

ABSTRACT
Macroautophagy/autophagy is generally regarded as a cytoprotective mechanism, and it remains a matter of controversy whether autophagy can cause cell death in mammals. Here, we show that chronic restraint stress suppresses adult hippocampal neurogenesis in mice by inducing autophagic cell death (ACD) of hippocampal neural stem cells (NSCs). We generated NSC-specific, inducible Atg7 conditional knockout mice and found that they had an intact number of NSCs and neurogenesis level under chronic restraint stress and were resilient to stress- or corticosterone-induced cognitive and mood deficits. Corticosterone treatment of adult hippocampal NSC cultures induced ACD via SGK3 (serum/glucocorticoid regulated kinase 3) without signs of apoptosis. Our results demonstrate that ACD is biologically important in a mammalian system in vivo and would be an attractive target for therapeutic intervention for psychological stress-induced disorders.
Abbreviations: AAV: adeno-associated virus; ACD: autophagic cell death; ACTB: actin, beta; Atg: autophagy-related; ASCL1/MASH1: achaete-scute family bHLH transcription factor 1; BafA1: bafilomycin A1; BrdU: Bromodeoxyuridine/5-bromo-2ʹ-deoxyuridine; CASP3: caspase 3; cKO: conditional knockout; CLEM: correlative light and electron microscopy; CORT: corticosterone; CRS: chronic restraint stress; DAB: 3,3ʹ–diaminobenzidine; DCX: doublecortin; DG: dentate gyrus; GC: glucocorticoid; GFAP: glial fibrillary acidic protein; HCN: hippocampal neural stem; i.p.: intraperitoneal; MAP1LC3B: microtubule-associated protein 1 light chain 3 beta; MKI67/Ki67: antigen identified by monoclonal antibody Ki 67; MWM: Morris water maze; Nec-1: necrostatin-1; NES: nestin; NR3C1/GR: nuclear receptor subfamily 3, group C, member 1; NSC: neural stem cell; PCD: programmed cell death; PFA: paraformaldehyde; PX: Phox homology; PtdIns3P: phosphatidylinositol-3-phosphate; RBFOX3/NeuN: RNA binding protein, fox-1 homolog (C. elegans) 3; SGK: serum/glucocorticoid-regulated kinases; SGZ: subgranular zone; SOX2: SRY (sex determining region Y)-box 2; SQSTM1: sequestosome 1; STS: staurosporine; TAM: tamoxifen; Ulk1: unc-51 like kinase 1; TUNEL: terminal deoxynucleotidyl transferase dUTP nick end labeling; VIM: vimentin; WT: wild type; ZFYVE1: zinc finger, FYVE domain containing 1; Z-VAD/Z-VAD-FMK: pan-caspase inhibitor
KEYWORDS: Atg7 knockout, autophagic cell death, corticosterone, hippocampal neurogenesis, serum/glucocorticoid regulated kinase 3, stress
Introduction
Macroautophagy/autophagy is a lysosome-dependent catabolic process characterized by increased formation of double-membraned autophagosomes for sequestration of cytoplasmic components. Autophagy is essential for normal development and physiology, and is generally considered as a cell survival mechanism that supplies nutrients and ensures turnover of obsolete cellular constituents [1]. However, accumulating evidence suggests that autophagy may cause or contribute to cell death under certain conditions [2]. Recent progress in the field of cell death indicates the importance of the modes of programmed cell death (PCD) other than apoptosis, such as autophagic cell death (ACD) or necroptosis in human physiology and diseases [3]. The best demonstration of the role of ACD in physiological cell death was presented in the model organism Drosophila, where ACD acts as a cell death mechanism independent of apoptosis in the removal of the larval midgut during development [4]. In mammals, authentic cases of ACD are rare. We have previously reported that adult hippocampal neural stem (HCN) cells isolated from adult rats undergo ACD following insulin withdrawal despite their intact apoptotic capabilities [5–8]. Autophagy flux is increased in insulin-deprived HCN cells without signs of apoptosis or necrosis, and genetic inactivation of autophagy protects HCN cells from cell death induced by insulin withdrawal. Hence, insulin-deprived HCN cells have been considered to be a genuine model of ACD that meets all criteria of ACD proposed by Shen and Codogno [9]. However, our previous studies have been restricted to in vitro cell cultures, and the physiological importance of ACD in vivo and relevant molecular mechanisms in mammals still remain to be demonstrated.
Hippocampus is one of the regions of the mammalian brain where neural stem cells (NSCs) reside and sustain the generation of new neurons throughout adulthood. Adult hippocampal neurogenesis is implicated in learning and memory, and mood regulation [10]. However, adult hippocampal neurogenesis is highly susceptible to stress and the major stress hormone, glucocorticoid (GC) [11–13]. Therefore, alteration in adult hippocampal neurogenesis is intimately involved in stress-induced psychological disorders, such as anxiety, depression, post-traumatic stress disorder, and panic disorder [14]. It is unclear whether PCD affects the balance between the survival and death of adult NSCs and thus mediates the suppressive effects of stress on adult neurogenesis. Previous studies suggested that apoptosis is not involved in stress-induced suppression of adult neurogenesis, as evidenced by the lack of DNA fragmentation or caspase-3 activation as an indicator of cell death [15,16].
Here, we report that chronic stress induces ACD of adult hippocampal NSCs, thereby suppressing adult hippocampal neurogenesis. Our study demonstrates the genuine in vivo case of ACD in a mammalian system. Our results also shed new light on the pathological mechanisms underlying detrimental effects of chronic stress on cognitive performance and may provide potential clues for the design of treatment of chronic stress-related neurological disorders.
Results
Adult hippocampal NSCs are protected from chronic restraint stress (CRS) in Atg7-NSC cKO mice
We first examined whether autophagy mediates neurogenesis decline during CRS by deleting a key autophagy-related (Atg) gene, Atg7, in a NSC-specific manner in adult mice. ATG7 is an E1-like enzyme for the two ubiquitin-like ATG8 and ATG12 conjugation systems, which are required for autophagosome formation [17]. Mice carrying a floxed Atg7 allele (Atg7fl/fl) [18] were bred with Nes/nestin-CreERT2 (Nes-Cre) mice to generate Atg7fl/fl + Nes-Cre or Atg7fl/+ + Nes-Cre mice (Figure 1A). Atg7 was deleted at the age of 7 weeks by daily injections of tamoxifen (TAM) for 3 days. TAM administration led to a gene dose-dependent loss of ATG7 immunoreactivity only in SOX2 (SRY [sex determining region Y]-box 2)-positive cells but not in the neighboring granule cells of dentate gyrus (DG), confirming successful deletion of Atg7 in an inducible, NSC-specific manner (Figure 1B). Quantification of ATG7-positive cells among the SOX2-positive cells shows that most of SOX2-positive cells already lost ATG7 immunoreactivity in heterozygous Atg7 knockout (KO) mice and homozygous KO mice shows slightly less SOX2-positive cells compared to WT and heterozygous KO mice. We designated these inducible, NSC-specific conditional knockout mice as Atg7-NSC cKO mice and subjected them to CRS for 1 week with 6 h (10:00–16:00 h) of daily restraint to examine whether Atg7 deficiency can prevent cell death and afford behavioral protection with intact neurogenesis (Figure 1C). To estimate NSC death, we determined the numbers of total (SOX2 staining) and proliferating NSCs (BrdU staining after BrdU injection for the last 3 days during CRS) 1 day after the termination of CRS (Figure 1C). CRS significantly decreased the number of NSCs in wild-type (WT) mice (Figure 1D). In contrast, Atg7 ablation prevented NSC death and blocked CRS-induced decrease in NSC number in both heterozygous (Figure 1D) and homozygous (Fig. S1) Atg7-NSC cKO mice. Since we observed similar levels of NSC protection in heterozygous and homozygous mice, hereafter we used heterozygous Atg7-NSC cKO mice, unless otherwise stated. A previous study that used the same Atg7 flox mice showed that the constitutive heterozygous Atg7 KO mice under the control of Nes-Cre developed normally to adulthood, but homozygous KO mice suffered developmental abnormalities [18]. Therefore, deletion in one allele not both can avoid any compounding effects of prolonged absence of autophagy. The percentage of BrdU-positive cells among the total NSCs in all four groups was similar (18–20%) and was only slightly lower in stressed mice (18%) than in the other groups (Figure 1D). Therefore, NSC proliferation rates were similar in the 4 groups, and the reduction in the number of NSCs in the stressed WT group was due to cell death rather than inhibition of proliferation. A reduction in the number of SOX2-positive cells by CRS was not due to a decrease in SOX2 expression, since staining for other NSC markers, such as NES (nestin) and VIM (vimentin), revealed a similar reduction in the number of NSCs (Figure 1E). Atg7-NSC cKO mice were also protected against more prolonged 3-week CRS (Fig. S2a–c).
Figure 1.
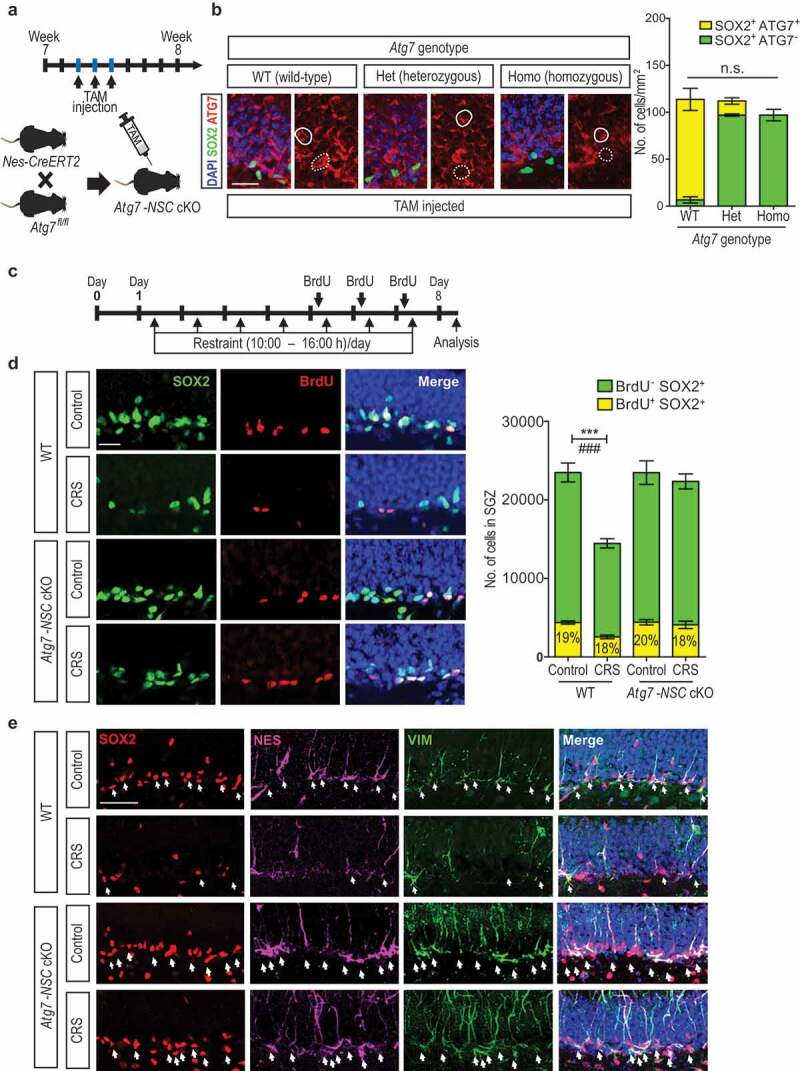
Chronic restraint stress (CRS)-induced reduction in the number of adult hippocampal NSCs is prevented by Atg7 deletion. (A) Scheme of mouse breeding and experimental time line for generation of tamoxifen (TAM)-inducible NSC-specific Atg7-NSC cKO mice. (B) Gene dose–dependence of ATG7 immunofluorescence intensities in the DG of the hippocampus. Scale bar: 50 μm. Solid and dotted circles indcate granule neurons and NSCs, respectively. The graph shows quantification of SOX2+ and ATG7+ cells (n = 6). (c) Timeline of the experiment. (D) Representative images of BrdU and SOX2 staining in the subgranular zone (SGZ) of the hippocampus. Scale bar: 20 μm. The graph on the right shows quantification of BrdU+ and SOX2+ cells (n = 6–8 per group). (E) Immunofluorescence of SOX2, NES and VIM in the SGZ of the DG of the hippocampus. Scale bar: 50 μm. Arrows indicate NSCs positive for each marker. ***P < 0.001 for the total SOX2+ cells. ###P < 0.001 for BrdU+ SOX2+ cells. n.s., not significant.
