

Abstract
Wool keratin is a natural material with excellent properties, which is considered as scaffold biomaterial for tissue engineering. Polyethylene glycol can improve the mechanical properties of keratin materials because of its excellent biocompatibility and plasticity. In the present work, poly (ethylene glycol) methyl ether methacrylate (MPEGMA) was grafted onto keratin by thiol‐ene photoclick reaction. The results of FTIR and SDS‐PAGE verified the successful reaction between MPEGMA and keratin. Compared with the keratin, circular dichroism and XRD results showed that the β‐sheet ratio increased in MPEG‐g‐keratin. Additionally, it can be found that the exposure of keratin hydrophobic amino acids increased quickly and the micelle size became larger due to the introduction of MPEG from the results of fluorescence spectroscopy and particle size analysis. The MPEG‐g‐ keratin was formed into a membrane to further study the application of the modified keratin. Compared with the keratin membrane, the flexibility and biocompatibility of modified keratin have been improved. This work provides an eco‐friendly and facile approach for preparation of the keratin biomaterials.
Keywords: biomaterial, keratin, MPEGMA, thiol‐ene photoclick
Abbreviations
- ANS
8‐anilino‐1‐naphthalenesulfonic acid ammonium salt
- CD
circular dichroism
- DLS
dynamic light scattering
- HMPP
2‐hydroxy‐2‐methylpropiophenone
- MPEGMA
poly (ethylene glycol) methyl ether methacrylate
- PEG
polyethylene glycol
- XRD
X‐ray diffraction
1. INTRODUCTION
Keratin is a renewable resource, which is widely distributed in wool, hair, feathers, hooves, and horns 1. Wool has been estimated worldwide by‐products from textile processing, hair and feathers from butchery more than 5 million tons per year 2, 3. Hence, various methods have been made to extract keratin from the wasted resources. Chemical methods mostly employ strong acids, alkali hydrolysis, and other chemicals containing peroxides, sulfites, dithiothreitol, and thiols 4. Wool keratin, a kind of unbranched polymer comprised of amino acids, exhibits a stable 3‐D conformation maintained by a range of non‐covalent interactions and covalent interactions, in addition to the peptide bonds between individual amino acids 5. Recent researches have reported that the regenerated keratin obtained by the reduction method has a unique Arg‐Gly‐Asp‐Ser (RGDS) structure at its end, which makes its biocompatibility better than other biomaterials such as collagen and silk fibroin 6, 7. Warwick J. Duncan et al. implanted keratin hydrogel into the distal femoral condyle of sheep and found that keratin hydrogel has the potential to enhance successful osseointegration of dental implants placed in inferior bone 8. Xiangyu Xu et al. found that the wool keratin adsorbed and stabilized MoS2 nanosheets provided excellent biocompatibility in the cultivation of osteoblasts 9.
However, there is a brittle problem in pure regenerated keratin materials, which leads to difficulties in the formation of keratin membranes or fibers 10. Thus, it is hard to meet the demand for tissue engineering materials which require high mechanical property. Fortunately, the keratin side chain contains a large number of amino, carboxyl, sulfhydryl, and hydroxyl groups, which can be used as sites for photocatalytic grafting reactions 10, 11. Among them, the sulfhydryl group has the highest reactivity, which participates in the formation of disulfide bonds to confer keratin stability 12. In addition, the disulfide bonds can be easily cleaved by reducing agents to form more thiols, which offer great opportunities for keratin to be modified and functionalized through the thiol‐ene reaction 13, 14, 15. There are many kinds of initiation mechanisms to initiate thiol‐ene click reaction, such as thermal, photo and redox, in which photo initiation becomes more and more popular because of its high efficiency, versatility, and simplicity for the preparation of new functional polymer materials 16, 17, 18, 19, 20. Chien‐Chi Lin et al. fabricated polyethylene glycol (PEG) hydrogels through thiol‐ene photoclick reaction for in situ cell encapsulation 21. Yu, Bing et al. prepared thiol‐ene photocrosslinked hybrid vesicles, which can be used to control the dispersion of both hydrophilic and hydrophobic dyes in water 22.
PRACTICAL APPLICATION
Wool keratin is known as a renewable resource around the world usable as potential sources for polyamides and also as a promising natural material for tissue engineering. Therefore, it is highly desirable to solve the brittleness problem of pure regenerative keratin materials. In this paper, MPEG soft segments were introduced onto keratin through thiol‐ene photoclick reaction. The modified membrane is non‐toxic without the introduction of metal ions. Meanwhile, the modified keratin membrane combines the advantages of keratin and MPEG with excellent biocompatibility and flexibility compared with the untreated keratin material. The method that is presented in this publication is eco‐friendly and facile for preparation of the keratin biomaterial
PEG, a linear or branched neutral polyether with a variety of molecular weights, is a very good choice to improve mechanical properties of keratin membrane 23. Despite its apparent simplicity, it has been extensively studied because of its biocompatibility, non‐toxicity, and low immunogenicity 24. In addition, it is easy to be chemically modified and attach to other molecules and surfaces. When PEG is combined with other molecules, there is little effect on their chemical properties but controls their solubility and increases their size 23. Therefore, PEGylation is becoming more and more popular for enhancing the biotechnological potential of keratin. Many researchers have reported that PEG has excellent plasticizing abilities 25, 26, which can reduce keratin chain‐to‐chain interaction and increase membrane flexibility.
