

Abstract
Very‐long‐chain fatty acids, with a chain length of >C20, are abundant in myelin sphingolipids. Recently, a de novo mutation in the ELOVL1 gene, which encodes fatty acid elongase, was identified in patients with neurocutaneous disorders involving skin ichthyosis and multiple neurological abnormalities, including hypomyelination, spastic paraplegia, and high‐frequency deafness. However, the consequences of ELOVL1 deficiency for lipid composition and detailed pathological changes in the brain remain unclear. Here, we analyzed Elovl1 mutant mice as a model of human ELOVL1 deficiency. The mice exhibited a decreased postnatal survival rate, and some died of startle epilepsy. The acyl chain length of sphingolipids such as galactosylceramides, sulfatides, sphingomyelins, and ceramides in the brains of these mice was markedly shortened. Moreover, the mice exhibited reduced levels of galactosylceramides, which are important for myelin formation and stability. Electron microscope analysis of the corpus callosum in Elovl1 mutant mice revealed modest hypomyelination, especially in large‐diameter axons. Furthermore, behavioral testing of the mice revealed deficits such as poorer motor coordination and reduced acoustic startle response to high‐intensity stimulus. These findings provide clues to the molecular mechanism of the neurological symptoms of patients with the ELOVL1 mutation.
Keywords: galactosylceramide, lipid, spastic paraplegia, very‐long‐chain fatty acids
Abbreviations
- 2‐OH
2‐hydroxy
- FA
fatty acid
- GalCer
galactosylceramide
- LC
liquid chromatography
- MS/MS
tandem mass spectrometry
- RT
reverse transcription
- TLC
thin‐layer chromatography
- VLC
very‐long‐chain
1. INTRODUCTION
Myelin, a multilayered stack of tightly apposed membranes around an axon, functions as an insulator and enables the saltatory propagation of action potentials for the rapid transmission of neural information.1 Myelin is essential for motor, sensory, and cognitive functions, as evidenced by the symptoms of many myelin‐related disorders, such as multiple sclerosis and leukodystrophy.2 Myelin is a highly lipid‐enriched structure, with its lipid content constituting 70‐80% of its dry weight.3 Sphingolipids are one of the most abundant lipids in myelin. They are composed of a hydrophobic backbone (ceramide) and a polar head group. Sphingolipids are classified into sphingophospholipids and glycosphingolipids according to differences in the polar head groups. Sphingomyelin, which has a phosphocholine moiety, is the primary mammalian sphingophospholipid. The most abundant sphingolipid in myelin is the glycosphingolipid galactosylceramide (GalCer), which has a galactose moiety.4 Myelin also contains sulfatide, which is sulfated at the C3 position of the galactose in GalCer, and which is 1/6 to 1/3 as abundant as GalCer.3 Mice deficient in the GalCer synthase Ugt8a lack both GalCer and sulfatide and exhibit splitting of the myelin lamellae, nerve‐conduction deficiency, and ataxia, demonstrating that these species are essential for myelin function and stability.5, 6
A remarkably large proportion of the fatty acid (FA) moieties of myelin sphingolipids are very‐long‐chain (VLC) FAs, with a carbon chain length of >C20.7, 8 VLCFAs are synthesized from long‐chain FAs (C11–C20) via the FA elongation cycle in the endoplasmic reticulum. Each FA elongation cycle is composed of four sequential reactions: condensation, reduction, dehydration, and reduction.7, 8 The first reaction is the condensation of acyl‐CoA with malonyl‐CoA, a donor of two carbon units, resulting in the synthesis of 3‐ketoacyl‐CoA. The subsequent three reactions convert 3‐ketoacyl‐CoA to acyl‐CoA in the following order: reduction to 3‐hydroxyacyl‐CoA, dehydration to trans‐2‐enoyl‐CoA, and finally reduction to acyl‐CoA. Thus, acyl‐CoA is elongated by two carbon units per cycle. The first condensation reaction is the rate‐limiting step of the cycle9, 10 and is catalyzed by the FA elongases called ELOVLs (elongation of very‐long‐chain FAs).7, 8 Mammals have seven ELOVL isozymes (ELOVL1 to ELOVL7); these exhibit characteristic but partially overlapping substrate specificities toward acyl‐CoAs, making it possible to synthesize acyl‐CoAs differing in chain length and the number of double bonds.11, 12 Of the seven isozymes, ELOVL1 is active toward saturated and monounsaturated C20‐ to C24‐CoAs in vitro.12, 13 The VLC acyl‐CoAs produced by ELOVL1 are mainly incorporated into sphingolipids throughout the body, although they are also used for the production of wax esters and cholesterol esters in the meibomian glands in the eyelids.13
To study the function of VLC lipids, we previously generated Elovl1 knockout (Elovl1 −/−) mice.14 These mice exhibited an epidermal permeability barrier defect due to a decrease in acylceramide (ω‐O‐acylceramide).14 Acylceramide contains a C30–C36 VLCFA esterified with a linoleic acid at ω‐carbon and is essential for epidermal barrier formation.15, 16 We also found that Elovl1 mutant mice showed a dry‐eye‐like phenotype due to the shortening of VLC wax and cholesterol esters secreted from the meibomian glands.13 These studies demonstrated the involvement of VLC lipids in barrier function on the body surface. However, the functions of VLC lipids in other tissues, such as the brain, are incompletely understood. Recently, a de novo mutation in the ELOVL1 gene was identified in two patients with a neurocutaneous disorder.17, 18 These patients exhibited similar symptoms and carried an identical heterozygous mutation, which changed the Ser residue at 165 to Phe in the ELOVL1 protein.17, 18 Although the mutation abolished ELOVL1 enzymatic activity,18 it is unclear whether the disorder is caused by haploinsufficiency or an autosomal dominant mode of inheritance. The patients exhibited ichthyosis symptoms, which are associated with epidermal permeability‐barrier dysfunction, resemble the phenotype observed in Elovl1 −/− mice and are likely explained by the reduction in VLC ceramides, especially acylceramides.14, 17, 18 The neurological abnormalities of these patients included hypomyelination in the brain, spastic paraplegia, and high‐frequency (>2000 Hz) hearing deficit.17, 18 One of the patients also exhibited photophobia. However, the link between the ELOVL1 mutation and these neurological abnormalities remained unclear.
In this study, we analyzed the effects of Elovl1 deficiency on the nervous system in mice. We found that the VLC acyl moieties in sphingolipids were significantly shortened in the brains of Elovl1 −/− mice, and that their GalCer levels were decreased. The mutant mice exhibited hypomyelination of large‐diameter axons in the corpus callosum, mild impairment of their motor functions, and a reduced acoustic startle response to high‐intensity stimulus. The changes in sphingolipid chain length and/or composition in their brains may be associated with these abnormalities. Our results suggest that VLC sphingolipids in myelin are necessary for the development of fully functional brain circuitry.
2. MATERIALS AND METHODS
2.1. Mice and behavioral testing
In this study we analyzed Elovl1 −/− Tg(IVL‐Elovl1) mice, in which the epidermal barrier defect and neonatal lethality was rescued by the transgenic expression of Elovl1 in the epidermis.13 Mice were housed in a room kept at 23 ± 1°C with a 12 hours light/dark cycle (lights on at 07:00) with food and water available ad libitum. Elovl1 +/− Tg(IVL‐Elovl1) mice were maintained by backcrossing with C57BL/6J mice. For behavioral testing, we generated male Elovl1 +/+ Tg(IVL‐Elovl1) and Elovl1 −/− Tg(IVL‐Elovl1) mice by in vitro fertilization using sperm from a homozygous transgenic male Elovl1 +/− Tg(IVL‐Elovl1) mouse and eggs from female Elovl1 +/− mice. Mice used in this study were from at least the eighth generation of backcrossing. All mice were 8 weeks old at the start of the behavioral testing. Behavioral tests were carried out using protocols identical to previous reports,19 except for the acoustic startle response test, in which mice were given acoustic stimuli with intensities of 70, 80, 85, 90, 95, 100, 105, 110, and 120 dB. Behavioral testing was performed between 09:00 and 18:00. The animal experiments performed in this study were approved by the institutional animal care and use committees of Hokkaido University and Fujita Health University.
2.2. Quantitative real‐time reverse transcription (RT)‐PCR
Total RNAs were isolated from the whole brain using the NucleoSpin RNA kit (Macherey‐Nagel) according to the manufacturer's instructions. First‐strand cDNAs were synthesized using oligo (dT) primer and the PrimeScript II 1st strand cDNA Synthesis Kit (Takara Bio) according to the manufacturer's instructions. Quantitative real‐time PCR was performed using first‐strand cDNAs and KOD SYBR qPCR Mix (Toyobo) on a CFX96 Touch real‐time PCR detection system (Bio‐Rad). Forward (‐F) and reverse (‐R) primers for the respective genes were used (Table 1). The mRNA levels were normalized with respect to those of GAPDH. The reactions were conducted by incubating the samples at 98°C for 2 minutes, followed by 40 cycles of 98°C for 10 seconds, 60°C for 15 seconds, and 68°C for 1 minute.
