

ABSTRACT
The immune checkpoint blockade (ICB) immunotherapy has prolonged overall survival for cancer patients but the response rates are low. The resistance to ICB is likely due to compensatory upregulation of additional immune inhibitory molecules. In this study, we first systematically examined Tim-3 expression in immune cells in mouse tumors and found that Tim-3 was specifically up-regulated in a large number of Treg, conventional CD4+, CD8+ T cells, dendritic cell 1 (DC1), and macrophage 1 (M1) in the tumor microenvironment (TME). Interestingly, Tim-3+ T cells in the TME were phenotypically effector but not “exhausted” T cells because Tim-3+ PD-1+ CD8+ T cells had a higher number of mitochondria, greater levels of glycolysis, and higher tumor-specific cytolytic activities compared to Tim-3− PD-1− CD8+ T cells. The combination treatment with Tim-3 and PD-1 mAbs resulted in a synergistic antitumor activity but also increased the expression of Lag-3 and GITR in TIL, demonstrating cross-regulation between multiple checkpoint molecules. Furthermore, we found that the antitumor efficacy with triple combination of Tim-3, PD-1, and Lag3 mAbs was much greater than any two antibodies. Mechanistically, we demonstrated that simultaneous targeting of Tim-3, PD-1, and Lag-3 cooperatively increased the levels of granzyme B and tumor-specific cytolytic activities of CD8+ TIL. Our data indicate that multiple checkpoint molecules are coordinately upregulated to inhibit the function of hyperactivated T cells in the TME and requirement for the simultaneous blockade of PD-1, Tim-3 and Lag3 for cancer treatment.
KEYWORDS: Tim-3, Lag-3, PD-1, exhaustion, tumor immunotherapy, combination therapy
Introduction
Cancer progression is associated with immune evasion and immune suppression. Although tumor antigens trigger adaptive antitumor immune responses, tumors evolve multitudes of mechanisms that suppress and eventually overcome antitumor immunity. Tumor cells undergo profound genetic adaptation to reduce their immunogenicity and produce immune suppressive factors to shape the tumor microenvironment (TME).1–4 The TME is also dominated by regulatory T cells (Treg), myeloid-derived suppressor cells (MDSC) and tumor-associated macrophages (TAM) which mediate immune suppression.5–7 As a consequence, tumor-infiltrating Th1 and CD8+ T cells fail to eradicate cancer cells but instead participate in a stalemate of chronic inflammation. Similar to chronically activated T cells during chronic viral infection, T cells in the TME exhibit characteristics of “exhausted” T cells, such as reduced proliferation and cytokine production as well as dysregulation of metabolism.8 Whether reversing the T cell exhaustion state can be an effective strategy for tumor immunotherapy remains to be determined.
Multiple immune regulatory receptors have been found in the tumor-infiltrating lymphocytes (TIL). It has been found that TIL co-expressing PD-1 and Tim-3 or PD-1 and Lag-3 are functionally “exhausted” compared to other TIL subsets.9–12 In fact, CD4+ and CD8+ TIL co-expressing multiple immune regulatory receptors such as PD-1, Tim-3, Lag-3, OX-40, GITR, and TIGIT have been reported, and the underlying genetic regulatory network has been delineated.13 TIL expressing PD-1, Tim-3, and Lag-3 have been shown to be the most functionally and metabolically “exhausted”.14 In addition, in vivo studies have demonstrated Tim-3 and Lag-3 regulate antitumor immunity.15–17 Whether, and how, these molecules have redundant roles in suppressing T cell function is not fully understood.
In order to help establish the combination immunotherapy and understand the intricate immune checkpoint network, we set out to study the expression and regulation of multiple immune checkpoint molecules, particularly Tim-3 and Lag-3, in the immune cells in the TME of syngeneic mouse tumor models. We further determined the underlying mechanisms mediating antitumor effects of PD-1, Tim-3/PD-1, and Tim-3/PD-1/Lag-3 combination therapy.
