

ABSTRACT
Lysosomal membrane permeabilization (LMP) is observed under many pathological conditions, leading to cellular dysfunction and death. However, the mechanisms by which lysosomal membranes become leaky in vivo are not clear. Our data demonstrate that LMP occurs in neurons following controlled cortical impact induced (CCI) traumatic brain injury (TBI) in mice, leading to impaired macroautophagy (autophagy) and neuronal cell death. Comparison of LC-MS/MS lysosomal membrane lipid profiles from TBI and sham animals suggested a role for PLA2G4A/cPLA2 (phospholipase A2, group IVA [cytosolic, calcium-dependent]) in TBI-induced LMP. Activation of PLA2G4A caused LMP and inhibition of autophagy flux in cell lines and primary neurons. In vivo pharmacological inhibition of PLA2G4A attenuated TBI-induced LMP, as well as subsequent impairment of autophagy and neuronal loss, and was associated with improved neurological outcomes. Inhibition of PLA2G4A in vitro limited amyloid-β-induced LMP and inhibition of autophagy. Together, our data indicate that PLA2G4A -mediated lysosomal membrane damage is involved in neuronal cell death following CCI-induced TBI and potentially in other neurodegenerative disorders.
Abbreviations: AACOCF3, arachidonyl trifluoromethyl ketone; ACTB/β-actin, actin, beta; AD, Alzheimer disease; ATG5, autophagy related 5; ATG7, autophagy related 7; ATG12, autophagy related 12; BECN1, beclin 1, autophagy related; C1P, ceramide-1-phosphate; CCI, controlled cortical impact; CTSD, cathepsin D; CTSL, cathepsin L; GFP, green fluorescent protein; IF, immunofluorescence; LAMP1, lysosomal-associated membrane protein 1; LAMP2, lysosomal-associated membrane protein 2; LC-MS/MS, liquid chromatography-tandem mass spectrometry; LMP, Lysosomal membrane permeabilization; LPC, lysophosphatidylcholine; LPE, lysophosphatidylethanolamine; MAP1LC3/LC3, microtuble-associated protein 1 light chain 3; NAGLU, alpha-N-acetylglucosaminidase (Sanfilippo disease IIIB); PC, diacyl glycerophosphatidylcholine; PE, diacyl glycerophosphatidylethanolamine; PE-O, plasmanyl glycerophosphatidylethanolamine; PE-P, plasmenyl glycerophosphatidylethanolamine; PLA2G4A/cPLA2, phospholipase A2, group IVA (cytosolic, calcium-dependent); RBFOX3, RNA binding protein, fox-1 homolog (C. elegans) 3; RFP, red fluorescent protein; ROS, reactive oxygen species; SQSTM1, sequestosome 1; TUBA1/α-tubulin, tubulin, alpha; TBI, traumatic brain injury; TFEB, transcription factor EB; ULK1, unc-51 like kinase 1.
KEYWORDS: Amyloid β, autophagy, cytosolic phospholipase A2 (cPLA2), lysosomal membrane permeabilization (LMP), membrane lipidomic analysis, traumatic brain injury (TBI)
Introduction
Traumatic brain injury (TBI) is a major cause of death and disability among young adults worldwide [1–5]. TBI is caused by acute physical impact to the brain due to causes such as vehicle accidents, falls, assaults or explosive blast waves. The initial mechanical impact (primary injury) initiates cascade of complex cellular and biochemical changes (secondary injury), which include glutamate excitotoxicity, oxidative stress, perturbation of calcium homeostasis, mitochondrial and metabolic dysfunction, inflammation and apoptosis, among others, that propagate the injury and contribute to progressive tissue damage and neuronal loss [1–3,5–7].
We recently demonstrated that autophagy, a lysosome-dependent intracellular protein and organelle degradation process, is inhibited after TBI in a mouse model, and contributes to secondary neuronal cell death [7,8]. Our data also indicated that disruption of neuronal autophagy after TBI is caused by impairment of lysosomal function leading to pathological accumulation of autophagosomes and their cargo. Similar disruption of autophagy and lysosomal function is observed in many neurodegenerative disorders including Alzheimer, Parkinson and Huntington diseases [9–14], suggesting that convergent mechanisms may contribute to acute (trauma) and chronic (age-related neurodegeneration) neuronal cell death.
Lysosomes are membrane bound organelles containing acid hydrolases involved in catabolism of intracellular biomolecules and organelles as well as of extracellular components delivered by, respectively, autophagy and the endosomal pathways [15–18]. Lysosomes also play a crucial role in cellular membrane repair, as well as serve as platforms for cellular signaling, making them essential for maintenance of cellular homeostasis [18,19]. Conversely, defects in lysosomal function and in particular increase in lysosomal membrane permeability (LMP) have been demonstrated in various neurodegenerative diseases and aging [20–25]. Preservation of lysosomal membrane integrity is of utmost importance not only for maintenance of lysosomal function, but also to protect cellular components from exposure to lysosomal luminal enzymes. Yet, the mechanisms by which the lysosomal lipid membrane is altered under pathological conditions remain poorly understood.
Like other cellular organelles, lysosomes are surrounded by a phospholipid containing membrane, making them vulnerable to the activation of phospholipases. There are three major phospholipases present in the brain, namely PLA2G2A/sPLA2 (phospholipase A2, group IIA), PLA2G4A/cPLA2 (phospholipase A2, group IVA [cytosolic, calcium-dependent]) and PLA2G6/iPLA2 (phospholipase A2, group VI) [26,27]. Among these, both activity and expression levels of PLA2G4A are upregulated in several neurodegenerative diseases [26,28–31] as well as following spinal cord trauma [32]. PLA2G4A cleaves the fatty acyl linkage at the sn-2 position of glycerophospholipids releasing predominantly arachidonic acid and leaving lysophospholipids in the membrane [26,27,33–36]. Arachidonic acid is then converted into eicosanoids-prostaglandins, leukotrienes, thromboxanes and other signaling lipid metabolites which may trigger inflammatory responses. Lysophospholids remaining in the membrane can alter its fluidity and permeability [33,34,36]. Although the function of PLA2G4A generated signaling metabolites has been widely studied, the consequences of its action on the properties of the native membranes and function of the organelles they enclose are less well understood. Hydrolysis of oxygenated cardiolipin at the mitochondrial membrane has been reported under pathological conditions [37,38] and attributed to the activation of PLA2G4A [39]. Amyloid-β-mediated PLA2G4A activation has also been suggested to contribute to the loss of mitochondrial membrane potential [40]. However, cardiolipin is uniquely enriched at mitochondria and largely absent from other cellular membranes. Neither the effects of PLA2G4A on lipid composition of cellular membranes devoid of cardiolipin (such as the lysosomes), nor how such alteration affect membrane integrity and organelle function have been elucidated.
Here we demonstrate that CCI-induced TBI leads to neuronal lysosomal membrane permeabilization (LMP) and use in vivo lysosomal lipidomics to suggest that this effect is mediated through the activation of PLA2G4A. Our data indicate that PLA2G4A-mediated LMP leads to release of lysosomal enzymes into the cytosol, inhibition of autophagy flux and neuronal cell death in vitro and in vivo. We also demonstrate that early administration of PLA2G4A inhibitor arachidonyl trifluoromethyl ketone (AACOCF3) [36] in mice reduces lysosomal damage, improves autophagy flux, limits brain cell death and improves functional outcomes following TBI. Finally, our data indicate that PLA2G4A is involved in amyloid-β-induced LMP and inhibition of autophagy in vitro, suggesting that this mechanism may contribute to lysosomal and autophagy defects in other neurodegenerative disorders.
Results
TBI causes an increase in lysosomal membrane permeability
We previously reported that autophagy flux is inhibited in the mouse cortex and hippocampus within 1 h after lateral controlled cortical impact (CCI) TBI and is associated with decreased expression and activity of the lysosomal protease CTSD (cathepsin D) [7,8]. To further examine effect of TBI on lysosomal function, we measured activity of lysosomal enzymes CTSD and NAGLU (alpha-N-acetylglucosaminidase) in sham and injured mouse cortices 1 day following CCI. Activity of both enzymes was significantly decreased in the injured cortex (Fig. S1A-B), confirming general decline in lysosomal function. Sub-cellular fractionation of cortical tissues revealed lower CTSD and NAGLU activities in the lysosomal fractions prepared from injured mice as compared to sham (Figure 1A-B). Interestingly we observed significantly higher activity for both enzymes in the cytosolic fractions from TBI cortices as compared to sham mice (Figure 1A-B). Presence of lysosomal enzymes in the cytosol was confirmed by immunofluorescence (IF) staining of brain sections from sham and day 1 injured mice with antibodies against lysosomal membrane protein LAMP2 and the soluble lysosomal enzyme CTSL (cathepsin L). While LAMP2 staining showed mostly punctate lysosomal pattern in all mice, CTSL staining appeared punctate in sham mouse cortex, but more diffuse and with lower co-localization with LAMP2 in the injured cortex (Figure 1C-D). A similar diffuse pattern in injured cortex was also observed for CTSD (Fig. S1C). The leakage of lysosomal enzymes at day 1 after TBI predominantly occurred in neurons, as revealed by colocalization of diffuse CTSL staining with staining for neuronal marker RBFOX3/NeuN, but not with the transgenic microglial marker CX3CR1-GFP (Fig. S1D-E). Taken together, these data indicate that lysosomal membrane permeability (LMP) increases in cortical neurons after TBI, allowing leakage of soluble lysosomal enzymes into the cytosol.
Figure 1.
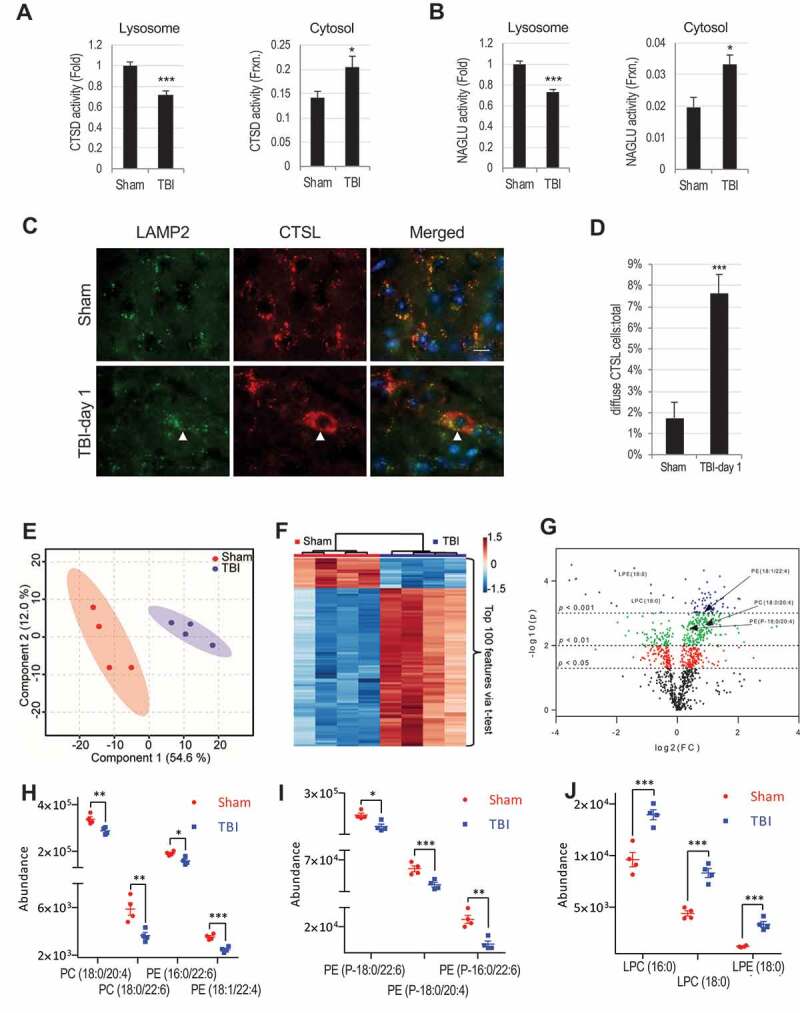
TBI leads to lysosomal membrane permeabilization and altered lysosomal membrane lipid composition. (A-B) Activity of lysosomal enzymes (A) CTSD (cathepsin D) and (B) NAGLU is decreased in lysosomal and increased in cytosolic fractions from sham and TBI mouse cortex. Data are mean ± SEM, n = 10 mice/group for CTSD and 5 for NAGLU; *p < 0.05, ***p < 0.001 (students’ t-test). (C) Images (60x) demonstrating leakage of cathepsin L (CTSL) (red) from lysosomes stained with antibody against lysosomal membrane protein LAMP2 (green) into the cytosol in the cortex of mice 24 h after TBI. Cells with diffused CTSL (cytosolic) staining are indicated with arrowheads in the injured brain sections. Scale bar:10 μm. (D) Quantification of cells with diffused CTSL. Data are mean ± SEM, n = 3 sham and 5 TBI mice; ***p < 0.001 (students’ t-test). (E-J) Results of LC-MS/MS lipid analysis of purified cortical lysosomal membranes from sham and TBI mice at 1 h after injury. (E) Partial Least Squares – Discriminate Analysis (PLS-DA) plot comparing sham (red) and TBI (blue) in positive ion mode UPLC-HDMS demonstrating separation of sham and TBI data; R2 = 0.98, Q2 = 0.81. Each point represents a data set from an individual animal. The 95% confidence intervals are indicated by elliptical shaded areas surrounding each group. Data were sum normalized, log transformed, and mean centered. (F) Heatmap displaying the top 100 differential abundance features based on t-test/ANOVA, euclidean distancing and ward clustering in positive ion mode UPLC-HDMS. (G) Volcano plot highlighting features that had a p < 0.05 (red), p < 0.01(green), and p < 0.001 (blue) when comparing Sham to TBI. Location of selected lipid species of interest is indicated. The x-axis is log2(FC) (FC = fold change) and the y-axis is – log10(p) (p = p-value based on t-test). Plots in E-G generated using Metaboanalyst; n = 4 mice/group. (H-J) Altered abundance of specific phospholipid classes in lysosomal membranes from cortices of sham (red) and TBI (blue) mice. Statistical significance was determined using t-test. (H) PC/PE abundance. Calculated p-values were 0.0080 (PC(18:0/20:4)), 0.0084 (PC(18:0/22:6)), 0.0112 (PE(16:0/22:6)), and 0.0006 (PE(18:1/22:4)). (I) Ether PE abundance. Calculated p-values were 0.0106 (PE(P-18:0/22:6)), 0.0050 (PE(P-18:0/20:4)), and 0.0026 (PE(P-18:0/22:6)). (J) LPC/LPE abundance. Calculated p-values were 0.0020 (LPC(16:0)), 0.0002 (LPC(18:0)), and 0.0003 (LPE(18:0)). Individual data points as well as mean ± SEM are indicated; n = 4 mice/group.