CRS-induced autophagy is blocked in Atg7-NSC cKO mice
Next, we focused on the issue whether CRS induced autophagy in the hippocampal NSCs in vivo. To address this question, we performed correlative light and electron microscopy (CLEM) combined with 3,3ʹ–diaminobenzidine (DAB) immunostaining for SOX2 in the subgranular zone (SGZ) of DG (Figure 2A). Autophagosomes were absent in control sets and modestly present in SOX2-positive cells after CRS (Figure 2B). MAP1LC3B/LC3B (microtubule-associated protein 1 light chain 3 beta; a mammalian homolog of yeast Atg8) is the best-known biochemical marker of autophagy [19]. To demonstrate an increase in autophagy flux in vivo, we generated lentivirus expressing a tandem fusion protein composed of monomeric RFP, GFP, and MAP1LC3B (mRFP-EGFP-MAP1LC3B) under the control of the Nes promoter. The quenching of the GFP, but not RFP signal at the acidic pH upon fusion of autophagosomes and lysosomes results in red puncta, while yellow puncta (merged red and green fluorescence signals) indicate autophagosomes before fusion with lysosomes [8]. Consistent with the results of CLEM, this tandem MAP1LC3B construct demonstrated a higher ratio of red puncta and an increased number of overall MAP1LC3B puncta in WT hippocampus following CRS, but these increases were not observed in Atg7-NSC cKO mice (Figure 2C). These findings show that CRS increases autophagy flux in the WT hippocampal NSCs, but genetic inactivation of Atg7 blocks this increase. In contrast to a robust induction of autophagy flux, we were unable to detect cleaved CASP3/caspase-3 (c.CASP3), the activated form of CASP3, following CRS, whereas CASP3 cleavage was induced by injection of staurosporine (STS), a prototypical apoptosis inducer (Figure 2D). We also performed terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay to detect DNA fragmentation and examine whether caspase-independent apoptosis occurs by CRS. Consistent with no CASP3 activation, we were not able to detect DNA fragmentation following CRS, while STS induced an increase in TUNEL-positive cells (Figure 2E). These results indicate an elevated level of autophagy flux but not apoptosis in adult hippocampal NSCs after CRS.
Figure 2.
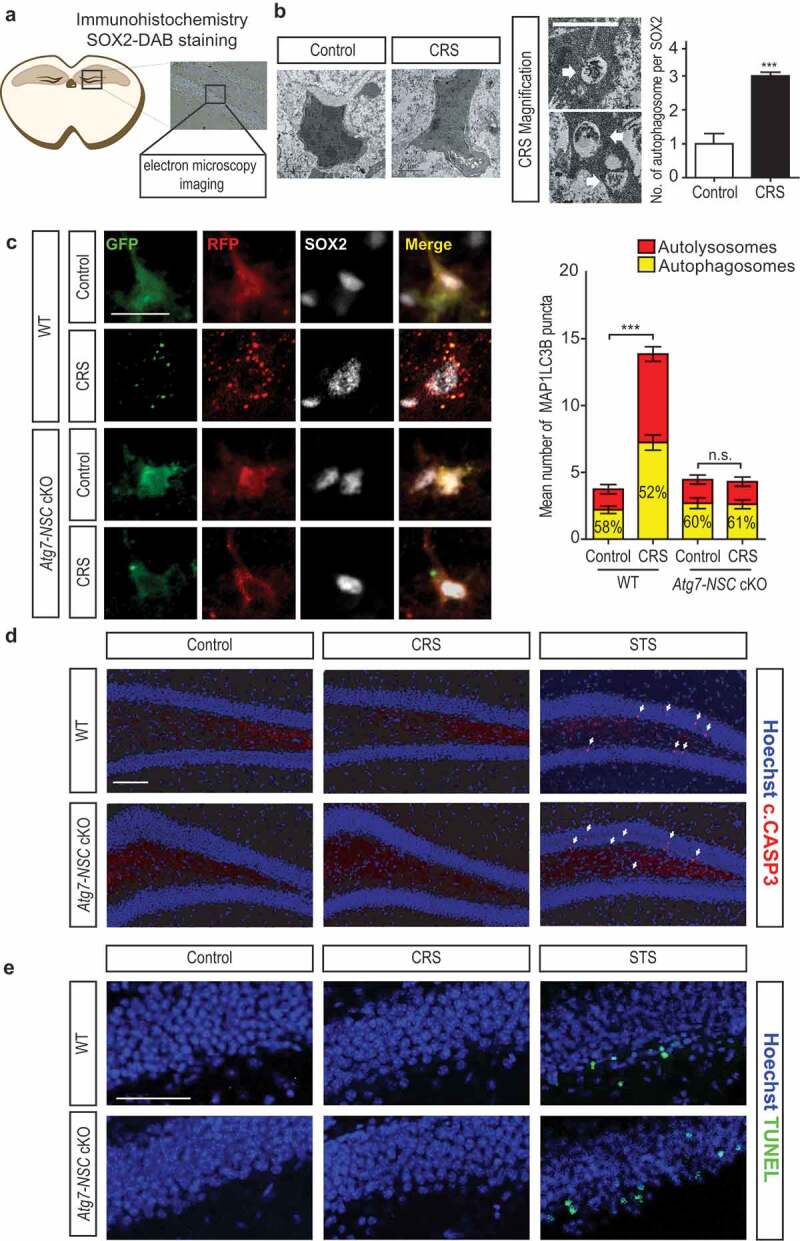
CRS induces autophagy in adult hippocampal NSCs. (A) Work flow of correlative light electron microscopy (CLEM) imaging. (B) CLEM imaging of SOX2-positive cells at day-4 of CRS (n = 13 cells per group). Representative images of autophagosomes (white arrows) in CRS group are shown at higher magnification. The number of autophagosomes was counted per 5 μm × 5 μm without counting the autophagosomes of whole cells using serial sections. Scale bars: 2 μm. (C) Nes promoter-driven lentiviral expression of mRFP-EGFP-MAP1LC3B in vivo. Virus was injected 4 days prior to CRS and analyzed at day 2 of CRS. Scale bar: 20 μm. The graph on the right shows quantification of autophagosomes and autolysosomes (n = 7 or 8 cells per group). (D and E) Cleaved CASP3 (c.CASP3) staining (D) and TUNEL assay (E) in the DG at day 7 of CRS. STS was injected 12 h before analysis. Arrows indicate c.CASP3-positive cells. Scale bar: 100 μm. ***P < 0.001. n.s., not significant.
Suppression of neurogenesis and cognitive deficits induced by CRS are prevented in Atg7-NSC cKO mice
Neurogenesis was analyzed 28 days after the end of CRS with BrdU injection for the first 5 days of CRS (Figure 3A). Prevention of stress-induced NSC death in Atg7-NSC cKO mice led to the same extent of neurogenesis as in unstressed mice, and there was no difference in the rate of RBFOX3/NeuN (RNA binding protein, fox-1 homolog [C. elegans] 3)-positive cells among BrdU-labeled cells of all groups in the DG of the hippocampus, suggesting intact differentiation of surviving NSCs during CRS (Figure 3B,C). Stress-induced suppression of adult hippocampal neurogenesis causes anxiety-like and depressive behaviors [20]; however, the effects of neurogenesis suppression by stress on hippocampal learning and memory have not been well elucidated. To investigate whether preservation of adult neurogenesis in Atg7-NSC cKO mice confers functional resilience to CRS, adult neurogenesis-dependent hippocampal functions were examined 28 days after the end of CRS. After CRS, WT mice spent shorter time in the center of an open field in the open field test (Figure 3D) and in the open arms with less entry into the open arms in the elevated plus maze test (Figure 3E). However, Atg7-NSC cKO mice were resistant to CRS and did not show anxiety-like behavioral alterations (Figure 3d,E). Stress-induced attenuation of sucrose preference was also not observed in Atg7-NSC cKO mice, demonstrating prevention of stress-induced depressive behavior (Figure 3F). Next, we assed spatial working memory by measuring the spontaneous alternation in the Y-maze test. Arm alternation rate was significantly reduced in the WT CRS group, indicating impaired spatial working memory, whereas the Atg7-NSC cKO mice did not show such impairment (Figure 3G). We also analyzed the 6-day performance in the Morris water maze (MWM) test. When we measured the escape latency during the acquisition phase of MWM, the WT CRS group exhibited learning deficits compared to control group, whereas the Atg7-NSC cKO CRS group performed as well as unstressed WT or Atg7-NSC cKO mice (Figure 3H). Measurement of the time spent in the probe-containing quarter confirmed that spatial learning and memory were protected in the Atg7-NSC cKO CRS group (Figure 3H). The same total distance in the open field test and total arm entries in the elevated plus maze test as well as the same swimming velocity in the MWM test demonstrated no difference in locomotive activity among the groups (Fig. S3a–c). Likewise, similar total volume of water was consumed across all the groups in the sucrose preference test (data not shown). These results demonstrate that Atg7-NSC cKO mice are protected from CRS-induced decrease in neurogenesis, and are also functionally resistant to learning and memory deficits, and to depressive and anxiety-like neurobehaviors.
Figure 3.
