

Abstract
Extracellular vesicles are emerging key actors in adipocyte communication. Notably, small extracellular vesicles shed by adipocytes stimulate fatty acid oxidation and migration in melanoma cells and these effects are enhanced in obesity. However, the vesicular actors and cellular processes involved remain largely unknown. Here, we elucidate the mechanisms linking adipocyte extracellular vesicles to metabolic remodeling and cell migration. We show that adipocyte vesicles stimulate melanoma fatty acid oxidation by providing both enzymes and substrates. In obesity, the heightened effect of extracellular vesicles depends on increased transport of fatty acids, not fatty acid oxidation‐related enzymes. These fatty acids, stored within lipid droplets in cancer cells, drive fatty acid oxidation upon being released by lipophagy. This increase in mitochondrial activity redistributes mitochondria to membrane protrusions of migrating cells, which is necessary to increase cell migration in the presence of adipocyte vesicles. Our results provide key insights into the role of extracellular vesicles in the metabolic cooperation that takes place between adipocytes and tumors with particular relevance to obesity.
Keywords: exosome, fatty acid oxidation, lipophagy, melanoma, obesity
Subject Categories: Cancer, Membrane & Intracellular Transport, Metabolism
Adipocyte‐derived extracellular vesicles laden with enzymatic machinery and lipid substrates promote fatty acid oxidation and aggressiveness of melanoma cells, a process that is amplified in obesity.
Introduction
As worldwide obesity rates continue to climb, excess body fat has emerged as a major public health issue, given its associated complications such as cardiovascular diseases, diabetes, and cancer (De Pergola & Silvestris, 2013; Park et al, 2014). Obesity is a recognized factor to increase cancer incidence and progression (Gallagher & LeRoith, 2015). This association occurs in melanoma, a skin cancer that develops from transformed melanocytes, pigment‐producing cells that reside at the junction between the epidermis and the dermis. Given their high propensity to invade adjacent tissues, including subcutaneous adipose tissue (AT), and to metastasize to distant organs, melanoma is the most aggressive cutaneous cancer. Studies in murine melanoma models and epidemiological data point to a positive correlation between obesity and melanoma incidence and progression (for a recent review, see Clement et al, 2017). Indeed, although controverted at first (Sergentanis et al, 2013), now, most epidemiological studies indicate that obesity increases the risk of developing melanoma, at least in men (Renehan et al, 2008; Dobbins et al, 2013). Although this association is not always observed in the female population (Olsen et al, 2008; Dobbins et al, 2013), this may be due to confounding factors. Indeed, adjustment for sunlight exposure (Shors et al, 2001; Gallus et al, 2006) or use of hormone replacement therapy and menopausal status (Reeves et al, 2007) reveal positive associations between melanoma risk and obesity in women. On the other hand, the association between obesity and melanoma aggressiveness has been demonstrated in epidemiological studies in both men and women (de Giorgi et al, 2013; Skowron et al, 2015; Stenehjem et al, 2018) and in murine models (Pandey et al, 2012; Jung et al, 2015; Malvi et al, 2016). Seemingly contrarily, recent data from McQuade and collaborators show increased BMI (body mass index) can increase progression‐free and overall survival in male patients with metastatic melanoma, but this association only concerns patients treated with immunotherapy or targeted therapy but not chemotherapy, showing that obesity favors response to certain treatments but not the aggressiveness of the disease itself (McQuade et al, 2018).
Adipocytes, the main component of AT, reside in many cancer microenvironments and contribute to tumor progression through soluble factors, such as leptin or interleukin 6, and extracellular matrix remodeling (Andarawewa et al, 2005; Dirat et al, 2011; Duong et al, 2017). Recent findings highlight a metabolic cooperation between adipocytes and tumor cells, which is proving to be a key process in their tumor‐promoting effects (Nieman et al, 2011; Balaban et al, 2017; Wang et al, 2017; Zhang et al, 2018). Cellular components in the tumor microenvironment are major regulators of tumor metabolism, driving cancer cells to favor certain metabolic pathways (Gouirand et al, 2018). In particular, adipocytes provide a local supply of fatty acids (FA) that can serve as an energy source for tumors. Indeed, tumor secretions trigger lipolysis in neighboring adipocytes (Dirat et al, 2011), and the released FA fuel fatty acid oxidation (FAO) in tumors, which increases tumor aggressiveness (Nieman et al, 2011; Balaban et al, 2017; Wang et al, 2017; Zhang et al, 2018). FAO has recently emerged as a pro‐tumoral pathway involved in cancer cell proliferation, stemness, and invasion (Carracedo et al, 2013; Kuo & Ann, 2018), but many of the molecular mechanisms behind these effects remain elusive.
Even though adipocytes have a heightened effect on tumor progression in obesity (Nieman et al, 2013; Duong et al, 2017), we still do not understand whether their metabolic cooperation with tumor cells is involved in this process. Until now, studies showed that metabolic exchanges between adipocytes and tumor cells require a close proximity between the two cell types in order for tumors to provoke adipocyte lipolysis. For most types of cancer, this process can only occur at the later stages of cancer progression, when tumors become invasive and penetrate local AT or metastasize to adipocyte‐rich environments (Nieman et al, 2011; Wang et al, 2017; Zhang et al, 2018; Laurent et al, 2019). However, whether adipocytes could also influence tumors at distance, for example, during the early stages of disease progression before cancer cells infiltrate surrounding adipose tissue, remains unknown.
We predict that extracellular vesicles (EV) are key contributors in such a process. They allow the transfer of biomolecules, including nucleic acids, proteins, and lipids, to distant cells, since they diffuse through tissues and circulate in body fluids (Shah et al, 2018). Previously, we demonstrated that adipocyte EV are key participants in melanoma progression (Lazar et al, 2016). Indeed, adipocytes secrete great quantities of EV that specifically carry proteins involved in lipid metabolism, including FAO enzymes. Melanoma cells internalize these EV, and this increases FAO, which promotes tumor migration and invasion but not proliferation. In obesity, adipocytes secrete more EV and, when used at equal concentrations, these EV elicit a heightened effect on melanoma migration, indicating qualitative changes in EV cargo. We predict these changes likely involve FAO actors, which remain to be identified. Although the increase in tumor aggressiveness in response to adipocyte EV is dependent on FAO, the cellular processes that link this metabolic remodeling to cell migration remain unknown.
Here, we reveal a mechanism by which naïve adipocytes (unaltered by tumor cells) influence tumor metabolism. EV secreted by these adipocytes transfer both protein machinery and FA substrates required for FAO. We further show that the heightened effect of adipocyte EV in obesity depends on increased FA levels, whereas FAO‐related protein levels remain unchanged. Cytoplasmic lipid droplets store FA transferred from adipocytes to melanoma cells by EV, which are then released by lipophagy to fuel FAO. Finally, we propose a mechanism that links adipocyte‐induced FAO to tumor cell migration, mitochondrial dynamics.
Results
Adipocyte EV transfer proteins involved in FA metabolism to melanoma cells
In a previous study, we have shown that both murine and human adipocyte EV increase human melanoma cell migration, invasion and metastasis (Lazar et al, 2016), and clonogenicity (Appendix Fig S1). Although we know that adipocyte EV are enriched in proteins involved in FA metabolism and that they increase melanoma migration through a process dependent on FAO (Lazar et al, 2016), the link remains correlative. So, we developed an experimental workflow using SILAC (Stable Isotope Labeling of Amino Acids in Cell Culture) to perform an unsupervised analysis of adipocyte proteins transferred to recipient tumor cells by EV (Fig 1A). We identified 2107‐labeled proteins in adipocyte EV (Table EV1), which equates to approximately 85% of total EV proteins (Fig 1B). In melanoma cells exposed to these EV, we detected 587 proteins containing heavy amino acids, which indicates they were transferred from adipocytes (Table EV1). In cells not exposed to EV, we only aberrantly detected five false‐positive “labeled” proteins, which underlines the robustness of this technique. Consequently, we eliminated these proteins from the list of labeled proteins. Our results indicated that approximately 30% of adipocyte EV proteins are effectively transferred to melanoma cells. Many abundant proteins within EV, such as FASN, were not transferred to melanoma cells, indicating a selective transfer and/or uptake of material. Among the proteins effectively transferred from adipocytes to melanoma cells by EV, we identified a large number of proteins involved in vesicle‐mediated transport and in energy metabolism, including mitochondrial FAO enzymes (Fig 1C and Table EV1). We also identified proteins involved in FA transport and storage, as well as oxidative phosphorylation (Table EV1). Thus, using an adapted SILAC technique, we conclude that FAO‐related proteins are not only present in adipocyte EV, but also efficiently transferred to melanoma cells.
Figure 1. Adipocyte EV transfer proteins involved in FA metabolism to melanoma cells.
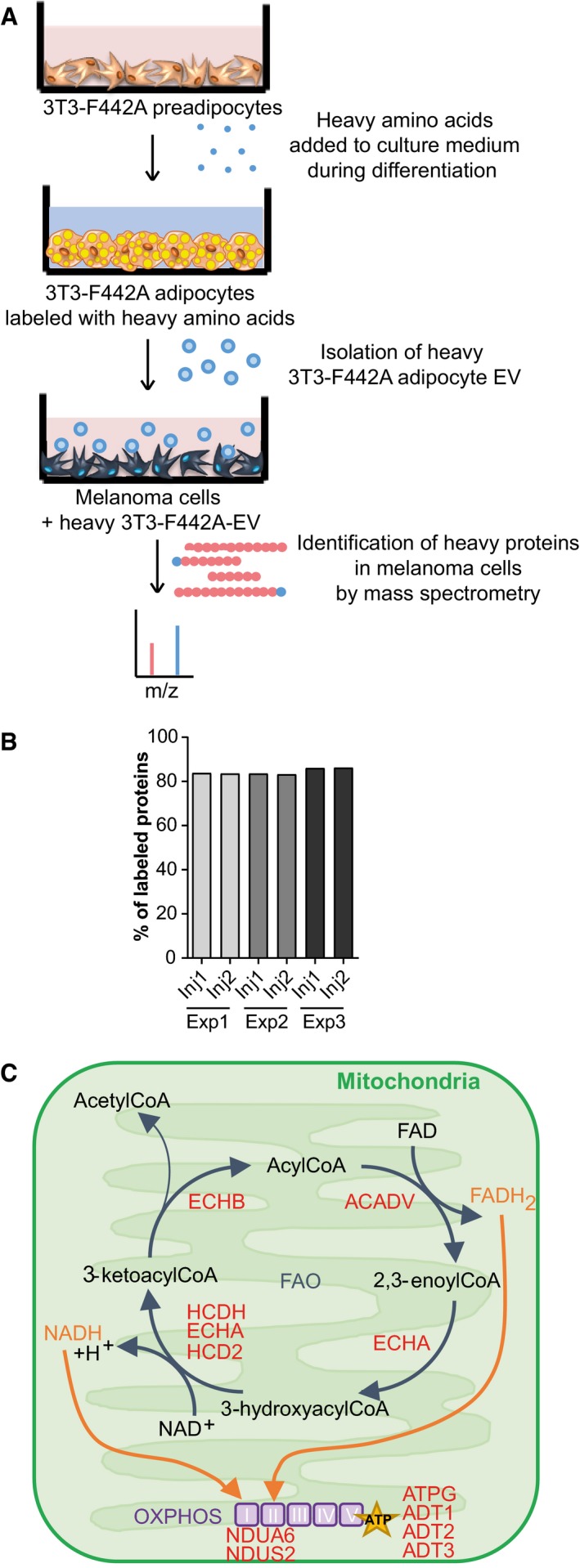
- Workflow of the SILAC approach. 3T3‐F442A cells were seeded and differentiated in the presence of heavy amino acids. After 14 days of differentiation, the EV secreted by the mature labeled adipocytes were isolated and analyzed by mass spectrometry to evaluate the presence of heavy amino acid‐containing proteins. These EV were also added to SKMEL28 cells for 12 h, and then, LC‐MS/MS analysis was performed to identify heavy amino acid‐containing proteins that had been transferred from adipocytes to melanoma cells via EV.
- Three independent samples (Exp 1–3) of EV secreted by labeled 3T3‐F442A cells were analyzed by mass spectrometry (in duplicate injections, Inj1/2). The percentage of proteins bearing at least one peptide containing a heavy amino acid is indicated.
- Proteins involved in FAO and oxidative phosphorylation (OXPHOS) that are transferred from adipocytes to melanoma cells via EV are shown in red.