Table 1.
Primers used in this study
| Primer | Sequence |
|---|---|
| Elovl1‐F | 5′‐CATGCTTTCCAAGGTCATTGAGCTG‐3′ |
| Elovl1‐R | 5′‐TCTCAGTTGGCCTTGACCTTGGTGG‐3′ |
| Ugt8a‐F | 5′‐CACTGCCAGAAGATCTGCAGAGGTG‐3′ |
| Ugt8a‐R | 5′‐GAGCTTAGTGTTGTTTCCTAGGTTC‐3′ |
| Fa2h‐F | 5′‐GATGGGCCACCTCACAGACACTCCG‐3′ |
| Fa2h‐R | 5′‐CTCGATGAGGTCTGAGTGGAAGAGG‐3′ |
| Cers2‐F | 5′‐AAGTTCCGAGAAGCCAGCTGGAGAT‐3′ |
| Cers2‐R | 5′‐CAATGCTGAAGAGCAGGGACCAGTA‐3′ |
| Gapdh‐F | 5′‐GAACGGGAAGCTCACTGGCATGGCC‐3′ |
| Gapdh‐R | 5′‐TGTCATACCAGGAAATGAGCTTGAC‐3′ |
2.3. Lipid analyses
Chopped brain tissue was suspended in 40 μl of chloroform/methanol (1:2, v/v) per mg tissue and homogenized in a homogenizer. The homogenate was cleared by centrifugation at 1100 × g at room temperature for 5 minutes. Lipids were extracted from 900 µl of the cleared homogenate by sequential addition of 300 μl of chloroform and 540 μl of water with vigorous mixing. After centrifugation at 10 000 × g for 3 minutes, the organic phase was recovered and dried. The dried lipids were suspended in chloroform/methanol (2:1, v/v), and lipids corresponding to 1 mg of brain tissue were resolved by thin‐layer chromatography (TLC) on silica gel 60 high‐performance TLC plates (Merck), using 1‐butanol/acetic acid/water (3:1:1, v/v) as the solvent system, and stained with a cupric acetate/phosphoric acid solution.
Lipids corresponding to 5 µg of brain tissue [for GalCer, 2‐hydroxy (2‐OH) GalCer, sulfatide, and ceramide] or 0.5 µg of brain tissue (for sphingomyelin) were subjected to liquid chromatography (LC)–tandem mass spectrometry (MS/MS) analysis using an ultra‐performance LC–triple‐quadrupole mass spectrometry system (Xevo TQ‐S; Waters), as described previously.20 Lipids were resolved on a reverse‐phase column (ACQUITY UPLC CSH C18 column; 1.7 µm particle size, 2.1 × 100 mm; Waters), ionized by electrospray ionization in positive ion mode, and detected using multiple reaction monitoring by selecting the specific m/z at quadrupole mass filters Q1 and Q3 and using the appropriate collision energy for each GalCer, 2‐OH GalCer, sulfatide, sphingomyelin, and ceramide species (Tables 2, 3, 4). Data were analyzed and quantified using MassLynx software (Waters).
Table 2.
Selected m/z values and collision energy for GalCer and 2‐OH GalCer species in LC‐MS/MS analyses
| Lipid species |
Precursor ions (Q1) [M−H2O+H]+ |
Product ion (Q3) | Collision energy (eV) |
|---|---|---|---|
| C12:0 GalCer* | 626.6 | 264.3 | 40 |
| C16:0 GalCer | 682.6 | 264.3 | 40 |
| C18:1 GalCer | 708.6 | 264.3 | 40 |
| C18:0 GalCer | 710.6 | 264.3 | 40 |
| C20:1 GalCer | 736.6 | 264.3 | 40 |
| C20:0 GalCer | 738.6 | 264.3 | 40 |
| C22:1 GalCer | 764.6 | 264.3 | 40 |
| C22:0 GalCer | 766.6 | 264.3 | 40 |
| C24:1 GalCer | 792.7 | 264.3 | 40 |
| C24:0 GalCer | 794.7 | 264.3 | 40 |
| C26:1 GalCer | 820.7 | 264.3 | 40 |
| C26:0 GalCer | 822.7 | 264.3 | 40 |
| 2‐OH C16:0 GalCer | 698.6 | 264.3 | 40 |
| 2‐OH C18:1 GalCer | 724.6 | 264.3 | 40 |
| 2‐OH C18:0 GalCer | 726.6 | 264.3 | 40 |
| 2‐OH C20:1 GalCer | 752.6 | 264.3 | 40 |
| 2‐OH C20:0 GalCer | 754.6 | 264.3 | 40 |
| 2‐OH C22:1 GalCer | 780.6 | 264.3 | 40 |
| 2‐OH C22:0 GalCer | 782.6 | 264.3 | 40 |
| 2‐OH C24:1 GalCer | 808.7 | 264.3 | 40 |
| 2‐OH C24:0 GalCer | 810.7 | 264.3 | 40 |
| 2‐OH C26:1 GalCer | 836.7 | 264.3 | 40 |
| 2‐OH C26:0 GalCer | 838.7 | 264.3 | 40 |
External control
Table 3.
Selected m/z values and collision energy for sulfatide and 2‐OH sulfatide species in LC‐MS/MS analyses
| Lipid species |
Precursor ions (Q1) [M−SO3−H2O+H]+ |
Product ion (Q3) | Collision energy (eV) |
|---|---|---|---|
| C16:0 sulfatide | 682.6 | 264.3 | 40 |
| C18:1 sulfatide | 708.6 | 264.3 | 40 |
| C18:0 sulfatide | 710.6 | 264.3 | 40 |
| C20:1 sulfatide | 736.6 | 264.3 | 40 |
| C20:0 sulfatide | 738.6 | 264.3 | 40 |
| C22:1 sulfatide | 764.6 | 264.3 | 40 |
| C22:0 sulfatide | 766.6 | 264.3 | 40 |
| C24:1 sulfatide | 792.7 | 264.3 | 40 |
| C24:0 sulfatide | 794.7 | 264.3 | 40 |
| C26:1 sulfatide | 820.7 | 264.3 | 40 |
| C26:0 sulfatide | 822.7 | 264.3 | 40 |
| 2‐OH C16:0 sulfatide | 698.6 | 264.3 | 40 |
| 2‐OH C18:1 sulfatide | 724.6 | 264.3 | 40 |
| 2‐OH C18:0 sulfatide | 726.6 | 264.3 | 40 |
| 2‐OH C20:1 sulfatide | 752.6 | 264.3 | 40 |
| 2‐OH C20:0 sulfatide | 754.6 | 264.3 | 40 |
| 2‐OH C22:1 sulfatide | 780.6 | 264.3 | 40 |
| 2‐OH C22:0 sulfatide | 782.6 | 264.3 | 40 |
| 2‐OH C24:1 sulfatide | 808.7 | 264.3 | 40 |
| 2‐OH C24:0 sulfatide | 810.7 | 264.3 | 40 |
| 2‐OH C26:1 sulfatide | 836.7 | 264.3 | 40 |
| 2‐OH C26:0 sulfatide | 838.7 | 264.3 | 40 |
Table 4.
Selected m/z values and collision energy for ceramide and sphingomyelin species in LC‐MS/MS analyses
| Lipid species |
Precursor ions (Q1) [M−H2O+H]+ |
Product ion (Q3) | Collision energy (eV) |
|---|---|---|---|
| C16:0 ceramide | 520.6 | 264.3 | 32 |
| C18:1 ceramide | 546.6 | 264.3 | 32 |
| C18:0 ceramide | 548.6 | 264.3 | 32 |
| C20:1 ceramide | 574.6 | 264.3 | 32 |
| C20:0 ceramide | 576.6 | 264.3 | 32 |
| C22:1 ceramide | 602.6 | 264.3 | 32 |
| C22:0 ceramide | 604.6 | 264.3 | 32 |
| C24:1 ceramide | 630.7 | 264.3 | 32 |
| C24:0 ceramide | 632.7 | 264.3 | 32 |
| C25:0 ceramide* | 646.7 | 264.3 | 32 |
| C26:1 ceramide | 658.7 | 264.3 | 32 |
| C26:0 ceramide | 660.7 | 264.3 | 32 |
| C12:0 sphingomyelin* | 647.6 | 181.4 | 60 |
| C16:0 sphingomyelin | 703.6 | 181.4 | 60 |
| C18:1 sphingomyelin | 729.6 | 181.4 | 60 |
| C18:0 sphingomyelin | 731.6 | 181.4 | 60 |
| C20:1 sphingomyelin | 757.6 | 181.4 | 60 |
| C20:0 sphingomyelin | 759.6 | 181.4 | 60 |
| C22:1 sphingomyelin | 785.6 | 181.4 | 60 |
| C22:0 sphingomyelin | 787.6 | 181.4 | 60 |
| C24:1 sphingomyelin | 813.7 | 181.4 | 60 |
| C24:0 sphingomyelin | 815.7 | 181.4 | 60 |
| C26:1 sphingomyelin | 841.7 | 181.4 | 60 |
| C26:0 sphingomyelin | 843.7 | 181.4 | 60 |
External control
2.4. Hematoxylin and eosin staining
Eyes of 16 week‐old mice were fixed with SUPER FIX (Kurabo) at 4°C for >24 hours. They were then processed for dehydration, embedding in paraffin, preparation of 4 µm sections, deparaffinization, rehydration, and staining with hematoxylin and eosin using an automated staining system (Tissue Tek DRS 2000, Sakura Finetek) as described previously.19 Bright‐field images were captured using a Leica DM5000B microscope equipped with a DFC295 digital color camera (Leica Microsystems).
