to an Rfree of 22.3 % with excellent stereochemistry as assessed by MolProbity [54]. Details for data collection and refinement statistics are summarized in Supplemental Table S1. Figures were generated using PyMOL (Schrödinger, LLC), the electrostatic potential was calculated with APBS [55]. Atomic coordinates and structure factors have been deposited with the Protein Data Bank under PDB: 6DNZ.
Multi-angle light scattering
Purified proteins were characterized by multi-angle light scattering following size-exclusion chromatography [56]. Protein at 50 μM was injected onto a Superdex 200 10/300 GL size-exclusion chromatography column (GE Healthcare) equilibrated in a buffer containing 20 mM HEPES pH 7.5, 150 mM NaCl, and 0.5 mM TCEP. The chromatography system was connected in series with an 18-angle light scattering detector (DAWN HELEOS) and refractive index detector (OptilabrEX) (Wyatt Technology). Data were collected every second at a flow rate of 0.25 mL/min at 25 °C. Data analysis was carried out using the program ASTRA, yielding the molar mass and mass distribution (polydispersity) of the sample.
Isothermal titration calorimetry
ITC measurements were performed at 25°C using a MicroCal auto-iTC200 calorimeter (MicroCal, LLC). Wild-type and mutant ENZ-PRO proteins as well as MBP-TbRGG1 protein were extensively dialyzed against a buffer containing 20 mM HEPES, pH 7.5, 150 mM NaCl, and 0.5 mM TCEP. 2 μL of 0.13 mM MBP-TbRGG1 was injected into 0.2 mL of 0.03 mM ENZ-PRO proteins in the chamber every 150 s. Baseline-corrected data were analyzed with ORIGIN software.
Methylation assays
To assay the activity of TbPRMT1 tetramerization mutants, 37.5 nM TbPRMT1 tetramer was mixed with 6 μM MBP-TbRGG1 substrate, 0.7 μM [3H]AdoMet (Adenosyl-L-Methionine, S-[methyl-3H]-, 55–85Ci (2.03–3.15TBq)/mmol; PerkinElmer), 9.3 μM unlabeled AdoMet, 2 mM DTT and 2mM PMSF in PBS in a total volume of 25 μl. Reactions were incubated at 26°C for 1.5 hour, stopped by addition of SDS loading dye and separated on SDS PAGE. Gels were Coomassie stained and soaked in EN3HANCE (PerkinElmer). Dried gels were then exposed to film for one week at −80°C. To assay the activity of TbPRMT1 containing N-terminal prozyme truncations, reactions were performed as above, except the amount of MBP-TbRGG1 substrate was lowered to 0.6 μM, unlabeled AdoMet was left out and 6 μg of MBP2* protein (NEB) was added to each reaction to increase molecular crowding.
Small-Angle X-ray Scattering
SEC–SAXS of wild-type and mutant TbPRMT1 ENZ-PRO proteins was performed at the G1 station at MacCHESS, which is equipped with an ÄKTA Pure FPLC system (GE Healthcare). Protein was loaded at concentrations ranging from 2 to 16 mg/ml on a Superdex 200 10/300 GL column (GE Healthcare) equilibrated in 20 mM HEPES, pH 7.5, 150 mM NaCl, and 0.5 mM TCEP. SAXS data were recorded on a Pilatus 100 K-S detector at 2 s per frame with a fixed camera length of 1.522 m and an energy of 9.91 keV, allowing the collection of the angular range q of 0.01–0.30 Å−1. Primary reduction of the SAXS data was performed using RAW [57]. A Guinier plot of the buffer-subtracted profile was linear to the lowest measured q value. GNOM [58] was used to calculate P(r) plots from the scattering data. The maximum diameter was chosen so that the P(r) function fell gradually to zero at r = Dmax unconstrained. Theoretical radii of gyration were calculated using CRYSOL [42]. SEC–SAXS data collection and analysis statistics are listed in Table 2.
