

Abstract
RNase J1 is the major 5′‐to‐3′ bacterial exoribonuclease. We demonstrate that in its absence, RNA polymerases (RNAPs) are redistributed on DNA, with increased RNAP occupancy on some genes without a parallel increase in transcriptional output. This suggests that some of these RNAPs represent stalled, non‐transcribing complexes. We show that RNase J1 is able to resolve these stalled RNAP complexes by a “torpedo” mechanism, whereby RNase J1 degrades the nascent RNA and causes the transcription complex to disassemble upon collision with RNAP. A heterologous enzyme, yeast Xrn1 (5′‐to‐3′ exonuclease), is less efficient than RNase J1 in resolving stalled Bacillus subtilis RNAP, suggesting that the effect is RNase‐specific. Our results thus reveal a novel general principle, whereby an RNase can participate in genome‐wide surveillance of stalled RNAP complexes, preventing potentially deleterious transcription–replication collisions.
Keywords: RNAP, RNase J1, stalling, torpedo, transcription–replication collision
Subject Categories: DNA Replication, Repair & Recombination; Microbiology, Virology & Host Pathogen Interaction; Transcription
Nascent RNA degradation by RNase J1 invokes disassembly of bacterial transcription complex upon collision with RNA polymerase.

Introduction
Ribonucleic acids (RNAs) are indispensable for living organisms. They are transcribed from DNA and function as templates for translation into proteins, while also serving regulatory, catalytic, and structural roles (Jimenez et al, 2015; Radhakrishnan & Green, 2016; Gimpel & Brantl, 2017; Sikova et al, 2018). The amount of any RNA in the cell is determined by the ratio between its synthesis and degradation rates (Arraiano et al, 2010).
Synthesis of RNA in bacteria is dependent on RNA polymerase (RNAP) that recognizes promoter DNA sequences where transcription begins (Ruff et al, 2015). After initiation, RNAP forms the elongation complex (EC) and proceeds in a step‐wise manner, functioning as a Brownian ratchet (Bar‐Nahum et al, 2005). During elongation, RNAP may encounter obstacles, such as thymidine dimers or DNA‐binding proteins that make it pause or even backtrack, and subsequently stall (He & Zalkin, 1992; Tornaletti & Hanawalt, 1999; Kireeva & Kashlev, 2009). These stalled complexes can be resolved by various factors that either allow RNAP to proceed with transcription or liberate RNAP from the stalled complex (Toulme et al, 2000; Peters et al, 2009; Epshtein et al, 2014; Fan et al, 2016). These factors include (i) the termination factor Rho (Epshtein et al, 2010), (ii) the transcription–repair coupling factor, Mfd, that recognizes stalled RNAPs and which recruits UvrA to initiate nucleotide excision repair (Le et al, 2018), and (iii) GreA, a translation elongation factor that induces hydrolysis of RNA by RNAP in backtracked complexes (Kusuya et al, 2011). These factors are vital for physiologically appropriate gene expression as well as for genome integrity (Nadkarni et al, 2016). At the ends of genes, transcription is terminated in a manner dependent on RNA hairpins and/or auxiliary proteins such as Rho (Larson et al, 2008; Epshtein et al, 2010).
Degradation of RNA is carried out by various ribonucleases (RNases) that can cleave RNA either endo‐ or exonucleolytically. Exoribonucleases can function either in the 3′‐to‐5′ or in the 5′‐to‐3′ direction (Lehnik‐Habrink et al, 2012). Until relatively recently, 5′‐to‐3′ exoribonucleases were believed to be exclusively eukaryotic. However, a bacterial 5′‐to‐3′ exoribonuclease, RNase J1, was discovered in the model soil‐dwelling organism Bacillus subtilis (Mathy et al, 2007) and shown to be widespread in bacteria and archaea (Phung et al, 2013; Condon et al, 2018). RNase J1 is a member of the β‐lactamase family of ribonucleases that also possesses endoribonucleolytic activity, at least in vitro (Even et al, 2005). In B. subtilis, RNase J1 associates with its paralog RNase J2 in the cell and although these two RNases act synergistically, formation of this complex is not necessary for its enzymatic activity (Mathy et al, 2010). RNase J1 is not essential, but its depletion results in significant changes in the transcriptome (Durand et al, 2012).
Here, using the B. subtilis model system, we describe a new phenomenon, the ability of RNase J1 to disassemble stalled transcription complexes, preventing transcription–replication collisions. Our initial goal was to determine the effect of the absence of RNase J1 on the transcriptome using an rnjA (encodes RNase J1) deletion strain, as previously this effect had been determined with a strain that allowed only incomplete depletion of this enzyme (Durand et al, 2012). We performed RNAseq and ChIPseq experiments and detected massive changes both in the levels of individual transcripts and in the distribution of DNA occupancy by RNAP, where RNAP accumulated on genes that have low transcript levels, suggesting the presence of stalled transcription complexes. Subsequently, we demonstrated that RNase J1 is close to, and physically linked to, RNAP through RNA in the cell. We present a model supported by experimental evidence in which RNase J1 resolves stalled RNAP complexes by degrading nascent RNA and disassembling the stalled complex after colliding with it, thereby likely acting as a “torpedo” in a manner analogous to a particular mode of eukaryotic transcription termination (Luo & Bentley, 2004).
Results
The absence of RNase J1 affects the transcriptome and DNA occupancy by RNAP
To characterize the global effects of a complete absence of RNase J1, we first created a B. subtilis 168 tryptophan prototrophic strain (BaSysBio; Nicolas et al, 2012) with a deletion of the rnjA gene. As previously reported for rnjA deletions, this strain displayed a markedly decreased growth rate (Fig EV1A) and altered cell morphology (Figaro et al, 2013; Cascante‐Estepa et al, 2016)—both with respect to cell shape and length (Fig EV1B; see Fig EV1C for distribution of non‐spiral cell lengths). We also measured the overall RNA synthesis rate, and, in agreement with the decreased growth rate of the ∆rnjA mutant, it displayed a strongly decreased RNA synthesis rate (Fig EV1D).
Figure EV1. Phenotypic characterization of the ΔrnjA strain.
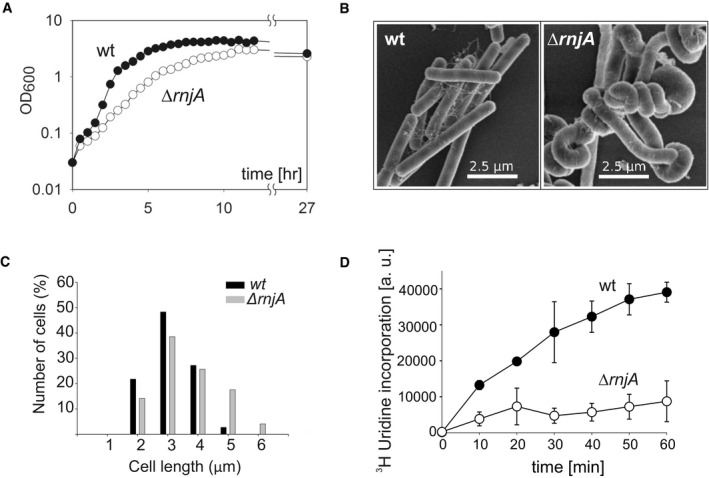
- Growth of Bacillus subtilis wt (LK1371) and ΔrnjA (LK1381) strains in LB medium at 37°C.
- Morphology of wt (LK1371) and ΔrnjA (LK1381) strains as captured by electron microscopy of exponential phase cells (OD600 0.5).
- Quantification of cells length of exponentially growing (OD600 0.5) wt (LK1371) and ΔrnjA (LK1381) cells stained with a membrane dye (NileRed, Sigma‐Aldrich). The analysis was performed on two sets consisting of 100 cells each. Due to technical considerations, the length of only non‐spiral cells was measured. The length of the cells was analyzed by Fiji ImageJ.
- Total RNA synthesis wt (LK1371) and ΔrnjA (LK1381) strains normalized to cell density (OD600). The data points are averages from two independent experiments (each done in technical duplicates); the error bars show the range.
Source data are available online for this figure.
Next, using RNAseq we determined the effects of the absence of RNase J1 on individual gene expression. The ΔrnjA mutant showed altered RNA levels for 1,740 genes (at least a twofold difference), of which 879 were upregulated (↑) and 861 were downregulated (↓) with respect to wt (Tables EV1 and EV2). Thirteen selected differentially expressed genes were validated by RT–qPCR (Appendix Fig S1). The similar distribution of up‐ and downregulated mRNAs applied to five out of six major gene ontology categories; the exceptions were mRNAs from prophages and mobile genetic elements where the gene expression was preferentially downregulated (Appendix Fig S2). Among the most upregulated mRNAs was mreBH (~ 16× ↑) that encodes a protein whose physiologically correct level is required for cell shape determination (Kawai et al, 2009). We have shown previously that the upregulation of the mreBH mRNA is primarily due to the strong stabilization of a non‐functional degradation intermediate whose 5′ end lies within the mreBH ORF (Durand et al, 2012). The perturbed expression of this and several other mRNAs encoding proteins involved in cell‐wall synthesis possibly explains, at least in part, the “spiral” phenotype [see (Figaro et al, 2013)].
Several alternative sigma factor‐encoding mRNAs, especially sigD [~ 8× ↓] but also sigB, sigM, sigW, sigX, and sigY were downregulated in cells lacking RNase J1 (Fig 1C and D). While most of the upregulated mRNAs are likely due to the direct stabilization of either full‐length mRNAs or long degradation intermediates (Durand et al, 2012), we anticipate that most of the downregulated mRNAs are due to indirect transcription effects. This is because stabilizing effects of RNase J1 5′‐to‐3′ exoribonuclease activity, the primary in vivo activity of this enzyme, are expected to be rare (if any). The downregulation of sigD expression likely explains the loss of motility as expression of the flagellar machinery is controlled by this sigma factor (Mirel et al, 1992). The long filaments of the ΔrnjA strain might be related to downregulation of the gene responsible for cell separation—cwlS (~ 18× ↓; Fukushima et al, 2006).
Figure 1. Global changes in ΔrnjA compared to wt.
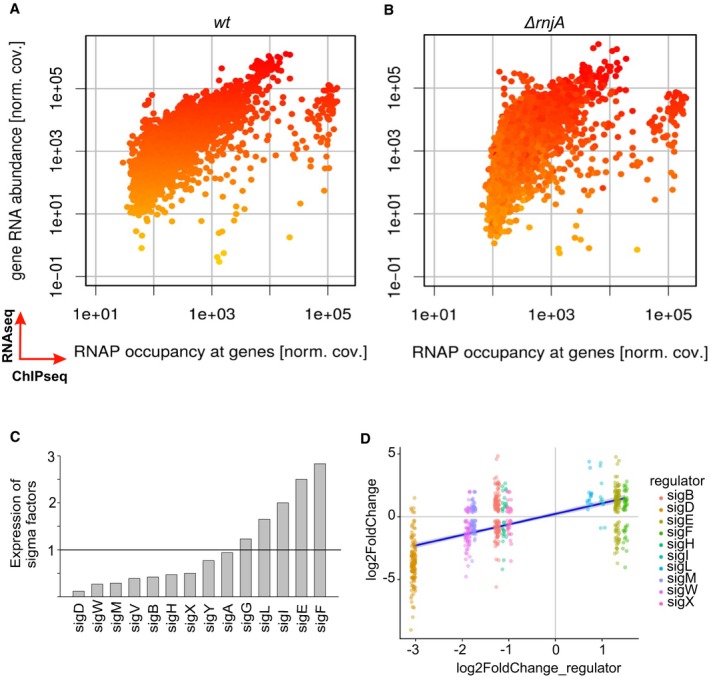
-
A, BComparisons of RNAseq and ChIPseq data. Wt strain (A) and ΔrnjA (B). The x‐axis shows the relative RNAP occupancy at a given gene (normalized ChIPseq coverage). The y‐axis shows the relative abundance of the transcript (normalized RNAseq coverage; each dot is one gene). The genes are color‐coded, ranging from yellow (low RNA abundance) to red (high RNA abundance) in wt. The ΔrnjA strain has higher RNAP occupancy mainly among the genes with less abundant transcripts. The color coding in (B) reveals no dramatic overall changes in the vertical direction (RNA abundance). If anything, some of the low abundance transcripts decreased further in level in the mutant strain. The main difference (mostly among the low abundance transcripts) is their shift in the horizontal direction to the right (toward higher occupancy with RNAP). Data represent mean values of three independent experiments.
-
CRelative expression of all sigma factors in wt (LK1371) vs. ΔrnjA (LK1381). Wt levels of each sigma factor were set as 1 (indicated with the horizontal line).
-
DSigma‐dependent genes and correlation with expression of sigma factors (significantly changed) in ΔrnjA (LK1381). The most downregulated sigma factor was sigD, and almost all sigD‐dependent genes were downregulated. A similar trend is visible for the rest of sigma factors and their respective dependent genes. The x‐axis shows expression of each sigma factor, and the y‐axis shows expression of genes for each sigma regulon. The violet (dark blue) line: regression line.
Source data are available online for this figure.
A comparison with the previously reported transcriptome data based on the depletion strain (Durand et al, 2012) revealed that the effect of the complete absence of RNase J1 significantly overlapped with the effect of its depletion. The depletion strain displayed changes in RNA accumulation of 1,261 genes (at least a twofold difference). 239 out of the 385 downregulated mRNAs (62%) in the RNase J1 depletion strain were also downregulated in the RNase J1 null strain. 504 out of the 876 upregulated mRNAs (58%) in the RNase J1 depletion strain were also upregulated in the RNase J1 null strain (Fig EV2). Nevertheless, using the deletion strain we identified ~ 1,000 new genes whose expression was affected by RNase J1 (Table EV3).
Figure EV2. Comparison of differentially regulated genes in RNase J1 depletion vs. deletion strains.