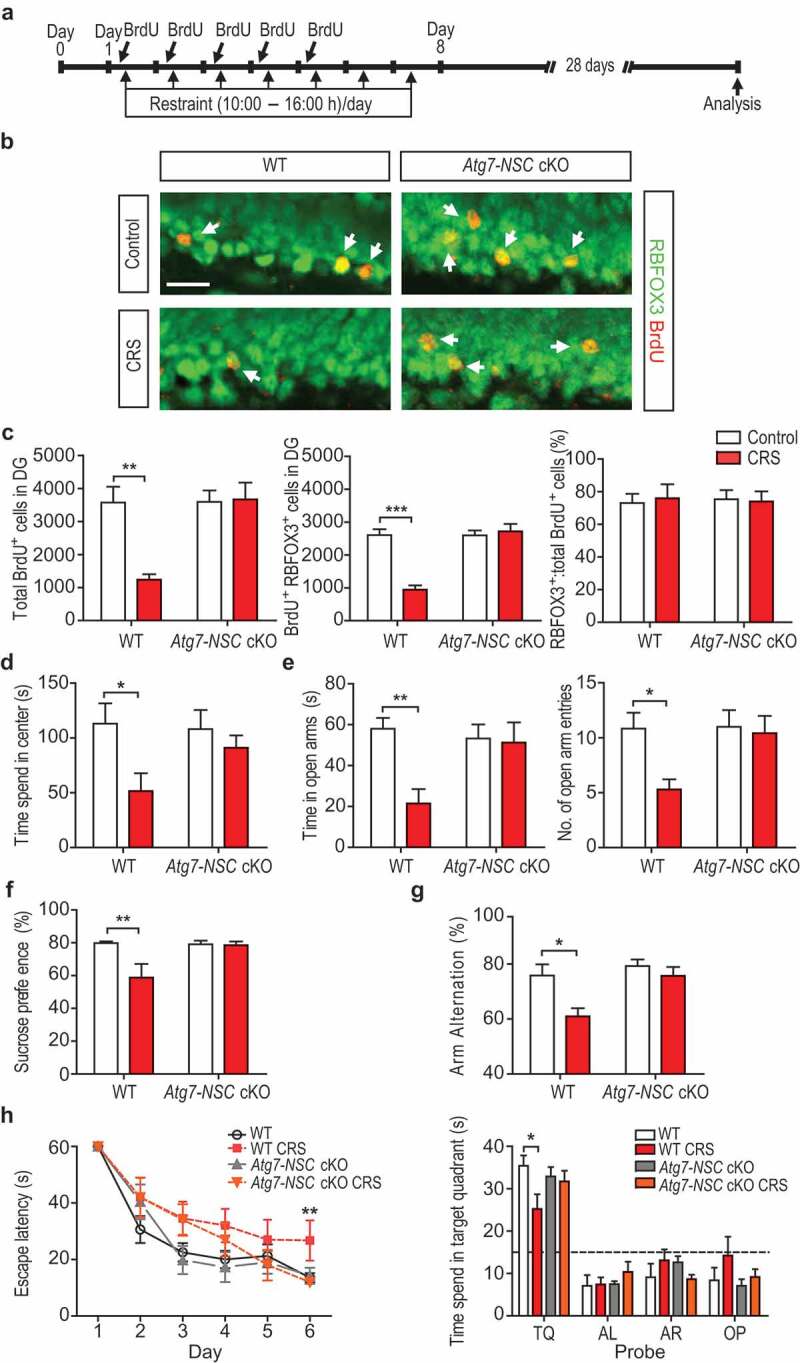
Suppression of hippocampal neurogenesis, anxiety-like and depressive behaviors, and spatial memory deficits induced by CRS are prevented in Atg7-NSC cKO mice. (A) Timeline for the measurement of the number of BrdU+ and RBFOX3+ double-positive NSCs. (B) Representative images of BrdU+ RBFOX3+ staining. Scale bar: 20 μm. Arrow indicate RBFOX3+ BrdU+ double-positive cells. (C) Quantification of BrdU+ RBFOX3+-positive cells (n = 6 or 7 per group). (D and E) Measurement of anxiety-like behaviors by the open field test (n = 6 or 7 per group) (D) and elevated plus maze test (n = 6–13 per group) (E). (F) Measurement of depressive behavior by the sucrose preference test (n = 6). (G) Assessment of spatial working memory by the Y-maze test (n = 6–8 per group). (H) Spatial learning and memory test using the acquisition phase (left graph) and probe test at day 6 (right graph) in MWM test (n = 6–8 per group). TQ, target quadrant; AL, adjacent left; AR, adjacent right; OP, opposite. *P < 0.05, **P < 0.01, ***P < 0.001.
Loss of NSCs activity after CRS is blocked by autophagy deficiency
In the extension of the previous observations, we characterized which subpopulations of NSCs are sensitive to ACD after CRS. NSCs in the SGZ of the adult DG are heterogeneous and different groups composed of early progenitor cells (type 1), intermediate progenitor cells (type 2a and 2b), and neuroblasts (type 3) can be distinguished by expression of stage-specific marker proteins [21]. Each NSC group is known to respond distinctly to physiological and pathological stimuli [22]. Type 1 GFAP+ SOX2+ cells were significantly reduced in WT stressed mice, but protected in Atg7-NSC cKO mice when analyzed 1 day after the termination of CRS (Figure 4A,B). Stress also significantly decreased proliferating population of NSCs (GFAP+ SOX2+ MKI67/KI67+ triple positive cells) in WT mice, but not in Atg7-NSC cKO mice (Figure 4A,B). Immunostaining analysis also revealed that type 2b DCX+ SOX2+ cells and type 3 DCX+ only cells were also vulnerable to CRS whereas type 2a ASCL1/MASH1+ SOX2+ cells were less affected than other types of NSCs (Figure 4A,B). We found that loss of each subtype by CRS was preserved until 28 days after CRS (Figure 4C). There was no difference in the ratio of MKI67-positive cells among GFAP and SOX2 co-labeled cells 28 days after CRS, although there was a modest decline 1 day after CRS (Figure 4B). These data suggest that both proliferating and type-2a populations of the NSCs are relatively less affected.
Figure 4.
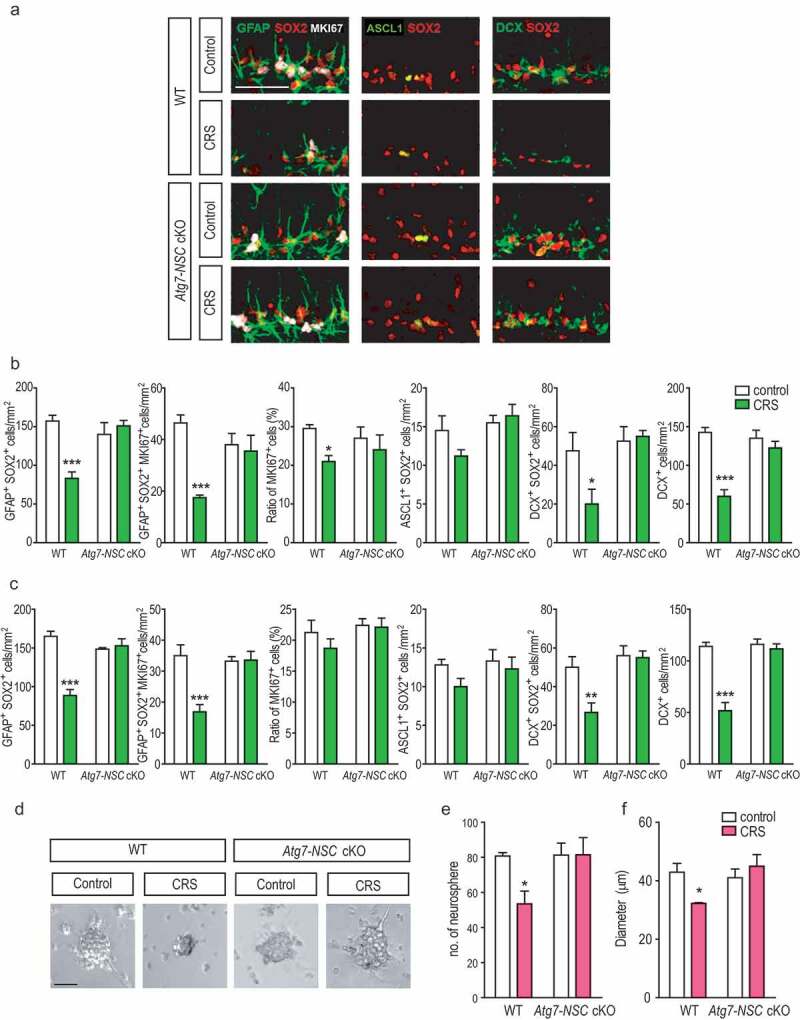
Atg7 deficiency prevents loss of subtypes of NSCs in the adult DG and the activity of NSCs in neurosphere cultures. (A) Representative image of each subtype of NSCs in the SGZ of the DG. (band c) Quantification of GFAP+ SOX2+ (type 1), GFAP+ SOX2+ MKI67+ (active type 1), ASCL1+ SOX2+ (type 2a), DCX+ SOX2+ (type 2b), and DCX+ (type 3) NSCs 1 day (n = 4 or 5 per group) (B) and 28 days after CRS (n = 5 or 6 per group) (C). (D) Representative image of neurospheres in culture for 7 days. Scale bar: 40 μm. (E) Quantification of neurospheres after 7 days in cultures (n = 3–5 per group). (F) Measurement of neurosphere size after 7 days in cultures (n = 3–5 per group). *P < 0.05, **P < 0.01, ***P < 0.001.
Neurosphere assays is frequently used to measure the activities of NSCs [23]. We assessed whether CRS reduces neurosphere number and size, and autophagy deficiency can regulate the NSC activity. The number of DG-derived neurospheres from WT CRS group was significantly lower compared to control group, but not in Atg7-NSC cKO CRS group (Figure 4D,E). The median size of neurospheres from WT CRS group was also significantly smaller than other groups including stressed Atg7-NSC cKO mice (Figure 4D,F), indicating that Atg7 deletion can preserve stem cell activity against CRS.
Chronic CORT injection-induced autophagy is blocked in Atg7-NSC cKO mice
Cortisol in humans and corticosterone (CORT) are the major forms of GCs released during stress. Circulating CORT level in rodents is higher in the afternoon than in the morning due to circadian regulation [24]. Therefore, we measured the blood CORT level 30 min after the termination of stress in the afternoon (4:30 PM; Fig. S4a). Atg7-NSC cKO and WT mice had similarly high CORT levels 1 day after stress, but CORT level returned to the normal level more slowly in WT than Atg7-NSC cKO mice (Fig. S4b). Adult hippocampal neurogenesis plays a role in regulation of the hypothalamic-pituitary-adrenal axis and buffers stress responses [20]. Consistent with this notion, preservation of NSCs in Atg7-NSC cKO mice seems to contribute to fast recovery of serum CORT level after stress. Atg7-NSC cKO mice also suffered body weight loss to the same extent as WT mice (Fig. S2b and S4c). These data suggest that the beneficial effects of NSC-specific autophagy attenuation are due to NSC protection from the detrimental effects of high CORT levels, but not to an alteration in the hypothalamic-pituitary-adrenal axis or release of CORT.
On the basis of these in vivo results with Atg7-NSC cKO mice, we hypothesized that CORT secreted during psychological stress reduces neurogenesis by inducing ACD of NSCs. To test this idea, we first examined whether CORT injection can recapitulate the effects of CRS on NSC death (Figure 5A). As expected, intraperitoneal (i.p.) injection of CORT for 7 days greatly increased blood CORT levels both in WT and Atg7-NSC cKO mice (Fig. S5a), but reduced the number of NSCs and induced autophagy flux only in WT mice (Figure 5B,C). When we analyzed neurogenesis 28 days after CORT injection (Figure 5D), total BrdU+ and RBFOX3+ BrdU+ positive cells were reduced only in WT mice (Figure 5E). In addition, Atg7-NSC cKO mice were resistant to CORT-induced anxiety like behavior and cognitive dysfunction, as illustrated by number and time in open arm entries of elevated plus maze test (Figure 5F) and spontaneous alternation of Y-maze test (Figure 5G). Analysis of subtypes of NSCs after 7-day CORT injection also revealed the results similar to CRS (Fig. S5b,c).
Figure 5.