Electron Microscopy
Negative stain transmission electron microscopy was performed on the wild-type TbPRMT1 ENZ-PRO complex and the TbPRMT1 PRO triple mutant Y215A/M219A/E223A. Samples at 0.03 mg/ml were stained using 2% uranyl formate on continuous carbon grids. Micrographs were collected on the JEOL-1230 transmission electron microscope with a Gatan US400 detector. Data were processed using the Appion pipeline and ISAC [59, 60]. Using Appion, a contrast transfer function estimation was performed using CTFFIND4 [61]. Automated particle picking was done using DoG Picker and FindEM [62]. An initial stack of particles was assembled in Appion. 2D Classification was performed in ISAC [60].
Accession numbers:
The x-ray structure (coordinates and structure files) of the TbPRMT1 ENZ-Δ52PRO complex with AdoHcy have been deposited in the PDB with accession number 6DNZ.
Supplementary Material
Highlights.
The crystal structure of Trypanosoma brucei protein arginine methyltransferase 1 (PRMT1) is reported
Two enzyme-prozyme heterodimers form a stable heterotetramer essential for catalysis
The catalytically dead prozyme lacks elements essential for AdoMet binding
The enzyme alone is unstable and cannot form a canonical dimer due to steric clashes
T. brucei PRMT1 prozyme adopts a chaperone function conserved across kinetoplastids
Acknowledgements
We thank David King (University of California, Berkeley) for mass spectrometry analysis, Igor Kourinov and Kay Perry for support during data collection at NE-CAT, Richard Gillilan for support during data collection at CHESS, the High-Throughput Screening and Spectroscopy Resource Center at Rockefeller University, and the X-Ray Crystallography & Molecular Characterization Facility at the Sidney Kimmel Cancer Center, which is supported in part by National Cancer Institute Cancer Center Support Grant P30 CA56036 and S10 OD017987. X-ray data were collected at the NE-CAT beamlines (GM103403) on a Pilatus detector (RR029205) at the APS (DE-AC02-06CH11357). CHESS is supported by the NSF award DMR-1332208, and the MacCHESS resource is supported by NIGMS award GM-103485. The electron microscopy work was performed at the Simons Electron Microscopy Center and National Resource for Automated Molecular Microscopy located at the New York Structural Biology Center, supported by grants from the Simons Foundation (SF349247), NYSTAR, and the NIH National Institute of General Medical Sciences (GM103310). This work was supported by National Institutes of Health Grant R01 AI060260 (to L. K. R.) and American Heart Association Predoctoral Fellowship 15PRE24480155 (to L. K.). We would like to thank the members of the Debler laboratory for helpful discussions and the reviewers for constructive criticism.
Abbreviations used
- ADMA
asymmetric dimethyl arginine
- AdoMet
S-Adenosyl-L-methionine
- AdoHcy
S-Adenosyl-L-homocysteine
- ENZ
TbPRMT1 enzyme
- ITC
isothermal titration calorimetry
- MMA
monomethyl arginine
- PRMT
protein arginine methyltransferase
- PRO
TbPRMT1 prozyme
- rmsd
root-mean-square deviation
- SDMA
symmetric dimethyl arginine
References
- [1].Keating J, Yukich JO, Sutherland CS, Woods G, Tediosi F. Human African trypanosomiasis prevention, treatment and control costs: a systematic review. Acta Trop. 2015;150:4–13. [DOI] [PubMed] [Google Scholar]
- [2].Gibson WC, Swinkels BW, Borst P. Post-transcriptional control of the differential expression of phosphoglycerate kinase genes in Trypanosoma brucei. J Mol Biol. 1988;201:315–25. [DOI] [PubMed] [Google Scholar]
- [3].Clayton CE. Gene expression in Kinetoplastids. Curr Opin Microbiol. 2016;32:46–51. [DOI] [PubMed] [Google Scholar]
- [4].Schulz D, Mugnier MR, Paulsen EM, Kim HS, Chung CW, Tough DF, et al. Bromodomain Proteins Contribute to Maintenance of Bloodstream Form Stage Identity in the African Trypanosome. PLoS Biol. 2015;13:e1002316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gary JD, Clarke S. RNA and protein interactions modulated by protein arginine methylation. Prog Nucleic Acid Res Mol Biol. 1998;61:65–131. [DOI] [PubMed] [Google Scholar]