In the present work, we demonstrated a facile approach for grafting poly (ethylene glycol) methyl ether methacrylate (MPEGMA) onto wool keratin. The disulfide bonds in the keratin were reduced to thiol groups and then the thiol groups reacted with MPEGMA through thiol‐ene photoclick reaction (Scheme 1). After the modification, the keratin membrane can be successfully prepared with better flexibility and biocompatibility, which shows a good inspiration for the modification of keratin biomaterials.
Scheme 1.
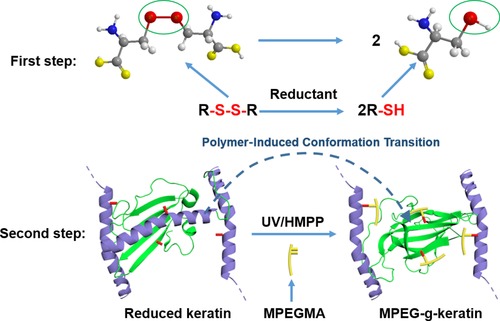
Synthetic scheme for the preparation of MPEG‐g‐keratin
2. MATERIALS AND METHODS
2.1. Materials
Wool fiber was provided by Xiexin Worsted Spinning Weaving and Dyeing (Wuxi, China). MPEGMA (Mn = 300) was purchased from Sigma‐Aldrich (Shanghai, China). 2‐Mercaptoethanol was provided by Xiya reagent (Shandong, China). Tris (2‐carboxyethyl) phosphine hydrochloride, 2‐Hydroxy‐2‐methylpropiophenone (HMPP) and 8‐anilino‐1‐naphthalenesulfonic acid ammonium salt (ANS) were purchased from Aladdin Reagent (Shanghai, China). All the other reagents used in this study were commercially available and were of analytical grade throughout.
2.2. Extraction of wool keratin
Keratin was extracted from wool according to the method reported previously 27. Wool fiber was cleaned by acetone in a Soxhlet extractor for 48 h. The degreased wool fiber (1 g) was dissolved in a solution (18 mL) containing 8 M urea, 0.5 M 2‐mercaptoethanol, 0.1 M SDS, and the pH was adjusted to 9.5 with concentrated sodium hydroxide. The system was shaken in a water bath at 90°C for 6 h. After removing part of the insoluble deposits by vacuum filtration, the keratin solution was dialyzed against deionized water to remove the residual salts and urea. The sample solution was obtained after removal of insoluble precipitate again by centrifugation. Finally, keratin powder was obtained by keeping the sample solution at −20°C for 8 h, followed by freeze drying at −50°C for 48 h.
2.3. Grafting procedure
The wool keratin (25 g/L) was dissolved in 8 M urea solution, and then the dissolved keratin was reduced with tris (2‐carboxyethyl) phosphine hydrochloride (6 g/L) at 70°C. After 1 h, HMPP (2.5 mg/L) and MPEGMA (25 g/L) were added to the solution, subsequently exposed to 254 nm UV light (band width ≤5 nm, intensity = 5 mW) for 15 min. After the reaction, the mixture solution was dialyzed for 72 h to remove unreacted MPEGMA, urea and other impurities. Furthermore, adding only MPEGMA without HMPP (control sample) as a control was also performed under the same condition.
2.4. Preparation of keratin membrane
Keratin solution (60 g/L) was first prepared. After thoroughly mixing with 0.5% w/v of glycerol, the solution was poured into a Teflon plate and naturally air‐dried at 20°C and a relative humidity of 62%.
2.5. Determination of free thiol (‐SH) content
The free thiol content was measured according to literature procedures 28. Fifteen milligrams of samples were placed in 0.8 mL buffer A (8 M urea, 3 mM EDTA, 1% SDS, and 0.2 M Tris‐HCl, pH 8.0). Subsequently, the samples were shaken at room temperature for 2.5 h. And then, added 0.2 mL of Buffer B (50 mM DTNB reagent, 0.2 M Tris‐HCl, pH 8.0). The samples were again shaken for 1 h, and the absorbance was measured at 412 nm. The blank control was buffer A (0.8 mL) and buffer B (0.2 mL). The free thiol content was calculated according to the molarextinction coefficient 13 600 L/(mol × cm).
2.6. FTIR analysis
FTIR analysis was performed using a Nicolet iS10 spectrometer (Thermo, Nicolet, USA), and spectrum was recorded in the wavelength range 4000–500 cm−1, at a resolution of 4 cm−1 and 32 scans per sample.