The heightened effect of adipocyte EV in obesity does not depend on increased FAO protein transfer
FAO levels are greater in melanoma cells treated with adipocyte EV from obese mice fed a high‐fat diet (HFD), which will be termed hereafter HFD‐EV, when compared to those treated with EV secreted by adipocytes from lean mice fed a normal diet (ND‐EV) (Fig 2A). To decipher the underlying mechanisms responsible for increased FAO induced by HFD‐EV, we performed a comparative quantitative proteomic analysis of adipocyte EV from lean and obese mice. We identified 1,557 proteins, of which 87 and 100 were, respectively, more or less abundant in obese samples (Table EV2). Twenty proteins involved in mitochondrial FAO were present in equal abundance in both samples in this analysis (Fig 2B, Table EV2, and Appendix Fig S2A), which we confirmed by Western blot for the two key FAO enzymes, ECHA (trifunctional enzyme subunit alpha, gene name HADHA) and HCDH (Hydroxyacyl‐coenzyme A dehydrogenase, gene name HADH), in murine and human samples (Fig 2C). Moreover, we found that HFD‐EV are not preferentially taken up by melanoma cells, when compared to ND‐EV (Appendix Fig S2B and C). This suggests that increased transfer of FAO enzymes is not responsible for the heightened effect of adipocyte EV in obesity. In accord, although FAO enzyme levels increased in the presence of primary adipocyte EV (Fig 2D), in align with a transfer of these proteins by EV, as demonstrated in the SILAC experiment (Table EV1 and Fig 1C), we found no further increase in cells treated with HFD‐EV (Fig 2D). Moreover, in melanoma cells treated with primary adipocyte EV from lean and obese mice, HADHA and HADH (respectively, coding for ECHA and HCDH) mRNA levels are unchanged, as well as mRNA levels of CPT1A, a rate‐limiting enzyme of FAO that controls FA entry in mitochondria (Fig 2E). Finally, cycloheximide, an inhibitor of protein synthesis, has no effect on the increase of FAO induced by adipocyte EV, demonstrating that this process does not depend on endogenous protein synthesis (Fig 2F).
Figure 2. Adipocyte EV‐induced FAO is increased by obesity, but this process is not dependent on increased protein transfer.
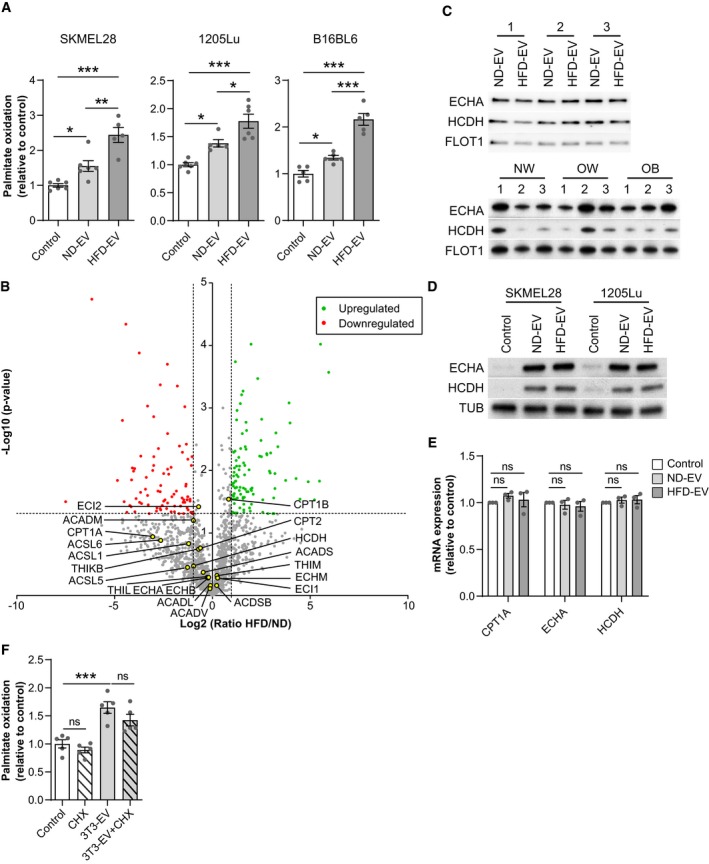
- Two human (SKMEL28 and 1205Lu) and a murine (B16BL6) melanoma cell lines were exposed, or not, to the indicated EV from primary murine adipocytes obtained from lean mice fed a normal diet (ND) or obese mice fed a high fat diet (HFD), and then, FAO was measured (n = 5 for B16BL6, SKMEL28 HFD‐EV and 1205Lu ND‐EV and n = 6 for other conditions).
- Volcano plot of mass spectrometry‐based quantitative proteomics results showing relative abundance of proteins in primary murine adipocyte EV from obese mice (HFD), as compared to those from lean mice (ND). The dashed lines indicate cutoff values and points colored in gray indicate proteins that display non‐significant fold‐change by Welch t‐test between both conditions (n = 3 for ND, 4 for HFD). Proteins involved in FAO are indicated by yellow dots.
- Western blot analysis of the indicated FAO enzymes in the EV secreted by primary adipocytes from lean (ND) and obese (HFD) mice (top panel) and from human individuals with varying BMI (normal weight, NW; overweight, OW; and obese, OB) (bottom panel). For each blot, extracts from three independent batches of murine samples or three independent individuals for human samples (1–3) are shown. Flotillin 1 (FLOT1) is used as a loading control.
- Western blot analysis of the indicated FAO enzymes in melanoma cells treated, or not, with EV from lean (ND) and obese (HFD) mice. Tubulin (TUB) is used as a loading control.
- RT–qPCR analysis of mRNAs for the indicated genes in 1205Lu cells treated or not with EV secreted by primary adipocytes from lean (ND) and obese (HFD) mice for 48 h. Results are expressed relative to the corresponding value for control cells (n = 3).
- Analysis of FAO levels in 1205Lu cells exposed to 3T3‐F442A EV and treated, or not, with cycloheximide (CHX) (n = 5).
Collectively, these results show that the heightened effect of adipocyte EV in obesity is not related to increased levels of FAO enzymes, whether transferred by adipocyte EV or expressed endogenously.
Adipocyte EV transfer FA to melanoma cells to fuel FAO, a process increased by obesity
Increased FAO requires both the protein machinery necessary to perform the enzymatic processes and the presence of the substrates. So, we postulated that adipocyte EV transfer FA to melanoma cells and that this process may be amplified in obesity, which could account for increased tumor cell FAO (Fig 2A) and consequently aggressiveness. EV secreted by mature 3T3‐F442A adipocytes contain high levels of FA compared to their precursors (Fig 3A). To determine whether these FA are subsequently transferred to tumor cells, we used a lipid pulse‐chase assay (Fig 3B). We loaded 3T3‐F442A adipocytes with BODIPY FL C16 (Appendix Fig S3A) and monitored the EV transfer of this fluorescent FA. We detected fluorescent FA in tumor cells exposed to adipocyte EV (Fig 3C, and Appendix Fig S3B and C). This fluorescent FA serves as a substrate for FAO in melanoma cells, as treatment with the FAO inhibitor, Etomoxir, leads to a further accumulation of fluorescent lipids (Fig 3D). To confirm that EV were responsible for this FA transfer, and not other structures such as lipoproteins or other particles that may have been co‐isolated by differential centrifugations, we fractioned the EV‐containing 100,000 g pellet by size exclusion chromatography and characterized their size and their FA, triglyceride, and protein content (Appendix Fig S4A–E). Our results show that only fractions containing EV (fractions 6–13) can reproduce lipid droplet accumulation in melanoma cells, but also their pro‐migratory effect (Appendix Fig S4F and G).
Figure 3. Adipocyte EV transfer FA to melanoma cells to fuel FAO, and this transfer is increased in obesity.
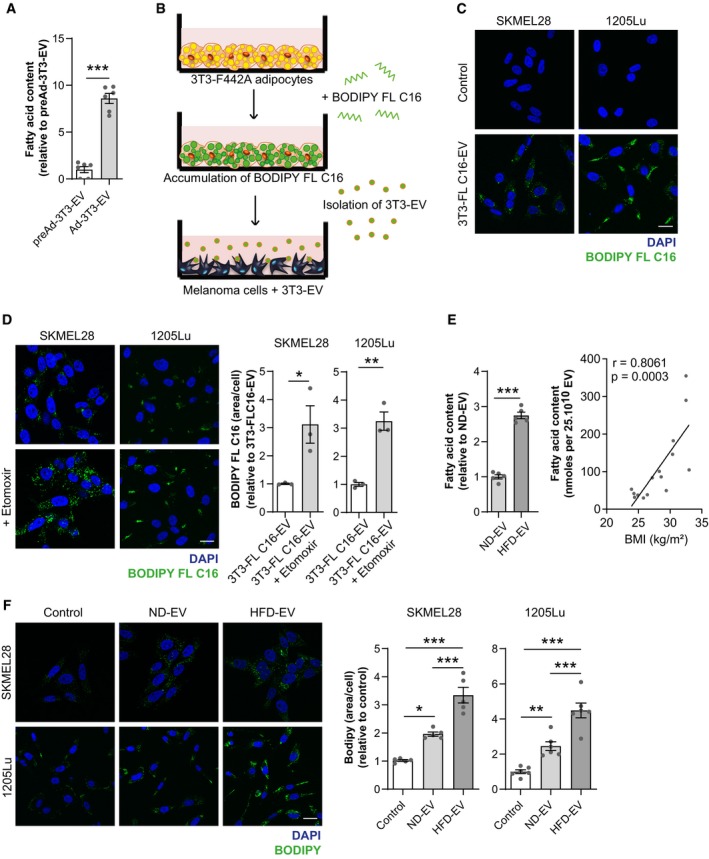
- Lipids were extracted from EV secreted by 3T3‐F442A preadipocytes and differentiated 3T3‐F442A adipocytes (respectively, preAd‐3T3‐EV and Ad‐3T3‐EV), and FA content was measured (n = 6).
- Workflow of the assay used to evaluate FA transfer by 3T3‐F442A adipocyte EV (3T3‐EV) to melanoma cells. Mature 3T3‐F442A adipocytes were loaded with BODIPY FL C16. Cells were then washed, and fresh medium was added. Seventy‐two hours later, conditioned medium was harvested, and 3T3‐F442A EV (3T3‐EV) were isolated and added to melanoma cells.
- Indicated melanoma cells were incubated with EV from 3T3‐F442A adipocytes previously loaded with BODIPY FL C16 (3T3‐FL C16‐EV) and, 24 h later, cells were fixed and nuclei were counterstained with DAPI before observation by confocal microscopy.
- Left panel, indicated cells were incubated with EV from 3T3‐F442A adipocytes previously loaded with BODIPY FL C16 and immediately treated, or not, with Etomoxir for 24 h. Then, cells were fixed and nuclei were counterstained with DAPI before observation by confocal microscopy. Right panel, quantification of BODIPY FL C16 staining area per cell (n = 3).
- Lipids were extracted from EV secreted by adipocytes from lean (ND) and obese (HFD) mice (n = 5) (left panel) or from human adipose tissue samples from patients with varying BMI (right panel) and fatty acid content was measured (n = 15).
- Indicated cells were exposed, or not, to adipocyte EV from primary adipocytes from lean mice fed a normal diet (ND) or obese mice fed a high fat diet (HFD) for 24 h. Then, cells were fixed, stained with BODIPY, and counterstained with DAPI. Left panel, confocal microscopy observation. Right panel, quantification of BODIPY staining area per cell (n = 5 for SKMEL28 and n = 6 for 1205Lu).
EV secreted by primary adipocytes also transfer FA to melanoma cells (Appendix Fig S5A). To determine whether this FA transfer increases in obesity, we evaluated FA content in EV secreted by primary murine and human adipocytes. FA content significantly increased in murine adipocyte EV in obesity (Fig 3E, left panel) and positively correlated with BMI in human samples (Fig 3E, right panel), although triglyceride content remained unchanged in obesity (Appendix Fig S5B), consistent with recent lipidomic data (Flaherty et al, 2019). Since hypertrophic adipocytes internalize less lipids than their smaller counterparts (Frayn, 2001; Hill et al, 2009; Appendix Fig S5C), we could not use the lipid pulse‐chase assay described in Fig 3B to compare FA transfer by ND‐EV and HFD‐EV. Nevertheless, HFD‐EV strongly increased the total neutral lipid content in melanoma cells compared to ND‐EV, which supports our hypothesis (Fig 3F and Appendix Fig S5D). Thus, these results demonstrate that adipocytes transfer FA to melanoma cells through EV, a process that is amplified by obesity.