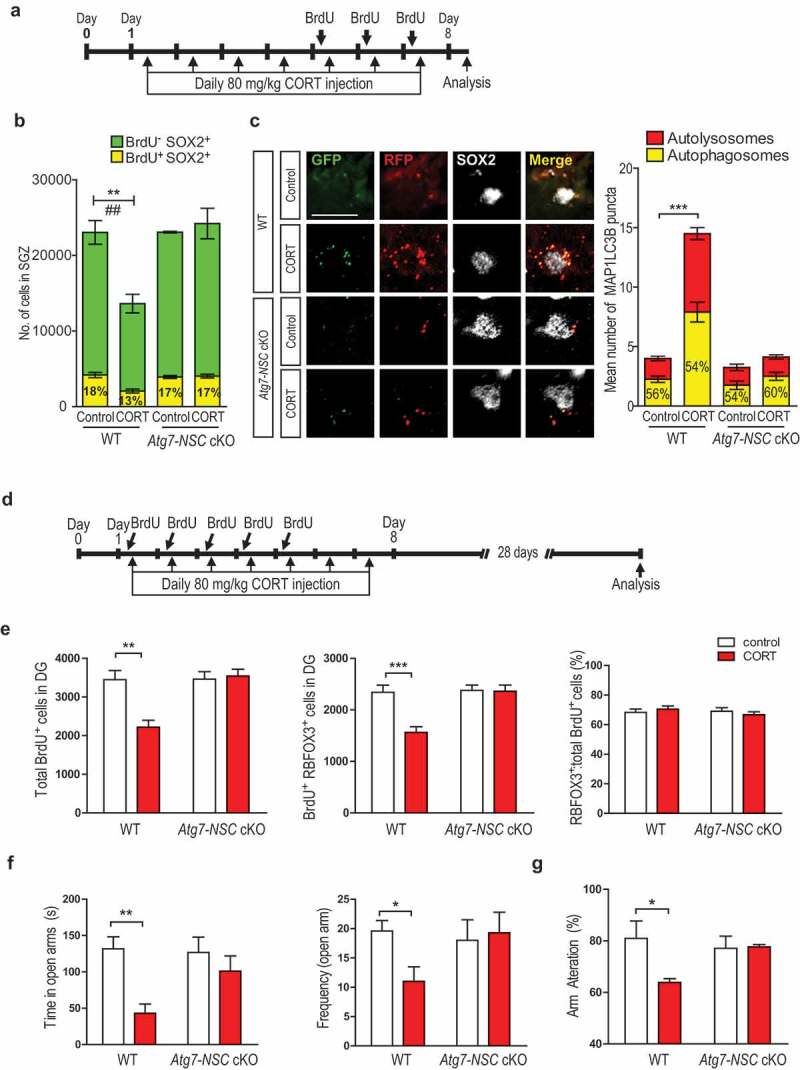
CORT-induced reduction of NSC number and hippocampal dysfuction is prevented in Atg7-NSC cKO mice. (A) Timeline of the CORT injection experiment. (B) Quantification of BrdU+ and SOX2+ cells (n = 4 or 5 per group). **P < 0.01 for the total SOX2+ cells ##P < 0.01 for BrdU+ SOX2+ cells. (C) Nes promoter-driven lentiviral expression of mRFP-EGFP-MAPLC3B in SOX2+ cells in vivo. Scale bar: 20 μm. The graph on the right shows quantification of autophagosomes and autolysosomes (n = 7 or 8 cells per group). (D) Timeline for the measurement of the number of BrdU+ RBFOX3+ double–positive NSCs. (E) Quantification of BrdU+ RBFOX3+ double-positive cells (n = 4 or 5 per group). (f) Elevated plus maze test (n = 4 per group). (g) Y-maze test (n = 4 per group). *P < 0.05, **P < 0.01, ***P < 0.001.
CORT induces ACD, but not apoptosis or necroptosis
To elucidate the molecular mechanisms of death of adult hippocampal NSCs induced by CORT, we used in vitro HCN cell cultures. Most of HCN cells were stained with NES (data not shown), confirming their neural stem cell-stage in accordance with the previous report [5]. CORT caused cell death in a dose- and time-dependent manner in HCN cells derived from both rat and mouse (Figure 6A and S6a). In the following experiments, 200 µM CORT was used unless otherwise stated. Representative images of the Hoechst-propidium iodide (PI) cell death assay were shown (Figure 6A). Nuclear condensation, a distinct morphological feature of apoptosis, was not seen in CORT-treated HCN cells (Figure 6B). Likewise, CASP3 activation by immunostaining (Figure 6C) or western blotting (Figure 6F), and nuclear fragmentation by the TUNEL staining (Figure 6D) or DNA electrophoresis in agarose gel (Figure 6E) were not observed after prolonged CORT treatment (48–72 h). On the other hand, STS efficiently induced all of these apoptosis features within 6–8 h, demonstrating intact apoptotic capability of HCN cells (Figure 6B–F). Furthermore, the apoptosis inhibitor Z-VAD/Z-VAD-FMK and the necrosis inhibitor necrostatin-1 (Nec-1) failed to prevent CORT-induced HCN cell death (Figure 6G). Z-VAD also could not alter autophagy flux in CORT-treated HCN cells (Figure 6H). On the other hand, Z-VAD and Nec-1 protected HCN cells from STS and H2O2, respectively (Fig. S6b,c). These data suggest that CORT-induced death of HCN cells does not occur through apoptosis or necroptosis.
Figure 6.
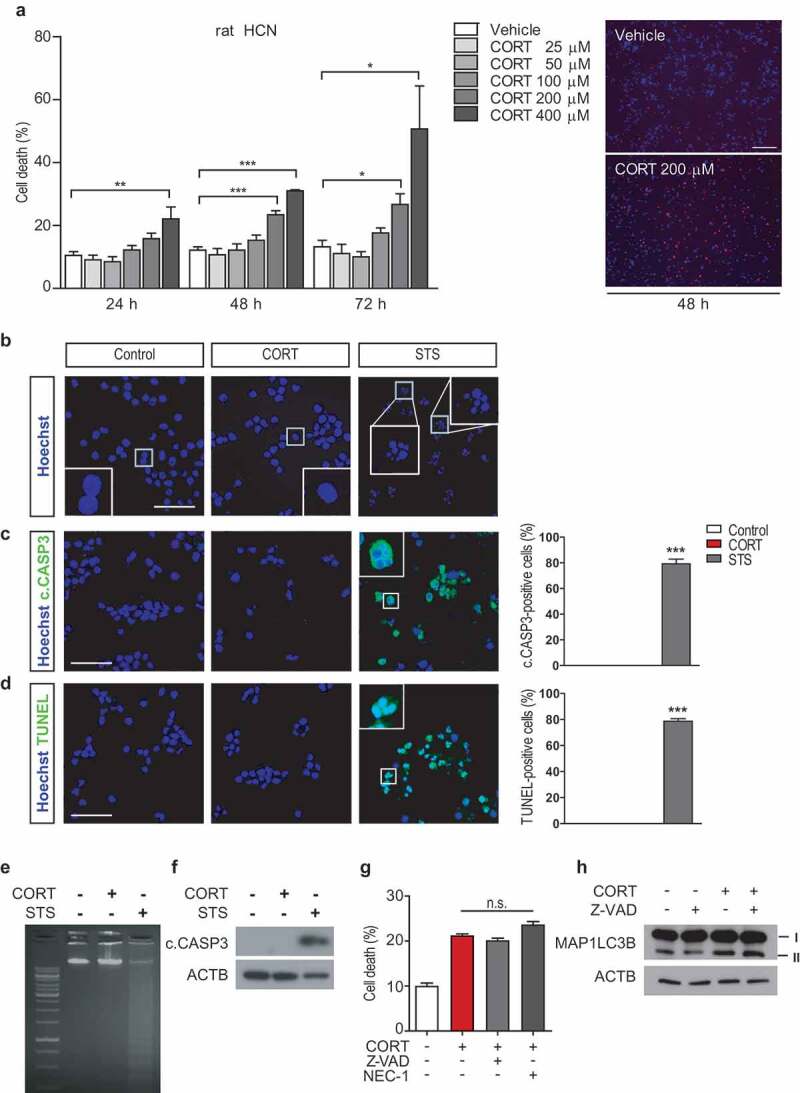
CORT treatment does not induce apoptosis or necroptosis in HCN cells. (A) Death rate of HCN cells after CORT treatment (n = 4). Right, representative image of Hoechst and PI staining 48 h after CORT treatment in HCN cells. (B) Nucleus condensation assay with Hoechst staining. (C) Immunostaining of cleaved CASP3 (c.CASP3). (D) Nuclear fragmentation assay by TUNEL staining. Scale bar: 40 μm for b-d. (E and F) Agarose gel electrophoresis of DNA fragmentation assay (E) and western blots of c.CASP3 (F) are representative of at least 3 experiments with similar results. All apoptotic markers were analyzed after CORT (200 µM for 48 h, except western blotting analysis of c.CASP3 with 72 h) or staurosporine (STS, 0.5 µM for 6 h) treatment. (G) Effects of Z-VAD (25 μM) or necrostatin-1 (NEC-1, 100 μM) on HCN cell death after CORT treatment for 48 h (n = 3). (H) Western blotting analysis of the effects of Z-VAD (25 μM) on autophagy flux after CORT treatment for 48 h. The blots are representative of 3 experiments with similar results. *P < 0.05, **P < 0.01, ***P < 0.001. n.s., not significant.
To determine whether ACD was induced by CORT, we examined whether CORT treatment increased autophagosome formation. Transmission electron microscopy analysis revealed an increased number of autophagosomes after CORT treatment in HCN cells (Figure 7A). Next, we assessed a conversion of the cytosolic MAP1LC3B-I (18 kDa) to lipidated, autophagosome-associated MAP1LC3B-II (16 kDa). Western blotting analyses showed an increase in MAP1LC3B-II level in CORT-treated HCN cells (Figure 7B). Blocking of autophagy flux with bafilomycin A1 (BafA1) led to more accumulation of MAP1LC3B-II in CORT-treated than in control HCN cells (Figure 7B). Also, the protein level of SQSTM1, an autophagy cargo receptor, was reduced selectively after CORT treatment and restored by BafA1 (Figure 7B). These results indicate an increase in autophagy flux, not impairment of autophagy, in CORT-treated HCN cells. We used mRFP-EGFP-MAP1LC3B as an alternative indicator of autophagy flux. Consistent with in vivo results, we observed an increase in the ratio of red puncta and the number of overall MA1LC3B puncta following CORT treatment, confirming elevated autophagy flux (Figure 7C).
Figure 7.
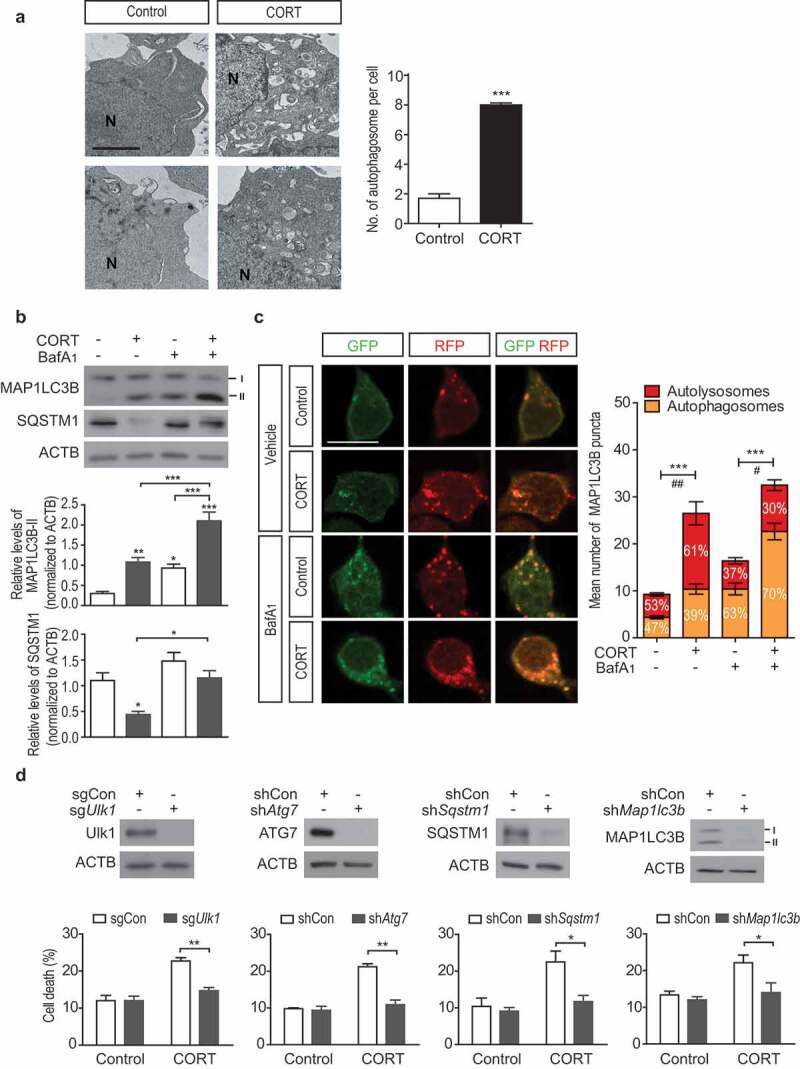
CORT treatment induces ACD in HCN cells. (A) Representative EM images of HCN cells treated with CORT for 48 h. Scale bar: 2 μm. N, nucleus. The graph on the right shows quantification of autophagosomes (n = 35 cells per group). (B) Western blotting analyses of MAP1LC3B and SQSTM1 levels after CORT treatment for 48 h. The graphs on the right show quantification of MAP1LC3B-II (n = 6) and SQSTM1 (n = 4) after normalization to ACTB. (C) Analysis of autophagy flux using mRFP-EGFP-MAP1LC3B after CORT treatment for 48 h. Scale bar: 10 μm. The graph on the right shows quantification of MAP1LC3B puncta (n = 8). ***P < 0.001 for the total MAP1LC3B puncta. #P < 0.05, ##P < 0.01 for yellow puncta. (D) Death rates of HCN cells after KO of Ulk1 (sgUlk1) or with stable knockdown of ATG7 (shAtg7), SQSTM1 (shSqstm1) or MAP1LC3B (shMap1lc3b) in comparison with control cells (sgCon or shCon) after CORT treatment for 48 h (n = 3). In all experiments, BafA1 (20 nM) was added 1 h before cell harvest. *P < 0.05, **P < 0.01, ***P < 0.001.
