

Abstract
Hindered ethers represent an underexplored area of chemical space due to the difficulty and inoperability associated with conventional reactions, despite the high-value of such structural motifs in a variety of societal applications1–2. For example, such motifs are highly coveted in medicinal chemistry, as extensive substitution about the ether bond prevents unwanted metabolic processes that can lead to rapid in vivo degradation. Demonstrated herein is an exceptionally simple solution to this problem that leverages the power of electrochemical oxidation to liberate high-energy carbocations from simple carboxylic acids. The controlled formation of these reactive intermediates takes place with low electrochemical potentials under non-acidic conditions to capture an alcohol donor thereby producing a range (>80) of ethers that would be extremely difficult to otherwise access. Simple nucleophiles can also intercept such cations, leading to hindered alcohols and even alkyl fluorides. This method has been field tested to solve the synthetic bottlenecks encountered on twelve real-world chemical scaffolds with documented societal impact, resulting in a dramatic reduction in step-count and labor required, accompanied with higher yields. Finally, the use of molecular probes coupled to kinetic studies support the proposed mechanism and role of additives in the conditions employed.
Students of organic chemistry are taught the Williamson ether synthesis3–4 as a classic way to make primary alkyl ethers via SN2 substitution (Fig. 1A), but in contexts involving secondary or tertiary alkyl halides the reaction often derails, leading to elimination byproducts or no reaction at all. Hindered ether 1, a key intermediate for the synthesis of an aurora kinase modulator, exemplifies this commonly-faced challenge. Despite the documented utility of hindered ethers1–2, very little progress has been registered to bridge this gap in reactivity. The alternative workhorse method, the Mitsunobu reaction, also fails in such settings due to steric demands of the SN2 process and pKa requirements of the nucleophile5. To the best of our knowledge, the most often-employed method for the synthesis of hindered dialkyl ether bonds still uses carbocation chemistry accessed from olefins (hydroalkoxylation) under strongly acidic conditions6. While this transformation has been known for nearly a century, its use is drastically limited in scope due to a lack of chemoselectivity and sluggish reactivity7. Indeed, in preparing hindered ether 1, a multi-step route via 4-hydroxyproline 2 (R = CO2Me) was employed, requiring over 6 days of reaction time (<4% overall yield), wherein the key C–O bond-forming reaction—the treatment of methylenecyclobutane with BF3•Et2O in the presence of the requisite secondary alcohol—provides the ether in only 11% yield8.
Figure 1.

(A) The synthesis of hindered ethers is a long-standing challenge in organic synthesis; (B) Historical context and prior strategies for decarboxylative etherification; and (C) Development and optimization of hindered ether synthesis depicted through electromechanistic analysis. aCompound 3 (0.2 mmol), 3.0 eq. alcohol 4 was used (except where designated). bGC yield. All entries performed in triplicate. cConditions: acid 3 (0.2 mmol), alcohol 4 (0.6 mmol), AgPF6 (0.3 mmol), 2,4,6-collidine (0.6 mmol), nBu4NPF6 (0.1 M), 3Å MS (150 mg), CH2Cl2 (3 mL), I = 10 mA, 3 h. dIsolated yield. DMF, N,N-dimethylformamide; DCM, dichloromethane; 3Å MS, 3Å molecular sieves.
Distinct from sterically-sensitive SN2 and strongly acidic carbocation pathways, a third class of ether synthesis has been known for many years, but has remained largely underexplored. This third reaction paradigm (Figure 1A, yellow inset) stems from the oldest synthetic organic electrochemical reaction, the Kolbe dimerization, discovered in 18479. In the so-called interrupted Kolbe variant, known as the Hofer-Moest reaction10, electrolytic oxidation of a carboxylic acid under mildly alkaline conditions generates a carbocation that can be captured by incipient nucleophiles10–18. A distinct advantage of this reactivity paradigm is the non-acidic generation of high energy carbocations directly from ubiquitous carboxylic acids.