Transferred FA are stored in lipid droplets and released by lipophagy
Although the FA transferred to melanoma cells by adipocyte EV fuel FAO, we also show an increase in neutral lipid storage within cytoplasmic lipid droplets (Fig 3D and F, and Appendix Figs S5D and S6), a process known to prevent lipotoxicity (Listenberger et al, 2003). We therefore examined whether these stored lipids are mobilized to drive FAO. We observed that melanoma cells incubated with adipocyte EV present double membrane structures, characteristic of autophagosomes that contain lipids within their lumen (Figs 4A and EV1A). Thus, we hypothesized that the transferred FA were degraded by lipophagy, an autophagic process that releases FA from lipid droplets (Singh et al, 2009). In accord with this theory, FA transferred by adipocyte EV colocalize with lysosomes (Figs 4B and EV1B). We found that inhibiting autophagy using Bafilomycin A1 prevented the degradation of lipid stores accumulated in response to adipocyte EV (Appendix Fig S7A) and blocked their pro‐migratory effect on melanoma cells (Appendix Fig S7B). We obtained similar results using the selective lysosomal acid lipase inhibitor, Lalistat 2 (Rosenbaum et al, 2010; Hamilton et al, 2012). This compound increased colocalization between fluorescent FA transferred by adipocyte EV and lysosomes (Fig 4B) and effectively inhibited degradation of transferred FA (Figs 4C and EV1C). This result was also reflected in the total neutral lipid stores accumulated in the presence of adipocyte EV (Fig EV1D). We also found that lipophagy is requisite to elicit the effect of adipocyte EV on melanoma migration and motility (Figs 4D and EV1E). As stored lipids could also be mobilized by cytosolic lipolysis (Lass et al, 2011), we studied the involvement of this process using 2‐(5,5‐Dimethyl‐1,3,2‐dioxaborinan‐2‐yl) benzoic acid ethyl ester (HSLi), a compound that targets HSL to effectively inhibit cytosolic lipolysis (Appendix Fig S8A). HSLi had no effect on the increase in lipid droplet accumulation and melanoma migration induced by adipocyte EV (Appendix Fig S8B and C). Furthermore, adipocyte EV have no effect on levels of cytosolic lipases ATGL, HSL (and its active phosphorylated form), and MAGL (Appendix Fig S8D).
Figure 4. FA transferred from adipocytes to melanoma cells by EV are released from lipid droplets by lipophagy.
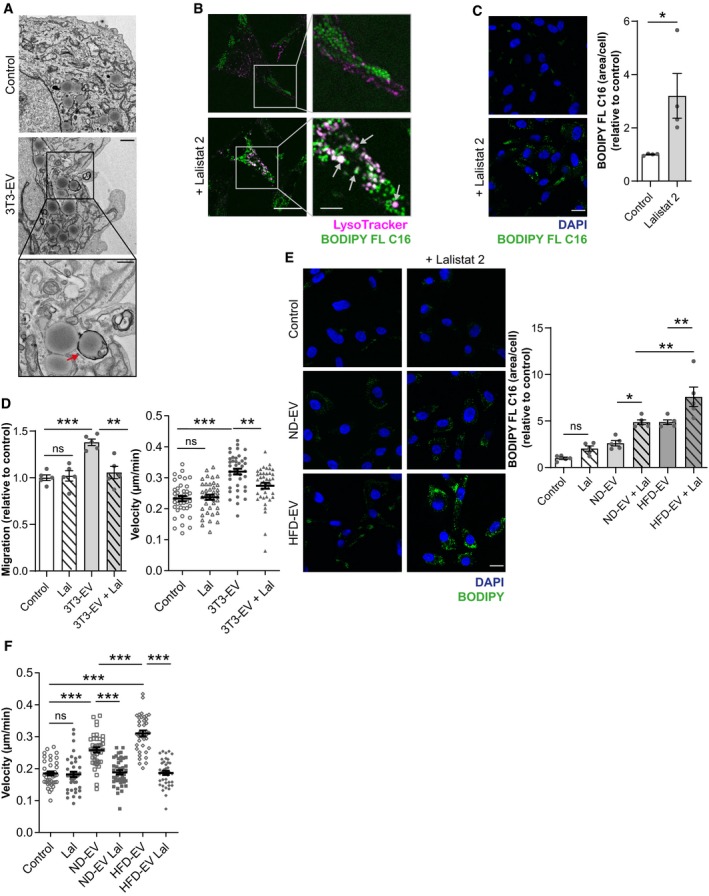
- Transmission electron micrographs of 1205Lu cells exposed, or not, to 3T3‐F442A EV (3T3‐EV). Scale bar represents 1 μm. A zoomed crop of the area with autophagic structures containing lipids (indicated by an arrow) is shown (scale bar: 0.5 μm).
- 1205Lu cells were incubated with EV from 3T3‐F442A adipocytes, previously loaded with BODIPY FL C16 in the presence, or not, of Lalistat 2. Then, live cells were stained with the LysoTracker probe and observed by confocal microscopy. Arrows indicate colocalization.
- 1205Lu were incubated with EV from 3T3‐F442A adipocytes, previously loaded with BODIPY FL C16 and treated, or not, with Lalistat 2. Then, cells were fixed and counterstained with DAPI before observation by confocal microscopy. Quantification of BODIPY FL C16 staining per cell is shown beside (n = 4).
- 1205Lu cells were exposed to 3T3‐F442A EV (3T3‐EV) and treated, or not, with Lalistat 2 (Lal). Cell migration was then evaluated in Boyden chamber assays (left panel; n = 5), or cell motility was tracked by video microscopy (right panel; n = 40 cells per group).
- 1205Lu cells were exposed, or not, to adipocyte EV from lean mice fed a normal diet (ND) or obese mice fed a high‐fat diet (HFD) with, or without, Lalistat 2 (Lal). Cells were then fixed, stained with BODIPY, and counterstain with DAPI before observation by confocal microscopy. Quantification of BODIPY staining per cell is shown on the right (n = 5).
- 1205Lu cells were exposed, or not, to adipocyte EV from ND or HFD mice with, or without, Lalistat 2 (Lal). Cell motility was then tracked by video microscopy (n = 40 cells per group).
Figure EV1. FA transferred from adipocytes to melanoma cells by EV are released from lipid droplets by lipophagy.
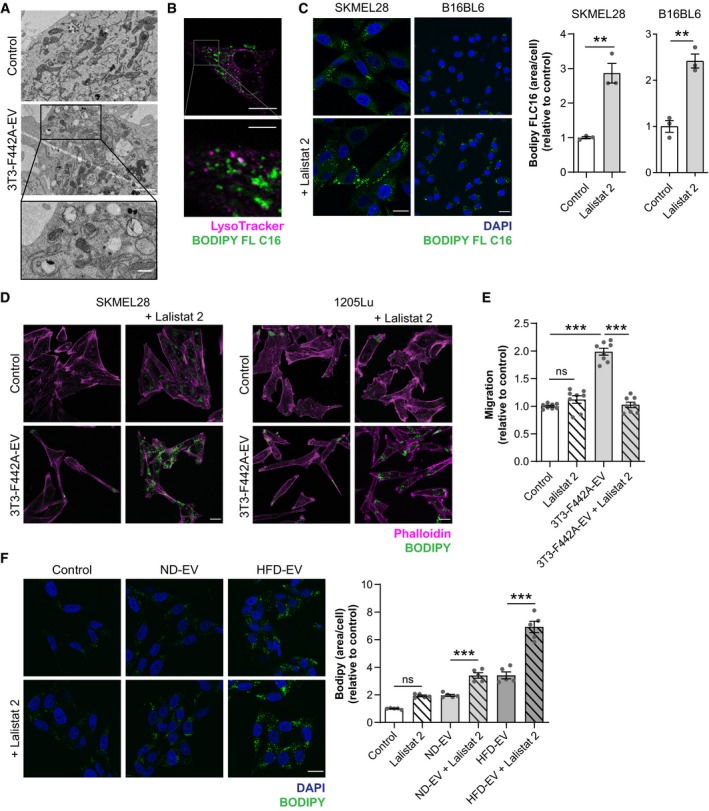
- Transmission electron micrographs of SKMEL28 cells exposed, or not, to 3T3‐F442A EV. Scale bar: 1 μm. A zoomed crop of the area with autophagic structures containing lipids is shown. Scale bar: 0.5 μm.
- SKMEL28 cells were incubated with EV secreted by 3T3‐F442A adipocytes previously loaded with BODIPY FL C16. Then, cells were washed and stained with the LysoTracker probe, and live cells were observed by confocal microscopy. A zoomed crop is shown below (scale bar: 5 μm).
- Indicated melanoma cells were exposed to 3T3‐F442A EV and treated, or not, with Lalistat 2. Then, cells were fixed, stained with BODIPY, and counterstained with DAPI for confocal microscopy observation. Quantification of BODIPY FL C16 staining area per cell is shown on the right (n = 3).
- SKMEL28 and 1205Lu cells were exposed to 3T3‐F442A EV in the presence, or not, of Lalistat 2. Cells were then fixed and stained with BODIPY and Phalloidin before observation by confocal microscopy.
- SKMEL28 cells were exposed to 3T3‐F442A EV and treated, or not, with Lalistat 2. Cell migration was then evaluated in Boyden chamber assays (n = 8).
- SKMEL28 cells were exposed, or not, to adipocyte EV from lean (ND) or obese (HFD) mice with, or without, Lalistat 2. Cells were then fixed, stained with BODIPY, and counterstained with DAPI before observation by confocal microscopy. Quantification of BODIPY staining per cell is shown on the right (n = 5).
Furthermore, inhibiting lipophagy also prevented degradation of stored lipids following exposure to both lean and obese primary murine adipocyte EV (Figs 4E and EV1F), as well as their effect on cell motility (Fig 4F). Our results from the SILAC experiment revealed that various autophagic and lysosomal proteins are transferred by adipocyte EV to melanoma cells (Table EV1). This suggests that EV may promote lipophagy by providing these proteins. Thus, our results show that adipocyte EV‐derived lipids stored by melanoma cells are released by lipophagy, which is crucial for the pro‐migratory effect of EV.
Adipocyte EV remodel the mitochondrial network to support melanoma migration
Increased lipid accumulation (Zhang et al, 2011) and subsequent increased FAO (Kuzmicic et al, 2014) can remodel the mitochondrial network. Interestingly, mitochondrial dynamics also play a crucial role in tumor cell migration (Altieri, 2017). Mitochondrial fission and redistribution toward cell extremities during tumor cell migration increases energy supply for actin polymerization in these zones (Zhao et al, 2013; Cunniff et al, 2016). Consistent with such a process in response to adipocyte EV, treated melanoma cells adopt an elongated morphology and present profound changes in mitochondrial distribution switching from a perinuclear localization to a distribution throughout the cytoplasm, particularly toward membrane protrusions (Fig 5A), and mitochondria size is decreased (Appendix Fig S9). In our SILAC experiment, we found that key regulators of mitochondrial dynamics, FIS1 and OPA1, are among the transferred proteins from adipocytes to melanoma cells by EV (Table EV1). In accord, DRP1 (DNM1L) protein levels in melanoma cells are unchanged by EV treatment, whereas FIS1 is increased in 1205Lu cells (Fig 5B). In SKMEL28 EV‐treated cells, FIS1 is only slightly increased but this result could be explained by the higher amount of endogenous FIS1 in SKMEL28 compared to 1205Lu cells (Fig 5B). Moreover, increased melanoma cell migration and motility following treatment with adipocyte EV is completely abrogated by the mitochondrial fission inhibitor Mdivi‐1 (Figs 5C and EV2A). We confirmed these findings using gene‐silencing experiments. Two different siRNAs targeting the key mitochondrial fission regulator DRP1 effectively reduced the expression of the protein in melanoma cells (Fig 5D, top panel) and strongly abrogated the pro‐migratory effect of adipocyte EV (Fig 5D, bottom panel). Finally, FAO was clearly required for mitochondrial redistribution in both lean and obese conditions, since mitochondria retain a perinuclear localization in the presence of Etomoxir (Fig EV2B). This mitochondrial redistribution is associated with an increase in mitochondrial activity that is dependent on FAO (Appendix Fig S10A). Indeed, in the presence of adipocyte EV, basal and maximal respiration, as well as respiration coupled to ATP production, are increased (Appendix Fig S10B). Moreover, after treatment with adipocyte EV, cellular respiration is more dependent on FAO (Appendix Fig S10C). In obesity, the amplified effect of adipocyte EV on cell motility also depends on mitochondrial fission, as Mdivi‐1 totally abrogates their effect (Fig 5E).
Figure 5. Adipocyte EV modify melanoma mitochondrial dynamics, a process that promotes melanoma aggressiveness and is exacerbated by obesity.
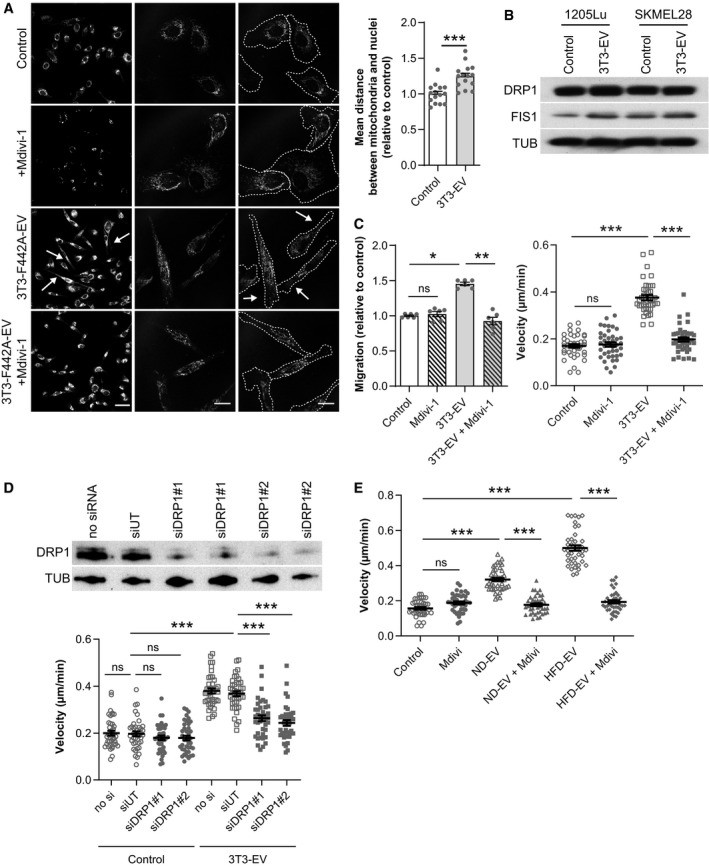
- 1205Lu cells exposed to 3T3‐F442A EV (3T3‐EV) and treated, or not, with Mdivi‐1, were stained with a MitoTracker probe, fixed, and observed by confocal microscopy. Arrows indicate the presence of mitochondria in membrane protrusions. On the images to the right, the outline of cells is shown in dotted lines. Scale bars: 20 μm. Right panel, quantification of the distance between mitochondria and nuclei (n = 15).
