

Abstract
Tuberculosis (TB), caused by Mycobacterium tuberculosis (Mtb), is a severe infectious disease in need of new chemotherapies especially for drug-resistant cases. To meet the urgent requirement of new TB drugs with novel modes of action, the TB research community has been validating numerous targets from several biosynthetic pathways. The methylerythritol phosphate (MEP) pathway is utilized by Mtb for the biosynthesis of isopentenyl pyrophosphate (IPP) and its isomer dimethylallyl pyrophosphate (DMAPP), the universal five-carbon building blocks of isoprenoids. While being a common biosynthetic pathway in pathogens, the MEP pathway is completely absent in humans. Due to its unique presence in pathogens as well as the essentiality of the MEP pathway in Mtb, the enzymes in this pathway are promising targets for the development of new drugs against tuberculosis. In this review, we discuss three enzymes in the MEP pathway: DXS, DXR, and IspF, which appear to be the most promising antitubercular drug targets. Structural and mechanistic features of these enzymes are reviewed, as well as selected inhibitors that show promise as antitubercular agents.
Keywords: Mycobacterium tuberculosis, MEP pathway, DXS, DXR, IspF, isoprenoid, IPP, DMAPP, drug discovery, antitubercular
Graphical Abstract
INTRODUCTION
Tuberculosis (TB) is now the world’s deadliest infectious disease, and 1.8 million people died from it alone in 2015.1 In addition to high mortality, a high morbidity rate worsens the situation caused by TB: 10.4 million people fell ill from the disease in 2015.1 TB is caused primarily by Mycobacterium tuberculosis (Mtb) and this organism is the main infectious agent in humans. TB can also be caused by the Mtb complex, consisting of Mtb, Mycobacterium bovis, and Mycobacterium africanum.2 Able to spread via a single, airborne bacillus from a TB patient, Mtb has made TB a pandemic.3–4 Around one in three individuals in the world has an established TB infection through such successful transmission of Mtb.5 Yet, most of these infections do not progress to active disease immediately since alveolar macrophages phagocytose the bacilli after arrival in the lung. Here, the bacilli stay inactive and do not replicate often.4 Although latent TB in the nonreplicating form is asymptomatic, the risk of activating it to active disease is increased by many factors including HIV infection, immunodeficiency disorders, and others.6 With the challenge of complete elimination of all bacilli and few treatment options against latent TB, this lifetime threat lingers.6 Current chemotherapy for drug-susceptible TB consists of an intensive phase for two months using the four first-line TB drugs (isoniazid, rifampicin, pyrazinamide and ethambutol) and a continuation phase for four months using isoniazid and rifampicin.7 While it is essential for patients to follow the regimen strictly, the pill burden over such a long duration makes treatment adherence very difficult. Misuse of drugs contributes significantly to the emergence of drug-resistant TB, which has very limited treatment options, often associated with safety and efficacy issues.8–9 In order to improve patient compliance, to shorten the duration of treatment, and to provide more treatment options for TB, new drugs with novel modes of action are urgently required.
Blocking the biosynthesis of various essential metabolites can be bactericidal and has drawn close attention from many research groups. The TB research community has worked to validate numerous targets from several biosynthetic pathways, propelling such a strategy to a great extent. These targets include ATP synthase, the target of bedaquiline (the first antitubercular drug approved by the US FDA in over 4 decades) used to treat multi-drugresistant tuberculosis10, several enzymes related to cholesterol catabolism11–15, isocitrate lyase16–17, malate synthase18, and fumarase19–20 in the glyoxylate cycle and related metabolic pathways, NAD+ synthetase21–22, adenylating enzymes such as biotin protein ligase23–24, siderophore biosynthesis25–29, and CoaBC in the coenzyme A biosynthesis pathway30. This review focuses on advances in drug discovery involving disruption of the methylerythritol phosphate (MEP) pathway (also known as the nonmevalonate pathway, NMP), and validation of the enzymes in this pathway as promising antitubercular drug targets.
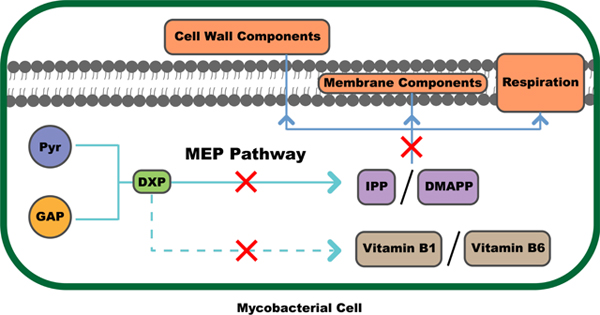
It was long believed that isopentenyl pyrophosphate (IPP) and its isomer dimethylallyl pyrophosphate (DMAPP), the C5 precursors of isoprenoids, are synthesized in all organisms via the mevalonate pathway since its discovery in eukaryotes by Conrad Bloch and Feodor Lynen.31–34 The mevalonate pathway uses two-carbon acetate as the starting material. In 1993, an alternative pathway, the MEP pathway, was found and shown to use completely different starting materials.35 The MEP pathway began with three-carbon pyruvate, rather than acetate. Mtb, among other organisms, depends solely on this pathway for the production of IPP and DMAPP, while humans exclusively utilize the alternate mevalonate pathway for such metabolites.34, 36–37 Isoprenoids generated from IPP and DMAPP are crucial for survival of Mtb and other microorganisms. They are used for production of many secondary metabolites including hopanoids which modify membrane properties38, menaquinone or vitamin K2 that acts in the respiratory electron transport system39, and polyprenyl phosphates that are crucial for bacterial cell wall biosynthesis40 (Figure 1). Additionally, DXP, an intermediate in the MEP pathway, feeds into the synthesis of vitamins B1 and B6. The importance of these and other downstream molecular products to Mtb survival, as well as the pathway’s absence in human metabolism, make the MEP pathway a promising target for developing antitubercular drugs.36–37
Figure 1.

The MEP pathway is essential in Mtb. The terminal products from IPP and DMAPP include hopanoids which modify membrane properties, menaquinones or vitamin K2 that play essential roles in the respiratory electron transport systems, and polyprenyl phosphates that are crucial for bacterial cell wall biosynthesis. DXP, the product of the first enzymatic reaction in the MEP pathway, is a precursor to vitamins B1 and vitamin B6.
The MEP pathway consists of seven enzymatic steps starting from pyruvate (Pyr) and Dglyceraldehyde-3-phosphate (D-GAP) to produce IPP and DMAPP (Figure 2). Thiamin diphosphate (ThDP)-dependent DXS catalyzes the first step of the biosynthesis, converting Pyr and D-GAP to DXP with release of carbon dioxide. DXP then undergoes an NADPH-dependent reduction to MEP mediated by IspC/DXR. The third step of the MEP pathway is catalyzed by IspD, transforming MEP to CDP-ME. IspE then catalyzes the ATP-dependent phosphorylation from CDP-ME to CDP-MEP. With release of CMP, the cyclic product MEcPP is generated from CDP-MEP by the enzyme IspF. In the sixth step of the pathway, HMBPP is obtained by the reductive deoxygenation of MEcPP catalyzed by IspG. Finally, IspH converts HMBPP to IPP and DMAPP, the isomeric C5 precursors of isoprenoids.41–44 Most of these enzymes play an essential role in MEP biosynthesis.43
Figure 2.

The MEP pathway for the biosynthesis of IPP and DMAPP
While each of the seven enzymes has potential as a drug target,42–43, 45 many of the enzymes present particular challenges with respect to inhibitor design and drug discovery. Despite reported crystal structures,46–47 demonstrated essentiality,43 and initial inhibitors,45 IspD and IspE suffer from other obstacles. The active site of IspD is very flexible, highly polar, and lies at a dimer interface.46 These factors can lead to challenges when designing successful inhibitors, particularly against mycobacteria with lipophilic cell walls. While IspD may hold promise as an antimalarial target,48 its potential for Mtb is less certain. IspE belongs to a family of ATP-binding enzymes.49 Selectivity among bacterial and mammalian enzymes is a potential issue.45 IspG and IspH, at the end of the MEP pathway, both contain an Fe-S cluster, notoriously difficult to screen and assay against.43, 50 While some crystal structures of IspG/H have been reported, none come from pathogenic organisms.45 Furthermore, inhibitors that have been described target the Fe-S cluster, raising concerns about selectivity.43, 45 For these reasons, we focus in this review on the most promising MEP pathway drug targets for development of antitubercular agents: DXS, DXR, and IspF.
1-DEOXY-D-XYLULOSE 5-PHOSPHATE SYNTHASE (DXS)
The Mtb genome contains two genes encoding for DXR, Rv2682c and Rv3379c. Only Rv2682c has synthase activity due to loss of a critical residue in Rv3379c resulting from an N-terminal truncation.51–53 DXS is the first enzyme of the MEP pathway and catalyzes one of two rate-determining reactions in the pathway.54 With the release of CO2, DXS catalyzes a condensation reaction joining Pyr and D-GAP to form DXP. DXS sits at a branch point in the pathway as, in addition to the production of isoprenoid precursors, it also plays an important role in the biosynthesis of vitamins B1 and B6 (Figure 1).55–56 Therefore, inhibition of DXS could block multiple metabolic pathways, which could be advantageous in eliminating the bacterium.
Although X-ray crystal structures of DXS from Escherichia coli (E. coli) and Deinococcus radiodurans (D. radiodurans) have been published,57 the Mtb DXS crystal structure has not yet been reported. Hence, design of selective inhibitors against Mtb DXS based directly on structural information of the Mtb target has been hindered to some extent. Other strategies, however, have assisted in the design of such inhibitors. Significant structural57–58 and mechanistic 53,57–59 understanding of DXS from E. coli, D. radiodurans, and P. falciparum have greatly impacted inhibitor design against these and other microorganisms, including Mtb.
Sequence alignment of DXS homologs shows that Mtb DXS shares 45% and 40% sequence identity compared with D. radiodurans and E. coli DXS, respectively.54 Based on crystal structures from the E. coli and D. radiodurans DXS homologs, it is known that the enzyme exists as a homodimer of three-domain monomers, similar to mammalian transketolase (TK).57 Interestingly, however, the domain arrangements of the two enzymes are significantly different, affecting the location of each enzyme’s active site. The active site of transketolase lies at the dimer interface, with active site residues stemming from two subunits. The active site of DXS, however, lies between domains of a single monomer. Residues comprising the DXS active site belong to a single subunit.57 The location difference noted here is impactful in terms of DXS inhibitor design. As also noted in this report, the ThDP and pyruvate binding sites are conserved across DXS homologs as well as residues outside these binding sites including Arg420 and Arg478, part of the DRAG sequence and suggested to play an important role in the mechanistic features of DXS.59–60
In the DXS active site, ThDP is activated by deprotonation of the C-2 carbon on the thiazolium ring to generate the ThDP ylide (Figure 3).61 The ThDP ylide, acting as a nucleophile, then attacks C-2 of Pyr to form the predecarboxylation intermediate, lactyl thiamin diphosphate (LThDP). Following a random sequential order, DXS, LThDP, and D-GAP form a ternary complex, which is required for decarboxylation.62–64 With the release of CO2, the product is a carbanion, in equilibrium with its enamine form. The carbanion attacks the aldehyde carbon of D-GAP bound to DXS. The generated tetrahedral intermediate is deprotonated, releasing the ketone product (DXP) as well as the regenerated ThDP ylide for the next round of catalysis.