2.5. Transmission electron microscopy
Brains isolated from 13‐month‐old mice were dissected and the parasagittal blocks (3 × 3 × 1 mm) containing the corpus callosum were fixed with 2% paraformaldehyde, 2% glutaraldehyde, 2% osmium tetroxide, and 2.5% sucrose in 0.1 M sodium cacodylate buffer (pH 7.4) at 4°C for 3 hours. Fixed samples were dehydrated in graded ethanol, infiltrated with propylene oxide, and embedded in Quetol‐812 resin (Nisshin EM). The samples were ultrathin‐sectioned at 70 nm on a microtome (Leica Ultracut UCT; Leica Microsystems), mounted on copper grids, and stained sequentially with 2% uranyl acetate and lead staining solution (Merck). They were observed with a transmission electron microscope (JEM‐1400Plus; JEOL) equipped with a digital camera (EM‐14830RUBY2; JEOL).
2.6. Quantification and statistical analyses
Data are presented as means ± SD or means ± SEM. Data analyses were performed using StatView 5.0 (SAS Institute, Cary, NC, USA) and JMP13.1 (SAS Institute). Between‐group differences were evaluated using Student's t test, one‐way ANOVA, or two‐way repeated measures ANOVA. P‐values of <.05 were considered significant.
3. RESULTS
3.1. Decreased acyl chain lengths and amounts of brain sphingolipids in Elovl1‐deficient mice
The Elovl1 −/− Tg(IVL‐Elovl1) mice, which are deficient in Elovl1 throughout the body except the epidermis, harbor the transgene Tg(IVL‐Elovl1) expressing 3×FLAG‐tagged ELOVL1 under the control of the epidermis‐specific human IVL (involucrin) promoter. We have previously shown, by immunoblot analysis, that 3×FLAG‐ELOVL1 protein is expressed in the epidermis but not in the brain.13 To confirm the absence of transgene expression in the brain of Elovl1 −/− Tg(IVL‐Elovl1) mice at the transcriptional level, we conducted quantitative real‐time RT‐PCR analysis using primers that detect both endogenous and transgene‐derived Elovl1 mRNAs. The levels of Elovl1 mRNA in Elovl1 −/− Tg(IVL‐Elovl1) mice were close to the detection limit and significantly lower than in control [Elovl1 +/+ Tg(IVL‐Elovl1)] mice (Figure 1A). Thus, Elovl1 −/− Tg(IVL‐Elovl1) mice are useful for studying the function of Elovl1 in the brain.
Figure 1.
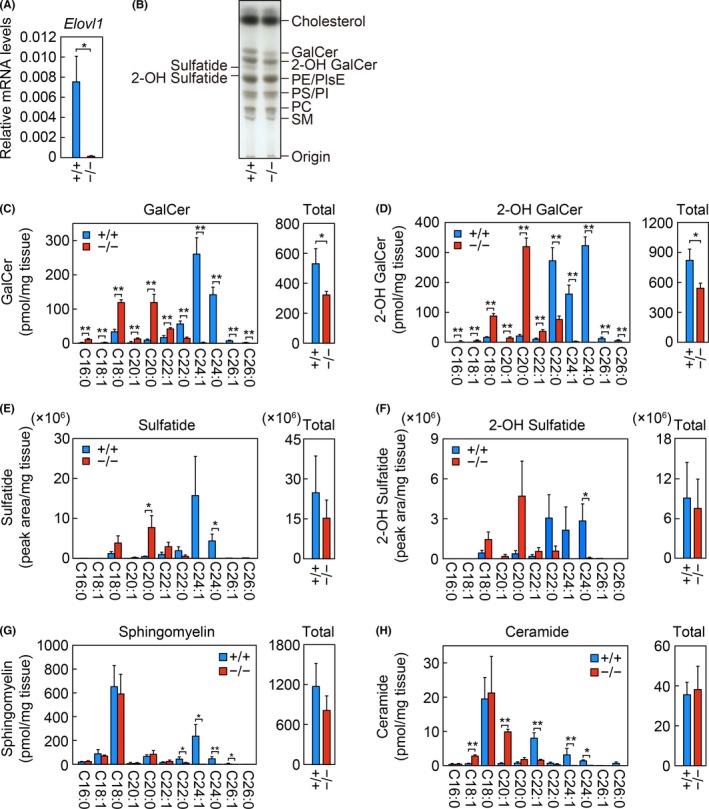
Decreased acyl chain lengths and levels of brain sphingolipids in Elovl1 −/− Tg(IVL‐Elovl1) mice. A) Total RNA prepared from the brains of two‐ to three‐month‐old control [Elovl1 +/+ Tg(IVL‐Elovl1)] (n = 3) and Elovl1 −/− Tg(IVL‐Elovl1) (n = 3) mice were subjected to RT, followed by SYBR Green‐based quantitative real‐time PCR using specific primers that detect both transgene‐derived and endogenous Elovl1 mRNAs. Values are the amount of total Elovl1 mRNA relative to that of Gapdh and represent the means ± SD. B) Lipids were extracted from the brains of control and Elovl1 −/− Tg(IVL‐Elovl1) mice, separated by TLC, and stained with a cupric acetate/phosphoric acid solution. PC, phosphatidylcholine; PI, phosphatidylinositol; PE, phosphatidylethanolamine; PlsE, plasmanyl/plasmenyl ethanolamine; PS, phosphatidylserine. C–H) Lipids were extracted from the brains of control (n = 3) and Elovl1 −/− Tg(IVL‐Elovl1) (n = 3) mice, and GalCer (C), 2‐OH GalCer (D), sulfatide (E), 2‐OH sulfatide (F), sphingomyelin (G), and ceramide (H) species were analyzed by LC‐MS/MS. The left‐hand graphs in (C–H) show the amount of each species containing an FA moiety of the indicated chain length and degree of saturation. The right‐hand graphs in (C–H) show the total amounts of all species. Values represent the means ± SD. Asterisks indicate significant differences based on Student's t test (*, P < .05; **, P < .01). +/+, Elovl1 +/+ Tg(IVL‐Elovl1) mice; −/−, Elovl1 −/− Tg(IVL‐Elovl1) mice
To examine the effect of diminished Elovl1 expression on lipid composition, we performed a TLC analysis of total lipids in the brain. The levels of both the non‐hydroxy and the 2‐OH forms of the glycosphingolipid GalCer were apparently decreased in Elovl1 −/− Tg(IVL‐Elovl1) mice, whereas those of other lipids were nearly unchanged (Figure 1B). Next, we performed LC‐MS/MS analyses to investigate the effect of Elovl1 deficiency on the chain length of sphingolipids. GalCer and 2‐OH GalCer mostly consisted of C22:0, C24:1, and C24:0 VLC species in control mice (Figure 1C, D). In Elovl1 −/− Tg(IVL‐Elovl1) mice, the levels of these VLC GalCer and 2‐OH GalCer species were substantially decreased, with ≥C24 species being almost undetected, whereas C22:1 and ≤C20 species were significantly increased. Consistent with the results of the TLC analysis, the total levels of GalCer and 2‐OH GalCer were reduced by 39% and 34%, respectively, in Elovl1 −/− Tg(IVL‐Elovl1) mice relative to control mice. We also quantified sulfatides and 2‐OH sulfatides (sulfated forms of GalCer and 2‐OH GalCer, respectively). Their FA composition resembled that of their respective non‐sulfated forms: they mostly consisted of C22:0, C24:1, and C24:0 VLC species in control mice, but were shortened to C22:1, C20:0, and C18:0 species in Elovl1 −/− Tg(IVL‐Elovl1) mice (Figure 1E, F). The total levels of sulfatides and 2‐OH sulfatides did not differ significantly between these two groups of mice, although there was a slight reduction tendency in the sulfatides in the latter group. We then quantified sphingomyelin and ceramide species. The levels of C22:0 and ≥C24 VLC sphingomyelin species were decreased in Elovl1 −/− Tg(IVL‐Elovl1) mice compared with control mice, while the levels of other species were almost unchanged (Figure 1G). Among ceramides, the levels of ≥C22 VLC species were decreased, whereas C18:1 and C20:1 species were increased (Figure 1H). The total levels of sphingomyelin and ceramide were comparable between control and Elovl1 −/− Tg(IVL‐Elovl1) mice. These results indicate that Elovl1 deficiency in the brain caused shortening of the acyl chain lengths of VLC sphingolipids to ≤C20 and decreases in the total amounts of GalCer and 2‐OH GalCer.