- Western blot analysis of the indicated mitochondrial fission proteins in melanoma cells treated, or not, with EV from 3T3‐F442A adipocytes (3T3‐EV). Tubulin (TUB) is used as a loading control.
- 1205Lu cells were exposed to 3T3‐F442A EV (3T3‐EV) and treated, or not, with Mdivi‐1. Cell migration was then evaluated in Boyden chamber assays (left panel; n = 6). Right panel, 3T3‐F442A EV were added to 1205Lu cells, which were immediately treated, or not, with Mdivi‐1. Cell motility was then tracked by video microscopy (n = 40 cells per group) (right panel).
- 1205Lu cells were transfected, or not, with two different siRNA targeted against DRP1 (siDRP1#1 or 2) or an untargeted siRNA (siUT). Top, 48 h after transfection, protein extracts were prepared and DRP1 expression was evaluated by Western blot. Bottom, 36 h post‐transfection, cells were exposed to 3T3‐F442A EV (3T3‐EV), and cell motility was tracked by video microscopy (n = 40 cells per group).
- 1205Lu cells were exposed to adipocyte EV from lean mice fed a normal diet (ND) or obese mice fed a high‐fat diet (HFD) and immediately treated, or not, with Mdivi‐1. Cell motility was then tracked by video microscopy (n = 40 cells per group).
Figure EV2. Adipocyte EV modify melanoma cell mitochondrial dynamics, a process that is dependent on FAO and promotes aggressiveness.
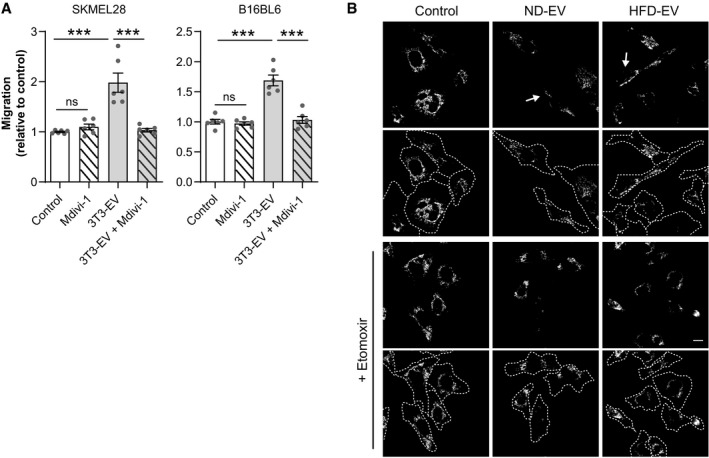
- Human SKMEL28 and murine B16BL6 cells were exposed to 3T3‐F442A EV (3T3‐EV) and treated, or not, with Mdivi‐1. Cell migration was then evaluated in Boyden chamber assays. Bars and error bars represent means ± SEM (n = 6); statistically significant determined by one‐way ANOVA with post hoc Tukey's test, ***P < 0.001, ns: non‐significant.
- 1205Lu cells were exposed to adipocyte EV from lean (ND) or obese (HFD) mice for 48 with, or without, Etomoxir for the last 24 h. Cells were then stained with a MitoTracker probe, fixed, and observed by confocal microscopy. Arrows indicate the presence of mitochondria in membrane protrusions. Underneath each original image, an image depicting the outline of cells in dotted lines is shown. Scale bar: 20 μm.
For FAO to efficiently take place in cell protrusions and supply energy for cell migration, we expected that both the mitochondrial machinery and the substrate would be present. In melanoma cells incubated with adipocyte EV, we found a striking lipid droplet redistribution toward cell protrusions (Fig 6A). Obesity further exacerbated this redistribution (Fig 6B). Moreover, lipid droplets are found in close proximity with mitochondria in these areas, as observed with confocal microscopy (Figs 6C and EV3A) and transmission electron microscopy (Figs 6D and E, and EV3B and C), and their size is increased (Figs 6F and EV3D). Lysosomes are also redistributed to cell protrusions after treatment with adipocyte EV (Figs 6G and EV3E). Finally, to study the specificity of these processes in response to adipocyte EV, we stimulated cell migration using TNF‐α (Appendix Fig S11A) and although this triggered mitochondrial redistribution, as expected (Zhao et al, 2013; Cunniff et al, 2016), TNF‐α had no effect on lipid droplet accumulation or redistribution in melanoma cells (Appendix Fig S11B).
Figure 6. Lipid droplets are found in membrane protrusions, at proximity to mitochondria, in melanoma cells exposed to adipocyte EV.
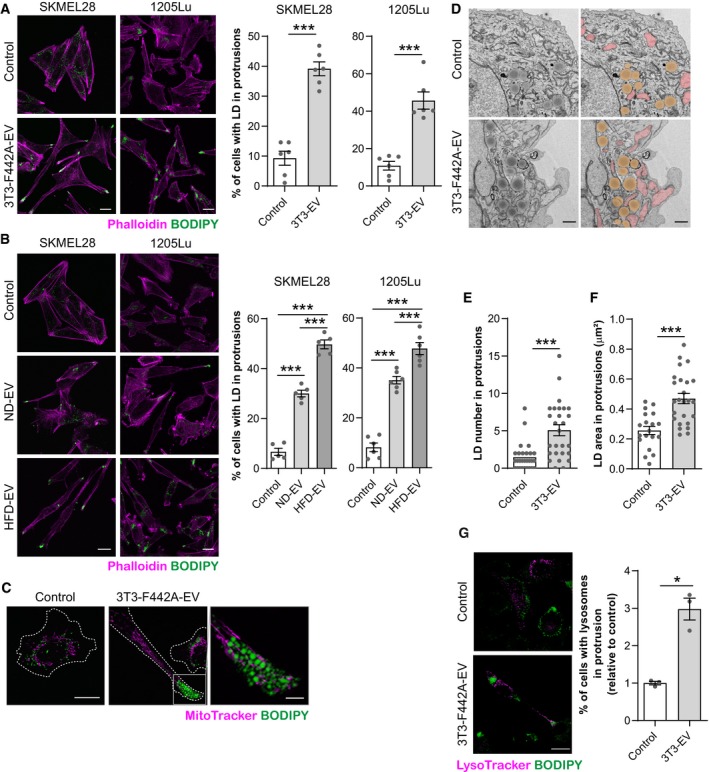
- Left panel, indicated melanoma cells exposed to 3T3‐F442A EV were fixed, stained with BODIPY and Phalloidin before observation by confocal microscopy. Right panel, quantification of the percentage of cells presenting lipid droplets (LD) within membrane protrusions (n = 6).
- Left panel, melanoma cells exposed to EV secreted by adipocytes from lean mice fed a normal diet (ND) or obese mice fed a high‐fat diet (HFD) were fixed, stained with BODIPY and Phalloidin before observation by confocal microscopy. Right panel, quantification of the percentage of cells presenting lipid droplets (LD) within membrane protrusions (n = 5 for SKMEL28 and n = 6 for 1205Lu).
- 1205Lu cells exposed to 3T3‐F442A EV were stained with a MitoTracker probe. Then, cells were fixed and stained with BODIPY before observation by confocal microscopy. A zoomed crop of the area containing mitochondria and lipid droplets in a membrane protrusion is shown (scale bar: 5 μm).
- Transmission electron microscope observations of 1205Lu cells exposed, or not, to 3T3‐F442A EV. Mitochondria are colored in pink and lipid droplets are colored in yellow on images on the right.
- Number of lipid droplets (LD) found within membrane protrusions on transmission electron microscopy images of 1205Lu cells exposed, or not, to 3T3‐F442A EV (3T3‐EV) (n = 28 for control and n = 27 for 3T3‐EV).
- Area of lipid droplets (LD) found within membrane protrusions on transmission electron microscopy images of 1205Lu cells exposed, or not, to 3T3‐F442A EV (n = 19 for control and n = 25 for 3T3‐EV).
- Left panel, 1205Lu melanoma cells were exposed, or not, to 3T3‐F442A EV. Then, live cells were stained with LysoTracker and BODIPY probes and observed by confocal microscopy. Right panel, quantification of the percentage of cells presenting lysosome within membrane protrusions (n = 3).
Figure EV3. Lipid droplets and lysosomes are found in membrane protrusions, at proximity to mitochondria, in melanoma SKMEL28 cells exposed to adipocyte EV.
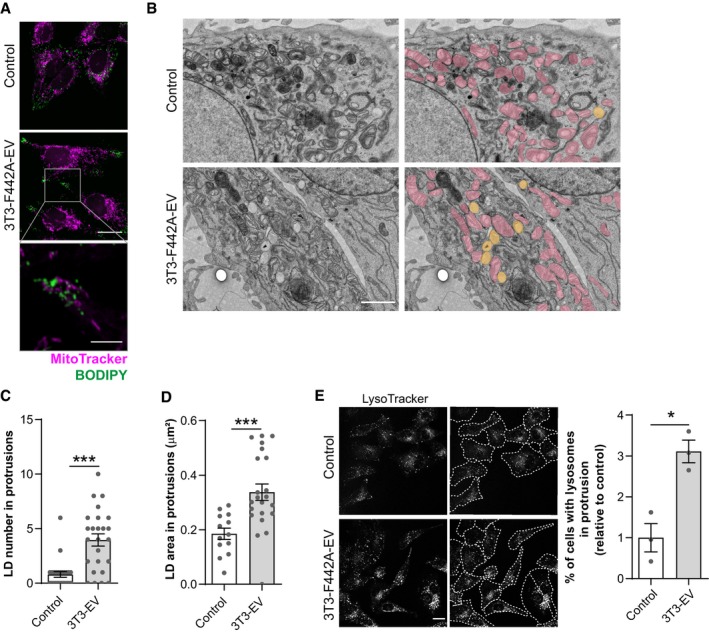
- SKMEL28 cells exposed to 3T3‐F442A EV were stained with a MitoTracker probe. Then, cells were fixed, stained with BODIPY, and observed by confocal microscopy. Scale bar: 20 μm. A zoomed crop of the area containing mitochondria and lipid droplets in a membrane protrusion is shown. Scale bar: 5 μm.
- Transmission electron microscope observations of SKMEL28 cells exposed, or not, to 3T3‐F442A EV. Mitochondria are colored in pink, and lipid droplets are colored in yellow on images on the right. Scale bar: 1 μm.
- Number of lipid droplets (LD) found within membrane protrusions in transmission electron microscopy images of SKMEL28 cells exposed, or not, to 3T3‐F442A EV (3T3‐EV) (n = 25).
- Area of lipid droplets (LD) found within membrane protrusions in transmission electron microscopy images of SKMEL28 cells exposed, or not, to 3T3‐F442A EV (3T3‐EV) (n = 13 for control and n = 22 for 3T3‐EV).
- Left panel, SKMEL28 melanoma cells were exposed, or not, to 3T3‐F442A EV. Then, live cells were stained with LysoTracker and observed by confocal microscopy. Beside each original image, an image depicting the outline of cells in dotted lines is shown. Right panel, quantification of the percentage of cells presenting lysosomes within membrane protrusions (n = 3). Scale bar: 20 μm.
All together, these results show that mitochondrial dynamics, and possibly lipid droplet dynamics, trigger cell migration in response to adipocyte EV, with a heightened effect in obesity.
FAO and mitochondrial dynamics correlate with melanoma aggressiveness
To assess the clinical relevance of our findings, we performed a targeted analysis of human melanoma sample data that is publicly available online in The Cancer Genome Atlas database (TCGA, http://www.cbioportal.org). Although this tumor collection is not annotated with biometric data to study obesity, we investigated whether the cellular processes we identified here are key to melanoma progression in the general population. This analysis revealed that increased expression of FAO enzymes correlated with poor overall survival (OS) in melanoma patients (Fig 7A). Accordingly, neutral lipid content and FAO progressively increased in melanoma cell lines with increasing migratory and metastatic potentials (Goodall et al, 2008; Lazar et al, 2015; Fig 7B and C). Finally, mitochondrial dynamics strongly correlated with melanoma progression, as heightened expression of genes involved in mitochondrial fission (FIS1) or fusion (MFN2 and OPA1) correlated with decreased OS in melanoma patients (Fig 7D).