2.7. SDS‐PAGE
According to the method of Pascal Bailon 29, the sample was subjected to SDS/polyacrylamide (8–16%) gel electrophoresis, and the keratin was stained with Coomassie blue dye.
The PEG part of the sample was specifically stained with barium iodide solution according to the method of literature 30. The SDS‐PAGE gel was rinsed with distilled water and placed in a 5% barium chloride solution. After 15 min, the gel was washed with distilled water and placed in a 0.1 M iodine solution for another 5 min. Iodine was washed off with distilled water.
2.8. Particle size analysis
Particle size and its distribution for each keratin solution were determined at 25°C by dynamic light scattering (DLS), using a Malvern Zeta sizer Nano ZS90 instrument (Malvern, British) with a fixed angle of 90° 31. The cumulate analysis was used for analyzing the DLS data of the different keratin samples.
2.9. Circular dichroism test
Circular dichroism measurements of the keratin solution were performed using a MOS‐450 Circular Dichroism instrument (Biologic, France), at a scanning rate of 100 nm/min from 190 to 250 nm. The samples were tested in a 1 mm quartz cuvette at 25°C.
2.10. Fluorescence spectroscopy
Fluorescence spectroscopy was used to determine the degree of exposure of the hydrophobic amino acids of the keratin, and the solubility of the sample was diluted to 0.1 mg/mL with a phosphate buffer solution (pH = 7.4, 0.01 M). Sample containing 4 mL of solution was added 20 µL of ANS (8 mM) solution 32. Then, the sample was tested in an F‐4600 fluorescence spectrophotometer (Hitachi, Japan) with an excitation wavelength of 380 nm, a voltage of 800 V, and a slit width of 5 nm. The fluorescence intensity of the sample at the emission wavelength of 400–600 nm was measured.
2.11. X‐ray diffraction
X‐ray diffraction (XRD) was performed using a Bruker D8 Focus with Cu target at 40 kV, 40 mA in the scanning range of 5–50° with a step size of 0.02°. The sample was evenly applied to the wafer surface when the test was conducted.
2.12. Biocompatibility
The biocompatibility of keratin samples were tested by ISO 10993‐5‐2009 using NIH/3T3 cells 33. After being sterilized by ethanol (75%) for 24 h, the samples were prepared using DMEM (containing 10% fetal bovine serum, 100 U/Ml penicillin and 100 µg/mL streptomycin) with a concentration of 5 mg/mL. 100 µL of the NIH/3T3 cell suspension and 50 µL of the sample were injected into the cell culture plate and diluted to 500 µL. Subsequently, the cell plate was kept at 37°C in a humidified atmosphere of 5% CO2 for 48 h. Then, 10 µL CCK‐8 solution was added and incubated for another 2 h. The absorbance at 450 nm was recorded by using an ELISA microplate readermicroplate reader. The viable cell percentage was calculated according to Eq. (1).
| (1) |
where, A 1 and A 0 are the absorbances for the sample and the blank, respectively.
3. RESULTS AND DISCUSSION
3.1. Determination of free thiol content
The degree of thiol reaction can be directly visualized by detecting the free thiol content of the sample. It can be seen from Figure 1 that the thiol group content of the control sample without adding photoinitiator (HMPP) was almost the same as that of keratin while the thiol group content of MPEG‐g‐keratin sample decreased dramatically. The result of free thiol content indicated that MPEGMA was successfully grafted onto keratin via thiol‐ene click reaction.
Figure 1.
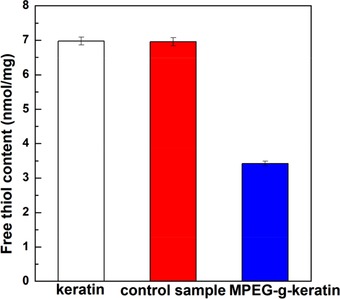
Free thiol content of keratin and MPEG‐g‐keratin
3.2. Characterization of the keratin and MPEG‐g‐keratin
3.2.1. FTIR spectroscopy
The FTIR spectra of keratin and MPEG‐g‐keratin were compared in Figure 2. The spectra contained characteristic peaks of peptide bonds (–CONH), and these characteristic peaks were classified into Amide A, Amide I, Amide II, and Amide III bands. An absorption peak at 3297 cm−1 was assigned to N–H stretching (Amide A). A strong absorption peak at 1655 cm−1 should be ascribed to the C=O (Amide I). A medium strong peak was observed at 1542 cm−1 and assigned to C–N stretching and N–H in‐plane bending vibrations (Amide II), while the weak band in the range of 1242 cm−1 was related to the C–N and C–O stretching vibrations (Amide III) 1. As for the spectra of the MPEGMA, a strong band at 1105 cm−1 was ascribed to the C–O–C ether stretch and a band at 1716 cm−1 assigned to the C=O stretch 34, 35. In Figure 2b, no additional bands were seen in the control sample spectra, which meant that most of the unreacted MPEMGA was removed by dialysis. As for Figure 2c, two characteristic absorption peaks of MPEGMA at 1729 and 1105 cm−1 can be observed, which confirmed the successful grafting of MPEGMA onto wool keratin via thiol‐ene click reaction.