To determine the causative role of autophagy in CORT-induced cell death, we blocked several different phases of autophagosome formation process by CRISPR/Cas9-mediated deletion of Ulk1 (unc-51 like kinase 1; initiation), or by stable knockdown of Atg7 (elongation), Sqstm1 (cargo recruitment), and Map1lc3b (maturation). Inactivation of these essential Atg genes efficiently reduced CORT-induced cell death (Figure 7D). CORT-induced autophagy flux was suppressed in shAtg7 HCN cells (Fig. S7). This approach of targeting multiple, distinct autophagy steps, as recommended by the Nomenclature Committee on Cell Death [25], demonstrates the primary role of autophagy in CORT-induced death of HCN cells.
SGK3 is a critical mediator of cort-induced ACD in HCN cells
GCs bind to the high-affinity mineralocorticoid receptor and the low-affinity NR3C1/GR (nuclear receptor subfamily 3, group C, member 1; glucocorticoid receptor). Due to its low affinity, NR3C1 is activated only when GC concentrations are high and NR3C1 activation is considered to be important for the regulation of hippocampal neurogenesis, subsequent anxiety-like and depressive behavior, and deficits in learning and memory [12,13,26]. SGK (serum/glucocorticoid regulated kinases) is a family of serine/threonine kinases consisting of 3 isoforms that play various physiological roles, including regulation of ion channels, neuroexcitability, metabolism, proliferation, and apoptosis [27]. In particular, SGK1 has been reported to be transcriptionally upregulated by NR3C1 activation and mediate the stress-induced suppression of hippocampal neurogenesis [26,28]. Therefore, we first examined the change in the level of SGK family proteins in response to CORT treatment in HCN cells. CORT increased the mRNA and protein levels of SGK1 and SGK3, but not SGK2 (Figure 8A,B). To investigate the molecular mechanism underlying stress-induced ACD, we generated Sgk1 KO HCN cells using CRISPR/Cas9 gene editing methods and designated them sgSgk1 HCN cells; we used sgCon HCN cells as a control (Figure 8C). Interestingly, sgSgk1 HCN cells were not protected from CORT-induced cell death (Figure 8D). Sgk2 KO was not protective either (Figure 8C,D). Among the SGK family members, only SGK3 contains a Phox homology (PX) domain, which binds to phosphatidylinositol 3-phosphate (PtdIns3P) [29]. PtdIns3P is the catalytic product of class III phosphatidylinositol-3 kinases (PtdIns3KC3) and plays an essential role in the induction of autophagy [30]. Therefore, we hypothesized that SGK3 rather than SGK1 or 2 may play an important role in ACD of HCN cells following CORT treatment. As expected, genetic ablation of Sgk3 (sgSgk3) substantially reduced CORT-induced cell death (Figure 8E,F). Of note, sgSgk3 cells were susceptible to STS-induced apoptosis or H2O2-induced necroptosis, suggesting the specific role of SGK3 in ACD (Figure 8G,H).
Figure 8.
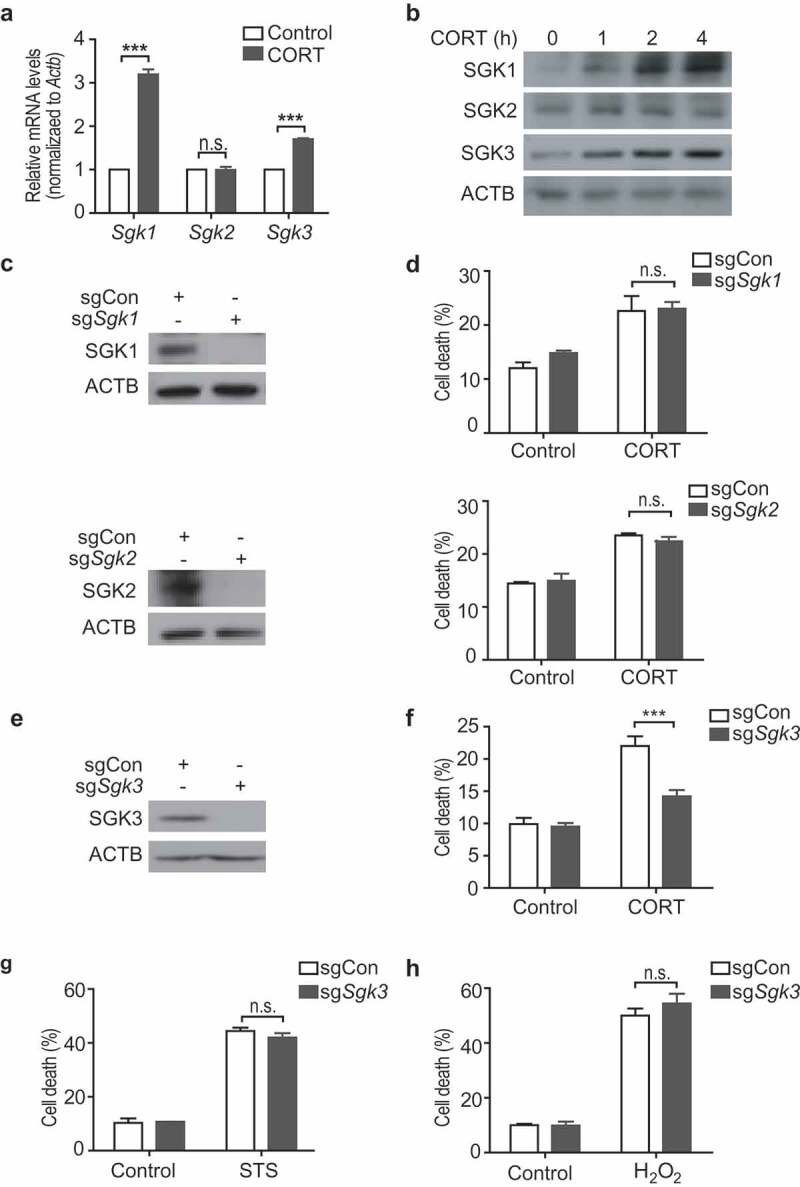
SGK3 is critical for CORT-induced cell death, but dispensable for apoptosis or necroptosis in HCN cells. (A and B) Changes in the expression levels of SGK1, 2, and 3 following CORT treatment in HCN cells. mRNA levels after CORT treatment for 24 h (n = 3) (A). Western blotting analyses of protein levels (B). Blots are representative of 3 experiments with similar results. (C) KO of Sgk1 (sgSgk1) and Sgk2 (sgSgk2). (D) Death rates of sgSgk1 and sgSgk2 cells after CORT treatment for 48 h (n = 3). (E) KO of Sgk3 (sgSgk3). (F) Death rate of sgSgk3 cells (n = 8). (G) Death rate of sgSgk3 cells after STS treatment (0.5 μM) for 24 h (n = 3). (G) Death rate of sgSgk3 cells after H2O2 treatment (100 μM) for 6 h (n = 3). ***P < 0.001. n.s., not significant.
Next, we examined the effects of SGK3 deficiency on CORT-induced autophagy. Deletion of Sgk3 gene blocked autophagy flux (Figure 9A). Furthermore, CORT-triggered formation of EGFP-ZFYVE1/DFCP1 and mRFP-MAP1LC3B puncta was prevented in sgSgk3 cells, suggesting impaired initiation or elongation of autophagosome formation (Figure 9B,C). The importance of the SGK3 PX domain in ACD induction was verified by mutating the critical Arg90 residue to Ala (Figure 9D). This mutation is known to block SGK3 binding to PtdIns3P [29]. This SGK3 mutant, unlike the WT form, failed to rescue cell death and autophagy flux when reintroduced into sgSgk3 HCN cells (Figure 9E,F). SGK3 WT showed colocalization with autophagosomes and restored MAP1LC3B puncta formation following CORT treatment in sgSgk3 cells, while SGK3R90A mutant failed to do so (Figure 9F).
Figure 9.
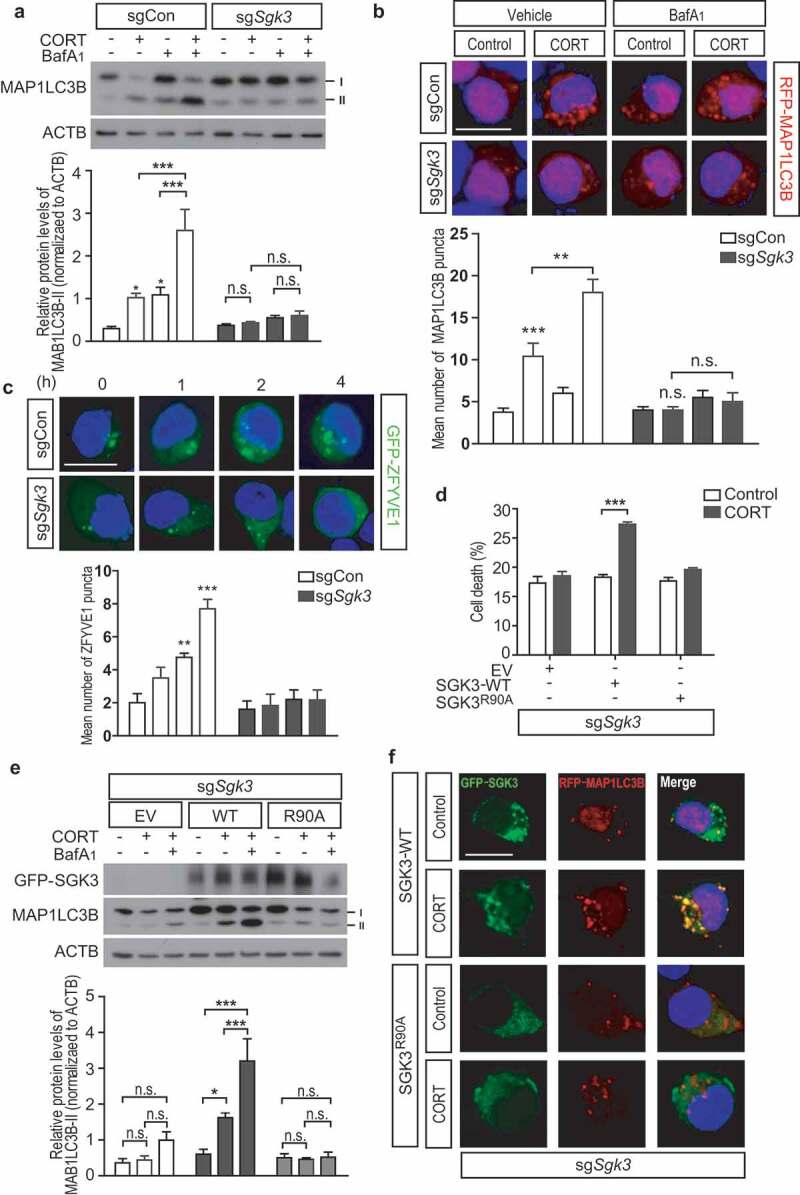
SGK3 mediates ACD in HCN cells following CORT treatment. (A) Analysis of autophagy flux by western blotting of MAP1LC3B. The graph shows quantification of MAP1LC3B-II after normalization to ACTB (n = 5). (B) Analysis of autophagosome formation using mRFP-MAP1LC3B. Scale bar: 10 μm. The graph shows quantification of autophagosomes (n = 4 or 5). (C) Time-course analysis of EGFP-ZFYVE1 puncta formation after CORT treatment using EGFP-ZFYVE1. Scale bar: 10 μm. The graph shows quantification of ZFYVE1 puncta (n = 4–6). (D) Domain diagrams of SGK1, 2, and 3 showing the critical Arg90 residue in the Phox homology (PX) domain in SGK3. SGK1 has an incomplete PX domain [38]. (E) Effects of SGK3 WT and SGK3R90A mutant on sgSgk3 cell death (n = 4). EV, empty vector. (F) Analysis of autophagy flux by western blotting of MAP1LC3B in sgSgk3 cells transfected with SGK3 WT or SGK3R90A mutant. The graph shows quantification of MAP1LC3B-II after normalization to ACTB (n = 3). (G) Effects of the SGK3R90A mutation on the MAP1LC3B puncta formation and colocalization of SGK3 with MAP1LC3B. sgSgk3 cells were co-transfected with EGFP-tagged SGK3 WT or SGK3R90A mutant with mRFP-MAP1LC3B. Scale bar: 10 μm. In all experiments, BafA1 (20 nM) was added 1 h before cell harvest. *P < 0.05, ***P < 0.001. n.s., not significant.