To confirm that the previously observed block of autophagy flux after TBI [8] is associated with the increase in lysosomal membrane permeability, we stained sections with antibodies against CTSL and the autophagy substrate SQSTM1 (sequestosome 1). At day 1 after TBI 60% of SQSTM1 signal colocalized in cells with diffuse CTSL staining (Fig. S1F-G). Therefore, block of autophagy flux after TBI is likely due to the increase in LMP and resulting loss of lysosomal function.
TBI causes alteration in lysosomal membrane lipid composition
In order to determine the mechanism of lysosomal membrane damage leading to LMP after TBI, we analyzed the lipid composition of isolated lysosomal membranes prepared from sham and injured cortices using liquid chromatography-tandem mass spectrometry (LC-MS/MS). Although autophagosome accumulation peaks at day 1 after injury, autophagic substrates start to accumulate 1 h after TBI [7,8], suggesting that lysosomal membrane damage is initiated early after injury. Accordingly, we purified lysosome enriched fraction from the cortices of sham and injured mice at 1 h after TBI. The total lipid extract of the lysosomal preparation was subjected to LC-MS/MS analysis (Schematically depicted in Fig. S2A-D). Our preparation was highly enriched in lysosomes/lysosomal content with almost undetectable levels of endoplasmic reticulum or mitochondrial proteins (Fig. S2B). The lipid composition of the lysosomal preparations from injured cortices showed significant differences when compared to sham, as visualized by multivariate and univariate analyses (Figure 1E-G; Fig. S2E-G). In total we identified 146 specific lipids that differed in abundance between the lysosomal membranes of TBI and sham brains (Table S1). A number of glycerophospholipids, including several species of diacyl glycerophosphatidylcholine (PC), diacyl glycerophosphatidylethanolamine (PE), and ether (plasmenyl and plasmanyl) glycerophosphatidylethanolamine (PE-P and PE-O, respectively) (Figure 1H-I; Table S1) were significantly less abundant in lysosomal preparations from TBI cortices as compared to sham. Lipids most significantly elevated in TBI lysosomal membranes included lysophospholipids (lysophosphatidylcholine [LPC] and lysophosphatidylethanolamine [LPE]) (Figure 1J; Table S1). Lysophospholipids are generated by the cleavage of one of the fatty acyl linkages in glycerophospholipids by phospholipases [33,36]. Therefore, lower abundance of glycerophospholipids and increased abundance of lysophospholipids in the lysosomal preparations from injured cortices strongly suggested that lysosomal membrane damage after TBI may be mediated by the activation of phospholipases.
PLA2G4A is activated and present at lysosomes after TBI
The calcium-dependent phospholipase PLA2G4A is upregulated in neurodegenerative disorders such as Alzheimer disease [26,28] and activated and implicated in neuronal cell death after spinal cord injury (SCI) [32]. PLA2G4A cleaves fatty acyl linkage in the sn-2 position of glycerophospholipids leaving lysophospholipids in the membrane and releasing fatty acids (mostly arachidonic acid) [33,41]. An increased level of lysophospholipids in the lysosomal membrane (Figure 1J) led us to hypothesize that PLA2G4A might be involved in lysosomal membrane damage after TBI. We observed a biphasic pattern of PLA2G4A activation after TBI, with activating PLA2G4A phosphorylation transiently increased within 1 h (Figure 2A-B), followed by a secondary activation at both the protein expression and phosphorylation level at day 3 (Figure 2A-B). We also detected higher levels of PLA2G4A enzymatic activity in brain lysates within 1 h after TBI (Fig. S3A). Despite lower phosphorylation levels of PLA2G4A, its enzymatic activity remained elevated at day 1 after TBI. This could be due to other mechanisms known to regulate PLA2G4A activity such as nitrosylation [42]. IF staining revealed that early (1 h) PLA2G4A phosphorylation occurred primarily in neurons (marked with RBFOX3/NeuN), while secondary activation (day 3) was predominantly microglial (marked by CX3CR1-GFP, Figure 2C-D and Fig. S3B-C).
Figure 2.
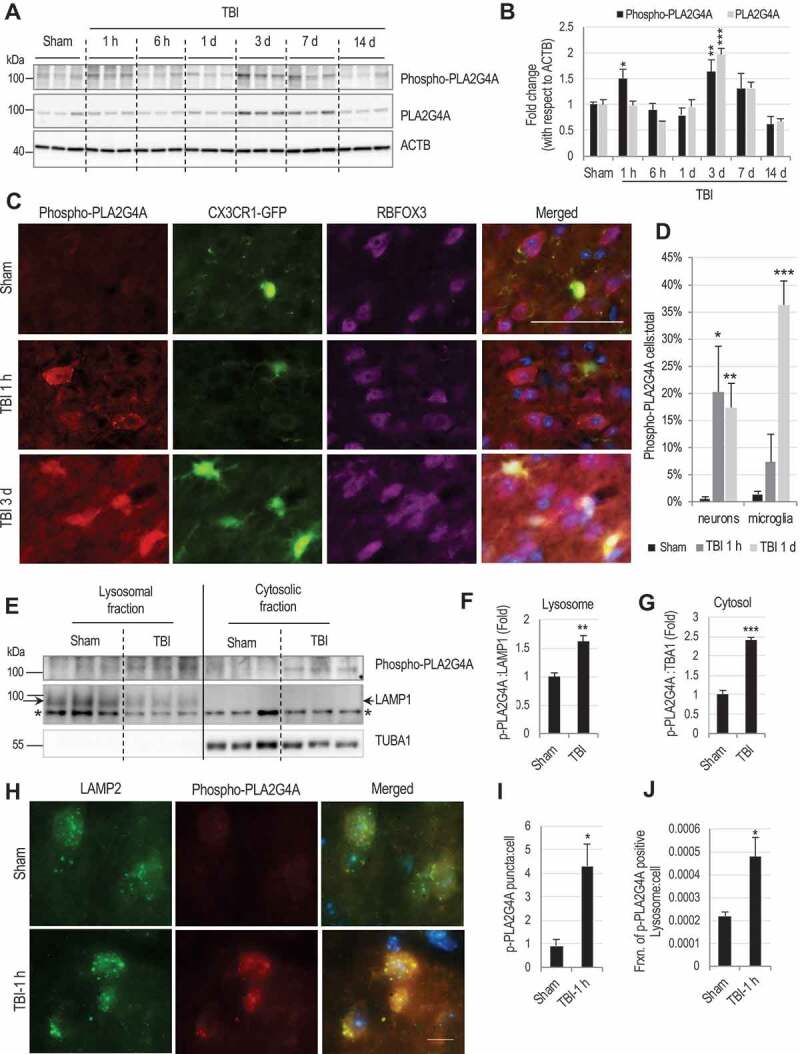
PLA2G4A is activated and present at lysosomal membranes after TBI.
(A) Western blot of phospho- and total PLA2G4A in cortical tissue lysates from sham and TBI animals. Each lane represents an individual animal. (B) Quantification of phosopho- (black bars) and total (gray bars) PLA2G4A normalized to loading control (ACTB). Data are mean ± SEM, n = 3 mice/group; *p < 0.05, **p < 0.01, ***p < 0.001 vs. sham. (Two-way ANOVA with Bonferroni posttests). (C and D) Time dependent and cell type specific activation of PLA2G4A after TBI. (C) Images (20×) of Cx3CR1-GFP (marks microglia) mouse cortical brain sections stained with antibodies against phospho- PLA2G4A (red) and neuronal marker RBFOX3/NeuN (purple). Scale bar: 50 μm. (D) Quantification of phospho-PLA2G4A positive neurons and microglial cells in sham (black bars) and injured brain sections at 1 h (dark gray) and 1 day (light gray) after TBI. Data are mean ± SEM, n = 3 mice/group; *p < 0.05, **p < 0.01, ***p < 0.001 vs. sham. (Two-way ANOVA with Bonferroni posttests); (E-J) Activated PLA2G4A is present at the lysosomes after TBI. (E) Western blot of phospho-PLA2G4A in lysosomal and cytosolic fractions from cortices of sham and TBI mice. *indicated non-specific band. (F-G) Quantification of phospho-PLA2G4A with respect to the loading controls (F) LAMP1 in lysosomal fractions and (G) TUBA1/α-tubulin in the cytosolic fraction. Data are mean ± SEM, n = 4 mice/group; **p < 0.01, ***p < 0.001 (Students’ t-test). (H) Images (60×) of cells in the cortex of sham and TBI (1 h) mice stained with antibodies against lysosomal membrane protein LAMP2 (green) and phospho-PLA2G4A (red). Scale bar: 10 μm. (I-J) Quantification of data from (H) demonstrating increased levels of phospho-PLA2G4A at lysosomes in the injured brain section as compared to sham. (I) Quantification of phospho-PLA2G4A puncta in sham and TBI (1 h) brain sections. (J) Quantification of fraction of phospho-PLA2G4A-positive lysosomes per cell in sham and TBI (1 h) brain sections. Data are presented as mean ± SEM, n = 3 sham and 4 TBI mice; *p < 0.05, (Students’ t-test).
Subcellular fractionation revealed increased level of PLA2G4A phosphorylation in both cytosolic and lysosomal fractions from the injured cortex at 1 h after injury as compared to sham (Figure 2E-G), suggesting presence of activated PLA2G4A at lysosomes after injury. This was supported by IF analysis where we observed higher level of co-localization of phospho- PLA2G4A with lysosomal marker LAMP2 in the injured cortex (Figure 2H-J). Thus, PLA2G4A is activated in mouse cortical neurons after injury and is present at the lysosomes, suggesting potential direct involvement in cleavage of lysosomal membrane phospholipids to generate lysophospholipids, leading to lysosomal membrane damage and LMP after TBI.
PLA2G4A can directly cause lysosomal damage
To determine if PLA2G4A can directly cause lysosomal damage, we treated human H4 neuroglioma cells with ceramide-1-phosphate (C1P), which activates PLA2G4A by interacting with its CalB/C2 domain [43,44]. In cells co-expressing GFP-tagged PLA2G4A and the lysosomal marker RFP-LAMP1, we observed higher GFP- PLA2G4A intensity at RFP-LAMP1+ lysosomes as compared to other cell regions in both treated and control cells (Fig. S4A). We did not observe significant increase in PLA2G4A translocation to the lysosomes following C1P treatment, suggesting that in H4 cells both activated and inactive PLA2G4A remains in close proximity to the lysosomal membranes. To determine if activated PLA2G4A can affect lysosomal membrane integrity, we extracted cytosolic and whole cell lysates based on differential sensitivity to digitonin [45], followed by assessment of lysosomal enzyme activity. C1P treatment resulted in dose-dependent increase in activity of lysosomal enzymes NAGLU and CTSD in the cytosolic extracts (Figure 3A-B), suggesting increase in LMP following PLA2G4A activation. To further confirm LMP, we loaded lysosomes of H4 cells expressing RFP-LAMP1, with 3 kDa Alexa Fluor 488-conjugated dextran and assessed its retention by fluorescence imaging. C1P treatment led to significant decrease in the number of dextran-loaded lysosomes (Figure 3C-E). In C1P-treated cells we also observed numerous abnormally enlarged RFP-LAMP1 positive lysosomes, many of which were entirely devoid of dextran fluorescence (Figure 3D, arrowheads). These data suggest accumulation of undigested materials within lysosomes leading to swelling and confirm that PLA2G4A/activation induces damage to the lysosomal membrane and LMP.
Figure 3.
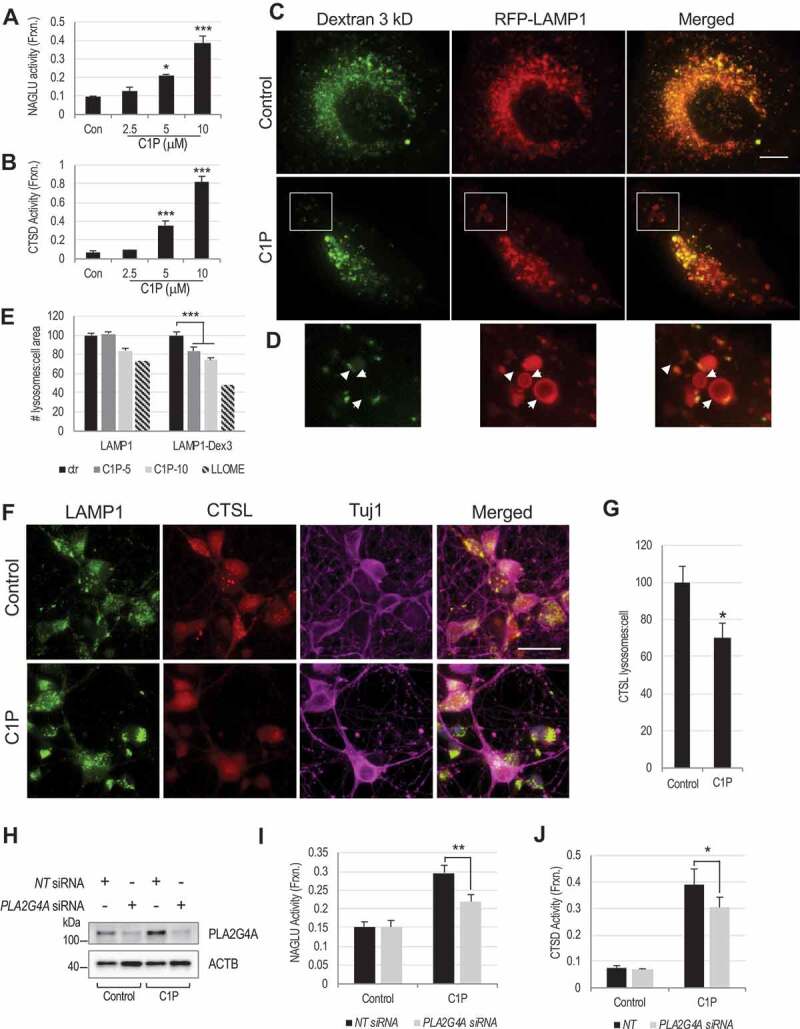
PLA2G4A activation leads to LMP in H4 neuroglioma cells and rat cortical neurons.