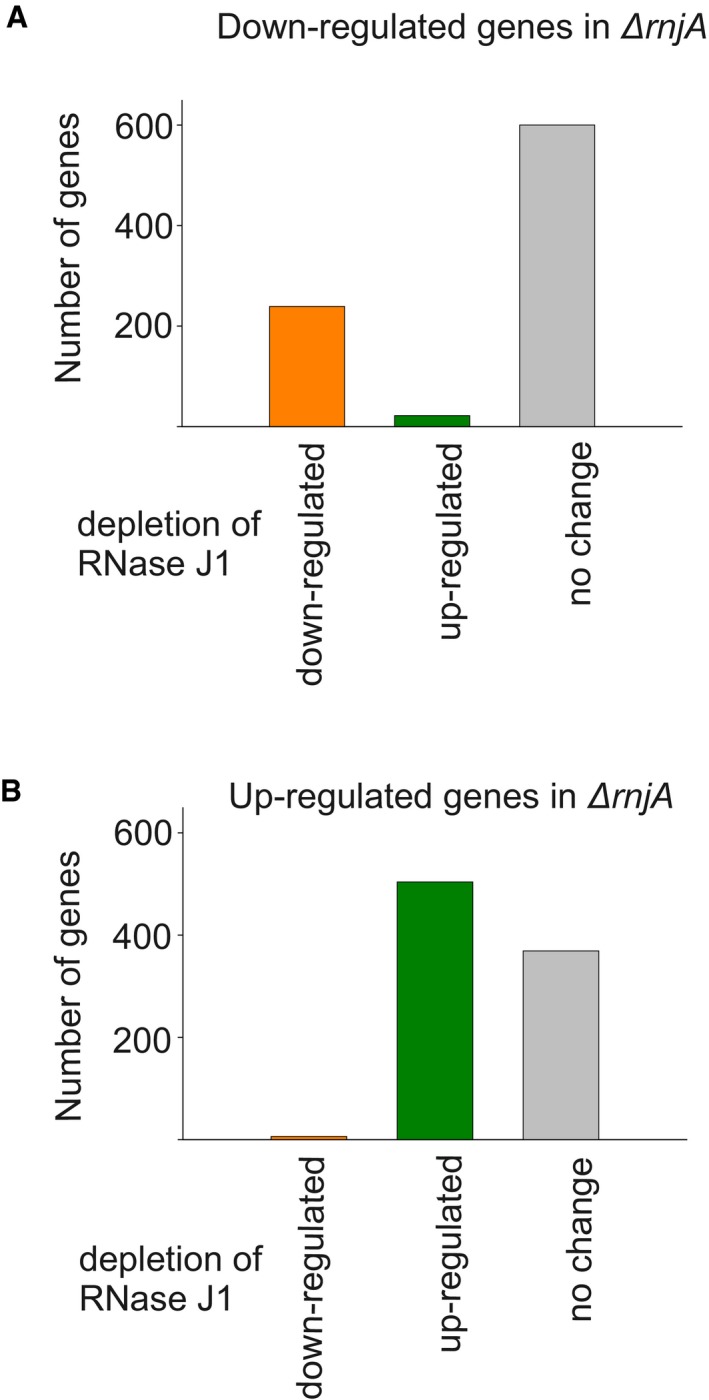
Source data are available online for this figure.
To see whether we could correlate the changes in gene expression with RNAP occupancy of the affected genes, we conducted ChIPseq experiments, comparing wt and RNase J1‐null strains, where we sequenced the DNA associated with RNAP in the cell. Surprisingly, we observed a major redistribution of RNAPs over the genome. Figure 1A and B shows an overall comparison of the RNAseq and ChIPseq data for wt and ΔrnjA.
For comparisons of RNAseq and ChIPseq data, we had anticipated and subsequently detected two types of effects: (i) correlated effects—the more or less occupied a gene with RNAP, the higher or lower the respective RNA level (classes I and II, showing transcriptional up or down effects, respectively), and (ii) effects where genes with increased RNA levels in the mutant displayed unchanged or possibly decreased occupancy with RNAP, reflecting an increased stability of these RNAs because they are direct targets of RNase J1 (class III, representing primarily post‐transcriptional effects). Indeed, the averaged effects of the rnjA deletion on both RNA abundance (RNAseq) and DNA occupancy with RNAP (ChIPseq) showed mRNAs belonging to these three classes (Fig 2).
Figure 2. Class I, II, III, and IV genes.
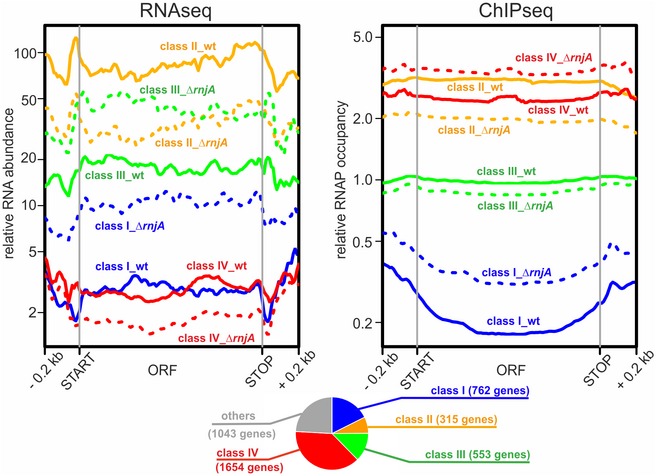
Average gene profiles of normalized RNAseq and RNAP ChIPseq coverages from wt (solid lines) and ΔrnjA (dashed lines) strains were plotted for gene classes I–IV (n = 762, 315, 553, and 1,654 genes, respectively). Open reading frames were rescaled to 1 kb; upstream and downstream regions of 0.2 kb were also included in the plots. Data represent mean values of three independent experiments. For the definition of class I–IV genes, see text and Materials and Methods section “Gene classification (classes I–IV)”. The pie chart shows the overall distribution of classes I–IV and other genes.Source data are available online for this figure.
Remarkably, we also detected another effect—a reciprocal phenomenon to class III, i.e., an increased RNAP occupancy and an equal or decreased mRNA level (Fig 2). This occurred especially in the case of genes with less abundant transcripts, although some genes, such as veg (Lei et al, 2013), with highly abundant transcripts were identified as well (Fig EV3, class IV). These genes were shifted to the right (in the ChIPseq dimension) in the graph in Fig 1, unlike highly expressed genes in the upper part of the graph. The increased occupancy of RNAP within these genes without a parallel RNA output suggests the presence of non‐productive or stalled elongation complexes. Genes of classes I–IV represented three quarters of all B. subtilis genes; a quarter of all genes was relatively unaffected or not detected by RNAseq (Fig 2). The list of genes sorted according to classes I–IV is provided in Table EV4. Examples of individual genes from these classes are shown in Fig EV3. Validations of the ChIPseq data (by qPCR) are shown in Appendix Fig S3.
Figure EV3. Detailed comparisons of the RNAseq and ChIPseq data for selected genes.
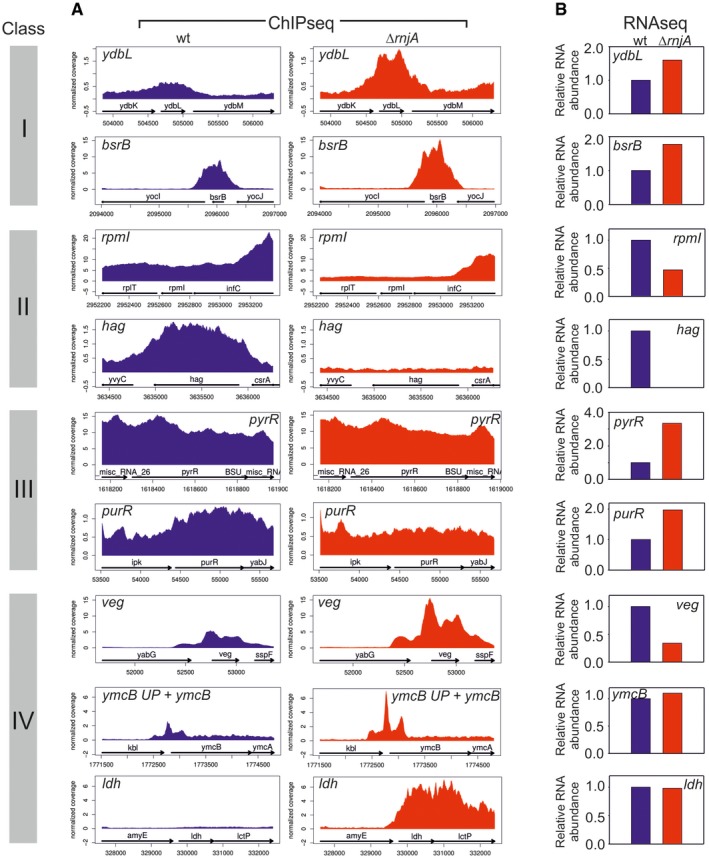
- RNAP occupancy at selected genes (gene names indicated) in wt (blue) and ΔrnjA (red) strains. ymcB UP—5′ untranslated region (UTR) of the ymcB gene. Data represent mean of normalized RNAP coverage from three independent ChIPseq experiments.
- RNA abundance data for genes shown in (A). The genes are grouped into classes (indicated with colored bars with roman numerals) according to trends they displayed between wt and ΔrnjA with respect to gene occupancy and relative expression. Class I: Increased gene occupancy in ΔrnjA is accompanied by increased gene expression; class II: decreased gene occupancy in ΔrnjA is accompanied by decreased gene expression; class III: decreased or equal gene occupancy in ΔrnjA is accompanied by increased gene expression; and class IV: increased gene occupancy in ΔrnjA is accompanied by decreased or equal gene expression. The relative RNA level in wt was set as 1. Data represent mean of normalized read coverage from three independent RNAseq experiments.
Source data are available online for this figure.
We checked whether the increased occupancy of DNA with RNAP on class IV genes might be explained by an elevated concentration of RNAP in the mutant; we determined the level of RNAP in both wt and ΔrnjA cells. In fact, we detected a lower level of RNAP in cells lacking RNase J1 (Fig EV4A–C). This correlated with the decreased levels of transcripts of the rpoA, rpoB, and rpoC RNAP subunit‐encoding genes, observed in the RNAseq experiments (Fig EV4D). Thus, the redistribution of RNAP over the genome in the rnjA mutant was not due to an increased abundance of RNAP.
Figure EV4. Amount of RNAP in wt and ∆rnjA strains.
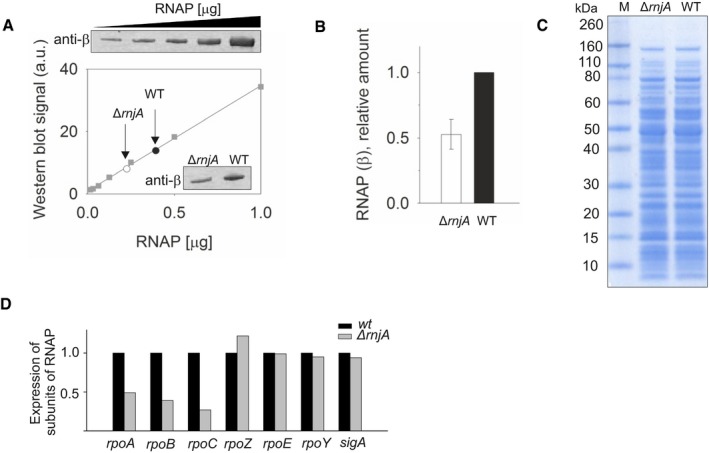
- Determination of the amount of RNAP in wt (black circle) and ΔrnjA (open circle) strains by Western blotting with monoclonal antibody against the β subunit of RNAP. The data for the calibration curve (squares) are shown above the graph; the data for the two strains are in the inset.
- Quantitation of the data shown in (A). The amount of RNAP in wt was set as 1. The experiment was performed three times on three different days. The bars show the average, the error bars ± SD.
- The amounts of cell lysates used to determine relative amounts of β. 5 µg of total protein was loaded per lane.
- Relative mRNA levels of three main RNAP subunits in wt [black bars, set as 1 (LK1371)] and ΔrnjA (gray bars; LK1381) strains. The main RNAP subunits (rpoA, rpoB, rpoC) had lower expression compared to wt. The expression of small subunits (ω, δ and ε) and sigA was unchanged.
Source data are available online for this figure.
These results prompted the following question: Could RNase J1 directly affect gene occupancy by RNAP? A possible scenario was that it might contribute to removing stalled RNAP complexes from the DNA. Hence, in the absence of RNase J1, the occupancy of some genes with RNAP would increase as they would contain more stalled RNAPs. To remove these stalled RNAPs, we speculated that RNase J1 might act on the RNA extruding from RNAP and degrade it in the 5′‐to‐3′ direction. Upon encountering RNAP, the resulting interaction would cause the transcription complex to disassemble.
RNase J1 and RNAP co‐localize in vivo and are associated through RNA
If our hypothesis is correct, it would require RNase J1 and RNAP to interact in the cell. However, RNase J1 has been reported to be localized either in the polar regions of the cell (Cascante‐Estepa et al, 2016) or associated with ribosomes (Even et al, 2005), and generally not present around/in the nucleoid. To resolve this issue, we used super‐resolution microscopy (SIM) and strain bearing RNase J1‐GFP and RNAP‐mCherry. Figure 3A shows that RNase J1 was present in other regions of the cell besides the poles. Moreover, the strong overlap of the two fluorophores, especially on the periphery of the nucleoid, supports the idea that these two enzymes could encounter each other in the cell. To view a larger number of cells, see Appendix Fig S4.
Figure 3. RNAP and RNase J1 may interact in the cell.
