

Abstract
The selection of aptamers represents a promising route in the development of high affinity ligands. In these processes the formation of by‐products is a common problem during the PCR‐based amplification of complex oligonucleotide libraries. One approach to overcome this drawback is to separate each template oligonucleotide into an individual reaction compartment provided by a droplet. This method, termed emulsion PCR (ePCR), has already emerged to a standard method in sample preparation for 2nd generation sequencing. In this work, we compare different literature protocols that have been developed to generate stable emulsions for ePCR. We investigate different emulsification methods and evaluate the importance of the initial template concentration. We demonstrate that emulsion stability is of utmost importance for the successful inhibition of by‐product formation and give an optimized protocol for generation of an emulsified PCR.
Keywords: Aptamer, By‐product formation, Emulsion PCR, Polymerase chain reaction, SELEX
Abbreviation
- SELEX
systematic evolution of ligands by exponential enrichment
1. Introduction
Modern biology and medicine rely heavily on the use of specific ligands for molecular recognition of biomarkers or other molecules. Historically, this field is dominated by the use of antibodies that are produced by either immunization of animals and subsequent harvesting of antibodies or by large‐scale cell culture. However, in the past 20 years the generation of artificial ligands has made substantial progress. One type of artificial ligands is represented by single‐stranded oligonucleotides, termed aptamers. Depending on their sequence and applied conditions (e.g. buffer composition) these aptamers fold into specific 3D structures. In their folded state aptamers are able to bind to a variety of targets such as ions, small molecules, and peptides as well as to proteins or cells 1. Aptamers are usually described as high affinity ligands with a distinct specificity for their corresponding target, which makes them excellent ligands for affinity purification 2, 3, 4 and offers great potential in sensing applications 5, 6, 7. The aptamer‐target binding is based on electrostatic interactions, van der Waals interactions, and hydrogen bonding that are nonspecific by nature, but the synergistic combination of these interactions may result in a high affinity binding. Consequently, aptamers do display high specificities for their target, due to an excellent structural match with the target.
Aptamers are generated via an iterative selection process called systematic evolution of ligands by exponential enrichment (SELEX). SELEX typically is initiated by incubation of a target molecule with an oligonucleotide library consisting of 1014–1015 different oligonucleotides. All oligonucleotides are composed of a central randomized region that is flanked by defined priming regions. After incubation of library and target, unbound oligonucleotides are washed away, while bound oligonucleotides are later recovered by elution. Subsequently, these recovered aptamer‐candidates are amplified using PCR. Following a strand separation, the enriched library of aptamer candidates is subjected to the next round of selection and amplification. Typically, it takes 8–15 rounds of selection to enrich high affinity aptamers from a randomized library 8. After sufficient enrichment the aptamer‐candidates are commonly identified by cloning and sequencing. Once identified, affinity and specificity can be assessed for each aptamer candidate individually. Despite employing only standard methods the success rate of conventional SELEX is only about 30% 9.
Since the beginning of aptamer development many technologies have been applied to overcome inherent barriers and limitations of SELEX. Here, we only give a short overview of major improvements, while in‐depth reviews on these topics can be found elsewhere 1, 9, 10. A core step in SELEX is the separation of bound and unbound aptamer‐candidates after incubation with the target. Today, highly efficient microfluidic, bead‐based, or electrophoretic partitioning techniques are most utilized in aptamer selection, while traditional SELEX relied on nitrocellulose filter binding 9. Another key factor for SELEX success is the starting library, as the structure of an aptamer and its possible interactions with the respective target directly depends on the aptamer's sequence. Therefore, an increasing effort has been put on modifying starting libraries. These modifications include optimized motif distribution, degenerated libraries of a known binding motif or secondary structure and the use of artificial nucleotides with an extended nucleotide alphabet 11, 12. Furthermore, downstream identification and characterization of aptamer‐candidates is greatly facilitated by 2nd generation sequencing and high‐throughput characterization 10. Altogether these improvements have raised the overall success rate of SELEX to ≈85%.
