

Summary
Inferring genome-scale metabolic networks in emerging model organisms is challenged by incomplete biochemical knowledge and partial conservation of biochemical pathways during evolution. Therefore, specific bioinformatic tools are necessary to infer biochemical reactions and metabolic structures that can be checked experimentally. Using an integrative approach combining genomic and metabolomic data in the red algal model Chondrus crispus, we show that, even metabolic pathways considered as conserved, like sterols or mycosporine-like amino acid synthesis pathways, undergo substantial turnover. This phenomenon, here formally defined as “metabolic pathway drift,” is consistent with findings from other areas of evolutionary biology, indicating that a given phenotype can be conserved even if the underlying molecular mechanisms are changing. We present a proof of concept with a methodological approach to formalize the logical reasoning necessary to infer reactions and molecular structures, abstracting molecular transformations based on previous biochemical knowledge.
Subject Areas: Bioinformatics, Genomics, Metabolomics
Graphical Abstract
Highlights
-
•
Combination of metabolite profiling and genome-scale metabolic networks analysis
-
•
New method to infer biochemical reactions and metabolites in promiscuous pathways
-
•
Red algal model for sterol and mycosporine-like amino acid biosynthesis pathways
-
•
Metabolic drift if pathway variation despite conserved initial and final metabolites
Bioinformatics; Genomics; Metabolomics
Introduction
Life is driven by a high diversity of metabolic processes, and each species or even strain may be characterized by its own metabolic particularities (e.g., Rhee et al., 2011). During evolutionary time and speciation processes, there are many ways that variations can be generated within metabolic pathways. Evolutionary models have been developed and experimentally tested to explain the arising of new pathways, but these efforts were focused on the activities of individual enzymes (Noda-Garcia et al., 2018). Changes in the metabolism may, however, also occur at a higher level of organization, notably an enzymatic replacement by non-orthologous genes encoding enzymes with identical biochemical function (Koonin et al., 1996; Figure 1, left side), or a change in enzyme order, which leads to different main biosynthetic intermediates (Figure 1, right side). This kind of variability, which we refer to as “metabolic drift” in this article, is possible due to substrate promiscuity of the enzymes and may be an important driver of evolution (Peracchi, 2018).
Figure 1.

The Hypothesis of Metabolic Pathway Drift Based on Two Possible Elementary Mechanisms
Starting from an ancestral promiscuous pathway (main pathway in teal; upper part, alternative pathway in olive green), changes can occur either by non-orthologous gene displacement (in orange, left side) or by change in main enzyme order, leading to a different intermediate metabolite (in olive green, right side). Substrate promiscuity enables the same molecular transformation to occur on different molecules, making the enzyme able to catalyze two different reactions. Promiscuity can be secondarily lost, as shown on the left side, leading to the impossibility to observe the star-shaped metabolite in contemporary metabolic pathway 1.
The concept of “drift” has previously been used in the field of animal comparative developmental biology, where it was already used to explain how morphologically similar structures can be maintained even if there are substantial variations in the molecular mechanisms underlying their formation (True and Haag, 2001). Here also, the use of “drift” is distinct from genetic drift, but, nevertheless, appropriate because chance and not selection explains how changes occur. The concept was more recently extended to plants, where such cases have been observed in leaf development (Townsley and Sinha, 2012). It was later exported to the fields of protein evolution (Hart et al., 2014) and gene expression evolution (Petit et al., 2016). Here, we hypothesize that the evolutionary concept of drift could also adequately explain the strict functional conservation of many metabolic pathways, despite variations in the underlying mechanisms (Figure 1).
To study metabolic drift by non-orthologous gene displacement (Figure 1, left part), classical comparative genomic approaches can generate hypotheses that can be experimentally checked using targeted metabolic profiling combined with inactivation of enzyme-encoding genes by reverse genetics (Markov et al., 2016, Sonawane et al., 2016). However, in case of drift by change in enzyme order (Figure 1, right part), it is necessary to combine both genomic and metabolomic data and to introduce a knowledge-based approach that implements reasoning in the manner of a biochemist. Such inter-disciplinary strategies have already been used to design experiments for the analysis of auxotrophic mutants in yeast (King et al., 2004) or for synthetic biology, where ab initio pathway inference is done to find a biosynthetic route that is not necessarily present in nature (Koch et al., 2017). We hypothesize that similar tools can also be used to group biochemical reaction variants based on shared ancestry and to infer undescribed pathways that may be present in emerging model species.
To test this hypothesis, we have implemented a semi-automatic analogy reasoning approach, which we use to study two distinct metabolic pathways, the sterol pathway and the mycosporine-like amino acid (MAA) pathway, in the red alga Chondrus crispus. Red algae are sufficiently distant from terrestrial plants to anticipate substantial metabolic drift compared with the known pathways in organisms such as Arabidopsis thaliana, yet C. crispus has been subject to biological studies for more than two centuries (Collén et al., 2014). Notably, its genome was sequenced and annotation was performed with a focus on metabolic features (Collén et al., 2013), and there is extensive literature available describing its metabolome (Young and Smith, 1958, Saito and Idler, 1966, Laycock and Craigie, 1977, Matsuhiro and Urzua, 1992, Karsten et al., 1998, Tasende, 2000, Kräbs et al., 2004, Gaquerel et al., 2007, Banskota et al., 2014, Pina et al., 2014, Melo et al., 2015, Alcaide et al., 1968, Goldberg et al., 1982, Kremer and Kirst, 1982, Pettit et al., 1989, van Ginneken et al., 2011, Santos et al., 2015, Robertson et al., 2015, Athukorala et al., 2016, Belghit et al., 2017, Guihéneuf et al., 2018, Lalegerie et al., 2019).
The sterol metabolism was chosen as one focus point, because there is extensive knowledge about this pathway at the comparative genomics level (Desmond and Gribaldo, 2009). Furthermore, analytical standards are available for different sterol molecules, enabling their identification by mass spectrometry (MS) (Sumner et al., 2007). MAA synthesis, on the other hand, involves combination of different building blocks, and analytical standards are lacking for this class of compounds, limiting metabolite identification to putative annotated compounds based on spectral similarity with spectral libraries (Sumner et al., 2007). Therefore the reconstruction of MAA pathway raised the problem of integrating unannotated compounds that were identified uniquely based on their m/z ratio.
By implementing a semi-automatic analogy reasoning approach that integrates both metabolite and genomic data, we here propose an exhaustive model for both metabolic pathways in C. crispus and provide strong indications for the importance of metabolic drift in shaping these pathways. We furthermore consolidated at least part of these hypotheses with targeted metabolite profiling of metabolic intermediates predicted in the models.
Results
Genome-Scale Metabolic Network Enriches the Inferred Basic Integrated Metabolism for C. crispus
Genome-scale metabolic networks (GSMNs) are graph-based representations of enzymatic reactions assumed to occur in a given organism. In this framework, an enzymatic reaction denotes a chemical reaction that transforms one or several metabolic substrates into one or several metabolic products under the control of an enzyme that can be associated with a gene in the considered species.
We used the tool AuReMe (Automatic Reconstruction of Metabolic models) dedicated to “à la carte” reconstruction of GSMNs (Aite et al., 2018) to reconstruct a GSMN of C. crispus. This GSMN comprised in total 2,024 enzymatic or transport reactions (Table 1). Among them, 595 reactions were recovered from annotation-based searches from the C. crispus genome annotation (Collén et al., 2013) with the Pathway Tools suite (Karp et al., 2002) and the MetaCyc database (Caspi et al., 2016). In addition, 1,429 reactions were included in the network according to orthology evidences with protein sequences encoding enzyme reactions in the Arabidopsis thaliana GSMN (de Oliveira Dal'Molin et al., 2010), the Galdieria sulphuraria GSMN (based on genome data from Schönknecht et al., 2013), or the Ectocarpus siliculosus GSMN (Cormier et al., 2017, Aite et al., 2018). A biomass reaction was established based on the previous E. siliculosus data, defining a list of 33 compounds to be produced to consider the network functional (Prigent et al., 2014). According to this biomass reaction, the network was manually gap-filled to unblock the production of L-alpha-alanine with an alanine dehydrogenase reaction whose associated gene had been incorrectly annotated in the C. crispus genome. The predicted maximal growth rate was then 2.43 g.gDW−1.h−1 (gram per gram dry weight per hour). As shown in Table 1, the C. crispus GSMN is comparable in size with the E. siliculosus GSMN. A total of 254 pathways are complete, including those involved in central metabolism of carbohydrates, fatty acids, and amino acids, as well as those related with photosynthesis. The greatest contributor to the inferred reaction set was the phylogenetically close red microalga G. sulphuraria, which provided 1,361 reactions.