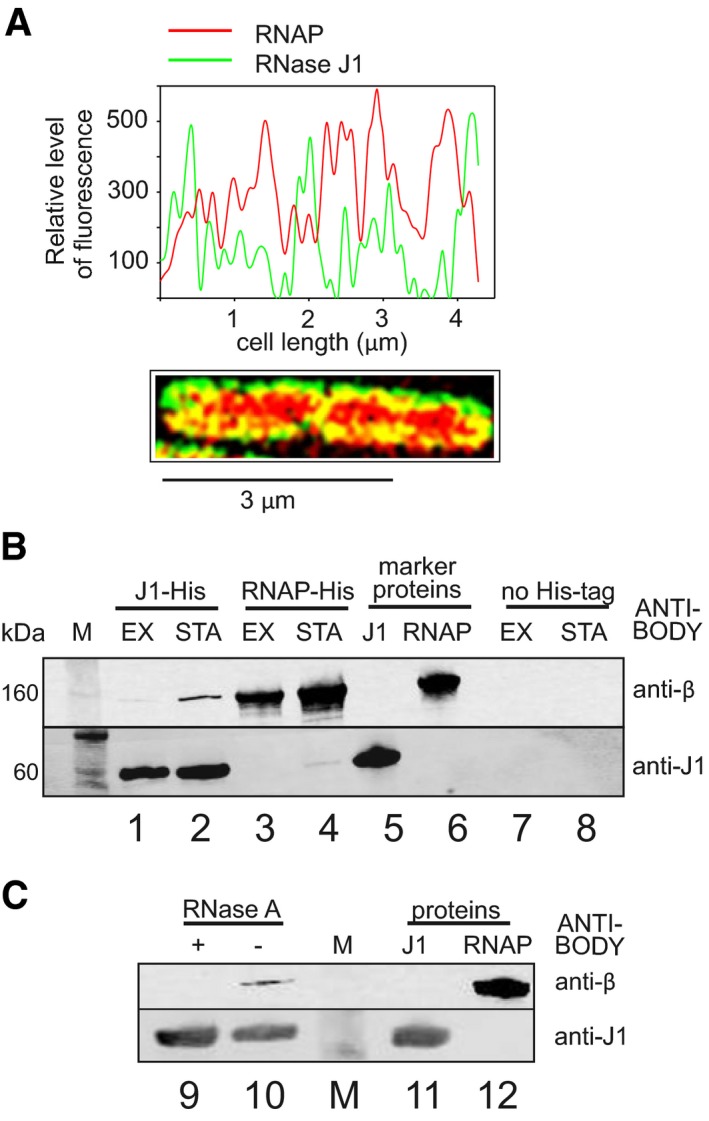
- SIM of exponential Bacillus subtilis cells. RNase J1 was fused to GFP (green), RNAP to mCherry (red). The graph shows relative fluorescence intensities at the cell midsection (along the long axis); SIM of the cell is below. Yellow indicates colocalization of the two proteins.
- Pull‐down with RNase J1‐8xHis tag and RNAP‐10xHis tag and detection of the proteins with antibodies. Lanes 1 and 2—RNase J1‐8xHis was used to pull down RNAP; lanes 3 and 4—RNAP‐10xHis was used to pull down RNase J1; lanes 5 and 6—purified proteins were used as markers; lanes 7 and 8—strains without His‐tagged proteins were used as negative controls to demonstrate the specificity of the interaction. M, molecular size marker; EX, exponential phase; STA, stationary phase.
- Pull‐down with RNase J1‐8xHis tag from stationary phase cells—the same conditions as in (B). The samples then either were (lane 9) or were not (lane 10) treated with RNase A to detect whether the interaction was via RNA. Lanes 11 and 12—purified proteins were used as markers. The experiment was performed three times (biological replicates) with the same result.
Source data are available online for this figure.
To test more directly whether RNase J1 is associated with RNAP, we pulled down RNase J1 via a His‐tag and used an antibody against the β subunit of RNAP to detect its presence in complex with RNase J1. Figure 3B shows that RNase J1 associates with RNAP in both exponential and stationary phases (lanes 1 and 2), although we retrieved larger amounts of RNase J1 in the latter. In a reciprocal experiment with His‐tagged RNAP, we detected RNase J1 in stationary phase only (lane 4). In a control (using a strain without His‐tagged proteins), neither RNase J1 nor RNAP was detected (lanes 7 and 8).
We next asked whether the observed complexes between RNase J1 and RNAP represented a direct protein–protein interaction, or whether this association depended on RNA. Figure 3C shows that RNase A treatment abolished the association (lanes 9 and 10), suggesting that RNase J1 and RNAP were linked via RNA.
RNase J1 dissociates transcription elongation complexes
To address directly whether RNase J1 is able to resolve stalled transcription complexes, we set up an in vitro system with purified components (Fig 4A). We assembled B. subtilis RNAP transcription elongation complex on a DNA scaffold with a 30‐nt RNA. The complex was attached to streptavidin beads via biotinylated DNA. Then, we utilized the enzymatic activity of RNAP to label the RNA with three consecutive Us (encoded in the DNA), by adding radioactive UTP to the complex (Fig 4B). Subsequently, we washed off the unincorporated UTP and added either RNase J1 or RNase R, a 3′‐to‐5′ exoribonuclease, as a negative control. Figure 4B (lane 2) shows that RNase J1 was able to degrade the extruding RNA up to RNAP, leaving 17–18 nt long stubs, consistent with the length of the RNA channel that protects the exiting RNA (~ 16 nt; Fig EV5A). Importantly, smaller RNA fragments, 1–5 nt in length, also appeared in the gel. RNase J1 is known to have processive 5′ exoribonuclease activity until RNA fragments are chewed down to < 5 nts in size, after which the enzyme behaves distributively (Dorleans et al, 2011). The fragments released in the in vitro degradation assay thus correspond to the expected sizes of the distributive products of RNA digestion by RNase J1, after the RNA was released from RNAP into the buffer. That these short RNA products are no longer associated with RNAP was confirmed by showing that they disappeared upon washing the beads after RNase J1 treatment (lane 6). The 17–18 nt long fragments, however, remained tightly associated with the beads after the wash step showing they were still in complex with RNAP and suggesting that not all RNAP complexes reached by RNase J1 release their RNAs immediately.
Figure 4. RNase J1 disassembles stalled transcription complexes (TC).
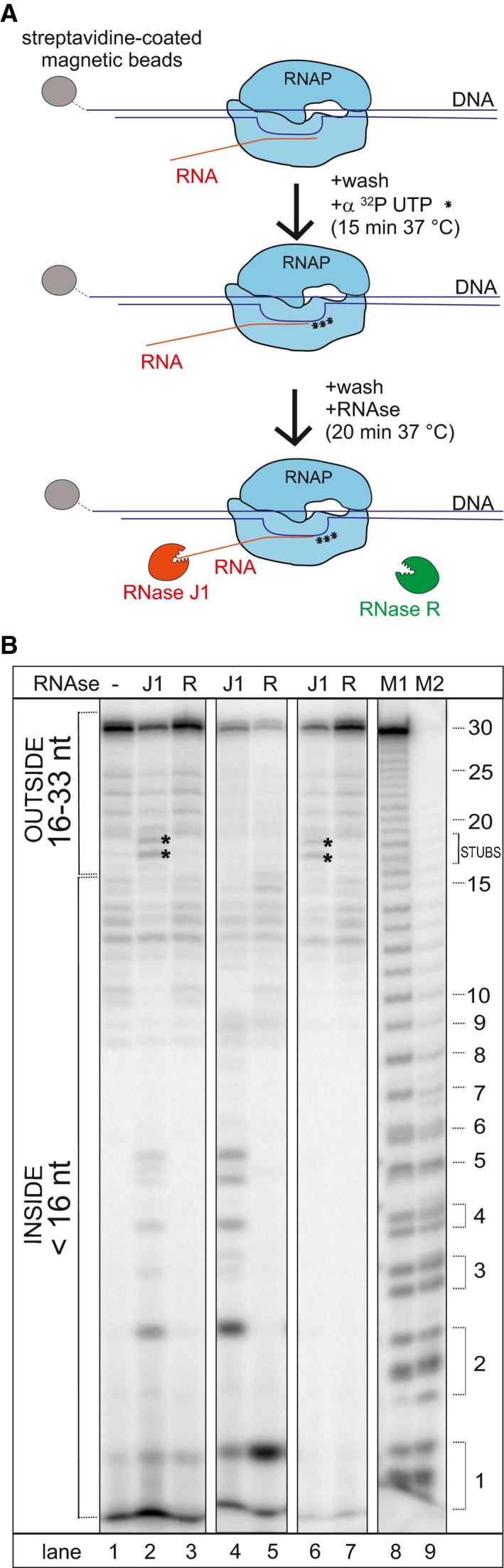
- Schematic representation of the flow of the experiment. TCs were assembled on DNA‐RNA scaffolds (DNA was biotinylated); RNA was labeled at 3′ with radioactive UTPs (asterisks). The RNA length (including label) was 33 nt.
- Representative primary data—polyacrylamide gel (the experiment was performed 3× with the same results). Lane 1, the full‐length (33 nt) labeled RNA; lane 2, the same as lane 1 but it included also incubation with RNase J1 (J1), 17–18 nt long fragments (RNase J1 stopped by RNAP; indicated with asterisks) and < 5 nt fragments (RNA released from TC into buffer—indicative of TC disassembly) are shown; lane 3, the same as lane 1 but included also incubation with RNase R (R); lanes 4 and 5, TCs were denatured by heat prior to RNase addition to demonstrate the activity and cleavage patterns of both enzymes; lanes 6 and 7, the same as lanes 2 and 3 but the buffer was washed off (TCs were retained by streptavidin beads) to demonstrate which RNA fragments were associated with TC; lanes 8 and 9 (M1, M2) Mw marker generated by treating the 30 nt RNA with alkali and formamide (M1—4‐min treatment, M2—7‐min treatment). As reported in Costanzo et al (2016) (and references therein), the cleavage by alkali or formamide leaves the phosphate group of the attacked phosphodiester bond bound at 3′, initially in the 2′,3′ cyclic form (upper band in the band couples). This successively opens (lower band in the band couples) yielding a double‐banded pattern for short oligoribonucleotides.
Source data are available online for this figure.
Figure EV5. Paths of RNA in RNAP, RNase J1, and Xrn1.
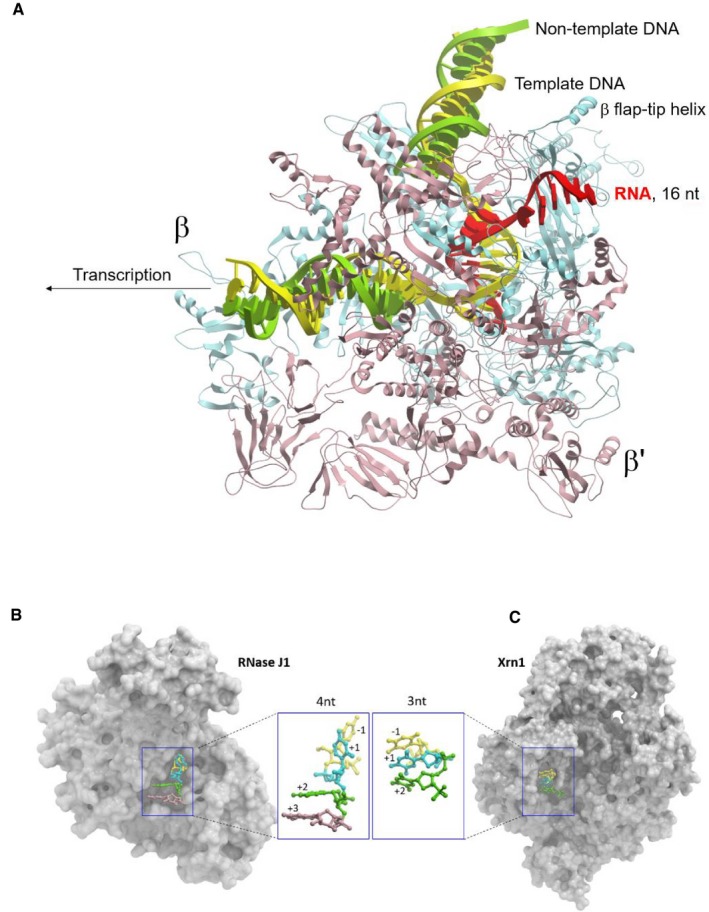
- Overall view of RNAP‐16nt‐mRNA (PDB id: 6flq). RNAP and bound nucleic acids are shown as cartoon (β—blue, β′—salmon, template DNA—yellow, non‐template DNA—green, mRNA—red).
- Overall view of RNaseJ1‐4nt‐RNA substrate complex (PDB id: 3t3o).
- Overall view of Xrn1‐3nt‐RNA substrate complex (PDB id: 2y35). RNaseJ1/Xrn1 are shown as surface representation. The nucleic acid substrates are shown in ball and stick format. The carbon atoms of individual nucleotides are colored relative to the position of the scissile bond: yellow (−1), cyan (+1), green (+2), and pink (+3).
The 3′ exoribonuclease RNase R was unable to digest the RNA, as expected, since the 3′ RNA end is protected inside RNAP (Fig 4B, lane 3). This control was important to show that no free RNA was present in the reaction and that the complexes did not spontaneously dissociate during the incubation period. Lanes 4 and 5 show reactions where the elongation complexes were heat‐denatured prior to nuclease treatment to show the patterns of free RNA digested with the two RNases. As expected, no 17–18 nt long fragments were detected with either RNase J1 or RNase R. Rather, the 1–5 nt distributive products of RNase J1 digestion were observed. The experiments clearly show that RNase J1 can digest RNA in stalled transcription complexes until it reaches RNAP and then trigger the release of the 17–18 nt RNA stub to digest it further to 1–5 nt end products.
Next, we wished to determine to what extent the effect of RNase J1 was specific. We assembled elongation complexes and treated them with either RNase J1 or the eukaryotic 5′‐to‐3′ exoribonuclease Xrn1 (Sun et al, 2013). Figure 5A–C shows that RNase J1 was significantly more efficient at degrading full‐length RNA than Xrn1, which was stopped more frequently by RNAP, as indicated by the greater quantities of RNA stubs still associated with RNAP. We also note that Xrn1‐generated stubs were more diverse than RNase J1‐generated stubs (Fig 5A—asterisks), likely reflecting differences in the behavior of these two RNases as they approach RNAP (Fig EV5B and C).
Figure 5. The effect of RNase J1 on TC is RNase‐specific.
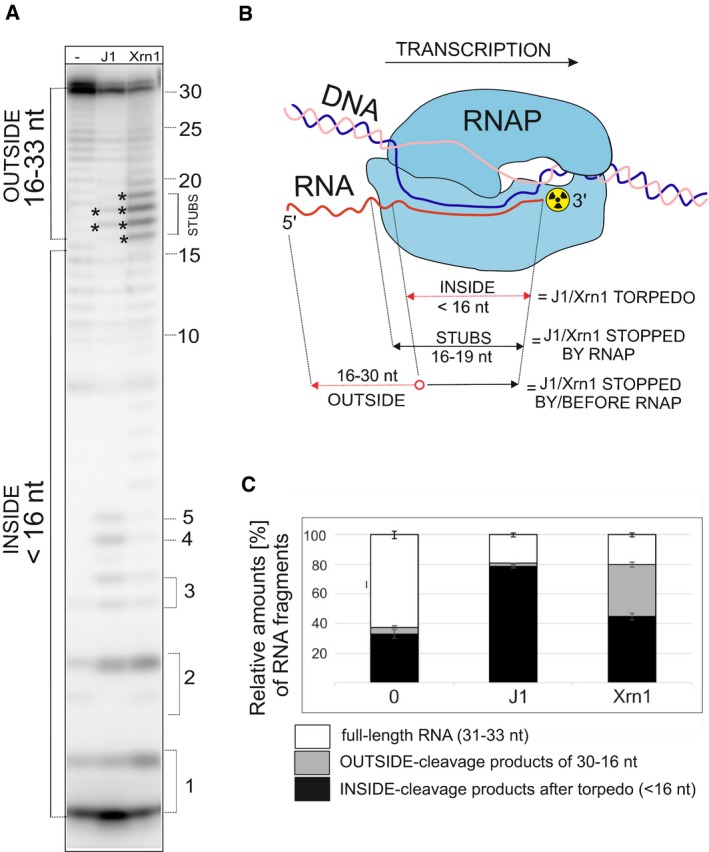
- Primary data—polyacrylamide gel—a representative result. The experimental setup was the same as in Fig 4. For the description of bands/fragments, see next panel legend. Asterisks indicate STUBS (16–19 nt).