Deletion of SGK3, but not SGK1, blocks crs-induced HCN cell death in vivo
Lastly, we investigated the role of SGK3 on the determination of NSC fate after CRS in vivo. To see the effects of SGK family deficiency on stressed NSCs, we created Sgk1- or Sgk3-targeting sgRNA expressing adeno-associated virus (AAV). We designed AAV construct which uses the Rnu6 promoter to drive Sgk1- or Sgk3-specific sgRNA expression and the Nes promoter to drive mCherry expression in an NSC-specific manner (Figure 10A). AAC viruses were injected to DG of S-Cas9 mice. S-Cas9 mice constitutively express Cas9 for CRISPR/Cas9-mediated gene editing in vivo [31]. Expression of AAV-sgSgk1 (data not shown) or -sgSgk3 (Figure 10B) exhibits the expected NSC-specific mCherry signal. Also, the efficiency of sgSgk1 and sgSgk3 viruses were validated in HCN cells after co-transfection with pRGEN-Cas9-Hyg/EGFP and western blotting analysis of SGK1 and SGK3 protein levels (Figure 10C). CRS was performed 2 weeks after injection, then we counted the mCherry- and SOX2- double-positive cells (Figure 10D). Sgk1 deletion in DG showed reduced number of mCherry- and SOX2-positive cells after CRS (Figure 10E,F). However, Sgk3 deletion in DG preserved the number of mCherry- and SOX2-positive cells after CRS (Figure 10E,F). These data demonstrate that SGK3 is essential for CRS-induced NSC cell death in adult hippocampus.
Figure 10.
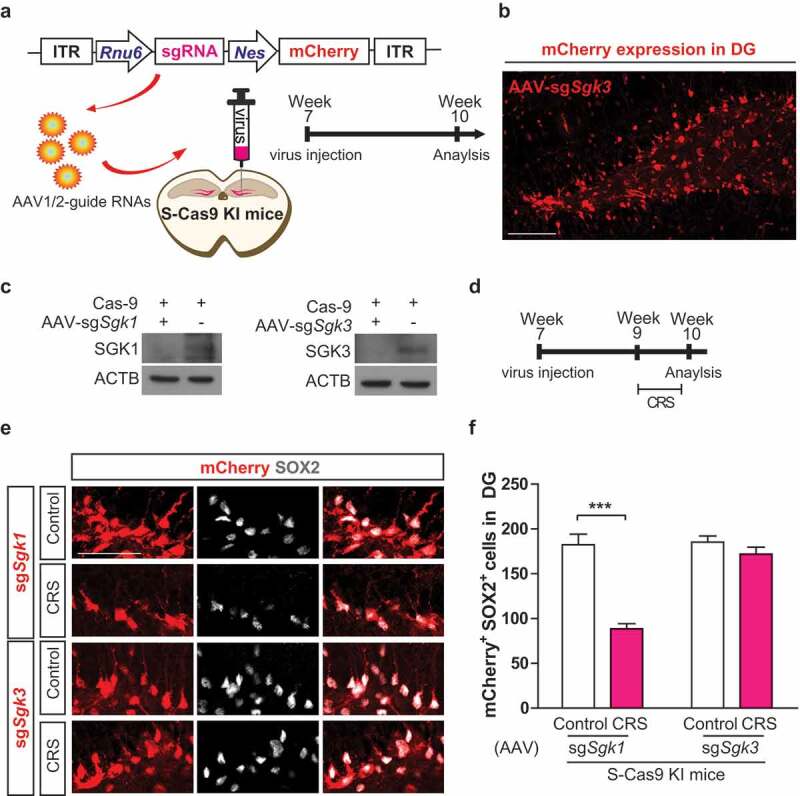
SGK3 silencing attenuates NSC reduction in the DG after CRS. (A) Experimental design illustrating stereotaxic injection of AAV1/2-guide RNAs into DG of S-Cas9 KI mice. (B) Image of AAV1/2-sgSGK3-mCherry expression in the SGZ of DG. Scale bar, 100 μm. (C) KO of Sgk1 (AAV-sgSgk1) and Sgk3 (AAV-sgSgk3). (D) Timeline of the experiment. (E) Representative images of mCherry and SOX2 co-labeling in the SGZ of DG 3 weeks after injection of AAV-sgSgk1 or AAV-sgSgk3 in S-Cas9 mice. Scale bar, 50 μm. (F) The graph shows quantification of SGK-mCherry and SOX2 double-positive cells. (n = 5). ***P < 0.001.
Discussion
ACD is seemingly opposed to a generally cytoprotective role of autophagy, and has been argued as a misconception resulting from the use of nonspecific pharmacological and genetic tools for autophagy manipulation, wrong interpretation of autophagy and cell death assay results, and lack of proper controls [32]. Therefore, it was contended that ACD may not exist in mammalian cells under physiologically relevant conditions [33]. Another criticism of ACD is that there is no evidence that autophagy-deficient cells show a resistance to cell death and survive for a long time in vivo [32]. Apropos of this, our study identifies an in vivo physiological situation where deletion of Atg7 in an NSC-specific and temporally regulatable manner attenuated an increase in autophagy flux and protected HCN cells against stress for a prolonged period (survival for more than 4 weeks after 1-week long stress). Therefore, these data strongly suggest that prevention of ACD preserves the pool of NSCs and intact neurogenesis, and mitigates the deleterious effects of stress on mood regulation and cognitive functions. Interestingly, Atg7-NSC cKO mice underwent the same distress as WT mice, as shown by reduced body weight and elevated circulating CORT levels. Accordingly, protection afforded by conditional Atg7 deletion is clearly specifically related to NSC death evoked by a high level of CORT rather than to altering the hypothalamic–pituitary-adrenal axis. Also, we found no difference in the proliferation and differentiation rates of hippocampal NSCs between stressed and unstressed groups, suggesting that the beneficial effect of Atg7 deletion was directly related to NSC survival and death. However, we observed reduced rates of BrdU+ SOX2+ cells after 3-week CRS. Therefore, more prolonged or severe stress may also reduce proliferation of adult hippocampal NSCs. To further study molecular mechanism, we used CORT to mimic psychological stress in vivo and in vitro at concentrations selected based on previous studies reported by others as well as our assays. Analyses of various apoptosis and autophagy markers in combination with deletion of several autophagy components clearly demonstrate that HCN cells have intact apoptotic capability; nevertheless, CORT shifts the mode of cell death towards ACD. Furthermore, we demonstrate that SGK3 mediates CORT-induced ACD in HCN cells. Suppressed autophagosome nucleation revealed by the lack of ZFYVE1 puncta in sgSgk3 cells suggests that SGK3 may be on the apex of a signal transduction pathway and a key instigator of CORT-induced autophagy. However, conventional Sgk3 KO adult mice have deficit in locomotion and space navigation [34]. These subtle defects suggest neural derangement that accumulates during development and exclude their use in psychological stress study at the adult stage. Also, to our knowledge, currently no isoform-specific SGK inhibitors are available. These limitations hinder further understanding of the roles of SGK3 and related molecular mechanisms, and warrant development of new genetic animal models and pharmacological tools targeting SGK3. Nonetheless, our results demonstrate that psychological stress or exposure to the stress hormone CORT induces ACD via SGK3.
Maintenance of the hippocampal NSC pool after chronic stress is important to prevent impairment of neurogenesis that affects anxiety-related and depressive-like behaviors, and learning and memory dysfunction. An increase in adult neurogenesis is one of the potential mechanisms through which antidepressants achieve their therapeutic effects. However, chronic administration of antidepressants, typically more than 2 weeks, is required for promoting neurogenesis, and their direct effects on NSC under stress conditions are not clearly defined. We examined whether fluoxetine, a widely used antidepressant, can mitigate CORT-induced ACD of HCN cells and found no protective effects on cell death (data not shown). The findings from our study have important implications, not only for understanding the regulatory mechanisms of adult hippocampal neurogenesis in the face of stress challenge, but also for an alternative design of interventional strategies by targeting PCD, especially SGK3-mediated ACD for the prevention or treatment of the consequences of psychological stress.
Materials and methods
Animals and CRS procedure
All procedures for the care and use of laboratory animals were approved by the Institutional Animal Care and Use Committee of DGIST. All animals were maintained in a specific pathogen free environment at the DGIST animal facility.
Nes-Cre/ERT2 transgenic mice and S-Cas9 mice were purchased from Jackson Laboratories and Atg7fl/fl mice were provided by Dr. Myung-Shik Lee (Yonsei University, Korea) with the permission of Dr. Masaaki Komatsu (Tokyo Metropolitan Institute of Medical Science, Japan). Nes-Cre/ERT2 mice were crossed with Atg7fl/fl mice to generate TAM-inducible, NSC-specific Atg7 heterozygous or homozygous cKO (Atg7-NSC cKO) mice. TAM (Sigma-Aldrich, T5648) was dissolved in corn oil (Sigma-Aldrich, C8267) and administered i.p. to mice at 180 mg/kg/d for 3 days at the age of 7 weeks [35]. CRS began 4 days after the last administration of TAM. Mice were housed under a 12 h light/dark cycle with lights on at 7 AM; food and water were provided ad libitum. Each mouse from the CRS group was placed in a flat-bottomed rodent restrainer from 10 AM to 4 PM daily for 7 or 21 days. The unstressed mice were left undisturbed in their home cages without access to food and water during the restraint period. The body weight of each mouse was recorded at 10 AM. Mice were analyzed 1 day (SOX2 and BrdU immunostaining) or 4 weeks (RBFOX3 and BrdU immunostaining and analyses of neurobehaviors) after the termination of CRS.
Reagents and antibodies
CORT (Sigma-Aldrich, 27840), hygromycin B (Duchefa, ALX-380–306-G001), puromycin (Invitrogen, ant-pr-1), BafA1 (BML-CM110-0100), Nec-1 (BML-AP309-0020) and Z-VAD (ALX-260–020-M005) from Enzo Life Science, BrdU (B9285) and STS (9953S) from Cell Signaling Technology were purchased from the indicated companies. Antibodies used for western blotting and immunofluorescence staining are summarized in Table 1.