Figure 2.
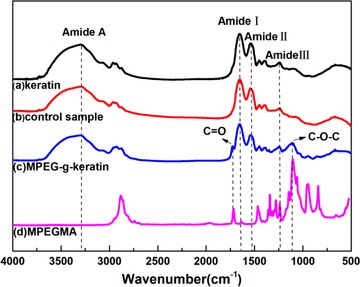
FTIR spectra of keratin and MPEG‐g‐keratin
3.2.2. SDS‐PAGE
Figure 3 showed SDS‐PAGE results of keratin (b), control sample (c), and MPEG‐g‐keratin (d). The three samples had a similar molecular weight distribution and mainly ranged between 40–70 kDa. This was mainly due to the small molecular weight of MPEGMA, which had little effect on the molecular weight of the grafted product. The results of PEG staining were shown in Figure 3 (b1, c1, and d1). The keratin (b1) was almost white after stained with barium iodide, because the sample does not contain MPEG and cannot react with barium iodide. While the control sample (c1) had a very light yellowish color maybe due to the small amount of MPEGMA adsorbed on the keratin by hydrogen bonding. The color of MPEG‐g‐keratin (d1) was deep yellow indicating that a large amount of MPEG was grafted onto keratin 30.
Figure 3.

SDS‐PAGE of keratin and MPEG‐g‐keratin (a: protein standard, b: keratin, c: control sample, d: MPEG‐g‐keratin; b1: keratin, c1: control sample, d1: MPEG‐g‐keratin)
3.2.3. Particle size
After the MPEG chains were grafted onto keratin, stronger hydrogen bonds were found between MPEG and keratin to shape a core‐shell structure. Hydrophobic segments were segregated from the aqueous exterior to form an inner core surrounded by a shell of hydrophilic segments, and thus formed micelles in the aqueous solution 36. The diffusion coefficient of these micelles can be measured by DLS 37.
The particle sizes and polydispersity index (PDI) of the keratin was identified and shown in Figure 4. As can be seen, the average particle size of keratin was found to be 63.7 dnm with the PDI of 0.235. The control sample had an average particle size of 75.66 dnm with the PDI of 0.276, since some of the MPEGMA in the control sample was covalently bonded and adsorbed on the keratin peptide chain, so the average particle size was slightly increased. As for MPEG‐g‐keratin solution, the average particle size was 97.7 dnm with the PDI of 0.388 due to the increase of the hydrophilic MPEG chains. Meanwhile, the high PDI values can be attributed to higher MPEG content in keratin that could reduce the interactions of hydrophobic chains and create a steric barrier around keratin 38. By introducing hydrophilic MPEG chains, the hydrophilicity of keratin can be increased, and the stability of keratin micelle in aqueous solution can be improved 39.
Figure 4.
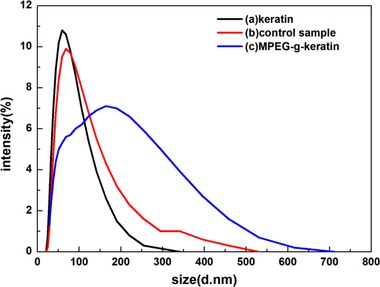
Particle size and its distributions of keratin and MPEG‐g‐keratin
3.2.4. Circular dichroism
The secondary structure of the keratin peptide chain follows the electronic transition caused by the peptide bond (–CONH–) and the modification induced by its supramolecular arrangement. Therefore, the changes in their photoconformation can be detected by circular dichroism (CD) spectroscopy 40.
The results of CD were shown in Figure 5. The α‐helical peptides presented a characteristic positive band at 193 nm and two negative bands at 208 and 222 nm 40. The curve trends of the three samples were consistent, indicating that the secondary conformation of the sample was not greatly affected by the introduction of MPEG. However, the proportion of the secondary conformation had changed as shown in Table 1, which was calculated from their CD spectra. The sample of keratin has the highest ratio of α‐helical structure and lowest ratio of β‐sheet structure, while the sample of MPEG‐g‐keratin has the lowest ratio of α‐helical structure and lowest ratio of β‐sheet structure. For the sample of MPEG‐g‐keratin, the ratio of β‐sheet, β‐turn, and random coil structure increased greatly, which was due to the keratin chain as a mold plate to stretch itself to form a β‐sheet structure according to the strong hydrogen bond between MPEG and keratin. This conformational change was the “Polymer‐Induced Conformation Transition” proposed by Xin Chen 41. The β‐sheet, β‐turn and random coil ratios of the control sample increased slightly due to the introduction and removal of MPEGMA. MPEGMA can form stronger hydrogen bonds with amino acids of keratin, causing the peptide chain to stretch, which reduced the α‐helix ratio and promoted the formation of other secondary structures. This effect was preserved even if MPEGMA was dialyzed away.