Based on this apparent advantage, we surveyed the literature, and were startled to observe an extremely limited application of the Hofer-Moest reaction in synthetic contexts (Figure 1B). Indeed, rather than using this reaction, much more complex catalytic systems that take advantage of photolytic conditions have been developed to make alkyl-aryl ethers19. There are likely two reasons for the limited exploration of electrolytic decarboxylative ether synthesis: First, the barrier to entry for electrosynthesis has traditionally been high for a practicing synthetic organic chemist20. Second, all Hofer-Moest systems known to date required solvent-quantities of the alcoholic nucleophile, which is untenable for complex ethereal substrates. The alcoholic solvent, in addition to functioning as a reagent, permits current to pass and also acts as an electron-sink to balance the electrochemical process (liberating H2 gas).
Herein, we report how the generation of carbocations from unactivated, aliphatic carboxylic acids, and their subsequent capture with heteroatom nucleophiles can be leveraged to provide a wide array of hindered C–X bonds. The reaction exhibits remarkable scope (>100 examples) across a range of valuable substrate classes (including previously inaccessible fluorinated ethers), is easily scalable in an undivided cell using inexpensive graphite electrodes, and has been studied in detail mechanistically.
The development of a practical electrochemical decarboxylative etherification was predicated on the valuable examples that preceded this work. Specifically, we set out to invent a set of Hofer-Moest conditions that were amenable to the needs of an organic chemist working on elaborate substrates (using a practical commercial setup with simple electrodes, mild conditions to tolerate sensitive functionality, and non-solvent quantities of alcohol donor). To achieve these goals, the extensive body of electrochemical literature10–18, 20 around this reaction gave us key clues for initial reaction development. Specifically, it was known that carbon-based electrodes favored the desired carbocation generation, while platinum electrodes favored unproductive radical (Kolbe-type) dimerization21. It was also known that inert, non-oxidizable anions (e.g., ClO4−) enhance cation-like reactivity in the Hofer-Moest reaction22. Third, literature precedent showed that mildly alkaline conditions would be beneficial for the desired carbocation formation22,23. Finally, simple undivided cells are generally used in this process, suggesting that cathodic reduction would not significantly interfere with the reaction.
It became immediately apparent that limiting the amount of alcohol posed a number of significant challenges: decomposition of carbocation due to the low nucleophilicity of alcohols, competitive trapping of carbocation by water, consumption of alcohols by anodic oxidation and necessity of external electron-acceptor to balance electrons. Figure 1C summarizes the results of ca. 1000 experiments (see Supplementary Information for an extensive sampling) to solve all of these problems. Not surprisingly, initial exploratory experiments using the proposed conversion of 3 and 4 to 5 as an example, based on the literature precedent available (vide supra)11,22, led to only trace amounts of product (entry 1). The conversion of the carboxylate was improved by choosing K2CO3 or 2,4,6-collidine as a non-oxidizable base (entries 2,3), though 6 and 7 were identified as major byproducts due to the presence of radical intermediate22 and elimination pathways, respectively. These problems were effectively suppressed by changing solvents to CH2Cl2 (entry 4), resulting in the large increase of the desired ether product. In CH2Cl2, hydration of the carbocation (leading to 8) persisted (entry 4), which was suppressed with the addition of 3Å molecular sieves (entry 5). It was also found that CH2Cl2 was apparently reduced at the cathode (see Figure S2 of Supplementary Information), acting as an electron-sink. Past approaches using water or simple alcohols as solvent did not need to address this issue, as the solvent can serve as the reagent and an electron sink (via proton reduction). Accordingly, adding a better sacrificial oxidant (AgPF6) significantly improved the yield of 5, leading to our optimized conditions (entry 6). Addition of AgPF6 also completely suppressed the formation of 6 and 7, though this effect varies by substrate and, in some cases (vide infra), silver additives are not necessary at all. Negative controls confirmed the necessity of a slight stoichiometric excess of the alcoholic partner (entry 7). In the absence of base, no desired product was observed, with the major products being the ketone 9 and the ester 10 (entry 8).