- [6].Lott K, Li J, Fisk JC, Wang H, Aletta JM, Qu J, et al. Global proteomic analysis in trypanosomes reveals unique proteins and conserved cellular processes impacted by arginine methylation. J Proteomics. 2013;91:210–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Thandapani P, O’Connor TR, Bailey TL, Richard S. Defining the RGG/RG motif. Mol Cell. 2013;50:613–23. [DOI] [PubMed] [Google Scholar]
- [8].Cheng X, Collins RE, Zhang X. Structural and sequence motifs of protein (histone) methylation enzymes. Annu Rev Biophys Biomol Struct. 2005;34:267–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Jain K, Warmack RA, Debler EW, Hadjikyriacou A, Stavropoulos P, Clarke SG. Protein Arginine Methyltransferase Product Specificity Is Mediated by Distinct Active-site Architectures. J Biol Chem. 2016;291:18299–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Debler EW, Jain K, Warmack RA, Feng Y, Clarke SG, Blobel G, et al. A glutamate/aspartate switch controls product specificity in a protein arginine methyltransferase. Proc Natl Acad Sci U S A. 2016;113:2068–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Fuhrmann J, Clancy KW, Thompson PR. Chemical biology of protein arginine modifications in epigenetic regulation. Chem Rev. 2015;115:5413–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Goulah CC, Pelletier M, Read LK. Arginine methylation regulates mitochondrial gene expression in Trypanosoma brucei through multiple effector proteins. RNA. 2006;12:1545–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Pelletier M, Pasternack DA, Read LK. In vitro and in vivo analysis of the major type I protein arginine methyltransferase from Trypanosoma brucei. Mol Biochem Parasitol. 2005;144:206–17. [DOI] [PubMed] [Google Scholar]
- [14].Kafkova L, Debler EW, Fisk JC, Jain K, Clarke SG, Read LK. The Major Protein Arginine Methyltransferase in Trypanosoma brucei Functions as an Enzyme-Prozyme Complex. J Biol Chem. 2017;292:2089–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kafková L, Tu C, Pazzo KL, Smith KP, Debler EW, Paul KS, Qu J, Read LK. 2018. Trypanosoma brucei PRMT1 is a nucleic acid binding protein with a role in energy metabolism and the starvation stress response. mBio 9:e02430–18. 10.1128/mBio.02430-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Goulah CC, Read LK. Differential effects of arginine methylation on RBP16 mRNA binding, guide RNA (gRNA) binding, and gRNA-containing ribonucleoprotein complex (gRNP) formation. J Biol Chem. 2007;282:7181–90. [DOI] [PubMed] [Google Scholar]
- [17].Zhang X, Cheng X. Structure of the predominant protein arginine methyltransferase PRMT1 and analysis of its binding to substrate peptides. Structure. 2003;11:509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhang X, Zhou L, Cheng X. Crystal structure of the conserved core of protein arginine methyltransferase PRMT3. EMBO J. 2000;19:3509–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Weiss VH, McBride AE, Soriano MA, Filman DJ, Silver PA, Hogle JM. The structure and oligomerization of the yeast arginine methyltransferase, Hmt1. Nat Struct Biol. 2000;7:1165–71. [DOI] [PubMed] [Google Scholar]
- [20].Tang J, Frankel A, Cook RJ, Kim S, Paik WK, Williams KR, et al. PRMT1 is the predominant type I protein arginine methyltransferase in mammalian cells. J Biol Chem. 2000;275:7723–30. [DOI] [PubMed] [Google Scholar]
- [21].Wang H, Huang ZQ, Xia L, Feng Q, Erdjument-Bromage H, Strahl BD, et al. Methylation of histone H4 at arginine 3 facilitating transcriptional activation by nuclear hormone receptor. Science. 2001;293:853–7. [DOI] [PubMed] [Google Scholar]
- [22].Tang J, Gary JD, Clarke S, Herschman HR. PRMT 3, a type I protein arginine N-methyltransferase that differs from PRMT1 in its oligomerization, subcellular localization, substrate specificity, and regulation. J Biol Chem. 1998;273:16935–45. [DOI] [PubMed] [Google Scholar]