Figure 3.

Proposed mechanism for the DXP formation catalyzed by DXS
Extensive studies have been conducted on DXS from D. radiodurans, E. coli, and Plasmodium falciparum (P. falciparum) clarifying the enzyme’s unique mechanism.62, 65 While the overall chemical reaction catalyzed by DXS combines features of a carboligase (like TK) and a decarboxylase (like pyruvate dehydrogenase, PDH), DXS follows a unique mechanism, impacting inhibitor development.42, 53, 58, 62, 66–67 The pyruvate decarboxylation catalyzed by DXS has a unique mechanism. Most ThDP-dependent enzymes such as TK and acetolactate synthase follow a canonical ping-pong kinetic mechanism.68 Contrary to this, a ternary complex is formed during DXS catalysis, involving D-GAP, LThDP, and DXS (Figure 3). This complex is formed through random sequential binding, distinguishing it from other enzymes in this class.53, 62–63, 65, 67, 69–70 As reported in recent studies, DXS, in the absence of D-GAP, stabilizes the predecarboxylation intermediate, LThDP (Figure 3), presumably via a closed conformation of the region near the active site.63–64 D-GAP-binding is thought to cause a conversion of the enzyme to an open conformation, which greatly accelerates the rate of decarboxylation by at least 600-fold.63–64 The unusually large active site volume present in DXS allows for the bulky ternary complex to form. This striking mechanistic feature of DXS, in contrast to TK and PDH, highlights the possibility of selective inhibitor design. A possible strategy would be using pyruvate mimics that selectively inhibit formation of the unique ternary complex, which is indispensable for DXS catalysis.
In addition to the structural and mechanistic features of DXS, feedback inhibition of recombinant DXS cloned from Populus trichocarpa by the ultimate products in the pathway, IPP and DMAPP, has been reported.71 IPP and DMAPP were found to compete with ThDP when binding to DXS, with Ki values in micromolar range (60–80 μM). Similar feedback regulation was also recently confirmed on DXS from poplar trees.72 Despite the species difference, the report indicates that the products from the MEP pathway could downregulate DXS, limiting production of isoprenoids as a common regulatory mechanism.73
A variety of DXS inhibitors have been reported (Figure 4). The first reported DXS inhibitor was ketoclomazone which demonstrated good activity against DXS from Haemophilus influenza (Ki = 23.3 μM) and modest activity against DXS from Chlamydomonas (IC50 = 0.1 mM).74–76 Freel Meyers and co-workers have reported a series of alkylacetylphosphonates (Figure 4) as DXS inhibitors against a variety of pathogens including Mtb.77–79 These alkylacetylphosphonates are mechanism-based pyruvate mimics that are unable to undergo decarboxylation, and the size of the alkyl substitution regulates the selectivity for DXS over other ThDP-dependent enzymes. Specifically, the butylacetylphosphonate analog shows single-digit micromolar activity against Mtb DXS (Ki = 4.0 μM), as well as an outstanding selectivity profile over mammalian ThDP-dependent enzymes.78 While these phosphonates at first showed modest antimicrobial activity against pathogenic bacteria (e.g., E. coli MIC90 = 2500 μM), activity improved 500-fold by altering growth medium (E. coli MIC90 = 5 μM).79 Additionally, the antimicrobial effects of butylacetylphosphonate were shown to be synergistic with fosmidomycin against E. coli.78 Finally, Hirsch et al. reported a series of thiamin derivatives as Mtb DXS inhibitors (Figure 4). The active compounds are closely related to ThDP and have sub-micromolar activity against the enzyme.80 Taken together, these compounds serve as a proof-of-concept that DXS inhibitors could be viable antitubercular agents.
Figure 4.

Selected inhibitors of DXS in Mtb
The risk of developing resistance is an important consideration when validating a new drug target. Drug resistance in Mtb can be developed intrinsically or extrinsically. As the product of a single gene copy, drug resistance due to a single point mutation in Mtb DXS is less likely to lead to resistance compared with enzymes that stem from several gene copies, offering possible redundancy.54 In addition, the high conservation of the active site residues gives confidence that the risk of endogenous resistance through active site mutation is low.60 The overexpression of DXS, as a second form of intrinsic drug resistance, could be downregulated by the downstream metabolites IPP and DMAPP, as mentioned above.71, 81 Interestingly, spontaneous mutations in E. coli were found to rescue strains depleted of DXS.82 In this report, the loss of DXS activity could be bypassed via a single point mutation in the gene encoding E. coli PDH complex E1 subunit.82 As with all microorganisms, resistance may develop extrinsically via many avenues including drug efflux.83
DXS is a promising drug target against tuberculosis due to its unique structural and mechanistic features that encourage selective inhibitor development. Further, with its unique mechanism, synergism remains a possible strategy against drug resistance.78, 84–85 Essentiality of DXS for bacterial viability, as well as its important role in the MEP and vitamin B1/B6 pathways encourage further pursuit of inhibitors. Report of an Mtb DXS crystal structure could greatly improve our understanding of the roles of active site residues and the ternary complex binding pocket, accelerating design of Mtb DXS inhibitors working through this novel mode of action.
1-DEOXY-D-XYLULOSE 5-PHOSPHATE REDUCTOISOMERASE (IspC/DXR)
DXR is the second enzyme in the MEP pathway and catalyzes the second rate-determining reaction that converts DXP to MEP with the assistance of NADPH.86–87 As DXP is also a biosynthetic intermediate for the biosynthesis vitamins B1 and B6, DXR catalyzes the first committed step in the MEP pathway. The DXR reaction product is unique in bacterial metabolism, giving the possibility to selectively block the biosynthesis of isoprenoids.43 By screening ~20,000 E. coli mutant colonies auxotrophic for 2-C-methylerythritol (ME), the free alcohol of MEP that E. coli can use as a permeable precursor to MEP, Seto and co-workers first identified the gene that encodes DXR and successfully expressed the recombinant protein.88 Homologs of this gene were detected in various bacterial genomes as well as in plants, accounting for the production of plastidic isoprenoids.88–89 The locus of Mtb IspC/DXR is Rv2870c and was shown to be essential for bacterial vitality.90 Interestingly, this result is in contrast to the transposon site hybridization (TraSH) analysis that predicted Rv2870c to be non-essential in Mtb.90–91 Thus, the individual analysis of Rv2870c helped to validate DXR as a promising drug target against Mtb.
DXR catalyzes a two-step reaction. In the first step, with the assistance of redox cofactor NADPH, DXR catalyzes the isomerization of DXP to 2C-methyl-D-erythrose 4-phosphate (MEsP) where a divalent metal cation is absolutely required.92 In the second step, MEsP is reduced to MEP.43 Significant effort has been spent understanding the mechanism of DXR catalysis. Two mechanisms have been proposed for the initial isomerization phase of DXR (Figure 5).93 In both mechanisms, the divalent metal cation acts as a Lewis acid to activate the carbonyl of substrate DXP and coordinate its C-3 hydroxyl group. In the first hypothesis, an α-ketol rearrangement occurs via a 1,2-alkyl shift. The C-2 carbonyl is reduced to a tertiary alcohol. Abstraction of the proton from the C-3 hydroxyl group yields a carbonyl at C-3 to produce isomerization intermediate MEsP.94–95 A second mechanistic hypothesis for the isomerization is a sequential retro-aldol/aldol mechanism.94–96 Upon deprotonation of the C-4 hydroxyl group, formation of a C-4 carbonyl leads to cleavage of the C-3-C-4 carbon-carbon bond. Two intermediates would be generated: hydroxylacetone enolate and glycoaldehyde phosphate.97 An aldol reaction with C-2 as the nucleophile then recombines these intermediates and forms intermediate MEsP. It remains controversial which mechanism is actually at work for the isomerization step. Kinetic isotope effect studies using selectively deuterated substrate (DXP) demonstrated the change in hybridization of both the C-3 and C-4 carbon atoms, arguing against the a-ketol rearrangement mechanism.95, 97 On the other hand, NMR studies aiming to detect the hydroxyacetone enolate intermediate were not successful, and exogenously added hydroxyacetone enolate or glycoaldehyde phosphate were not converted to MEP by the enzyme. These data argue against the retro-aldol/aldol mechanism.94, 98 It is, however, possible that the putative fragments in the retro-aldol/aldol mechanism are strictly held in the active site, thereby being recalcitrant to detection or being replaced by exogenous sources. The two putative mechanisms were also examined via kinetic studies, where a C-1 mono-fluorinated DXP analog was synthesized.99 Thus, the carbonyl group on C-2 is made more electron deficient, which should accelerate a 1,2-migration if the reaction proceeds through the α-ketol rearrangement mechanism.99 However, the fluorinated DXP analog appeared to be a poor substrate with a higher Km value (100 μM) and a lower kcat value (4.5 s−1), when compared to DXP (Km = 61μM, kcat = 21.3 s−1).99 Hence, a retro-aldol/aldol mechanism is probably more likely to occur.
Figure 5.

Two proposed mechanisms for the MEsP formation catalyzed by DXR
Contrary to the isomerization step, the reduction mechanism of the DXR catalyzed reaction is more straightforward. The Mtb DXR catalyzed reduction reaction likely follows a random, sequential reaction mechanism where DXP and NADPH bind to the enzyme in a random order.96 This mechanism is in slight contrast to the sequential mechanism suggested for E. coli ortholog where NADPH binds before DXP.92 Mtb DXR has an ordered product release of NADP+ first, followed by MEP dissociation, which is rate-limiting for enzyme turnover.100 Monitoring NADPH oxidation has been a useful means of studying DXR enzyme kinetics as well as inhibitory activity of novel ligands.101–105
Fosmidomycin (FR-31564) was isolated from Streptomyces lavendulae and showed activity against many Gram-negative bacteria as well as the protozoan parasite P. falciparum.48, 106–109 Fosmidomycin was evaluated for the treatment of urinary tract infections and in Phase I and II clinical trials in 1985, before it was demonstrated to inhibit DXR specifically and block isoprenoid biosynthesis a decade later.87, 110–114 Because of the significance of fosmidomycin, research interest in DXR greatly increased and thus DXR has become the most studied enzyme in the MEP pathway. The first apo crystal structure was reported in 2002 by Reuter et al. using E. coli DXR.115 Soon after in 2006, the crystal structure of Mtb DXR was solved by Henriksson et al.116 These early structures revealed that the enzyme is a homodimer, with each subunit consisting of three domains including an N-terminal domain for cofactor binding, a central catalytic domain, and a C-terminal helical domain. Sequence alignments of DXR homologs from Mtb, E. coli, and Zymomonas mobilis showed that the central catalytic domain of the enzymes is highly conserved with 45–50% sequence identity, and the residues interacting with substrate DXP are even more strictly conserved.117 Thus, DXR inhibitors resembling DXP analogs are expected to have great potential against a broad spectrum of bacteria. Nonetheless, when ligands bind strongly to DXR, a dramatic conformational change occurs where a loop migrates, covering the active site. This is known as the “loop-open” to “loop-closed” conformation change.118 Thus, the complexity in predicting the binding mechanism of designed DXR inhibitors has made novel development of such inhibitors challenging despite the availability of sufficient crystallographic information.