3.2. Decreased expression of the GalCer synthase Ugt8a in the brains of Elovl1‐deficient mice
To elucidate the mechanism of the reduction of GalCer and 2‐OH GalCer levels, the mRNA levels of genes involved in their synthesis in the brain were examined by quantitative real‐time RT‐PCR. We examined the mRNA levels of Ugt8a, Cers2, and Fa2h. Ugt8a encodes the GalCer synthase UDP galactosyltransferase 8.6, 21 Cers2 encodes one of the six mammalian ceramide synthases and is involved in the synthesis of VLC ceramides.22, 23 Fa2h encodes FA 2‐hydroxylase, which produces 2‐OH FA,24, 25 and mutations in the human FA2H gene cause hereditary spastic paraplegia (SPG35).26 These genes are highly expressed in oligodendrocytes in the brain.5, 25, 27 There was a significant reduction in the mRNA levels of Ugt8a in Elovl1 −/− Tg(IVL‐Elovl1) mice of 38% relative to control mice, while the levels of Cers2 and Fa2h were unchanged (Figure 2A–C). Ugt8a +/− mice had reduced levels of GalCer and 2‐OH GalCer compared with Ugt8a +/+ mice,6 indicating that the mRNA levels of Ugt8a are correlated with the levels of GalCer and 2‐OH GalCer. Therefore, reductions in GalCer and 2‐OH GalCer levels in Elovl1 −/− Tg(IVL‐Elovl1) mice may be partially attributable to the reduced mRNA levels of Ugt8a.
Figure 2.
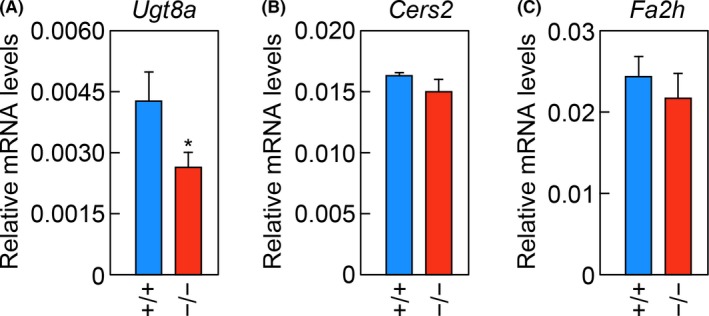
Decreased levels of Ugt8a mRNA in the brains of Elovl1 −/− Tg(IVL‐Elovl1) mice. A–C) Total RNA prepared from the brains of two‐ to three‐month‐old male/female Elovl1 +/+ Tg(IVL‐Elovl1) (n = 3) and Elovl1 −/− Tg(IVL‐Elovl1) (n = 3) mice were subjected to RT, followed by SYBR Green‐based quantitative real‐time PCR using specific primers that detect Ugt8a (A), Cers2 (B), and Fa2h (C) mRNAs. Values are the amount of each mRNA relative to that of Gapdh, and represent the means ± SD. The asterisk indicates a significant difference based on Student's t test (*, P < .05). +/+, Elovl1 +/+ Tg(IVL‐Elovl1) mice; −/−, Elovl1 −/− Tg(IVL‐Elovl1) mice
3.3. Thinning of myelin sheath in large‐diameter axons in Elovl1‐deficient mice
We performed transmission electron microscopy analysis of the corpus callosum to examine whether the changes in brain sphingolipids described above were associated with white matter abnormalities like the hypomyelination seen in patients with the ELOVL1 mutation.17, 18 There was no apparent change in the density of axons (Figure 3A), and the average axon diameter in control and Elovl1 −/− Tg(IVL‐Elovl1) mice was similar (Figure 3B). The thickness of myelin increased in proportion to axon diameter in control mice (Figure 3C); however, in Elovl1 −/− Tg(IVL‐Elovl1) mice such proportionality was less obvious, and the myelin was significantly thinner in axons with diameters of <0.4 µm or ≥1.6 µm, the latter being more severely affected (Figure 3A, C).
Figure 3.
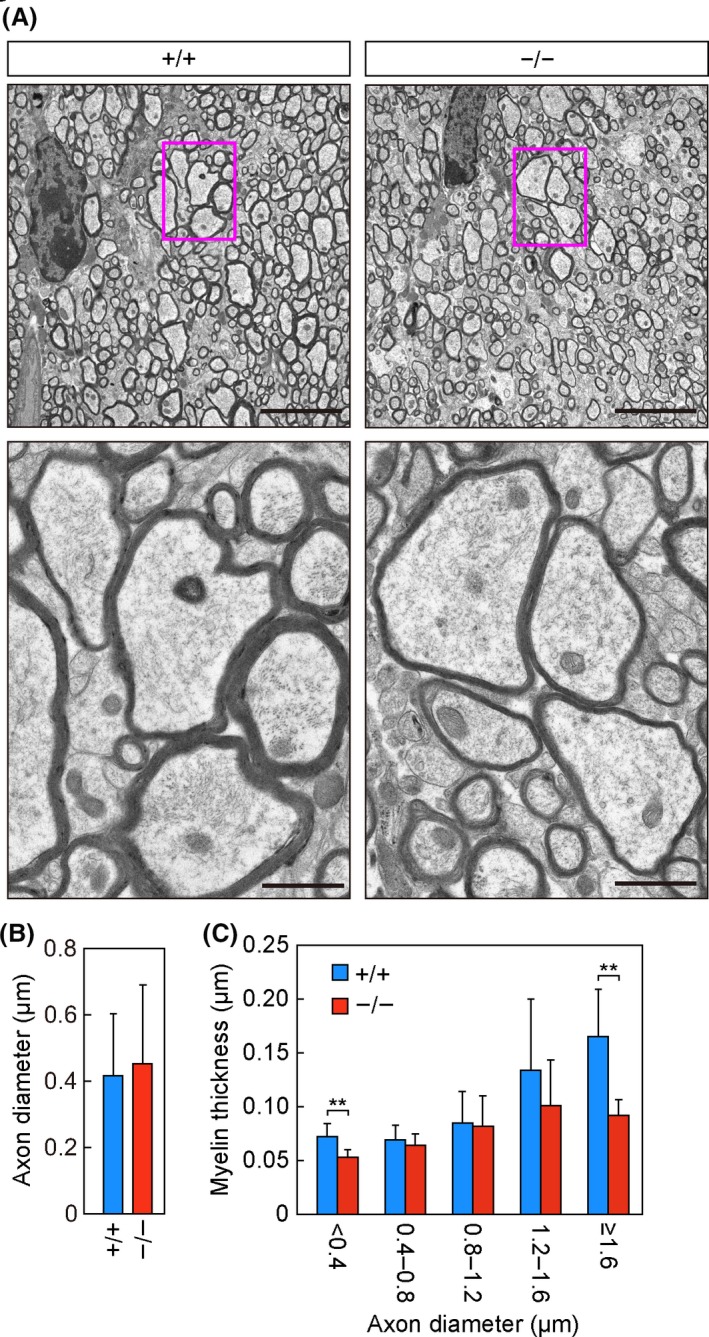
Thinning of the myelin sheath in Elovl1 −/− Tg(IVL‐Elovl1) mice. A–C) Transmission electron microscopy analysis of the corpus callosum in Elovl1 +/+ Tg(IVL‐Elovl1) and Elovl1 −/− Tg(IVL‐Elovl1) mice at 13 months of age. A) Representative images of the sagittal sections of the corpus callosum at low (5 µm, upper) or high (1 µm, lower) magnification. Red rectangles in upper panels indicate the area enlarged in lower panels. B) Average diameters ± SD of myelinated axons: n = 96 [Elovl1 +/+ Tg(IVL‐Elovl1)] and n = 73 [Elovl1 −/− Tg(IVL‐Elovl1)] total axons counted in 13 different fields of view. C) Myelin thickness of axons counted in (B) was measured and presented according to axon diameter. Values represent the means ± SD. Asterisks indicate significant differences based on Student's t test (**, P < .01). +/+, Elovl1 +/+ Tg(IVL‐Elovl1) mice; −/−, Elovl1 −/− Tg(IVL‐Elovl1) mice
3.4. Reduced postnatal viability, body weight, motor function, and acoustic startle response in Elovl1‐deficient mice
Elovl1 −/− Tg(IVL‐Elovl1) mice exhibited myelin thinning, which is related to the hypomyelination seen in patients with the ELOVL1 mutation; however, these mice did not exhibit any noticeable signs of movement abnormalities related to spastic paraplegia. Therefore, we conducted a battery of behavioral tests to assess their motor functions in detail. To obtain a sufficient number of age‐matched Elovl1 +/+ Tg(IVL‐Elovl1) and Elovl1 −/− Tg(IVL‐Elovl1) mice for these behavioral analyses, we performed in vitro fertilization using sperm from a homozygous transgenic male Elovl1 +/− Tg(IVL‐Elovl1) mouse and eggs from female Elovl1 +/− mice, and we obtained 265 newborn mice. Three weeks later, when genotyping was performed, the number of mice had decreased to 229 (Table 5). Of these, the genotype was successfully determined for 224. There were 22 Elovl1 −/− Tg(IVL‐Elovl1) mice, which was one‐third of the expected number of 66 (= 265/4) based on the Mendelian ratio. There were 66 and 136 Elovl1 +/+ Tg(IVL‐Elovl1) and Elovl1 +/− Tg(IVL‐Elovl1) mice, respectively. These numbers were close to the expected totals of 66 and 133, indicating that most of the mice lost during the three weeks after birth were Elovl1 −/− Tg(IVL‐Elovl1) individuals. Three more of these mice were lost by the age of eight weeks, and two of them died from startle epilepsy. Although the exact mechanism of this lethality during the juvenile period is unclear, these results suggest that Elovl1 plays an important role in survival.