(A and B) Activity of lysosomal enzymes (A) NAGLU and (B) CTSD in the digitonin extracted cytosolic fractions of H4 neuroglioma cells treated with PLA2G4A activator, ceramide-1-phosphate (C1P). Data are mean ± SEM, n = 6 for NAGLU and 3 for CTSD; *p < 0.05, ***p < 0.001 (One-way ANOVA with Turkey’s multiple comparison test). (C-E) C1P mediated PLA2G4A activation causes leakage of Alexa Fluor 488 dextran from lysosomes. (C) Images (60×) of H4 cells expressing RFP-LAMP1 and loaded with Alexa Fluor 488-dextran (3 kDa) treated with C1P or vehicle for 4 h. Scale bar: 10 μm (D) Magnified image of inset in (C). Many RFP-LAMP1 positive lysosomes are partially or fully devoid of Alexa Fluor 488 dextran (arrowheads) in C1P-trated cells. (E) Quantification of Alexa Fluor 488-dextran positive lysosomes (LAMP1-Dex3) in H4 cells treated with C1P (5 and 10 μM) or LLOME positive control for 4 h. Data are mean ± SEM, n = 9 for control and cells treated with 10 μM C1P and 6 for cells treated with 5 μM C1P; ***p < 0.001. (Two-way ANOVA with Bonferroni posttests). (F-G) C1P treatment causes leakage of lysosomal enzyme CTSL into the cytosol in rat cortical neurons. (F) Images (60×) of C1P (2.5 μM) treated rat cortical neurons stained with antibodies against lysosomal membrane protein LAMP1, soluble lysosomal enzyme CTSL and neuronal marker TUBB3/Tuj1. Scale bar: 20 μm. (G) Quantification of CTSL positive lysosomes per cell. Data are mean ± SEM, n = 12; *p < 0.05, vs. control. (Students’ t-test). (H-J) Lysosomal enzyme activity in the cytosolic fraction of C1P treated H4 is attenuated following siRNA-mediated knock down of PLA2G4A as compared to non-targeting (NT) controls. (H) Western blot confirming knock down of PLA2G4A in PLA2G4A siRNAs transfected cells. (I-J) Activity of lysosomal enzymes (I) NAGLU and (J) CTSD in the digitonin extracted cytosolic fractions of C1P treated or control H4 cells transfected with either non-targeting (NT) or PLA2G4A siRNAs. Data are presented as mean ± SEM, n = 4; *p < 0.05, **p < 0.01 (Two-way ANOVA with Bonferroni posttests).
We confirmed the ability of PLA2G4A to mediate lysosomal damage in rat cortical neurons. IF staining with antibodies against lysosomal membrane protein LAMP1, revealed punctate staining in both C1P treated and control cells. However, in C1P treated cells CTSL staining became diffuse, indicating cytosolic distribution of this enzyme (Figure 3F-G). To confirm that lysosomal damage following C1P treatment is mediated through PLA2G4A activation, we knocked down PLA2G4A in H4 cells using siRNA (Figure 3H), leading to approximately 60% reduction in PLA2G4A protein level (Fig. S4B). This was sufficient to attenuate C1P-induced increase in NAGLU and CTSD activity in the cytosolic fraction as compared to cells transfected with non-targeting siRNAs (Figure 3I-J). Both pro and mature CTSD protein level remained unchanged (Fig. S4C). Together these data indicate that isolated PLA2G4A activation in cell lines and primary neurons can lead to increase in LMP and release of soluble lysosomal enzymes into the cytosol, an effect similar to what we observed in vivo after TBI.
Activation of PLA2G4A can cause impairment of autophagy flux
Lysosomal damage and LMP after TBI are associated with inhibition of autophagy flux and accumulation of dysfunctional autophagosomes in cortical neurons. We detected dose-dependent increase in LC3-II in both HeLa and H4 cells treated with C1P (Fig. S4D-E). Upstream autophagy regulators phospho-ULK1, ULK1, PI3KC3/Vps34, ATG12–ATG5 conjugate and BECN1 remained unaltered. Increase in GFP-LC3 puncta was similarly observed in stable GFP-LC3 expressing H4 cells treated with C1P (Fig. S4F). We also detected dose-dependent accumulation of LC3-II in H4 cells treated with another PLA2G4A activator, phosphatidylinositol-4,5-bisphosphate (PtdIns [4,5]P2/PIP2) [46], suggesting that it is likely the result of PLA2G4A activity rather than off target effects of a specific activator (Fig. S4G). No further increase in accumulation of LC3-II was observed following addition of C1P to H4 cells treated with lysosomal inhibitors, chloroquine or bafilomycin A1 (Figure 4A), suggesting that accumulation of autophagosomes in C1P treated cells was mainly caused by impairment of their degradation. This was further supported by autophagy flux analysis using cells expressing a tandem mCherry-EGFP-LC3 reporter. This assay takes advantage of differential pH sensitivity of EGFP (acid labile) and mCherry (acid resistant) fluorophores to assess acidification of autophagosomes (green and red) upon fusion with lysosomes (red only). In cells treated with C1P, we observed accumulation of red/green autophagosomes, but not red-only autolysosomes as compared to controls (Figure 4B-C; Fig. S4H). These data confirm inhibition of autophagy flux following activation of PLA2G4A by C1P treatment as well as suggest impairment in autophagosome-lysosome fusion, similar to what we previously reported in vivo after TBI [8].
Figure 4.
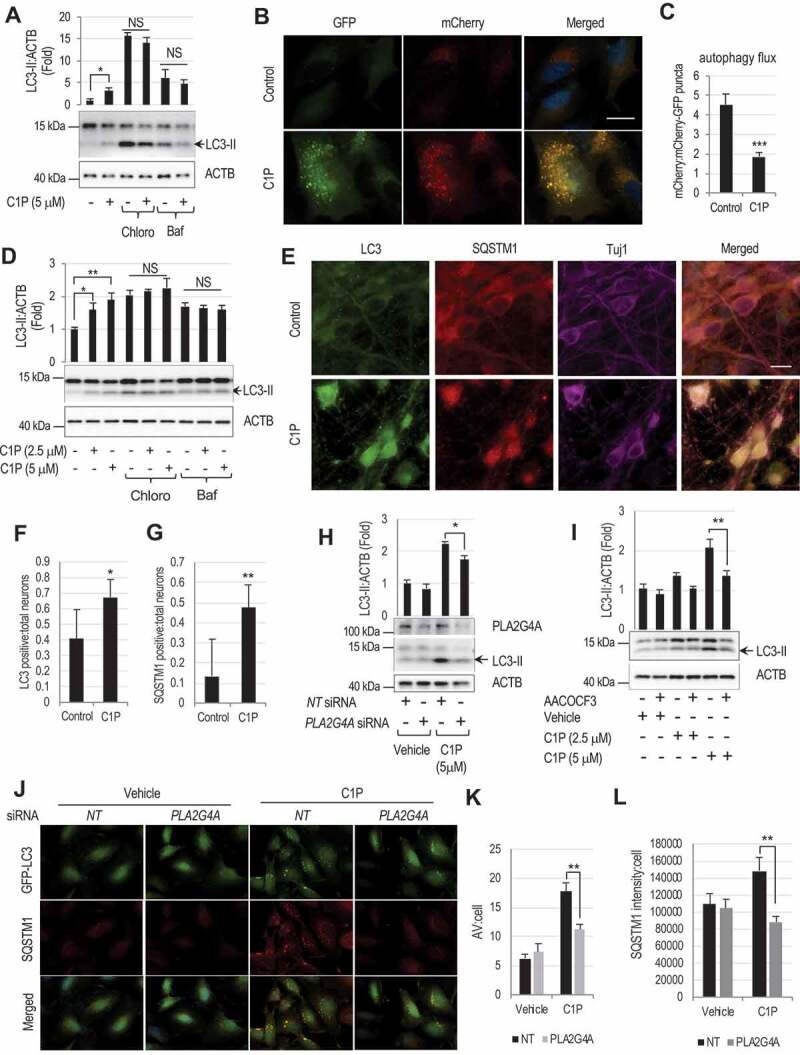
PLA2G4A -mediated lysosomal damage causes inhibition of autophagy flux.
(A) Western blot and corresponding quantification of LC3-II levels in H4 cells treated with lysosomal inhibitors chloroquine or bafilomycin in the presence or absence of C1P (5 μM) for 4 h. Data are mean ± SEM, n = 4; *p < 0.05, (Two-way ANOVA with Bonferroni posttests). (B) Images (60×) of C1P (5 μM) treated H4 cells expressing mCherry-GFP-LC3 reporter and (C) quantification of mCherry to mCherry-GFP puncta ratio, indicating inhibition of autophagy flux in C1P treated cells. Scale bar: 20 μm. Data are mean ± SEM, n = 4; *p < 0.05, (Students’ t-test). (D) Western blot and corresponding quantification of LC3-II levels in rat cortical neurons treated with chloroquine or bafilomycin in the presence or absence of C1P (2.5 or 5 μM) for 4 h. Data are mean ± SEM, n = 3; *p < 0.05, **p < 0.01, (Two-way ANOVA with Bonferroni posttests). (E-G) Images (60×) of C1P (5 μM) treated rat cortical neurons stained with antibodies against LC3, SQSTM1 and TUBB3/Tuj1 (E) and quantification of LC3 (F) and SQSTM1 (G) positive neurons. Scale bar: 10 μm. Data are mean ± SEM, n = 6; *p < 0.05, **p < 0.01 (Students’ t-test). (H-I) Knock down or inhibition of PLA2G4A prevent C1P induced accumulation of autophagosomes. (H) Western blot and corresponding quantification of LC3-II in C1P (5 μM) treated H4 cells transfected either with non-targeting (NT) or PLA2G4A (PLA2G4A) siRNAs. Data are mean ± SEM, n = 4; *p < 0.05, (Two-way ANOVA with Bonferroni posttests). (I) Western blot and corresponding quantification of LC3-II in rat cortical neurons pretreated with AACOCF3 (20 μM) for 90 min and then treated with C1P (2.5 μM) for 4 h. Data are mean ± SEM, n = 4; **p < 0.01, (Two-way ANOVA with Bonferroni posttests). (J-L) PLA2G4A knockdown prevents inhibition of autophagy flux in H4 cells. Images (60×) of C1P (5 μM) treated GFP-LC3 expressing H4 cell stained with antibodies against SQSTM1 (J) and quantification of GFP-LC3-positive autophagosomes (AV) per cell (K) and SQSTM1 intensity per cell (L). Scale bar: 20 μm. Data are mean ± SEM, n = 3; **p < 0.01, (Two-way ANOVA with Bonferroni posttests).
We observed a similar dose dependent increase in LC3-II levels in primary rat cortical neurons treated with C1P while the upstream autophagy regulators remained unchanged (Fig. S5A-B). This was primarily due to impairment of lysosomal degradation as evidenced by failure of C1P to further increase LC3-II levels in the presence of chloroquine or bafilomycin (Figure 4D). Consistently, we observed accumulation of both LC3 and the autophagy substrate SQSTM1 by IF in neurons treated with C1P as compared to controls (Figure 4E-G). Similar accumulation of autophagosomes and SQSTM1 was observed in cortical neurons from GFP-LC3 transgenic mice (Fig. S5C-E). Thus, activation of PLA2G4A by C1P treatment leads to inhibition of autophagy flux in neuronal cells.
To confirm that impairment of autophagy flux after C1P treatment is dependent on PLA2G4A activation we knocked down PLA2G4A in H4 cells. We observed significant attenuation in LC3-II accumulation after C1P treatment in cells transfected with PLA2G4A siRNA as compared to non-targeting siRNA controls (Figure 4H). This was confirmed in rat cortical neurons, where treatment with PLA2G4A inhibitor arachidonyl trifluoromethyl ketone (AACOCF3), attenuated C1P-induced increase in LC3-II level as compared to vehicle controls (Figure 4I). AACOCF3 treatment was also able to decrease LC3-II accumulation in control H4 cells treated with C1P but not in PLA2G4A knockdown cells (Fig. S5F), confirming that the inhibitor function is dependent on the presence of PLA2G4A. Finally, PLA2G4A knockdown decreased accumulation of autophagosomes and SQSTM1 in GFP-LC3 expressing H4 cells treated with C1P (Figure 5J-L). Therefore, even partial knockdown of PLA2G4A is sufficient to substantially attenuate impairment of autophagy flux following C1P treatment.
Figure 5.
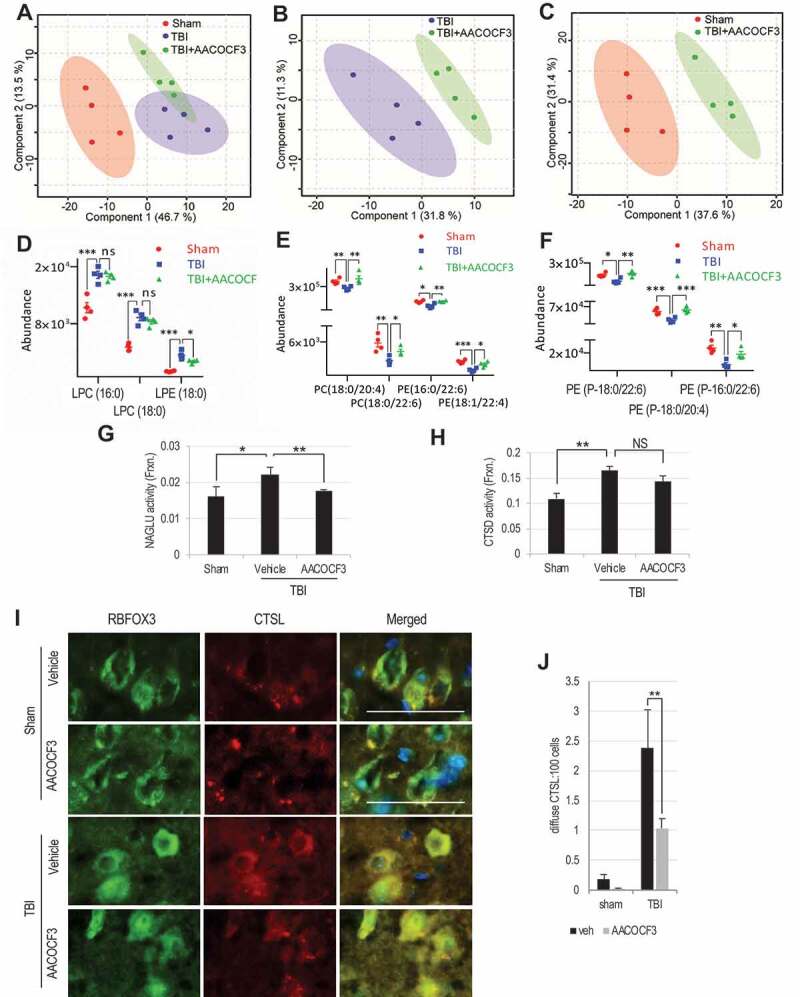
PLA2G4A inhibition normalizes lysosomal membrane lipid composition and attenuates lysosomal membrane permeabilization after TBI.