Although, this suggests an overall improvement of SELEX processes, little attention has been given to the particular field of aptamer‐candidate amplification. Researches frequently report the formation of by‐products in conventional PCR, but only a few publications address this issue. In recent years, it has been discovered that different PCR mechanisms apply to complex libraries in comparison to PCR of a single amplicon. Nonspecific primer annealing and the formation of primer dimers, which are the main source of by‐products in conventional PCR, seem to be outpaced by product–product hybridization (transpriming) of partially homologous sequences that occurs during amplification of complex libraries once a critical template concentration is reached 13, 14, 15. In further investigations Tolle et al. discovered mechanisms that lead to the formation of “ladder‐type” and “nonladder‐type” by‐products by sequence analysis 16. Beside the formation of by‐products, the product formation in PCR is biased toward shorter or structurally less stable sequences and sequences that match the polymerase sequence preference (PCR bias) 10, 17. Overall, studies demonstrate that conventional PCR results in a library diversity loss of ≈50% within each PCR 18.
To overcome these drawbacks, conventional PCR has been adapted to SELEX conditions by careful optimization of reaction parameters 13, 19. Furthermore, limitation of PCR cycles and real‐time PCR also has been applied to avoid overamplification 20. Other approaches include multiple partitioning steps between each amplification step (non‐SELEX 21) or reduction of selection cycles by using powerful partitioning methods, such as CE, ultimately leading to a one‐step selection 22, 23. Although these optimizations are able to reduce the amount of by‐products, the formation of by‐products and the PCR bias still are a major problem in SELEX processes.
However, in past years another PCR variation has emerged in the preparation of 2nd generation sequencing libraries, which is also prone to by‐product formation and PCR bias. In order to prevent transpriming each member of the initial library is encapsulated in a separate PCR droplet that is surrounded by a hydrophobic organic phase. This method is called emulsion PCR (ePCR). Due to this separation each target is amplified individually as each droplet only contains a single species. Consequently, ePCR has been found to significantly reduce the PCR bias and the formation of by‐products to a nondetectable level while preserving library diversity at the same time 17, 18. Thus ePCR should be considered the standard method for amplification of aptamer‐candidates during SELEX.
The current literature features two different protocols for emulsification of a PCR mixture: While Williams et al. describe the formulation of an emulsion PCR based on a mixture composed of mineral oil supplemented with the detergents Triton‐X‐100 and Tween 80 14, another organic phase for ePCR was developed by Diehl et al. using the emollient Tegosoft DEC mixed with mineral oil and the emulsifier ABIL WE 09 24. Both methods aim to create a stable emulsion that is a challenging task considering the high temperature applied during PCR. Based on the protocol of Williams, Shao et al. evaluated and optimized different parameters for ePCR, e.g. the starting template concentration, the annealing temperature, the primer concentration, and the polymerase concentration 15. They demonstrated that the initial target concentration is the most important parameter of an emulsion PCR besides the emulsion stability. Following the Poisson distribution, the starting concentration defines the number of different oligonucleotides that will be encapsulated in the same droplet. Reaching a critical number, templates will start to form by‐products within the droplet again 15. Additionally, PCR bias will be inevitable at high target concentration. Another important parameter is the concentration of BSA that is supplemented to the PCR mixture. Williams stated that BSA is essential in ePCR to saturate the aqueous/organic interface with a “bulk protein” to protect the polymerase from getting inactivated at that interface 14. Shao et al. required a high amount of DNA polymerase as they did not add BSA to the aqueous PCR phase 15. However, BSA and DNA polymerase concentration should be optimized for each PCR and emulsification protocol individually 25.
In this study, we compare the most common protocols for PCR emulsification with focus on emulsion stability, by‐product formation, and ease of preparation.
2. Materials and methods
2.1. Chemicals and materials
Mineral oil, ethyl acetate, Tween 80, and Span 80 were purchased from Sigma‐Aldrich (Munich, Germany). Platinum® Pfx DNA Polymerase, and dNTPs were purchased from Thermo Scientific (Rockford, USA). Tegosoft DEC and ABIL WE 09 were kindly supplied by Evonik (Essen, Germany). Diethly ether, agarose, and Roti®‐GelStain were purchased from Carl Roth (Karlsruhe, Germany). Tween 20 was purchased from AppliChem PanReac (Darmstadt, Germany). Isobutanol was purchased from Honeywell Riedel‐de Haën (Seelze, Germany). DNA purifications were performed with QIAquick® PCR Purification Kit from Qiagen (Hilden, Germany) according to the manufacturer's protocol.