Table 5.
Numbers of Elovl1+/+ Tg(IVL‐Elovl1), Elovl1+/− Tg(IVL‐Elovl1), and Elovl1−/− Tg(IVL‐Elovl1) mice at birth and three weeks after birth
| at birth | 265 | |||||
|---|---|---|---|---|---|---|
| at three weeks | genotype | Elovl1+/+ Tg(IVL‐Elovl1) | Elovl1+/− Tg(IVL‐Elovl1) | Elovl1−/− Tg(IVL‐Elovl1) | unknown | total |
| male | 41 | 61 | 12 | 2 | 116 | |
| female | 25 | 75 | 10 | 3 | 113 | |
| total | 66 | 136 | 22 | 5 | 229 | |
The behavioral tests were conducted on male control [Elovl1 +/+ Tg(IVL‐Elovl1)] and Elovl1 −/− Tg(IVL‐Elovl1) mice from when they were eight weeks old until they were 16 weeks old, well before the development of corneal opacity, as observed previously.13 The body weight of Elovl1 −/− Tg(IVL‐Elovl1) mice was about 15% smaller than that of control mice (Figure 4A). There were no significant differences between the two groups of mice in terms of body temperature, food consumption, grip strength, muscular endurance in the wire hang test, or pain sensitivity in the hot plate test (Figure 4B–F). In the open‐field test, which measures locomotor activity and emotionality in a novel environment, Elovl1 −/− Tg(IVL‐Elovl1) mice exhibited comparable responses with control mice in terms of total travel distance, time spent in the central area, number of vertical movements, and number of stereotypic behaviors (Figure 4G–J). However, the time course of time spent in the central area was significantly different between the two groups of mice, with Elovl1 −/− Tg(IVL‐Elovl1) mice showing a tendency to spend less time in this area during the first 45 minutes of the test (Figure 4H). These results suggest that the basic motor functions are normal in Elovl1 −/− Tg(IVL‐Elovl1) mice, but that they exhibit a behavioral preference for corners, which could be related to the photophobia seen in one patient with the ELOVL1 mutation.
Figure 4.
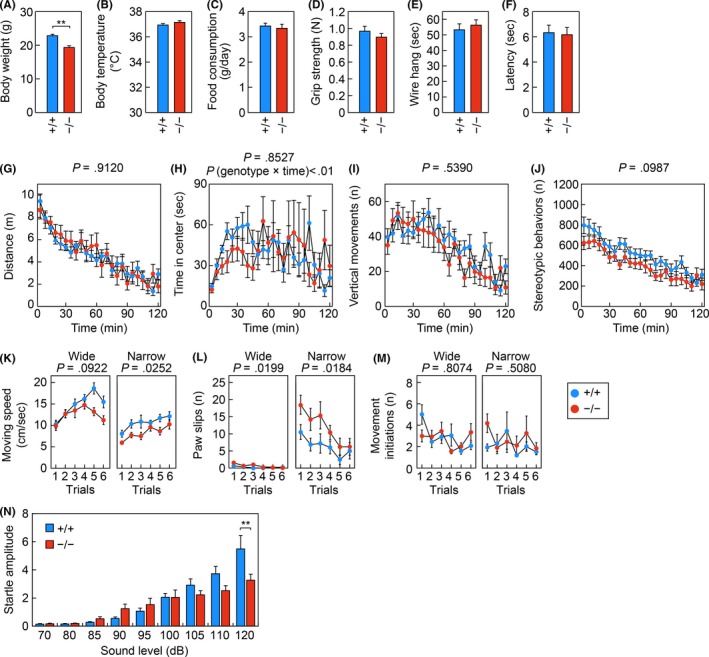
Reduced body weight, impaired motor function, and reduced acoustic startle response in Elovl1 −/− Tg(IVL‐Elovl1) mice. Elovl1 +/+ Tg(IVL‐Elovl1) and Elovl1 −/− Tg(IVL‐Elovl1) mice were assessed using the following metrics and tests: body weight (A), body temperature (B), food consumption (C), grip strength (D), wire hang latency (E), hot plate latency (F), the open‐field test (G–J), the balance beam test (K–M), and the acoustic startle response test (N). Open‐field test: distance traveled (G), time spent in the central area (H), number (n) of vertical movements (I), and number (n) of stereotypic behaviors (J). Balance beam test: travel speed (K), number (n) of paw slips (L), and number (n) of movement initiations (M) on a wide (left) or narrow (right) beam. Error bars represent SEM. Asterisks indicate significant differences based on one‐way ANOVA (**, P < .01; A–F, N). P‐values correspond to the genotype effect in two‐way repeated measures ANOVA (G–M). P (genotype × time) in (H) corresponds to the P‐value for the interaction of genotype and time in a two‐way repeated measures ANOVA. Number of mice analyzed: n = 12 for both genotypes (A–E), n = 12 for Elovl1 +/+ Tg(IVL‐Elovl1) and n = 11 for Elovl1 −/− Tg(IVL‐Elovl1) mice (F–M), n = 12 for Elovl1 +/+ Tg(IVL‐Elovl1) and n = 10 for Elovl1 −/− Tg(IVL‐Elovl1) mice (N). +/+, Elovl1 +/+ Tg(IVL‐Elovl1) mice; −/−, Elovl1 −/− Tg(IVL‐Elovl1) mice
The mice were then subjected to the balance beam test. This test is useful for detecting mild deficits in motor balance and coordination. Each mouse was placed on one end of a wide or narrow beam (2.5 or 0.8 cm in diameter, respectively), and their movement toward a dark box at the opposite end was observed. On the wide beam, there were no significant differences between control and Elovl1 −/− Tg(IVL‐Elovl1) mice; however, on the narrow beam, Elovl1 −/− Tg(IVL‐Elovl1) mice moved significantly more slowly and exhibited more paw slips (Figure 4K, L). No significant difference was observed between the two groups of mice in the number of movement initiations on the beam (Figure 4M). These results suggest that Elovl1 −/− Tg(IVL‐Elovl1) mice exhibit mild deficits in motor function.
The mice were next subjected to the acoustic startle response test. The mice were given a pulse (50 milliseconds) of white noise with various sound intensities, and their startle responses to the stimuli were measured. Control mice increased the amplitude of their startle response in proportion to the sound intensity (Figure 4N); however, the startle amplitude of Elovl1 −/− Tg(IVL‐Elovl1) mice was less responsive to increases in sound intensity, at 120 dB it was significantly smaller than that of control mice (Figure 4N). These results suggest that Elovl1 −/− Tg(IVL‐Elovl1) mice may exhibit defective audition.
3.5. Reduced activity in the light–dark transition test in Elovl1‐deficient mice
In addition to spastic paraplegia of the lower limbs, hypomyelination in the brain, and a hearing deficit at high frequencies, patients with the ELOVL1 mutation exhibit thinning of the nerve fiber layer in the retina, decreased visual acuity, and astigmatism; one patient also exhibited photophobia.17, 18 Therefore, we performed other tests that evaluate neurological functions not limited to motor and auditory functions. The mice were subjected to the light–dark transition test to examine their response to light. In this test, each mouse was first placed in the dark chamber, then allowed to move freely between the light and dark chambers. Elovl1 −/− Tg(IVL‐Elovl1) mice exhibited normal anxiety behavior in response to light, and there were no significant differences between control and Elovl1 −/− Tg(IVL‐Elovl1) mice in terms of latency before entering the light chamber for the first time or the amount of time spent in the light chamber (Figure 5A, 5B). However, Elovl1 −/− Tg(IVL‐Elovl1) mice were less active in the test regardless of the luminance; they exhibited fewer transitions between the two chambers and reduced travel distance in both chambers (Figure 5C, 5D). As another test of anxiety‐like behavior, we performed the elevated plus‐maze test. Elovl1 −/− Tg(IVL‐Elovl1) mice did not display any differences from control mice in the number of entries into open arms, the total number of entries into open or closed arm, the amount of time spent in open arms, or the travel distance (Figure 5E–H).