The patent literature is replete with examples of hindered ethers of use in a variety of pharmaceutical and materials applications. While carbocation-based routes from olefins predominate in literature, electrochemical access to such compounds has notable advantages on the time, step count and overall yield to access such valuable entities. Illustrated in Table 1 is an abbreviated depiction of six such applications as well as the 80+ ethers prepared (see Supplementary Information for full listing of substrates as well as comparison to prior routes). Primary carboxylic acids and certain secondary systems are not compatible with electrochemical etherification, because the resultant carbocations are not sufficiently stable. However, this limitation is inconsequential from a synthetic perspective, as those ethers can be easily prepared through standard SN2-type approaches3–5. Acids bearing a variety of functional groups are tolerated such as Boc-protected amine (17), aryl and alkyl halides (25, 27, 30), olefins (26), esters (29, 39), enones (39), ethers (35, 36, 62), and even oxidation-prone boronic esters (28). Similarly, the scope of alcohol coupling partners is vast and includes acid-labile and oxidation-prone chiral secondary benzylic alcohols (17), deuterated systems (49), azetidinyl (50), protected sugars (53), olefin-containing (48, 51, 59), acetals and esters (44), halides (15), nitriles (45), nitro groups (see Supplementary Information), and even Lewis-basic heterocycles (42, 43, 45). The ability to tolerate chiral, ionizable secondary alcohols is worth emphasizing, as acidic methods for ether synthesis would lead to elimination or racemization (17 is formed in 95% ee). Electrogenerated cations bearing fluorine atoms can also be intercepted, opening up a range of new organofluorine-containing ether systems that were either difficult to access or unknown (see Supplementary Information for full listing of fluorinated ethers). Finally, the synthesis of polyethyleneglycol (PEG) ethers, historically laborious, can be modularly produced through this process (no PEG ethers analogous to 66–69 are known). To ensure the robustness of the process, ten randomly selected examples in Table 1 were run in triplicate and exhibited no more than 5% yield variance between runs.