2.2. Oligonucleotides
The Oligonucleotide library design was taken from the US patent U.S.7329742 26 and was composed of a 40 nt randomized region flanked by upstream and downstream priming sites (78 nt total): 5′‐GGTATTGAGGGTCGCATC‐NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN‐GATGGCTCTAACTCTCCTCT‐3′. For size comparison 6H7 aptamer was chosen (78 nt), which also was published in the same US patent: 5′‐GGTATTGAGGGTCGCATC‐GCTATGGGTGGTCTGGTTGGGATTGGCCCCGGGAGCTGGC‐GATGGCTCTAACTCTCCTCT‐3′. The sequence of forward primer was 5′‐GGTATTGAGGGTCGCATC‐3′, while the oligonucleotide 5′‐AGAGGAGAGTTAGAGCCATC‐3′ was used as the reverse primer during PCR. All oligonucleotides were supplied by IDT (Coralville, USA).
2.3. Polymerase chain reaction
PCR was performed in a Life ECO instrument from Bioer (Hangzhou, China) utilizing Thermo Scientific's Platinum® Pfx DNA Polymerase. The final concentration of reagents in the aqueous PCR phase was as follows: 1× Pfx amplification buffer, 1 mM magnesium sulfate, 0.2 mM of each dNTP, 1 μM forward and reverse primer each, Platinum® Pfx DNA Polymerase 0.02 U/μL. BSA and DNA template concentration were varied as specified.
For emulsion PCR, organic phases were prepared following the primary literature. In brief, Diehl's organic phase was composed of 7% ABIL WE09, 20% mineral oil and 73% Tegosoft DEC and therefore is referred to as Tegosoft‐based 24. For the organic phase Williams et al. used, mineral oil is supplemented with 4.5% Span 80, 0.4% Tween 80, and 0.05% Triton X‐100 and is referred to as mineral oil‐based 14. To generate an emulsion, 100 μL of aqueous PCR phase were emulsified with 200 μL of the corresponding organic phase by shaking in a Mixer Mill MM 400 from Retsch (Haan, Germany) at 30 Hz for 30 s. Mastermixes of PCR phase were used during the preparation of ePCR mixtures to guarantee uniform distribution of reagents within each series. A nonemulsified control sample of 50 μL was drawn from each mastermix to ensure proper reaction setup (openPCR; oPCR).
To ensure proper temperature distribution within the reactions, each PCR reaction was divided into 50 μL aliquots prior to PCR, which were rejoined after thermocycling. After initial denaturation at 95°C for 5 min, samples were subjected to 20 cycles of denaturation at 95°C, primer annealing at 56°C, each for 30 s, and elongation at 68°C for 15 s. Following the final elongation at 68°C for 5 min, samples were stored at 4°C.
After PCR, products were pooled, extracted with 2 × 1 mL water‐saturated diethyl ether; 1 × 1 mL water‐saturated ethyl acetate and finally 2 × 1 mL water‐saturated diethyl ether. Subsequently, PCR products were purified using the QIAquick® PCR Purification Kit from Qiagen (Hilden, Germany). Purified PCR products were subjected to agarose gel electrophoresis and corresponding images were analyzed with AlphaEaseFC 6.0.0.14 after staining with Roti®‐GelStain.
3. Results and discussion
During our attempt to amplify a diverse oligonucleotide library, we noticed the formation of longer by‐products. Consistently, this by‐product formation was reproduced using fresh reagents and oligonucleotide library. The question arose if this was a library inherited matter, so we compared the amplification of the oligonucleotide library to the amplification of a single defined sequence (6H7) (Fig. 1).
Figure 1.