- A schematic representation of quantitation of fragments. OUTSIDE (16–30 nt) are RNA fragments that originated by digestion of the full‐length RNA by RNases that were stopped either before reaching or at the point of reaching RNAP; STUBS (16–19 nt) are RNA fragments that originated by digestion of the full‐length RNA that were stopped at the point of reaching RNAP; INSIDE (< 16 nt) are RNA fragments (oligonucleotides) that could be only generated after the disassembly of the complex by the torpedo mechanism. The red color indicates parts of RNA that were digested.
- Quantitation of three independent experiments. “Full length“indicates the remaining undigested RNA. “OUTSIDE” and “INSIDE” are fragments as explained in (B) and indicated in (A). The bars represent 100% (all fragments). The black‐gray‐white boxes indicate the percentage of each fragment group (in %). The error bars indicate ± SEM for each group of fragments calculated from three biological replicates.
Source data are available online for this figure.
As in the previous experiments we used the degradation of RNA in its entirety as an indirect indicator that the EC had been dissociated, we asked whether RNase J1 truly dissociates RNAP from DNA. We assembled ECs as in the previous experiment and challenged them either with buffer (mock treatment), RNase J1, or Xrn1. By Western blotting with anti‐β (subunit of RNAP) antibody, we then detected the amounts of RNAP retained on beads (in complex with DNA) and released in the buffer (dissociated). Figure 6A shows that RNase J1 was able to dislodge RNAP from DNA and was more efficient in this regard than Xrn1, consistent with the results from the previous experiment.
Figure 6. RNase J1 specifically dissociates RNAP from DNA in stalled TCs.
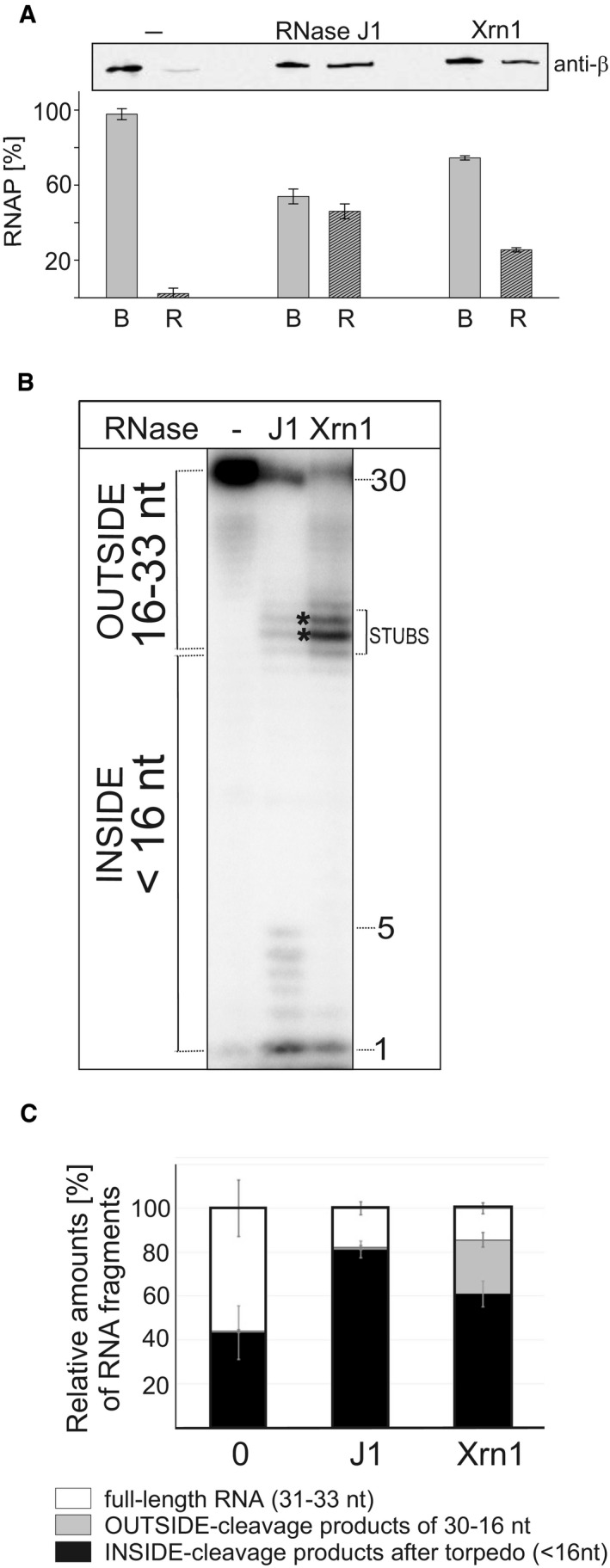
- TCs were assembled on DNA‐RNA scaffolds (DNA was biotinylated and attached to streptavidin‐coated magnetic beads); TCs were then divided into three tubes and challenged with buffer (“−”, mock treatment) or RNase J1 of Xrn1. Dissociation of RNAP from DNA was monitored by detecting RNAP with anti‐β antibody in two fractions: B—beads (RNAP still bound to DNA) and R—released (free in buffer). The gel strip shows representative primary data (Western blot). The graph shows averages (the bars) of two experiments (biological replicates), and the error bars show the range. The combined signal for B+R for each treatment was set as 100%.
- Primary data—polyacrylamide gel. The experimental setup was the same as in Fig 5 but with RNAP from Escherichia coli. Lane 1, the full‐length (33 nt) labeled RNA; lane 2, the same as lane 1 but it included also incubation with RNase J1 (J1); lane 3, the same as lane 1 but included also incubation with Xrn1.
- Quantitation of three independent experiments. “Full length” indicates the remaining undigested RNA. “OUTSIDE” and “INSIDE” are fragments as explained in Fig 5. The bars represent 100% (all fragments). The black‐gray‐white boxes indicate the percentage of each fragment group (in %). The error bars indicate ± SEM for each group of fragments.
Source data are available online for this figure.
To further pursue the question of specificity, we asked whether RNase J1 and Xrn1 would function similarly with RNAP from Escherichia coli. We performed the same experiments as in Fig 5 and obtained similar results (Fig 6B and C), suggesting that the effects of these two RNases are relatively non‐specific with respect to RNAP.
Finally, we tested whether ε (encoded by rpoY), a small, non‐essential subunit of B. subtilis RNAP, which is organized in a two‐gene operon with rnjA (Keller et al, 2014), had an effect on dissociation of ECs in the in vitro assay, but did not detect any impact (data not shown).
Effect of Rho
Of the factors involved in the resolution of stalled RNAP complexes, Rho was significantly downregulated in ΔrnjA (Fig 7B). A decreased level of Rho could conceivably contribute to the observed effect found in class IV genes. Another factor whose activity significantly changed (fivefold ↑) was HelD. HelD is a helicase‐like protein that associates with RNAP and helps with transcription recycling, possibly helping with RNAP release from DNA (Wiedermannova et al, 2014). This increased expression may help the cell to compensate for the absence of RNase J1. To address a possible role for Rho or HelD, we compared wt cells with single and double deletion mutants in RNase J1 and Rho or HelD under normal conditions and after UV irradiation. UV irradiation increases RNAP stalling as it creates changes in DNA (e.g., cyclobutane pyrimidine dimers and 6,4 pyrimidine–pyrimidones; Goodsell, 2001) that form obstacles to transcription. Consistent with the role of RNase J1 in disassembly of stalled RNAP complexes, the ΔrnjA mutant displayed increased sensitivity to UV irradiation compared to wt although the result was not statistically significant (P = 0.06; Fig 7). Interestingly, the absence of neither Rho nor HelD alone negatively impacted cell viability in response to UV treatment. In combination with the ∆rnjA mutation, the absence of Rho, but not HelD, appeared to lead to a further exacerbation of the UV‐sensitive phenotype (Fig 7).
Figure 7. Effect of RNase J1, Rho, and HelD in UV sensitivity assays.
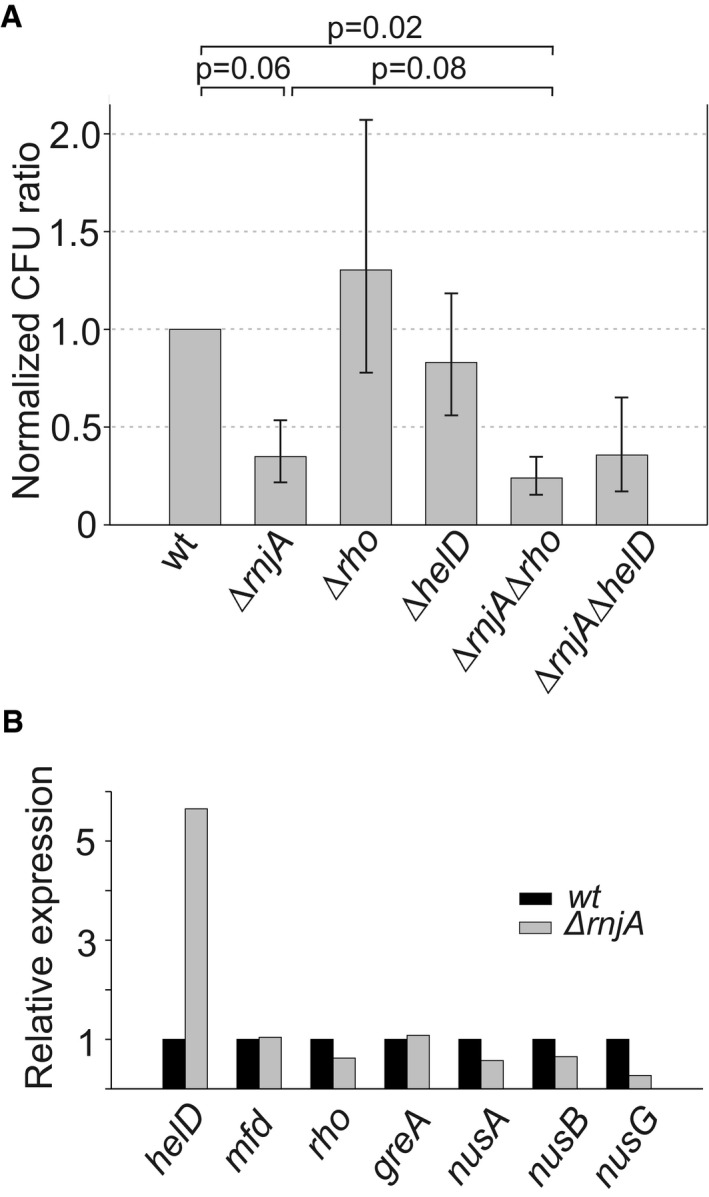
- Exponential cells of indicated strains (below the bars) were plated onto LB agar and either were or were not UV‐irradiated. After overnight incubation, CFU were counted. UV sensitivity of the mutant strains (KO) was then calculated as the ratio between irradiated vs. non‐irradiated cells and normalized to this ratio from the wt strain. As a consequence, the wt ratio is 1. The experiment was conducted 4× (biological replicates), and the bars show the geometric mean. The P‐values, shown above the bars, were computed using two‐tailed unpaired t‐test for logarithms of the ratios. The error bars show ± SEM (computed on the log scale).
- Relative expression (mRNA) of helD, mfd, rho, greA, nusA, nusB, and nusG in the ΔrnjA strain [normalized to wt (set as 1)].
Source data are available online for this figure.
Discussion
In this study, we have characterized the effect of the absence of B. subtilis RNase J1 on the transcriptome and DNA occupancy with RNAP. Moreover, we identified a novel concept linking RNA transcription and degradation: a 5′‐to‐3′ exoribonuclease (RNase J1 in B. subtilis), in addition to its canonical role in mRNA turnover, helps disassemble stalled transcription complexes, thereby contributing to the smooth functioning of the transcription machinery, and preventing transcription–replication collisions (Fig 8). Parallels can be drawn to the effects of prokaryotic RNA/DNA translocases such as Rho and Mfd, and even more closely to eukaryotic 5′‐to‐3′ exonucleases (see the second part of Discussion).
Figure 8. Model of RNase J1 functioning as the torpedo, dissociating stalled complexes of RNAP from DNA .
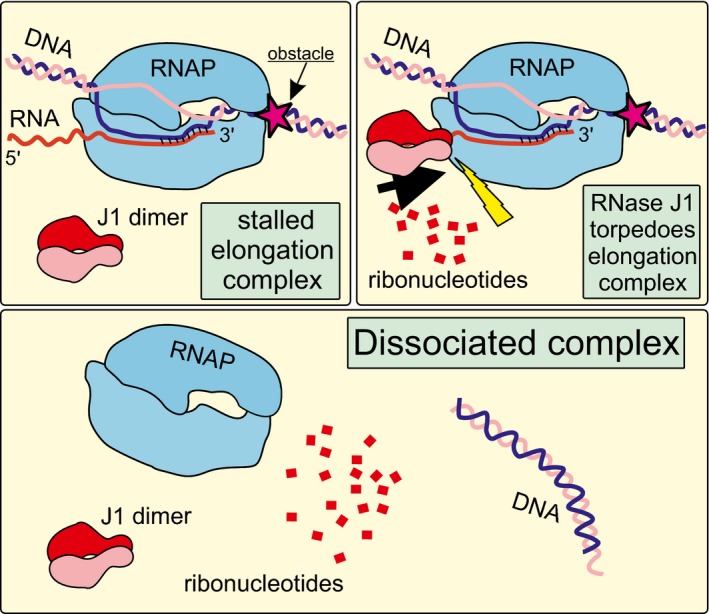
RNase J1 is depicted as a dimer. RNase J1, after the nascent transcript has been endonucleolytically cleaved, degrades RNA in the 5′‐to‐3′ direction. Upon encounter with RNAP, conformational changes are induced, likely resulting in dissociation of the complex, and liberating RNAP from DNA thereby removing obstacles potentially causing transcription–replication clashes.