Table 1. Applications, and partial scope of hindered ether synthesis via electrochemical decarboxylation (see Supplementary Information for full scope).
aAgSbF6 (0.3 mmol) instead of AgPF6. bDBU (0.6 mmol) instead of 2,4,6-collidine. cKSbF6 (0.3 mmol) instead of AgPF6. dAlcohol as limiting reagent, conditions: alcohol (0.15 mmol), carboxylic acid (0.45 mmol), AgClO4 (0.6 mmol), 2,4,6-collidine (0.675 mmol), nBu4NClO4 (0.2 M), 3Å MS (100 mg), CH2Cl2 (2 mL), I = 10 mA, 3 h. eAgClO4 (0.6 mmol) instead of AgPF6, nBu4NClO4 (0.1 M) instead of nBu4NPF6. f4.0 or 6.0 eq. alcohol. g1.5 mL CH2Cl2, I = 7.5 mA, 4 h. hnBu4NClO4 (0.1 M) instead of nBu4NPF6, no AgPF6. iReaction performed in triplicate; yield is average of three runs.
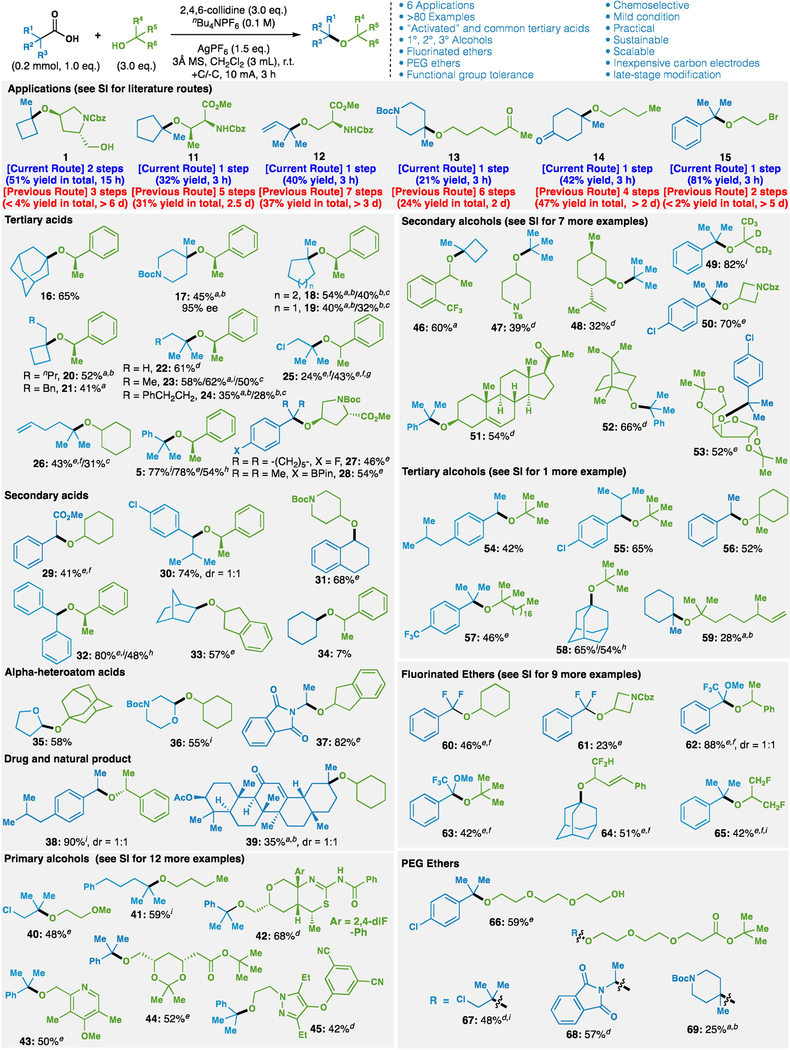
|
As mentioned at the outset, the use of simple alcohols such as methanol and water itself is already known in electrochemical decarboxylative processes (Figure 1B)10–18. However, the scope of such processes is quite limited. The conditions developed above were therefore adapted for these related reactions (see Supplementary Information). In order to render this reaction general, the choice of electrolyte (nBu4NPF6) and base (2,4,6-collidine) was crucial while silver additives were found to be unnecessary. As with hindered ether synthesis, the most convincing case for the use of this reaction stems from its ability to dramatically truncate synthetic pathways. Six such examples are illustrated in Table 2 (see Supplementary Information).
Table 2. Applications and partial scope of trapping electrogenerated carbocations with other nucleophiles along with scalability demonstration (see Supplementary Information for full scope).
aH2O (0.1 mL) as nucleophile. bCarboxylic acid or phenylacetonitrile as nucleophile (0.6 mmol, 3 eq.), conditions: AgClO4 (0.6 mmol, 3 eq.), 2,4,6-collidine (0.6 mmol, 3 eq.), nBu4NClO4 (0.1 M), 3Å MS (150 mg), CH2Cl2 (3 mL). cKF (0.72 mmol, 3.6 eq.) as nucleophile, conditions: 18-Crown-6 (0.72 mmol, 3.6 eq), AgClO4 (0.6 mmol, 3 eq.), 2,4,6-collidine (0.6 mmol, 3 eq.), nBu4NPF6 (0.1 M), 3Å MS (150 mg), CH2Cl2 (3 mL).