Figure 5.
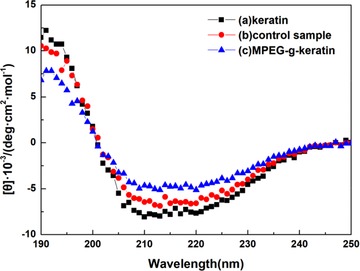
CD of keratin and MPEG‐g‐keratin
Table 1.
Secondary structure content of keratin and MPEG‐g‐keratin
| Sample | α‐helix% | β‐sheet% | β‐turn% | Random coil% |
|---|---|---|---|---|
| Keratin | 60.58 | 6.32 | 11.66 | 21.44 |
| Control sample | 54.21 | 8.71 | 13.41 | 23.68 |
| MPEG‐g‐keratin | 40.94 | 14.27 | 19.19 | 25.60 |
3.2.5. Fluorescence Spectroscopy
There are both hydrophilic amino acids and hydrophobic amino acids in the structure of keratin, and itself can constitute a biological macromolecule having a spatial structure with a hydrophobic amino acid as a core and a hydrophilic amino acid as a periphery 42. Any change will affect the stability of keratin in aqueous solution. In order to study the effect of the introduction of MPEG on the hydrophobicity of keratin, the fluorescent probe method was used to analyze some hydrophobic amino acid residues contained in keratin, such as phenylalanine, tryptophan and tyrosine 43. The change in fluorescence emission intensity can be used to analyze the effect of MPEGMA modification on the exposure of keratin hydrophobic amino acids.
As was shown in Figure 6, the fluorescence intensity of pure ANS at 530 nm was very weak. When the keratin was combined with ANS, a significant fluorescence peak appeared at 500 nm, indicating that some of the hydrophobic amino acids of keratin were exposed outside the peptide chain. However, the fluorescence emission intensity of the control sample was slightly lower than that of keratin, which may be due to that the steric hindrance of MPEGMA adsorbed on the keratin hindered the binding of the hydrophobic amino acid to the ANS. The fluorescence emission intensity of MPEG‐g‐keratin was higher than that of keratin, which indicated that the degree of exposure of hydrophobic amino acids was increased. The reason for this phenomenon was that the keratin was more hydrophilic after being grafted by MPEGMA, which caused the peptide chain to stretch and increased the exposure of hydrophobic amino acids.
Figure 6.
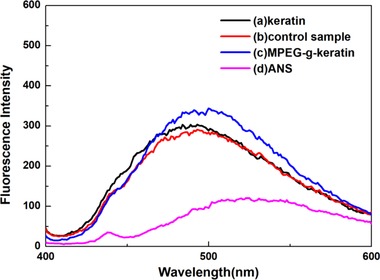
Fluorescence spectroscopy of keratin and MPEG‐g‐keratin
3.3. XRD
The XRD spectra of the keratin, control sample and MPEG‐g‐keratin were shown in Figure 7, all of these samples had two distinct peaks 44. The α‐helix structure generally appeared at 2θ = 9° and 17.8° and the β‐sheet structure peaks appeared at 2θ = 9° and 19°. However, due to the overlapping signals from the α‐helix and the β‐sheet at about 2θ = 17.8° and 19°, both peaks cannot be clearly assigned. The peaks of the MPEG‐g‐keratin at 2θ = 9° and 19° was stronger than that of keratin, indicating an increase in the number of the β‐sheet structure. While the peaks of the control sample 2θ = at 9° and 19° only increases a little bit, which was in consistence with the result of CD.
Figure 7.
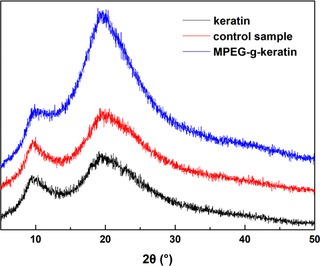
XRD patterns of keratin and MPEG‐g‐keratin
3.4. Properties of the keratin and MPEG‐g‐keratin membrane
The air‐dried keratin membrane was shown in Figure 8. The keratin membrane (A and B) had been broken due to brittleness of the material when it was air‐dried. While the membrane of MPEG‐g‐keratin (C and D) had a high toughness in the peptide chain segment, due to the introduction of MPEGMA, the forming property of membrane was improved. Biocompatibility of the keratin membrane was investigated using NIH/3T3 cell viability assay. As shown in Figure 9, cell viability of the keratin membrane was 96.2%, indicating that the material was non‐cytotoxic and allowed cell attachment and proliferation. The cell viability of the MPEG‐g‐keratin membrane increased to 107.5%, which indicated that the keratin was not only non‐toxic, but also had a proliferative effect on cells after grafted with MPEGMA. The testing data further proved that grafting of MPEGMA would help the improvement of the biocompatibility activity of keratin.