Figure 7. FA metabolism and mitochondrial dynamics are associated with melanoma aggressiveness.
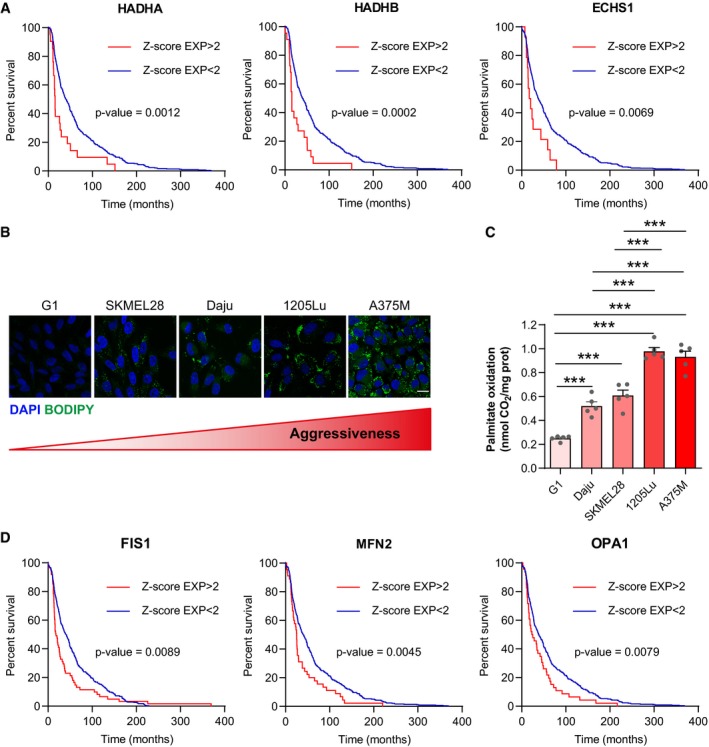
- TCGA data were analyzed to reveal the effect of high mRNA levels of key FAO enzymes on the overall survival of patients with melanoma. Statistical significance of survival was evaluated with a Log‐rank (Mantel‐Cox) test (n = 449, 450, and 459 for HADHA, HADHB, and ECHS1, respectively).
- BODIPY staining of neutral lipid stores in the indicated melanoma cell lines, which present increasing degrees of aggressiveness (from left to right). DAPI was used to counterstain nuclei. Scale bar: 20 μm.
- FAO in the indicated melanoma cell lines was measured. Bars and error bars represent means ± SEM (n = 5); statistically significant by one‐way ANOVA with post hoc Tukey's test, ***P < 0.001. If not indicated, non‐significant.
- TCGA data were analyzed to reveal the effect of high mRNA levels of key actors in mitochondrial dynamics on the overall survival of patients with melanoma. Statistical significance of survival was evaluated with a Log‐rank (Mantel‐Cox) test (n = 459).
Discussion
In summary, we reveal the importance of EV in the metabolic dialog between adipocytes and melanoma cells, which promotes FAO and, ultimately, tumor aggressiveness. Previous studies showed such a process occurred at the invasive front of tumors where secretions from cancer cells provoke adipocyte lipolysis, which provides tumors with the necessary substrate for FAO. However, our findings demonstrate that EV secreted by naïve adipocytes provoke a similar process and, in fact, provide melanoma cells with both the required machinery (proteins) and the substrate (FA) for FAO.
We determined the proteins transferred from adipocytes to tumor cells by EV using a groundbreaking approach based on the SILAC technique, which permitted widespread labeling of adipocyte proteins for subsequent identification within tumor cells by mass spectrometry. To our knowledge, this is the first time that a comprehensive analysis of all of the proteins transferred between two cell types by EV has been performed. We also illustrate the utility of this technique for any cultured cell types to study EV‐mediated intercellular protein transport. Using this approach, we found that only approximately 30% of labeled adipocyte EV proteins are detected in recipient melanoma cells, with some highly abundant EV proteins excluded from transfer. This process suggests a highly selective EV‐mediated protein transfer between adipocytes and melanoma cells. This could be the result of some populations of EV not being internalized by melanoma cells or some EV or the proteins they carry being rapidly sorted for degradation after internalization. While we do not fully understand how EV are selectively internalized and/or processed in recipient cells, our approach provides a foundation to address these questions in future studies. Despite this selective transfer, melanoma cells internalize adipocyte proteins involved in FAO, mitochondrial respiration, and ATP production through EV. Importantly, we also identified key contributors in other cellular processes induced in melanoma cells in response to adipocyte EV that help regulate their pro‐migratory effects, lipophagy, and mitochondrial dynamics (Table EV1).
In addition to this protein transfer, we demonstrated that adipocyte EV also convey FA to tumor cells to drive FAO. Increases in this process heightened the effect of adipocyte EV in obesity. Shuttling of metabolic substrates from the tumor microenvironment to cancer cells by EV has previously been described. Indeed, CAF‐derived EV transport metabolites, such as amino acids, lipids, and TCA cycle intermediates, to tumor cells, which serves as triggers for central carbon metabolism (Zhao et al, 2016). Even though we know FA are released by adipocytes in response to tumor secretions for transfer to melanoma cells (Kwan et al, 2014; Zhang et al, 2018), we reveal that naïve adipocytes, that have never encountered tumor secretions, can also convey lipids to cancer cells through EV. Previous studies had focused on the transfer of free FA from adipocytes to tumor cells and the role of FA transporters in their uptake. For example, in an ovarian cancer model (Nieman et al, 2011) and a melanoma model (Zhang et al, 2018), inhibition of FA transporters (respectively, FABP4 and FATP1) decreases the FA transfer from adipocytes to tumor cells during co‐culture. However, our findings clearly demonstrate that EV secreted by naïve adipocytes can also be responsible for FA transfer and that these EV alone are sufficient to remodel melanoma metabolism and favor aggressiveness.
Thus, adipocyte EV mediate a metabolic cooperation between adipocytes and melanoma cells, acting as shuttles to convey both the protein machinery and the lipid substrate required for FAO. Interestingly, upon stimulation of adipocyte lipolysis using isoproterenol (Appendix Fig S12A), EV secretion and EV FA levels are increased (Appendix Fig S12B and C). Thus, it is plausible that tumor‐induced lipolysis may also promote such processes. Following on from our work, future studies should focus on the effect of the bidirectional crosstalk that takes place between cancer cells and adipocytes on EV.
In this study, we focused on lipid and protein actors that regulate melanoma metabolism and aggressiveness in response to adipocyte EV, but it is important to note that EV also transport nucleic acids including mRNA, microRNA, and other non‐coding RNA (Tkach & Thery, 2016; van Niel et al, 2018) that could also participate in their pro‐tumoral effect. Moreover, adipocyte EV contain adipokines, in particular, leptin (Table EV2), which has been shown to possess pro‐tumoral properties in melanoma models (Amjadi et al, 2016) and be associated with an increased risk of lymph node metastasis in melanoma patients (Oba et al, 2016).
Analysis of TCGA data revealed that high levels of FAO enzymes correlate with lower OS in melanoma patients (Fig 7A). Consistent with this, studies found upregulated genes associated with FAO in aggressive melanoma (Xu et al, 2012; Rodrigues et al, 2016). Our work demonstrates that FAO depends on both the endogenous traits of melanoma cells and a metabolic cooperation with adipocytes mediated by EV. Although we did not observe an increased transfer of proteins involved in FAO in obese conditions, we cannot exclude the relevance of these enzymes under physiological conditions. Obese adipocytes do secrete larger quantities of EV compared to lean counterparts, which may further increase their effect in cancer patients (Lazar et al, 2016; Flaherty et al, 2019). In experimental conditions using equal quantities of vesicles, HFD‐EV convey higher levels of FA, which increases lipid accumulation in melanoma cells. In obesity, although stimulated lipolysis induced by activation of adrenergic receptors decreases, basal lipolysis is heightened (Duncan et al, 2007; Verboven et al, 2018). This process may increase FA release and, consequently, a higher availability of these substrates for tumor cells. Our results show that these FA are at least partially secreted through EV.
Understanding the mechanisms that promote tumor progression in obese patients is crucial to develop relevant therapies. Here, we show that a metabolic cooperation exists between adipocytes and tumor cells and is amplified in obesity, suggesting that obese cancer patients would likely benefit from FAO inhibitors and/or molecules that inhibit EV uptake. As EV diffuse through tissues and circulate throughout the organism, they may influence tumors not only at proximity to adipose depots, but also at distance. For example, we hypothesize that adipocyte EV may fuel circulating tumor cells to provide them with the nutrients they require to reach and colonize metastatic niches, and future studies should investigate such a process. As this study focuses on naïve adipocyte EV, unmodified by tumor secretions, our findings suggest these vesicles likely play significant roles in metabolic processes in a physiological context. In accord, elegant studies demonstrated the importance of EV in the dialog between adipocytes and endothelial cells (Crewe et al, 2018), as well as AT macrophages (Flaherty et al, 2019). We speculate that adipocyte EV could also regulate energy metabolism of other cell types; for example, one could imagine they may provide muscles with the necessary molecules for FAO during physical activity.
We also identified the cellular mechanisms induced in melanoma cells that process lipids transferred by adipocyte EV that then trigger metabolic remodeling and cell migration. We found that FA transferred by adipocyte EV are stored in lipid droplets and are then mobilized by melanoma cells through lipophagy. Lipophagy inhibitors prevented the degradation of stored lipids in response to adipocyte EV in both lean and obese conditions. Although the involvement of lipophagy in melanoma metabolism remains unclear, a recent study found that melanospheres containing cancer stem cells store lipids mobilized by lipophagy during differentiation of these cells (Giampietri et al, 2017).
Transferred FA that fuel FAO may remodel the mitochondrial network in melanoma cells to redistribute these organelles to the cell extremities, which promotes cell migration. The importance of mitochondrial dynamics in tumor cell migration is increasingly recognized (Altieri, 2017). Although mitochondrial fission is a focus of many studies, both fission and fusion events are critical to redistribute mitochondria toward the leading cell edge during migration (Desai et al, 2013). However, little is known about mitochondrial dynamics in melanoma progression. The limited data available suggest that the mitochondrial network is reorganized during B16‐F10 migration (Cunniff et al, 2016). Consistent with this, our analysis of TCGA data revealed that increased mRNA expression of key actors involved in mitochondrial dynamics correlates with lower OS in melanoma patients. Inhibiting mitochondrial fission abrogates the increase in cell motility observed after treatment of melanoma cells with adipocyte EV, which highlights the importance of this process. Importantly, we linked mitochondrial redistribution to EV‐induced FAO, as Etomoxir prevented mitochondrial relocalization to membrane protrusions. We identified key regulators of mitochondrial dynamics among the proteins transferred from adipocytes to melanoma cells by EV. Although the changes in the mitochondrial network that we found in the presence of adipocyte EV require FAO, we acknowledge other substrates may facilitate this process. Interestingly, we also showed that lipid droplets and lysosomes also relocalize toward membrane protrusions in melanoma cells exposed to adipocyte EV. This suggests that substrate is brought directly to specific sites to continue to fuel FAO during melanoma migration. Such a proximity can occur in other models, in which it increases lipid droplet and mitochondria interactions to facilitate lipid transfer between the two organelles (Herms et al, 2015; Rambold et al, 2015).
Our results also raise questions concerning the effect of drugs used to combat the metabolic complications associated with obesity on the dialog between adipocytes and tumor cells. For instance, glitazone drugs activate PPARγ in adipocytes, leading to increased insulin sensitivity and, ultimately, adipogenesis and lipid storage (Tontonoz & Spiegelman, 2008). This may also reduce the amount of FA released by these cells in EV. However, glitazone drugs have also been shown to activate AMPK (Saha et al, 2004), leading to heightened FAO through inhibition of ACC (Jeon, 2016). In tumor cells, glitazone drugs could therefore promote FA metabolism and exacerbate the effect of adipocyte EV. Similarly, metformin also influences different aspects of FA metabolism, decreasing lipid synthesis and increasing FAO in a number of different cell types but has also been shown to possess anti‐tumoral properties in pre‐clinical studies (Morales & Morris, 2015). Therefore, it is challenging to foresee what the effects of such treatments would be in obese cancer patients and future studies should focus on evaluating this.
In summary, our findings demonstrate that adipocytes EV provide both the machinery (enzymes) and substrate (FA) required for FAO to melanoma cells. The FA transferred by these EV are stored in lipid droplets and mobilized by lipophagy to fuel FAO. We identified mitochondrial dynamics as a process that links adipocyte‐induced FAO and tumor cell migration. We observed a mitochondrial redistribution toward cell protrusions that requires mitochondrial fission, which increases cell migration induced by adipocyte EV. In obesity, a higher supply of FA through EV amplifies this entire process. These results demonstrate a metabolic cooperation occurs between adipocytes and tumor cells. Further, we reveal the underlying cellular processes in this interaction. We propose these pathways could comprise novel therapeutic targets to treat obese melanoma patients.