Fosmidomycin is a potent inhibitor of E. coli DXR (IC50 = 8.2 nM),112 and its binding is competitive with substrate DXP and noncompetitive with cofactor NADPH.96 As would be expected given such high sequence identity, fosmidomycin is also a potent inhibitor of Mtb DXR, with an IC50 value of 80 nM, and a quaternary co-crystal structure with the enzyme, NADPH, and Mn2+ was resolved with high resolution.116 Despite the efficacy of fosmidomycin against various pathogens including E. coli (MIC90 = 0.78 μg/mL)109, Proteus mirabilis (MIC90 = 1.56 μg/mL),109 and P. falciparum (IC50 = 1 μM),119 as well as its potent inhibition against Mtb DXR, it is completely inactive against Mtb.120 As shown by Brown and Parish,90 its lack of whole cell Mtb activity is because the highly polar fosmidomycin cannot penetrate the waxy mycobacterial cell wall, a common cause of mycobacterial resistance to many antibiotics. In addition, Mtb lacks a glycerol 3-phosphate transporter (GlpT) used by fosmidomycin and other highly polar compounds to enter nonmycobacteria.120–121 Thus, the design of novel TB drug candidates via inhibition of DXR presents the following challenge: A polar inhibitor is required for binding to the hydrophilic substrate binding pocket. Such an inhibitor must also be lipophilic enough to penetrate through the greasy bacterial cell wall. The balance between these two physical characteristics represents the backbone of successful design of antitubercular DXR inhibitors.
Some time ago, lipophilic prodrugs of fosmidomycin and its acetyl analog FR900098 were synthesized and examined as antimalarial agents.45, 122–123 We adopted this strategy to ask if, as a proof-of-concept, these prodrug esters could display antitubercular activities better than that of the parent compounds. Lipophilic esters of the phosphonate moiety on fosmidomycin/FR900098 were subsequently synthesized, and tested against the cell growth of Mtb.101 These esters mask the polar phosphonate of fosmidomycin with nonpolar groups, gaining the lipophilicity required to penetrate the bacterial cell wall. The esters then undergo hydrolysis to yield the polar phosphonates. Regenerated fosmidomycin/FR900098 then inhibits DXR inside the bacilli to block the biosynthesis of essential isoprenoids and ultimately result in killing the bacterial cell. Examination of several prodrug esters of fosmidomycin/FR900098 showed inhibition of Mtb cell growth, albeit with weak MIC values (25–500 μg/mL, Figure 6).101 Research efforts were then dedicated to the development of more effective Mtb DXR inhibitors.
Figure 6.

Selected fosmidomycin analogs as Mtb DXR inhibitors
Because of the relatively clear mechanism of action of fosmidomycin, its low toxicity, its progress in clinical trials, as well as its ability to work synergistically with other drugs yielding enhanced bactericidal activities,84 it is not surprising that abundant research has focused on the synthesis of fosmidomycin analogs (Figure 6). These analogs aim at providing more options as novel Mtb DXR inhibitors, as well as improving antibacterial activity against Mtb. Conspicuously, most of the fosmidomycin analogs reported retained the retro-hydroxamate or hydroxamate moiety to mimic the crucial interaction of fosmidomycin with the divalent metal ion, as well as the phosphonate moiety, which favorably anchors the inhibitor in the active site, enabling potent enzymatic inhibition.
Since the adjacent adenosine-binding pocket of NADPH is druggable,50, 116 fosmidomycin analogs with aromatic groups extending from the N-acyl or N-hydroxyl moieties have been synthesized. This bisubstrate strategy aims to occupy both the DXP and NADPH sites in an attempt to gain more potent inhibition.102–103, 105, 124 While data supports the bisubstrate binding behavior of this strategy,105 fosmidomycin so far remains the most potent Mtb DXR inhibitor among these analogs.
Fosmidomycin analogs with modifications on different positions of the scaffold were also examined as Mtb DXR inhibitors. These changes primarily involve substitution on the alpha or beta carbon atom, relative to the phosphorus.104, 124–125 Some of these analogs showed more potent inhibition against Mtb Dxr when compared with the parent compound (e.g., an α-aryl-β-thia analog has an IC50 value of 9.2 nM,125 Figure 6). However, prodrug modifications to improve Mtb cell penetration have been reported for only a few of these analogs. The best of these prodrug compounds, bearing unsaturation along the propyl chain, shows significant of Mtb cell growth with an MIC value of 9.4 μg/mL.103, 105 Additional analogs, with low micromolar activity against the enzyme have unreported Mtb whole cell activity.104, 124 Hence, poor uptake of fosmidomycin analogs by Mtb cells remains a challenge, and the corresponding intracellular delivery/fate of these compounds is still underexplored.
Similar to DXS, DXR is encoded by a single gene copy, and thus has a lower risk of developing intrinsic drug resistance clinically due to a single point mutation.90 Endogenous resistance through active site mutation is improbable due to high conservation of the central catalytic domain.88, 117 Although mutation in E. coli dxr has been reported,87 the likelihood of this mutation occurring clinically is unknown. Fosmidomycin resistance was found in strains of bacteria deficient in GlpT,126–128 the transporter used to take up the drug. As noted above, GlpT is absent in Mtb. Further, lipophilic prodrugs of fosmidomycin analogs are active against GlpT mutants, bypassing this mechanism of resistance.126 Fosmidomycin resistance in Burkholderia was also noted due to upregulated efflux,127 a potential issue for an antitubercular agent. More recently, P. falciparum strains without Pfhad1 were found to be resistant to fosmidomycin.119, 129 The had1 gene encodes for a sugar phosphatase that regulates the MEP pathway.
2C-METHYL-D-ERYTHRITOL 2,4-CYCLODIPHOSPHATE SYNTHASE (IspF)
IspF is the fifth enzyme in the MEP pathway, catalyzing the cyclization and dephosphorylation reactions converting CDP-MEP to MEcPP and releasing CMP concomitantly.43 IspF is encoded by the Rv3581c gene, which was reported to be essential for the viability of Mtb, showing its potential as a valuable target.91, 130 Despite over 50 crystal structures of IspF appearing in the literature from different organisms, including many bacteria and P. falciparum, the structure of Mtb IspF remains undetermined.130–131 Nevertheless, most IspF orthologs share high sequence and structural identity, especially regarding key interactions in the active site.45 Moreover, the known crystal structure of IspF from Mycobacterium smegmatis (M. smegmatis) is a reliable homology model to adopt for structure-based inhibitor design against Mtb IspF because of the superior 73% sequence identity between the two enzymes.130
The reported crystal structures revealed that IspF, in general, forms a bell-shaped homotrimer, which is tightly associated and contains three active sites at each monomer interface.132 Each monomer consists of a four-stranded β-sheet and a flexible two-stranded β-sheet on one side, as well as three α- and two 310 helices on the other.132 This flexible two-stranded β-sheet caps the pocket involved in binding the phosphate moiety of the substrate, and becomes ordered upon substrate binding. Interestingly, this conformational change leaves the remaining substrate-binding pockets unaffected.133 Notably, two divalent ions are also present as they contribute to substrate alignment and are required for facilitating catalysis. They include Zn2+, with tetrahedral coordination, and Mn2+ or, under physiological conditions, Mg2+, with octahedral coordination.43 These metal cofactors act as Lewis acids to activate the diphosphate group of CDP-MEP. This activation facilitates nucleophilic attack by the terminal phosphate (from MEP) onto the β-phosphorus atom (Figure 7). A pentacoordinate transition state is subsequently generated, stabilized by the metal cations. This intermediate then collapses by (re)formation of a phosphoryl bond to produce the product MEcPP, with concomitant release of CMP.37
Figure 7.

Proposed mechanism for the MEcPP formation catalyzed by IspF
The flexibility of IspF complicates inhibitor design. Despite this, IspF has been predicted to be the most druggable target in the MEP pathway due to the lipophilic character of its active site compared with many other enzymes in the pathway.50 IspD, for example, has an active site with low lipophilicity and is rather solvent-exposed.50 Even with very potent activity against their target enzymes, polar inhibitors can rapidly lose efficacy when introduced to the lipid-rich cell wall of mycobacteria.121 Thus, the lipophilic character of the IspF active site renders this enzyme notably advantageous as a drug target against Mtb. Thus, rational design of nonpolar Mtb IspF inhibitors is feasible and could show promise in eliminating the bacillus.
IspF is an interesting target against Mtb also because of its involvement in metabolic regulation. Downstream products of the MEP pathway, such as IPP/DMAPP, geranyl diphosphate (GDP), and farnesyl diphosphate (FDP), are reported to bind to a hydrophobic cavity of IspF, indicating a possible feedback regulatory role of this enzyme.132, 134 In addition, IspF is stabilized and activated by MEP, the upstream intermediate produced by DXR.67, 127 This data outlines an MEP-specific regulatory mechanism using feedforward activation.73, 135 MEcPP, the product of IspF, appears to play an important role in general bacterial metabolism as well. It has been suggested that MEcPP acts as an antistressor signal in bacteria that accumulates when bacteria are under oxidative stresses,136–138 and has a resuscitating effect on non-culturable M. smegmatis.139 Efflux of MEcPP in E. coli was measured when certain enzymes were overexpressed.140 These results suggest the possibility of MEcPP being a branch point in bacterial metabolism, a role that necessitates more elucidation. In terms of drug resistance, the risk of a single point mutation is low in Mtb IspF, which is encoded by a single gene copy.130 High sequence and structural identities of the active site residues of most IspF orthologs decreased the chance of endogenous resistance via active site mutation.45 Further studies on drug resistance related to IspF remains unreported to date.
Despite the promise of IspF as a drug target against Mtb and some inhibitors reported against the orthologs from different organisms,141–144 inhibitor design targeting Mtb IspF is relatively underexplored. Part of the challenge toward discovery of an IspF inhibitor stems from the instability of the enzyme substrate, CDP-MEP, the lack of an assay for direct measurement of IspF substrate turnover,145 and the instability/insolubility of the enzyme itself.144 To the best of our knowledge, only a few non-cytidine-like thiazolopyrimidine derivatives144 were tested against Mtb IspF (Figure 8). These show potent activity against the enzyme with the most active compound displaying an IC50 value of 2.1 μM.144 While the Mtb MIC value of this compound has not been reported, its calculated clogP value is 5.96, indicating high lipophilicity that generally favors cell penetration in mycobacteria.146 This analysis supports the potential druggability of IspF as a target against Mtb as mentioned above, encouraging more research in rational inhibitor design via this promising antitubercular drug target.
Figure 8.

Selected inhibitors of IspF in Mtb
CONCLUSIONS
Various metabolic biosynthetic pathways have been explored to fight the pandemic threat caused by tuberculosis. The MEP pathway, which is used by Mtb to produce five carbon isoprenoids, stands out as a promising target toward developing antitubercular agents. MEP enzymes DXS, DXR, and IspF appear to be promising drug targets for tuberculosis in our view. Each of these enzymes is reported to be essential for the bacterial viability in Mtb and possesses druggable binding pockets. DXS catalyzes the first reaction in the MEP pathway to synthesize DXP, which is a branch point that is also involved in the biosynthesis of vitamins B1 and B6. Inhibitor design against Mtb DXS is attractive due to its importance in Mtb metabolism and unique mechanistic/structural features. With X-ray crystal structures available for Mtb DXR, the design of Mtb DXR inhibitors is greatly facilitated. The Mtb DXR inhibitor fosmidomycin, with an unambiguous mode of action and outstanding safety profile, is a promising parent structure in design of Mtb DXR inhibitors and antitubercular agents. IspF could be the most druggable target in the pathway, due to the lipophilic character of its active site. Reported inhibitors with favorable Mtb IspF inhibition possess clogP values that appear promising for mycobacterial cell wall penetration. These three enzymes in the MEP pathway show great potential as drug targets in fighting tuberculosis. Designing potent and selective inhibitors against these enzymes in Mtb should be of great significance and may facilitate the development of new TB drugs via a novel mode of action.