Figure 5.
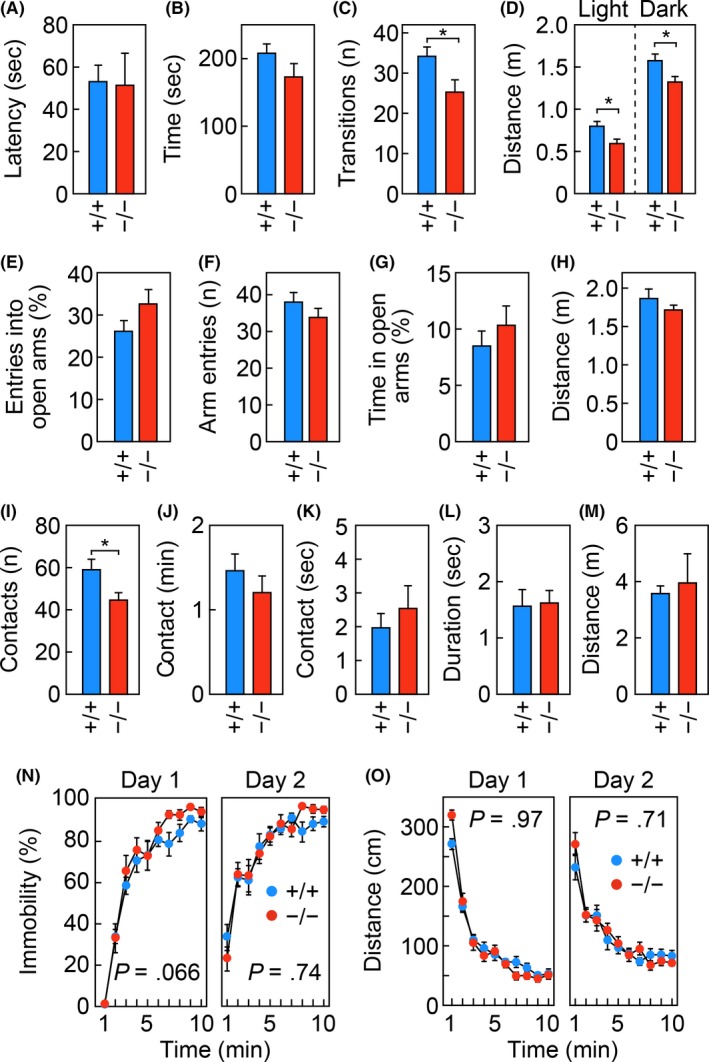
Reduced activity in the light–dark transition test in Elovl1 −/− Tg(IVL‐Elovl1) mice. Elovl1 +/+ Tg(IVL‐Elovl1) (n = 12) and Elovl1 −/− Tg(IVL‐Elovl1) (n = 11) mice were subjected to the light–dark transition test (A–D), the elevated plus‐maze test (E–H), the social‐interaction test in a novel environment (I–M), and the Porsolt forced‐swim test (N, O). Light–dark transition test: latency in entering the light chamber (A), time spent in the light chamber (B), number (n) of light–dark transitions (C), and distance traveled in the light and dark chambers (D). Elevated plus‐maze test: percentage of entries into open arms (E), number (n) of arm entries (F), percentage of time spent in open arms (G), and total distance traveled (H). Social‐interaction test in a novel environment: number (n) of contacts (I), total duration of contacts (J), total duration of active contacts (K), mean duration of each contact (L), and total distance traveled (M). Porsolt forced‐swim test: percentage of time spent immobile (N) and distance traveled (O). Error bars represent SEM. Asterisks indicate significant differences based on one‐way ANOVA (*, P < .05) (A–M). P‐values correspond to the genotype effect in a two‐way repeated measures ANOVA (N, O). +/+, Elovl1 +/+ Tg(IVL‐Elovl1) mice; −/−, Elovl1 −/− Tg(IVL‐Elovl1) mice
In the test of social interaction in a novel environment, two mice with the same genotype were placed together in the same box, and their behaviors were monitored. Elovl1 −/− Tg(IVL‐Elovl1) mice exhibited a slight but significant decrease in the number of contacts compared with control mice (Figure 5I), whereas there were no significant differences between the two groups of mice in the total duration of contact, duration of active contact, mean duration per contact, or travel distance (Figure 5J–M). Finally, the mice were subjected to the Porsolt forced‐swim test, in which they were placed in a cylinder filled with water. Elovl1 −/− Tg(IVL‐Elovl1) mice exhibited normal depression‐like responses, including the percentage of time spent immobile and travel distance (Figure 5N, O). These results suggest that Elovl1 −/− Tg(IVL‐Elovl1) mice may dislike changes in luminance more than control mice, and that they have a subtle defect in sociality.
3.6. Normal retinal and corneal architecture in Elovl1‐deficient mice
After completion of the behavioral tests, when the mice were 16 weeks old we performed histological analyses of the retina and cornea, because patients with the ELOVL1 mutation exhibit retinal pathology such as thinning of the nerve fiber layer in the retina,17, 18 and aged Elovl1 −/− Tg(IVL‐Elovl1) mice exhibit corneal opacity.13 The histological architecture of the retina, including the nerve fiber layer, did not differ detectably between control and Elovl1 −/− Tg(IVL‐Elovl1) mice (Figure 6A). We also found no discernible differences between the corneas of the two groups of mice (Figure 6B). These results suggest the absence of ocular abnormalities in Elovl1 −/− Tg(IVL‐Elovl1) mice.
Figure 6.
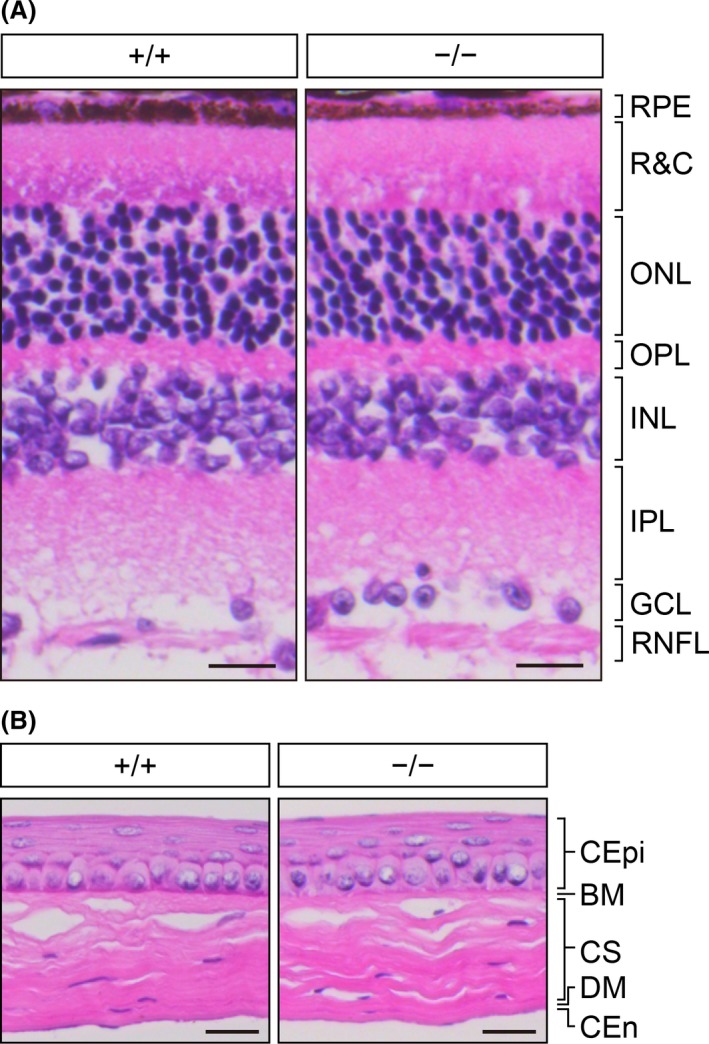
Normal retinal and corneal histology in Elovl1 −/− Tg(IVL‐Elovl1) mice at 16 weeks of age. Representative hematoxylin and eosin staining images of paraffin sections of the retina (A) and the cornea (B) from 16‐week‐old Elovl1 +/+ Tg(IVL‐Elovl1) and Elovl1 −/− Tg(IVL‐Elovl1) mice. Scale bars, 20 μm. Abbreviations: (A) GCL, ganglion cell layer; INL, inner nuclear layer; IPL, inner plexiform layer; ONL, outer nuclear layer; OPL, outer plexiform layer; R&C, layer of rods and cones; RNFL, retinal nerve fiber layer; RPE, retinal pigment epithelium. (B) BM, Bowman's membrane; CEn, corneal endothelium; CEpi, corneal epithelium; CS, corneal stroma; DM, Descemet's membrane. +/+, Elovl1 +/+ Tg(IVL‐Elovl1) mice; −/−, Elovl1 −/− Tg(IVL‐Elovl1) mice
4. DISCUSSION
In their brain tissue, Elovl1 −/− Tg(IVL‐Elovl1) mice had sphingolipids with shortened acyl moieties, with C24 sphingolipids being almost entirely lost (Figure 1). These results are consistent with the substrate specificity of ELOVL1 determined in vitro: ELOVL1 is highly active toward saturated and monounsaturated C20‐ and C22‐CoAs.12, 13 In addition to the shortening of acyl moieties, the total levels of GalCer and 2‐OH GalCer were significantly reduced in Elovl1 −/− Tg(IVL‐Elovl1) mice (Figure 1C, D). These reductions may be explained by a decrease in the mRNA levels of Ugt8a, encoding GalCer synthase (Figure 2A), although how the lack of Elovl1 affects the expression of Ugt8a remains unclear.