Table 1.
Comparison of Global Features of Genome-Scale Metabolic Networks from Macroalgae and Other Chlorophyllian Eukaryotes
| Species | Reactions | Enzymes | Metabolites | Pathways | Reference |
|---|---|---|---|---|---|
| C. crispus | 2,024 | 2,006 | 2,196 | 1,108 | This study, before curation |
| E. siliculosus | 1,977 | 2,281 | 2,132 | 1,101 | Aite et al. (2018) |
| Ectocarpus subulatus | 2,074 | 2,445 | 2,173 | 1,083 | Dittami et al., 2020 |
| A. thaliana | 1,567 | 1,419 | 1,748 | 796 | de Oliveira Dal'Molin et al. (2010) |
| Chlamydomonas reinhardtii | 3,083 | 1,355 | 1,133 | 522 | Imam et al., 2015 |
Building a Catalog of Evidenced Metabolites in C. crispus Using Metabolomic Data
To better understand the specificities of C. crispus, we built a catalog of metabolic compounds attested to be produced by the alga. To that goal, we reviewed the literature to collect experimental evidence of presence of all reported metabolites. To have more species-specific data on sterol and MAA synthesis we also experimentally tested for the presence of these compounds and their precursors in C. crispus using MS analysis. In this way, we assembled a set of 142 metabolites that are reported in Tables S1 and S2. Those metabolites broadly cover various classes of amino acids, carbohydrates, and lipids. We divided this dataset into two main categories: 85 database metabolites, which are already indexed into Metacyc (Table S1), and 57 orphan metabolites, not yet indexed (Table S2). In addition, we have acquired additional experimental data on two pathways for which molecules are in both categories: the sterol and the MAA pathways.
Our MS data confirmed previous findings and also pointed out possible additional precursors and intermediary metabolites in sterol and MAA biosynthesis pathways. More precisely, the results of these analyses of sterols are found in Table 2. In addition to confirming the presence of eight previously identified sterols (brassicasterol, campesterol, cholesterol, 7-dehydrocholesterol, desmosterol, lathosterol, β-sitosterol, and stigmasterol), we identified in C. crispus an immediate precursor of sterols, i.e., squalene (Figure S1). However, we did not find evidence for cycloeucalenol, ergosterol, fucosterol, and zymosterol, which are intermediates present in other eukaryotes (Desmond and Gribaldo, 2009, Sonawane et al., 2016). We also did not find cycloartenol, contrary to a previous report in C. crispus using thin-layer chromatography (Alcaide et al., 1968). This negative finding is strengthened by the fact that we are able to identify the cycloartenol standard when added in algal extract (Figure S2).
Table 2.
List of Sterols Profiled in This Study, and Comparisons with Previous Studies
| Analyzed Compounds | Molecular Formula | Found in This Study | Previous Evidence |
|---|---|---|---|
| Brassicasterol | C28H46O | Yes | Saito and Idler (1966) (GC-MS), Tasende (2000) (TLC, GC-MS) |
| Campesterol | C28H48O | Yes | Tasende (2000) (TLC, GC-MS) |
| Cholesterol | C27H46O | Yes | Saito and Idler (1966) (TLC, GC-MS), Tasende (2000) (TLC, GC-MS) |
| Cycloartanol | C30H52O | No | Not reported |
| Cycloartenol | C30H50O | No | Saito and Idler (1966) (TLC), Alcaide et al. (1968) (TLC) |
| Cycloeucalenol | C30H50O | No | Not reported |
| 7-Dehydrocholesterol | C27H44O | Yes | Tasende (2000) (TLC, GC-MS) |
| Desmosterol | C27H44O | Yes | Saito and Idler (1966) (TLC, GC-MS), Goldberg et al. (1982) (GC-MS) |
| Ergosterol | C28H44O | No | Not reported |
| Fucosterol | C29H48O | No | Not reported |
| Lanosterol | C30H50O | No | Saito and Idler (1966) (TLC) |
| Lathosterol | C27H46O | Yes | Goldberg et al. (1982) (GC-MS) |
| β-Sitosterol | C29H50O | Yes | Saito and Idler (1966) (GC-MS), Tasende (2000) (TLC, GC-MS) |
| Squalene | C30H50 | Yes | Not reported |
| Stigmasterol | C29H48O | Yes | Saito and Idler (1966) (GC-MS), Tasende (2000) (TLC, GC-MS) |
| Zymosterol | C27H44O | No | Not reported |
TLC, thin-layer chromatography
For each compound, analytical parameters (retention time and m/z ratio) are given in Table S1.
Similar experiments were carried out for the MAA pathway, and corresponding data are summed up in Table 3. Using liquid chromatography-MS (LC-MS) profiling we confirmed the presence of six MAAs in C. crispus (see references in Table 3): asterina-330, palythene, palythine, palythinol, porphyra-334, and shinorine. In addition, we identified mycosporine-glycine in C. crispus. The relative abundance of MAAs varied between sampling dates (Figure 2), which is consistent with a report of MAA variation in the Galway Bay, Ireland (Guihéneuf et al., 2018). We noticed that two unknown peaks potentially corresponding to MAAs were detected in our samples. These peaks exhibit m/z ratios consistent with peaks reported in a study on 40 red algae by Lalegerie et al. (2019). Specifically, we found a peak at m/z = 270.3 that does not match with any already identified candidate MAA, which we named MAA1, and a second one at m/z = 302.3, which we named MAA2.
Table 3.
List of Mycosporine-like Amino Acids Identified in This Study and Comparisons with Previous Studies
| Analyzed Compounds | Molecular Formula | Found in This Study | Previous Evidence |
|---|---|---|---|
| Asterina-330 | C12H20N2O6 | Yes | Athukorala et al. (2016) (LC-MS-MS); Guihéneuf et al. (2018) (LC-MS) |
| MAA1 | compatible with m/z 270.272 | Yes | Lalegerie et al. (2019) (HPLC) |
| MAA2 | compatible with m/z = 302.3117 | Yes | Lalegerie et al. (2019) (HPLC) |
| Mycosporine-glycine | C10H15NO6 | Yes | not reported |
| Palythine | C10H16N2O5 | Yes | Karsten et al. (1998) (UV + LC-MS); Athukorala et al. (2016) (LC-MS-MS); Guihéneuf et al. (2018) (LC-MS) |
| Usujirene/Palythene | C13H20N2O5 | Yes | Karsten et al. (1998) (UV + LC-MS) |
| Palythinol | C13H22N2O6 (m/z = 302.3117) | No | Karsten et al. (1998) (UV + LC-MS), Athukorala et al. (2016) (LC-MS-MS) |
| Porphyra-334 | C14H22N2O8 | Yes | Athukorala et al. (2016) (LC-MS-MS) |
| Shinorine | C13H20N2O8 | Yes | Karsten et al. (1998) (UV + LC-MS), Athukorala et al. (2016) (LC-MS-MS) |
HPLC, high-performance liquid chromatography.
Figure 2.

Composition and Seasonal Variation (MS Relative Quantification) of MAAs in C. crispus
The four most abundant compounds, including the unknown MAA1, are on the left panel. The four less abundant compounds, including MAA2, are on the right panel.