ACKNOWLEDGEMENTS
We are grateful for support from the National Institutes of Health (NIAID R01 AI123433) and the George Washington University Department of Chemistry.
ABBREVIATIONS
- ATP
adenosine triphosphate
- CoA
coenzyme A
- CoaBC
the bifunctional enzyme catalyzing the second and third steps of CoA biosynthesis
- CDP-ME
4-diphosphocytidyl-2C-methyl-D-erythritol
- CDP-MEP
4-diphosphocytidyl-2C-methyl-D-erythritol 2-phosphate
- CMP
cytidine monophosphate
- D. radiodurans
Deinococcus radiodurans
- DMAPP
dimethylallyl pyrophosphate
- DXP
1-deoxy-D-xylulose-5-phosphate
- DXS
1-deoxy-D-xylulose-5-phosphate synthase
- E. coli
Escherichia coli
- FDP
farnesyl diphosphate
- D-GAP
D-glyceraldehyde-3-phosphate
- GDP
geranyl diphosphate
- H. influenza
Haemophilus influenza
- HIV
human immunodeficiency virus
- HMBPP
1-hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate
- IC50
inhibitory concentration where the response (or binding) is reduced by half
- IPP
isopentenyl pyrophosphate
- IspC/DXR
1-deoxy-D-xylulose-5-phosphate reductoisomerase
- IspD
4-diphosphocytidyl-2C-methyl-D-erythritol synthase
- IspE
4-diphosphocytidyl-2C-methyl-D-erythritol kinase
- IspF
2Cmethyl-D-erythritol 2,4-cyclodiphosphate synthase
- IspG
1-hydroxy-2-methyl-2(E)butenyl-4-diphosphate Synthase
- IspH
1-hydroxy-2-methyl-2(E)-butenyl-4-diphosphate reductase
- Km
Michaelis constant
- kcat
turnover number
- LThDP
lactyl thiamin diphosphate
- M. smegmatis
Mycobacterium smegmatis
- MEcPP
2C-methyl-D-erythritol-2,4-cyclodiphosphate
- MEP
methylerythritol phosphate
- MEsP
2C-methyl-D-erythrose 4-phosphate
- MIC90 or MIC99
the lowest concentration of the antibiotic at which 90% or 99% of the isolates were inhibited
- Mtb
Mycobacterium tuberculosis
- NAD+
nicotinamide adenine dinucleotide (oxidized)
- NADP+
nicotinamide adenine dinucleotide phosphate (oxidized)
- NADPH
nicotinamide adenine dinucleotide phosphate (reduced)
- P. falciparum
Plasmodium falciparum
- PDH
pyruvate dehydrogenase
- Pyr
pyruvate
- TB
tuberculosis
- ThDP
thiamin diphosphate
- TK
transketolase
- TraSH
transposon site hybridization
Footnotes
Notes
The authors declare no competing financial interest.
REFERENCES
- 1.Global Tuberculosis Report 2016, World Health Organization. http://www.who.int/tb/publications/globalreport/en/.
- 2.Garcia-Monco JC, Chapter 100 - Tuberculosis In Handbook of Clinical Neurology, Biller J; Ferro JM, Eds. Elsevier: 2014; Vol. 121, pp 1485–1499. DOI: 10.1016/B978-0-7020-4088-7.00100-0. [DOI] [PubMed] [Google Scholar]
- 3.Zumla A; Squire SB; Chintu C; Grange JM, The tuberculosis pandemic: implications for health in the tropics. Transactions of the Royal Society of Tropical Medicine and Hygiene 1999, 93 (2), 113–117. DOI: 10.1016/S0035-9203199j90278-X. [DOI] [PubMed] [Google Scholar]
- 4.Russell DG; Cardona P-J; Kim M-J; Allain S; Altare F, Foamy macrophages and the progression of the human tuberculosis granuloma. Nat Immunol 2009,10 (9), 943–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tuberculosis fact sheets, World Health Organization. http://www.who.int/mediacentre/factsheets/fs104/en/.
- 6.Barry CE; Boshoff HI; Dartois V; Dick T; Ehrt S; Flynn J; Schnappinger D; Wilkinson RJ; Young D, The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat Rev Micro 2009, 7 (12), 845–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guidelines for treatment of drug-susceptible tuberculosis and patient care (2017. update), World Health Organization. http://www.who.int/tb/publications/2017/dstb_guidance2017/en/.
- 8.Cox E; Laessig K, FDA Approval of Bedaquiline — The Benefit-Risk Balance for Drug-Resistant Tuberculosis. New England Journal of Medicine 2014, 371 (8), 689–691. DOI: 10.1056/NEJMp1314385. [DOI] [PubMed] [Google Scholar]
- 9.Field SK, Bedaquiline for the treatment of multidrug-resistant tuberculosis: great promise or disappointment? Therapeutic Advances in Chronic Disease 2015, 6 (4), 170–184. DOI: 10.1177/2040622315582325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Osborne R, First novel anti-tuberculosis drug in 40 years. Nat Biotech 2013, 31 (2), 89–91. DOI: 10.1038/nbt0213-89. [DOI] [PubMed] [Google Scholar]
- 11.Wipperman MF; Sampson NS; Thomas ST, Pathogen roid rage: Cholesterol utilization by Mycobacterium tuberculosis. Critical Reviews in Biochemistry and Molecular Biology 2014, 49 (4), 269–293. DOI: 10.3109/10409238.2014.895700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang M; Lu R; Guja KE; Wipperman MF; Clair JR St.; Bonds AC; Garcia-Diaz M; Sampson NS, Unraveling Cholesterol Catabolism in Mycobacterium tuberculosis: ChsE4-ChsE5 α 2 β 2 Acyl-CoA Dehydrogenase Initiates β-Oxidation of 3-Oxo-cholest-4-en-26-oyl CoA. ACS Infectious Diseases 2015, 1 (2), 110–125. DOI: 10.1021/id500033m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nesbitt NM; Yang X; Fontán P; Kolesnikova I; Smith I; Sampson NS; Dubnau E, A Thiolase of Mycobacterium tuberculosis Is Required for Virulence and Production of Androstenedione and Androstadienedione from Cholesterol. Infection and Immunity 2010, 78 (1), 275–282. DOI: 10.1128/iai.00893-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yam KC; D’Angelo I; Kalscheuer R; Zhu H; Wang J-X; Snieckus V; Ly LH; Converse PJ; Jacobs WR Jr.; Strynadka N; Eltis LD, Studies of a Ring-Cleaving Dioxygenase Illuminate the Role of Cholesterol Metabolism in the Pathogenesis of Mycobacterium tuberculosis. PLOS Pathogens 2009, 5 (3), e1000344. DOI: 10.1371/journal.ppat.1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang JC; Miner MD; Pandey AK; Gill WP; Harik NS; Sassetti CM; Sherman DR, igr Genes and Mycobacterium tuberculosis Cholesterol Metabolism. Journal of Bacteriology 2009, 191 (16), 5232–5239. DOI: 10.1128/jb.00452-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McKinney JD; zu Bentrup KH; Munoz-Elias EJ; Miczak A; Chen B; Chan W-T; Swenson D; Sacchettini JC; Jacobs WR; Russell DG, Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature 2000, 406 (6797), 735–738. [DOI] [PubMed] [Google Scholar]
- 17.Munoz-Elias EJ; McKinney JD, Mycobacterium tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence. Nat Med 2005, 11 (6), 638–644. DOI: http://www.nature.com/nm/journal/v11/n6/suppinfo/nm1252_S1.html. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krieger Inna V.; Freundlich Joel S.; Gawandi Vijay B.; Roberts Justin P.; Gawandi Vidyadhar B.; Sun Q; Owen Joshua L.; Fraile Maria T.; Huss Sofia I.; Lavandera J-L; loerger Thomas R.; Sacchettini James C., Structure-Guided Discovery of Phenyl-diketo Acids as Potent Inhibitors of M. tuberculosis Malate Synthase. Chemistry & Biology 2012, 19 (12), 1556–1567. DOI: 10.1016/j.chembiol.2012.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ahn Y-M; Boshoff HI, Elevation of Fumarate Levels Compromise Redox Control and Viability in Mycobacterium tuberculosis. Cell Chemical Biology 2017, 24 (3), 243–245. DOI: 10.1016/j.chembiol.2017.03.003. [DOI] [PubMed] [Google Scholar]
- 20.Ruecker N; Jansen R; Trujillo C; Puckett S; Jayachandran P; Piroli GG; Frizzell N; Molina H; Rhee KY; Ehrt S, Fumarase Deficiency Causes Protein and Metabolite Succination and Intoxicates Mycobacterium tuberculosis. Cell Chemical Biology 2017, 24 (3), 306–315. DOI: 10.1016/j.chembiol.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim J-H; O’Brien KM; Sharma R; Boshoff HIM; Rehren G; Chakraborty S; Wallach JB; Monteleone M; Wilson DJ; Aldrich CC; Barry CE; Rhee KY; Ehrt S; Schnappinger D, A genetic strategy to identify targets for the development of drugs that prevent bacterial persistence. Proceedings of the National Academy of Sciences 2013, 110 (47), 19095–19100. DOI: 10.1073/pnas.1315860110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang X; Ahn Y-M; Lentscher AG; Lister JS; Brothers RC; Kneen MM; Gerratana B; Boshoff HI; Dowd CS, Design, synthesis, and evaluation of substituted nicotinamide adenine dinucleotide (NAD+) synthetase inhibitors as potential antitubercular agents. Bioorganic & Medicinal Chemistry Letters 2017, 27 (18), 4426–4430. DOI: https://doi.org/10.1016Zj.bmcl.2017.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duckworth Benjamin P.; Geders Todd W.; Tiwari D; Boshoff Helena I.; Sibbald Paul A.; Barry Clifton E.; Schnappinger D; Finzel Barry C.; Aldrich Courtney C., Bisubstrate Adenylation Inhibitors of Biotin Protein Ligase from Mycobacterium tuberculosis. Chemistry & Biology 2011, 18 (11), 1432–1441. DOI: 10.1016/j.chembiol.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi C; Tiwari D; Wilson DJ; Seiler CL; Schnappinger D; Aldrich CC, Bisubstrate Inhibitors of Biotin Protein Ligase in Mycobacterium tuberculosis Resistant to Cyclonucleoside Formation. ACS Medicinal Chemistry Letters 2013, 4 (12), 1213–1217. DOI: 10.1021/ml400328a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qiao C; Gupte A; Boshoff HI; Wilson DJ; Bennett EM; Somu RV; Barry CE; Aldrich CC, 5’-O-[(N-Acyl)sulfamoyl]adenosines as Antitubercular Agents that Inhibit MbtA: An Adenylation Enzyme Required for Siderophore Biosynthesis of the Mycobactins. Journal of Medicinal Chemistry 2007, 50 (24), 6080–6094. DOI: 10.1021/jm070905o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Somu RV; Wilson DJ; Bennett EM; Boshoff HI; Celia L; Beck BJ; Barry CE; Aldrich CC, Antitubercular Nucleosides That Inhibit Siderophore Biosynthesis: SAR of the Glycosyl Domain. Journal of Medicinal Chemistry 2006, 49 (26), 7623–7635. DOI: 10.1021/jm061068d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Somu RV; Boshoff H; Qiao C; Bennett EM; Barry CE; Aldrich CC, Rationally Designed Nucleoside Antibiotics That Inhibit Siderophore Biosynthesis of Mycobacterium tuberculosis. Journal of Medicinal Chemistry 2006, 49 (1), 31–34. DOI: 10.1021/jm051060o. [DOI] [PubMed] [Google Scholar]