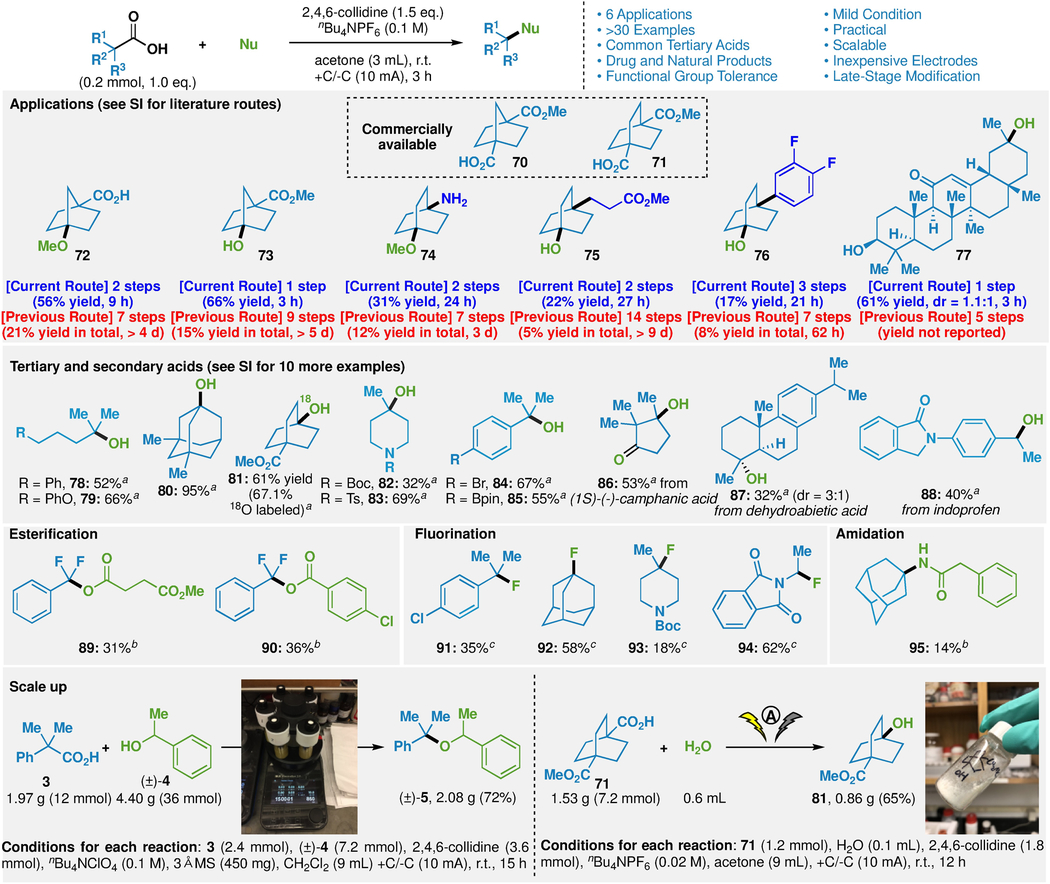
|
The addition of water to generate various tertiary and secondary alcohols is a broadly applicable process with selected examples depicted in Table 2 (for additional examples, see Supplementary Information). Again, the chemoselectivity is on par with that observed above; aryl bromides (84), boronic esters (85), electron rich aromatics (87), lactams (88), ethers (79), and esters (81) are tolerated. In preliminary studies, the potential of adding other nucleophiles to the putative electrogenerated carbocations was also studied. Carboxylates not capable of decarboxylation under these conditions could act as nucleophiles, to provide hindered esters (89 and 90)24. Useful organofluorine building blocks could also be accessed (91-94) in a process that might be of use in radiolabeling studies, since the fluorine source utilized (inexpensive KF) is preferred in such situations25. Finally, using benzonitrile as a nucleophile led to the expected Ritter-type product (95), albeit in lower yield26. The robust and practical nature of this process was also demonstrated with the gram-scale preparation of 5 and 81 through etherification and hydroxylation processes. In the case of large-scale etherification, the silver salt additive could be left out with a minimal effect on yield (72 vs 78% yield).
Extensive mechanistic studies were also undertaken to understand the role of additives and the nature of the reactive intermediate. In summary, the mechanism is likely to be the rate-limiting oxidation of a carboxylate on the anode to generate a carbocation, followed by nucleophilic attack by an alcohol to afford the ether product (See Supplementary Information for full details).
It is anticipated that the mild electrogeneration of carbocations reported herein will find use in numerous settings where standard SN2 and carbocation-based approaches for forming hindered functionalized carbogenic frameworks fail.
METHODS
Here we describe a typical procedure for the decarboxylative etherification, and further experimental details are provided in Supplementary Information.
General procedure for decarboxylative etherification.