Effect of the absence of RNase J1 on the transcriptome
More than one‐third of all genes were affected in the B. subtilis ΔrnjA mutant compared to wt, a moderately more pronounced effect than the one observed previously with an RNase J1 depletion strain (1,740 vs. 1,261 affected genes; Durand et al, 2012). In our previous study with an RNase J1‐depletion strain, we showed that most upregulated mRNAs were due to an increase in RNA stability, as one would expect for a loss of RNase activity, while most downregulated RNAs predicted to be due to indirect transcriptional effects (Durand et al, 2012). Although more genes belonged to class I (showing transcriptional up effects) than class III (post‐transcriptional up effects) in this study (Fig 2), some class I genes could have both increased transcription levels and increased stability. It is also important to note that “upregulated” mRNAs may not necessarily result in upregulated protein levels. In many cases, only non‐functional RNA fragments, corresponding to the 3′ products of endonucleolytic cleavages that are normally degraded by RNase J1, are overexpressed (Durand et al, 2012). Since the RNAseq reads were averaged over the whole open reading frame, if the degradation intermediate was reasonably long, it would result in an “overexpressed” candidate mRNA but not in more protein. For the following discussion, we will therefore only consider upregulated mRNA candidates whose full‐length, and presumably functional, mRNAs accumulate.
Transcription and translation machineries
We detected lower amounts of mRNAs encoding the core RNAP subunits (α, β, β′), which may be correlated with the slower growth rate of the ∆rnjA mutant. We did not detect a difference in the amount of the primary sigma factor mRNA, sigA (Nicolas et al, 2012; Ramaniuk et al, 2017). However, we observed differences (in both directions) for most alternative sigma factors (Fig 1C). Sigma factors involved in sporulation were generally upregulated as was the master regulator for entry into sporulation, spo0A (Molle et al, 2003). It should be noted, however, that the ∆rnjA mutant fails to sporulate (Figaro et al, 2013). On the other side of the spectrum, SigD, required for the motility of the cell (Helmann et al, 1988; Serizawa et al, 2004; Cozy & Kearns, 2010), was the most downregulated sigma factor, followed by extracytoplasmic function sigma factors, SigW (Turner & Helmann, 2000; Zweers et al, 2012) and SigM (Jervis et al, 2007; Eiamphungporn & Helmann, 2008). The upregulation/downregulation of sigma factors correlated to various degrees with upregulation/downregulation of the genes in their respective regulons, with the best correlation observed for SigD (Fig 1D). In general, the altered expression of sigma factors, especially those that were downregulated, appears to have contributed to the shaping of the transcriptome in the mutant strain. Interestingly, transcription elongation factors that stimulate pausing, NusA and NusG (Ma et al, 2015; Yakhnin et al, 2016), were downregulated (Fig 7B), which might be beneficial for the cell in light of the already high number of RNAPs stalled over the genome.
Many mRNAs transcribed from genes encoding the translation machinery were also downregulated, especially translation elongation factors (Appendix Fig S6), including tufA, encoding the most abundant protein in exponentially growing cells (Krasny et al, 2000). This might reflect the decreased growth rate of the ΔrnjA strain and may result in an increased stalling of ribosomes which could in turn increase stalling of RNAPs through uncoupling of the transcription–translation machineries (Nudler, 2012; Buskirk & Green, 2017).
RNases
Out of 22 RNases, half of them changed gene expression in the rnjA mutant. mRNAs for six RNases were upregulated (including rny, yhaM) and five RNases were downregulated. Of the upregulated RNases, RNase Y (rny) is a key RNase that is involved in maturation of RNase P RNA, small cytoplasmic RNA, and many mRNAs (Durand et al, 2012; Gilet et al, 2015). Upregulation of these RNases may be the result of the cell attempting to increase RNA turnover in the absence of RNase J1. The most downregulated RNase was rnhB, encoding RNase HII which is responsible for removing incorporated rNMP from DNA strand during replication (Randall et al, 2017). An absence of this RNase increases mutation rate in B. subtilis (Schroeder et al, 2017) and this could cause replication pausing, leading to replication–transcription collisions. Overall, alteration of expression of these RNases suggests that some of the effects of the absence of RNase J1 on RNA accumulation may be indirect, mediated by other RNases.
DNA repair and replication
A number of proteins involved in DNA repair were upregulated in the mutant strain (23 genes Table EV5), which may, at least in part, stem from feedback mechanisms increasing the amount of these proteins to help the cell cope with increased number of transcription–replication collisions with mutagenic/DNA damaging effects. These upregulated genes included genes for mismatch repair mutS, mutL (Liu et al, 2016; LeBlanc et al, 2018), nucleotide excision repair pcrA (Sanders et al, 2017), base excision repair mutM, mutT, ung, processing of abasic sites yqfS, exoA, yshC (Lenhart et al, 2012), and genes for restart after replication–transcription collision addA, addB, recA (Shepanek et al, 1989; Krajewski et al, 2014). Interestingly, genes involved in DNA repair after UV damage (UvrABC; Lenhart et al, 2012) were either unchanged or upregulated.
Genes involved in DNA replication were either up‐ or downregulated (see Table EV6). Conceivably, this imbalance in their expression could have contributed to the slow growth of the RNase J1‐null strain. Interestingly, ts (thermosensitive) mutants in the downregulated mRNAs DnaE/G/C form long filaments similar to the phenotype observed in the ΔrnjA strain (Janniere et al, 2007; Figaro et al, 2013). This phenotype is induced, e.g., when perturbations in DNA chain elongation lead to the generation of ssDNA.
RNase J1 and its role in disassembly of transcription complexes
The ChIPseq experiments revealed that genes, especially those with relatively low expression, displayed increased accumulation of RNAP in the ∆rnjA mutant, without a concomitant increase in RNA levels. This indicates an increase in the number of stalled RNAP complexes at these loci. Such complexes create obstacles for the replication machinery, and their collision may result in mutations in DNA and have an adverse effect on genome integrity. Pausing and stalling of RNAP is widespread and affects gene expression (Kang et al, 2019). The observed accumulation of RNAP on weakly transcribed genes is consistent with the finding that trailing RNAPs on heavily transcribed genes help the translocation of leading RNAPs, allowing them transcribe through regions that are more pause‐prone, or through possible obstacles on the DNA. On weakly transcribed genes, this phenomenon is absent (Epshtein & Nudler, 2003) and correlates with increased accumulation of RNAP on these genes in the absence of RNase J1.
Collectively, the experiments presented in this study reveal that RNase J1 is present in the cell in the vicinity of DNA, associates with RNAP, and is able to disassemble stalled transcription complexes.
Transcription and translation are often coupled when the leading ribosome helps push RNAP forward (Kohler et al, 2017). However, if these two processes become uncoupled, RNAP may be more prone to pausing and possibly backtracking, remaining in an inactive form (Zhang et al, 2014). RNase J1 access to primary transcripts is known to be inhibited by the presence of the 5′ triphosphate group (Mathy et al, 2007). We envision that either deprotection of the 5′ end by RNA pyrophosphohydrolase (RppH) activity (Hsieh et al, 2013; Frindert et al, 2018) or prior endonucleolytic cleavage allows RNase J1 access to the mRNA to degrade it to the outer edge of the RNA exit channel. Subsequently, the remaining part of RNA, initially protected by RNAP, becomes available for degradation by action of RNase J1. The action may be mediated by the “torpedo” effect. The torpedo effect was first described in eukaryotes, and it is mediated by the 5′‐to‐3′ exonucleases Rat1/Xrn2 or CPSF‐73 acting on eukaryotic RNAP II to terminate its transcription (Kim et al, 2004; Yang et al, 2009; Pearson & Moore, 2013; Baejen et al, 2017). This occurs after the nascent RNA has been cleaved at the polyadenylation signal when RNAP continues synthesizing non‐coding RNA and needs to be stopped. The mechanistic details of the torpedo effect are poorly understood. We do not know whether RNase J1 first dissociates RNAP and then degrades the RNA, or if it degrades the RNA first, which would cause collapse of the transcription bubble and subsequent dissociation of RNAP. Alternatively, shortening the RNA may induce backtracking and destabilize the complex, resulting in its disassembly. However, the fact that Xrn1 was also capable of shortening RNA to the RNAP‐protected stubs but unable to degrade the remaining RNA as efficiently as RNase J1 argues against this possibility.
We also considered the possibility that the endoribonuclease activity of RNase J1 might contribute to the results observed in this study, but a number of arguments favor the idea that the torpedo effect is primarily related to its 5′‐exoribonuclease activity. First, while RNase J1 does have endoribonuclease activity in vitro, it is primarily thought to act as an exoribonuclease in vivo. Indeed, most of the endonucleolytic cleavage sites previously ascribed to RNase J1 in vivo are now thought to be performed by RNase Y, which has a similar specificity (Condon, 2010). The enzyme's preference for exonucleolytic activity has been further confirmed by the crystal structure of RNase J1 bound to RNA (Dorleans et al, 2011). While RNA can easily be threaded through an entry channel to reach the catalytic site in exonucleolytic mode, endonucleolytic cleavage requires dissociation of dimers and then additional separation of the β‐CASP from the β‐lactamase domain to allow the RNA to lie across the catalytic site. This likely explains why endonuclease activity is only observed in the presence of a large excess of enzyme over RNA, i.e., by simple probability, only a few isolated RNase J1 molecules are likely to be in a conformation capable of performing endonucleolytic cleavage. Lastly, RNase J1 acts more processively as an exoribonuclease with increasing length of RNA (Dorleans et al, 2011). Thus, if RNase J1 were to first shorten the RNA endonucleolytically before acting as an exoribonuclease in our torpedo assay, this would likely decrease the efficiency of degradation of the short RNA (< 5 nts) buried within RNAP and an underestimate the torpedo effect.
The length of the stubs and the path of RNA in RNase J1 and RNAP (Fig EV5A and B) suggest that RNase J1 and RNAP are likely in contact at this point in the process. The region on the surface of RNAP around the RNA exit channel contains elements important for the stability of RNAP complexes. These elements include the ω subunit (Weiss & Shaw, 2015) and the β‐flap helix that interacts also with other proteins, such as NusA (Twist et al, 2011; Tagami et al, 2014; Ma et al, 2015; Guo et al, 2018). Whether and how RNase J1 interacts with these elements is currently unknown, and the details of the RNase J1‐RNAP contacts will be studied in future experiments.
Regardless of the mechanistic details, it appears that the efficiency of the effect is RNase‐specific as RNase J1 and Xrn1 acted with different efficiencies to provoke the release of the RNA and dissociate the EC. Therefore, different 5′‐to‐3′ RNases likely possess different abilities to disassemble stalled transcription complexes. This is consistent with the previously reported observation that yeast Rat1/Rai1 does not terminate E. coli RNAP, probably due to the divergent structure of the yeast enzyme (Park et al, 2015). In contrast, the effect does not appear to discriminate between E. coli and B. subtilis RNAPs as both RNase J1 and Xrn1 exerted similar effects on complexes assembled with these enzymes. We stress, however, that despite the large phylogenetic distance between these two species, the relevant regions (RNA exit channel region) in β and β′ are highly homologous. It is possible that more divergent RNAPs, such as single‐peptide enzymes (e.g., E. coli T7 phage RNAP) may behave differently toward RNase “torpedoes”.
Other prokaryotic proteins that function in a “torpedo”‐like manner are Rho and Mfd. These factors are ATP‐dependent (Epshtein et al, 2010; Le et al, 2018). Their mechanisms of dissociating the transcription complex differ from each other and are likely also different from that of RNase J1. Rho was reported to decrease non‐specific, pervasive transcription (Bidnenko et al, 2017), and its mRNA level was decreased in the ΔrnjA strain. We tested the effect of its absence in UV sensitivity assay. While deletion of the rho gene alone had no negative effect, deletion of rho and rnjA appeared to exacerbate the phenotype, consistent with the hypothesis that these two proteins function in an analogous manner, but in different pathways. We compared RNAseq data from a B. subtilis Δrho strain (Nicolas et al, 2012) with our RNAseq data from ΔrnjA, focusing on class IV genes. Appendix Fig S5 reveals virtually no correlation between these two datasets, indicating that RNase J1 and Rho act on different sets of genes.
5′‐to‐3′ exoribonucleases are widely present in eukaryotes where transcription and replication also clash (Hamperl & Cimprich, 2016). Therefore, we envision that in eukaryotic cells 5′‐to‐3′ exoribonucleases may also be involved in the resolution of stalled RNAP complexes to prevent transcription–replication clashes that could result in mutations in DNA with undesirable consequences. This could be linked to diseases, such as polyglutamine diseases (neurodegenerative diseases) where, e.g., 5′‐to‐3′ exoribonuclease Xrn1 is sequestered and inactive in nuclear inclusions, a characteristic of the pathological state of the cell (Mori et al, 2018).
Materials and Methods
Cloning and strain construction
To prepare a ΔrnjA strain in a widely used B. subtilis genetic background, chromosomal DNA from a ΔrnjA strain (LK1191 = CCB434 in Figaro et al, 2013) was transformed into B. subtilis BaSysBio (Nicolas et al, 2012), yielding strain LK1381. Strains ΔrnjA Δrho (LK2082) and ΔrnjA ΔhelD (LK2336) were prepared with transformation of chromosomal DNA from the ΔrnjA strain (LK1190) into B. subtilis Δrho (LK2058) (Bidnenko et al, 2017) or ΔhelD (LK2329) strain (Wiedermannova et al, 2014).
To prepare a strain for pull‐out experiments, RNase J1 was first amplified by PCR from gDNA of B. subtilis (LK566) and primers #1647/#1648 with Expand High Fidelity PCR System (Roche) and ligated into the expression vector pMUTIN4 (LK957) encoding a N‐terminal 8xHis‐tag via HindIII (Takara) and BamHI (Takara) restriction sites and transformed into E. coli DH5α (RLG6911), yielding strain LK1647. The plasmid was isolated with Wizard Plus Midipreps DNA purification system (Promega), verified by sequencing, and transformed into B. subtilis BaSysBio, resulting in strain LK1651.