- 28.Neres J; Labello NP; Somu RV; Boshoff HI; Wilson DJ; Vannada J; Chen L; Barry CE; Bennett EM; Aldrich CC, Inhibition of Siderophore Biosynthesis in Mycobacterium tuberculosis with Nucleoside Bisubstrate Analogues: Structure-Activity Relationships of the Nucleobase Domain of 5’-O-[N- (Salicyl)sulfamoyl]adenosine. Journal of Medicinal Chemistry 2008, 51 (17), 5349–5370. DOI: 10.1021/jm800567v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vannada J; Bennett EM; Wilson DJ; Boshoff HI; Barry CE; Aldrich CC, Design, Synthesis, and Biological Evaluation of β-Ketosulfonamide Adenylation Inhibitors as Potential Antitubercular Agents. Organic Letters 2006, 8 (21), 4707–4710. DOI: 10.1021/ol0617289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Evans JC; Trujillo C; Wang Z; Eoh H; Ehrt S; Schnappinger D; Boshoff HIM; Rhee KY; Barry CE; Mizrahi V, Validation of CoaBC as a Bactericidal Target in the Coenzyme A Pathway of Mycobacterium tuberculosis. ACS Infectious Diseases 2016, 2 (12), 958–968. DOI: 10.1021/acsinfecdis.6b00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spurgeon SL a. P., J. W., Biosynthesis of lsoprenoid Compounds, vol. I (Porter JW and Spurgeon SL, eds), pp. 146 Wiley, New York: 1981. [Google Scholar]
- 32.Lichtenthaler HK; Rohmer M; Schwender J, Two independent biochemical pathways for isopentenyl diphosphate and isoprenoid biosynthesis in higher plants. Physiologia Plantarum 1997, 101 (3), 643–652. DOI: 10.1111/j.1399-3054.1997.tb01049.x. [DOI] [Google Scholar]
- 33.Lichtenthaler HK, THE 1-DEOXY-D-XYLULOSE-5-PHOSPHATE PATHWAY OF ISOPRENOID BIOSYNTHESIS IN PLANTS. Annual Review of Plant Physiology and Plant Molecular Biology 1999, 50, 47–65. DOI: 10.1146/annurev.arplant.50.1.47. [DOI] [PubMed] [Google Scholar]
- 34.Goldstein JL; Brown MS, Regulation of the mevalonate pathway. Nature 1990, 343 (6257), 425–430. [DOI] [PubMed] [Google Scholar]
- 35.Rohmer M; Knani M; Simonin P; Sutter B; Sahm H, Isoprenoid biosynthesis in bacteria: a novel pathway for the early steps leading to isopentenyl diphosphate. Biochemical Journal 1993, 295 (2), 517–524. DOI: 10.1042/bj2950517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eoh H; Brennan PJ; Crick DC, The Mycobacterium tuberculosis MEP (2C-methyl-d-erythritol 4-phosphate) pathway as a new drug target. Tuberculosis 2009,89 (1), 1–11. DOI: 10.1016/j.tube.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hunter WN, The Non-mevalonate Pathway of Isoprenoid Precursor Biosynthesis. Journal of Biological Chemistry 2007, 282 (30), 21573–21577. DOI: 10.1074/jbc.R700005200. [DOI] [PubMed] [Google Scholar]
- 38.Sandoval-Calderón M; Guan Z; Sohlenkamp C, Knowns and unknowns of membrane lipid synthesis in streptomycetes. Biochimie 2017, 141 (Supplement C), 21–29. DOI: https://doi.org/10.1016Zj.biochi.2017.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Upadhyay A; Fontes FL; Gonzalez-Juarrero M; McNeil MR; Crans DC; Jackson M; Crick DC, Partial Saturation of Menaquinone in Mycobacterium tuberculosis: Function and Essentiality of a Novel Reductase, MenJ. ACS Central Science 2015, 1 (6), 292–302. DOI: 10.1021/acscentsci.5b00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crick DC; Schulbach MC; Zink EE; Macchia M; Barontini S; Besra GS; Brennan PJ, Polyprenyl Phosphate Biosynthesis inMycobacterium tuberculosis and Mycobacterium smegmatis. Journal of Bacteriology 2000, 182 (20), 5771–5778. DOI: 10.1128/jb.182.20.5771-5778.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murkin AS; Manning KA; Kholodar SA, Mechanism and inhibition of 1-deoxy-d-xylulose-5-phosphate reductoisomerase. Bioorganic Chemistry 2014, 57, 171–185. DOI: 10.1016/hbioorg.2014.06.001. [DOI] [PubMed] [Google Scholar]
- 42.Obiol-Pardo C; Rubio-Martinez J; Imperial S, The Methylerythritol Phosphate (MEP) Pathway for Isoprenoid Biosynthesis as a Target for the Development of New Drugs Against Tuberculosis. Current Medicinal Chemistry 2011,18 (9), 1325–1338. DOI: 10.2174/092986711795029582. [DOI] [PubMed] [Google Scholar]
- 43.Frank A; Groll M, The Methylerythritol Phosphate Pathway to Isoprenoids. Chemical Reviews 2017, 117 (8), 5675–5703. DOI: 10.1021/acs.chemrev.6b00537. [DOI] [PubMed] [Google Scholar]
- 44.Emily RJ; Cynthia SD, Inhibition of 1-Deoxy-D-Xylulose-5-Phosphate Reductoisomerase (Dxr): A Review of the Synthesis and Biological Evaluation of Recent Inhibitors. Current Topics in Medicinal Chemistry 2012, 12 (7), 706–728. DOI: 10.2174/156802612799984599. [DOI] [PubMed] [Google Scholar]
- 45.Masini T; Hirsch AKH, Development of Inhibitors of the 2C-Methyl-d-erythritol 4-Phosphate (MEP) Pathway Enzymes as Potential Anti-Infective Agents. Journal of Medicinal Chemistry 2014, 57 (23), 9740–9763. DOI: 10.1021/jm5010978. [DOI] [PubMed] [Google Scholar]
- 46.Bjorkelid C; Bergfors T; Henriksson LM; Stern AL; Unge T; Mowbray SL; Jones TA, Structural and functional studies of mycobacterial IspD enzymes. Acta Crystallographica Section D 2011, 67 (5), 403–414. DOI: doi: 10.1107/S0907444911006160. [DOI] [PubMed] [Google Scholar]
- 47.Shan S; Chen X, Crystallization and preliminary X-ray analysis of 4--diphosphocytidyl-2-C-methyl-d-erythritol kinase (IspE) from Mycobacterium tuberculosis. Acta Crystallographica Section F: Structural Biology and Crystallization Communications 2011, 67 (Pt 7), 821–823. DOI: 10.1107/S1744309111019567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Imlay LS; Armstrong CM; Masters MC; Li T; Price KE; Edwards RL; Mann KM; Li LX; Stallings CL; Berry NG; O’Neill PM; Odom AR, Plasmodium IspD (2-C-Methyl-d-erythritol 4-Phosphate Cytidyltransferase), an Essential and Druggable Antimalarial Target. ACS Infectious Diseases 2015, 1 (4), 157–167. DOI: 10.1021/id500047s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shan S; Chen X; Liu T; Zhao H; Rao Z; Lou Z, Crystal structure of 4-diphosphocytidyl-2-C-methyl-d-erythritol kinase (IspE) from Mycobacterium tuberculosis. The FASEB Journal2011,25 (5), 1577–1584. DOI: 10.1096/fj.10-175786. [DOI] [PubMed] [Google Scholar]
- 50.Masini T; Kroezen BS; Hirsch AKH, Druggability of the enzymes of the non-mevalonate-pathway. Drug Discovery Today 2013, 18 (23), 1256–1262. DOI: 10.1016/j.drudis.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 51.Querol J; Rodriguez-Concepcion M; Boronat A; Imperial S, Essential Role of Residue H49 for Activity of Escherichia coli 1-Deoxy-d-xylulose 5-Phosphate Synthase, the Enzyme Catalyzing the First Step of the 2-C-Methyl-d-erythritol 4-Phosphate Pathway for Isoprenoid Synthesis. Biochemical and Biophysical Research Communications 2001, 289 (1), 155–160. DOI: 10.1006/bbrc.2001.5957. [DOI] [PubMed] [Google Scholar]
- 52.Bailey AM; Mahapatra S; Brennan PJ; Crick DC, Identification, cloning, purification, and enzymatic characterization of Mycobacterium tuberculosis 1-deoxy-d-xylulose 5-phosphate synthase. Glycobiology 2002,12 (12), 813–820. DOI: 10.1093/glycob/cwf100. [DOI] [PubMed] [Google Scholar]
- 53.Sprenger GA; Schorken U; Wiegert T; Grolle S; de Graaf AA; Taylor SV; Begley TP; Bringer-Meyer S; Sahm H, Identification of a thiamin-dependent synthase in Escherichia coli required for the formation of the 1-deoxy-d-xylulose 5-phosphate precursor to isoprenoids, thiamin, and pyridoxol. Proceedings of the National Academy of Sciences 1997, 94 (24), 12857–12862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gierse RM; Redeem E; Diamanti E; Wrenger C; Groves MR; Hirsch AK, DXS as a target for structure-based drug design. Future Medicinal Chemistry 2017. DOI: 10.4155/fmc-2016-0239. [DOI] [PubMed] [Google Scholar]
- 55.Hill RE; Himmeldirk K; Kennedy IA; Pauloski RM; Sayer BG; Wolf E; Spenser ID, The Biogenetic Anatomy of Vitamin B6: A 13C NMR INVESTIGATION OF THE BIOSYNTHESIS OF PYRIDOXOL IN ESCHERICHIA COLI. Journal of Biological Chemistry 1996, 271 (48), 30426–30435. DOI: 10.1074/jbc.271.48.30426. [DOI] [PubMed] [Google Scholar]
- 56.Brown AC; Eberl M; Crick DC; Jomaa H; Parish T, The Nonmevalonate Pathway of Isoprenoid Biosynthesis in Mycobacterium tuberculosis Is Essential and Transcriptionally Regulated by Dxs. Journal of Bacteriology 2010,192 (9), 2424–2433. DOI: 10.1128/jb.01402-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xiang S; Usunow G; Lange G; Busch M; Tong L, Crystal Structure of 1-Deoxy-d-xylulose 5-Phosphate Synthase, a Crucial Enzyme for Isoprenoids Biosynthesis. Journal of Biological Chemistry 2007, 282 (4), 2676–2682. DOI: 10.1074/jbc.M610235200. [DOI] [PubMed] [Google Scholar]