(A-C) Comparison of positive ion mode UPLC-HDMS of lysosomal membrane prepared from the cortices of sham, TBI and TBI+AACOCF3 using Partial Least Squares – Discriminate Analysis (PLS-DA) plot. Each point represents data set from an individual animal; sham (red), TBI (blue), TBI+AACOCF3 (green). The 95% confidence intervals are indicated by elliptical shaded areas surrounding each group. (A) PLS-DA plot comparing Sham, TBI, and TBI+AACOCF3 in positive ion mode UPLC-HDMS; R2 = 0.97, Q2 = 0.32. (B) PLS-DA plot comparing TBI and TBI+AACOCF3 in positive ion mode UPLC-HDMS; R2 = 0.92, Q2 = 0.34. (C) PLS-DA plot comparing Sham and TBI+AACOCF3 in positive ion mode UPLC-HDMS; R2 = 0.95, Q2 = 0.64. Data were sum normalized, log transformed, and mean centered. n = 4 animals/group. Plots generated using MetaboAnalyst. (D-F) Abundance of specific classes of phospholipids is normalized in the lysosomal membranes from AACOCF3 treated TBI mice as compared to vehicle treated TBI controls. Individual data points as well as mean ± SEM are indicated. Sham (red), TBI (blue), and TBI+AACOCF3 (green). n = 4 animals/group. (D) LPC/LPE abundance. Calculated p-values for TBI to TBI+AACOCF3 were 0.7124 (LPC(16:0)), 0.3140 (LPC(18:0)), and 0. 0366 (LPE(18:0)). (E) PC/PE abundance. Calculated p-values for TBI to TBI+AACOCF3 were 0.0253 (PC(18:0/20:4)), 0.0224 (PC(18:0/22:6)), 0.0071 (PE(16:0/22:6)), and 0.0130 (PE(18:1/22:4)). (F) Ether PE abundance. Calculated p-values for TBI to TBI+AACOCF3 were 0.0074 (PE(P-18:0/22:6)), 0.0020 (PE(P-18:0/20:4)), and 0.0209 (PE(P-18:0/22:6)). (G and H) Activity of lysosomal enzymes (G) NAGLU and (H) CTSD in the cytosolic fraction from sham and TBI mouse cortices. Data are mean ± SEM, n = 8–10 animals/group; *p < 0.05, **p < 0.01 (One-way ANOVA with Turkey’s multiple comparison test). (I-J) IF analysis demonstrating decreased cytoplasmic leakage of CTSL in TBI+AACOCF3 as compared to TBI cortex. (I) Images (20×) of cortical brain sections from sham, sham+AACOCF3, TBI and TBI+AACOCF3 mice stained with antibodies against neuronal marker RBFOX3/NeuN (green) and CTSL (red). Scale bar: 50 μm (J) Corresponding quantification of cells with diffused (cytosolic) CTSL staining. Data are mean ± SEM, n = 3 for vehicle or AACOCF3 treated sham and 5 for vehicle or AACOCF3 treated TBI mice; *p < 0.05, (Two-way ANOVA with Bonferroni posttests).
PLA2G4A activity releases arachidonic acid from the membrane, which is then converted into eicosanoids like prostaglandins (PG) or leukotrienes (LT) by the downstream enzymes, cyclooxygenase and lipoxygenase, respectively. To determine whether C1P-induced block of autophagy flux is mediated through these eicosanoids, we treated H4 cells with arachidonic acid, HPGDS/PGD2, PTGER3/PGE2, LTB4R2/LTB4, LTD4 or LTE4. We did not detect any changes in LC3-II levels in the cells treated with these eicosanoids as compared to the corresponding controls (Fig. S5G-L), suggesting that downstream products generated following PLA2G4A activation are not involved in autophagy impairment. Hence PLA2G4A activation leads to the impairment of autophagy flux by directly increasing LMP rather than through action of its downstream metabolites.
Inhibition of PLA2G4A attenuates TBI-induced lysosomal membrane damage
To determine if inhibition of PLA2G4A function could attenuate lysosomal membrane damage and increase in LMP in vivo, we pre-treated mice before TBI or sham surgery with AACOCF3. As expected [32,47], AACOCF3 treatment significantly decreased PLA2G4A activity in cortical tissue lysates of TBI mice as compared to vehicle treated controls (Fig. S6A). At 1 h after surgery we purified cortical lysosomes and analyzed lipid content of isolated lysosomal membranes by LC-MS/MS. Our data revealed clear differences in the lysosome lipid profiles between sham, TBI treated with vehicle and TBI treated with AACOCF3 groups (Figure 5A-C; Fig. S6B-D; Table S2 & S3). Importantly, significant differences were detected between lysosomal lipid profiles of AACOCF3 versus vehicle treated TBI groups (Figure 5B; Fig. S6C; Table S2). Conversely, AACOCF3 treatment decreased the number of differences between lysosomal lipid profiles of sham and TBI animals (146 differences between sham and TBI in Table S1 and 24 differences between sham and TBI+AACOCF3 in Table S3). However, some differences between sham and TBI+AACOCF3 lysosomal lipid profiles persisted (Figure 5C; Fig. S6D; Table S3), suggesting that PLA2G4A inhibitor provides partial but not total protection to the lysosomal membranes following TBI.
PLA2G4A cleaves glycerophospholipid fatty acyl linkages resulting in increased lysophospholipid membrane abundance. AACOCF3 treatment of TBI mice led to a significant decrease in LPE (Figure 5D) in cortical lysosomal membranes as compared to vehicle treated TBI controls. We also detected slightly lower lysosomal LPC levels in the TBI+AACOCF3 mice, this however failed to reach significance as compared to TBI+vehicle controls (Figure 5D), suggesting potential involvement of additional phospholipases (Table S2 & S3). On the other hand, we observed normalization in the levels of glycerophospholipids following AACOCF3 treatment (Figure 5E-F). In particular, the abundance of PC(18:0/20:4) and PE(16:0/22:6) in the TBI+AACOCF3 cohort was indistinguishable from sham, as opposed to the significant decrease observed between sham and TBI+vehicle mice (Figure 5E). Furthermore, PLA2G4A inhibitor resulted in significantly higher abundance of ether PE as compared to TBI+vehicle mice (Figure 5F). Ether lipids have been suggested to play a role as sacrificial antioxidants, preventing peroxidation of other membrane lipids, and a membrane reservoir for poly-unsaturated fatty acids (e.g., arachidonic acid) [48,49]. Decreased lysosomal ether PE abundance as observed in lysosomes from TBI mice potentially increases vulnerability of membranes to oxidative stress. The fact that AACOCF3 treatment restored lysosomal ether PE levels to those similar to sham mice suggests that PLA2G4A inhibitor not only prevents a general decrease in lysosomal membrane glycerophospholipids but also provides protection to specific lipids such as ether lipids after TBI.
Next, we assessed if PLA2G4A inhibition can prevent TBI-induced increase in LMP. We measured activity of the lysosomal enzymes in isolated lysosomal and cytosolic fraction from AACOCF3 or vehicle treated sham and TBI mouse cortices. AACOCF3 treatment did not alter CTSD or NAGLU activity in sham mice (Fig. S6E-F). Total and lysosomal activity of NAGLU and CTSD was also not affected by AACOCF3 treatment of TBI mice (Fig. S6G-J). However, the TBI-induced increase in cytosolic activity of NAGLU was significantly lower in TBI+AACOCF3 group as compared to TBI+vehicle (Figure 5G). Although the decrease in cytosolic CTSD activity did not reach significance (Figure 5H), we observed significantly less diffuse CTSL staining in the neuronal cytoplasm in TBI+AACOCF3 mice as compared to TBI+vehicle controls (Figure 5I-J). Therefore, PLA2G4A inhibition by AACOCF3 treatment can significantly attenuate lysosomal membrane damage and LMP after TBI.
Inhibition of PLA2G4A attenuates autophagy impairment and improves functional outcomes after TBI
We assessed whether PLA2G4A inhibition can prevent block of autophagy flux following TBI. While LC3-II levels increased in the brain of both AACOCF3 and vehicle treated TBI mice, AACOCF3 significantly attenuated accumulation of SQSTM1 (Figure 6A-B, Fig. S6K), suggesting increase in autophagy flux. AACOCF3 treatment did not affect LC3-II or SQSTM1 levels in sham mice (Fig. S6L-M). Consistent with the western blot data, IF analysis revealed lower SQSTM1 levels in cortical neurons of AACOCF3 treated TBI mice as compared with TBI+vehicle controls (Figure 6C-D). While LC3-II levels are indicative of the number of autophagosomes, a decrease in SQSTM1 level reflects the extent of autophagy flux. Thus, these data suggest that AACOCF3 mediated PLA2G4A inhibition attenuates block of autophagy flux after TBI.
Figure 6.
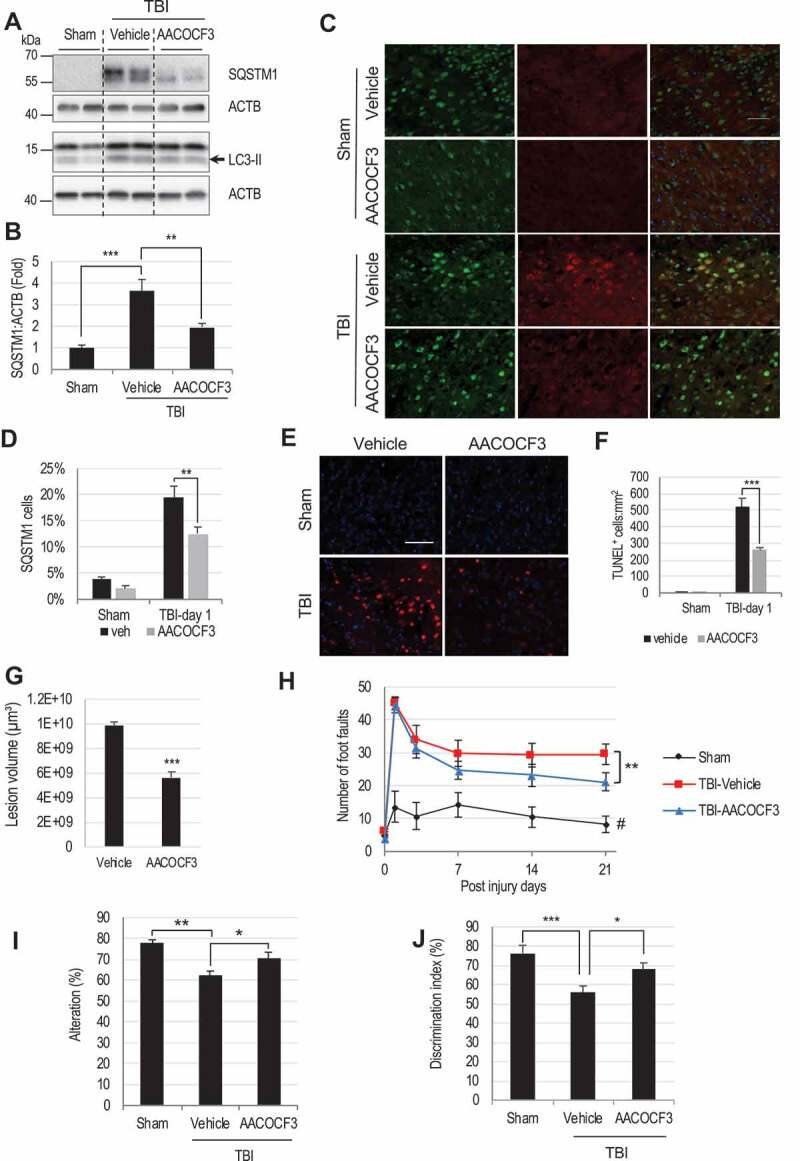
PLA2G4A inhibition restores autophagy flux, attenuates cortical cell death and improves motor and cognitive function in mice after TBI.
(A) Western blot of SQSTM1 and LC3 in sham, TBI and TBI+AACOCF3 cortical lysates and (B) Quantification of SQSTM1 level. Data are mean ± SEM, n = 6 for sham and 10 for vehicle or AACOCF3 treated TBI mice; **p < 0.01, ***p < 0.001, (One-way ANOVA with Turkey’s multiple comparison test). (C and D) IF analysis demonstrating decreased SQSTM1 accumulation in AACOCF3 treated TBI cortical neurons as compared to TBI controls. (C) Images (20×) of cortical brain sections of sham, sham+AACOCF3, TBI and TBI+AACOCF3 mice stained with antibodies against neuronal marker RBFOX3/NeuN and SQSTM1. Scale bar: 50 μm. (D) Quantification of SQSTM1 positive cells. Data are mean ± SEM, n = 3 for vehicle or AACOCF3 treated sham and 5 for vehicle or AACOCF3 treated TBI mice; *p < 0.05, ***p < 0.001, (Two-way ANOVA with Bonferroni posttests). (E) Images (20×) demonstrating decreased cell death (TUNEL) in cortical brain sections from TBI+AACOCF3 as compared to TBI mice. Scale bar: 50 μm (F) Quantification of TUNEL positive cells. Data are mean ± SEM, n = 3 for vehicle or AACOCF3 treated sham and 5 for vehicle or AACOCF3 treated TBI mice; ***p < 0.001, (Two-way ANOVA with Bonferroni posttests). (G) Quantification of lesion volume (Cavalieri method) at 28 days post injury in TBI and TBI+AACOCF3 mouse cortices. Representative images are included in the Fig. S6N. Data are mean ± SEM, n = 7 for vehicle treated and 8 for AACOCF3 treated TBI mice; ***p < 0.001, vs. vehicle treated TBI group. (Students’ t-test). (H-J) AACOCF3 treatment leads to improved functional outcomes after TBI. (H) Assessment of sensorimotor function of sham, TBI+vehicle and TBI+AACOCF3 mice at days 1, 3, 7, 14 and 21 post injury using the beam-walk test. Day 0 represents baseline prior to injury. AACOCF3 treatment of TBI mice led to improvement in motor function vs. TBI+vehicle controls, starting from day 14 (p = 0.0513) and increasing through day 21 (**p = 0.0047). Data are mean ± SEM, n = 11 sham, 14 TBI+vehicle and 14 TBI+AACOCF3 mice. Significant effects of injury and AACOCF3 treatment [F(2,26) = 18.41; p < 0.0001] were detected across all time points [F(5,65) = 105.8, p < 0.0001] except day 0. (Two-way repeated-measures ANOVA with Turkey’s multiple comparison test). (I) Spatial memory assessment using Y-maze spontaneous alternation test at 7 day after TBI. TBI led to significantly reduced percentages of spontaneous alternation (**p < 0.01 vs. sham). AACOCF3 treatment significantly increased percentage of spontaneous alternation compared with the vehicle treated TBI group (*p < 0.05). Data are mean ± SEM, n = 11 sham and 14 TBI and TBI+AACOCF3 mice, (One-way ANOVA, with Turkey’s multiple comparison test). (J) Memory retention assessment by novel object recognition (NOR) in mice at day 22 after TBI. All groups spent equal time with the two identical objects during the sample phase (Fig. S6O). AACOCF3 treated TBI mice spent significantly more time with the novel vs. familiar object as compared with vehicle treated TBI group (*P < 0.05). Significant differences were also observed between the sham and vehicle treated TBI group (**P < 0.01). Data are mean ± SEM, n = 11 sham and 14 TBI and TBI+AACOCF3 mice, (One-way ANOVA with Turkey’s multiple comparison test).
We previously demonstrated that disruption of autophagy is associated with neuronal cell death in the cortex and hippocampus following TBI [8]. To determine if PLA2G4A inhibition can prevent cortical cell death at 1 day after injury, we performed TUNEL assay. As expected, markedly higher numbers of TUNEL-positive cells were detected in all TBI brains as compared to sham controls (Figure 6E-F). However, the AACOCF3 treated TBI brains had significantly fewer TUNEL-positive cells than TBI+vehicle controls (Figure 6E-F), confirming attenuation of cell death. Long-term decrease in cell loss was further supported by significantly lower lesion volume in the AACOCF3 treated TBI mice as compared to vehicle treated groups at 28 days after injury (Figure 6G; Fig. S6N). Thus, PLA2G4A inhibition by AACOCF3 treatment significantly restricted cell loss after TBI.