With no precautions to exclude air or moisture, the ElectraSyn vial (5 mL) with a stir bar was charged with carboxylic acid (0.2 mmol, 1.0 equiv.), alcohol (0.6 mmol, 3.0 equiv.), 2,4,6-collidine (0.6 mmol, 3.0 equiv.), nBu4NPF6 (0.3 mmol, 1.5 equiv.), 3 Å molecular sieves (150 mg), AgPF6 (0.3 mmol, 1.5 equiv.), and CH2Cl2 (3.0 mL). The ElectraSyn vial cap equipped with anode (graphite) and cathode (graphite) were inserted into the mixture. After pre-stirring for 15 minutes, the reaction mixture was electrolyzed at a constant current of 10 mA for 3 hours. The ElectraSyn vial cap was removed, and electrodes were rinsed with Et2O (2 mL), which was combined with the crude mixture. Then, the crude mixture was further diluted with Et2O (30 mL). The resulting mixture was washed with 2N HCl (20 mL) and NaHCO3 (aq) (20 mL), dried over Na2SO4, and concentrated in vacuo. The crude material was purified by preparative thin-layer chromatography (PTLC) to furnish the desired product. Full experimental details and characterization of new compounds can be found in the Supplementary Information.
Data availability.
The data that support the findings of this study are available within the paper and its Supplementary Information. Metrical parameters for the structures of (2R)-77 and (11R)-138 are available free of charge from the Cambridge Crystallographic Data Centre (https://www.ccdc.cam.ac.uk/) under reference numbers 1918528 and 1903823, respectively.
Supplementary Material
Acknowledgments
Financial support for this work was provided by Pfizer, Inc., NSF (CCI Phase 1 grant 1740656), and the National Institutes of Health (grant number GM-118176). China Scholarship Council and Jilin University supported a fellowship to J.X., Zhejiang Yuanhong Medicine Technology Co. Ltd. supported a fellowship to M.S., The Hewitt Foundation supported a fellowship to Y.K., The Swedish Research Council supported a fellowship to H.L., Fulbright Fellowship supported a fellowship to P.M., S.H.R. acknowledges an NSF GRFP (#2017237151) and a Donald and Delia Baxter Fellowship. We thank D.-H. Huang and L. Pasternack (Scripps Research) for assistance with NMR spectroscopy; J. Chen (Automated Synthesis Facility, Scripps Research); A. Rheingold, C. E. Moore, and M. A. Galella (UCSD) for x-ray crystallographic analysis.
Footnotes
Supplementary information is available in the online version of the paper.
Competing interests The authors declare no competing interests.
Readers are welcome to comment on the online version of the paper.
References:
- 1.Roughley SD & Jordan AM The medicinal chemist’s toolbox: An analysis of reactions ssed in the pursuit of drug candidates. J. Med. Chem 54, 3451–3479 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Fischer J & Ganellin CR Analogue-based drug discovery, pp 206–217 (Wiley, 2006). [Google Scholar]
- 3.Williamson W Ueber die theorie der aetherbildung. Justus Liebigs Ann. Chem 77, 37–49 (1851) [Google Scholar]
- 4.Kürti L & Czakó B Strategic applications of named reactions in organic synthesis, pp 484–485. (Elsevier, 2005). [Google Scholar]
- 5.Swamy KCK, Kumar NNB, Balaraman E & Kumar KVPP Mitsunobu and related reactions: Advances and applications. Chem. Rev 109, 2551–2651 (2009). [DOI] [PubMed] [Google Scholar]
- 6.Beyerman HC & Heiszwolf GJ Reaction of steroidal alcohols with isobutene. Usefulness of t-butyl as a hydroxyl-protecting group in a synthesis of testosterone. Rec. Trav. Chim. Pays-Bas 84, 203–212 (1965). [Google Scholar]
- 7.Smith MB & March J March’s advanced organic chemistry, pp 1037–1041 (Wiley, 2007). [Google Scholar]
- 8.Abraham S et al. Aurora kinase compounds and methods of their use. WO2011088045 A1 (2011).