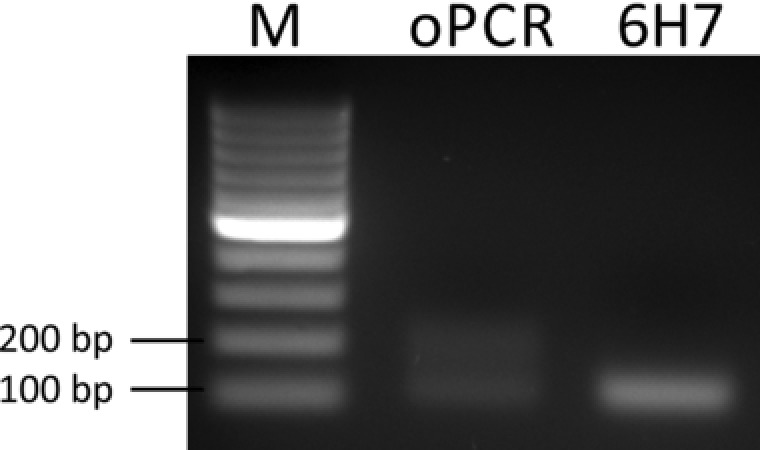
Formation of by‐products during PCR of a complex oligonucleotide library in open PCR. For comparison: PCR product of 6H7‐aptamer (defined sequence of the same length) shows no detectable by‐products.
As expected the amplification of the oligonucleotide library lead to by‐products, while the amplification product of the defined sequence did not show any by‐products. However, the phenomenon of by‐product formation has also been described by other researchers [15], who were amplifying highly complex oligonucleotide libraries and it is believed to be due to product‐product‐transpriming during the annealing phase in PCR. Emulsion PCR (ePCR) aims to inhibit the formation of by‐products by separation of individual templates. Most publications on the field of ePCR were based on the work of either Williams et al. or Diehl et al. 14, 24. As both publications described the generation of an emulsion for PCR, we focused on comparing these protocols in regard to effectiveness, reliability, and ease of integration into the lab workflow.
3.1. Oil‐surfactant mixtures for ePCR
To compare the protocols of Diehl and Williams, each ePCR reaction was prepared following the primary literature. However, for better comparability the initial template concentration was set to 0.2 nM and all emulsions were simultaneously generated by shaking in a bead mill for 30 s at 30 Hz. After 20 PCR cycles, the organic phase was extracted by diethyl ether/ethyl acetate and PCR products were analyzed on a 1.5% agarose gel (Fig. 2).
Figure 2.
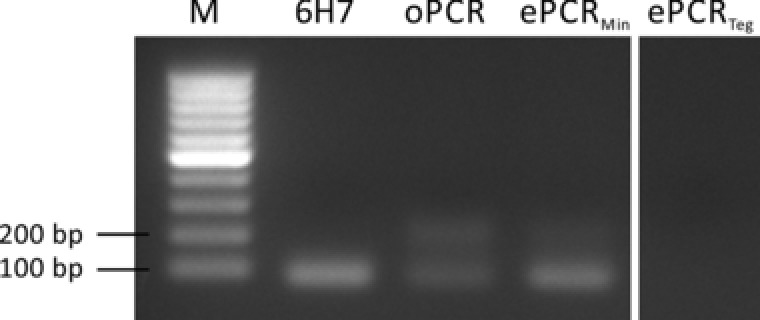
Comparison of organic phases for the emulsification of PCR (mineral oil mixture; ePCR Min/Tegosoft DEC mixture; ePCR Teg). 6H7 was amplified as a length standard. For clarification, only selected samples of the whole gel are shown.
The mineral oil‐based emulsion broke down during PCR, resulting in partial segregation into separate phases. Consequently, by‐products were formed during PCR, as target‐target‐hybridization is no longer prevented by droplet barriers. Despite the emulsion being unstable, less by‐product were produced than in the open PCR control. On the other hand, the emulsion generated with Tegosoft remained white/creamy after thermocycling, indicating a stable emulsion. Surprisingly, no PCR product was produced in the Tegosoft emulsion PCR. However, both Williams et al. 14 and Schütze et al. 25 stated that the polymerase may become trapped at the emulsion interface. Therefore, they advise the addition of BSA and the optimization of its concentration. Considering the poor emulsion stability using the mineral oil mixture we focused on optimizing the Tegosoft protocol for ePCR. The effect of BSA was investigated by applying different BSA concentrations of 0 mg/mL, 0.5 mg/mL, or 1 mg/mL during PCR in a Tegosoft‐based emulsion. Again the PCR products were degreased with diethyl ether/ethyl acetate, purified using the Qiaquick PCR purification kit (Qiagen) and analyzed via agarose gel electrophoresis (Fig. 3).