Secondary Metabolism Evidenced by Metabolomic Data Is Only Partially Accurately Described in the C. crispus GSMN
To investigate the accuracy of GSMNs to describe the synthesis pathway of secondary metabolites of importance, we studied the capability of the C. crispus GSMN to describe synthesis pathways of metabolic compounds whose presence have been evidenced in the alga. Among those 142 metabolites, only 85 (60%) were indexed in the MetaCyc database version 20.5 (Table S1). The GSMN could provide synthesis pathways for only 59 metabolites already indexed in the Metacyc database. Those metabolites were amino acids (19), fatty acids (12), aldehydes (6), aliphatic alcohols (3), carotenoids (3), ketones (2), carboxylic acids (2), a halocarbon, a galactolipid, an oxylipin, a tetrapyrrole, an MAA, a nucleotide sugar, a methylketone, and a polyol. The synthesis of the 29 remaining metabolites indexed in the MetaCyc database could not be explained by any addition of enzymatic reactions in the database (failure of the exhaustive gap-filling procedure). The metabolites belonged to the following classes: fatty acids (8), sterols (8), alkanes (2), carotenoids (2), carragheens (2), aliphatic alcohols (2), an aldehyde, and a halocarbon. For the 57 metabolites that had not yet been indexed in MetaCyc (Table S2), it was impossible to generate hypotheses on their synthesis pathways. There were galactolipids (12), oxylipins (9), fatty acids (8), MAAs (6), alcohols (6), unconventional amino acids (3), tetrapyrroles (3), alkanes (2), carboxylic acids (2), ketones (2), a heteroside, a sterol, a phospholipid, and an aldehyde.
Building a Knowledge Base of Enzymatic Reactions and Molecules in Sterol and MAA Synthesis
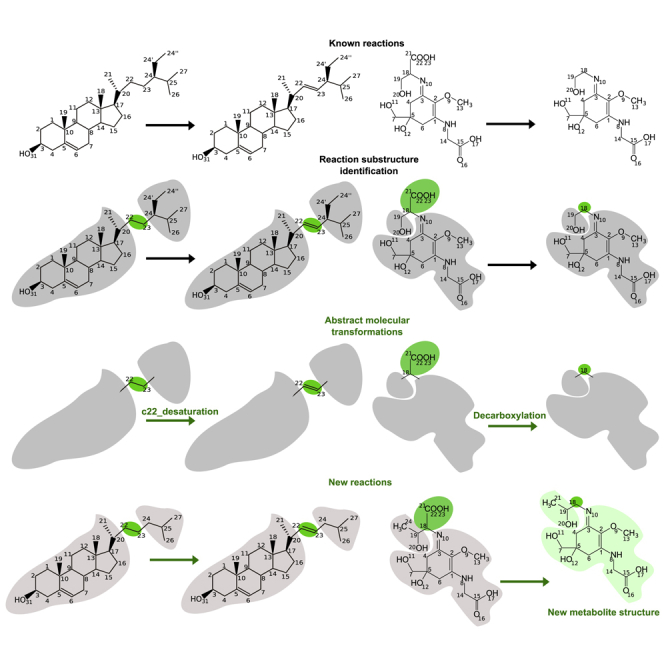
To enable the incorporation of the orphan metabolites that were not yet in MetaCyc and derive possible synthesis pathways for sterols and MAAs, we built two knowledge bases describing the existing knowledge about known enzymatic reactions and molecules involving sterols or MAAs (available on https://github.com/pathmodel/pathmodel). They are encoded in the Pathmodel datafiles sterol_pwy.lp and MAA_pwy.lp, both accessible in the pathmodel/data folder of the Github repository. In these knowledge bases, molecules are described by atoms (identified by a number and atom types) and bonds (identified by atom numbers and bond type) as highlighted in green on Figure 3. Atom numbers were assigned manually to ensure consistency between molecules from the same family and followed IUPAC conventions when existing (Moss, 1989). Molecules can be automatically associated with a theoretical m/z ratio, calculated using their chemical formula, as described in the program MZComputation.lp.
Figure 3.

The Two Reasoning Methods Implemented in Pathmodel
(A) Inference of a reaction between two known molecules.
(B) Inference of reaction and metabolite structure corresponding to an unassigned m/z peak. Input data encoded in the knowledge base are in black; inferred reactions and metabolite structures are in green. In both cases, reaction substructure identification followed by inference of molecular transformations are common intermediate steps.
Enzymatic reactions, denoted by reaction, model the link between two molecules (a reactant and a product, e.g., reaction(rxn_4243, "sitosterol", "stigmasterol")). They are associated with cross-references to MetaCyc pathway IDs when possible.
The MAA knowledge base (Figure 4) contained 13 enzymatic reactions involving 12 molecules. It first contained the shinorine biosynthesis pathway (MetaCyc: PWY-7751), corresponding to the best understood part of the pathway (Shick and Dunlap, 2002). We also encoded in the knowledge base an extended version of the amino acid C3-transfer reaction (MetaCyc: RXN-17371) to incorporate the hypothesis that the enzyme performing the C3-transfer of serine can also perform the C3-transfer of threonine, leading to porphyra-334 (Brawley et al., 2017). We also incorporated additional reactions hypothesized in the literature (Carreto and Carignan, 2011), for which either the substrate or the product was an MAA described in C. crispus. We finally added to the database the four orphan MAAs listed in Table S2, as well as two unknown molecules (MAA1 and MAA2) for which we had an experimental support for peaks corresponding to unassigned m/z ratios, as well as mycosporine-glycine, which was here identified in C. crispus.
Figure 4.

A Model for MAA Biosynthesis Pathway in C. crispus
PWY-7751: shinorine biosynthesis pathway. The figure legend details the various data sources integrated to infer the pathways. Stars indicate reactions, and squares indicate molecules. In light blue boxes: reactions and metabolites inferred through Pathmodel.
The sterol knowledge base (Figure 5) contained 15 enzymatic reactions involving 24 molecules, including the eight unproducible sterols and the orphan molecule 22-dehydrocholesterol, which was not linked to any reaction. We encoded the «early side-chain reductase» (early SSR) pathway based on the model previously published for tomato (Sonawane et al., 2016), which was added in MetaCyc upon our request (MetaCyc: PWY18C3-1). It also included portions of the canonical plant sterol biosynthesis pathway (MetaCyc: PWY-2541; Benveniste, 2004), as well as portions of the animal sterol synthesis pathway (MetaCyc: PWY66-4, Mitsche et al., 2015).
Figure 5.

A Model for Sterol Biosynthesis in C. crispus
Early SSR: pathway involving an early sterol side-chain reduction (SSR), also present in solanacean plants (PWY18C3-1). In light blue box: late SSR pathway, involving a late sterol SSR, so far only described in C. crispus. Portions identical with the plant sterol biosynthesis pathway (PWY-2541) are also boxed. Ovals indicate molecular transformations.
Inferring Molecular Transformations from a Database of Enzymatic Reactions
Based on these examples, we hypothesize that these orphan molecules challenge the ability of the GSMN to produce them because of lacks in secondary metabolism synthesis pathways that enable the description of all possible molecular transformations between compounds catalyzed by a small family of enzymes. First, we define a molecular transformation to be a chemical reaction transforming a metabolite into another metabolite by operating on a specific part of the metabolite structure, called a substructure. We define a substructure to be a set of one of several atoms associated with one or several bonds in a given molecule. For instance, substructure (“simple bond 22-23”) denotes a simple bond between atoms 22 and 23, such as in sitosterol on Figure 3A.
Importantly, an enzymatic reaction involving a single reactant and a single product can be defined as a molecular transformation by assuming that the enzyme enables the transformation of a metabolite site into another. Therefore, for each reaction involving a single reactant and a single product, we call pair of transformed substructures the substructure of the reactant (for instance, as shown in Figure 3A, simple bond between atoms 22 and 23 in sitosterol) and the substructure of the reaction product (for instance, as shown in Figure 3A, double bond 22-23 in stigmasterol).
Pairs of transformed substructures can be computed by removing all the atoms and bonds that are common to both reactant and product molecules, all atoms and bonds being previously ordered. For the sterol and MAA cases, we identified pairs of transformed substructures with a reasoning-based approach (see Methods) and then annotated them to describe a catalog of molecular transformations associated with known enzymatic reactions. For instance, the transformation replacing a simple bond between atoms 22 and 23 (as exhibited in sitosterol) into a double bond 22-23 (as exhibited in stigmasterol) is a c22 desaturation, described by the term transformation(c22_desaturation, simple bond 22-23, double-bond 22-23).
The complete list of molecular transformations for the sterol and MAA reactions is given in Table S5. The sterol enzymatic reaction database (15 reactions) yields 12 molecular transformations, whereas the MAA enzymatic reaction database (13 reactions) yields 9 molecular transformations. Reasoning on molecular transformations instead of enzymatic reactions, we reduce the complexity of the reaction set by abstracting a library of molecular transformations that can be applied to other known molecules belonging to the same chemical family.