- [23].Siegel TN, Hekstra DR, Wang X, Dewell S, Cross GA. Genome-wide analysis of mRNA abundance in two life-cycle stages of Trypanosoma brucei and identification of splicing and polyadenylation sites. Nucleic Acids Res. 2010;38:4946–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kolev NG, Franklin JB, Carmi S, Shi H, Michaeli S, Tschudi C. The transcriptome of the human pathogen Trypanosoma brucei at single-nucleotide resolution. PLoS Pathog. 2010;6:e1001090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Albanes D, Brown C. Relative weight, height and risk of breast cancer. JAMA. 1990;263:3148. [PubMed] [Google Scholar]
- [26].Shimogawa MM, Saada EA, Vashisht AA, Barshop WD, Wohlschlegel JA, Hill KL. Cell Surface Proteomics Provides Insight into Stage-Specific Remodeling of the Host-Parasite Interface in Trypanosoma brucei. Mol Cell Proteomics. 2015;14:1977–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lott K, Zhu L, Fisk JC, Tomasello DL, Read LK. Functional interplay between protein arginine methyltransferases in Trypanosoma brucei. Microbiologyopen. 2014;3:595–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Willert E, Phillips MA. Regulation and function of polyamines in African trypanosomes. Trends Parasitol. 2012;28:66–72. [DOI] [PubMed] [Google Scholar]
- [29].Volkov OA, Kinch L, Ariagno C, Deng X, Zhong S, Grishin N, et al. Relief of autoinhibition by conformational switch explains enzyme activation by a catalytically dead paralog. Elife. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Nguyen S, Jones DC, Wyllie S, Fairlamb AH, Phillips MA. Allosteric activation of trypanosomatid deoxyhypusine synthase by a catalytically dead paralog. J Biol Chem. 2013;288:15256–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Afanador GA, Tomchick DR, Phillips MA. Trypanosomatid Deoxyhypusine Synthase Activity Is Dependent on Shared Active-Site Complementation between Pseudoenzyme Paralogs. Structure. 2018;26:1499–512 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].McDermott SM, Carnes J, Stuart K. Editosome RNase III domain interactions are essential for editing and differ between life cycle stages in Trypanosoma brucei. RNA. 2019;25:1150–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Pils B, Schultz J. Inactive enzyme-homologues find new function in regulatory processes. J Mol Biol. 2004;340:399–404. [DOI] [PubMed] [Google Scholar]
- [34].Murphy JM, Mace PD, Eyers PA. Live and let die: insights into pseudoenzyme mechanisms from structure. Curr Opin Struct Biol. 2017;47:95–104. [DOI] [PubMed] [Google Scholar]
- [35].Rossmann MG, Moras D, Olsen KW. Chemical and biological evolution of nucleotide-binding protein. Nature. 1974;250:194–9. [DOI] [PubMed] [Google Scholar]
- [36].Cheng X, Roberts RJ. AdoMet-dependent methylation, DNA methyltransferases and base flipping. Nucleic Acids Res. 2001;29:3784–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].FAUMAN EB, BLUMENTHAL RM, CHENG X. STRUCTURE AND EVOLUTION OF ADOMET-DEPENDENT METHYLTRANSFERASES. S-Adenosylmethionine-Dependent Methyltransferases: WORLD SCIENTIFIC; 2011. p. 1–38. [Google Scholar]
- [38].Rambo RP, Tainer JA. Accurate assessment of mass, models and resolution by small-angle scattering. Nature. 2013;496:477–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Pawlak MR, Banik-Maiti S, Pietenpol JA, Ruley HE. Protein arginine methyltransferase I: substrate specificity and role in hnRNP assembly. J Cell Biochem. 2002;87:394–407. [DOI] [PubMed] [Google Scholar]
- [40].Goulet I, Gauvin G, Boisvenue S, Cote J. Alternative splicing yields protein arginine methyltransferase 1 isoforms with distinct activity, substrate specificity, and subcellular localization. J Biol Chem. 2007;282:33009–21. [DOI] [PubMed] [Google Scholar]