Figure 3.
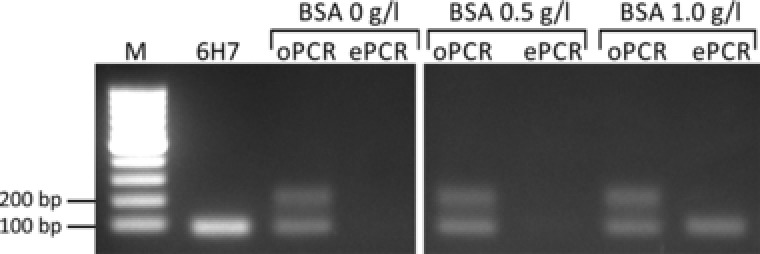
Effect of BSA concentration on amplification results of Tegosoft DEC‐based emulsion PCR. For clarification, only selected samples of the gel are shown.
As observed before, no PCR product was formed using BSA concentrations of 0 mg/mL. Also a BSA concentration of 0.5 mg/mL was not sufficient to successfully perform ePCR. However, increasing the BSA concentration to 1 mg/mL led to the formation of PCR products without any detectable amounts of by‐products. Therefore, all further emulsification of PCR mixtures was performed using the Tegosoft‐based organic phase and BSA at the concentration of 1 mg/mL.
3.2. Preparation of emulsions‐methods and important parameters
Besides the formulation of the organic phase, the emulsification technique is a key factor for the generation of a stable emulsion. Comparing the literature protocols, many emulsions have been generated using magnetic stirrers. In general, during this method the organic phase is continuously stirred while the PCR mixture is successively added in small portions. The resulting emulsion is then usually stirred for additional 5 min before performing PCR. Despite the emulsification of PCR with a magnetic stirrer seems to be the standard procedure, we instead focused on using a mixer mill (tissue lyser) as it is not only time efficient but also could be used for high parallel processing of many reactions. To assay the effect of mixing time on the emulsion stability, different Tegosoft DEC‐based PCR emulsions were prepared by variation of the emulsification time in a ball mill at 30 Hz. The generated emulsions were subjected to 20 cycles of PCR, and PCR products were subsequently purified and analyzed by agarose gel electrophoresis (Fig. 4). Phase contrast microscopy was used to determine the mean droplet diameter for the emulsions prepared by mixing for 10, 30, and 60 s (see Supporting Information Fig. 1).
Figure 4.
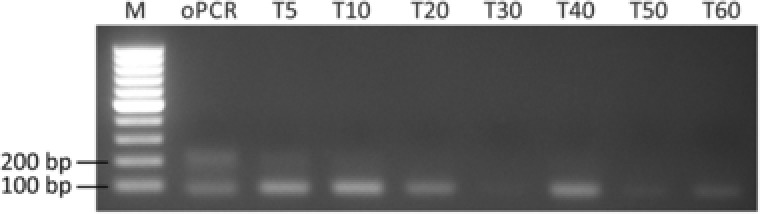
Formation of products and by‐products in emulsion PCR, depending on time of emulsification. Emulsification times of 5, 10, 20, 30, 40, 50, and 60 s were tested and analyzed on 1.5% agarose gel with Roti®‐GelStain.
After thermocycling the emulsions of 5–20 s have partially separated into the designated phases indicating that these mixing times are insufficient for the generation of stable emulsions. The other investigated emulsions have been stable. Generally, mixing times between 30 and 40 s yielded in the highest product concentration without any detectable by‐products. Here, droplet size is about 6 μm resulting in about 109 compartments. However, in this particular experiment emulsification of PCR for 30 s resulted in minimal amplification. Elongating the mixing time to 60 s lead to a decreased droplet diameter of 5 μm and increased the numbers of droplets by 75%. As the droplet diameter decreased, the amount of formed product is decreasing as well. This is consistent with theory considering that smaller droplets carry less PCR reagents, which will deplete earlier. This is not balanced out by the higher droplet amount, since many droplets remain empty with regard to target sequence. These experiments demonstrated that emulsions are best prepared in a mixer mill shaking for 40 s.