Results
Tim-3 is differentially expressed along with multiple activation and inhibitory molecules in tumor-infiltrating immune cells
In order to help define the role of Tim-3 in antitumor immune responses, we systematically characterized the Tim-3 expression in multiple immune cells in the TME using the MC38 tumor model, which is well known to be responsive to checkpoint immunotherapy. Consistent with previous studies,9–11 Tim-3 was highly expressed on approximately 81% Tregs, 30% Foxp3−CD4+, and 47% CD8+ tumor-infiltrating T cells in this model (Figure 1a). In addition, Tim-3 was also expressed on a small fraction of B cells, NK cells and γδT cells (Figure 1a). Besides the MC38 tumor model, we also confirmed that Tim-3 was expressed on TIL in ID8 and B16 tumors (Figure S1A). Besides lymphocytes, we also examined the Tim-3 expression on tumor-associated myeloid cells.18 We found that Tim-3 was expressed on about 39% CD45+CD11b+GR1−MHCII+CD24+ F4/80−CD103+ DC1 and 30% CD45+CD11b+GR1−MHCII+CD24−F4/80+CD206− type 1 macrophages (M1) (Figure 1b). In contrast, Tim-3 was expressed on smaller portions of CD45+CD11b+GR1−MHCII+CD24+ F4/80−CD103− DC2 (about 13.4%), CD45+CD11b+GR1−MHCII+CD24−F4/80+CD206+ type 2 macrophages (M2, about 14.6%), and MDSC (about 6.89%) (Figure 1b). We detected minimal expression on lymphocyte subsets in peripheral immune cells (Figure S1B). Thus, Tim-3 expression was differentially up-regulated in immune cells in the TME.
Figure 1.

Characterization of Tim-3+ tumor-infiltrating immune cells.
Tumors were isolated from MC38 tumor-bearing mice and immune cells were analyzed by flow cytometry. (a). Representative flow plots of Tim3 expression on tumor-infiltrating lymphocytes. (Foxp3−CD4+: CD45+CD8−CD4+Foxp3−, Foxp3+CD4+: CD45+CD8−CD4+Foxp3+, CD8+: CD45+CD4−CD8+, B cell: CD45+CD4−CD8−B220+NK1.1−γδTCR−, NK cell: CD45+CD4−CD8−γδTCR−NK1.1+ γδT cell: CD45+CD4−CD8+NK1.1− γδTCR+). (b). Representative flow plots of Tim-3 expression on tumor-infiltrating myeloid cells. (CD103+DC1: CD45+CD11b+Gr1−MHCII+F4/80−CD24+CD103+, CD103−DC2: CD45+CD11b+Gr1−MHCII+F4/80−CD24+CD103−, CD206−M1: CD45+CD11b+Gr1−MHCII+F4/80+CD24−CD206−, CD206+M2: CD45+CD11b+Gr1−MHCII+F4/80+CD24−CD206+, MDSC: CD45+CD11b+Gr1+). (c). Representative flow plots of Tim-3 vs IL7R, CD62L, CD44, OX40, Ki67, PD-1, GITR and Lag-3 expressions on Treg cells (top row), conventional CD4+ T cells (middle row) and CD8+ T cells (bottom row). (d) Statistics of IL7R, CD62L, CD44, OX40, Ki67, PD-1, GITR and Lag-3 expressions in Tim-3− and Tim-3+ subsets depicted in C, data were presented as mean ± SEM (n = 4–5). *P < .05, **P < .005, ***P < .0005, ****P < .0001, Student’s t test was performed.
We further characterized Tim-3+ tumor-infiltrating T cells using multi-color flow cytometry. We found that all Tim-3+ T cells were CD62L− CD44+, suggesting these cells are effector/memory T cells (Figure 1c-d). The percentage of IL7R+ T cells in Tim-3+ CD4+Foxp3− and Tim-3+CD8+ T cells was lower compared to Tim-3− subsets (Figure 1c-d), which was also consistent with an effector T cell status for Tim-3+ CD4+ and CD8+ TIL. In addition, OX-40, another T cell activation marker, was also upregulated in Tim-3+ CD4+ T cells and Treg cells compared to the Tim-3− TIL (Figure 1c-d). Surprisingly, Ki67, a cell proliferation marker, was positive for most Tim-3+ T cells (>90%), suggesting these cells are proliferative but not exhausted (Figure 1c-d).