Inferring Molecular Compounds and Enzymatic Reactions from a Database of Molecular Transformations
We used these databases of molecular transformations to infer putative new molecules and reactions.
To that goal, we assumed that for any molecular transformation from a substructure S1 to a substructure S2, and any pair of metabolites A and B, a putative enzymatic reaction can occur from the reactant A to the product B as soon as (1) the molecule A contains the substructure S1, (2) the molecule B contains the substructure S2, and (3) the molecules A and B have identical structures (e.g., same numbered atoms and bonds) for all atoms and bonds to the exception of S1 and S2 substructures. An example is shown in Figure 3A: cholesterol and 22-dehydrocholesterol share a sterane skeleton. The only difference between these molecules is that cholesterol has a simple bond between atoms 22 and 23, whereas 22-dehydrocholesterol has a double bond between atoms 22 and 23. As the transformation from a simple bond to a double bond between atoms 22 and 23 has been evidenced in the reaction database (reaction from sitosterol to stigmasterol, Figure 3), we assume that a putative enzymatic reaction exists between cholesterol and 22-dehydrocholesterol and that it should be a c22_desaturation.
In addition, we assumed that, for a given molecular transformation, a given metabolite A and an experimental mass-to-charge ratio m/z corresponding to an unassigned peak, a putative metabolite B can be produced as soon as (1) the theoretical mass-to-charge ratio of B equals the experimental m/z and (2) A can be transformed into B according to a putative enzymatic reaction associated with the selected molecular transformation. An example is depicted in Figure 3B. We consider that the molecule MAA2 is a putative derived metabolite of porphyra-334 because its mass-to-charge m/z = 302.3117 corresponds to an observed peak in our measurements and that it can be obtained from porphyra-334 by removing a carboxyl group from carbon 18, a transformation named decarboxylation, which occurs in the enzymatic reaction from shinorine to asterina-330.
With these two assumptions, we introduce the concept of putative synthesis pathway computed from a source molecule A, a list of target molecules, a list of molecular transformations, a list of putative metabolites, a list of corresponding theoretical mass-to-charge m/z, and a list of forbidden molecules, for which we have analytical standards that did not match experimental peaks. From these inputs, a putative synthesis pathway is a family of putative reactions connecting the source to all targets metabolites such that (1) all reactions are consistent with the database of molecular transformations, (2) all reactants and products of the reactions are allowed either metabolites or putative metabolites matching with the allowed theoretical mass-to-charge ratio m/z, (3) no transformation associated with the pathway can produce any of the forbidden molecules, and (4) a minimal number of reactions is used to connect the source to the targets.
The Pathmodel method was developed as a prototype implementation of a semi-automatic analogy reasoning approach (Figure 3). Its aim is to infer putative synthesis pathways, to connect orphan metabolites, not yet indexed in metabolic databases, according to the known enzymatic reactions. The Pathmodel method takes as input a knowledge base including a set of known metabolites, a set of known enzymatic reactions, a set of observed mass-to-charge (m/z) ratios for unknown metabolites, an initial source metabolite, a family of targeted metabolites, and a list of forbidden molecules. Metabolites are described by their numbered atoms and bounds. Pathmodel then computes the list of molecular transformations associated with the database of enzymatic reactions and putative synthesis pathways of the targeted metabolites from the source metabolite. More precisely, for each pair of metabolites not linked by a reaction in the knowledge base, the method checks whether a molecular transformation can occur between them (deductive reasoning) and further derives from known reactions candidate metabolites corresponding to observed unassigned m/z ratios (analogical reasoning). These are the bases for iterating the selection of potential reactants or products and the inference by a reasoning component of new reaction occurrences or new metabolites. It was implemented using a logic programming approach known as Answer Set Programming (Lifschitz, 2008, Gebser et al., 2012, see Methods). An example of this reasoning is given in Figure 3.
Application of Pathmodel to the MAA Synthesis Pathway
We first used Pathmodel to compute the synthesis pathway for MAA1 and palythine from sedoheptulose-7-phosphate according to the MAA enzymatic reaction database described above. We also assumed that Z-palythenic acid was defined as a forbidden molecule according to the absence of a corresponding peak in C. crispus extracts.
The putative pathway for MAA synthesis obtained by our method is shown in Figure 4. Thanks to this approach, the knowledge base of MAA biosynthesis could be enriched with two putative molecule structures and three putative enzymatic reactions. In addition, the reactions that we compiled from the literature but are not yet indexed in MetaCyc will be submitted to the next release.
A first output of this approach is a pathway leading to a molecule structure compatible with our measured mass-to-charge ratio m/z = 270.3. It was therefore possible to infer the hypothetical structure shown in Figure 4 for this unassigned compound, named MAA1, in MS data. The predicted transformation leading from asterina-330 to MAA1 is a dehydration (in purple), a molecular transformation already observed between other MAAs such as porphyra-334 and Z-palythenic acid, a compound not identified in C. crispus (Figure 4).
Another output of this approach is to suggest a putative involvement of decarboxylation and dehydration transformations in MAA biosynthesis pathways. As no candidate enzymes were mentioned so far in the literature related to MAA biosynthesis pathways, we performed a semantic search on the GSMN from C. crispus, to identify other enzymes that may perform those molecular transformations on a serine coupled with other chemical building blocks. Serine decarboxylation indeed occurs in phospholipid metabolism and was inferred in the C. crispus GSMN based on orthology with G. sulphuraria. The candidate gene is CHC_T00008892001, annotated as phosphatidylserine decarboxylase. Interestingly, there is some evidence of catalytic promiscuity for this enzyme, enabling it to also decarboxylate a threonine residue (Heikinheimo and Somerharju, 2002). Therefore, we hypothesize that this enzyme in C. crispus may also perform serine/threonine decarboxylation on a serine/threonine linked to a mycosporine-glycin.
A final interesting feature of the MAA synthesis pathways is the inferred enzymatic reaction required to decarboxylate shinorine and porphyra-334 and further dehydrate their derivatives. These reactions were added to the pathway to take into account the fact that Z-palythenic acid was absent in C. crispus extracts. Such an absence does not support dehydration occurring before decarboxylation, as proposed in other species (Carreto and Carignan, 2011), and therefore does not allow finding any chains of reaction producing palythinol, a compound previously considered to be present in C. crispus based on UV + LC-MS or LC-tandem MS data (Karsten et al., 1998, Athukorala et al., 2016). We noticed, however, that there is no synthesis-based analytical standard available for verifying this prediction. To identify alternative routes for the production of porphyra-334, we ran Pathmodel by allowing the production of putative metabolites with m/z ratio of 302.3177 (the one of palythinol). Pathmodel then predicted an additional intermediate with m/z ratio of 302.3177, named MAA2 on Figure 4. From a genomic viewpoint, switching palythinol with MAA2 does not necessitate a candidate enzyme to perform hydrogenation and demethylation on an MAA-like substrate (Figure 4) and thus reduces the number of unassigned enzymatic activities to candidate genes. We thus included this alternative route in the MAA synthesis pathway that is consistent with the possible absence of Z-palythenic acid.
Application of Pathmodel to the Sterol Synthesis Pathway
The Pathmodel approach was also applied to the sterol biosynthetic pathway in C. crispus (Figure 5). To that goal, we used it as an enzymatic reaction database described below. The source metabolite was cycloartenol. The targeted metabolites were 22-dehydrocholesterol, brassicasterol, and stigmasterol. In addition, forbidden metabolites were ergosterol, fucosterol, and zymosterol, which are compounds with no detection result using targeted profiling with analytical standards.
We decided to use cycloartenol even if we did not find it by gas chromatography (GC)-MS for the two following reasons. First, a cycloartenol synthase from the red alga Laurencia dendroidea was cloned and expressed in yeast cells, where it is able to transform squalene into cycloartenol, even if the authors did not report cycloartenol identification in the whole alga by GC-MS (Calegario et al., 2016). Second, unambiguous cycloartenol derivatives are known in another florideophyte red alga, Tricleocarpa fragilis (Horgen et al., 2000). Therefore, we considered more parsimonious to hypothesize that cycloartenol is present and below the limit of experimental detection rather than considering that this step is performed via an unknown intermediate.
We propose two alternative synthesis pathways from cycloartenol to cholesterol, depending on when the side-chain reductase (SSR) enzyme is acting (Figure 5).