- 58.Jurgenson CT; Begley TP; Ealick SE, The Structural and Biochemical Foundations of Thiamin Biosynthesis. Annu. Rev. Biochem. 2009, 78, 569–603. DOI: 10.1146/annurev.biochem.78.072407.102340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brammer Basta LA; Patel H; Kakalis L; Jordan F; Freel Meyers CL, Defining critical residues for substrate binding to 1-deoxy-d-xylulose 5-phosphate synthase -active site substitutions stabilize the predecarboxylation intermediate C2 α-lactylthiamin diphosphate. FEBS Journal 2014, 281 (12), 2820–2837. DOI: 10.1111/febs.12823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hahn FM; Eubanks LM; Testa CA; Blagg BSJ; Baker JA; Poulter CD, 1-Deoxy-d-Xylulose 5-Phosphate Synthase, the Gene Product of Open Reading Frame (ORF) 2816 and ORF 2895 inRhodobacter capsulatus. Journal of Bacteriology 2001,183 (1), 1–11. DOI: 10.1128/jb.183.1.1-11.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.White JK; Handa S; Vankayala SL; Merkler DJ; Woodcock HL, Thiamin Diphosphate Activation in 1-Deoxy-d-xylulose 5-Phosphate Synthase: Insights into the Mechanism and Underlying Intermolecular Interactions. The Journal of Physical Chemistry B 2016, 120 (37), 9922–9934. DOI: 10.1021/acs.jpcb.6b07248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brammer LA; Smith JM; Wade H; Meyers CF, 1-Deoxy-d-xylulose 5-Phosphate Synthase Catalyzes a Novel Random Sequential Mechanism. Journal of Biological Chemistry 2011, 286 (42), 36522–36531. DOI: 10.1074/jbc.M111.259747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Patel H; Nemeria NS; Brammer LA; Freel Meyers CL; Jordan F, Observation of Thiamin-Bound Intermediates and Microscopic Rate Constants for Their Interconversion on 1-Deoxy-d-xylulose 5-Phosphate Synthase: 600-Fold Rate Acceleration of Pyruvate Decarboxylation by d-Glyceraldehyde-3-phosphate. Journal of the American Chemical Society 2012,134 (44), 18374–18379. DOI: 10.1021/ja307315u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhou J; Yang L; DeColli A; Freel Meyers C; Nemeria NS; Jordan F, Conformational dynamics of 1-deoxy-d-xylulose 5-phosphate synthase on ligand binding revealed by H/D exchange MS. Proceedings of the National Academy of Sciences 2017, 114 (35), 9355–9360. DOI: 10.1073/pnas.1619981114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Battistini MR; Shoji C; Handa S; Breydo L; Merkler DJ, Mechanistic binding insights for 1-deoxy-d-Xylulose-5-Phosphate synthase, the enzyme catalyzing the first reaction of isoprenoid biosynthesis in the malaria-causing protists, Plasmodium falciparum and Plasmodium vivax. Protein Expression and Purification 2016, 120 (Supplement C), 16–27. DOI: https://doi.org/10.1016Zj.pep.2015.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schneider G; Lindqvist Y, Crystallography and mutagenesis of transketolase: mechanistic implications for enzymatic thiamin catalysis. Biochimica et Biophysica Acta (BBA) - Protein Structure and Molecular Enzymology 1998, 1385 (2), 387–398. DOI: 10.1016/S0167-4838(98)00082-X. [DOI] [PubMed] [Google Scholar]
- 67.Eubanks LM; Poulter CD, Rhodobacter capsulatus 1-Deoxy-d-xylulose 5-Phosphate Synthase: Steady-State Kinetics and Substrate Binding. Biochemistry 2003, 42 (4), 1140–1149. DOI: 10.1021/bi0205303. [DOI] [PubMed] [Google Scholar]
- 68.Frank RAW; Leeper FJ; Luisi BF, Structure, mechanism and catalytic duality of thiamine-dependent enzymes. Cell. Mol. Life Sci. 2007, 64, 892 DOI: 10.1007/s00018-007-6423-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sisquella X; de Pourcq K; Alguacil J; Robles J; Sanz F; Anselmetti D; Imperial S; Fernàndez-Busquets X, A single-molecule force spectroscopy nanosensor for the identification of new antibiotics and antimalarials. The FASEB Journal 2010, 24 (11), 4203–4217. DOI: 10.1096/fj.10-155507. [DOI] [PubMed] [Google Scholar]
- 70.Morris F; Vierling R; Boucher L; Bosch J; Freel Meyers CL, DXP Synthase-Catalyzed CN Bond Formation: Nitroso Substrate Specificity Studies Guide Selective Inhibitor Design. ChemBioChem 2013, 14 (11), 1309–1315. DOI: 10.1002/cbic.201300187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Banerjee A; Wu Y; Banerjee R; Li Y; Yan H; Sharkey TD, Feedback Inhibition of Deoxy-d-xylulose-5-phosphate Synthase Regulates the Methylerythritol 4-Phosphate Pathway. Journal of Biological Chemistry 2012, 288 (23), 16926–16936. DOI: 10.1074/jbc.M113.464636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ghirardo A; Wright LP; Bi Z; Rosenkranz M; Pulido P; Rodríguez-Concepción M; Niinemets Ü; Brüggemann N; Gershenzon J; Schnitzler J-P, Metabolic Flux Analysis of Plastidic Isoprenoid Biosynthesis in Poplar Leaves Emitting and Nonemitting Isoprene. Plant Physiology 2014, 165 (1), 37–51. DOI: 10.1104/pp.114.236018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Banerjee A; Sharkey TD, Methylerythritol 4-phosphate (MEP) pathway metabolic regulation. Natural Product Reports 2014, 31 (8), 1043–1055. DOI: 10.1039/C3NP70124G. [DOI] [PubMed] [Google Scholar]
- 74.Mueller C; Schwender J; Zeidler J; Lichtenthaler HK, Properties and inhibition of the first two enzymes of the non-mevalonate pathway of isoprenoid biosynthesis. Biochemical Society Transactions 2000, 28 (6), 792. [PubMed] [Google Scholar]
- 75.Ferhatoglu Y; Barrett M, Studies of clomazone mode of action. Pesticide Biochemistry and Physiology 2006, 85 (1), 7–14. DOI: https://doi.org/10.1016Zj.pestbp.2005.10.002. [Google Scholar]
- 76.Matsue Y; Mizuno H; Tomita T; Asami T; Nishiyama M; Kuzuyama T, The herbicide ketoclomazone inhibits 1-deoxy-D-xylulose 5-phosphate synthase in the 2-C-methyl-D-erythritol 4-phosphate pathway and shows antibacterial activity against Haemophilus influenzae. The Journal Of Antibiotics 2010, 63, 583 DOI: 10.1038/ja.2010.10010.1038/ja.2010.100https://www.nature.com/articles/ja2010100#supplementary-informationhttps://www.nature.com/articles/ja2010100#supplementary-information . [DOI] [PubMed] [Google Scholar]
- 77.Smith JM; Vierling RJ; Meyers CF, Selective inhibition of E coli1-deoxy-d-xylulose-5-phosphate synthase by acetylphosphonates. MedChemComm 2012, 3 (1), 65–67. DOI: 10.1039/C1MD00233C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Smith JM; Warrington NV; Vierling RJ; Kuhn ML; Anderson WF; Koppisch AT; Freel Meyers CL, Targeting DXP synthase in human pathogens: Enzyme inhibition and antimicrobial activity of butylacetylphosphonate. Journal of Antibiotics 2014, 67 (1), 77–83. DOI: 10.1038/ja.2013.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sanders S; Vierling RJ; Bartee D; DeColli AA; Harrison MJ; Aklinski JL; Koppisch AT; Freel Meyers CL, Challenges and Hallmarks of Establishing Alkylacetylphosphonates as Probes of Bacterial 1-Deoxy-d-xylulose 5-Phosphate Synthase. ACS Infectious Diseases 2017, 3 (7), 467–478. DOI: 10.1021/acsinfecdis.6b00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Masini T; Lacy B; Monjas L; Hawksley D; de Voogd AR; Illarionov B; Iqbal A; Leeper FJ; Fischer M; Kontoyianni M; Hirsch AKH, Validation of a homology model of Mycobacterium tuberculosis DXS: rationalization of observed activities of thiamine derivatives as potent inhibitors of two orthologues of DXS. Organic & Biomolecular Chemistry 2015, 13 (46), 11263–11277. DOI: 10.1039/C5OB01666E. [DOI] [PubMed] [Google Scholar]
- 81.Silver LL; Bostian KA, Discovery and development of new antibiotics: the problem of antibiotic resistance. Antimicrobial Agents and Chemotherapy 1993, 37 (3), 377–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sauret-Güeto S; Urós EM; Ibáñez E; Boronat A; Rodríguez-Concepción M, A mutant pyruvate dehydrogenase E1 subunit allows survival of Escherichia coli strains defective in 1-deoxy-d-xylulose 5-phosphate synthase. FEBS Letters 2006, 580 (3), 736–740. DOI: 10.1016/jfebslet.2005.12.092. [DOI] [PubMed] [Google Scholar]
- 83.Davies J; Davies D, Origins and Evolution of Antibiotic Resistance. Microbiology and Molecular Biology Reviews 2010, 74 (3), 417–433. DOI: 10.1128/mmbr.00016-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Leon A; Liu L; Yang Y; Hudock MP; Hall P; Yin F; Studer D; Puan K-J; Morita CT; Oldfield E, Isoprenoid Biosynthesis as a Drug Target: Bisphosphonate Inhibition of Escherichia coli K12 Growth and Synergistic Effects of Fosmidomycin. Journal of Medicinal Chemistry 2006, 49 (25), 7331–7341. DOI: 10.1021/jm060492b. [DOI] [PubMed] [Google Scholar]
- 85.Walsh CT; Wencewicz TA, Prospects for new antibiotics: a molecule-centered perspective. J. Antibiot. 2014, 67, 7–22. DOI: 10.1038/ja.2013.49. [DOI] [PubMed] [Google Scholar]
- 86.Hasunuma T; Takeno S; Hayashi S; Sendai M; Bamba T; Yoshimura S; Tomizawa K-I; Fukusaki E; Miyake C, Overexpression of 1-Deoxy-d-xylulose-5-phosphate reductoisomerase gene in chloroplast contributes to increment of isoprenoid production. Journal of Bioscience and Bioengineering 2008, 105 (5), 518–526. DOI: 10.1263/jbb.105.518. [DOI] [PubMed] [Google Scholar]