To prepare strains for super‐resolution microscopy (SIM), chromosomal DNA from B. subtilis expressing GFP‐RNase J1 (LK1728 = 3,565 in Hunt et al, 2006) and plasmid from E. coli mCherry‐RNAP (LK2320 = pNG622 in Doherty et al, 2010), a gift from P. Lewis] were transformed into B. subtilis BaSysBio.
For a complete list of strains (and their sources), see Table EV7.
Media and antibiotics
All experiments were performed in the rich LB medium, unless indicated otherwise. When required, the medium was supplemented with antibiotics: ampicillin 100 μg/ml (for E. coli), kanamycin 5 μg/ml, lincomycin 12.5 μg/ml, erythromycin 0.5 μg/ml, chloramphenicol 5 μg/ml, spectinomycin 100 μg/ml, and phleomycin 2 μg/ml (for B. subtilis). The expression of GFP and mCherry fusion proteins was induced with 0.5% xylose (final concentration).
3H incorporation
This experiment was conducted as described previously (Panova et al, 2015). Wt and ΔrnjA strains were grown in defined MOPS medium to OD600 0.3 (early exponential phase). RNA in cells was labeled with 3H‐uridine (1 μCi/ml), and cold uridine was added to a final concentration of 100 μM. At 0, 10, 20, 30, 40, 50, and 60 min, 100 μl and 250 μl of cells were withdrawn to measure cell density and determine 3H incorporation, respectively. The 250 μl of cells was mixed with 1 ml of 10% trichloroacetic acid (TCA) and kept on ice for at least 1 h. Thereafter, each sample was vacuum‐filtered and washed twice with 1 ml of 10% TCA and three times with 1 ml of ethanol. The filters were dried, scintillation liquid was added, and the radioactivity was measured. The signal was normalized to cell density.
Scanning electron microscopy
Exponential cultures of B. subtilis WT and ΔrnjA strains (OD600 0.5) were prefixed with 1.5% glutaraldehyde for 1 h at room temperature. The cells were then washed with cacodylate buffer (Thermo Fisher Scientific) and fixed with 3% glutaraldehyde in cacodylate buffer overnight at 4°C. The extensively washed cells were allowed to sediment overnight at 4°C onto poly‐l‐lysine‐treated circular glass coverslips. The coverslips were dehydrated in a graded ethanol series (25, 50, 75, 90, 96, 100, and 100%) followed by absolute acetone and critical point dried in a K850 Critical Point Dryer (Quorum Technologies Ltd, Ringmer, UK). The dried samples were sputter‐coated with 3 nm of platinum in a Q150T Turbo‐Pumped Sputter Coater (Quorum Technologies Ltd, Ringmer, UK). The final samples were examined in a FEI Nova NanoSEM scanning electron microscope (FEI, Brno, Czech Republic) at 5 kV using ETD, CBS, and TLD detectors.
Pull‐down of RNase J1 and RNAP and Western blotting
Bacillus subtilis RNAP with a His10‐tagged β′ subunit (LK1275) or His8‐RNase J1 (LK1651) was pulled out from exponential and stationary phase cells (EXP, OD600 0.5; STA OD600 ~ 3; 2 h after entry into STA) via Ni‐NTA beads. RNase A (200 ng/ml) was or was not added to the lysates. To determine the amount of RNAP, exponentially growing wt and ΔrnjA cells were sonicated and 5 μg of proteins was analyzed by sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS–PAGE) and detected by Western blotting using mouse monoclonal antibodies against the β subunit of RNA polymerase (clone name 8RB13) or rabbit polyclonal antibodies against RNase J1 and secondary antibodies conjugated with a fluorophore dye [WesternBright™ MCF‐IR, Advansta, 700 nm anti‐rabbit (RNase J1) or 800 nm anti‐mouse (RNAP) antibody], and the interactions were quantified with an Odyssey reader (LI‐COR Biosciences).
Purification of proteins
Bacillus subtilis RNAP from wild‐type strain with a His10‐tagged β′ subunit (LK1275), σA (LK1365), and RNase J1 was purified. The purification of RNAP was performed as described previously (Qi & Hulett, 1998), and σA was purified as described previously (Chang & Doi, 1990; Juang & Helmann, 1994). Purification of RNase J1 was performed as in Condon et al (2008).
Super‐resolution microscopy
One milliliter of exponentially growing cells (OD600 0.5) with GFP‐RNase J1 and mCherry‐RNAP (LK2328) was washed and resuspended in 1× PBS. To measure the cell length, exponentially growing wt and ΔrnjA cells (OD600 0.5) were incubated with membrane dye NileRed (5 μg/ml, Sigma‐Aldrich) for 10 min at RT, washed, and resuspended in 1× PBS. Strains were analyzed with super‐resolution microscopy DeltaVision OMX™ equipped with a 60 × 1.42, PlanApo N, oil immersion objective, and softWoRx™ Imaging Workstation software. GFP‐tagged proteins were imaged using 488 nm excitation; mCherry‐RNAP and NileRed were imaged using 568 nm excitation. 3D‐SIM resolution in XY was 130 ± 5 nm; 3D‐SIM resolution in Z was 340 ± 10 nm. Cell length was analyzed with Fiji ImageJ.
Chromatin immunoprecipitation, ChIPseq, and qPCR validation
Bacillus subtilis wt (LK1371) and ΔrnjA (LK1381) cells were grown at 37°C in LB to exponential phase (OD600 0.4–0.5). The culture was crosslinked with formaldehyde at a final concentration of 1% (30 min, 37°C). Then, 150 mM glycine was added and the cultures were incubated for additional 5 min at 37°C to stop the crosslinking process. Subsequently, cells were collected by centrifugation, washed with lysis buffer A (20 mM Tris–HCl, 150 mM NaCl, pH 8), resuspended in lysis buffer B (50 mM HEPES, 150 mM NaCl, 1 mM EDTA, 1% Triton X‐100, 0.1% natrium deoxycholate, protease inhibitor cocktail, Calbiochem), and sonicated to obtain 200–500 bp long DNA fragments. Concentrations of proteins were measured with the Bradford method (Bradford, 1976). 20 μl of DynaBeads protein A (Thermo Fisher Scientific) was incubated with 3 μg of antibody against the β subunit of RNAP (8RB13, Santa Cruz) for 2 h at 4°C. Cell lysates (1 mg of proteins) were then mixed with the complex of antibody‐DynaBeads and incubated overnight at 4°C. The beads were then washed 2× with lysis buffer B, 2× with lysis buffer 500 (50 mM HEPES, 1 mM EDTA, 500 mM NaCl, 1% Triton X‐100, 0.1% natrium deoxycholate, pH 7.6), 1× with LiCl buffer (10 mM Tris–HCl, 1 mM EDTA, 250 mM LiCl, 1% Triton X‐100, 1% natrium deoxycholate, pH 8), and 1× with TE buffer (10 mM Tris–HCl, 1 mM EDTA, pH 8). DNA–protein complexes were eluted from the beads with elution buffer (50 mM Tris–HCl, 10 mM EDTA, 1% SDS, pH 8) for 10 min at 65°C, dissociated at 65°C for 6 h in the presence of 200 mM NaCl and treated with 20 μg of proteinase K for 30 min at 37°C. Finally, DNA was purified by QIAquick Nucleotide Removal Kit (QIAGEN). Sequencing libraries were prepared in EMBL Heidelberg with NEBNext ChIP‐Seq Library Prep Master Mix Set for Illumina (BioLabs) according to the manufacturer's instructions. Pooled barcoded libraries (two samples in biological triplicates) were sequenced in a single lane at Illumina HiSeq 2000 in the 50 bp single‐end regime at EMBL Genomics Core Facility (Heidelberg, Germany). Read quality and potential adapter contamination were checked with FastQC v0.11.8 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Reads were aligned to the B. subtilis subsp. subtilis strain 168 genome (NCBI Nucleotide acc. no. NC_000964) using HISAT 2.0.5 (Kim et al, 2015)] and SAMtools 1.4 (Li et al, 2009). Only uniquely mapped reads (MAPQ ≥ 10) were kept. Alignment quality was checked visually using IGV 2.6.3 (Thorvaldsdottir et al, 2013). Read statistics for each sample can be found in Table EV8. Using deepTools 3.3.0 (Ramirez et al, 2016), sample data were first normalized to library size; then, IP sample coverage was normalized to the corresponding input samples, and finally, mean coverage from the three independent replicates was calculated. Per‐gene coverage values were then obtained using genome annotation obtained from NCBI (GCF_000009045.1; downloaded 15/Nov/2015), and coverage for each gene was also normalized to gene length. The ChIPseq data are available from the ArrayExpress database (http://www.ebi.ac.uk/arrayexpress) under accession number E‐MTAB‐5659. The scripts used for ChIPseq data processing and analysis are available from https://github.com/mprevorovsky/krasny-torpedo. To validate the ChIPseq data, we used quantitative PCR (qPCR) in a LightCycler 480 System (Roche Applied Science) containing LightCycler® 480 SYBR Green I Master and 0.5 μM primers (each). Primers were designed with Primer3 software, and their sequences are in Table EV7. The data were normalized to input.
RNAseq and RT–qPCR validation
Two milliliters of exponentially growing cells [wt (LK1371), ΔrnjA (LK1381); OD600 0.5] was treated with RNAprotect Bacteria Reagent (QIAGEN), pelleted, and immediately frozen. RNA was isolated with RNeasy Mini Kit (QIAGEN). Finally, RNA was DNase‐treated (TURBO DNA‐free Kit, Ambion). Five micrograms of total RNA was rRNA‐depleted with Ribo‐Zero Magnetic Kit; Gram‐positive bacteria (Epicentre) and strand‐specific libraries were then prepared with Illumina compatible NEXTflex Rapid Directional RNA‐Seq Kit (Bioo Scientific) according to the manufacturer′s instructions. Pooled barcoded library (two samples in biological triplicates) was sequenced in a single lane at Illumina HiSeq 2000 in 50 bp single‐end regime at EMBL Genomics Core Facility (Heidelberg, Germany). Read quality and potential adapter contamination were checked with FastQC v0.11.8 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Reads were aligned to the B. subtilis subsp. subtilis strain 168 genome (NCBI Nucleotide acc. no. NC_000964) using HISAT 2.0.5 (Kim et al, 2015) and SAMtools 1.4 (Li et al, 2009). Only uniquely mapped reads (MAPQ ≥ 10) were kept. Alignment quality was checked visually using IGV 2.6.3 (Thorvaldsdottir et al, 2013). Reads mapping to 30 ribosomal RNA genes (BSU_rRNA_1 – BSU_rRNA_30) amounted to only < 0.14% of total reads for each genotype, confirming a high efficiency of rRNA depletion. Read statistics for each sample can be found in Table EV8. Using deepTools 3.3.0. (Ramirez et al, 2016), sample data were first normalized to library size, and mean coverage from the three independent replicates was calculated. Per‐gene coverage values were then obtained using genome annotation obtained from NCBI (GCF_000009045.1; downloaded 15/Nov/2015), and coverage for each gene was also normalized to gene length. The analysis of differential gene expression was performed using unnormalized BAM files and the GenomicAlignments and DESeq2 packages in R/Bioconductor, with FDR set to 0.05 (Love et al, 2014). RNAseq data are available in the ArrayExpress database (http://www.ebi.ac.uk/arrayexpress) under accession number E‐MTAB‐5660. The scripts used for RNAseq data processing and analysis are available from https://github.com/mprevorovsky/krasny-torpedo. Gene Ontology terms are according to SubtiWiki (Zhu and Stulke, 2018). For Figs 1C, D, 7B, EV3, and EV4 and Appendix Figs S1 and S5, the log2(fold change) gene expression values determined by DESeq2 are shown. Since DESeq2 only reports wt‐normalized relative expression values, normalized gene coverage values determined by deepTools are shown in Figs 1A and B, and 2 to visualize gene expression levels separately for wt and the rnjA mutant. The two methods of calculating gene expression levels were in good agreement (Pearson's R = 0.95). Validation of RNAseq data: First, new RNA purifications were performed under identical conditions as those for RNAseq experiments. Prior to RNA extraction, recovery marker RNA was added [a fragment of 16S rRNA from M. smegmatis (amplified by primers #1281 and #1282, see Table EV7)] and total RNA was then extracted. 2 μg of total RNA was reverse‐transcribed to cDNA with reverse transcriptase (SuperScript™ III Reverse Transcriptase, Invitrogen). This was followed by qPCR, as described for ChIPseq validation. The data were then normalized to the recovery marker and the amount of cells.
Gene classification (classes I–IV)
Based on comparisons of mean normalized RNAP occupancy (ChIPseq) and transcript differential expression (DESeq2 results) between the rnjA mutant (ΔrnjA) and wt, 3,288 B. subtilis genes were assigned into four classes (I–IV). Class I: ≥ 120% RNAP occupancy and significantly upregulated in the rnjA mutant; class II: 0–80% RNAP occupancy and significantly downregulated in the rnjA mutant; class III: 0–120% RNAP occupancy in wt and significantly upregulated in the rnjA mutant; and class IV: ≥ 120% RNAP occupancy, and no significant change or significantly downregulated in the rnjA mutant. Average gene analyses of ChIPseq and RNAseq coverage for each class were performed using deepTools 3.3.0. (Ramirez et al, 2016).
In vitro effect of RNases J1/R/Xrn1 on elongation complexes
Transcription‐competent ECs, containing a fully complementary transcription bubble, were assembled with wild‐type RNA polymerase (RNAP) from B. subtilis (LK1275) as described before (Komissarova et al, 2003). DNA and RNA oligonucleotides were purchased and are listed in Table EV7. The RNA (#‐pRNA) was monophosphorylated at the 5′ end by the manufacturer. A twofold molar excess of RNA was mixed with template DNA (#632) in water and annealed in a cycler (45°C 2 min, 42–27°C: T decreasing by 3°C every 2 min, 25°C 10 min). RNAP (2 pmol per sample) was incubated with a twofold molar excess of the annealed hybrid in 10 μl of reaction buffer (40 mM Tris–HCl pH 8.0, 10 mM MgCl2, 1 mM dithiothreitol) for 15 min at room temperature while shaking. A fourfold molar excess of non‐template DNA (#631), containing biotin at the 5′‐end, was added, and the mixture was incubated at 37°C for 10 min.