If C. crispus uses the « early SSR » pathway (Sonawane et al., 2016), the metabolic intermediates would be identical to tomato, but there would be an important difference concerning the enzymes. Indeed, the genes encoding SSR are duplicated in Solanaceae (tomato and potato) but not in the C. crispus genome or in any red algal genome and in other plants, analyzed so far (Figure S3). The unduplicated SSR from non-solanaceous plants is known to be catalytically promiscuous, and indeed Pathmodel suggested that SSR could act on all possible intermediates (Figure 5).
Another alternative synthesis pathway inferred by Pathmodel consists in producing methylated sterols through C24-methylation on desmosterol (Figure 5). This is in agreement with the identification of a methylated sterol, 24-methylenecholesterol, in C. crispus (Tasende, 2000) and builds on with other reports about methyltransferase catalytic promiscuity across land plants and green algae (Neelakandan et al., 2009, Haubrich et al., 2015). This option highly reduces the number of non-identified methylated intermediates. Indeed, in land plants, a first methylation, involving methyltransferases, occurs directly on cycloartenol, whereas the second one occurs later on 24-methylenelophenol, to produce methylated sterols like campesterol or brassicasterol (Benveniste, 2004) with intermediates such as cycloeucalenol or fucosterol, both compounds for which we did not find any evidence of presence. By specifying in our model not to enable the production of fucosterol, we naturally omitted this possibility. This model seems also more relevant from a quantitative viewpoint with respect to the formation of cholesterol as the main sterol, because this late methylation step would enable the production of methylated sterols using the late SSR pathway.
A Complete Set of Candidate Enzymes for the C. crispus Sterol Synthesis Pathway
To identify the enzymes associated with the sterol synthesis pathways, we carried out a comparative genomic analysis. Results are summed up in Table S6. In line with previous analyses on sterol biosynthesis gene families in eukaryotes (Desmond and Gribaldo, 2009) or more specifically in green plants (Sonawane et al., 2016), the candidate sterol synthesis enzyme set shows a mixture of conservation and divergence. Seven enzymes are encoded by genes that are conserved as 1:1 orthologs, whereas four of them either underwent lineage-specific duplications (squalene epoxidase and C-4 demethylase) or were lost or may have been replaced by distant paralogs (C24 and C24′ methylases and C22 desaturases). In one case, we found no homolog of known plant or animal enzymes performing delta-7/delta-8 isomerization in the C. crispus genome, but we found a 1:1 ortholog of ERG2, the gene that secondarily took up this function in yeast (Desmond and Gribaldo, 2009). Another similar case in the sterol synthesis pathway occurs in diatoms, where the epoxisqualene cyclase, otherwise conserved in eukaryotes, was secondarily lost and replaced by a protein belonging to the fatty acid hydroxylase superfamily (Pollier et al., 2019). We consider the ERG2 ortholog to be the best candidate for delta-7/delta-8 isomerization in C. crispus, but it is also possible that this reaction is performed by an enzyme encoded by a taxonomically restricted orphan gene (Khalturin et al., 2009).
A Complete View of Algal Primary Metabolism Associated with MAA and Sterol Synthesis
The final GSMN associated with C. crispus is constituted of the initial GSMN, enriched with models curated in detail for the sterol and the MAA synthesis pathways. It was therefore reconstructed from both the genome data and the metabolomic data, using the extraction of molecular transformations implemented in Pathmodel (Figure 6). The final GSMN contains 2,207 metabolites, including the eight initial target sterols that are now producible based on Pathmodel inferences as well as seven formerly orphan metabolites (six MAAs and 22-dehydrocholesterol). In this way, we increased the proportion of producible targets from 69.4% (59/85) up to 80% (74/92). The final GSMN is available as a wiki website at the following address: https://gem-aureme.genouest.org/ccrgem/index.php/Main_Page. This website is open for further community curation and will serve as the central point to later integrate additional biochemical knowledge about C. crispus. More widely, the curation effort on those pathways will also benefit the entire GSMN community. As already done for the early SSR pathway (now MetaCyc: PWY18C1-3), we will systematically suggest the inclusion of biochemical data used in Pathmodel in the next versions of MetaCyc.
Figure 6.

Reconstruction Scheme for the Genome-Scale Metabolic Network of C. crispus
Green boxes indicate starting data and resulting knowledge. Blue boxes indicate the tools that were used to analyze and integrate genomic and metabolomic data. The part overshadowed in gray indicates tools that are already integrated in the AuReMe workflow (Aite et al., 2018).
Discussion
In this study we present an extensive analysis of the sterol and MAA biosynthetic pathways in the red alga C. crispus, integrating automatic metabolic network reconstruction, manual curation, metabolite profiling, and semi-automatic analogy reasoning approach based on molecular similarity and dissimilarity to generate hypotheses on metabolic pathways associated with secondary metabolism and metabolites, which are not described in GSMN reference databases such as Metacyc. Three main conclusions can be derived from the presented research. (1) Our findings underline the usefulness of semi-automatic analogy reasoning approaches to link orphan metabolites to existing pathways through the prediction of molecular transformations to infer reactions based on molecular similarity and dissimilarity. (2) These models support our hypothesis of drift as an evolutionary mechanism shaping the metabolism of living organisms. (3) We propose models of sterol and MAA biosynthesis in C. crispus and partially validate these models by metabolite profiling.
Semi-automatic Analogy Reasoning Approaches to Integrate Orphan Metabolites
Our study demonstrates that data on metabolite occurrence can be explicitly incorporated into the quality criteria for evaluating a GSMN. To deal with metabolic drift during metabolic network reconstruction, the incorporation of metabolite data is essential, because it puts further constraints on partially known pathways. Putting more emphasis on metabolites, especially the missing ones, creates methodological challenges regarding ab initio inferences of pathways when enzymes are not yet known, and we have shown that it is now possible to build tools to specifically address those challenges. The next issue is about the scalability of our approach. The Pathmodel version we present here is a working prototype that can already be applied to other metabolic pathways in C. crispus or in other organisms where genomic and metabolomic data are available.
Application of the Pathmodel Approach to Other Studies
The results presented in this study are a first step toward the further development of the underlying bioinformatic tools and their application to additional model biosynthesis pathways. Indeed, the Pathmodel tool was developed to support reasoning based on the metabolic pathway drift hypothesis to automatically infer reactions and metabolites. A key feature of the successful application of this strategy was the precision and the quality of the biochemical and biological knowledge encoded in the reaction and metabolite databases used as entries to Pathmodel. In particular, all metabolites have to be described with a homogeneous ordering of atoms to predict molecular transformations. Generalizing this approach to any other application will similarly require interactions between chemists, biologists, and computer scientists. Further improvements should be made to minimize the burden in manually entering molecular structures. It is not yet possible to fully automate the atom numbering during metabolic reaction, due to the intrinsic complexity of metabolic pathways. For example, the split of molecules during biochemical processes can generate inconsistent numbering (Figure S5A) that can only be handled by resetting atom numbering from one step to another. This is already done in MetaCyc, but in a way that does not make possible to automatically number in the same manner atoms from different reactions that share a molecular transformation (Figure S5B). Other methods, like the CLCA approach (Kumar and Maranas, 2014) already implemented ways to compare reactions sharing molecular transformations, and thus would provide lists of candidate reactions for molecular transformations, but the atom numbering during the comparisons does not allow simultaneous atom mapping between reactant and product (Figure S5C) that we need in Pathmodel to abstract the molecular transformation. Therefore, following IUPAC atom encoding as much as possible seems to be the best way to combine atom mapping and abstraction of molecular transformations (Figure S5D).
The second key feature of Pathmodel is to be focused on a selected pathway rather than on a complete genome-scale metabolic network. The selection of the relevant pathway to be considered, for instance, from preliminary evidences extracted from metabolomic analysis, is therefore a key pre-processing step to combine and filter the predictions of Pathmodel with genomic and metabolomic data. Practically, Pathmodel can already be used on any type of incomplete biochemical pathway on a molecule class for which there is knowledge about some biochemical reactions and orphan molecules not yet connected to each other by any reaction. The main limitation will be the user time necessary to properly encode the starting knowledge base.