Figure 8.

The membrane of keratin and MPEG‐g‐keratin. (A) The keratin membrane after air‐dried. (B) The keratin membrane after being squeezed. (C) The MPEG‐g‐keratin membrane after air‐dried. (D) The MPEG‐g‐keratin after being squeezed)
Figure 9.
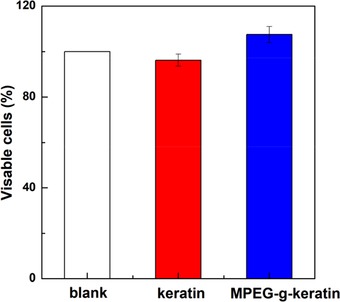
Biocompatibility of keratin and MPEG‐g‐keratin
4. CONCLUDING REMARKS
MPEG was grafted onto keratin through thiol‐ene photoclick reaction and modified keratin membrane can be successfully prepared without any reduction in biocompatibility. The flexibility of the modified keratin was improved because MPEG has the effect of “Polymer‐ Induced Conformation Transition”. The interaction between keratin and molecular chain‐to‐chain was waked, and PEG played a plasticizing role. As a result, the introduction of MPEG can induce the conformation of keratin chains and promote the formation of β‐sheet. The combined effect of these factors improved the flexibility of membrane. It provided an effective way for producing keratin biomaterials. However, 2‐mercaptoethanol and UV light employed in the paper are not eco‐friendly, though the efficiency is relatively high. In the future, alternative methods such as L‐cysteine and visible light can be employed to achieve a greener process.
CONFLICT OF INTEREST
The authors have declared no conflict of interest.
ACKNOWLEDGMENTS
This work was financially supported by the National Natural Science Foundation of China (31771039, 31300785, 51673087), Fundamental Research Funds for the Central Universities (JUSRP51717A), the National Key R&D Program of China (2017YFB0309200), the Open Project Program of Key Laboratory of Eco‐textiles, Ministry of Education, Jiangnan University (No.·KLET1504).
Ye X, Yuan J, Jiang Z, et al. Thiol‐ene photoclick reaction: An eco‐friendly and facile approach for preparation of MPEG‐g‐keratin biomaterial. Eng Life Sci. 2020;20:17–25. 10.1002/elsc.201900105
REFERENCES
- 1. Wang, K. , Li, R. , Ma, J. , Jian, Y. , Che, J. , Extracting keratin from wool by using L‐cysteine. Green Chem. 2016, 18, 476–481. [Google Scholar]
- 2. Zoccola, M. , Aluigi, A. , Tonin, C. , Characterisation of keratin biomass from butchery and wool industry wastes. J. Mol. Struct. 2009, 938, 35–40. [Google Scholar]
- 3. Eslahi, N. , Dadashian, F. , Nejad, N. H. , An investigation on keratin extraction from wool and feather waste by enzymatic hydrolysis. Prep. Biochem. Biotech. 2013, 43, 624–648. [DOI] [PubMed] [Google Scholar]
- 4. Idris, A. , Vijayaraghavan, R. , Rana, U. A. , Fredericks, D. et al., Dissolution of feather keratin in ionic liquids. Green Chem. 2013, 15, 525–534. [Google Scholar]
- 5. Xie, H. , Li, S. , Zhang, S. , Ionic liquids as novel solvents for the dissolution and blending of wool keratin fibers. Green Chem. 2005, 7, 606–608. [Google Scholar]
- 6. Yamauchi, K. , Hojo, H. , Yamamoto, Y. , Tanabe, T. , Enhanced cell adhesion on RGDS‐carrying keratin film. Mat. Sci. Eng. C‐Mater. 2003, 23, 467–472. [Google Scholar]
- 7. Andreia, V. , Artur, C. P. , The use of keratin in biomedical applications. Curr. Drug Targets 2013, 14, 612–619. [DOI] [PubMed] [Google Scholar]
- 8. Duncan, W. J. , Greer, P. F. C. , Lee, M.‐H. , Loch, C. , Gay, J. H. A. , Wool‐derived keratin hydrogel enhances implant osseointegration in cancellous bone. J. Biomed. Mater. Res. B 2018, 106, 2447–2454. [DOI] [PubMed] [Google Scholar]
- 9. Xu, X. , Wu, J. , Meng, Z. , Li, Y. et al., Enhanced exfoliation of biocompatible MoS2 nanosheets by wool keratin. ACS Appl. Nano Mater. 2018, 1, 5460–5469. [Google Scholar]
- 10. Sarac, A. S. , Redox polymerization. Prog. Polym. Sci. 1999, 24, 1149–1204. [Google Scholar]
- 11. Tsukada, M. , Shiozaki, H. , Freddi, G. , Crighton, J. S. , Graft copolymerization of benzyl methacrylate onto wool fibers. J. Appl. Polym. Sci. 1997, 64, 343–350. [Google Scholar]