Materials and Methods
Reagents and antibodies
BODIPY® 493/503 (hereafter referred to simply as BODIPY), BODIPY™ FL C16, MitoTracker Red CMXRos, LysoTracker Deep Red, DAPI, and rhodamine phalloidin were obtained from Molecular Probes‐Thermo Fisher Scientific (Eugene, OR, USA). [1‐14C] palmitate was obtained from PerkinElmer (Waltham, MA, USA). Etomoxir, Mdivi‐1, cycloheximide, bovine serum albumin (BSA), and insulin were obtained from Sigma‐Aldrich (Saint Louis, MO, USA). Lalistat 2 was obtained from Tocris Cookson (Bristol, UK). Antibodies are described in Appendix Table S1. DharmaFECT1 transfection reagent and siRNA were obtained from Dharmacon™ (Cambridge, UK). siRNA sequences are as follows: Untargeted (UT) siRNA: 5′ UAGCGACUAAACACAUCAA‐3′; siRNA DRP1#1: 5′ GGAGCCAGCUAGAUAUUAA; siRNA DRP1#2: 5′ CAUCAGAGAUUGUUUACCA.
Cell lines, culture, and treatments
SKMEL28 cells are an established line that originate from a human metastatic lymphonodal lesion (Carey et al, 1976). 1205Lu were derived from the vertical growth phase primary lesion‐derived human cell line WM793 by serial passage through athymic mice and selection of cells from lung metastases (Kath et al, 1991). Both melanoma lines were provided by Dr Lionel Larue (Institut Curie, Orsay, France). The murine B16BL6 melanoma cell line is derived from the B16F10 line by serial passages in tumor cell invasion assays through murine bladders in vitro (Poste et al, 1980). B16BL6 cells were kindly gifted by Dr Gilles Favres, originally purchased from AcceGen Biotechnology. G1 and Daju are established lines derived from resected human metastatic lymphonodal lesions (respectively, Dufour et al, 1997; Moore et al, 2004). A375M were obtained by clonal selection of the human metastatic cell line A375 (Kozlowski et al, 1984). All were provided by Dr Lionel Larue (Institut Curie, Orsay, France).
The murine 3T3‐F442A preadipocyte cell line was purchased from the European Collection of Cell Cultures.
All cell lines were cultured in DMEM supplemented with 10% FCS, 125 mg/ml streptomycin, and 125 UI/ml penicillin (all purchased from GIBCO‐Thermo Fisher Scientific, Eugene, OR, USA) and maintained at 37°C in a humidified atmosphere with 5% CO2. Cells were used within 2 months after resuscitation of frozen aliquots and regularly tested for mycoplasma contamination.
To generate mature 3T3‐F442A adipocytes, 9 × 104 3T3‐F442A preadipocytes were seeded in 6‐well plates in DMEM and, 3 days later, once cells had reached confluence, differentiation was induced by supplementing medium with 50 nM insulin for 14 days. During this time, medium was refreshed twice a week. The term “adipocyte” refers to cells that were differentiated for at least 14 days that present adipocyte characteristics and express specific markers (Djian et al, 1985; MacDougald & Lane, 1995). To condition medium, 1.5 ml of ultracentrifugated DMEM (protocol for preparation below) was added per well for 72 h.
For treatment of melanoma cells with EV in 6‐well plates, for example, 1 × 105 melanoma cells were seeded and, 24 h later, 5 × 1010 3T3‐F442A or primary adipocyte EV were added per well. Cells were incubated with EV for 24 h or 48 h for experiments to study lipid accumulation, 48 h for experiments to study cell metabolism and mitochondrial dynamics, and 72 h for experiments to evaluate cell migration. Where indicated, Etomoxir (50 μM) or Mdivi‐1 (20 μM) was added 24 h before the end of incubation with EV, unless specified otherwise. Cycloheximide (10 μM) was added 36 h before the end of incubation with EV, and Lalistat 2 (50 μM) was added from the beginning of EV treatment.
For siRNA treatment, cells were transfected with 50 nM siRNA in the presence of DharmaFECT1, as indicated by the supplier; 6 h post‐transfection, fresh media, and, if needed, EV, were added. Cells were used 48 h post‐transfection for Western blot analysis and 36 h post‐transfection to track cell motility.
Animals and primary cell isolation
Mice were handled in accordance with National Institute of Medical Research (INSERM) principles and guidelines. All experiments were approved by the local committee on ethics of animal experimentation. Mice were housed according to national and institutional guidelines for animals, in a controlled environment with a 12:12‐h light–dark cycle. Eight‐week‐old C57BL/6J male mice (Janvier, Le Genest St Isle, France) were fed a normal or high fat diet (ND and HFD) for 15 weeks [ND (PicoLab Rodent Diet 20, Purina Mills Inc., Brentwood, MO, USA) and HFD (Research Diets Inc., New Brunswick, New Jersey, USA)]. The energy contents of the diets were as follows: 16% protein, 80% carbohydrate, and 4% fat for the ND; and 20% protein, 20% carbohydrate, and 60% fat for the HFD. After 15 weeks of diet, the average weight of ND mice was 29.6 g (± 1.2 g) and of HFD mice was 48.9 g (± 3.8 g). Mice were then sacrificed to collect inguinal subcutaneous AT. AT was weighed and placed in DMEM supplemented with 1% BSA (2.5 ml/g of tissue). Adipocytes were isolated as previously described (Lazar et al, 2016). To condition medium, one million adipocytes per ml were suspended in ultracentrifugated DMEM overnight (protocol for preparation below).
Preparation of human adipocytes
Human AT samples were collected from abdominal dermolipectomies in accordance with the recommendations of the ethics committee of the Rangueil Hospital (Toulouse, France). All patients gave their informed consent in accordance with the Declaration of Helsinki Principles as revised in 2000. All of them were drug free and healthy (lean patients: 20 < BMI (body mass indexes); < 25; overweight patients 25 < BMI < 30; obese patients BMI > 30). Tissues were processed within 1 h of surgical resection. Briefly, AT was separated from skin and large blood vessels, glandular tissue, and fascia were discarded. AT was then weighed and cut into < 1 mm3 pieces, and adipocytes were isolated as previously described (Lazar et al, 2016). To condition medium, one million adipocytes per ml were suspended in ultracentrifugated DMEM overnight (protocol for preparation below).
Preparation of ultracentrifugated DMEM, EV isolation and Nanoparticle Tracking Analysis
To prepare medium to be conditioned for EV isolation, DMEM supplemented with 10% FCS was depleted of contaminating vesicles by overnight centrifugation at 100,000 g. The conditioned medium was obtained from 3T3‐F442A or primary adipocytes as described above, and small EV, enriched in exosomes, were purified using differential centrifugations. First, conditioned medium was centrifuged at 3,000 g for 30 min to remove cell debris and most “large” EV. The resulting supernatant was further centrifuged for 60 min at 10,000 g to remove most “medium” EV, and finally, small EV were enriched by ultracentrifugation at 100,000 g for 90 min (Kowal et al, 2016). The pellets were suspended in EV‐depleted DMEM medium (100,000 g for at least 18 h) for functional assays. For biochemical and molecular studies, small EV were suspended in PBS and re‐centrifuged at 100,000 g for 90 min. All steps were carried out at 4°C. Using this protocol, EV characterization revealed the obtained vesicles presented the expected morphological and biochemical characteristics of small EV (Lazar et al, 2016), in accordance with MISEV (Minimal information for studies of extracellular vesicles) 2018 (Thery et al, 2018).
We previously referred to the population of EV that we study as exosomes (Lazar et al, 2016), since we use a high‐speed spin to isolate small EV, which are enriched in this type of vesicle (Thery et al, 2006). However, due to guidelines from the International Society for Extracellular Vesicles (Thery et al, 2018), we will refer to this same population simply as “EV” in the present study. EV number and size distribution were analyzed by Nanoparticle Tracking Analysis (NTA) with a NanoSight LM10‐HS (Malvern, Orsay, France), equipped with a 405 nm laser as previously described (Lazar et al, 2015).
Stable isotope labeling with amino acids in cell culture
3T3‐F442A preadipocytes were grown and differentiated in a modified DMEM designed for isotope labeling in cell culture (Sigma Aldrich; Saint Louis, MO, USA) supplemented with 10% dialyzed FCS (GIBCO‐Thermo Fisher Scientific, Eugene, OR, USA), 125 mg/ml streptomycin, 125 UI/ml penicillin, and 50 nM of insulin. The following amino acids were also added to the culture medium (200 mg/l of each): l‐lysine:2HCl (13C6), l‐arginine:HCl (13C6) (both purchased from Cambridge Isotope laboratories Inc, Tewksbury, MA, USA), l‐proline, and l‐leucine (Sigma). At maturation, adipocytes were incubated in ultracentrifugated DMEM for 72 h before collecting conditioned medium for EV isolation. SKMEL28 cells were incubated with these EV for 12 h. Labeled EV and cells treated or not with these EV were lysed in PBS containing 1% SDS, and protein concentration was determined using the DC™ Protein Assay from Bio‐Rad. Samples were then processed and analyzed using nano‐LC‐MS/MS, as described in Appendix Supplementary Methods.
Comparative proteomic analysis of EV from lean and obese adipocytes
EV released by adipocytes from lean or obese mice were isolated and purified as described earlier. EV were lysed in PBS containing 1% SDS, and protein concentration was determined using the DC™ Protein Assay from Bio‐Rad. Samples were then processed and analyzed using nano‐LC‐MS/MS, as described in Appendix Supplementary Methods. Three ND and four HFD independent EV preparations were analyzed with technical duplicates for each.
BODIPY, rhodamine phalloidin, and DAPI staining in fixed cells
Cells were seeded on glass coverslips and, after the indicated treatments, were fixed with 3.7% paraformaldehyde for 15 min at room temperature and permeabilized with 0.2% Triton X‐100 for 5 min. Then, cells were incubated with BODIPY (used at 1 μg/ml) for 15 min, the rhodamine phalloidin probe (used at 6.6 μM) for 1 h and/or DAPI (used at 1 μM) for 5 min. Coverslips were mounted using VECTASHIELD® Antifade Mounting Medium (Vector Laboratories, Burlingame, CA, USA).
BODIPY, MitoTracker, and LysoTracker staining in live cells
Cells were seeded in Lab‐Tek® chamber slides, or glass coverslips for analysis of mitochondrial redistribution, and treated as indicated. Cells were then washed twice with pre‐warmed PBS (with 1% BSA for BODIPY and LysoTracker staining) and incubated with BODIPY (1 μg/ml in culture medium with no FCS, 15 min), MitoTracker Red CMXRos (75 nM in PBS, for 45 min), or LysoTracker Deep Red (50 nM in PBS with 1% BSA, for 1 h) at 37°C. Cells were then washed three times with pre‐warmed PBS (with 1% BSA after BODIPY and LysoTracker staining) then fixed (for analysis of mitochondrial redistribution) or incubated in pre‐warmed medium with appropriate treatments for analyses in live cells.
FA extraction and dosage
For lipid extraction, 25 × 1010 EV were resuspended in 200 μl of PBS, and then, 1.5 ml of methanol was added and samples were vortexed. Five ml of Tert‐Butyl methyl ether (MTBE) was added before vortexing again, and then, samples were incubated for 1 h at room temperature under mixing. Finally, 1.2 ml of water was added to separate aqueous and organic phases. The organic phase was evaporated under liquid nitrogen, and lipids were resuspended in 50 μl of fatty acid assay buffer (Free Fatty Acid Assay Kit, Abcam, Cambridge, MA, USA). FA (contained in 5 μl of buffer) were dosed using this kit, as indicated by the supplier.
FA transfer by 3T3‐F442A adipocyte EV
3T3‐F442A mature adipocytes were washed twice with pre‐warmed PBS with 0.1% BSA, then incubated with BODIPY FL C16 at 5 μM in DMEM with no FCS and with 0.1% BSA and 50 nM insulin for 6 h. Adipocytes were washed twice with pre‐warmed PBS with 0.1% BSA, then incubated in ultracentrifugated DMEM with 50 nM insulin for 72 h before collecting conditioned medium. EV were isolated from the medium as described above and then incubated with unstained melanoma cells.
Measurement of FAO
Cells, treated or not with adipocyte EV, were incubated for 3 h with 1 ml of warmed (37°C), modified Krebs Ringer buffer pH 7.4 containing 1.5% FA‐free BSA, 5 mM glucose, 1 mM palmitate (Sigma Aldrich), and 0.5 μCi/ml 14C‐palmitate (PerkinElmer, Waltham, MA, USA) as previously described (Wang et al, 2017). Following this treatment, 1 ml of incubation media was transferred to a glass tube and a microtube containing 300 μl of NaOH 1N was placed in the vial to capture the 14CO2. The buffer was acidified with 1 ml of 1 M H2SO4, and the vial was sealed and left overnight before counting the radioactivity (CytoScint, MP Biomedicals, Illkirch Graffenstaden, France). Cells were lysed and protein concentration was determined using the DC™ Protein Assay from Bio‐Rad (Hercules, CA, USA) for data normalization.