Metabolic Drift as a Driver for Pathway Evolution
Whatever the actual topology of the sterol and MAA pathways in C. crispus, each discussed hypothesis has implications regarding metabolic pathway drift. All possible sterol pathways provide further strong candidate case studies for drift by non-homologous enzyme replacement, and the pathways inferred by Pathmodel provide candidate case studies for drift by enzyme inversion. The unresolved point with the sterol pathways is that, among eukaryotes, there is no consensus yet about the ancestral order of enzymatic reactions. Experimental data are too disparate across the tree of life to enable firm conclusions on this. In that respect, the MAA pathway is interesting, because if our hypothesis about decarboxylation of porphyra-334 before dehydration is true, this would mean that an enzymatic inversion took place in other lineages where porphyra-334 is first dehydrated to Z-palythenic acid and then decarboxylated to palythene. Here the limit is that, to date, enzymes are unknown for both reactions, so the system is not yet genomically tractable. Identifying close enzymatic inversions would be important to provide a mechanism for gradual divergence of pathways. Indeed, experimental analyses on E. coli have shown that drastic pathway rewiring by enzyme knockout or gene overexpression can led to toxic intermediates (Kim et al., 2010).
Limitations of the Study
Our arguments for the metabolic pathway drift hypothesis rely on comparisons between pathways from C. crispus and from distantly related species belonging to a few other phyla, such as land plants or animals. Additional support should become available from detailed comparisons based on additional eukaryotic lineages and, when possible, from multiple species in the same phylum. Finally, it remains to be determined to what extent chance and selection contributed in generating metabolic pathway diversity.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Cécile Hervé for help in collecting field samples from C. crispus, Gaëlle Correc for help in sample preparation, Karine Cahier for help during GC-MS analyses on the MetaboMer-Corsaire platform, Jeanne Got and Marie Chevallier for help in using preliminary versions of AuReMe, and Clémence Frioux for help in analyzing FBA artifacts. G.V.M. is also grateful to Ralf J. Sommer for giving him the possibility to start developing Pathmodel during a previous postdoctoral stay at the Max-Planck Institute for Developmental Biology in Tübingen. We thank Alessio Peracchi for commenting on the putative role of PLP-enzymes in the MAA biosynthesis pathway and the two anonymous reviewers for their improvement suggestions. This work benefited from the support of the French Government via the National Research Agency investment expenditure program IDEALG (ANR-10-BTBR-04) and from Région Bretagne via the grant « SAD 2016 - METALG (9673) ».
Author Contributions
G.V.M., L.D., S.M.D., P.S., E.C., J.N., C.B., C. Leblanc, A.S., and J.C. conceived the project. J.G. conducted the sterol profiling with help from L.D., C. Leroux, C. Leblanc, J.C., and G.V.M. P.S. conduced the MAA profiling with help from C.M. G.V.M. performed the genome-scale metabolic network reconstruction with help from M.A., C.T., S.M.D, J.C., and A.S. A.B., G.V.M., and J.N. wrote the Pathmodel software. A.B., J.G., L.D., and G.V.M. curated the biosynthetic pathway models. G.V.M. wrote the manuscript with input and edits from all authors.
Declaration of Interests
The authors declare no competing interests.
Published: February 21, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.100849.
Supporting Citations
The following reference appears in the Supplemental Information: Anisimova and Gascuel, 2006, Gouy et al., 2010, Guindon and Gascuel, 2003, Kanehisa et al., 2017, King et al., 2016, Le and Gascuel, 2010, Loira et al., 2015, Moretti et al., 2016, Prigent et al., 2017, Sievers and Higgins, 2014.
Supplemental Information
References
- Aite M., Chevallier M., Frioux C., Trottier C., Got J., Cortés M.P., Mendoza S.N., Carrier G., Dameron O., Guillaudeux N. Traceability, reproducibility and wiki-exploration for “à-la-carte” reconstructions of genome-scale metabolic models. PLoS Comput. Biol. 2018;14:e1006146. doi: 10.1371/journal.pcbi.1006146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcaide A., Devys M., Barbier M. Remarques sur les stérols des algues rouges. Phytochemistry. 1968;7:329–330. [Google Scholar]
- Anisimova M., Gascuel O. Approximate likelihood-ratio test for branches: a fast, accurate, and powerful alternative. Syst. Biol. 2006;55:539–552. doi: 10.1080/10635150600755453. [DOI] [PubMed] [Google Scholar]
- Athukorala Y., Trang S., Kwok C., Yuan Y.V. Antiproliferative and antioxidant activities and mycosporine-like amino acid profiles of wild-harvested and cultivated edible Canadian marine red macroalgae. Molecules. 2016;21:E119. doi: 10.3390/molecules21010119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banskota A.H., Stefanova R., Sperker S., Lall S., Craigie J.S., Hafting J.T. Lipids isolated from the cultivated red alga Chondrus crispus inhibit nitric oxide production. J. Appl. Phycol. 2014;26:1565–1571. [Google Scholar]
- Belghit I., Rasinger J.D., Heesch S., Biancarosa I., Liland N., Torstensen B., Waagbø R., Lock E.-J., Bruckner C.G. In-depth metabolic profiling of marine macroalgae confirms strong biochemical differences between brown, red and green algae. Algal Res. 2017;26:240–249. [Google Scholar]
- Benveniste P. Biosynthesis and accumulation of sterols. Annu. Rev. Plant Biol. 2004;55:429–457. doi: 10.1146/annurev.arplant.55.031903.141616. [DOI] [PubMed] [Google Scholar]
- Brawley S.H., Blouin N.A., Ficko-Blean E., Wheeler G.L., Lohr M., Goodson H.V., Jenkins J.W., Blaby-Haas C.E., Helliwell K.E., Chan C.X. Insights into the red algae and eukaryotic evolution from the genome of Porphyra umbilicalis (Bangiophyceae, Rhodophyta) Proc. Natl. Acad. Sci. U S A. 2017;114:E6361–E6370. doi: 10.1073/pnas.1703088114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calegario G., Pollier J., Arendt P., de Oliveira L.S., Thompson C., Soares A.R., Pereira R.C., Goossens A., Thompson F.L. Cloning and functional characterization of cycloartenol synthase from the red seaweed Laurencia dendroidea. PLoS One. 2016;11:e0165954. doi: 10.1371/journal.pone.0165954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreto J.I., Carignan M.O. Mycosporine-like amino acids: relevant secondary metabolites. Chemical and ecological aspects. Mar. Drugs. 2011;9:387–446. doi: 10.3390/md9030387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi R., Billington R., Ferrer L., Foerster H., Fulcher C.A., Keseler I.M., Kothari A., Krummenacker M., Latendresse M., Mueller L.A. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2016;44:D471–D480. doi: 10.1093/nar/gkv1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collén J., Porcel B., Carré W., Ball S.G., Chaparro C., Tonon T., Barbeyron T., Michel G., Noel B., Valentin K. Genome structure and metabolic features in the red seaweed Chondrus crispus shed light on evolution of the Archaeplastida. Proc. Natl. Acad. Sci. U S A. 2013;110:5247–5252. doi: 10.1073/pnas.1221259110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collén J., Cornish M.L., Craigie J., Ficko-Blean E., Hervé C., Krueger-Hadfield S.A., Leblanc C., Michel G., Potin P., Tonon T., Boyen C. Chondrus crispus – a present and historical model organism for red seaweeds. Adv. Bot. Res. 2014;71:53–90. [Google Scholar]