Elovl1 −/− Tg(IVL‐Elovl1) mice exhibited mild myelin thinning in the corpus callosum (Figure 3). This abnormality may be associated with the hypomyelination observed in the corpus callosum and other brain regions in patients with the ELOVL1 mutation. In Ugt8a‐knockout mice, in which both GalCer and 2‐OH GalCer and the sulfatides were absent, thinner and split myelin sheaths were observed in the white matter of the spinal cord,5, 6 suggesting that reductions in GalCer and 2‐OH GalCer underlie the observed myelin thinning in Elovl1 −/− Tg(IVL‐Elovl1) mice. These reductions in GalCer and 2‐OH GalCer in Elovl1 −/− Tg(IVL‐Elovl1) mice may impair the recruitment of PLP1, the major myelin protein essential to myelin function, into lipid rafts rich in GalCer, 2‐OH GalCer, and cholesterol, and this may in turn impair myelin function.28, 29 Alternatively, myelin membranes with reduced GalCer and 2‐OH GalCer content may have fewer hydrogen bonds between and/or within the myelin layers, which would render them less stable. It is possible that the shortening of acyl moieties in myelin sphingolipids also contributes to the myelin thinning. Giant unilamellar vesicles prepared from microsomal lipids in Cers2‐knockout mice have reduced VLC sphingolipids and exhibit morphological changes such as the presence of non‐spherical vesicles with different membrane curvature, small vesicles inside bigger vesicles, and the formation of aggregates.30 These changes in the biophysical properties of membranes may affect the formation and/or stability of myelin. In contrast to the deficit in myelin, we did not detect signs of axonal abnormalities. In fact, axon diameters and densities in the corpus callosum were comparable between Elovl1 +/+ Tg(IVL‐Elovl1) and Elovl1 −/− Tg(IVL‐Elovl1) mice (Figure 3A, B). Thus, the hypomyelination in patients with the ELOVL1 mutation may be caused by autonomous cellular defects in oligodendrocytes.
To identify neurological phenotypes related to the neural symptoms seen in patients with the ELOVL1 mutation, we performed a battery of behavioral tests and identified several abnormalities in Elovl1 −/− Tg(IVL‐Elovl1) mice: reduced body weight, slower travel speed and more paw slips in the balance beam test, decreased acoustic startle response to high‐intensity stimulus, reduced travel distance and fewer transitions between light and dark chambers in the light–dark transition test, and fewer contacts in the social‐interaction test (Figures 4 and 5). Considering that body temperature and food consumption are unaffected in Elovl1 −/− Tg(IVL‐Elovl1) mice (Figure 4B, C), it is possible that their reduced body weight is associated with reduced nutrient absorption from foods. The defects in the balance beam test (slower travel speed and more paw slips on a narrow beam; Figure 4K, L) are presumably related to spastic paraplegia. The balance beam test is a powerful analysis for identifying a subtle defect in motor function even in mice with no apparent spasticity or ambulation difficulty. In fact, we have previously identified similar defects in Aldh3a2‐knockout mice using this test.19 Aldh3a2‐knockout mice serve as a model for Sjögren‐Larsson syndrome,31 an autosomal recessive neurocutaneous disorder caused by mutations in the ALDH3A2 gene and characterized by spastic paraplegia and ichthyosis32—symptoms also observed in patients with the ELOVL1 mutation. ALDH3A2 is a fatty aldehyde metabolizing enzyme, and Aldh3a2‐knockout mice exhibit a decrease in 2‐OH GalCer due to inactivation of FA2H, which synthesizes 2‐OH FAs for 2‐OH GalCer production.19 These correlations further support our hypothesis that decreases in 2‐OH GalCer are associated with the hypomyelination and spastic paraplegia observed in patients with the ELOVL1 mutation.
Whereas patients with this mutation suffered from spasticity of the lower legs and became wheelchair‐dependent,17, 18 the defect in motor function in Elovl1 −/− Tg(IVL‐Elovl1) mice did not involve paraplegia but was instead rather mild (Figure 4K, L). This difference in the extent of motor dysfunction between human and mouse may result from the degree of hypomyelination. Patients with the ELOVL1 mutation exhibit widespread hypomyelination, as observed in magnetic resonance imaging (MRI),17, 18 whereas Elovl1 −/− Tg(IVL‐Elovl1) mice exhibit subtle hypomyelination—specifically, myelin thinning in a subpopulation of axons (Figure 3). Thus, in humans, oligodendrocytes may depend more on VLC sphingolipids for myelination formation and maintenance than they do in mice. Another possibility is that the oligodendrocytes themselves may require VLC sphingolipids equally in human and mouse, but that human nerves are more likely to be affected due to their longer axons. Because there is more myelin sheathing along the length of longer axons, human nerves such as the motor nerves that innervate the lower limbs may be more susceptible to the cumulative effect exerted by each defective myelin sheath.
Patients with the ELOVL1 mutation exhibited abnormalities related to vision, such as decreased visual acuity, astigmatism, photophobia, and thinning of the nerve fiber layer in the retina.17, 18 Elovl1 −/− Tg(IVL‐Elovl1) mice exhibited a tendency to avoid the central area during the first 45 minutes of the open‐field test and lower activity levels in the light–dark transition test, despite the lack of detectable retinal histologic abnormality (Figures 4, 5, 6). These defects are likely related to differences in luminance, and may be partially relevant to the photophobia observed in one of the two patients. Although aged Elovl1 −/− Tg(IVL‐Elovl1) mice exhibit corneal opacity because of chronic dry‐eye symptoms,13 we confirmed that the corneas of these mice exhibited no pathological abnormalities at 16 weeks of age, when the behavioral analyses were completed (Figure 6B). Thus, the deficiency in motor function identified in the balance beam test is likely associated with a defect in the brain rather than in the eye.
The defect in the acoustic startle response test in Elovl1 −/− Tg(IVL‐Elovl1) mice may be related to the high‐frequency hearing deficit in patients with the ELOVL1 mutation. Mice lacking St3gal5, which encodes the enzyme involved in the synthesis of ganglioside GM3 (one of the glycosphingolipids), exhibit hearing deficiency due to degeneration of the organ of Corti in the cochlea.33 Thus, VLC gangliosides may be necessary for the detection of high‐frequency sound.
The neurography of the peripheral motor and sensory nerves was normal in patients with the ELOVL1 mutation.17, 18 Similarly, Elovl1 −/− Tg(IVL‐Elovl1) mice exhibited normal nociceptive responses in the hot plate test (Figure 4F). These mice also exhibited normal anxiety‐like responses in the elevated plus‐maze test, mostly normal scores except for a slight decrease in the number of contacts in the social‐interaction test, and normal depression‐like responses in the Porsolt forced‐swim test (Figure 5). These results may correlate with the normal development of sociality in patients with the ELOVL1 mutation.
Combined, the results of this study show that Elovl1 −/− Tg(IVL‐Elovl1) mice exhibit changes in myelin sphingolipid composition and abundance, reduced myelin thickness, and subtle but significant impairment of motor and auditory functions. These results demonstrate the importance of VLC sphingolipids for myelin function, as well as the suitability of Elovl1 −/− Tg(IVL‐Elovl1) mice as a model for the study of human ELOVL1 deficiency.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
T. Sassa and A. Kihara designed the research and wrote the paper; M. Isokawa, T. Sassa, and S. Hattori performed the experiments and analyzed the data. T. Miyakawa provided technical support for behavioral analyses. All the authors reviewed and approved the final version of the manuscript.