- [41].Yue WW, Hassler M, Roe SM, Thompson-Vale V, Pearl LH. Insights into histone code syntax from structural and biochemical studies of CARM1 methyltransferase. EMBO J. 2007;26:4402–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Svergun D, Barberato C, Koch MHJ. CRYSOL - a Program to Evaluate X-ray Solution Scattering of Biological Macromolecules from Atomic Coordinates. Journal of Applied Crystallography. 1995;28:768–73. [Google Scholar]
- [43].Antonysamy S, Bonday Z, Campbell RM, Doyle B, Druzina Z, Gheyi T, et al. Crystal structure of the human PRMT5:MEP50 complex. Proc Natl Acad Sci U S A. 2012;109:17960–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Rho J, Choi S, Seong YR, Cho WK, Kim SH, Im DS. Prmt5, which forms distinct homo-oligomers, is a member of the protein-arginine methyltransferase family. J Biol Chem. 2001;276:11393–401. [DOI] [PubMed] [Google Scholar]
- [45].Eyers PA, Murphy JM. The evolving world of pseudoenzymes: proteins, prejudice and zombies. BMC Biol. 2016;14:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lijnzaad P, Argos P. Hydrophobic patches on protein subunit interfaces: characteristics and prediction. Proteins. 1997;28:333–43. [PubMed] [Google Scholar]
- [47].Ekstrom JL, Mathews II, Stanley BA, Pegg AE, Ealick SE. The crystal structure of human S-adenosylmethionine decarboxylase at 2.25 A resolution reveals a novel fold. Structure. 1999;7:583–95. [DOI] [PubMed] [Google Scholar]
- [48].Tolbert WD, Ekstrom JL, Mathews II, Secrist JA 3rd, Kapoor P, Pegg AE, et al. The structural basis for substrate specificity and inhibition of human S-adenosylmethionine decarboxylase. Biochemistry. 2001;40:9484–94. [DOI] [PubMed] [Google Scholar]
- [49].Wang C, Zhu Y, Chen J, Li X, Peng J, Chen J, et al. Crystal structure of arginine methyltransferase 6 from Trypanosoma brucei. PLoS One. 2014;9:e87267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Otwinowski Z, Minor W. Processing of X-Ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997;276:307–26. [DOI] [PubMed] [Google Scholar]
- [51].Adams PD, Afonine PV, Bunkóczi G, Chen VB, Echols N, Headd JJ, et al. The Phenix software for automated determination of macromolecular structures. Methods. 2011;55:94–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Jones TA, Zou J-Y, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47:110–9. [DOI] [PubMed] [Google Scholar]
- [53].Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta crystallographica Section D, Biological crystallography. 2010;66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic acids research. 2007;35:W375–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:10037–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Wyatt PJ. Multiangle light scattering: The basic tool for macromolecular characterization. Instrum Sci Technol. 1997;25:1–18. [Google Scholar]
- [57].Hopkins JB, Gillilan RE, Skou S. BioXTAS RAW: improvements to a free open-source program for small-angle X-ray scattering data reduction and analysis. J Appl Crystallogr. 2017;50:1545–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Svergun DI, Semenyuk AV, Feigin LA. Small-angle-scattering-data treatment by the regularization method. Acta Crystallogr. A 1988;44:244–250. [Google Scholar]
- [59].Lander GC, Stagg SM, Voss NR, Cheng A, Fellmann D, Pulokas J, et al. Appion: an integrated, database-driven pipeline to facilitate EM image processing. J Struct Biol. 2009;166:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Yang Z, Fang J, Chittuluru J, Asturias FJ, Penczek PA. Iterative stable alignment and clustering of 2D transmission electron microscope images. Structure. 2012;20:237–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Rohou A, Grigorieff N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol. 2015;192:216–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Voss NR, Yoshioka CK, Radermacher M, Potter CS, Carragher B. DoG Picker and TiltPicker: software tools to facilitate particle selection in single particle electron microscopy. J Struct Biol. 2009;166:205–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.