Loss of both cortical and hippocampal neurons following moderate or severe TBI contributes to long-term functional impairment. To determine if PLA2G4A inhibition can improve functional outcomes in the injured mice, we assessed motor coordination by beam walk test immediately before and on days 1, 3, 7, 14 and 21 post-injury. We observed a trend towards improvement in beam walk performance in AACOCF3 treated as compared to vehicle treated TBI mice starting from day 14 (p = 0.0513) and reaching significance by day 21 after injury (p = 0.0047) (Figure 6H), suggesting that AACOCF3 treatment improved motor function in injured mice. Cognitive function was assessed by the Y-maze alternation test to measure spatial working memory, and the novel object recognition (NOR) test to measure non-spatial learning and memory. Spontaneous alternation between the arms of the Y-maze were significantly increased in AACOCF3 as compared to vehicle treated TBI mice suggesting better recovery of spatial working memory (Figure 6I). In the novel object recognition test we observed that AACOCF3 treated mice spent considerably longer time with the novel object versus familiar object as compared to the vehicle treated TBI group (Figure 6J; Fig. S6O). Therefore, PLA2G4A inhibitor AACOCF3 can improve memory retention among injured mice. AACOCF3 treatment did not have any effect on functional outcomes in sham mice (Fig. S6P-R).
Taken together these results indicate that PLA2G4A inhibitor can prevent lysosomal damage and subsequent autophagy disruption after TBI, thus restricting brain cell loss and improving neurological function in injured mice.
PLA2G4A contributes to amyloid-β-induced lysosomal damage
Lysosomal damage, inhibition of autophagy and PLA2G4A activation have been reported in age-related neurodegenerative diseases including Alzheimer disease (AD), neurodegenerative lysosomal storage diseases and following spinal cord injury [9,10,14,22–24,26,29]. In particular, increased levels of PLA2G4A phosphorylation have been demonstrated in a mouse models of AD and in post mortem human AD brain [28]. To determine if PLA2G4A may be involved in lysosomal damage induced by the Alzheimer disease associated pathogenic peptide amyloid-β, we treated H4 and HeLa cells with amyloid-β1–40. We observed rapid increase in PLA2G4A phosphorylation, followed by accumulation of LC3-II and SQSTM1 indicative of autophagy flux inhibition (Figure 7A; Fig. S7A-D). While levels of pro and mature CTSD remained unaltered (Fig. S7E), Amyloid-β treatment led to lysosomal membrane damage and an increase in LMP as indicated by a significant decrease in co-localization of dextran to lysosomes in amyloid-β treated H4 cells (Figure 7B-C). Similar to PLA2G4A inducer C1P, amyloid-β treatment also caused accumulation of abnormally enlarged lysosomes, many completely devoid of dextran staining (Figure 7B and Fig. S7F-G). Lysosomal dextran leakage induced by amyloid-β was attenuated in cells transfected with PLA2G4A siRNA as compared to non-targeting siRNA (Figure 7B-C), confirming involvement of PLA2G4A in amyloid-β induced LMP. Furthermore, accumulation of autophagosomes and SQSTM1 following amyloid-β treatment was attenuated in GFP-LC3 H4 cells transfected with PLA2G4A siRNA as compared to non-targeting siRNA controls (Figure 7D-F). Similarly, PLA2G4A inhibitor AACOCF3 decreased accumulation of LC3-II in amyloid-β treated rat cortical neurons (Figure 7G-H). Taken together, these data demonstrate that lysosomal membrane damage and inhibition of autophagy flux induced by amyloid-β is mediated at least in part through PLA2G4A activation and suggest that PLA2G4A may contribute to lysosomal dysfunction not only in TBI but also in other neurodegenerative diseases.
Figure 7.
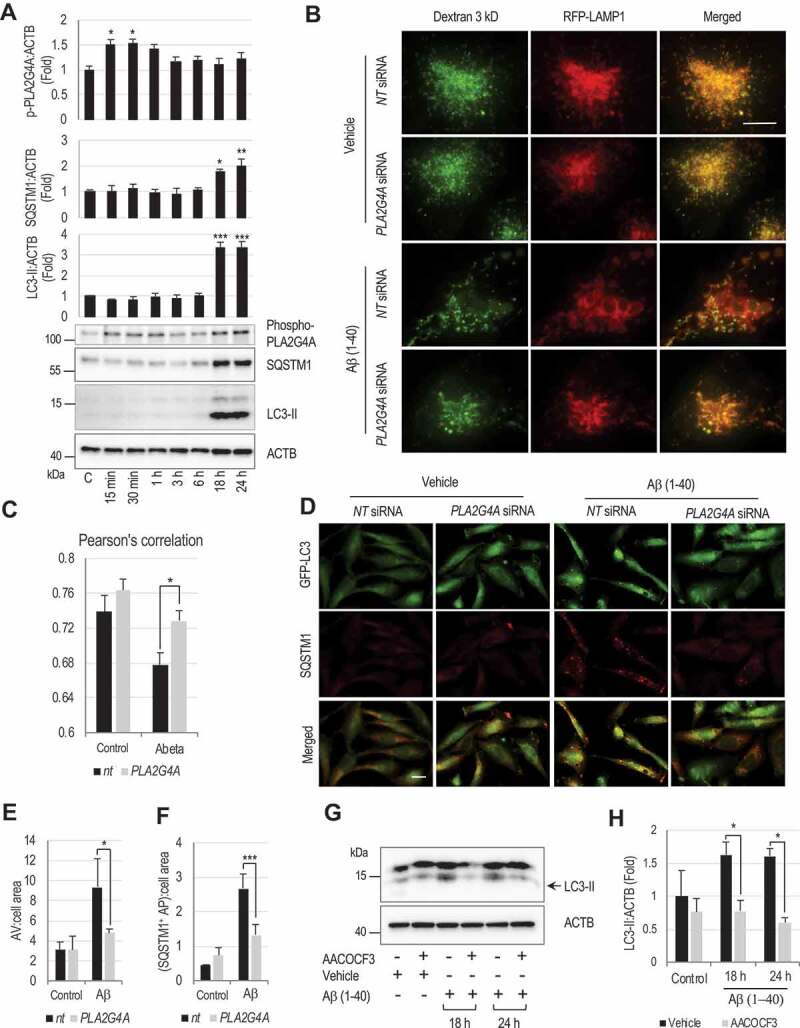
Amyloid-β activates PLA2G4A leading to LMP and subsequent autophagy impairment.
(A) Western blot demonstrating activating phosphorylation of PLA2G4A, and accumulation of SQSTM1 and LC3-II in amyloid-β(1–40) (5 μM) treated H4 neuroglioma cells and corresponding quantification. Data are mean ± SEM, n = 3, *p < 0.05, **p < 0.01, ***p < 0.001, (One-way ANOVA with Turkey’s multiple comparison test). (B and C) Amyloid-β-induced PLA2G4A activation causes lysosomal abnormalities and LMP. (B) Images (60×) of H4 cells expressing RFP-LAMP1 treated with amyloid-β(1–40) (5 μM) or vehicle control. Cells were transfected with either non-targeting (NT) or PLA2G4A siRNA for 48 h, loaded with Alexa Fluor 488-dextran (3 kDa) and then treated with amyloid β(1–40) for 18 h. Enlarged lysosomes with low dextran levels were detected in amyloid-β treated cells. Lysosomal abnormalities were attenuated in cells transfected with PLA2G4A siRNA. Scale bar: 10 μm. (C) Pearson’s correlation analysis of Alexa fluor 488 dextran co-localization to RFP-LAMP1 lysosomes. (*p < 0.05), n = 10. (D-F) Knock down of PLA2G4A attenuated inhibition of autophagy flux induced by amyloid-β treatment. (D) Images (20×) of GFP-LC3 expressing H4 cells treated amyloid-β for 24 h and stained with antibody against SQSTM1. Cells were transfected with either NT or PLA2G4A siRNA 48 h prior to treatment. Scale bar: 20 μm. (E-F) Quantification of (E) GFP-LC3 positive autophagic vesicles (AV) and (F) SQSTM1 positive cells. Data are mean ± SEM, n = 3; *p < 0.05, ***p < 0.001, (Two-way ANOVA with Bonferroni posttests). (G and H) PLA2G4A/inhibition prevents amyloid-β-induced autophagosome accumulation in rat cortical neurons. (G) Western blot of LC3 in rat cortical neuron cells treated with amyloid β in presence or absence of AACOCF3 for 18 or 24 h. (H) Corresponding quantification of LC3-II. Data are presented as mean ± SEM, n = 4; *p < 0.05, (Two-way ANOVA with Bonferroni posttests).
Discussion
Lysosomes are major sites of intracellular degradation, with both autophagosomal and endosomal pathways converging to deliver intracellular and extracellular contents to the highly acidic lysosomal compartment. This highly digestive environment is surrounded by a single membrane which allows both maintenance of the unique environment necessary for lysosomal function and isolation of potentially toxic lysosomal contents from the rest of the cell. The barrier function of lysosomal membrane is impaired under many pathological conditions, leading to the release of lysosomal contents into the cytosol and allowing neutral cytosolic solution to enter into the lysosomal lumen [20–24]. Although various processes, including membrane oxidation, accumulation of sphingosine and insertion of pore forming proteins such as BAX [23,50], have been suggested to participate in LMP, the molecular mechanisms how the phospholipid membrane of lysosome is destabilized under pathological stress remain unclear. In this study we demonstrate that activated cytosolic phospholipases, and in particular PLA2G4A, can directly hydrolyze the fatty acyl linkage of the lysosomal membrane phosopholipids, thereby destabilizing the lysosomal membrane. Our data also show that PLA2G4A is a major contributor to LMP in the mouse cortex following TBI as well as is involved in mediation of amyloid-β-induced LMP.
PLA2G4A -mediated hydrolysis of membrane phospholipids can alter membrane lipid composition, straining the membrane structural integrity and leading to LMP in several ways. Lysophospholipids generated following PLA2G4A -mediated hydrolysis of membrane phospholipids have detergent-like properties and can directly lead to pore formation [34]. PLA2G4A activation following TBI is also associated with decreased levels of both ether glycerophospholipids and diacyl-glycerophospholipids in the lysosomal membrane. Whereas perturbation of both types of glycerophospholipids can affect membrane structure and fluidity, decrease in ether lipids may be particularly detrimental [49]. Moreover, ether lipids are known to have free radical scavenging properties [48,49]. Lower levels of ether glycerophospholipids would likely make the lysosomal membrane more vulnerable to additional insults, such as reactive oxygen species (ROS) and other oxidative stressors generated at the injury site. Thus, PLA2G4A activation after brain injury may not only cause direct loss of lysosomal membrane integrity, but also facilitate further damage by making the membrane more vulnerable to oxidation. We hypothesize that similar mechanisms may contribute to potentiation of lysosomal damage and LMP in other conditions associated with both activation of PLA2G4A and increased levels of ROS, such as AD and spinal cord injury [26,28–30,32].
Although our investigation focuses on the influence of TBI-induced lysosomal damage on the inhibition of autophagy flux, the cellular impact of LMP is much broader. In addition to the impact on lysosome-dependent cellular processes such as autophagy and endocytosis, release of lysosomal contents per se can be highly toxic to the cell. Although the optimum pH for lysosomal protease activity is acidic, many can remain functional in the neutral pH of the cytosol and cause severe damage to the cytoplasmic components including other organelles, thus potentiating cellular dysfunction and promoting cell death. Furthermore, damaged cellular organelles are normally removed through the autophagy-lysosomal system. When lysosomal function is impaired cells fail to eliminate their damaged components, which in turn can exert additional insult. Lysosomes also participate in plasma membrane repair through exocytosis [51,52], which would similarly be predicted to be negatively affected by LMP. The lysosomal membranes also serve as docking platforms for many signaling molecules including MTOR. Thus, an alteration in the lysosomal membrane lipid composition could affect membrane affinity and/or activity of MTOR and other lysosome-associated signaling components.
PLA2G4A shows cell type specific activation in the cortex following TBI. Our data indicate that the initial neuron-specific activation of PLA2G4A is involved in the lysosomal damage and subsequent neuronal cell death, whereas its later activation (day 3) is predominantly associated with inflammatory responses in microglial cells. Early activation of PLA2G4A is likely mediated by the upstream kinases like ERK1/2 and p38 MAPK [53], which are rapidly activated in response to injury-induced glutamate excitotoxicity and ROS accumulation [2,5,7,53]. Additionally, a rapid increase in intracellular calcium occurs soon after injury and also likely contributes to the activation and membrane translocation of PLA2G4A. On the other hand, activation of PLA2G4A in microglia and/or infiltrating macrophages at the later time (3 days) after TBI is likely a response to inflammatory signaling [3,5].
LMP and subsequent cytosolic leakage of lysosomal enzymes after TBI was attenuated by treatment with PLA2G4A inhibitor AACOCF3. AACOCF3 inhibits PLA2G4A by forming covalent bond at Serine 228 of is active site. It can also weakly inhibit PLA2G2A/sPLA2 and PLA2G6/iPLA2. However, it is 4 orders of magnitude less potent against these phospholipases as compared to PLA2G4A [54]. Additionally, C57/BL6 mice used in this study have a null mutation in the gene encoding PLA2G2A/sPLA2 [55], thus eliminating the possibility of its involvement in the lysosomal damage. While PLA2G6/iPLA2 is expressed in the mouse brain, its function is downregulated following TBI [56,57]. Thus, the protective effect of AACOCF3 in TBI mice is likely the result of inhibition of PLA2G4A, although we cannot exclude the possibility of additional off target effects. As AACOCF3 did not increase the overall lysosomal enzyme activity in the injured cortex, other mechanisms affecting lysosomal function must also occur after TBI. One possibility, suggested by the partial protection against TBI-induced alteration in lysosomal membrane lipid composition by AACOCF3 treatment, is potential involvement of other phospholipases and/or other mechanisms of lipid damage such as ROS, in lysosomal membrane damage following injury. Another possibility could be lower expression of lysosomal enzymes after injury. Lower lysosomal biogenesis has been reported in neurodegenerative diseases due to decrease in expression levels and activity of the TFEB (transcription factor EB), a master regulator of lysosomal gene expression. TFEB function is negatively regulated by MTOR, which activity is increased after TBI [58,59]. Since mTOR regulation occurs at the lysosome [60], lysosomal damage after TBI could contribute to this dysregulation, thus lowering activity of TFEB and expression of lysosomal proteins in the injured cortex through a regulatory feedback mechanism. Lysosomes are also involved in membrane repair through exocytosis. Thus, another potential explanation for lower lysosomal enzyme levels could be increased lysosomal exocytosis for plasma membrane repair following AACOCF3 treatment. Nevertheless, our data indicate that PLA2G4A inhibition is able to sufficiently improve lysosomal function to allow restoration of autophagy flux and attenuation of cell death after TBI.