- 12. Tachibana, A. , Furuta, Y. H. , Tanabe, T. , Yamauchi, K. , Fabrication of wool keratin sponge scaffolds for long‐term cell cultivation. J. Biotechnol. 2002, 93, 165–170. [DOI] [PubMed] [Google Scholar]
- 13. Yu, D. , Cai, J. Y. , Church, J. S. , Wang, L. , Modifying surface resistivity and liquid moisture management property of keratin fibers through thiol–ene click reactions. Acs Appl. Mater. Inter. 2014, 6, 1236–1242. [DOI] [PubMed] [Google Scholar]
- 14. Liu, P. , O'mara, B. W. , Warrack, B. M. , Wu, W. et al., A tris (2‐carboxyethyl) phosphine (TCEP) related cleavage on cysteine‐containing proteins. J. Am. Soc. Mass Spectr. 2010, 21, 837–844. [DOI] [PubMed] [Google Scholar]
- 15. Burns, J. A. , Butler, J. C. , Moran, J. , Whitesides, G. M. , Selective reduction of disulfides by tris (2‐carboxyethyl) phosphine. J. Org. Chem. 1991, 22, 2648–2650. [Google Scholar]
- 16. Garg, S. M. , Xiong, X. , Lu, C. , Lavasanifar, A. , Application of click chemistry in the preparation of poly(ethylene oxide)‐block‐poly(ε‐caprolactone) with hydrolyzable cross‐links in the micellar core. Macromolecules 2011, 39, 2058–2066. [Google Scholar]
- 17. Liu, Y. , Hou, W. , Sun, H. , Cui, C. et al., Thiol–ene click chemistry: a biocompatible way for orthogonal bioconjugation of colloidal nanoparticles, Chem. Sci. 2017, 8, 6182–6187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang, S. , He, X. , Chen, L. , Zhang, Y. , Boronic acid functionalized magnetic nanoparticles via thiol–ene click chemistry for selective enrichment of glycoproteins, New J. Chem. 2014, 38, 4212–4218. [Google Scholar]
- 19. Wu, J. T. , Huang, C. H. , Liang, W. C. , Wu, Y. L. et al., Reactive polymer coatings: a general route to thiol‐ene and thiol‐yne click reactions. Macromol. Rapid Comm. 2012, 33, 922–927. [DOI] [PubMed] [Google Scholar]
- 20. Wendeln, C. , Rinnen, S. , Schulz, C. , Arlinghaus, H. F. , Ravoo, B. J. , Photochemical microcontant printing by thiol‐ene and thiol ‐yne click chemistry. Langmuir the Acs J. Surfaces & Colloids 2010, 26, 15966–15971. [DOI] [PubMed] [Google Scholar]
- 21. Lin, C. C. , Raza, A. , Shih, H. , PEG hydrogels formed by thiol‐ene photo‐click chemistry and their effect on the formation and recovery of insulin‐secreting cell spheroids. Biomaterials 2011, 32, 9685–9695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yu, B. , Jiang, X. , Qin, N. , Jie, Y. , Thiol–ene photocrosslinked hybrid vesicles from co‐assembly of POSS and poly(ether amine) (PEA). Chem. Commun. 2011, 47,12110‐12112. [DOI] [PubMed] [Google Scholar]
- 23. Harris, J. M. , Introduction to Biotechnical and Biomedical Applications of Poly(Ethylene Glycol), Springer, Boston, MA: 1992. [Google Scholar]
- 24. Slavin, S. , Khoshd, E. , Haddleton, D. M. , PEGylation of surface protein filaments: coverage and impact on denaturation. RSC Adv. 2011, 1, 58–66. [Google Scholar]
- 25. Yu, Y. , Yan, C. , Ren, J. , Cao, E. et al., Plasticizing effect of poly(ethylene glycol)s with different molecular weights in poly(lactic acid)/starch blends. J. Appl. Polym. Sci. 2015, 132, 41808. [Google Scholar]
- 26. DomjáN, A. , Bajdik, J. N. , Pintye‐HóDi, K. R. , Understanding of the plasticizing effects of glycerol and PEG 400 on chitosan films using solid‐state NMR spectroscopy. Macromolecules 2009, 42, 4667–4673. [Google Scholar]
- 27. Yamauchi, K. , Yamauchi, A. , Kusunoki, T. , Kohda, A. , Konishi, Y. , Preparation of stable aqueous solution of keratins, and physiochemical and biodegradational properties of films. J. Biomed. Mater. Res. B 1996, 31, 439–444. [DOI] [PubMed] [Google Scholar]
- 28. Chan, K. Y. , Wasserman, B. P. , Direct colorimetric assay of free thiol groups and disulfide bonds in suspensions of solubilized and particulate cereal proteins. Cereal Chem. 1993, 70, 22–26. [Google Scholar]