Table 1.
Antibodies used in this study.
| Target | Species | Application | Dilution | Company and catalog number |
|---|---|---|---|---|
| ACTB-HRP | Mouse | WB | 1:20000 | Santa Cruz Biotechnology |
| sc-47778 | ||||
| ATG7 | Rabbit | WB | 1:1000 | Cell Signaling Technology 8558 |
| ATG7 | Mouse | IHC | 1:50 | R&D Systems MAB6608 |
| CASP3 | Rabbit | WB/IHC | 1:1000 | Cell Signaling Technology 9664 |
| MAB1LC3B | Rabbit | WB | 1:20000 | Novus NB100-2220 |
| SQSTM1/p62 | Rabbit | WB | 1:1000 | Sigma-Aldrich P0067 |
| SGK1 | Mouse | WB | 1:1000 | Abcam ab63527 |
| SGK2 | Rabbit | WB | 1:1000 | Proteintech 11185–1 |
| SGK3 | Rabbit | WB | 1:1000 | Proteintech 12699–1 |
| ULK1 | Rabbit | WB | 1:1000 | Cell Signaling Technology 8554 |
| Mouse IgG, HRP | Mouse | WB | 1:5000 | Cell Signaling Technology 7076S |
| Rabbit IgG, HRP | Rabbit | WB | 1:5000 | Thermo Fisher Scientific 31460 |
| BrdU | Mouse | IHC | 1:400 | Sigma-Aldrich B2531 |
| DCX | Rabbit | IHC | 1:1000 | Abcam ab18723 |
| GFAP | Chicken | IHC | 1:1000 | Novus NBP1-05198 |
| MKI67 | Rabbit | IHC | 1:500 | Abcam ab1558 |
| ASCL1/MASH1 | Mouse | IHC | 1:500 | BD Pharmingen 556604 |
| NES | Mouse | IHC | 1:500 | Abcam ab196693 |
| RBFOX3 | Rabbit | IHC | 1:1000 | Abcam ab104225 |
| SOX2 | Rabbit | IHC | 1:500 | Abcam ab97959 |
| SOX2 | Mouse | IHC | 1:500 | Abcam ab79351 |
| VIM | Chicken | IHC | 1:500 | Millipore AB5733 |
| Chicken IgG, | Chicken | IHC | 1:500 | Jackson Laboratories |
| Alexa Fluor 488 | 703-545-155 | |||
| Mouse IgG, | Mouse | IHC | 1:500 | Jackson Laboratories |
| Alexa Fluor 488 | 715-545-151 | |||
| Mouse IgG, | Mouse | IHC | 1:500 | Jackson Laboratories |
| Alexa Fluor 647 | 711-605-150 | |||
| Rabbit IgG, | Rabbit | IHC | 1:500 | Jackson Laboratories |
| Alexa Fluor 488 | 711-545-152 | |||
| Rabbit IgG, | Rabbit | IHC | 1:500 | Thermo Fisher Scientific |
| Alexa Fluor 555 | A-31572 | |||
| Rabbit IgG, | Rabbit | IHC | 1:500 | Jackson Laboratories |
| Alexa Fluor 647 | 711-605-152 |
CORT injection
CORT was dissolved on the first day of injection in 10% ethanol in corn oil at a concentration of 8 mg/ml. Each mouse received daily i.p. injections of 80 mg/kg CORT for 7 consecutive days at 10 AM.
Brdu injection
To count the proliferating cells in the DG, BrdU (50 mg/kg of body weight) was injected i.p. 30 min before restraint for the last 3 days of CRS and mice were perfused 1 day after the termination of CRS. To examine the neurogenesis, the same amount of BrdU was injected for 5 days 30 min before restraint in the beginning of CRS and mice were analyzed 4 weeks after the termination of CRS.
CORT measurement
Submandibular blood samples were collected 30 min after stress, using animal lancets (MEDIpoint International, GR-5MM). Blood samples were centrifuged (2,000 × g, 10 min), and serum was collected. Serum CORT concentration was measured using Corticosterone ELISA kit (Enzo Life Sciences, ADI-901–097).
Immunofluorescence staining
For immunocytochemistry analyses, cells were fixed with 4% paraformaldehyde (PFA, Sigma-Aldrich, P6178), permeabilized with 0.2% Triton X-100 (Sigma-Aldrich, X100) in antibody diluent solution (Invitrogen, 003218r) and blocked with antibody diluent solution for 10 min. After blocking, samples were incubated with appropriate primary antibodies diluted in antibody diluent solution at 4°C overnight and incubated with secondary antibodies for 1 h. The cells were then stained with Hoechst33342 (Invitrogen, H3570) diluted in PBS (1:1000) for 10 min before mounting. Samples were examined under an LSM700 confocal laser scanning microscope (Carl Zeiss, Oberkochen, Germany).
For immunohistochemistry analyses, mice were deeply anesthetized with an injection of Zoletil (Virbac, 6FX7; 50 mg/kg) and Rompun (Bayer; 10 mg/kg) and perfused with PBS, followed by 4% PFA. The brains were removed, post-fixed in 4% PFA for 12 h and cryoprotected in 30% sucrose (Duchefa, S0809) until they sank to the bottom of the tube. Brains were frozen in optimal cutting temperature compound and cut into 40-μm thick coronal sections. Samples were free-floated in PBS and blocked with PBS containing 1% bovine serum albumin (BIOWORLD, 22070008–3) and 0.3% Triton X-100 for 1 h. Samples were then incubated with the appropriate primary antibodies for 36 h at 4°C. Appropriate secondary antibodies (Table 1) were used and the samples were examined under an LSM780 confocal laser scanning microscope (Carl Zeiss).
TUNEL assay was performed using TUNEL assay kit (Promega, G3250) according to the manufacturer’s instruction.
Stereological cell counting
The dentate gyrus (bregma −1.2 mm to −2.8 mm) was sectioned and immunoreactive cells from both hemispheres were counted on a 1/8 series of 40-μm coronal sections. The cells were quantified using the optical fractionator method of Stereo Investigator software (MBF Bioscience). For the representative images, medial section of DG (bregma −2.0 mm) was imaged with LSM 780 confocal microscope (Carl Zeiss).
Elevated plus maze test
The maze consisted of two open arms without walls and two enclosed arms with 30 cm high walls, and was positioned 30 cm above the floor and illuminated evenly at 5 lux. Equivalent arms were arranged opposite to one another. Mice were placed at the center of the maze facing one of the open arms and allowed to explore freely for 5 min. The time spent in the open arms were measured. Data were collected using EthoVision Observer software (Noldus Information Technology).
Open field test
The apparatus consisted of a square-shaped arena (40 × 40 cm2). Mice were placed facing the center of one of the walls and allowed to explore the apparatus for 20 min. The time spent exploring the central region (20 × 20 cm2 area) and total distance traveled were measured. Data were collected using EthoVision Observer.
Y-maze test
The Y-maze was constructed of white plastic with three arms that extended from a central platform at a 120° angle. Each mouse was placed in the center of the Y-maze and was allowed to explore freely through the maze for 6 min. The sequence and total number of arms entered were recorded using Ethovision Observer. Arm entry was considered to be complete when the whole body of the mouse was completely placed within the arm.
Morris water maze test
The Morris water maze consisted of a circular pool filled with water at 27°C (diameter 100 cm, height 35 cm), situated in a room. Mice were trained to find a platform hidden 1.5 cm below the water surface, at a fixed location in one of the maze’s quadrants. The mouse was placed in the quadrant opposite to the target platform and allowed to find the platform for 60 s. Mice were given 3 trials/day for 6 days with a 2 min inter-trial interval. After the last trial, the mice were placed in the pool without the platform for 60 s for the probe test. Data were collected using EthoVision Observer.
Sucrose preference test
Each mouse was given 2 water bottles, one containing water and the other with 1% sucrose balanced across animals for 3 days of habituation. Following habituation, bottles were removed for 6 h and then returned for testing but the left and right bottles were swapped. The bottles were weighed before testing and after 12 h. Total consumption was measured and sucrose preference was calculated as (sucrose consumption) ÷ (sucrose consumption + water consumption) ×100 [20].
Constructs and virus production
mRFP-EGFP-MAP1LC3B (21074, Tamotsu Yoshimori’s lab), mRFP-MAP1LC3B (21075, Tamotsu Yoshimori’s lab), and EGFP-DFCP1 (38269, Noboru Mizushima’s lab) were purchased from Addgene. EGFP-SGK3 (RG214835) were purchased from Origen. For transfection, Lipofectamine 2000 (Invitrogen, 11668019) was used according to the manufacturer’s instructions. Plasmids encoding rep and cap (VPK-424) for AAV serotype 1 and 2 were purchased from Cell Biolabs. For lentiviral gene expression in the DG of hippocampal tissue, mRFP-EGFP-MAP1LC3B was cloned in pEZX-LPM02 (GeneCopoeia, HPRM13080) containing the human Nes promoter. Lentivirus production followed a published protocol [36].
For Sgk1 and Sgk3 deletion in the DG of hippocampus, pAAV1/2-Rnu6-sgSgk1-Nes-mCherry and pAAV1/2- Rnu6-sgSgk3-Nes-mCherry were produced by a combination of PCR and restriction endonuclease enzyme-based directional cloning. Rnu6 promoter and sgSgk1 or sgSgk3 purchased from Toolgen were cloned into pAAV-hSyn-EGFP (Addgene, 50465, Bryan Roth’s lab) vector where hSyn-EGFP-WPRE fragment was replaced to Nes-mCherry of pEZX-LPM02 (GeneCopoeia). AAV virus was produced, as described previously [37].
Stereotaxic injection
Mice were anesthetized and stereotaxic surgery was performed to deliver 2 μl of lentivirus or 1 μl of AAV bilaterally in the DG according to the following coordinates: AP −2.2 mm, ML +/−1.5 mm, DV −2.1 mm from the bregma. Virus was injected at 0.25 μl/min over a period of 20 min by using a 10 μl Hamilton syringe and 27G needle. Mice were allowed to recover in a heated chamber.
Cell culture
Rat HCN cells were prepared from 2-month-old female Sprague-Dawley rats and cultured, as described previously [5] in a chemically defined serum-free medium containing Dulbecco’s modified Eagle’s medium/F-12 (Thermo Fisher Scientific, 12400–024) supplemented with N2 components and basic fibroblast growth factor (20 ng/ml; PeproTech, 100-18B-500). Mouse HCN cells were isolated from 2-month-old C57BL/6 mice and cultured in the same way as rat HCN cells, except for an extra addition of epidermal growth factor (20 ng/ml; PeproTech, AF-100-15).
Neurosphere assay
Neurosphere assay was performed following a previously published protocol [23]. DG from each group of mice was collected and dissociated using the mixture of the enzymes containing 0.8 U/ml dispase (Stem cell, 07923), 2.85 U/ml papain (Worthington, LK003176) and 250 U/ml DNase (Stem cell, 07900) for 30 min at 37°C. Dissociated cells were plated in fresh DMEM/F12 with 100 ng/ml basic fibroblast growth factor and epidermal growth factor at a density of 1 × 104 cells per well of 96-well plate. Culture medium was added every 2 days during the entire culture period. To determine the NSC activity, the number and size of the spheres were determined 7 days after plating. Image of neurospheres were recorded using a digital camera attached to an Axiovert fluorescent microscope (Carl Zeiss) at a 20× magnification. Four images were randomly acquired from each plate, and the diameter of the neurospheres in these images was measured with a minimum cutoff diameter of 20 μm using the Image J software.
Cell death assay
Cells were stained with Hoechst 33342 and propidium iodide (PI; Sigma-Aldrich, P4170), and images were taken using a fluorescence microscope (Axiovert 40 CFL; Carl Zeiss) as we previously described [5]. The percentage of cell death was calculated as follows:
Cell death (%) = (PI [red] positive cell number ÷ Hoechst [blue] positive cell number) × 100.