Both PLA2G4A activation and increased LMP are observed in many neurodegenerative disorders, including AD [40]. In the current study we demonstrate that PLA2G4A activation is involved in LMP not only in TBI but also in response to amyloid-β treatment, a cellular model relevant to AD. This suggests that PLA2G4A -mediated LMP could contribute to autophagy-lysosomal defects observed in AD and potentially other age-related neurodegenerative diseases. Inhibition of PLA2G4A has been previously reported to be beneficial in a mouse model of AD by suppressing pathological arachidonic acid-mediated signaling [28]. Our data suggest that attenuation of lysosomal membrane damage and inhibition of autophagy flux could also contribute to the benefits of PLA2G4A inhibition in this model. More generally, our data support existence of common mechanisms contributing to neuronal cell damage and death triggered by both acute (such as TBI) and chronic insults (such as amyloid-β accumulation in AD). The defined insult followed by acute and coordinated wave of neuronal cell damage and death in the mouse TBI model made it possible to identify in vivo changes in lysosomal membrane lipid composition and to dissect events leading from PLA2G4A activation to LMP to inhibition of autophagy and neuronal cell death. Thus, studying the mechanism of acute neuronal death after TBI might also provide insight to understand progressive neurodegeneration in chronic neurodegenerative diseases.
Materials and methods
Controlled cortical impact
All surgical procedures and animal experiments were performed in accordance with the protocols approved by the Animal Care and Use Committee of the University of Maryland. Controlled cortical impact (CCI) induced TBI was performed in male C57BL6/J mice (20–25 g) or transgenic C57BL6/J mice expressing GFP-LC3 [61] or Cx3cr1-GFP (The Jackson Laboratory, 005582) [62] reporters as described previously [63]. A 10-mm midline incision was made over the skull, the skin and fascia were retracted, and a 4-mm craniotomy was made on the central aspect of the left parietal bone of mice under surgical anesthesia (2–3% isoflurane evaporated in a gas mixture containing 70% N2O and 30% O2). Moderate injury was induced by a custom microprocessor-controlled and compressed air driven pneumatic impactor of 3.5-mm diameter tip with the impact velocity of 6 m/s and a deformation depth of 2 mm. In sham animals, same procedure was performed except for the impact.
AACOCF3 injection
AACOCF3 (Enzo, BML-ST335-0050) was dissolved in ethanol to make 40-mM stock solution which was then diluted to 4-mM concentration in saline containing 0.1% DMSO (American Bio, AB00435). AACOCF3 solution (200 μl of 4 mM) was administered intraperitoneally in mice 1 h before, immediately and 3 h after TBI. DMSO (0.1%) containing saline with same volume of ethanol as AACOCF3 solution used to prepare diluted solution, was used as vehicle. For longer study mice were injected three times as mentioned here on the day of surgery followed by once a day on day 2 and 3 and then once in every alternate day till day 7.
Lesion volume
Lesion volume was assessed in injured mouse brain at 28 days after injury as previously described [63]. Briefly, 60-μm sections located approximately 250 μm apart across the entire lesion volume were stained with cresyl violet (FD NeuroTechnologies, Columbia, MD, USA, PS102-2); images were acquired on Leica DM4000 B TL (BF) microscope and lesion volume was quantified by Cavalieri method of unbiased stereology using Stereologer 2000 program software (Systems Planning and Analysis, Alexandria, VA, USA).
Behavioral assessment
Beam walk test: Motor co-ordination was evaluated in sham and vehicle or AACOCF3 treated mice using beam walk test as described previously [63]. Test was performed at day 0 (before injury), 1, 3, 7, 14 and 21 days after TBI.
Novel object recognition (NOR) test: Hippocampal spatial memory was assessed using Novel object recognition (NOR) test as per the method described previously [63]. The test was performed on 21 and 22 post injury days. Relative time spent with the novel object over total time spent with novel and familiar object is expressed as discrimination index.
Y-maze test: Y-maze test was performed to assess exploratory behavior and spatial temporary memory in sham and vehicle or AACOCF3 treated TBI mice. This test was performed on day 7 as per the method described previously [63].
Subcellular fractionation and preparation of lysosome enriched fraction
Lysosomal and cytosolic fractions were prepared from cortices of sham and injured mice as described previously [8]. Highly enriched lysosomal fraction was prepared from the mouse cortical tissues using Lysosomal purification kit (Thermo Fisher Scientific, 89839) as per the manufacturer’s instruction.
Lysosomal enzyme assay
Activities of lysosomal enzymes CTSD (cathepsin D) and NAGLU (alpha-N-acetylglucosaminidase [Sanfilippo disease IIIB]) were determined using fluorometric cathepsin D assay kit from Biovision Inc. (K143-100) and NAGLU assay kit from Sigma-Aldrich (CS0780) as per the manufacturers’ instruction. Enzyme activities in the lysosomal fraction and whole cell or tissue lysate were estimated as change in absorbance or fluorescence per μg of protein. In cytosolic lysosomal enzyme activities were determined as fractions of their activities in total tissue or cell lysate per μg protein of whole tissue or cell lysate.
Immunohistochemistry
Mouse brains fixed with 4% paraformaldehyde (PFA, pH 7.4) and protected in 30% sucrose were cut into 20-μm frozen sections as described previously [8]. In each IF study, for each mouse we used 4 coronal brain sections 1–1.2 mm apart across the lesion area. Sections were blocked with 5% goat or donkey serum (Millipore, S30-100) in 1(X) phosphate-buffered saline (PBS; Quality Biological, INC., 119–069-101) containing 0.025% Triton X-100 (Sigma, T8787), incubated. For lysosomal staining sections were treated with 0.1 M glycine in PBS for 30 min, permeabilized with 0.2% saponin (Sigma, S7900) in PBS for 30 min and blocked with 5% goat or donkey serum and 0.05% BSA (Sigma, S7906) in 0.04% saponin containing PBS [64]. Sections were incubated overnight with primary antibodies at 4ºC and then with secondary antibodies in the blocking solution for 2 h at room temperature. Nuclei were stained with DAPI.
Primary antibodies used include: SQSTM1 (1:200; Progen, GP62-C), RBFOX3/NeuN (1:500; Millipore Sigma, MAB377), CTSD/CathepsinD (1:100, Santa Cruz Biotechnology, sc-6486), CTSL (1:250; R&D Systems, AF1515 and MAB9521), phospho-PLA2G4A (1:400; Sigma-Aldrich, SAB4503812) LAMP2 (1:100; GL2A7, developed by Granger, B.L. and obtained from Developmental Studies Hybridoma Bank, developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biology,Iowa City, IA 52242). Secondary antibodies: Alexa Fluor 488 goat anti-rabbit (A11034), Alexa Fluor 546 anti-rabbit (A11035), Alexa Fluor 546 goat anti-mouse (A11030), Alexa Fluor 568 goat anti-guinea pig (A11075), Alexa Fluor 633 goat anti-mouse (A21052) and Alexa Fluor 546 donkey anti-goat (A11056) were from Invitrogen; Cf633 donkey anti-rat (SAB4600133) and Cf633 donkey anti-guinea pig (SAB4600129) were from Sigma-Aldrich. TUNEL assay was performed on frozen brain sections using ApopTag In Situ Apoptosis Detection Kit (Millipore Sigma, S7165) as per the manufacturer’s protocol.
Immunocytochemistry
Cells cultured on glass coverslip were fixed with 4% PFA, treated with 0.1 M glycine in PBS for 30 min, followed by permeabilization with 0.2% saponin in PBS for 30 min and blocked with 5% goat or donkey serum and 0.05% BSA in 0.04% saponin containing PBS [64]. Coverslips were incubated overnight with primary antibodies at 4ºC and then with secondary antibodies in the blocking solution for 2 h at room temperature. Nuclei were stained with DAPI.
Primary antibodies include: SQSTM1 (1:200; Progen, GP62-C), TUBB3/Tuj1 (1:500; Biolegend, 801202), CTSL (1:250, R&D Systems, AF-1515), LAMP1 (1:200; Abcam, 24170). Secondary antibodies: Alexa Fluor 488 goat anti-rabbit (A11034), Alexa Fluor 546 anti-rabbit (A11035), Alexa Fluor 546 goat anti-mouse (A11030), Alexa Fluor 568 goat anti-guinea pig (A11075), Alexa Fluor 633 goat anti-mouse (A21052) and Alexa Fluor 546 donkey anti-goat (A11056) were from Invitrogen.
Image acquisition and quantification
Images following immunohistochemistry and immunocytochemistry were acquired using a fluorescent Nikon Ti-E inverted microscope, at 20X (CFI Plan APO VC 20X NA 0.75 WD 1 mm) or 60X (CFI Plan APO VC 60X NA 1.4 Oil) magnification and processed and quantified by Nikon Element software as described previously [8]. All 60X images were acquired as z-stacks and focused using Extended Depth of Focus (EDF) module. All images were quantified using custom automated macros in Elements: nuclei were identified using Spot Detection algorithm; cells positive for IF markers were identified using Detect Regional Maxima algorithm, followed by global thresholding. Number of positive cells was normalized to the total number of cells imaged. Intracellular puncta were detected using Spot Detection and normalized to the number of cells imaged. Median cell numbers of ≈ 3,000 (cortical sections) and ≈ 200 (in vitro) were quantified at 20x; ≈ 250 (cortical sections) and ≈ 50 (in vitro) at 60x, per group per condition.
LC-MS
Materials: LC-MS grade acetonitrile (A955-500), methanol (A456-500), water (W6500), isopropanol (A461-500), formic acid (A117-50) and ammonium formate (A11550) were from Fisher Scientific and HPLC grade tert-butyl methyl ether (MTBE) was from Sigma-Aldrich (650560). Lipid standards: PC(17:0/20:4) (lm1002), PE(17:0/20:4) (lm1102), PG(17:0/20:4) (lm1202), PI(17:0/20:4) (lm1502), PS(17:0/20:4) (lm1302), LPC(17:0) (8503600), LPE(17:1) (856707), Cer(d18:1/12:0) (lm22120), and SM(d18:1/12:0) (860583) from Avanti Polar Lipids, Inc. were used as internal standards.
Sample Preparation: Isolated lysosomes were stored at −80°C. Total lipid extracts from the lysosome samples were prepared using MTBE lipid extraction protocol [65] with slight modifications. Briefly, 400 µL of methanol:water (3:1, v/v) was added to the lysosome fraction and homogenized with ceramic beads (Precellys®24-Dual; PeqLab, P000669-PR240-A). Internal standards and 1 mL of MTBE were added followed by room temperature shaking for 1 hour. 250 µL of water was added and left to sit at room temperature for 15 minutes. Phase separation was completed by centrifugation at 10 minutes for 9400 × g. The upper phase was removed and dried at room temperature under nitrogen. The aqueous phase was re-extracted with 200 µL of MTBE. The recovered lipids were reconstituted in 500 µL of chloroform:methanol (2:1, v/v). Prior to analysis samples were further diluted with isopropanol:acetonitrile:water (2:1:1, v/v/v).
Ultra-Performance Liquid Chromatography (UPLC): UPLC was performed on a Waters ACQUITY UPLC system (Milford, MA, 176015000). The separation was achieved using a Waters C18 CSH (1.7 µm; 2.1 × 100 mm) column (Milford, MA, 176850035). UPLC parameters were adopted with slight modifications [66]. Mobile phase A was 10 mM ammonium formate with 0.1% formic acid in water/acetonitrile (40:60, v/v) and mobile phase B was 10 mM ammonium formate with 0.1% formic acid in acetonitrile/isopropanol (10:90, v/v). The gradient was ramped from 40% to 43% B in 2 min, ramped to 50% in 0.1 min, ramped to 54% B in 9.9 minutes, ramped to 70% in 0.1 min, and ramped to 99% B in 5.9 min. The gradient was returned to initial conditions in 0.5 min and held for 1.9 min for column equilibration. The flow rate was 0.4 mL/min. The column was maintained at 55°C and the auto-sampler was kept at 5 °C. A 2 µL injection was used for all samples.
Data-Independent Tandem Mass Spectrometry with Traveling Wave Ion Mobility (HDMS): HDMS experiments were performed with a traveling wave ion mobility-enabled hybrid quadrupole orthogonal acceleration time-of-flight mass spectrometer (SYNAPT G2-S, Waters Corporation, Wilmslow, United Kingdom). HDMS parameters were adopted with slight modifications [67]. The instrument was operated in positive and negative ion mode electrospray ionization. The capillary voltage was 2.0 kV and sampling cone voltage was 30 V. Nitrogen at a flow of 650 L/h was used as the desolvation gas with a constant desolvation temperature of 400°C. The source temperature was set at 125°C. Data were acquired over the m/z range of 100–1800. The mass spectrometer was operated in ion mobility, data-independent (MSE) acquisition for both positive and negative ion modes. During acquisition, the first quadrupole was set to pass all precursor ions with the ‘trap’ T-wave operated at 4 V (T-wave velocity of 315 m/s). The precursor ions were transferred to the traveling wave ion mobility cell and subjected to a nitrogen flow of 80 ml/min (T-wave velocity of 600 m/s). As the ions excited the ion mobility cell they were transferred to the ‘transfer’ T-wave and underwent two discrete and alternating scan functions (T-wave velocity of 190 m/s). The first scan was set at low-collision energy (4 eV) and used to collect precursor ion spectra. The second scan was set at high-collision energy and ramped from 30–55 eV which was used for generation of product ion spectra. Argon gas was used for collision-induced dissociation (CID). Leucine Enkephalin (0.1 mg/mL) (Waters Corporation, 186006013) at a flow rate of 7.5 µL/min was used as the lock-mass to ensure high mass accuracy data acquisition. Poly-DL-alanine (Waters Corporation, 186008113) was used for collisional cross section calibration at a concentration of 10 µg/mL. Data were acquired with MassLynx v4.1 (Waters Corporation, Wilmslow, United Kingdom).
Data Processing/Bioinformatics: UPLC-HDMS data was analyzed with MSE Data Viewer v1.2 (Waters), DriftScope HDMS v2.7 (Waters), Progenesis QI v2.2 (Nonlinear Dynamics, Newcastle, United Kingdom), MetaboAnalyst 3.0 [68,69], and Prism 6 (GraphPad, La Jolla, CA). Raw data files were directly imported into Progenesis QI where retention time alignment, peak picking, deconvolution of adducts, relative abundance, and preliminary identification were performed. Preliminary identification involved accurate mass correlation at a threshold of 10 ppm to LIPIDMAPS (http://www.lipidmaps.org). The processed data generated from Progenesis QI which included peak area and m/z value was exported into MetaboAnalyst for multivariate analysis. Multivariate analysis included principal component analysis (PCA) and partial least square discriminate anlaysis (PLS-DA). Univariate analysis via Prism 6 was performed using normalized values generated from Progenesis QI. Putative and confirmatory structure assignments relied on chromatographic retention time, HDMS, positive and negative ion mass spectral correlation, and for a number of selected lipids, authentic standard verification.