- Cormier A., Avia K., Sterck L., Derrien T., Wucher V., Andres G., Monsoor M., Godfroy O., Lipinska A., Perrineau M.M. Re-annotation, improved large-scale assembly and establishment of a catalogue of noncoding loci for the genome of the model brown alga Ectocarpus. New Phytol. 2017;214:219–232. doi: 10.1111/nph.14321. [DOI] [PubMed] [Google Scholar]
- de Oliveira Dal'Molin C.G., Quek L.E., Palfreyman R.W., Brumbley S.M., Nielsen L.K. AraGEM, a genome-scale reconstruction of the primary metabolic network in Arabidopsis. Plant Physiol. 2010;152:579–589. doi: 10.1104/pp.109.148817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmond E., Gribaldo S. Phylogenomics of sterol synthesis: insights into the origin, evolution, and diversity of a key eukaryotic feature. Genome Biol. Evol. 2009;1:364–381. doi: 10.1093/gbe/evp036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittami S.M., Corre E., Brillet-Guéguen L., Lipinska A.P., Pontoizeau N., Aite M., Avia K., Caron C., Cho C.H., Collén J. The genome of Ectocarpus subulatus highlights unique mechanisms for stress tolerance in brown algae. Mar. Genomics. 2020 doi: 10.1016/j.margen.2020.100740. [DOI] [PubMed] [Google Scholar]
- Gaquerel E., Hervé C., Labrière C., Boyen C., Potin P., Salaün J.P. Evidence for oxylipin synthesis and induction of a new polyunsaturated fatty acid hydroxylase activity in Chondrus crispus in response to methyljasmonate. Biochim. Biophys. Acta. 2007;1771:565–575. doi: 10.1016/j.bbalip.2007.02.007. [DOI] [PubMed] [Google Scholar]
- Gebser M., Kaminski R., Kaufmann B., Schaub T. Answer set solving in practice. Synth. Lect. Artif. Intell. Mach. Learn. 2012;6:1–238. [Google Scholar]
- Goldberg A., Hubby C., Cobb D., Millard P., Ferrara N., Galdi G., Premuzic E.T., Gaffney J.S. Sterol distribution in red algae from the waters of eastern Long Island. Bot. Mar. 1982;25:351–355. [Google Scholar]
- Gouy M., Guindon S., Gascuel O. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2010;27:221–224. doi: 10.1093/molbev/msp259. [DOI] [PubMed] [Google Scholar]
- Guihéneuf F., Gietl A., Stengel D.B. Temporal and spatial variability of mycosporine-like amino acids and pigments in three edible red seaweeds from western Ireland. J. Appl. Phycol. 2018;30:2573–2586. [Google Scholar]
- Guindon S., Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Hart K.M., Harms M.J., Schmidt B.H., Elya C., Thornton J.W., Marqusee S. Thermodynamic system drift in protein evolution. PLoS Biol. 2014;12:e1001994. doi: 10.1371/journal.pbio.1001994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haubrich B.A., Collins E.K., Howard A.L., Wang Q., Snell W.J., Miller M.B., Thomas C.D., Pleasant S.K., Nes W.D. Characterization, mutagenesis and mechanistic analysis of an ancient algal sterol C24-methyltransferase: implications for understanding sterol evolution in the green lineage. Phytochemistry. 2015;113:64–72. doi: 10.1016/j.phytochem.2014.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heikinheimo L., Somerharju P. Translocation of phosphatidylthreonine and -serine to mitochondria diminishes exponentially with increasing molecular hydrophobicity. Traffic. 2002;3:367–377. doi: 10.1034/j.1600-0854.2002.30506.x. [DOI] [PubMed] [Google Scholar]
- Horgen F.D., Sakamoto B., Scheuer P.J. New triterpenoid sulfates from the red alga Tricleocarpa fragilis. J. Nat. Prod. 2000;63:210–216. doi: 10.1021/np990448h. [DOI] [PubMed] [Google Scholar]
- Imam S., Schäuble S., Valenzuela J., López García de Lomana A., Carter W., Price N.D., Baliga N.S. Refined genome-scale reconstruction of Chlamydomonas metabolism provides a platform for systems-level analyses. Plant J. 2015;84:1239–1256. doi: 10.1111/tpj.13059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M., Furumichi M., Tanabe M., Sato Y., Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45:D353–D361. doi: 10.1093/nar/gkw1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp P., D., Paley, Romero The Pathway Tools software. Bioinformatics. 2002;18:S225–S232. doi: 10.1093/bioinformatics/18.suppl_1.s225. [DOI] [PubMed] [Google Scholar]
- Karsten U., Franklin L.A., Lüning K., Wiencke C. Natural ultraviolet radiation and photosynthetically active radiation induce formation of mycosporine-like amino acids in the marine macroalga Chondrus crispus (Rhodophyta) Planta. 1998;205:257–262. [Google Scholar]
- Khalturin K., Hemmrich G., Fraune S., Augustin R., Bosch T.C.G. More than just orphans: are taxonomically-restricted genes important in evolution? Trends Genet. 2009;25:404–413. doi: 10.1016/j.tig.2009.07.006. [DOI] [PubMed] [Google Scholar]
- Kim J., Kershner J.P., Novikov Y., Shoemaker R.K., Copley S.D. Three serendipitous pathways in E. coli can bypass a block in pyridoxal-5'-phosphate synthesis. Mol. Syst. Biol. 2010;6:436. doi: 10.1038/msb.2010.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King R.D., Whelan K.E., Jones F.M., Reiser P.G.K., Bryant C.H., Muggleton S.H., Kell D.B., Oliver S.G. Functional genomic hypothesis generation and experimentation by a robot scientist. Nature. 2004;427:247–252. doi: 10.1038/nature02236. [DOI] [PubMed] [Google Scholar]
- King Z.A., Lu J., Dräger A., Miller P., Federowicz S., Lerman J.A., Ebrahim A., Palsson B.O., Lewis N.E. BiGG Models: a platform for integrating, standardizing and sharing genome-scale models. Nucleic Acids Res. 2016;44:D515–D522. doi: 10.1093/nar/gkv1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch M., Duigou T., Carbonell P., Faulon J.L. Molecular structures enumeration and virtual screening in the chemical space with RetroPath2.0. J. Cheminform. 2017;9:64. doi: 10.1186/s13321-017-0252-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin E.V., Mushegian A.R., Bork P. Non-orthologous gene displacement. Trends Genet. 1996;12:334–336. [PubMed] [Google Scholar]
- Kräbs G., Watanabe M., Wiencke C. A monochromatic action spectrum for the photoinduction of the UV-absorbing mycosporine-like amino acid shinorine in the red alga Chondrus crispus. Photochem. Photobiol. 2004;79:515–519. doi: 10.1562/2003-12-14-ra.1. [DOI] [PubMed] [Google Scholar]
- Kremer B.P., Kirst G.O. Biosynthesis of photosynthates and taxonomy of algae. Z. Naturforsch. 1982;37c:761–771. [Google Scholar]
- Kumar A., Maranas C.D. CLCA: maximum common molecular substructure queries within the MetRxn Database. J. Chem. Inf. Model. 2014;54:3417–3438. doi: 10.1021/ci5003922. [DOI] [PubMed] [Google Scholar]
- Lalegerie F., Lajili S., Bedoux G., Taupin L., Stiger-Pouvreau V., Connan S. Photo-protective compounds in red macroalgae from Brittany: considerable diversity in mycosporine-like amino acids (MAAs) Mar. Environ. Res. 2019;147:37–48. doi: 10.1016/j.marenvres.2019.04.001. [DOI] [PubMed] [Google Scholar]
- Laycock M.V., Craigie J.S. The occurrence and seasonal variation of gigartinine and L-citrullinyl-L-arginine in Chondrus crispus Stackh. Can. J. Biochem. 1977;55:27–30. doi: 10.1139/o77-004. [DOI] [PubMed] [Google Scholar]
- Le S.Q., Gascuel O. Accounting for solvent accessibility and secondary structure in protein phylogenetics is clearly beneficial. Syst. Biol. 2010;59:277–287. doi: 10.1093/sysbio/syq002. [DOI] [PubMed] [Google Scholar]
- Lifschitz V. What is answer set programming? In: Cohn A., editor. AAAI’08: Proceedings of the 23rd national conference on Artificial intelligence. AAAI Press; Chicago: 2008. pp. 1594–1597. [Google Scholar]
- Loira N., Zhukova A., Sherman D.J. Pantograph: a template-based method for genome-scale metabolic model reconstruction. J. Bioinform. Comput. Biol. 2015;13:1550006. doi: 10.1142/S0219720015500067. [DOI] [PubMed] [Google Scholar]