RNA extraction and RT–qPCR analysis
RNA extraction, reverse transcription, and quantitative PCR (RT–qPCR) were conducted as previously described (Steunou et al, 2013). Results were normalized to HPRT and GAPDH mRNA. All the primers used in this study are presented in the following table.
| Genes | Species | Primer forward | Primer reverse |
|---|---|---|---|
| HADHA | Human | 5′ AAGGTAGAGCCCACTGCTCA | 5′ GGCAAAGATGCTGACACAGA |
| HADH | Human | 5′ AGTTCGGCAAGAAGACTGGA | 5′ TGTGAGGGAATGTCTGACCA |
| CPT1A | Human | 5′ CGTCTTTTGGGATCCACGATT | 5′ TGTGCTGGATGGTGTCTGTCTC |
| GAPDH | Human | 5′ TGCACCACCAACTGCTTAGC | 5′ GGCATGGACTGTGGTCATGAG |
| HPRT | Human | 5′ TGGCCATCTGCCTAGTAAAGC | 5′ GGACGCAGCAACTGACATTTC |
Western blotting analysis
Cells or EV were lysed in PBS containing 1% SDS, and protein concentration was determined using the DC™ Protein Assay from Bio‐Rad. 2 μg of proteins was electrophoresed on SDS–PAGE. WB analysis was performed as previously described (Lazar et al, 2015).
Confocal microscopy
A confocal microscope FV‐1000 was used to observe cells with a 60× oil PLAPON OSC objective (Olympus, Hamburg, Germany). Images were processed to filter the noise with ImageJ software (National Institute of Health, Bethesda, MD), and a similar filter was used to analyze all acquisitions for the experiment. For the lipid accumulation quantification, total lipid droplet area per cell was quantified. For analysis of intracellular distribution of organelles, the presence of lipid droplets or lysosomes was evaluated in different subcellular regions. Both analyses were conducted using ImageJ software, and for each independent experiment, at least four images were analyzed to obtain a mean value. Graphs represent pooled means from at least three independent experiments. The distance between nuclei and mitochondria was measured using ImageJ software after segmentation of the objects of interest. To note, perinuclear mitochondria were excluded from measurements. At least 15 cells were analyzed per condition, equating to over 300 mitochondria.
Migration assays
Equal numbers of cells (20 × 104) in serum‐free DMEM were added to the upper compartments of Transwell chambers (ThinCerts®, 12 wells, 8 μm pores, Greiner Bio‐One). DMEM supplemented with 10% FCS was added to the bottom chamber as a chemoattractant. Cells were then incubated at 37°C for 12 and 24 h for 1205Lu and SKMEL28 cells, respectively. Migrating cells were evaluated as previously described (Lazar et al, 2015).
Video microscopy and analysis
Images of cells were acquired every 30 min over 24 h with an automated Leica DMIRB microscope equipped with a CoolSnap Ez camera (Roper Scientific, Lisses, France) with a 10× objective, programmed using MetaMorph software (Molecular Devices, San José, CA, USA). All images were processed using the same filter to reduce noise with ImageJ software. Then, cells were tracked in time‐lapse image stacks using the Manual Tracking plug‐in in ImageJ to determine velocity.
Transmission electron microscopy
Specimens were prepared as previously described (Lazar et al, 2015). Grids were examined with a transmission electron microscope (Jeol JEM‐1400, Akishima, Tokyo, Japan) at 80 kV. Lipid droplet area was measured on these images using ImageJ software.
The Cancer Genome Atlas analysis
Analysis for the correlations between overall survival (OS) and mRNA expression of indicated genes was performed using the cBioPortal for Cancer Genomics Web site, http://www.cbioportal.org/public-portal/. The data set “Skin Cutaneous Melanoma (TCGA, Provisional)”, containing a total of 479 samples, was used to analyze the RNA Seq V2 RSEM z‐score for each gene, which represents a normalized relative expression level. The z‐score cutoff used was a 2‐fold increase in mRNA expression. Data were downloaded, and association of OS with increased gene expression was represented using Kaplan–Meier survival analysis, and significance was assessed by Log‐rank (Mantel‐Cox) test with Prism 5 software. Data were accessed from the cBioPortal in September 2018.
Statistical analysis
Values are means ± standard error of the mean. The statistical significance of differences between the means (of at least three independent assays) was evaluated using Student's t‐tests, if two groups are compared, or one‐way ANOVA, if more than two groups are compared, with the indicated associated post hoc tests using GraphPad Prism software. To determine the appropriate post hoc test to apply, normality of samples was determined using a Kolmogorov–Smirnov test. P values below 0.05 (*), < 0.01 (**), and < 0.001 (***) were deemed as significant, ns: non‐significant. Spearman's rank correlation was used to evaluate the correlation between FA content in EV and BMI.
Author contributions
EC and IL performed most of the experiments, with the help of CA for metabolic studies, LC for lipophagy experiments, MM and SLG for AT collection, DE for immunofluorescence, and SDau for confocal and videomicroscopy experiments. MD‐P and TM performed the proteomic studies under the supervision of OB‐S. EC, IL, LN, and CM analyzed the data with the help of SDal, PV, and OB‐S. EC, IL, and LN conceived the idea for this project and wrote the manuscript with significant inputs from all authors. LN supervised the study.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Source Data for Appendix
Review Process File
Source Data for Figure 2
Source Data for Figure 5
Acknowledgements
The authors thank Drs Lionel Larue and Gilles Favre for donating melanoma lines and Pascale Belenguer for insights and guidance on experiments relating to mitochondrial dynamics. Studies were supported by the “Ligue Régionale Midi‐Pyrénées contre le Cancer”, “Fondation ARC pour la recherche sur le cancer”, and “Société Française de Dermatologie” to LN and CM. The work was also supported by the Région Midi‐Pyrénées, European Found (FEDER), and “Toulouse Métropole”, and the French Ministry of Research with the program “Investment for the Future, National Infrastructures for Biology and Health” (ProFI, Proteomics French Infrastructure project, ANR 10‐INBS‐08) to OBS. We thank “Région Midi‐Pyrénées” for supporting the “Toulouse Réseaux Imagerie” platform. This work benefited of the assistance of the Multiscale Electron Imaging platform (METi) of the FRBT (Federation de Recherche en Biologie de Toulouse). EC is a recipient of a PhD fellowship from “Ligue nationale contre le Cancer”, and EC and IL are recipients of PhD fellowships from “Fondation ARC pour la recherche sur le cancer”. CA benefits from a grant from “Fondation de France” (00081132). We thank Life Science Editors for editing assistance.
The EMBO Journal (2020) 39: e102525
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez‐Riverol et al, 2019) partner repository with the dataset identifier PXD014487 (http://www.ebi.ac.uk/pride/archive/projects/PXD014487).
References
- Altieri DC (2017) Mitochondria on the move: emerging paradigms of organelle trafficking in tumour plasticity and metastasis. Br J Cancer 117: 301–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amjadi F, Mehdipoor R, Zarkesh‐Esfahani H, Javanmard SH (2016) Leptin serves as angiogenic/mitogenic factor in melanoma tumor growth. Adv Biomed Res 5: 127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andarawewa KL, Motrescu ER, Chenard MP, Gansmuller A, Stoll I, Tomasetto C, Rio MC (2005) Stromelysin‐3 is a potent negative regulator of adipogenesis participating to cancer cell‐adipocyte interaction/crosstalk at the tumor invasive front. Can Res 65: 10862–10871 [DOI] [PubMed] [Google Scholar]
- Balaban S, Shearer RF, Lee LS, van Geldermalsen M, Schreuder M, Shtein HC, Cairns R, Thomas KC, Fazakerley DJ, Grewal T et al (2017) Adipocyte lipolysis links obesity to breast cancer growth: adipocyte‐derived fatty acids drive breast cancer cell proliferation and migration. Cancer Metab 5: 1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey TE, Takahashi T, Resnick LA, Oettgen HF, Old LJ (1976) Cell surface antigens of human malignant melanoma: mixed hemadsorption assays for humoral immunity to cultured autologous melanoma cells. Proc Natl Acad Sci USA 73: 3278–3282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carracedo A, Cantley LC, Pandolfi PP (2013) Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer 13: 227–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement E, Lazar I, Muller C, Nieto L (2017) Obesity and melanoma: could fat be fueling malignancy? Pigment Cell Melanoma Res 30: 294–306 [DOI] [PubMed] [Google Scholar]
- Crewe C, Joffin N, Rutkowski JM, Kim M, Zhang F, Towler DA, Gordillo R, Scherer PE (2018) An endothelial‐to‐adipocyte extracellular vesicle axis governed by metabolic state. Cell 175: 695–708 e613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunniff B, McKenzie AJ, Heintz NH, Howe AK (2016) AMPK activity regulates trafficking of mitochondria to the leading edge during cell migration and matrix invasion. Mol Biol Cell 27: 2662–2674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Pergola G, Silvestris F (2013) Obesity as a major risk factor for cancer. J Obes 2013: 291546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai SP, Bhatia SN, Toner M, Irimia D (2013) Mitochondrial localization and the persistent migration of epithelial cancer cells. Biophys J 104: 2077–2088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirat B, Bochet L, Dabek M, Daviaud D, Dauvillier S, Majed B, Wang YY, Meulle A, Salles B, Le Gonidec S et al (2011) Cancer‐associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Can Res 71: 2455–2465 [DOI] [PubMed] [Google Scholar]
- Djian P, Phillips M, Green H (1985) The activation of specific gene transcription in the adipose conversion of 3T3 cells. J Cell Physiol 124: 554–556 [DOI] [PubMed] [Google Scholar]
- Dobbins M, Decorby K, Choi BC (2013) The association between obesity and cancer risk: a meta‐analysis of observational studies from 1985 to 2011. ISRN Prev Med 2013: 680536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour E, Carcelain G, Gaudin C, Flament C, Avril MF, Faure F (1997) Diversity of the cytotoxic melanoma‐specific immune response: some CTL clones recognize autologous fresh tumor cells and not tumor cell lines. J Immunol 158: 3787–3795 [PubMed] [Google Scholar]
- Duncan RE, Ahmadian M, Jaworski K, Sarkadi‐Nagy E, Sul HS (2007) Regulation of lipolysis in adipocytes. Annu Rev Nutr 27: 79–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duong MN, Geneste A, Fallone F, Li X, Dumontet C, Muller C (2017) The fat and the bad: mature adipocytes, key actors in tumor progression and resistance. Oncotarget 8: 57622–57641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty SE III, Grijalva A, Xu X, Ables E, Nomani A, Ferrante AW Jr (2019) A lipase‐independent pathway of lipid release and immune modulation by adipocytes. Science 363: 989–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frayn KN (2001) Adipose tissue and the insulin resistance syndrome. Proc Nutr Soc 60: 375–380 [DOI] [PubMed] [Google Scholar]
- Gallagher EJ, LeRoith D (2015) Obesity and diabetes: the increased risk of cancer and cancer‐related mortality. Physiol Rev 95: 727–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallus S, Naldi L, Martin L, Martinelli M, La Vecchia C (2006) Anthropometric measures and risk of cutaneous malignant melanoma: a case‐control study from Italy. Melanoma Res 16: 83–87 [DOI] [PubMed] [Google Scholar]
- Giampietri C, Petrungaro S, Cordella M, Tabolacci C, Tomaipitinca L, Facchiano A, Eramo A, Filippini A, Facchiano F, Ziparo E (2017) Lipid storage and autophagy in melanoma cancer cells. Int J Mol Sci 18: E1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Giorgi V, Gori A, Papi F, Grazzini M, Rossari S, Verdelli A, Crocetti E, Savarese I, Giorgi L, Massi D (2013) Excess body weight and increased Breslow thickness in melanoma patients: a retrospective study. Eur J Cancer Prev 22: 480–485 [DOI] [PubMed] [Google Scholar]
- Goodall J, Carreira S, Denat L, Kobi D, Davidson I, Nuciforo P, Sturm RA, Larue L, Goding CR (2008) Brn‐2 represses microphthalmia‐associated transcription factor expression and marks a distinct subpopulation of microphthalmia‐associated transcription factor‐negative melanoma cells. Can Res 68: 7788–7794 [DOI] [PubMed] [Google Scholar]
- Gouirand V, Guillaumond F, Vasseur S (2018) Influence of the tumor microenvironment on cancer cells metabolic reprogramming. Front Oncol 8: 117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton J, Jones I, Srivastava R, Galloway P (2012) A new method for the measurement of lysosomal acid lipase in dried blood spots using the inhibitor Lalistat 2. Clin Chim Acta 413: 1207–1210 [DOI] [PubMed] [Google Scholar]