Streptavidin‐coated magnetic beads (25 μl per sample; Sigma S‐2415) were washed with 500 μl of binding buffer (20 mM Tris, pH 8.0, 0.15 M NaCl) and resuspended in the same volume of fresh binding buffer. Assembled elongation complexes were then mixed with washed beads, and this was followed by incubation for 30 min at RT with continuous gentle shaking. Unbound complexes were removed by subsequent washing with 500 μl of binding buffer, 500 μl of washing buffer (20 mM Tris–HCl pH 8.0, 0.5 M NaCl, 2 mM MgCl2, 1 mM dithiothreitol), and 500 μl of reaction buffer (Dengl & Cramer, 2009). Beads were resuspended in 10 μl of reaction buffer with 150 mM final concentration of KCl and 0.1 mg/ml BSA. RNA in the elongation complex was labeled at the 3′‐end by RNAP (E. coli (BioLabs) or B. subtilis) activity by adding 0.1 μl of [α32P] UTP (10 mCi/ml) per reaction, followed by incubation at 37 °C for 15 min. Unincorporated nucleotides were washed off by applying 500 μl of reaction buffer, two times 500 μl of washing buffer. Beads with bound ECs were resuspended in RNAse R reaction buffer (20 mM Tris–HCl, pH 7.0; 100 mM KCl, 1.1 mM MgCl2), and 80 pmol of RNase J1 or 1U of RNase Xrn1 (New England Biolabs) or 1U of RNase R (Epicentre Biotechnologies) was added to the reaction. Samples were incubated for 20 min at 37°C. When indicated, samples were denatured for 3 min in 95°C and cooled down prior to the addition of RNases, or the cleavage products were washed off with 500 μl of binding buffer and 2 × 500 μl of washing buffer and resuspended in 10 μl of RNase R buffer after the cleavage. All the reactions were stopped by adding 10 μl of 2× loading buffer [95% formamide and 20 mM EDTA (pH 8.0)].
The RNA ladder was generated as follows: We phosphorylated a 30 nt RNA (the same as #‐pRNA but without the 5′ phosphate) with 32P by T4 polynucleotide kinase following the manufacturer's instructions. This RNA was then subjected to alkaline hydrolysis (15 μl reaction containing: 1 μg of yeast RNA, 0.1–1 μg of radiolabeled RNA, 1× alkaline hydrolysis buffer: 50 mM sodium carbonate pH 9.2; 1 mM EDTA; incubated at 95°C for 4 or 7 min, and then, equal amounts of 2× loading buffer [95% formamide and 20 mM EDTA (pH 8.0)] were added.
Samples were resolved on 20% polyacrylamide sequencing gels, and radioactively labeled RNA was detected by exposing the gels to a storage phosphor screen (Fujifilm) overnight. Scanning of the storage screens was done with a Molecular Imager FX (Bio‐Rad). In quantitative analyses, background of the appropriate lane was subtracted from the specific signal.
RNAP release assay
Transcription elongation complexes were assembled, and the reaction conditions were as described in the previous in vitro experiment. TCs were bound to magnetic streptavidin‐coated beads, divided into three tubes, and treated with either buffer (mock treatment) or 80 pmol RNase J1 or 1 U Xrn1 (New England Biolabs) for 20 min at 37°C. The bound (in complex with DNA) and released (free in buffer) RNAPs were separated by using a DYNAL Invitrogen bead separation device. Subsequently, the fractions were analyzed with SDS–PAGE, and RNAPs were detected by Western blotting using mouse monoclonal antibodies against the β subunit of RNA polymerase (clone name 8RB13) and secondary antibodies conjugated with a fluorophore dye (WesternBrightTM MCF‐IR, Advansta, 800 nm anti‐mouse antibody) and scanned with an Odyssey reader (LI‐COR Biosciences). The analysis was done with the Quantity One software (Bio‐Rad). The experiment was conducted in two biological replicates.
In silico models
Figures were created using the ICM Molsoft software package (ICM Molsoft (http://www.molsoft.com/icm_browser.html). The PDB codes are in the Figure legends.
UV sensitivity phenotype
Exponential cells of wt (LK1371), ΔrnjA (LK1381), Δrho (LK2058), ΔhelD (LK2329), ΔrnjAΔrho (LK2082), and ΔrnjAΔhelD (LK2336) strains (OD600 ~ 0.5) were serially diluted (10‐fold dilutions) and plated (100 μl) on duplicate LB agar plates (two sets of plates) without antibiotics. One set of plates was irradiated by UVT‐20M (Herolab) at 312 nm for 15 s (0.12 W/cm2); the other set was not irradiated. Plates were incubated at 37°C overnight. Plates were then analyzed: Colony‐forming units (CFU) were counted. UV sensitivity of the mutant strains (KO) was then calculated as the ratio between irradiated vs. non‐irradiated cells and normalized to this ratio from the wt strain. As a consequence, the wt ratio is 1. The experiment was conducted 4× (biological replicates). All analyses were done on the log scale. The P‐values were computed using two‐tailed unpaired t‐test.
Author contributions
CC and LK conceptualized the study and designed the experiments. MŠ, JW, MP, PS, OB, OK, and HŠ performed the experiments. MŠ, MP, and LK analyzed the data. IB performed in silico modeling. LK and CC wrote the manuscript; all the other authors contributed to the final revised version.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Table EV6
Table EV7
Table EV8
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We would like to acknowledge the Czech Science Foundation—originally by grant P305/12/G034 and subsequently by 19‐12956S (to LK); Czech Research Infrastructure for Systems Biology C4SYS (project LM2015055); Light Microscopy Core Facility, IMG ASCR, Prague, Czech Republic (MEYS, LM2015062), OPPK (CZ.2.16/3.1.00/21547), and LO1419; and electron microscopy facility (LO1509, Ministry of Education, Youth, and Sports of the Czech Republic). MP was supported by Charles University grants PRIMUS/MED/26 and UNCE 204013. CC was supported by the Agence Nationale de la Recherche (ARNr‐QC) and the Labex (Dynamo) program. The authors thank Dr. M. Modrák for the discussion on statistical analyses.
The EMBO Journal (2020) 39: e102500
See also: https://doi.org/10.15252/embj.2019104112 (February 2020)
Data availability
The datasets produced in this study are available in the following databases:
RNAseq data: E‐MTAB‐5660 (http://www.ebi.ac.uk/arrayexpressexperiments/E-MTAB-5660)
ChIPseq data: E‐MTAB‐5659 (http://www.ebi.ac.uk/arrayexpressexperiments/E-MTAB-5659)
References
- Arraiano CM, Andrade JM, Domingues S, Guinote IB, Malecki M, Matos RG, Moreira RN, Pobre V, Reis FP, Saramago M et al (2010) The critical role of RNA processing and degradation in the control of gene expression. FEMS Microbiol Rev 34: 883–923 [DOI] [PubMed] [Google Scholar]
- Baejen C, Andreani J, Torkler P, Battaglia S, Schwalb B, Lidschreiber M, Maier KC, Boltendahl A, Rus P, Esslinger S et al (2017) Genome‐wide analysis of RNA polymerase II termination at protein‐coding genes. Mol Cell 66: 38–49 e36 [DOI] [PubMed] [Google Scholar]
- Bar‐Nahum G, Epshtein V, Ruckenstein AE, Rafikov R, Mustaev A, Nudler E (2005) A ratchet mechanism of transcription elongation and its control. Cell 120: 183–193 [DOI] [PubMed] [Google Scholar]
- Bidnenko V, Nicolas P, Grylak‐Mielnicka A, Delumeau O, Auger S, Aucouturier A, Guerin C, Repoila F, Bardowski J, Aymerich S et al (2017) Termination factor Rho: from the control of pervasive transcription to cell fate determination in Bacillus subtilis . PLoS Genet 13: e1006909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein‐dye binding. Anal Biochem 72: 248–254 [DOI] [PubMed] [Google Scholar]
- Buskirk AR, Green R (2017) Ribosome pausing, arrest and rescue in bacteria and eukaryotes. Philos Trans R Soc Lond B Biol Sci 372: 20160183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascante‐Estepa N, Gunka K, Stulke J (2016) Localization of components of the RNA‐degrading machine in Bacillus subtilis . Front Microbiol 7: 1492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang BY, Doi RH (1990) Overproduction, purification, and characterization of Bacillus subtilis RNA polymerase sigma A factor. J Bacteriol 172: 3257–3263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condon C, Pellegrini O, Mathy N, Benard L, Redko Y, Oussenko IA, Deikus G, Bechhofer DH (2008) Assay of Bacillus subtilis ribonucleases in vitro . Methods Enzymol 447: 277–308 [DOI] [PubMed] [Google Scholar]
- Condon C (2010) What is the role of RNase J in mRNA turnover? RNA Biol 7: 316–321 [DOI] [PubMed] [Google Scholar]
- Condon C, Piton J, Braun F (2018) Distribution of the ribosome associated endonuclease Rae1 and the potential role of conserved amino acids in codon recognition. RNA Biol 15: 683–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo G, Pino S, Timperio AM, Sponer JE, Sponer J, Novakova O, Sedo O, Zdrahal Z, Di Mauro E (2016) Non‐enzymatic oligomerization of 3′, 5′ cyclic AMP. PLoS One 11: e0165723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozy LM, Kearns DB (2010) Gene position in a long operon governs motility development in Bacillus subtilis . Mol Microbiol 76: 273–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dengl S, Cramer P (2009) Torpedo nuclease Rat1 is insufficient to terminate RNA polymerase II in vitro . J Biol Chem 284: 21270–21279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty GP, Fogg MJ, Wilkinson AJ, Lewis PJ (2010) Small subunits of RNA polymerase: localization, levels and implications for core enzyme composition. Microbiology 156: 3532–3543 [DOI] [PubMed] [Google Scholar]
- Dorleans A, Li de la Sierra‐Gallay I, Piton J, Zig L, Gilet L, Putzer H, Condon C (2011) Molecular basis for the recognition and cleavage of RNA by the bifunctional 5′‐3′ exo/endoribonuclease RNase J. Structure 19: 1252–1261 [DOI] [PubMed] [Google Scholar]
- Durand S, Gilet L, Bessieres P, Nicolas P, Condon C (2012) Three essential ribonucleases‐RNase Y, J1, and III‐control the abundance of a majority of Bacillus subtilis mRNAs. PLoS Genet 8: e1002520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eiamphungporn W, Helmann JD (2008) The Bacillus subtilis sigma(M) regulon and its contribution to cell envelope stress responses. Mol Microbiol 67: 830–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epshtein V, Nudler E (2003) Cooperation between RNA polymerase molecules in transcription elongation. Science 300: 801–805 [DOI] [PubMed] [Google Scholar]
- Epshtein V, Dutta D, Wade J, Nudler E (2010) An allosteric mechanism of Rho‐dependent transcription termination. Nature 463: 245–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epshtein V, Kamarthapu V, McGary K, Svetlov V, Ueberheide B, Proshkin S, Mironov A, Nudler E (2014) UvrD facilitates DNA repair by pulling RNA polymerase backwards. Nature 505: 372–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Even S, Pellegrini O, Zig L, Labas V, Vinh J, Brechemmier‐Baey D, Putzer H (2005) Ribonucleases J1 and J2: two novel endoribonucleases in B. subtilis with functional homology to E. coli RNase E. Nucleic Acids Res 33: 2141–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Leroux‐Coyau M, Savery NJ, Strick TR (2016) Reconstruction of bacterial transcription‐coupled repair at single‐molecule resolution. Nature 536: 234–237 [DOI] [PubMed] [Google Scholar]
- Figaro S, Durand S, Gilet L, Cayet N, Sachse M, Condon C (2013) Bacillus subtilis mutants with knockouts of the genes encoding ribonucleases RNase Y and RNase J1 are viable, with major defects in cell morphology, sporulation, and competence. J Bacteriol 195: 2340–2348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frindert J, Zhang Y, Nubel G, Kahloon M, Kolmar L, Hotz‐Wagenblatt A, Burhenne J, Haefeli WE, Jaschke A (2018) Identification, biosynthesis, and decapping of NAD‐capped RNAs in B. subtilis . Cell Rep 24: 1890–1901 e1898 [DOI] [PubMed] [Google Scholar]
- Fukushima T, Afkham A, Kurosawa S, Tanabe T, Yamamoto H, Sekiguchi J (2006) A new D, L‐endopeptidase gene product, YojL (renamed CwlS), plays a role in cell separation with LytE and LytF in Bacillus subtilis . J Bacteriol 188: 5541–5550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilet L, DiChiara JM, Figaro S, Bechhofer DH, Condon C (2015) Small stable RNA maturation and turnover in Bacillus subtilis . Mol Microbiol 95: 270–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimpel M, Brantl S (2017) Dual‐function small regulatory RNAs in bacteria. Mol Microbiol 103: 387–397 [DOI] [PubMed] [Google Scholar]
- Goodsell DS (2001) The molecular perspective: ultraviolet light and pyrimidine dimers. Oncologist 6: 298–299 [DOI] [PubMed] [Google Scholar]
- Guo X, Myasnikov AG, Chen J, Crucifix C, Papai G, Takacs M, Schultz P, Weixlbaumer A (2018) Structural basis for NusA stabilized transcriptional pausing. Mol Cell 69: 816–827 e814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamperl S, Cimprich KA (2016) Conflict resolution in the genome: how transcription and replication make it work. Cell 167: 1455–1467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B, Zalkin H (1992) Repression of Escherichia coli purB is by a transcriptional roadblock mechanism. J Bacteriol 174: 7121–7127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmann JD, Masiarz FR, Chamberlin MJ (1988) Isolation and characterization of the Bacillus subtilis sigma 28 factor. J Bacteriol 170: 1560–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh PK, Richards J, Liu Q, Belasco JG (2013) Specificity of RppH‐dependent RNA degradation in Bacillus subtilis . Proc Natl Acad Sci USA 110: 8864–8869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt A, Rawlins JP, Thomaides HB, Errington J (2006) Functional analysis of 11 putative essential genes in Bacillus subtilis . Microbiology 152: 2895–2907 [DOI] [PubMed] [Google Scholar]
- Janniere L, Canceill D, Suski C, Kanga S, Dalmais B, Lestini R, Monnier AF, Chapuis J, Bolotin A, Titok M et al (2007) Genetic evidence for a link between glycolysis and DNA replication. PLoS One 2: e447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jervis AJ, Thackray PD, Houston CW, Horsburgh MJ, Moir A (2007) SigM‐responsive genes of Bacillus subtilis and their promoters. J Bacteriol 189: 4534–4538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez RM, Polanco JA, Luptak A (2015) Chemistry and biology of self‐cleaving ribozymes. Trends Biochem Sci 40: 648–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juang YL, Helmann JD (1994) A promoter melting region in the primary sigma factor of Bacillus subtilis. Identification of functionally important aromatic amino acids. J Mol Biol 235: 1470–1488 [DOI] [PubMed] [Google Scholar]