Generation of KO or stable knockdown HCN cells
Rat HCN cells were transduced with lentiviruses expressing small hairpin RNA (shRNA) targeting Atg7 (TRCN0000092164, TRCN0000369085, Sigma-Aldrich), Sqstm1 (TRCN0000238135, TRCN000257058, Sigma-Aldrich) or Map1lc3b (TRCN0000120800, Sigma-Aldrich). Stable cell lines were generated after puromycin (5 μg/ml) selection and named shAtg7, shSqstm1, and shMap1lc3b cells, respectively. CRSPR/Cas9-mediated KO of Ulk1, Sgk1, Sgk2 or Sgk3 was performed by transfecting HCN cells with pRGEN-cas9-Hyg/EGFP-CMV and dRGEN-Ulk1 (GATTGGACACGGCGCCTTCGCGG),-Sgk1 (GCTTCTGAATAAAATCGTTCAGG), -Sgk2 (GTCATTGGCAAAGGGAACTATGG) or Sgk3 (GAAGCGAATGGTTCGTCTTCAGG) sgRNA, all purchased from ToolGen. After incubation for 24 h, cells were selected using hygromycin (300 μg/ml) for 72 h.
Electron microscopy
After perfusion with 4% PFA, 80 μm sections of the hippocampus were cut with vibratome were used for SOX2 immunostaining. SOX2-positive cells were visualized with DAB (Vector Laboratories, SK-4100), and the area containing DAB–stained cells were fixed with 2% PFA and 2.5% glutaraldehyde in 0.1 M PBS for 1 h at 4°C. Then, the samples were post-fixed with 2% osmium tetroxide for 1 h at 4°C, followed by en bloc staining with uranyl acetate. The tissues were dehydrated in a graded series of acetone, and infiltrated with Spurr’s resin. Samples were sectioned at 60 nm using an ultramicrotome (MTX-L, RMC Boeckeler). The sections were double stained with 2% uranyl acetate for 20 min and lead citrate for 10 min, and then viewed with a Tecnai G2 transmission electron microscope operating at 200 kV (Thermo Fisher Scientific).
HCN cells with or without CORT were fixed with 2% PFA and 2.5% glutaraldehyde in 0.1 M PBS (pH 7.0) for 1 h, and followed by 2% osmium tetroxide for 2 h at 4°C. Then, dehydration, embedding, and sectioning were performed, as described above, and the cells were viewed under a Tecnai G2 microscope at 200 kV.
Western blotting analysis
Cells were lysed in radioimmunoprecipitation assay buffer (Sigma-Aldrich, 89901) containing 1 × protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific, 78441) for 15 min on ice. Following centrifugation (12,000 × g, 15 min), the supernatant was collected and protein concentration was measured using the BCA protein assay reagent (Thermo Fisher Scientific, 23225). Total proteins (10 – 20 μg per lane) were loaded and separated by electrophoresis, and electrotransferred to polyvinylidene fluoride membranes in a semi-dry electrophoretic transfer cell (Bio-Rad). Membranes were blocked for 1 h at room temperature in PBS with 5% nonfat dry milk, 0.1% Tween 20 (Duchefa, P1362). Then the membranes were incubated overnight with the appropriate primary antibodies, followed by 1 h incubation with peroxidase-conjugated secondary antibodies. Protein bands were visualized using a Supersignal chemiluminescence detection kit (Thermo Fisher Scientific, 34080).
Statistical analysis
Data were expressed as mean ± standard error of the mean (SEM) and were the results of at least 3 independent experiments. All analyses were performed with the experimenter blinded to the groups of mice and cultured cells. Statistical significance was determined using the unpaired t-test for two-group experiments. For comparison among 3 or more groups, two-way analysis of variance (ANOVA) and Bonferroni post-tests were used. Sample sizes were selected on the basis of preliminary results to ensure an adequate power. Differences were considered statistically significant at P < 0.05.
Supplementary Material
Funding Statement
This work was supported by the National Research Foundation of Korea (NRF) grants (2017R1A2B4004289, 2018M3C7A1056275), the KBRI basic research program (19-BR-01-08), and the DGIST Convergence Science Center Program (19-BD-04) of the Ministry of Science and ICT of Korea; National Research Foundation of Korea [2018M3C7A1056275]; National Research Foundation of Korea [2017R1A2B4004289]; Ministry of Science and ICT of Korea [19-BR-01-08]; Ministry of Science and ICT of Korea [19-BD-04].
Acknowledgments
We thank Drs. Masaaki Komatsu (Tokyo Metropolitan Institute of Medical Science, Japan) and Myung-Shik Lee (Yonsei University, Korea) for Atg7fl/fl mice.
Disclosure statement
No potential conflict of interest was reported by the authors.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
References
- [1].Mizushima N, Levine B, Cuervo AN, et al. Autophagy fights disease through cellular self-digestion. Nature. 2008;451(7182):1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Tsujimoto Y, Shimizu S.. Another way to die: autophagic programmed cell death. Cell Death Differ. 2005;12(Suppl 2):1528–1534. [DOI] [PubMed] [Google Scholar]
- [3].Tait SW, Ichim G, Green DR.. Die another way–non-apoptotic mechanisms of cell death. J Cell Sci. 2014;127(Pt 10):2135–2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Denton D, Shravage B, Simin R, et al. Autophagy, not apoptosis, is essential for midgut cell death in Drosophila. Curr Biol. 2009;19(20):1741–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Yu S-W, Baek S-H, Brennan RT, et al. Autophagic death of adult hippocampal neural stem cells following insulin withdrawal. Stem Cells. 2008;26(10):2602–2610. [DOI] [PubMed] [Google Scholar]
- [6].Ha S, Ryu HY, Chung KM, et al. Regulation of autophagic cell death by glycogen synthase kinase-3beta in adult hippocampal neural stem cells following insulin withdrawal. Mol Brain. 2015;8:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chung KM, Jeong EJ, Park H, et al. Mediation of autophagic cell death by type 3 ryanodine receptor (RyR3) in adult hippocampal neural stem cells. Front Cell Neurosci. 2016;10:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ha S, Jeong S-H, Yi K, et al. Phosphorylation of p62 by AMP-activated protein kinase mediates autophagic cell death in adult hippocampal neural stem cells. J Biol Chem. 2017;292(33):13795–13808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Shen HM, Codogno P. Autophagic cell death: loch Ness monster or endangered species?. Autophagy. 2011;7(5):457–465. [DOI] [PubMed] [Google Scholar]
- [10].Zhao C, Deng W, Gage FH. Mechanisms and functional implications of adult neurogenesis. Cell. 2008;132(4):645–660. [DOI] [PubMed] [Google Scholar]
- [11].Gould E, Cameron HA, Daniels DC, et al. Adrenal hormones suppress cell division in the adult rat dentate gyrus. J Neurosci. 1992;12(9):3642–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lehmann ML, Brachman RA, Martinowich K, et al. Glucocorticoids orchestrate divergent effects on mood through adult neurogenesis. J Neurosci. 2013;33(7):2961–2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Egeland M, Zunszain PA, Pariante CM. Molecular mechanisms in the regulation of adult neurogenesis during stress. Nat Rev Neurosci. 2015;16(4):189–200. [DOI] [PubMed] [Google Scholar]
- [14].Kheirbek MA, Klemenhagen KC, Sahay A, et al. Neurogenesis and generalization: a new approach to stratify and treat anxiety disorders. Nat Neurosci. 2012;15(12):1613–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lagace DC, Donovan MH, DeCarolis NA, et al. Adult hippocampal neurogenesis is functionally important for stress-induced social avoidance. Proc Natl Acad Sci U S A. 2010;107(9):4436–4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Koo JW, Russo SJ, Ferguson D, et al. Nuclear factor-kappaB is a critical mediator of stress-impaired neurogenesis and depressive behavior. Proc Natl Acad Sci U S A. 2010;107(6):2669–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol. 2001;2(3):211–216. [DOI] [PubMed] [Google Scholar]
- [18].Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441(7095):880–884. [DOI] [PubMed] [Google Scholar]
- [19].Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J. 2000;19(21):5720–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Snyder JS, Soumier A, Brewer M, et al. Adult hippocampal neurogenesis buffers stress responses and depressive behaviour. Nature. 2011;476(7361):458–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Imayoshi I, Sakamoto M, Kageyama R. Genetic methods to identify and manipulate newly born neurons in the adult brain. Front Neurosci. 2011;5:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lugert S, Basak O, Knuckles P, et al. Quiescent and active hippocampal neural stem cells with distinct morphologies respond selectively to physiological and pathological stimuli and aging. Cell Stem Cell. 2010;6(5):445–456. [DOI] [PubMed] [Google Scholar]
- [23].Jongbloets BC, Lemstra S, Schellino R, et al. Stage-specific functions of Semaphorin7A during adult hippocampal neurogenesis rely on distinct receptors. Nat Commun. 2017;8:14666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yoshida M, Koyanagi S, Matsuo A, et al. Glucocorticoid hormone regulates the circadian coordination of micro-opioid receptor expression in mouse brainstem. J Pharmacol Exp Ther. 2005;315(3):1119–1124. [DOI] [PubMed] [Google Scholar]
- [25].Galluzzi L, Vitale I, Abrams JM, et al. Molecular definitions of cell death subroutines: recommendations of the nomenclature committee on cell death 2012. Cell Death Differ. 2012;19(1):107–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Schoenfeld TJ, Gould E. Stress, stress hormones, and adult neurogenesis. Exp Neurol. 2012;233(1):12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lang F, Böhmer C, Palmada M, et al. (Patho)physiological significance of the serum- and glucocorticoid-inducible kinase isoforms. Physiol Rev. 2006;86(4):1151–1178. [DOI] [PubMed] [Google Scholar]
- [28].Anacker C, Cattaneo A, Musaelyan K, et al. Role for the kinase SGK1 in stress, depression, and glucocorticoid effects on hippocampal neurogenesis. Proc Natl Acad Sci U S A. 2013;110(21):8708–8713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Tessier M, Woodgett JR. Role of the Phox homology domain and phosphorylation in activation of serum and glucocorticoid-regulated kinase-3. J Biol Chem. 2006;281(33):23978–23989. [DOI] [PubMed] [Google Scholar]
- [30].Yu X, Long YC, Shen HM. Differential regulatory functions of three classes of phosphatidylinositol and phosphoinositide 3-kinases in autophagy. Autophagy. 2015;11(10):1711–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Platt RJ, Chen S, Zhou Y, et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell. 2014;159(2):440–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008;9(12):1004–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Shen S, Kepp O, Kroemer G. The end of autophagic cell death?. Autophagy. 2012;8(1):1–3. [DOI] [PubMed] [Google Scholar]
- [34].Lang UE, Wolfer DP, Grahammer F, et al. Reduced locomotion in the serum and glucocorticoid inducible kinase 3 knock out mouse. Behav Brain Res. 2006;167(1):75–86. [DOI] [PubMed] [Google Scholar]
- [35].Lagace DC, Whitman MC, Noonan MA, et al. Dynamic contribution of nestin-expressing stem cells to adult neurogenesis. J Neurosci. 2007;27(46):12623–12629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Tiscornia G, Singer O, Verma IM. Production and purification of lentiviral vectors. Nat Protoc. 2006;1(1):241–245. [DOI] [PubMed] [Google Scholar]
- [37].Grieger JC, Choi VW, Samulski RJ. Production and characterization of adeno-associated viral vectors. Nat Protoc. 2006;1(3):1412–1428. [DOI] [PubMed] [Google Scholar]
- [38].Pao AC, McCormick JA, Li H, et al. NH2 terminus of serum and glucocorticoid-regulated kinase 1 binds to phosphoinositides and is essential for isoform-specific physiological functions. Am J Physiol Renal Physiol. 2007;292(6):F1741–F1750. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.