ACKNOWLEDGEMENTS
This work was supported by the Advanced Research and Development Programs for Medical Innovation (AMED‐CREST) Grant Number JP19gm0910002h0005 (to AK) from the Japan Agency for Medical Research and Development (AMED), by a Grant‐in‐Aid for Scientific Research on Innovative Areas “Platform of Advanced Animal Model Support” Grant number 16H06276 (to TM) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), and by JSPS KAKENHI Grant Numbers JP18H03976 (to AK), JP18H04664 (to AK), and JP16K08220 (to TS) from the Japan Society for the Promotion of Science (JSPS). Behavioral analysis was carried out at the Institute for Comprehensive Medical Science, Fujita Health University (Joint Usage/Research Center for Genes, Brain and Behavior, accredited by MEXT).
Isokawa M, Sassa T, Hattori S, Miyakawa T, Kihara A. Reduced chain length in myelin sphingolipids and poorer motor coordination in mice deficient in the fatty acid elongase Elovl1 . FASEB BioAdvances. 2019;1:747–759. 10.1096/fba.2019-00067
Contributor Information
Takayuki Sassa, Email: tasasa@pharm.hokudai.ac.jp.
Akio Kihara, Email: kihara@pharm.hokudai.ac.jp.
REFERENCES
- 1. Aggarwal S, Yurlova L, Simons M. Central nervous system myelin: Structure, synthesis and assembly. Trends Cell Biol. 2011;21:585‐593. [DOI] [PubMed] [Google Scholar]
- 2. Duncan ID, Radcliff AB. Inherited and acquired disorders of myelin: The underlying myelin pathology. Exp Neurol. 2016;283:452‐475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Norton WT, Cammer W. Isolation and characterization of myelin In: Morell P, ed. Myelin. New York: Plenum; 1984:147‐195. [Google Scholar]
- 4. Schmitt S, Castelvetri LC, Simons M. Metabolism and functions of lipids in myelin. Biochim Biophys Acta. 2015;1851:999‐1005. [DOI] [PubMed] [Google Scholar]
- 5. Bosio A, Binczek E, Stoffel W. Functional breakdown of the lipid bilayer of the myelin membrane in central and peripheral nervous system by disrupted galactocerebroside synthesis. Proc Natl Acad Sci U. S. A. 1996;93:13280‐13285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Coetzee T, Fujita N, Dupree J, et al. Myelination in the absence of galactocerebroside and sulfatide: Normal structure with abnormal function and regional instability. Cell. 1996;86:209‐219. [DOI] [PubMed] [Google Scholar]
- 7. Kihara A. Very long‐chain fatty acids: Elongation, physiology and related disorders. J Biochem. 2012;152:387‐395. [DOI] [PubMed] [Google Scholar]
- 8. Sassa T, Kihara A. Metabolism of very long‐chain fatty acids: Genes and pathophysiology. Biomol Ther. 2014;22:83‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nugteren DH. The enzymic chain elongation of fatty acids by rat‐liver microsomes. Biochim Biophys Acta. 1965;106:280‐290. [DOI] [PubMed] [Google Scholar]
- 10. Bernert JT Jr, Sprecher H. An analysis of partial reactions in the overall chain elongation of saturated and unsaturated fatty acids by rat liver microsomes. J Biol Chem. 1977;252:6736‐6744. [PubMed] [Google Scholar]
- 11. Jakobsson A, Westerberg R, Jacobsson A. Fatty acid elongases in mammals: Their regulation and roles in metabolism. Prog Lipid Res. 2006;45:237‐249. [DOI] [PubMed] [Google Scholar]
- 12. Ohno Y, Suto S, Yamanaka M, et al. ELOVL1 production of C24 acyl‐CoAs is linked to C24 sphingolipid synthesis. Proc Natl Acad Sci U. S. A. 2010;107:18439‐18444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sassa T, Tadaki M, Kiyonari H, Kihara A. Very long‐chain tear film lipids produced by fatty acid elongase ELOVL1 prevent dry eye disease in mice. FASEB J. 2018;32:2966‐2978. [DOI] [PubMed] [Google Scholar]
- 14. Sassa T, Ohno Y, Suzuki S, et al. Impaired epidermal permeability barrier in mice lacking Elovl1, the gene responsible for very‐long‐chain fatty acid production. Mol Cell Biol. 2013;33:2787‐2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kihara A. Synthesis and degradation pathways, functions, and pathology of ceramides and epidermal acylceramides. Prog Lipid Res. 2016;63:50‐69. [DOI] [PubMed] [Google Scholar]
- 16. Hirabayashi T, Murakami M, Kihara A. The role of PNPLA1 in ω‐O‐acylceramide synthesis and skin barrier function. Biochim Biophys Acta. 2019;1864:869‐879. [DOI] [PubMed] [Google Scholar]
- 17. Kutkowska‐Kazmierczak A, Rydzanicz M, Chlebowski A, et al. Dominant ELOVL1 mutation causes neurological disorder with ichthyotic keratoderma, spasticity, hypomyelination and dysmorphic features. J Med Genet. 2018;55:408‐414. [DOI] [PubMed] [Google Scholar]
- 18. Mueller N, Sassa T, Morales‐Gonzalez S, et al. De novo mutation in ELOVL1 causes ichthyosis, acanthosis nigricans, hypomyelination, spastic paraplegia, high frequency deafness and optic atrophy. J Med Genet. 2019;56:164‐175. [DOI] [PubMed] [Google Scholar]
- 19. Kanetake T, Sassa T, Nojiri K, et al. Neural symptoms in a gene knockout mouse model of Sjögren‐Larsson syndrome are associated with a decrease in 2‐hydroxygalactosylceramide. FASEB J. 2019;33:928‐941. [DOI] [PubMed] [Google Scholar]
- 20. Ohno Y, Kamiyama N, Nakamichi S, Kihara A. PNPLA1 is a transacylase essential for the generation of the skin barrier lipid ω‐O‐acylceramide. Nat Commun. 2017;8:14610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schulte S, Stoffel W. Ceramide UDPgalactosyltransferase from myelinating rat brain: Purification, cloning, and expression. Proc Natl Acad Sci U. S. A. 1993;90:10265‐10269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Imgrund S, Hartmann D, Farwanah H, et al. Adult ceramide synthase 2 (CERS2)‐deficient mice exhibit myelin sheath defects, cerebellar degeneration, and hepatocarcinomas. J Biol Chem. 2009;284:33549‐33560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mizutani Y, Kihara A, Igarashi Y. Mammalian Lass6 and its related family members regulate synthesis of specific ceramides. Biochem J. 2005;390:263‐271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Alderson NL, Rembiesa BM, Walla MD, Bielawska A, Bielawski J, Hama H. The human FA2H gene encodes a fatty acid 2‐hydroxylase. J Biol Chem. 2004;279:48562‐48568. [DOI] [PubMed] [Google Scholar]
- 25. Zoller I, Meixner M, Hartmann D, et al. Absence of 2‐hydroxylated sphingolipids is compatible with normal neural development but causes late‐onset axon and myelin sheath degeneration. J Neurosci. 2008;28:9741‐9754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dick KJ, Eckhardt M, Paisan‐Ruiz C, et al. Mutation of FA2H underlies a complicated form of hereditary spastic paraplegia (SPG35). Hum Mutat. 2010;31:E1251‐1260. [DOI] [PubMed] [Google Scholar]
- 27. Becker I, Wang‐Eckhardt L, Yaghootfam A, Gieselmann V, Eckhardt M. Differential expression of (dihydro)ceramide synthases in mouse brain: Oligodendrocyte‐specific expression of CerS2/Lass2. Histochem Cell Biol. 2008;129:233‐241. [DOI] [PubMed] [Google Scholar]
- 28. Klugmann M, Schwab MH, Puhlhofer A, et al. Assembly of CNS myelin in the absence of proteolipid protein. Neuron. 1997;18:59‐70. [DOI] [PubMed] [Google Scholar]
- 29. Simons M, Kramer EM, Thiele C, Stoffel W, Trotter J. Assembly of myelin by association of proteolipid protein with cholesterol‐ and galactosylceramide‐rich membrane domains. J Cell Biol. 2000;151:143‐154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Silva LC, Ben David O, Pewzner‐Jung Y, et al. Ablation of ceramide synthase 2 strongly affects biophysical properties of membranes. J Lipid Res. 2012;53:430‐436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Naganuma T, Takagi S, Kanetake T, et al. Disruption of the Sjögren‐Larsson syndrome gene Aldh3a2 in mice increases keratinocyte growth and retards skin barrier recovery. J Biol Chem. 2016;291:11676‐11688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rizzo WB. Sjögren‐Larsson syndrome: molecular genetics and biochemical pathogenesis of fatty aldehyde dehydrogenase deficiency. Mol Genet Metab. 2007;90:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yoshikawa M, Go S, Takasaki K, et al. Mice lacking ganglioside GM3 synthase exhibit complete hearing loss due to selective degeneration of the organ of Corti. Proc Natl Acad Sci U. S. A. 2009;106:9483‐9488. [DOI] [PMC free article] [PubMed] [Google Scholar]