3.3. Overloading droplets‐Impact of initial template concentration
The initial template concentration is another important parameter to consider during ePCR. Emulsion PCR aims to circumvent product–product hybridization by separation of the templates into individual droplets. However, the number of compartments is limited. Therefore, high target concentrations can lead to droplets that initially contain more than one single template. Increasing the initial template number per droplet raises the likelihood of product–product hybridization drastically and facilitates by‐product formation. In contrast, too low template concentrations might result in low PCR efficiency due to loss of PCR reagents in droplets not containing a template molecule.
Here, we demonstrate that overloading of droplets with templates lead to by‐product formation and therefore limit the ePCR's effectiveness. For this purpose, we performed ePCR and open PCR with an initial target concentration range between 0.02 and 2000 nM and compared the amounts of formed by‐products (Fig. 5).
Figure 5.
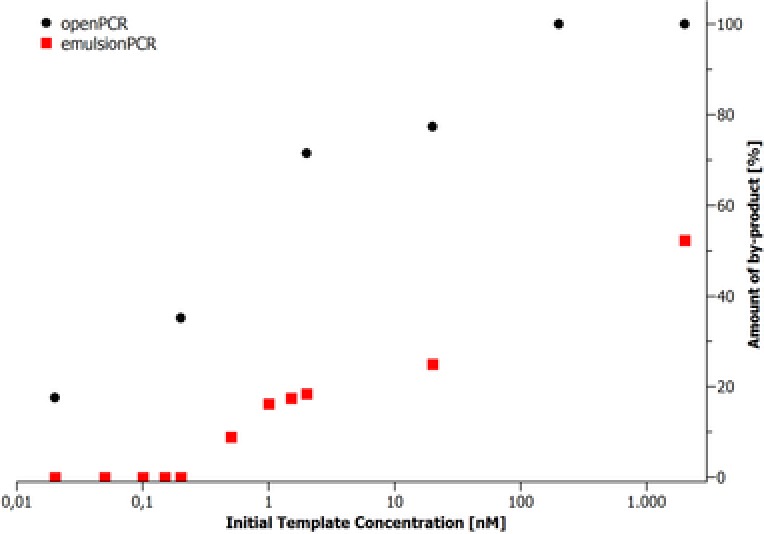
Concentration‐dependent formation of by‐products in open PCR and emulsion PCR. Amounts were determined by agarose gel electrophoresis and subsequent densitometric analysis of the corresponding band‐pattern.
In the open PCR by‐products are formed, even at low template concentrations. The amount of by‐products steadily increases in the concentration range between 0.2 and 20 nM. Above the initial concentration of 20 nM nearly all oPCR product is transformed into by‐product. For the ePCR, by‐product formation starts at the concentration above 0.2 nM with a reduced degree of by‐product formation in comparison to the open PCR control. At the highest investigated concentration of 2000 nM the amount of by‐product reaches ≈50%. However, by‐product formation is totally inhibited for starting concentrations up to 0.2 nM. This demonstrates the advantages of ePCR for the amplification of highly diverse oligonucleotide libraries.
4. Concluding remarks
PCR bias and the formation of by‐products are common problems during the amplification of complex oligonucleotide libraries. To overcome these drawbacks, ePCR, which relies on the separation of templates into individual droplets, has been applied. We compared different literature protocols that have been developed to generate stable emulsions for PCR. During our experiments we found that emulsions based on Tegosoft were far more stable than emulsions based on mineral oil. However, initial ePCR attempts with Tegosoft‐based emulsions failed. Successful amplification was achieved by increasing BSA concentration to 1 mg/mL. Therefore, we strongly advise the optimization of BSA concentration. In addition to oil‐surfactant comparison, different emulsification methods have been evaluated. In most literature protocols emulsions were generated by stirring with a magnetic rod. To allow parallel reaction setup and shorten the emulsification times we employed a bead mill instead. Notably, incubation time is a key parameter as short emulsification lead to instable emulsions but longer emulsification times decreased the obtained product amount. This is due to an increased droplet number, which do not contain a target sequence to be amplified. Beside these parameters being key factors for the emulsion stability, the initial template concentration is critical to circumvent the formation of by‐products. High template concentrations result in the entrapment of multiple templates per droplet which then may form by‐products. Therefore, ePCR's effectiveness is significantly reduced at high template concentrations. Our findings suggest that the tolerable template concentration can be determined by titration.