Western blot analysis
For in vivo analysis, 5-mm tissue of ipsilateral cortex around the injury area from TBI mice or the same region and volume from sham mice were dissected and processed as described [8]. Cell lysates were prepared by lysing the H4 neuroglioma, HeLa and rat cortical neuron cultured in 24-well plates with SDS-PAGE buffer. Cell or tissue lysates were resolved on 4–20% SDS-PAGE gels (Bio-Rad Laboratories, 5671095) and transferred to PVDF membrane (Millipore Sigma, IPVH00010) using semi-dry transfer (Bio-Rad Laboratories). Membranes were blocked with 5% nonfat milk in TBST (Tris-buffered saline with 0.05% Tween 20 [National Diagnostics, 9005–64–5]), probed with primary antibodies in 1% BSA in TBST overnight at 4°C and incubated with HRP-conjugated secondary antibodies (KPL, 474–1506, 474–1806, 14–16–06 and 14–13–06) in blocking solution at room temperature for 1 h. Protein bands were detected using SuperSignal West Dura Extended Duration Substrate (Thermo Fisher Scientific, 34076) or SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific, 34080) and visualized using Chemi-doc system (Bio-Rad Laboratories, Universal Hood II). Band intensity was analyzed using Image Lab software (Bio-Rad Laboratories) and normalized to loading control.
Primary antibodies: LC3 (1:1000; Novus Biologicals, NB100-2220), PIK3C3/Vps34 (1:1000; Invitrogen, 382100), BECN1/beclin1 (1:1000; Santa Cruz Biotechnology, sc-11427), SQSTM1 (1:1000; BD Bioscience, 610832), ATG5 (1:1000; Sigma-Aldrich, A0731), ACTB/β-actin (1:10,000; Sigma-Aldrich, A1978), phospho-PLA2G4A (1:1000; Sigma-Aldrich, SAB4503812), LAMP1 (1:1000; Abcam, 24170), PLA2G4A (1:1000; 2832), ULK1 (1:1000; Cell Signaling Technology, 4773), phospho-UK1(1:1000; Cell Signaling Technology, 5869) and α-tubulin (1:500; AA4.3-s, developed by Walsh, C. and obtained from Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biology, Iowa City, IA 52,242).
Cell culture and treatment
Human H4 neuroglioma cells and HeLa cells were used in this study. H4-GFP-LC3 and RFP-LAMP1 cells have been previously described [70]. mCherry-GFP-LC3 expressing H4 cells were generated by transducing H4 neuroglioma cells with mCherry-EGFP-LC3 reporter (Addgene 22418, deposited by Dr. Jayanta Debnath) cloned into lentiviral pLESIP vector. Cells were cultured and maintained in DMEM (Gibco, 11995065) with 10% heat inactivated Newborn Calf Serum (Sigma-Aldrich, N4762-500ML) and 1% antibiotic-antimycotic (Thermo Fisher Scientific, 15240062) at 37°C in humidified atmosphere with 5% CO2. Cells were plated in 24-well plate at density of 4-5 × 104 cells per well for western blot analysis and lysosomal enzyme activity determination and for immuonocytochemistry 2 × 104 cells were plated on coverslips in 24-well plates.
Primary neurons were cultured from 15- to 16-days-old rat or GFP-LC3 transgenic heterozygote mouse embryos as described previously [71]. Neurons were seeded at a density of 3–5 × 105 cells per well in poly-D-lysine coated 24-well plate. For immunocytochemistry neurons were plated at a density of 3 × 105 cells on poly-D-lysine coated coverslip in 24-well plate. Neurons were maintained in serum free neurobasal media (Invitrogen, 21103049) supplemented with 2% B-27 (Invitrogen, 17504044). All the treatments in neurons were done using preconditioned B-27 containing neurobasal media.
For PLA2G4A activation cells were treated with PLA2G4A activator ceramide-1-phosphate (C1P) (Sigma-Aldrich, C4832) or 1,2-dipalmitoylphosphatidylinositol 4,5-diphosphate triammonium salt (Sigma-Aldrich, P7115) for 4 h. C1P was dissolved in a mixture of methanol (Sigma-Aldrich, 179337) and dodecane (Sigma-Aldrich, 297879) (49:1) to obtain a stock concentration of 4 mM which was aliquoted and stored in −20°C. Stock solution of C1P was diluted in the culture media to prepare required concentration before treatment. Control cells were treated with vehicle only. For PLA2G4A inhibition, cells were pretreated for one and half hours with 20 μM of AACOCF3 (Enzo, BML-ST335-0050) prepared by diluting 40 mM stock solution (in ethanol) in the culture media to working concentration. Cells were treated with 5 μM Amyloid β(1-40) (Bachem, 4014442.1000) prepared in aqueous buffer as described previously [72].
Sirna transfection
H4 cells were transfected with siRNA specific for PLA2G4A (Sigma-Aldrich, EHU009531) or not-targeting (nt) siRNA control (Sigma-Aldrich, 45-SIC001). Cells were reverse transfected in 12-well tissue culture plates with 2 μL of 20 μM siRNA plus 202 μL of the master mix containing 200 μL OptiMEM media (Invitrogen, 31985070) and 2 uL HiPerfect (Invitrogen, 301705) following manufacturer’s instructions. Cells were added at 1.5 × 105 per well. After 24 h cells were re-plated in 24-well plates at 2 × 104 per well on coverslips for IF and at 4 × 104 for western blot analysis. After additional 48 h cells were treated with C1P or amyloid-β(1-40).
Digitonin extraction assay
Lysosomal membrane permeability in H4 cells was assessed by digitonin extraction assay as per the method describe previously [45]. Briefly, cells were plated in 24-well plates and next day incubated with different concentration of digitonin in extraction buffer [45] at room temperature on shaker for 15 min. Cell extracts from each well were collected and activities of cytosolic enzyme LDH and lysosomal enzyme NAGLU were determined using LDH assay kit (Promega Corp., G1780) and NAGLU assay kit respectively to determine optimum digitonin concentration required to permeabilize cytosolic membrane with minimum or negligible impact on lysosomal membrane. 7.5 μg/ml concentration of digitonin was found to be optimum for H4 cells to cause cytosolic membrane permeabilization thereby releasing cytosolic content with no or minimum effect on lysosomal membrane. Digitonin (200 μg/ml; Sigma, D141) was used to lyse cell and lysosomal membrane. Activities of lysosomal enzymes NAGLU and CTSD were determined in these extracts. LDH activity was also determined as an internal standard and was used to normalize lysosomal enzyme activity in these extracts. Extent of lysosomal enzyme release was expressed as a fraction of LDH corrected cytosolic lysosomal enzyme activity over LDH corrected total lysosomal enzyme activity in the whole cell lysate.
Dextran release assay
Dextran release assay was performed in H4 cells expressing RFP-LAMP1 as described previously [45]. 5 × 104 cells were seeded in glass bottom plates (Mattek Corp., P35G-1.0–14-C). After 24 h media was replaced with media containing 40 μg/ml of 3 kDa Alexa fluor 488-dextran (Thermo Fisher Scientific, D34682); after 16–18 h cells were washed twice with PBS and incubated with fresh phenol red free media for 2 h to achieve maximum uptake of dextran into the lysosomes. Cells were then treated with C1P or amyloid β in phenol red free media and visualized using fluorescent Nikon Ti-E inverted microscope at 60x at the end of the incubation. Cells were also treated with 2 mM L-leucyl-L-leucine-methyl ester (LLOME) (Sigma-Aldrich, L7393) for 2 h as a positive control. Images were processed and quantified using Nikon Elements software as above. Pearson’s correlation was used to quantify co-localization between lysosomes (RFP) and dextran (Alexa Fluor 488 in the FITC channel). To further assess lysosomal size and dextran loading, intensity profiles across selected cells were analyzed: lysosomes were identified as areas with RFP intensity > 1.5 average; length of continuous RFP > 1.5 profile segments was used to estimate the size of lysosomes. Dextran loaded lysosomes were defined as lysosomes with GFP intensity > 2.0 average.
Statistical analysis
All data are presented as mean ± standard error of the mean (SEM). For in vivo studies n represents number of mice used per group, and was determined by power analysis (power of 0.8, alpha value 0.05) based on our past experience with the CCI model [8]. For in vitro studies n represents total number of repeats from at least 3 independent experiments. All n values are specified in the figure legends. Graphs were generated using Microsoft Excel or MetaboAnalyst software. Statistical analyses were performed using GraphPad Prism (version 7). For experiments with only 2 groups unpaired 2-tailed Student’s t-test was performed. For experiment with multiple groups one-way, two-way or repeated-measures two-way ANOVA was performed followed by appropriate post-hoc test (Tukey’s, Newman-Keuls or Bonferroni) as specified in the figure legends. A p value ≤ 0.05 was considered statistically significant.
Funding Statement
This work was supported by R01NS 091218 and R01NS 094527 from National Institutes of Health to MML and MSCRFE-2747 from Maryland Stem Cell Research Fund to CS. [
Acknowledgments
We thank Dr. Noboru Mizushima (Tokyo Medical and Dental University, Tokyo, Japan) and Dr. Beth Levine (UT Southwestern Medical center, Dallas TX) for the GFP-LC3 mice; Drs. Junfang Wu, David Loane, Bogdan Stoica and Boris Sabirzhanov for technical assistance and advice; Dr. Shuo Liu for help with animal husbandry, Ms. Victoria Meadow for help with behavioral studies and Mr. Robert A. Brown for help with in vitro cell culture.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental material
Supplemental data for this article can be accessed here.
References
- [1].Xiong Y, Mahmood A, Chopp M.. Animal models of traumatic brain injury. Nat Rev Neurosci. 2013. February;14(2):128–142.nrn3407 [pii]. PubMed PMID: 23329160; PubMed Central PMCID: PMC3951995. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Werner C, Engelhard K.. Pathophysiology of traumatic brain injury. Br J Anaesth. 2007. July;99(1):4–9. 99/1/4 [pii] PubMed PMID: 17573392; eng. [DOI] [PubMed] [Google Scholar]
- [3].Loane DJ, Faden AI. Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol Sci. 2010. December;31(12):596–604.S0165-6147(10)00168-9 [pii]. PubMed PMID: 21035878; PubMed Central PMCID: PMC2999630. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gardner AJ, Zafonte R. Neuroepidemiology of traumatic brain injury. Handb Clin Neurol. 2016;138:207–223. B978-0-12-802973-2.00012-4 [pii]. PubMed PMID: 27637960; eng. [DOI] [PubMed] [Google Scholar]
- [5].Jassam YN, Izzy S, Whalen M, et al. Neuroimmunology of traumatic brain injury: time for a paradigm shift. Neuron. 2017. September 13;95(6):1246–1265. [pii]10.1016/j.neuron.2017.07.010. PubMed PMID: 28910616; PubMed Central PMCID: PMC5678753. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Faden AI. Neuroprotection and traumatic brain injury: theoretical option or realistic proposition. Curr Opin Neurol. 2002. December;15(6):707–712.PubMed PMID: 12447109; eng. [DOI] [PubMed] [Google Scholar]
- [7].Lipinski MM, Wu J, Faden AI, et al. Function and mechanisms of autophagy in brain and spinal cord trauma. Antioxid Redox Signal. 2015. August 20;23(6):565–577. PubMed PMID: 25808205; PubMed Central PMCID: PMC4545370. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sarkar C, Zhao Z, Aungst S, et al. Impaired autophagy flux is associated with neuronal cell death after traumatic brain injury. Autophagy. 2014;10(12):2208–2222. PubMed PMID: 25484084; PubMed Central PMCID: PMC4502690. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013. August;19(8):983–997.nm.3232 [pii]. PubMed PMID: 23921753; eng. [DOI] [PubMed] [Google Scholar]
- [10].Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007. December 1;120(Pt 23):4081–4091. [pii]10.1242/jcs.019265. PubMed PMID: 18032783; eng. [DOI] [PubMed] [Google Scholar]
- [11].Lee JH, Yu WH, Kumar A, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010. June 25;141(7):1146–1158. S0092-8674(10)00544-1 [pii]. PubMed PMID: 20541250; PubMed Central PMCID: PMC3647462. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Menzies FM, Fleming A, Caricasole A, et al. Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron. 2017. March 8;93(5):1015–1034. [pii]10.1016/j.neuron.2017.01.022. PubMed PMID: 28279350; eng. [DOI] [PubMed] [Google Scholar]
- [13].Menzies FM, Fleming A, Rubinsztein DC. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci. 2015. June;16(6):345–357.nrn3961 [pii]. PubMed PMID: 25991442; eng. [DOI] [PubMed] [Google Scholar]
- [14].Wong E, Cuervo AM. Autophagy gone awry in neurodegenerative diseases. Nat Neurosci. 2010. July;13(7):805–811.nn.2575 [pii]. PubMed PMID: 20581817; PubMed Central PMCID: PMC4038747. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Xu H, Ren D. Lysosomal physiology. Annu Rev Physiol. 2015;77:57–80. PubMed PMID: 25668017; PubMed Central PMCID: PMC4524569. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kilpatrick BS, Eden ER, Hockey LN, et al. Methods for monitoring lysosomal morphology. Methods Cell Biol. 2015;126:1–19. S0091-679X(14)00019-3 [pii]. PubMed PMID: 25665438; eng. [DOI] [PubMed] [Google Scholar]
- [17].de Duve D. The peroxisome: a new cytoplasmic organelle. Proc R Soc Lond B Biol Sci. 1969. April 15;173(1030):71–83. PubMed PMID: 4389354; eng. [PubMed] [Google Scholar]
- [18].Luzio JP, Pryor PR, Bright NA. Lysosomes: fusion and function. Nat Rev Mol Cell Biol. 2007. August;8(8):622–632. nrm2217 [pii]. PubMed PMID: 17637737; eng. [DOI] [PubMed] [Google Scholar]
- [19].Walker MW, Lloyd-Evans E. A rapid method for the preparation of ultrapure, functional lysosomes using functionalized superparamagnetic iron oxide nanoparticles. Methods Cell Biol. 2015;126:21–43. S0091-679X(14)00020-X [pii]. PubMed PMID: 25665439; eng. [DOI] [PubMed] [Google Scholar]
- [20].Gomez-Sintes R, Ledesma MD, Boya P. Lysosomal cell death mechanisms in aging. Ageing Res Rev. 2016. December;32:150–168. [pii]10.1016/j.arr.2016.02.009. PubMed PMID: 26947122; eng. [DOI] [PubMed] [Google Scholar]