- 87.Armstrong CM; Meyers DJ; Imlay LS; Freel Meyers C; Odom AR, Resistance to the Antimicrobial Agent Fosmidomycin and an FR900098 Prodrug through Mutations in the Deoxyxylulose Phosphate Reductoisomerase Gene (dxr). Antimicrobial Agents and Chemotherapy 2015, 59 (9), 5511–5519. DOI: 10.1128/aac.00602-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Takahashi S; Kuzuyama T; Watanabe H; Seto H, A 1-deoxy-d-xylulose 5-phosphate reductoisomerase catalyzing the formation of 2-C-methyl-d-erythritol 4-phosphate in an alternative nonmevalonate pathway for terpenoid biosynthesis. Proceedings of the National Academy of Sciences 1998, 95 (17), 9879–9884. DOI: 10.1073/pnas.95.17.9879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Schwender J; Müller C; Zeidler J; Lichtenthaler HK, Cloning and heterologous expression of a cDNA encoding 1-deoxy-D-xylulose-5-phosphate reductoisomerase of Arabidopsis thaliana11The sequence reported in this paper has been deposited in the EMBL database under accession number AJ242588. FEBS Letters 1999, 455 (1), 140–144. DOI: 10.1016/S0014-5793(99)00849-2. [DOI] [PubMed] [Google Scholar]
- 90.Brown AC; Parish T, Dxr is essential in Mycobacterium tuberculosisand fosmidomycin resistance is due to a lack of uptake. BMC Microbiology 2008, 8 (1), 78 DOI: 10.1186/1471-2180-8-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sassetti CM; Boyd DH; Rubin EJ, Genes required for mycobacterial growth defined by high density mutagenesis. Molecular Microbiology 2003, 48 (1), 77–84. DOI: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 92.Argyrou A; Blanchard JS, Kinetic and Chemical Mechanism of Mycobacterium tuberculosis 1-Deoxy-d-xylulose-5-phosphate Isomeroreductase. Biochemistry 2004,43 (14), 4375–4384. DOI: 10.1021/bi049974k. [DOI] [PubMed] [Google Scholar]
- 93.Proteau PJ; Woo Y-H; Williamson RT; Phaosiri C, Stereochemistry of the Reduction Step Mediated by Recombinant 1-Deoxy-d-xylulose 5-Phosphate Isomeroreductase. Organic Letters 1999,1 (6), 921–923. DOI: 10.1021/ol990839n. [DOI] [PubMed] [Google Scholar]
- 94.Hoeffler J-F; Tritsch D; Grosdemange-Billiard C; Rohmer M, Isoprenoid biosynthesis via the methylerythritol phosphate pathway. European Journal of Biochemistry 2002, 269 (18), 4446–4457. DOI: 10.1046/j.1432-1033.2002.03150.x. [DOI] [PubMed] [Google Scholar]
- 95.Wong U; Cox RJ, The Chemical Mechanism of D-1-Deoxyxylulose-5-phosphate Reductoisomerase from Escherichia coli. Angewandte Chemie International Edition 2007, 46 (26), 4926–4929. DOI: 10.1002/anie.200700647. [DOI] [PubMed] [Google Scholar]
- 96.Koppisch AT; Fox DT; Blagg BSJ; Poulter CD, coli MEP Synthase E: Steady-State Kinetic Analysis and Substrate Binding. Biochemistry 2002, 41 (1), 236–243. DOI: 10.1021/bi0118207. [DOI] [PubMed] [Google Scholar]
- 97.Munos JW; Pu X; Mansoorabadi SO; Kim HJ; Liu H. w., A Secondary Kinetic Isotope Effect Study of the 1-Deoxy-d-xylulose-5-phosphate Reductoisomerase-Catalyzed Reaction: Evidence for a Retroaldol-Aldol Rearrangement. Journal of the American Chemical Society 2009, 131 (6), 2048–2049. DOI: 10.1021/ja807987h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lauw S; Illarionova V; Bacher A; Rohdich F; Eisenreich W, Biosynthesis of isoprenoids – studies on the mechanism of 2C-methyl-d-erythritol-4-phosphate synthase. FEBS Journal 2008, 275 (16), 4060–4073. DOI: 10.1111/j.1742-4658.2008.06547.x. [DOI] [PubMed] [Google Scholar]
- 99.Wong A; Munos JW; Devasthali V; Johnson KA; Liu H. w., Study of 1-Deoxy-d-xylulose-5-phosphate Reductoisomerase: Synthesis and Evaluation of Fluorinated Substrate Analogues. Organic Letters 2004, 6 (20), 3625–3628. DOI: 10.1021/ol048459b. [DOI] [PubMed] [Google Scholar]
- 100.Liu J; Murkin AS, Pre-Steady-State Kinetic Analysis of 1-Deoxy-d-xylulose-5-phosphate Reductoisomerase from Mycobacterium tuberculosis Reveals Partially Rate-Limiting Product Release by Parallel Pathways. Biochemistry 2012, 51 (26), 5307–5319. DOI: 10.1021/bi300513r. [DOI] [PubMed] [Google Scholar]
- 101.Uh E; Jackson ER; San Jose G; Maddox M; Lee RE; Lee RE; Boshoff HI; Dowd CS, Antibacterial and antitubercular activity of fosmidomycin, FR900098, and their lipophilic analogs. Bioorganic & Medicinal Chemistry Letters 2011, 21 (23), 6973–6976. DOI: http://dx.doi.org/10.10167i.bmcl.2011.09.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.San Jose G; Jackson ER; Uh E; Johny C; Haymond A; Lundberg L; Pinkham C; Kehn-Hall K; Boshoff HI; Couch RD; Dowd CS, Design of potential bisubstrate inhibitors against Mycobacterium tuberculosis (Mtb) 1-deoxy-d-xylulose 5-phosphate reductoisomerase (Dxr)-evidence of a novel binding mode. MedChemComm 2013, 4 (7), 1099–1104. DOI: 10.1039/C3MD00085K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jackson ER; San Jose G; Brothers RC; Edelstein EK; Sheldon Z; Haymond A; Johny C; Boshoff HI; Couch RD; Dowd CS, The effect of chain length and unsaturation on Mtb Dxr inhibition and antitubercular killing activity of FR900098 analogs. Bioorganic & Medicinal Chemistry Letters 2014, 24 (2), 649–653. DOI: 10.1016/j.bmcl.2013.11.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chofor R; Sooriyaarachchi S; Risseeuw MDP; Bergfors T; Pouyez J; Johny C; Haymond A; Everaert A; Dowd CS; Maes L; Coenye T; Alex A; Couch RD; Jones TA; Wouters J; Mowbray SL; Van Calenbergh S, Synthesis and Bioactivity of β-Substituted Fosmidomycin Analogues Targeting 1-Deoxy-d-xylulose-5-phosphate Reductoisomerase. Journal of Medicinal Chemistry 2015, 58 (7), 2988–3001. DOI: 10.1021/jm5014264. [DOI] [PubMed] [Google Scholar]
- 105.San Jose G; Jackson ER; Haymond A; Johny C; Edwards RL; Wang X; Brothers RC; Edelstein EK; Odom AR; Boshoff HI; Couch RD; Dowd CS, Structure–Activity Relationships of the MEPicides: N-Acyl and O-Linked Analogs of FR900098 as Inhibitors of Dxr from Mycobacterium tuberculosis and Yersinia pestis. ACS Infectious Diseases 2016, 2 (12), 923–935. DOI: 10.1021/acsinfecdis.6b00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gräwert T; Groll M; Rohdich F; Bacher A; Eisenreich W, Biochemistry of the non-mevalonate isoprenoid pathway. Cellular and Molecular Life Sciences 2011, 68 (23), 3797–3814. DOI: 10.1007/s00018-011-0753-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mine Y; Kamimura T; Nonoyama S; Nishida M; Goto S; Kuwahara S, In vitro and in vivo antibacterial activities of FR-31564, a new phosphonic acid antibiotic. J. Antibiot. (Tokyo) 1980, 33, 36–43. [DOI] [PubMed] [Google Scholar]
- 108.Yokota Y; Murakawa T; Nishida M, In vitro synergism of Fr-31564, a new phosphonic acid antibiotic. J. Antibiot. (Tokyo) 1981, 34, 876–883. [DOI] [PubMed] [Google Scholar]
- 109.Neu HC; Kamimura T, In vitro and in vivo antibacterial activity of FR-31564, a phosphonic acid antimicrobial agent. Antimicrobial Agents and Chemotherapy 1981, 19 (6), 1013–1023. DOI: 10.1128/aac.19.6.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kuemmerle HP; Murakawa T; Soneoka K; Konishi T, Fosmidomycin: a new phosphonic acid antibiotic. Part I: Phase I tolerance studies. International journal of clinical pharmacology, therapy, and toxicology 1985, 23 (10), 515–20. [PubMed] [Google Scholar]
- 111.Kuemmerle HP; Murakawa T; Sakamoto H; Sato N; Konishi T; De Santis F, Fosmidomycin, a new phosphonic acid antibiotic. Part II: 1. Human pharmacokinetics. 2. Preliminary early phase IIa clinical studies. International journal of clinical pharmacology, therapy, and toxicology 1985, 23 (10), 521–8. [PubMed] [Google Scholar]
- 112.Kuzuyama T; Shimizu T; Takahashi S; Seto H, Fosmidomycin, a specific inhibitor of 1-deoxy-d-xylulose 5-phosphate reductoisomerase in the nonmevalonate pathway for terpenoid biosynthesis. Tetrahedron Letters 1998, 39 (43), 7913–7916. DOI: 10.1016/S0040-4039(98)01755-9. [DOI] [Google Scholar]
- 113.Shigi Y, Inhibition of bacterial isoprenoid synthesis by fosmidomycin, a phosphonic acid-containing antibiotic. Journal of Antimicrobial Chemotherapy 1989, 24 (2), 131–145. DOI: 10.1093/jac/24.2.131. [DOI] [PubMed] [Google Scholar]
- 114.Zeidler J; Schwender J; Müller C; Wiesner J; Weidemeyer C; Beck E; Jomaa H; Lichtenthaler Hartmut K, Inhibition of the Non-Mevalonate 1-Deoxy-D-xylulose-5-phosphate Pathway of Plant Isoprenoid Biosynthesis by Fosmidomycin. In Zeitschrift für Naturforschung C, 1998; Vol. 53, p 980 DOI: 10.1515/znc-1998-11-1208. [DOI] [Google Scholar]
- 115.Reuter K; Sanderbrand S; Jomaa H; Wiesner J; Steinbrecher I; Beck E; Hintz M; Klebe G; Stubbs MT, Crystal Structure of 1-Deoxy-d-xylulose-5-phosphate Reductoisomerase, a Crucial Enzyme in the Non-mevalonate Pathway of Isoprenoid Biosynthesis. Journal of Biological Chemistry 2002, 277 (7), 5378–5384. DOI: 10.1074/jbc.M109500200. [DOI] [PubMed] [Google Scholar]
- 116.Henriksson LM; Unge T; Carlsson J; Åqvist J; Mowbray SL; Jones TA, Structures of Mycobacterium tuberculosis 1-Deoxy-D-xylulose-5-phosphate Reductoisomerase Provide New Insights into Catalysis. Journal of Biological Chemistry 2007, 282 (27), 19905–19916. DOI: 10.1074/jbc.M701935200. [DOI] [PubMed] [Google Scholar]
- 117.Henriksson LM; Bjorkelid C; Mowbray SL; Unge T, The 1.9 A resolution structure of Mycobacterium tuberculosis 1-deoxy-d-xylulose 5-phosphate reductoisomerase, a potential drug target. Acta Crystallographica Section D 2006, 62 (7), 807–813. DOI: doi: 10.1107/S0907444906019196. [DOI] [PubMed] [Google Scholar]