Overall, in this work we demonstrated the advantages of ePCR over open PCR for the amplification of complex libraries in regard to by‐product formation and PCR bias. Moreover, we present key parameters for successful ePCR along with corresponding methods to optimize each parameter.
Practical Application
Amplification of complex oligonucleotide libraries via PCR is essential, e.g. for the selection of aptamers. This amplification is hampered by the formation of by‐products and PCR‐bias. To overcome these limitations, we have compared different protocols for emulsion PCR and optimized conditions for emulsion PCR. Besides providing optimized protocols, this study can also be used as a guideline for systematic optimization of emulsion PCR.
The authors have declared no conflicts of interest.
Supporting information
Supplementary information
Aknowledgments
This project was funded by Biofabrication for NIFE (supported by the german Lower Saxonian Ministry for Science and Culture and the VolkswagenStiftung). Tegosoft DEC and ABIL WE 09 were kindly provided by Evonik Industries AG.
5 References
- 1. Stoltenburg, R. , Reinemann, C. , Strehlitz, B. , SELEX—a (r)evolutionary method to generate high‐affinity nucleic acid ligands. Biomol. Eng. 2007, 24, 381–403. [DOI] [PubMed] [Google Scholar]
- 2. Lönne, M. , Bolten, S. , Lavrentieva, A. , Stahl, F. et al., Development of an aptamer‐based affinity purification method for vascular endothelial growth factor. Biotechnol. Rep. 2015, 8, 16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schax, E. , Lönne, M. , Scheper, T. , Belkin, S. et al., Aptamer‐based depletion of small molecular contaminants: a case study using ochratoxin a. Biotechnol. Bioprocess Eng. 2015, 20, 1016–1025. [Google Scholar]
- 4. Walter, J.‐G. , Stahl, F. , Scheper, T. , Aptamers as affinity ligands for downstream processing. Eng. Life Sci. 2012, 12, 496–506. [Google Scholar]
- 5. Urmann, K. , Walter, J.‐G. , Scheper, T. , Segal, E. , Label‐free optical biosensors based on aptamer‐functionalized porous silicon scaffolds. Anal. Chem. 2015, 87, 1999–2006. [DOI] [PubMed] [Google Scholar]
- 6. Urmann, K. , Arshavsky‐Graham, S. , Walter, J.‐G. , Scheper, T. et al., Whole‐cell detection of live lactobacillus acidophilus on aptamer‐decorated porous silicon biosensors. Analyst 2016, 141, 5432–5440. [DOI] [PubMed] [Google Scholar]
- 7. Urmann, K. , Reich, P. , Walter, J.‐G. , Beckmann, D. et al., Rapid and label‐free detection of protein a by aptamer‐tethered porous silicon nanostructures. J. Biotechnol. 2017. 10.1016/j.jbiotec.2017.01.005. Available online: https://www.sciencedirect.com/science/article/pii/S0168165617300226. [DOI] [PubMed] [Google Scholar]
- 8. Hamula, C. , Guthrie, J. , Zhang, H. , Li, X. et al., Selection and analytical applications of aptamers. TrAC Trends Anal. Chem. 2006, 25, 681–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ozer, A. , Pagano, J. M. , Lis, J. T. , New technologies provide quantum changes in the scale, speed, and success of SELEX methods and aptamer characterization. Mol. Ther. Nucleic Acids 2014, 3, e183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Blind, M. , Blank, M. , Aptamer selection technology and recent advances. Mol. Ther. Nucleic Acids 2015, 4, e223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rohloff, J. C. , Gelinas, A. D. , Jarvis, T. C. , Ochsner, U. A. et al., Nucleic acid ligands with protein‐like side chains: modified aptamers and their use as diagnostic and therapeutic agents. Mol. Ther. Nucleic Acids 2014, 3, e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Diafa, S. , Hollenstein, M. , Generation of aptamers with an expanded chemical repertoire. Molecules 2015, 20, 16643–16671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Musheev, M. U. , Krylov, S. N. , Selection of aptamers by systematic evolution of ligands by exponential enrichment: addressing the polymerase chain reaction issue. Anal. Chim. Acta 2006, 564, 91–96. [DOI] [PubMed] [Google Scholar]