- Markov G.V., Meyer J.M., Panda O., Artyukhin A.B., Claaen M., Witte H., Schroeder F.C., Sommer R.J. Functional conservation and divergence of daf-22 paralogs in Pristionchus pacificus dauer development. Mol. Biol. Evol. 2016;33:2506–2514. doi: 10.1093/molbev/msw090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuhiro B., Urzua C. Heterogeneity of carrageenans from Chondrus crispus. Phytochemistry. 1992;31:531–534. doi: 10.1016/0031-9422(92)90032-l. [DOI] [PubMed] [Google Scholar]
- Melo T., Alves E., Azevedo V., Martins A.S., Neves B., Domingues P., Calado R., Abreu M.H., Domingues M.R. Lipidomics as a new approach for the bioprospecting of marine macroalgae – Unraveling the polar lipid and fatty acid composition of Chondrus crispus. Algal Res. 2015;8:181–191. [Google Scholar]
- Mitsche M.A., McDonald J.G., Hobbs H.H., Cohen J.C. Flux analysis of cholesterol biosynthesis in vivo reveals multiple tissue and cell-type specific pathways. Elife. 2015;4:e07999. doi: 10.7554/eLife.07999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti S., Martin O., Van Du Tran T., Bridge A., Morgat A., Pagni M. MetaNetX/MNXref – reconciliation of metabolites and biochemical reactions to bring together genome-scale metabolic networks. Nucleic Acids Res. 2016;44:D523–D526. doi: 10.1093/nar/gkv1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss G.P. IUPAC-IUB Joint Commission on Biochemical Nomenclature. Nomenclature of steroids (Recommendations 1989) Eur. J. Biochem. 1989;61:1783–1822. [PubMed] [Google Scholar]
- Neelakandan A.K., Song Z., Wang J., Richards M.H., Wu X., Valliyodan B., Nguyen H.T., Nes W.D. Cloning, functional expression and phylogenetic analysis of plant sterol 24C-methyltransferases involved in sitosterol biosynthesis. Phytochemistry. 2009;70:1982–1998. doi: 10.1016/j.phytochem.2009.09.003. [DOI] [PubMed] [Google Scholar]
- Noda-Garcia L., Liebermeister W., Tawfik D.S. Metabolite-enzyme coevolution: from single enzymes to metabolic pathways and networks. Annu. Rev. Biochem. 2018;87:187–216. doi: 10.1146/annurev-biochem-062917-012023. [DOI] [PubMed] [Google Scholar]
- Peracchi A. The limits of enzyme specificity and the evolution of metabolism. Trends Biochem. Sci. 2018;43:984–996. doi: 10.1016/j.tibs.2018.09.015. [DOI] [PubMed] [Google Scholar]
- Pettit T., Jones A., Harwood J. Lipid metabolism in the red marine algae Chondrus crispus and Polysiphonia lanosa as modified by temperature. Phytochemistry. 1989;28:2053–2089. [Google Scholar]
- Pina A., Costa A., Lage-Yusty M., López-Hernández J. An evaluation of edible red seaweed (Chondrus crispus) components and their modification during the cooking process. LWT - Food Sci. Technol. 2014;56:175–180. [Google Scholar]
- Pollier J., Vancaester E., Kuzhiumparambil U., Vickers C.E., Vandepoele K., Goossens A., Fabris M. A widespread alternative squalene epoxidase participates in eukaryote steroid biosynthesis. Nat. Microbiol. 2019;4:226–233. doi: 10.1038/s41564-018-0305-5. [DOI] [PubMed] [Google Scholar]
- Prigent S., Collet G., Dittami S.M., Delage L., Ethis de Corny F., Dameron O., Eveillard D., Thiele S., Cambefort J., Boyen C. The genome-scale metabolic network of Ectocarpus siliculosus (EctoGEM): a resource to study brown algal physiology and beyond. Plant J. 2014;80:367–381. doi: 10.1111/tpj.12627. [DOI] [PubMed] [Google Scholar]
- Prigent S., Frioux C., Dittami S.M., Thiele S., Larhlimi A., Collet G., Gutknecht F., Got J., Eveillard D., Bourdon J. Meneco, a topology-based gap-filling tool applicable to degraded genome-wide metabolic networks. PLoS Comput. Biol. 2017;13:e1005276. doi: 10.1371/journal.pcbi.1005276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee K.Y., de Carvalho L.P.S., Bryk R., Ehrt S., Marrero J., Park S.W., Schnappinger D., Venugopal A., Nathan C. Central carbon metabolism in Mycobacterium tuberculosis: an unexpected frontier. Trends Microbiol. 2011;19:307–314. doi: 10.1016/j.tim.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson R.C., Guihéneuf F., Bahar B., Schmid M., Stengel D.B., Fitzgerald G.F., Ross R.P., Stanton C. The anti-inflammatory effect of algae-derived lipid extracts on lipopolysaccharide (LPS)-stimulated human THP-1 macrophages. Mar. Drugs. 2015;13:5402–5424. doi: 10.3390/md13085402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito A., Idler D.R. Sterols in Irish moss (Chondrus crispus) Can. J. Biochem. 1966;44:1195–1199. doi: 10.1139/o66-136. [DOI] [PubMed] [Google Scholar]
- Santos S.A., Vilela C., Freire C.S., Abreu M.H., Rocha S.M., Silvestre A.J. Chlorophyta and Rhodophyta macroalgae: a source of health promoting phytochemicals. Food Chem. 2015;183:122–128. doi: 10.1016/j.foodchem.2015.03.006. [DOI] [PubMed] [Google Scholar]
- Schönknecht G., Chen W.H., Ternes C.M., Barbier G.G., Shrestha R.P., Stanke M., Bräutigam A., Baker B.J., Banfield J.F., Garavito R.M. Gene transfer from bacteria and archaea facilitated evolution of an extremophilic eukaryote. Science. 2013;339:1207–1210. doi: 10.1126/science.1231707. [DOI] [PubMed] [Google Scholar]
- Shick J.M., Dunlap W.C. Mycosporine-like amino acids and related gadusols: biosynthesis, accumulation, and UV-protective functions in aquatic organisms. Annu. Rev. Physiol. 2002;64:223–262. doi: 10.1146/annurev.physiol.64.081501.155802. [DOI] [PubMed] [Google Scholar]
- Sievers F., Higgins D.G. Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol. Biol. 2014;1079:105–116. doi: 10.1007/978-1-62703-646-7_6. [DOI] [PubMed] [Google Scholar]
- Sonawane P.D., Pollier J., Panda S., Szymanski J., Massalha H., Yona M., Unger T., Malitsky S., Arendt P., Pauwels L. Plant cholesterol biosynthetic pathway overlaps with phytosterol metabolism. Nat. Plants. 2016;3:16205. doi: 10.1038/nplants.2016.205. [DOI] [PubMed] [Google Scholar]
- Sumner L.W., Amberg A., Barrett D., Beale M.H., Beger R., Daykin C.A., Fan T.W., Fiehn O., Goodacre R., Griffin J.L. Proposed minimum reporting standards for chemical analysis chemical analysis working group (CAWG) metabolomics standards initiative (MSI) Metabolomics. 2007;3:211–221. doi: 10.1007/s11306-007-0082-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasende M. Fatty acid and sterol composition of gametophytes and sporophytes of Chondrus crispus (Gigartinaceae, Rhodophyta) Sci. Mar. 2000;64:421–426. [Google Scholar]
- Townsley B.T., Sinha N.R. A new development: evolving concepts in leaf ontogeny. Annu. Rev. Plant Biol. 2012;63:535–562. doi: 10.1146/annurev-arplant-042811-105524. [DOI] [PubMed] [Google Scholar]
- True J.R., Haag E.S. Developmental system drift and flexibility in evolutionary trajectories. Evol. Dev. 2001;3:109–119. doi: 10.1046/j.1525-142x.2001.003002109.x. [DOI] [PubMed] [Google Scholar]
- van Ginneken V.J., Helsper J.P., de Visser W., van Keulen H., Brandenburg W.A. Polyunsaturated fatty acids in various macroalgal species from north Atlantic and tropical seas. Lipids Health Dis. 2011;10:104. doi: 10.1186/1476-511X-10-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young E.G., Smith D.G. Amino acids, peptides, and proteins of Irish moss, Chondrus crispus. J. Biol. Chem. 1958;233:406–410. [PubMed] [Google Scholar]
- Petit C., Rey C., Lambert A., Peltier M., Pantalacci S., Sémon M. Comparing transcriptomes to probe into the evolution of developmental program reveals an extensive developmental system drift. In: Perrière G., Picard F., editors. JOBIM2016: Conference Proceedings of the 17èmes Journées Ouvertes en Biologie, Informatique et Mathématiques. Published online by the Société Française de Bioinformatique (SFBI; 2016. pp. 118–120. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.