- 9.Kolbe H Beobachtungen über die oxydirende Wirkung des Sauerstoffs, wenn derselbe mit Hülfe einer elektrischen Säule entwickelt wird. J. Prakt. Chem 41, 137–139 (1847). [Google Scholar]
- 10.Hofer H & Moest M Mittheilung aus dem elektrochemischen Laboratorium der Königl, Technischen Hochschule zu München. Justus Liebigs Ann. Chem 323, 284–323 (1902). [Google Scholar]
- 11.Corey EJ, Bauld NL, La Londe RT, Casanova J Jr & Kaiser ET Generation of cationic carbon by anodic oxidation of carboxylic acids. J. Am. Chem. Soc 82, 2645–2646 (1960). [Google Scholar]
- 12.Luo X, Ma X, Lebreux F, Markó IE & Lam K Electrochemical methoxymethylation of alcohols – A new, green and safe approach for the preparation of MOM ethers and other acetals. Chem. Commun 54, 9969–9972 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Bunyan PJ & Hey DH The electrolysis of some aryl-substituted, aliphatic acids. J. Chem. Soc 1360–1365 (1962). [Google Scholar]
- 14.Iwasaki T, Hrorikawa H, Matsumoto K & Miyoshi M Electrochemical synthesis and reactivity of α-alkoxy α-amino acid derivatives. Bull. Chem. Soc. Japan 52, 826–830 (1979). [Google Scholar]
- 15.Tajima T, Kurihara H & Fuchigami T Development of an electrolytic system for non-Kolbe electrolysis based on the acid−base reaction between carboxylic acids as a substrate and solid-supported bases. J. Am. Chem. Soc 129, 6680–6681 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Shtelman AV & Becker JY Electrochemical synthesis of 1,2-disilylethanes from α-silylacetic acids. J. Org. Chem 76, 4710–4714 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Torii S, Inokuchi T, Mizuguchi K & Yamazaki M Electrolytic decarboxylation reactions. 4. Electrosyntheses of 3-alkyl-2-cycloalken-1-ol acetates from 1-alkyl-2-cycloalkene-1-carboxylic acids. Preparation of dl-muscone from cyclopentadecanone. J. Org. Chem 44, 2303–2307 (1979). [Google Scholar]
- 18.Coleman JP, Lines R, Utley JHP & Weedon BCL Electro-organic reactions. Part II. Mechanism of the kolbe electrolysis of substituted phenylacetate ions. J. Chem. Soc., Perkin Trans. 2, 0, 1064–1069 (1974). [Google Scholar]
- 19.Mao R, Balon J & Hu X Decarboxylative C(sp3)–O cross-coupling. Angew. Chem. Int. Ed 57, 13624–13628 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Yan M, Kawamata Y & Baran PS Synthetic organic electrochemical methods since 2000: On the verge of a renaissance. Chem. Rev 117, 13230–13319 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ross SD & Finkelstein M Anodic oxidations. V. The Kolbe oxidation of phenylacetic acid and 1-methylcyclohexaneacetic acid at platinum and at carbon. J. Org. Chem 34, 2923–2927 (1969). [Google Scholar]
- 22.Schäfer HJ Recent contributions of Kolbe electrolysis to organic synthesis. Top. Curr. Chem 152, 91–151 (1990). [Google Scholar]
- 23.Koehl WJ Anodic oxidation of aliphatic acids at carbon anodes, J. Am. Chem. Soc 86, 4686–4690 (1964). [Google Scholar]
- 24.Iwasaki T, Horikawa H, Matsumoto K & Miyoshi M An electrochemical synthesis of 2-acetoxy-2-amino acid and 3-acetoxy-3-amino acid derivatives. J. Org. Chem 42, 2419–2423 (1977). [DOI] [PubMed] [Google Scholar]
- 25.Huang X, Liu W, Hooker JM & Groves JT Targeted fluorination with the fluoride ion by manganese-catalyzed decarboxylation. Angew. Chem. Int. Ed 54, 5241–5245 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Eberson L & Nyberg K Studies on the Kolbe electrolytic synthesis. V. An electrochemical analogue of the Ritter reaction. Acta Chem. Scand 18, 1567–1568 (1964). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available within the paper and its Supplementary Information. Metrical parameters for the structures of (2R)-77 and (11R)-138 are available free of charge from the Cambridge Crystallographic Data Centre (https://www.ccdc.cam.ac.uk/) under reference numbers 1918528 and 1903823, respectively.