- Herms A, Bosch M, Reddy BJ, Schieber NL, Fajardo A, Ruperez C, Fernandez‐Vidal A, Ferguson C, Rentero C, Tebar F et al (2015) AMPK activation promotes lipid droplet dispersion on detyrosinated microtubules to increase mitochondrial fatty acid oxidation. Nat Commun 6: 7176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MJ, Metcalfe D, McTernan PG (2009) Obesity and diabetes: lipids, ‘nowhere to run to’. Clin Sci (Lond) 116: 113–123 [DOI] [PubMed] [Google Scholar]
- Jeon SM (2016) Regulation and function of AMPK in physiology and diseases. Exp Mol Med 48: e245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung JI, Cho HJ, Jung YJ, Kwon SH, Her S, Choi SS, Shin SH, Lee KW, Park JH (2015) High‐fat diet‐induced obesity increases lymphangiogenesis and lymph node metastasis in the B16F10 melanoma allograft model: roles of adipocytes and M2‐macrophages. Int J Cancer 136: 258–270 [DOI] [PubMed] [Google Scholar]
- Kath R, Jambrosic JA, Holland L, Rodeck U, Herlyn M (1991) Development of invasive and growth factor‐independent cell variants from primary human melanomas. Can Res 51: 2205–2211 [PubMed] [Google Scholar]
- Kowal J, Arras G, Colombo M, Jouve M, Morath JP, Primdal‐Bengtson B, Dingli F, Loew D, Tkach M, Thery C (2016) Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci USA 113: E968–E977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlowski JM, Hart IR, Fidler IJ, Hanna N (1984) A human melanoma line heterogeneous with respect to metastatic capacity in athymic nude mice. J Natl Cancer Inst 72: 913–917 [PubMed] [Google Scholar]
- Kuo CY, Ann DK (2018) When fats commit crimes: fatty acid metabolism, cancer stemness and therapeutic resistance. Cancer Commun (Lond) 38: 47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmicic J, Parra V, Verdejo HE, Lopez‐Crisosto C, Chiong M, Garcia L, Jensen MD, Bernlohr DA, Castro PF, Lavandero S (2014) Trimetazidine prevents palmitate‐induced mitochondrial fission and dysfunction in cultured cardiomyocytes. Biochem Pharmacol 91: 323–336 [DOI] [PubMed] [Google Scholar]
- Kwan HY, Fu X, Liu B, Chao X, Chan CL, Cao H, Su T, Tse AK, Fong WF, Yu ZL (2014) Subcutaneous adipocytes promote melanoma cell growth by activating the Akt signaling pathway: role of palmitic acid. J Biol Chem 289: 30525–30537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lass A, Zimmermann R, Oberer M, Zechner R (2011) Lipolysis – a highly regulated multi‐enzyme complex mediates the catabolism of cellular fat stores. Prog Lipid Res 50: 14–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent V, Toulet A, Attane C, Milhas D, Dauvillier S, Zaidi F, Clement E, Cinato M, Le Gonidec S, Guerard A et al (2019) Periprostatic adipose tissue favors prostate cancer cell invasion in an obesity‐dependent manner: role of oxidative stress. Mol Cancer Res 17: 821–835 [DOI] [PubMed] [Google Scholar]
- Lazar I, Clement E, Ducoux‐Petit M, Denat L, Soldan V, Dauvillier S, Balor S, Burlet‐Schiltz O, Larue L, Muller C et al (2015) Proteome characterization of melanoma exosomes reveals a specific signature for metastatic cell lines. Pigment Cell Melanoma Res 28: 464–475 [DOI] [PubMed] [Google Scholar]
- Lazar I, Clement E, Dauvillier S, Milhas D, Ducoux‐Petit M, LeGonidec S, Moro C, Soldan V, Dalle S, Balor S et al (2016) Adipocyte exosomes promote melanoma aggressiveness through fatty acid oxidation: a novel mechanism linking obesity and cancer. Can Res 76: 4051–4057 [DOI] [PubMed] [Google Scholar]
- Listenberger LL, Han X, Lewis SE, Cases S, Farese RV Jr, Ory DS, Schaffer JE (2003) Triglyceride accumulation protects against fatty acid‐induced lipotoxicity. Proc Natl Acad Sci USA 100: 3077–3082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDougald OA, Lane MD (1995) Transcriptional regulation of gene expression during adipocyte differentiation. Annu Rev Biochem 64: 345–373 [DOI] [PubMed] [Google Scholar]
- Malvi P, Chaube B, Singh SV, Mohammad N, Pandey V, Vijayakumar MV, Radhakrishnan RM, Vanuopadath M, Nair SS, Nair BG et al (2016) Weight control interventions improve therapeutic efficacy of dacarbazine in melanoma by reversing obesity‐induced drug resistance. Cancer Metab 4: 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuade JL, Daniel CR, Hess KR, Mak C, Wang DY, Rai RR, Park JJ, Haydu LE, Spencer C, Wongchenko M et al (2018) Association of body‐mass index and outcomes in patients with metastatic melanoma treated with targeted therapy, immunotherapy, or chemotherapy: a retrospective, multicohort analysis. Lancet Oncol 19: 310–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore R, Champeval D, Denat L, Tan SS, Faure F, Julien‐Grille S, Larue L (2004) Involvement of cadherins 7 and 20 in mouse embryogenesis and melanocyte transformation. Oncogene 23: 6726–6735 [DOI] [PubMed] [Google Scholar]
- Morales DR, Morris AD (2015) Metformin in cancer treatment and prevention. Annu Rev Med 66: 17–29 [DOI] [PubMed] [Google Scholar]
- van Niel G, D'Angelo G, Raposo G (2018) Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol 19: 213–228 [DOI] [PubMed] [Google Scholar]
- Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell‐Gutbrod R, Zillhardt MR, Romero IL, Carey MS, Mills GB, Hotamisligil GS et al (2011) Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med 17: 1498–1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieman KM, Romero IL, Van Houten B, Lengyel E (2013) Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochem Biophys Acta 1831: 1533–1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oba J, Wei W, Gershenwald JE, Johnson MM, Wyatt CM, Ellerhorst JA, Grimm EA (2016) Elevated serum leptin levels are associated with an increased risk of sentinel lymph node metastasis in cutaneous melanoma. Medicine (Baltimore) 95: e3073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen CM, Green AC, Zens MS, Stukel TA, Bataille V, Berwick M, Elwood JM, Gallagher R, Holly EA, Kirkpatrick C et al (2008) Anthropometric factors and risk of melanoma in women: a pooled analysis. Int J Cancer 122: 1100–1108 [DOI] [PubMed] [Google Scholar]
- Pandey V, Vijayakumar MV, Ajay AK, Malvi P, Bhat MK (2012) Diet‐induced obesity increases melanoma progression: involvement of Cav‐1 and FASN. Int J Cancer 130: 497–508 [DOI] [PubMed] [Google Scholar]
- Park J, Morley TS, Kim M, Clegg DJ, Scherer PE (2014) Obesity and cancer–mechanisms underlying tumour progression and recurrence. Nat Rev Endocrinol 10: 455–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Riverol Y, Csordas A, Bai J, Bernal‐Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M et al (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47: D442–D450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poste G, Doll J, Hart IR, Fidler IJ (1980) In vitro selection of murine B16 melanoma variants with enhanced tissue‐invasive properties. Can Res 40: 1636–1644 [PubMed] [Google Scholar]
- Rambold AS, Cohen S, Lippincott‐Schwartz J (2015) Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev Cell 32: 678–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves GK, Pirie K, Beral V, Green J, Spencer E, Bull D (2007) Cancer incidence and mortality in relation to body mass index in the Million Women Study: cohort study. BMJ 335: 1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renehan AG, Tyson M, Egger M, Heller RF, Zwahlen M (2008) Body‐mass index and incidence of cancer: a systematic review and meta‐analysis of prospective observational studies. Lancet 371: 569–578 [DOI] [PubMed] [Google Scholar]
- Rodrigues MF, Obre E, de Melo FH, Santos GC Jr, Galina A, Jasiulionis MG, Rossignol R, Rumjanek FD, Amoedo ND (2016) Enhanced OXPHOS, glutaminolysis and beta‐oxidation constitute the metastatic phenotype of melanoma cells. Biochem J 473: 703–715 [DOI] [PubMed] [Google Scholar]
- Rosenbaum AI, Cosner CC, Mariani CJ, Maxfield FR, Wiest O, Helquist P (2010) Thiadiazole carbamates: potent inhibitors of lysosomal acid lipase and potential Niemann‐Pick type C disease therapeutics. J Med Chem 53: 5281–5289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha AK, Avilucea PR, Ye JM, Assifi MM, Kraegen EW, Ruderman NB (2004) Pioglitazone treatment activates AMP‐activated protein kinase in rat liver and adipose tissue in vivo . Biochem Biophys Res Comm 314: 580–585 [DOI] [PubMed] [Google Scholar]
- Sergentanis TN, Antoniadis AG, Gogas HJ, Antonopoulos CN, Adami HO, Ekbom A, Petridou ET (2013) Obesity and risk of malignant melanoma: a meta‐analysis of cohort and case‐control studies. Eur J Cancer 49: 642–657 [DOI] [PubMed] [Google Scholar]
- Shah R, Patel T, Freedman JE (2018) Circulating extracellular vesicles in human disease. N Engl J Med 379: 958–966 [DOI] [PubMed] [Google Scholar]
- Shors AR, Solomon C, McTiernan A, White E (2001) Melanoma risk in relation to height, weight, and exercise (United States). Cancer Causes Control 12: 599–606 [DOI] [PubMed] [Google Scholar]
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ (2009) Autophagy regulates lipid metabolism. Nature 458: 1131–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skowron F, Berard F, Balme B, Maucort‐Boulch D (2015) Role of obesity on the thickness of primary cutaneous melanoma. J Eur Acad Dermatol Venereol 29: 262–269 [DOI] [PubMed] [Google Scholar]
- Stenehjem JS, Veierod MB, Nilsen LT, Ghiasvand R, Johnsen B, Grimsrud TK, Babigumira R, Stoer NC, Rees JR, Robsahm TE (2018) Anthropometric factors and Breslow thickness: prospective data on 2570 cases of cutaneous melanoma in the population‐based Janus Cohort. Br J Dermatol 179: 632–641 [DOI] [PubMed] [Google Scholar]
- Steunou AL, Ducoux‐Petit M, Lazar I, Monsarrat B, Erard M, Muller C, Clottes E, Burlet‐Schiltz O, Nieto L (2013) Identification of the hypoxia‐inducible factor 2alpha nuclear interactome in melanoma cells reveals master proteins involved in melanoma development. Mol Cell Proteomics 12: 736–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thery C, Amigorena S, Raposo G, Clayton A (2006) Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol Chapter 3: Unit 3.22 [DOI] [PubMed] [Google Scholar]
- Thery C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, Antoniou A, Arab T, Archer F, Atkin‐Smith GK et al (2018) Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles 7: 1535750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkach M, Thery C (2016) Communication by extracellular vesicles: where we are and where we need to go. Cell 164: 1226–1232 [DOI] [PubMed] [Google Scholar]
- Tontonoz P, Spiegelman BM (2008) Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem 77: 289–312 [DOI] [PubMed] [Google Scholar]
- Verboven K, Wouters K, Gaens K, Hansen D, Bijnen M, Wetzels S, Stehouwer CD, Goossens GH, Schalkwijk CG, Blaak EE et al (2018) Abdominal subcutaneous and visceral adipocyte size, lipolysis and inflammation relate to insulin resistance in male obese humans. Sci Rep 8: 4677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YY, Attane C, Milhas D, Dirat B, Dauvillier S, Guerard A, Gilhodes J, Lazar I, Alet N, Laurent V et al (2017) Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight 2: e87489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Mao X, Mehta M, Cui J, Zhang C, Xu Y (2012) A comparative study of gene‐expression data of basal cell carcinoma and melanoma reveals new insights about the two cancers. PLoS One 7: e30750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Jiang L, Hu W, Zheng Q, Xiang W (2011) Mitochondrial dysfunction during in vitro hepatocyte steatosis is reversed by omega‐3 fatty acid‐induced up‐regulation of mitofusin 2. Metabolism 60: 767–775 [DOI] [PubMed] [Google Scholar]
- Zhang M, Di Martino JS, Bowman RL, Campbell NR, Baksh SC, Simon‐Vermot T, Kim IS, Haldeman P, Mondal C, Yong‐Gonzales V et al (2018) Adipocyte‐derived lipids mediate melanoma progression via FATP proteins. Cancer Discov 8: 1006–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Zhang J, Yu M, Xie Y, Huang Y, Wolff DW, Abel PW, Tu Y (2013) Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 32: 4814–4824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Yang L, Baddour J, Achreja A, Bernard V, Moss T, Marini JC, Tudawe T, Seviour EG, San Lucas FA et al (2016) Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife 5: e10250 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Source Data for Appendix
Review Process File
Source Data for Figure 2
Source Data for Figure 5
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez‐Riverol et al, 2019) partner repository with the dataset identifier PXD014487 (http://www.ebi.ac.uk/pride/archive/projects/PXD014487).