- [21].Rodriguez-Muela N, Hernandez-Pinto AM, Serrano-Puebla A, et al. Lysosomal membrane permeabilization and autophagy blockade contribute to photoreceptor cell death in a mouse model of retinitis pigmentosa. Cell Death Differ. 2015. March;22(3):476–487. cdd2014203 [pii]. PubMed PMID: 25501597; PubMed Central PMCID: PMC4326579. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Aits S, Jaattela M. Lysosomal cell death at a glance. J Cell Sci. 2013. May 1;126(Pt 9):1905–1912. 126/9/1905 [pii]. PubMed PMID: 23720375; eng. [DOI] [PubMed] [Google Scholar]
- [23].Boya P, Kroemer G. Lysosomal membrane permeabilization in cell death. Oncogene. 2008. October 27;27(50):6434–6451. onc2008310 [pii]. PubMed PMID: 18955971; eng. [DOI] [PubMed] [Google Scholar]
- [24].Yamashima T, Oikawa S. The role of lysosomal rupture in neuronal death. Prog Neurobiol. 2009. December;89(4):343–358.S0301-0082(09)00140-3 [pii]. PubMed PMID: 19772886; eng. [DOI] [PubMed] [Google Scholar]
- [25].Cuervo AM, Dice JF. When lysosomes get old. Exp Gerontol. 2000. March;35(2):119–131. S0531-5565(00)00075-9 [pii]. PubMed PMID: 10767573; eng. [DOI] [PubMed] [Google Scholar]
- [26].Lee JC, Simonyi A, Sun AY, et al. Phospholipases A2 and neural membrane dynamics: implications for Alzheimer’s disease. J Neurochem. 2011. March;116(5):813–819. PubMed PMID: 21214562; PubMed Central PMCID: PMC3058290. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Burke JE, Dennis EA. Phospholipase A2 structure/function, mechanism, and signaling. J Lipid Res. 2009. April;50(Suppl):S237–42.R800033-JLR200 [pii]. PubMed PMID: 19011112; PubMed Central PMCID: PMC2674709. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sanchez-Mejia RO, Newman JW, Toh S, et al. Phospholipase A2 reduction ameliorates cognitive deficits in a mouse model of Alzheimer’s disease. Nat Neurosci. 2008. November;11(11):1311–1318. nn.2213 [pii]. PubMed PMID: 18931664; PubMed Central PMCID: PMC2597064. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Leslie CC. Cytosolic phospholipase A(2): physiological function and role in disease. J Lipid Res. 2015. August;56(8):1386–1402.jlr.R057588 [pii]. PubMed PMID: 25838312; PubMed Central PMCID: PMC4513982. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Phillis JW, O’Regan MH. A potentially critical role of phospholipases in central nervous system ischemic, traumatic, and neurodegenerative disorders. Brain Res Brain Res Rev. 2004. January;44(1):13–47. S0165017303002522 [pii]. PubMed PMID: 14739001; eng. [DOI] [PubMed] [Google Scholar]
- [31].Zhang Z, Lee YC, Kim SJ, et al. Production of lysophosphatidylcholine by cPLA2 in the brain of mice lacking PPT1 is a signal for phagocyte infiltration. Hum Mol Genet. 2007. April 1;16(7):837–847. ddm029 [pii] PubMed PMID: 17341491; eng. [DOI] [PubMed] [Google Scholar]
- [32].Liu NK, Deng LX, Zhang YP, et al. Cytosolic phospholipase A2 protein as a novel therapeutic target for spinal cord injury. Ann Neurol. 2014. May;75(5):644–658. PubMed PMID: 24623140; PubMed Central PMCID: PMC4320750. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Farooqui AA, Horrocks LA. Brain phospholipases A2: a perspective on the history. Prostaglandins Leukot Essent Fatty Acids. 2004. September;71(3):161–169.S0952327804000535 [pii]. PubMed PMID: 15253885; eng. [DOI] [PubMed] [Google Scholar]
- [34].Gijon MA, Leslie CC. Regulation of arachidonic acid release and cytosolic phospholipase A2 activation. J Leukoc Biol. 1999. March;65(3):330–336. PubMed PMID: 10080535; eng. [DOI] [PubMed] [Google Scholar]
- [35].Murakami M, Taketomi Y, Miki Y, et al. Recent progress in phospholipase A(2) research: from cells to animals to humans. Prog Lipid Res. 2011. April;50(2):152–192. S0163-7827(10)00065-2 [pii]. PubMed PMID: 21185866; eng. [DOI] [PubMed] [Google Scholar]
- [36].Farooqui AA, Ong WY, Horrocks LA. Inhibitors of brain phospholipase A2 activity: their neuropharmacological effects and therapeutic importance for the treatment of neurologic disorders. Pharmacol Rev. 2006. September;58(3):591–620. 58/3/591 [pii] PubMed PMID: 16968951; eng. [DOI] [PubMed] [Google Scholar]
- [37].Chu CT, Bayir H, Kagan VE. LC3 binds externalized cardiolipin on injured mitochondria to signal mitophagy in neurons: implications for Parkinson disease. Autophagy. 2014. February;10(2):376–378.27191 [pii]. PubMed PMID: 24351649; PubMed Central PMCID: PMC5396091. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Monteiro-Cardoso VF, Oliveira MM, Melo T, et al. Cardiolipin profile changes are associated to the early synaptic mitochondrial dysfunction in Alzheimer’s disease. J Alzheimers Dis. 2015;43(4):1375–1392. D1N5364751107022 [pii]. PubMed PMID: 25182746; eng. [DOI] [PubMed] [Google Scholar]
- [39].Tyurina YY, Poloyac SM, Tyurin VA, et al. A mitochondrial pathway for biosynthesis of lipid mediators. Nat Chem. 2014. June;6(6):542–552. nchem.1924 [pii]. PubMed PMID: 24848241; PubMed Central PMCID: PMC4201180. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zhu D, Lai Y, Shelat PB, et al. Phospholipases A2 mediate amyloid-beta peptide-induced mitochondrial dysfunction. J Neurosci. 2006. October 25;26(43):11111–11119. 26/43/11111 [pii] PubMed PMID: 17065451; eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Farooqui AA, Horrocks LA. Phospholipase A2-generated lipid mediators in the brain: the good, the bad, and the ugly. Neuroscientist. 2006. June;12(3):245–260. 12/3/245 [pii] PubMed PMID: 16684969; eng. [DOI] [PubMed] [Google Scholar]
- [42].Slomiany BL, Slomiany A. Ghrelin protection against cytotoxic effect of ethanol on rat salivary mucin synthesis involves cytosolic phospholipase A2 activation through S-nitrosylation.Int J Biomed Sci. 2010. March;6(1):37–44. PubMed PMID: 23675174; PubMed Central PMCID: PMC3614730. eng. [PMC free article] [PubMed] [Google Scholar]
- [43].Lamour NF, Subramanian P, Wijesinghe DS, et al. Ceramide 1-phosphate is required for the translocation of group IVA cytosolic phospholipase A2 and prostaglandin synthesis. J Biol Chem. 2009. September 25;284(39):26897–26907. M109.001677 [pii]. PubMed PMID: 19632995; PubMed Central PMCID: PMC2785377. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Pettus BJ, Bielawska A, Subramanian P, et al. Ceramide 1-phosphate is a direct activator of cytosolic phospholipase A2. J Biol Chem. 2004. March 19;279(12):11320–11326. M309262200 [pii]. PubMed PMID: 14676210; eng. [DOI] [PubMed] [Google Scholar]
- [45].Aits S, Jaattela M, Nylandsted J. Methods for the quantification of lysosomal membrane permeabilization: a hallmark of lysosomal cell death. Methods Cell Biol. 2015;126:261–285. S0091-679X(14)00033-8 [pii]. PubMed PMID: 25665450; eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Mosior M, Six DA, Dennis EA. Group IV cytosolic phospholipase A2 binds with high affinity and specificity to phosphatidylinositol 4,5-bisphosphate resulting in dramatic increases in activity. J Biol Chem. 1998. January 23;273(4):2184–2191. PubMed PMID: 9442060; eng. [DOI] [PubMed] [Google Scholar]
- [47].Kalyvas A, David S. Cytosolic phospholipase A2 plays a key role in the pathogenesis of multiple sclerosis-like disease. Neuron. 2004. February 5;41(3):323–335. S0896627304000030 [pii]. PubMed PMID: 14766173; eng. [DOI] [PubMed] [Google Scholar]
- [48].Wallner S, Schmitz G. Plasmalogens the neglected regulatory and scavenging lipid species. Chem Phys Lipids. 2011. September;164(6):573–589.S0009-3084(11)00216-7 [pii]. PubMed PMID: 21723266; eng. [DOI] [PubMed] [Google Scholar]
- [49].Dean JM, Lodhi IJ. Structural and functional roles of ether lipids. Protein Cell. 2017. May 18. 10.1007/s13238-017-0423-5 [pii]. PubMed PMID: 28523433; eng. DOI: 10.1007/s13238-017-0423-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ullio C, Casas J, UT B, et al. Sphingosine mediates TNFalpha-induced lysosomal membrane permeabilization and ensuing programmed cell death in hepatoma cells. J Lipid Res. 2012. June;53(6):1134–1143. jlr.M022384 [pii]. PubMed PMID: 22454477; PubMed Central PMCID: PMC3351820. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Corrotte M, Castro-Gomes T, Koushik AB, et al. Approaches for plasma membrane wounding and assessment of lysosome-mediated repair responses. Methods Cell Biol. 2015;126:139–158. S0091-679X(14)00043-0 [pii]. PubMed PMID: 25665445; PubMed Central PMCID: PMC4739812. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].McNeil PL. Repairing a torn cell surface: make way, lysosomes to the rescue. J Cell Sci. 2002. March 1;115(Pt 5):873–879. PubMed PMID: 11870206; eng. [DOI] [PubMed] [Google Scholar]
- [53].Syrbu SI, Waterman WH, Molski TF, et al. Phosphorylation of cytosolic phospholipase A2 and the release of arachidonic acid in human neutrophils. J Immunol. 1999. February 15;162(4):2334–2340. PubMed PMID: 9973512; eng. [PubMed] [Google Scholar]
- [54].Street IP, Lin HK, Laliberte F, et al. Slow- and tight-binding inhibitors of the 85-kDa human phospholipase A2. Biochemistry. 1993. June 15;32(23):5935–5940. PubMed PMID: 8018213; eng. [DOI] [PubMed] [Google Scholar]
- [55].Kennedy BP, Payette P, Mudgett J, et al. A natural disruption of the secretory group II phospholipase A2 gene in inbred mouse strains. J Biol Chem. 1995. September 22;270(38):22378–22385. PubMed PMID: 7673223; eng. [DOI] [PubMed] [Google Scholar]
- [56].Sharma S, Ying Z, Gomez-Pinilla F. A pyrazole curcumin derivative restores membrane homeostasis disrupted after brain trauma. Exp Neurol. 2010. November;226(1):191–199.S0014-4886(10)00326-2 [pii]. PubMed PMID: 20816821; PubMed Central PMCID: PMC3225197. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Wu A, Ying Z, Gomez-Pinilla F. The salutary effects of DHA dietary supplementation on cognition, neuroplasticity, and membrane homeostasis after brain trauma. J Neurotrauma. 2011. October;28(10):2113–2122.PubMed PMID: 21851229; PubMed Central PMCID: PMC3191367. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Nikolaeva I, Crowell B, Valenziano J, et al. beneficial effects of early mTORC1 inhibition after traumatic brain injury. J Neurotrauma. 2016. January 15;33(2):183–193. PubMed PMID: 26122481; PubMed Central PMCID: PMC4722609. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Zhu X, Park J, Golinski J, et al. Role of Akt and mammalian target of rapamycin in functional outcome after concussive brain injury in mice. J Cereb Blood Flow Metab. 2014. September;34(9):1531–1539. jcbfm2014113 [pii]. PubMed PMID: 24938400; PubMed Central PMCID: PMC4158669. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Zoncu R, Bar-Peled L, Efeyan A, et al. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science. 2011. November 4;334(6056):678–683. 334/6056/678 [pii]. PubMed PMID: 22053050; PubMed Central PMCID: PMC3211112. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Mizushima N, Yamamoto A, Matsui M, et al. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004. March;15(3):1101–1111. E03-09-0704 [pii]. PubMed PMID: 14699058; PubMed Central PMCID: PMC363084. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Jung S, Aliberti J, Graemmel P, et al. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000. June;20(11):4106–4114. PubMed PMID: 10805752; PubMed Central PMCID: PMC85780. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Zhao Z, Loane DJ, Murray MG 2nd. et al. Comparing the predictive value of multiple cognitive, affective, and motor tasks after rodent traumatic brain injury. J Neurotrauma. 2012. October 10;29(15):2475–2489. PubMed PMID: 22924665; PubMed Central PMCID: PMC3471125. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Kreuzaler PA, Staniszewska AD, Li W, et al. Stat3 controls lysosomal-mediated cell death in vivo. Nat Cell Biol. 2011. March;13(3):303–309. ncb2171 [pii]. PubMed PMID: 21336304; eng. [DOI] [PubMed] [Google Scholar]
- [65].Matyash V, Liebisch G, Kurzchalia TV, et al. Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J Lipid Res. 2008. May;49(5):1137–1146. D700041-JLR200 [pii]. PubMed PMID: 18281723; PubMed Central PMCID: PMC2311442. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Damen CW, Isaac G, Langridge J, et al. Enhanced lipid isomer separation in human plasma using reversed-phase UPLC with ion-mobility/high-resolution MS detection. J Lipid Res. 2014. August;55(8):1772–1783. jlr.D047795 [pii]. PubMed PMID: 24891331; PubMed Central PMCID: PMC4109771. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Paglia G, Astarita G. Metabolomics and lipidomics using traveling-wave ion mobility mass spectrometry. Nat Protoc. 2017. April;12(4):797–813.nprot.2017.013 [pii]. PubMed PMID: 28301461; eng. [DOI] [PubMed] [Google Scholar]
- [68].Xia J, Sinelnikov IV, Han B, et al. MetaboAnalyst 3.0–making metabolomics more meaningful. Nucleic Acids Res. 2015. July 1;43(W1):W251–7. gkv380 [pii]. PubMed PMID: 25897128; PubMed Central PMCID: PMC4489235. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Xia J, Wishart DS. Using Metaboanalyst 3.0 for comprehensive metabolomics data analysis. Curr Protoc Bioinformatics. 2016. September 7;55:14 10 1–14 10 91. [DOI] [PubMed] [Google Scholar]
- [70].Lipinski MM, Hoffman G, Ng A, et al. A genome-wide siRNA screen reveals multiple mTORC1 independent signaling pathways regulating autophagy under normal nutritional conditions. Dev Cell. 2010. June 15;18(6):1041–1052. S1534-5807(10)00213-3 [pii]. PubMed PMID: 20627085; PubMed Central PMCID: PMC2935848. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Yakovlev AG, Ota K, Wang G, et al. Differential expression of apoptotic protease-activating factor-1 and caspase-3 genes and susceptibility to apoptosis during brain development and after traumatic brain injury. J Neurosci. 2001. October 1;21(19):7439–7446. 21/19/7439 [pii]. PubMed PMID: 11567033; eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Ofengeim D, Shi P, Miao B, et al. Identification of small molecule inhibitors of neurite loss induced by Abeta peptide using high content screening. J Biol Chem. 2012. March 16;287(12):8714–8723. M111.290957 [pii]. PubMed PMID: 22277654; PubMed Central PMCID: PMC3308767. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.