- 118.Yajima S; Nonaka T; Kuzuyama T; Seto H; Ohsawa K, Crystal Structure of 1-Deoxy-D-xylulose 5-phosphate Reductoisomerase Complexed with Cofactors: Implications of a Flexible Loop Movement upon Substrate Binding. The Journal of Biochemistry 2002, 131 (3), 313–317. [DOI] [PubMed] [Google Scholar]
- 119.Edwards RL; Brothers RC; Wang X; Maron MI; Ziniel PD; Tsang PS; Kraft TE; Hruz PW; Williamson KC; Dowd CS; John ARO, MEPicides: potent antimalarial prodrugs targeting isoprenoid biosynthesis. Scientific Reports 2017, 7 (1), 8400 DOI: 10.1038/s41598-017-07159-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dhiman RK; Schaeffer ML; Bailey AM; Testa CA; Scherman H; Crick DC, 1-Deoxy-d-Xylulose 5-Phosphate Reductoisomerase (IspC) from Mycobacterium tuberculosis: towards Understanding Mycobacterial Resistance to Fosmidomycin. Journal of Bacteriology 2005, 187 (24), 8395–8402. DOI: 10.1128/jb.187.24.8395-8402.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hett EC; Rubin EJ, Bacterial Growth and Cell Division: a Mycobacterial Perspective. Microbiology and Molecular Biology Reviews 2008, 72 (1), 126–156. DOI: 10.1128/mmbr.00028-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sparr C; Purkayastha N; Kolesinska B; Gengenbacher M; Amulic B; Matuschewski K; Seebach D; Kamena F, Improved Efficacy of Fosmidomycin against Plasmodium and Mycobacterium Species by Combination with the Cell-Penetrating Peptide Octaarginine. Antimicrobial Agents and Chemotherapy 2013, 57 (10), 4689–4698. DOI: 10.1128/aac.00427-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kurz T; Schlüter K; Kaula U; Bergmann B; Walter RD; Geffken D, Synthesis and antimalarial activity of chain substituted pivaloyloxymethyl ester analogues of Fosmidomycin and FR900098. Bioorganic & Medicinal Chemistry 2006, 14 (15), 5121–5135. DOI: 10.1016/j.bmc.2006.04.018. [DOI] [PubMed] [Google Scholar]
- 124.Jansson AM; Więckowska A; Björkelid C; Yahiaoui S; Sooriyaarachchi S; Lindh M; Bergfors T; Dharavath S; Desroses M; Suresh S; Andaloussi M; Nikhil R; Sreevalli S; Srinivasa BR; Larhed M; Jones TA; Karlen A; Mowbray SL, DXR Inhibition by Potent Mono- and Disubstituted Fosmidomycin Analogues. Journal of Medicinal Chemistry 2013, 56 (15), 6190–6199. DOI: 10.1021/jm4006498. [DOI] [PubMed] [Google Scholar]
- 125.Kunfermann A; Lienau C; Illarionov B; Held J; Gräwert T; Behrendt CT; Werner P; Hähn S; Eisenreich W; Riederer U; Mordmüller B; Bacher A; Fischer M; Groll M; Kurz T, IspC as Target for Antiinfective Drug Discovery: Synthesis, Enantiomeric Separation, and Structural Biology of Fosmidomycin Thia Isosters. Journal of Medicinal Chemistry 2013, 56 (20), 8151–8162. DOI: 10.1021/jm4012559. [DOI] [PubMed] [Google Scholar]
- 126.McKenney ES; Sargent M; Khan H; Uh E; Jackson ER; Jose GS; Couch RD; Dowd CS; van Hoek ML, Lipophilic Prodrugs of FR900098 Are Antimicrobial against Francisella novicida In Vivo and In Vitro and Show GlpT Independent Efficacy. PLOS ONE 2012, 7 (10), e38167. DOI: 10.1371/journal.pone.0038167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Messiaen A-S; Verbrugghen T; Declerck C; Ortmann R; Schlitzer M; Nelis H; Van Calenbergh S; Coenye T, Resistance of the Burkholderia cepacia complex to fosmidomycin and fosmidomycin derivatives. International Journal of Antimicrobial Agents 2011, 38 (3), 261–264. DOI: 10.1016/j.ijantimicag.2011.04.020. [DOI] [PubMed] [Google Scholar]
- 128.Sakamoto Y; Furukawa S; Ogihara H; Yamasaki M, Fosmidomycin resistance in adenylate cyclase deficient (cya) mutants of Escherichia coli. Biosci Biotechnol Biochem 2003, 67 DOI: 10.1271/bbb.67.2030. [DOI] [PubMed] [Google Scholar]
- 129.Guggisberg AM; Park J; Edwards RL; Kelly ML; Hodge DM; Tolia NH; Odom AR, A sugar phosphatase regulates the methylerythritol phosphate (MEP) pathway in malaria parasites. Nature Communications 2014, 5, 4467 DOI: 10.1038/ncomms546710.1038/ncomms5467https://www.nature.com/articles/ncomms5467#supplementary-informationhttps://www.nature.com/articles/ncomms5467#supplementary-information . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Buetow L; Brown AC; Parish T; Hunter WN, The structure of Mycobacteria 2C-methyl-D-erythritol-2,4-cyclodiphosphate synthase, an essential enzyme, provides a platform for drug discovery. BMC Structural Biology 2007, 7 (1), 68 DOI: 10.1186/1472-6807-7-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.O’Rourke PE; Kalinowska-Tłuścik J; Fyfe PK; Dawson A; Hunter WN, Crystal structures of IspF from Plasmodium falciparum and Burkholderia cenocepacia: comparisons inform antimicrobial drug target assessment. BMC Structural Biology 2014, 14 (1), 1 DOI: 10.1186/1472-6807-14-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kemp LE; Alphey MS; Bond CS; Ferguson MA; Hecht S; Bacher A; Eisenreich W; Rohdich F; Hunter WN, The identification of isoprenoids that bind in the intersubunit cavity of Escherichia coli 2C-methyl-D-erythritol-2,4-cyclodiphosphate synthase by complementary biophysical methods. Acta Crystallogr D Biol Crystallogr 2005, 61 DOI: 10.1107/s0907444904025971. [DOI] [PubMed] [Google Scholar]
- 133.Steinbacher S; Kaiser J; Wungsintaweekul J; Hecht S; Eisenreich W; Gerhardt S; Bacher A; Rohdich F, Structure of 2C-methyl-D-erythritol-2,4-cyclodiphosphate synthase involved in mevalonate-independent biosynthesis of isoprenoids. J Mol Biol 2002, 316 DOI: 10.1006/jmbi.2001.5341. [DOI] [PubMed] [Google Scholar]
- 134.Ni S; Robinson H; Marsing GC; Bussiere DE; Kennedy MA, Structure of 2C-methyl-d-erythritol-2,4-cyclodiphosphate synthase from Shewanella oneidensis at 1.6 Å: identification of farnesyl pyrophosphate trapped in a hydrophobic cavity. Acta Crystallographica Section D 2004, 60 (11), 1949–1957. DOI: 10.1107/S0907444904021791. [DOI] [PubMed] [Google Scholar]
- 135.Bitok JK; Meyers CF, 2C-Methyl-d-erythritol 4-Phosphate Enhances and Sustains Cyclodiphosphate Synthase IspF Activity. ACS Chemical Biology 2012, 7 (10), 1702–1710. DOI: 10.1021/cb300243w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ostrovsky D; Diomina G; Lysak E; Matveeva E; Ogrel O; Trutko S, Effect of oxidative stress on the biosynthesis of 2-C-methyl-d-erythritol-2,4-cyclopyrophosphate and isoprenoids by several bacterial strains. Archives of Microbiology 1998, 171 (1), 69–72. DOI: 10.1007/s002030050680. [DOI] [PubMed] [Google Scholar]
- 137.Ostrovsky D; Shipanova I; Sibeldina L; Shashkov A; Kharatian E; Malyarova I; Tantsyrev G, A new cyclopyrophosphate as a bacterial antistressor? FEBS Letters 1992, 298 (2–3), 159–161. DOI: 10.1016/0014-5793(92)80045-I. [DOI] [PubMed] [Google Scholar]
- 138.Ostrovsky D; Kharatian E; Malarova I; Shipanova I; Sibeldina L; Shashkov A; Tantsirev G, Synthesis of a new organic pyrophosphate in large quantities is induced in some bacteria by oxidative stress. Biofactors 1992, 3 (4), 261–264. [PubMed] [Google Scholar]
- 139.Goncharenko AV; Ershov YV; Salina EG; Wiesner J; Vostroknutova GN; Sandanov AA; Kaprelyants AS; Ostrovsky DN, The role of 2-C-Methylerythritol-2,4-cyclopyrophosphate in the resuscitation of the “nonculturable” forms of Mycobacterium smegmatis. Microbiology 2007, 76 (2), 147–152. DOI: 10.1134/S0026261707020038. [DOI] [PubMed] [Google Scholar]
- 140.Zhou K; Zou R; Stephanopoulos G; Too H-P, Metabolite Profiling Identified Methylerythritol Cyclodiphosphate Efflux as a Limiting Step in Microbial Isoprenoid Production. PLOS ONE 2012, 7 (11), e47513. DOI: 10.1371/journal.pone.0047513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zhang Z; Jakkaraju S; Blain J; Gogol K; Zhao L; Hartley RC; Karlsson CA; Staker BL; Edwards TE; Stewart LJ; Myler PJ; Clare M; Begley DW; Horn JR; Hagen TJ, Cytidine derivatives as IspF inhibitors of Burkolderia pseudomallei. Bioorganic & Medicinal Chemistry Letters 2013, 23 (24), 6860–6863. DOI: https://doi.org/10.1016Zj.bmcl.2013.09.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Thelemann J; Illarionov B; Barylyuk K; Geist J; Kirchmair J; Schneider P; Anthore L; Root K; Trapp N; Bacher A; Witschel M; Zenobi R; Fischer M; Schneider G; Diederich F, Aryl Bis-Sulfonamide Inhibitors of IspF from Arabidopsis thaliana and Plasmodium falciparum. ChemMedChem 2015, 10 (12), 2090–2098. DOI: 10.1002/cmdc.201500382. [DOI] [PubMed] [Google Scholar]
- 143.Kipchirchir Bitok J; Meyers CF, Synthesis and evaluation of stable substrate analogs as potential modulators of cyclodiphosphate synthase IspF. MedChemComm 2013, 4 (1), 130–134. DOI: 10.1039/C2MD20175E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Geist JG; Lauw S; Illarionova V; Illarionov B; Fischer M; Gräwert T; Rohdich F; Eisenreich W; Kaiser J; Groll M; Scheurer C; Wittlin S; Alonso-Gomez JL; Schweizer WB; Bacher A; Diederich F, Thiazolopyrimidine Inhibitors of 2-Methylerythritol 2,4-Cyclodiphosphate Synthase (IspF) from Mycobacterium tuberculosis and Plasmodium falciparum. ChemMedChem 2010, 5 (7), 1092–1101. DOI: 10.1002/cmdc.201000083. [DOI] [PubMed] [Google Scholar]
- 145.Li Z; Sharkey TD, Metabolic profiling of the methylerythritol phosphate pathway reveals the source of post-illumination isoprene burst from leaves. Plant, Cell & Environment 2013, 36 (2), 429–437. DOI: 10.1111/j.1365-3040.2012.02584.x. [DOI] [PubMed] [Google Scholar]
- 146.Pomona College and BioByte, I. CLOGP, Copyright, Claremont, CA. 1988-2011.