- 14. Williams, R. , Peisajovich, S. G. , Miller, O. J. , Magdassi, S. et al., Amplification of complex gene libraries by emulsion PCR. Nat. Methods 2006, 3, 545–550. [DOI] [PubMed] [Google Scholar]
- 15. Shao, K. , Ding, W. , Wang, F. , Li, H. et al., Emulsion PCR: a high efficient way of PCR amplification of random DNA libraries in aptamer selection. PLoS One 2011, 6, e24910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tolle, F. , Wilke, J. , Wengel, J. , Mayer, G. , By‐product formation in repetitive PCR amplification of DNA libraries during SELEX. PLoS One 2014, 9, e114693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yufa, R. , Krylova, S. M. , Bruce, C. , Bagg, E. A. et al., Emulsion PCR significantly improves nonequilibrium capillary electrophoresis of equilibrium mixtures‐based aptamer selection: allowing for efficient and rapid selection of aptamer to unmodified ABH2 protein. Anal. Chem. 2015, 87, 1411–1419. [DOI] [PubMed] [Google Scholar]
- 18. Levay, A. , Brenneman, R. , Hoinka, J. , Sant, D. et al., Identifying high‐affinity aptamer ligands with defined cross‐reactivity using high‐throughput guided systematic evolution of ligands by exponential enrichment. Nucleic Acids Res. 2015, 43, e82–e82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ji, Y. , Wang, Q.‐Q. , Fu, J. , Gao, X. et al., Optimization of polymerase chains reaction amplification for ssdeoxyribonucleic acid library using capillary electrophoresis with laser‐induced fluorescence detector. Chin. J. Anal. Chem. 2010, 38, 622–626. [Google Scholar]
- 20. Ruff, P. , Pai, R. B. , Storici, F. , Real‐time PCR‐coupled CE‐SELEX for DNA aptamer selection. ISRN Mol. Biol. 2012, 2012, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Berezovski, M. V , Musheev, M. U. , Drabovich, A. P. , Jitkova, J. V. et al., Non‐SELEX: selection of aptamers without intermediate amplification of candidate oligonucleotides. Nat. Protoc. 2006, 1, 1359–1369. [DOI] [PubMed] [Google Scholar]
- 22. Lauridsen, L. H. , Shamaileh, H. A. , Edwards, S. L. , Taran, E. et al., Rapid one‐step selection method for generating nucleic acid aptamers: development of a DNA aptamer against α‐bungarotoxin. PLoS One 2012, 7, e41702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wilson, R. , Bourne, C. , Chaudhuri, R. R. , Gregory, R. et al., Single‐step selection of bivalent aptamers validated by comparison with SELEX using high‐throughput sequencing. PLoS One 2014, 9, e100572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Diehl, F. , Li, M. , He, Y. , Kinzler, K. W. et al., BEAMing: single‐molecule PCR on microparticles in water‐in‐oil emulsions. Nat. Methods 2006, 3, 551–559. [DOI] [PubMed] [Google Scholar]
- 25. Schütze, T. , Rubelt, F. , Repkow, J. , Greiner, N. et al., A streamlined protocol for emulsion polymerase chain reaction and subsequent purification. Anal. Biochem. 2011, 410, 155–157. [DOI] [PubMed] [Google Scholar]
- 26. Doyle, S. A. , Murphy, M. B. , Aptamers and methods for their in vitro selection and uses thereof . US Patent 7329742, 2008. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information