- 29. Bailon, P. , Palleroni, A. , Schaffer, C. A. , Spence, C. L. et al., Rational design of a potent, long‐lasting form of interferon: a 40 kDa branched polyethylene glycol‐conjugated interferon α‐2a for the treatment of hepatitis C. Bioconjug. Chem. 2001, 12, 195–202. [DOI] [PubMed] [Google Scholar]
- 30. Kurfürst, M. M. , Detection and molecular weight determination of polyethylene glycol‐modified hirudin by staining after sodium dodecyl sulfate‐polyacrylamide gel electrophoresis. Anal. Biochem. 1992, 200, 244–248. [DOI] [PubMed] [Google Scholar]
- 31. Xu, Y. , Liu, J. , Du, C. , Fu, S. , Liu, X. , Preparation of nanoscale carbon black dispersion using hyper‐branched poly(styrene‐ alt ‐maleic anhydride). Prog Org Coat. 2012, 75, 537–542. [Google Scholar]
- 32. Haskard, C. A. , Li‐Chan, E. C. Y. , Hydrophobicity of bovine serum albumin and ovalbumin determined using uncharged (PRODAN) and anionic (ANS‐) fluorescent probes. J. Agr. Food Chem. 1998, 46, 2671–2677. [Google Scholar]
- 33. Zhou, Q. , Cui, L. , Ren, L. , Wang, P. et al., Preparation of a multifunctional fibroin‐based biomaterial via laccase‐assisted grafting of chitooligosaccharide. Int. J. Biol. Macromol. 2018, 113, 1062–1072. [DOI] [PubMed] [Google Scholar]
- 34. Fan, Q.‐L. , Neoh, K.‐G. , Kang, E.‐T. , Shuter, B. , Wang, S.‐C. , Solvent‐free atom transfer radical polymerization for the preparation of poly (poly (ethyleneglycol) monomethacrylate)‐grafted Fe3O4 nanoparticles: synthesis, characterization and cellular uptake. Biomaterials 2007, 28, 5426–5436. [DOI] [PubMed] [Google Scholar]
- 35. Shi, Y. , Liu, Z. , Yang, Y. , Xu, X. et al., Design of poly(mPEGMA‐ co ‐MAA) hydrogel‐based mPEG‐b‐PCL nanoparticles for oral meloxicam delivery. Mat. Sci. Eng. C‐Mater. 2017, 76, 975–984. [DOI] [PubMed] [Google Scholar]
- 36. Shi, Y. , Sun, F. , Dan, W. , Zhang, R. et al., Enhancement of bioavailability by formulating rhEPO ionic complex with lysine into PEG–PLA micelle. J. Nanopart. Res. 2013, 15, 1–10. [Google Scholar]
- 37. Takahashi, K. , Kato, H. , Kinugasa, S. , Development of a standard method for nanoparticle sizing by using the angular dependence of dynamic light scattering. Anal. Sci. 2011, 27, 751–756. [DOI] [PubMed] [Google Scholar]
- 38. Zhao, J. , Wang, H. , Liu, J. , Deng, L. et al., Comb‐like amphiphilic copolymers bearing acetal‐functionalized backbones with the ability of acid‐triggered hydrophobic‐to‐hydrophilic transition as effective nanocarriers for intracellular release of curcumin, Biomacromolecules 2013, 14, 3973–3984. [DOI] [PubMed] [Google Scholar]
- 39. Logie, J. , Owen, S. C. , Mclaughlin, C. K. , Shoichet, M. S. , PEG‐graft density controls polymeric nanoparticle micelle stability, Chem. Mater. 2014, 26, 2847–2855. [Google Scholar]
- 40. Gattuso, H. , García‐Iriepa, C. , Sampedro, D. , Monari, A. , Marazzi, M. , Simulating the electronic circular dichroism spectra of photoreversible peptide conformations, J. Chem. Theory Comput. 2017, 13, 3290–3296. [DOI] [PubMed] [Google Scholar]
- 41. Xin, C. , Li, W. , Yu, T. , Conformation transition of silk fibroin induced by blending chitosan, J. Polym. Sci. Pol. Phys. 1997, 35, 2293–2296. [Google Scholar]
- 42. Meirovitch, H. , Rackovsky, S. , Scheraga, H. A. , Empirical studies of hydrophobicity. 1. Effect of protein size on the hydrophobic behavior of amino acids, Macromolecules 1980, 13, 1398–1405. [Google Scholar]
- 43. Alizadeh‐Pasdar, N. , Li‐Chan, E. C. , Comparison of protein surface hydrophobicity measured at various pH values using three different fluorescent probes, J. Agr. Food Chem. 2000, 48, 328–334. [DOI] [PubMed] [Google Scholar]
- 44. Jiang, Z. , Yuan, J. , Wang, P. , Fan X. et al., Dissolution and regeneration of wool keratin in the deep eutectic solvent of choline chloride‐urea, Int. J. Biol. Macromol. 2018, 119, 423–430. [DOI] [PubMed] [Google Scholar]