- Kang JY, Mishanina TV, Landick R, Darst SA (2019) Mechanisms of transcriptional pausing in bacteria. J Mol Biol 431: 4007–4029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai Y, Asai K, Errington J (2009) Partial functional redundancy of MreB isoforms, MreB, Mbl and MreBH, in cell morphogenesis of Bacillus subtilis . Mol Microbiol 73: 719–731 [DOI] [PubMed] [Google Scholar]
- Keller AN, Yang X, Wiedermannova J, Delumeau O, Krasny L, Lewis PJ (2014) epsilon, a new subunit of RNA polymerase found in gram‐positive bacteria. J Bacteriol 196: 3622–3632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Krogan NJ, Vasiljeva L, Rando OJ, Nedea E, Greenblatt JF, Buratowski S (2004) The yeast Rat1 exonuclease promotes transcription termination by RNA polymerase II. Nature 432: 517–522 [DOI] [PubMed] [Google Scholar]
- Kim D, Langmead B, Salzberg SL (2015) HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12: 357–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kireeva ML, Kashlev M (2009) Mechanism of sequence‐specific pausing of bacterial RNA polymerase. Proc Natl Acad Sci USA 106: 8900–8905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler R, Mooney RA, Mills DJ, Landick R, Cramer P (2017) Architecture of a transcribing‐translating expressome. Science 356: 194–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komissarova N, Kireeva ML, Becker J, Sidorenkov I, Kashlev M (2003) Engineering of elongation complexes of bacterial and yeast RNA polymerases. Methods Enzymol 371: 233–251 [DOI] [PubMed] [Google Scholar]
- Krajewski WW, Fu X, Wilkinson M, Cronin NB, Dillingham MS, Wigley DB (2014) Structural basis for translocation by AddAB helicase‐nuclease and its arrest at chi sites. Nature 508: 416–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasny L, Vacik T, Fucik V, Jonak J (2000) Cloning and characterization of the str operon and elongation factor Tu expression in Bacillus stearothermophilus. J Bacteriol 182: 6114–6122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusuya Y, Kurokawa K, Ishikawa S, Ogasawara N, Oshima T (2011) Transcription factor GreA contributes to resolving promoter‐proximal pausing of RNA polymerase in Bacillus subtilis cells. J Bacteriol 193: 3090–3099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson MH, Greenleaf WJ, Landick R, Block SM (2008) Applied force reveals mechanistic and energetic details of transcription termination. Cell 132: 971–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le TT, Yang Y, Tan C, Suhanovsky MM, Fulbright RM Jr, Inman JT, Li M, Lee J, Perelman S, Roberts JW et al (2018) Mfd dynamically regulates transcription via a release and catch‐up mechanism. Cell 172: 344–357 e315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBlanc SJ, Gauer JW, Hao P, Case BC, Hingorani MM, Weninger KR, Erie DA (2018) Coordinated protein and DNA conformational changes govern mismatch repair initiation by MutS. Nucleic Acids Res 46: 10782–10795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnik‐Habrink M, Lewis RJ, Mader U, Stulke J (2012) RNA degradation in Bacillus subtilis: an interplay of essential endo‐ and exoribonucleases. Mol Microbiol 84: 1005–1017 [DOI] [PubMed] [Google Scholar]
- Lei Y, Oshima T, Ogasawara N, Ishikawa S (2013) Functional analysis of the protein Veg, which stimulates biofilm formation in Bacillus subtilis . J Bacteriol 195: 1697–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenhart JS, Schroeder JW, Walsh BW, Simmons LA (2012) DNA repair and genome maintenance in Bacillus subtilis . Microbiol Mol Biol Rev 76: 530–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R Genome Project Data Processing (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Hanne J, Britton BM, Bennett J, Kim D, Lee JB, Fishel R (2016) Cascading MutS and MutL sliding clamps control DNA diffusion to activate mismatch repair. Nature 539: 583–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Bentley D (2004) A ribonucleolytic rat torpedoes RNA polymerase II. Cell 119: 911–914 [DOI] [PubMed] [Google Scholar]
- Ma C, Mobli M, Yang X, Keller AN, King GF, Lewis PJ (2015) RNA polymerase‐induced remodelling of NusA produces a pause enhancement complex. Nucleic Acids Res 43: 2829–2840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathy N, Benard L, Pellegrini O, Daou R, Wen T, Condon C (2007) 5′‐to‐3′ exoribonuclease activity in bacteria: role of RNase J1 in rRNA maturation and 5′ stability of mRNA. Cell 129: 681–692 [DOI] [PubMed] [Google Scholar]
- Mathy N, Hebert A, Mervelet P, Benard L, Dorleans A, Li de la Sierra‐Gallay I, Noirot P, Putzer H, Condon C (2010) Bacillus subtilis ribonucleases J1 and J2 form a complex with altered enzyme behaviour. Mol Microbiol 75: 489–498 [DOI] [PubMed] [Google Scholar]
- Mirel DB, Lustre VM, Chamberlin MJ (1992) An operon of Bacillus subtilis motility genes transcribed by the sigma D form of RNA polymerase. J Bacteriol 174: 4197–4204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molle V, Fujita M, Jensen ST, Eichenberger P, Gonzalez‐Pastor JE, Liu JS, Losick R (2003) The Spo0A regulon of Bacillus subtilis . Mol Microbiol 50: 1683–1701 [DOI] [PubMed] [Google Scholar]
- Mori F, Tanji K, Miki Y, Toyoshima Y, Sasaki H, Yoshida M, Kakita A, Takahashi H, Wakabayashi K (2018) Immunohistochemical localization of exoribonucleases (DIS3L2 and XRN1) in intranuclear inclusion body disease. Neurosci Lett 662: 389–394 [DOI] [PubMed] [Google Scholar]
- Nadkarni A, Burns JA, Gandolfi A, Chowdhury MA, Cartularo L, Berens C, Geacintov NE, Scicchitano DA (2016) Nucleotide excision repair and transcription‐coupled DNA repair Abrogate the impact of DNA damage on transcription. J Biol Chem 291: 848–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas P, Mader U, Dervyn E, Rochat T, Leduc A, Pigeonneau N, Bidnenko E, Marchadier E, Hoebeke M, Aymerich S et al (2012) Condition‐dependent transcriptome reveals high‐level regulatory architecture in Bacillus subtilis . Science 335: 1103–1106 [DOI] [PubMed] [Google Scholar]
- Nudler E (2012) RNA polymerase backtracking in gene regulation and genome instability. Cell 149: 1438–1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panova N, Zborníková E, Šimák O, Pohl R, Kolář M, Bogdanová K, Večeřová R, Seydlová G, Fišer R, Hadravová R et al (2015) Insights into the mechanism of action of bactericidal lipophosphonoxins. PLoS One 10: e0145918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Kang M, Kim M (2015) Unraveling the mechanistic features of RNA polymerase II termination by the 5′‐3′ exoribonuclease Rat1. Nucleic Acids Res 43: 2625–2637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson EL, Moore CL (2013) Dismantling promoter‐driven RNA polymerase II transcription complexes in vitro by the termination factor Rat1. J Biol Chem 288: 19750–19759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM, Mooney RA, Kuan PF, Rowland JL, Keles S, Landick R (2009) Rho directs widespread termination of intragenic and stable RNA transcription. Proc Natl Acad Sci USA 106: 15406–15411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phung DK, Rinaldi D, Langendijk‐Genevaux PS, Quentin Y, Carpousis AJ, Clouet‐d'Orval B (2013) Archaeal beta‐CASP ribonucleases of the aCPSF1 family are orthologs of the eukaryal CPSF‐73 factor. Nucleic Acids Res 41: 1091–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y, Hulett FM (1998) PhoP‐P and RNA polymerase sigmaA holoenzyme are sufficient for transcription of Pho regulon promoters in Bacillus subtilis: PhoP‐P activator sites within the coding region stimulate transcription in vitro . Mol Microbiol 28: 1187–1197 [DOI] [PubMed] [Google Scholar]
- Radhakrishnan A, Green R (2016) Connections underlying translation and mRNA stability. J Mol Biol 428: 3558–3564 [DOI] [PubMed] [Google Scholar]
- Ramaniuk O, Cerny M, Krasny L, Vohradsky J (2017) Kinetic modelling and meta‐analysis of the B. subtilis SigA regulatory network during spore germination and outgrowth. Biochim Biophys Acta Gene Regul Mech 1860: 894–904 [DOI] [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, Manke T (2016) deepTools2: a next generation web server for deep‐sequencing data analysis. Nucleic Acids Res 44: W160–W165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall JR, Hirst WG, Simmons LA (2017) Substrate specificity for bacterial RNase HII and HIII is influenced by metal availability. J Bacteriol 200: e00401‐17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruff EF, Record MT Jr, Artsimovitch I (2015) Initial events in bacterial transcription initiation. Biomolecules 5: 1035–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders K, Lin CL, Smith AJ, Cronin N, Fisher G, Eftychidis V, McGlynn P, Savery NJ, Wigley DB, Dillingham MS (2017) The structure and function of an RNA polymerase interaction domain in the PcrA/UvrD helicase. Nucleic Acids Res 45: 3875–3887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder JW, Randall JR, Hirst WG, O'Donnell ME, Simmons LA (2017) Mutagenic cost of ribonucleotides in bacterial DNA. Proc Natl Acad Sci USA 114: 11733–11738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serizawa M, Yamamoto H, Yamaguchi H, Fujita Y, Kobayashi K, Ogasawara N, Sekiguchi J (2004) Systematic analysis of SigD‐regulated genes in Bacillus subtilis by DNA microarray and Northern blotting analyses. Gene 329: 125–136 [DOI] [PubMed] [Google Scholar]
- Shepanek NA, Smith RF, Tyer ZE, Royall GD, Allen KS (1989) Behavioral and neuroanatomical sequelae of prenatal naloxone administration in the rat. Neurotoxicol Teratol 11: 441–446 [DOI] [PubMed] [Google Scholar]
- Sikova M, Janouskova M, Ramaniuk O, Palenikova P, Pospisil J, Bartl P, Suder A, Pajer P, Kubickova P, Pavlis O et al (2018) Ms1 RNA increases the amount of RNA polymerase in Mycobacterium smegmatis . Mol Microbiol 111: 354–372 [DOI] [PubMed] [Google Scholar]
- Sun M, Schwalb B, Pirkl N, Maier KC, Schenk A, Failmezger H, Tresch A, Cramer P (2013) Global analysis of eukaryotic mRNA degradation reveals Xrn1‐dependent buffering of transcript levels. Mol Cell 52: 52–62 [DOI] [PubMed] [Google Scholar]
- Tagami S, Sekine S, Minakhin L, Esyunina D, Akasaka R, Shirouzu M, Kulbachinskiy A, Severinov K, Yokoyama S (2014) Structural basis for promoter specificity switching of RNA polymerase by a phage factor. Genes Dev 28: 521–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdottir H, Robinson JT, Mesirov JP (2013) Integrative Genomics Viewer (IGV): high‐performance genomics data visualization and exploration. Brief Bioinform 14: 178–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornaletti S, Hanawalt PC (1999) Effect of DNA lesions on transcription elongation. Biochimie 81: 139–146 [DOI] [PubMed] [Google Scholar]
- Toulme F, Mosrin‐Huaman C, Sparkowski J, Das A, Leng M, Rahmouni AR (2000) GreA and GreB proteins revive backtracked RNA polymerase in vivo by promoting transcript trimming. EMBO J 19: 6853–6859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner MS, Helmann JD (2000) Mutations in multidrug efflux homologs, sugar isomerases, and antimicrobial biosynthesis genes differentially elevate activity of the sigma(X) and sigma(W) factors in Bacillus subtilis . J Bacteriol 182: 5202–5210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twist KA, Campbell EA, Deighan P, Nechaev S, Jain V, Geiduschek EP, Hochschild A, Darst SA (2011) Crystal structure of the bacteriophage T4 late‐transcription coactivator gp33 with the beta‐subunit flap domain of Escherichia coli RNA polymerase. Proc Natl Acad Sci USA 108: 19961–19966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss A, Shaw LN (2015) Small things considered: the small accessory subunits of RNA polymerase in Gram‐positive bacteria. FEMS Microbiol Rev 39: 541–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedermannova J, Sudzinova P, Koval T, Rabatinova A, Sanderova H, Ramaniuk O, Rittich S, Dohnalek J, Fu Z, Halada P et al (2014) Characterization of HelD, an interacting partner of RNA polymerase from Bacillus subtilis . Nucleic Acids Res 42: 5151–5163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakhnin AV, Murakami KS, Babitzke P (2016) NusG is a sequence‐specific RNA polymerase pause factor that binds to the non‐template DNA within the paused transcription bubble. J Biol Chem 291: 5299–5308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XC, Sullivan KD, Marzluff WF, Dominski Z (2009) Studies of the 5′ exonuclease and endonuclease activities of CPSF‐73 in histone pre‐mRNA processing. Mol Cell Biol 29: 31–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Mooney RA, Grass JA, Sivaramakrishnan P, Herman C, Landick R, Wang JD (2014) DksA guards elongating RNA polymerase against ribosome‐stalling‐induced arrest. Mol Cell 53: 766–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu B, Stulke J (2018) SubtiWiki in 2018: from genes and proteins to functional network annotation of the model organism Bacillus subtilis. Nucleic Acids Res 46: D743–D748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweers JC, Nicolas P, Wiegert T, van Dijl JM, Denham EL (2012) Definition of the sigma(W) regulon of Bacillus subtilis in the absence of stress. PLoS One 7: e48471 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Table EV6
Table EV7
Table EV8
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Data Availability Statement
The datasets produced in this study are available in the following databases:
RNAseq data: E‐MTAB‐5660 (http://www.ebi.ac.uk/arrayexpressexperiments/E-MTAB-5660)
ChIPseq data: E‐MTAB‐5659 (http://www.ebi.ac.uk/arrayexpressexperiments/E-MTAB-5659)