Tumoral Tim-3+ T cells are highly activated effector cells
In addition to activation and proliferative markers, Tim-3+ T cells in the TME also consisted of higher percentages of cells that expressed effector molecules such as IFN-γ and granzyme B (Figure 2a-b). These data further showed that Tim-3 marked effector T cells in the TME in the MC38 tumor model. It has been shown that Tim-3+PD-1+ T cells are “exhausted” in cancer patients and chronically infected individuals.8–11 We found multiple immune regulatory receptors such as PD-1, GITR, and Lag-3 were upregulated in Tim-3+ T cells compared to the Tim-3− TIL (Figure 1c-d). Surprisingly, we detected that similar percentages of IFN-γ+ and granzyme B+ were present in PD1+, PD1−, Lag3+, and Lag-3− subsets among Tim-3+ CD8+ T cells (Figure 2a-b). These data suggest that CD8+ TIL expressing multiple immune inhibitory receptors are equally capable of producing effector molecules. Recent studies have established that reduced mitochondrial biogenesis as a hallmark of T cell exhaustion in the TME.14 We found a slightly but significantly higher numbers of mitochondria in the Tim3+PD-1+ CD8+ T cells compared to the Tim3−PD-1− CD8+ T cell subset in MC38 tumors (Figure 2c). Despite a slight increase in the numbers of mitochondria, seahorse assay demonstrated that no difference in oxygen consumption rates between Tim-3+PD-1+ and Tim-3−PD-1− CD8+ TIL (Figure 2d). Strikingly, Tim-3+PD-1+ CD8+ TIL had a higher glycolysis level compared to Tim3−PD-1− CD8+ TIL (Figure 2d). To further determine whether Tim3+PD-1+ CD8+ T cells were “exhausted” T cells, we performed an ex vivo tumor cytolytic assay using the CD8+ TIL isolated from tumors (Figure 2e). Our data showed that Tim3+PD-1+ CD8+ TIL had higher tumor-specific cytolytic activities than Tim-3−PD-1− CD8+ TIL (Figure 2e). Collectively, these data indicated that, besides PD-1, multiple surface molecules were upregulated in effector T cells rather than “exhausted” T cells in the TME, potentially regulating their function.
Figure 2.
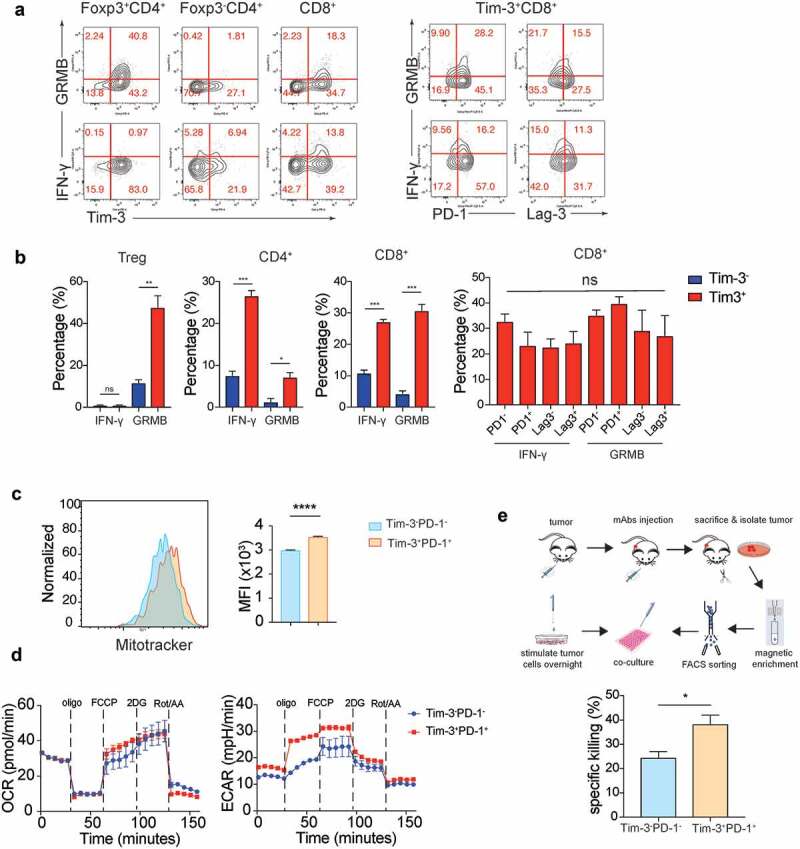
Tim-3+ cells were highly activated but not exhausted T cells.
Tumors were isolated from MC38 tumor-bearing mice and TILs analyzed by flow cytometry and CD8+ TIL subsets were sorted for Seahorse assay and ex vivo cytolytic assay. (a). (left panel) Representative flow plots of expression of Tim-3 vs granzyme B (top row) and IFN-γ (bottom row) on Treg cells, conventional CD4+ T cells and CD8+ T cells. (right panel) Representative flow plots show expression of PD-1, Lag-3 vs granzyme B, IFN-γ in Tim3+CD8+T cells. (b). Statistical analysis of granzyme B and IFN-γ depicted in A. (c). Representative histograms (left) and MFI (right) of mitochondrial levels in Tim-3−PD-1− and Tim-3+PD-1+ subsets in CD8+ T cells. (d). Oxygen consumption rate (OCR, left) and extracellular acidification rate (ECAR, right) traces of Tim-3+PD-1+ and Tim-3−PD-1− CD8+ T cells isolated from MC38 tumors. (e) Graphic scheme of ex vivo cytolytic assay and the statistical analysis of specific cytotoxicity of the Tim-3−PD-1− and Tim-3+PD-1+ CD8 T cell subsets. Data were presented as mean ± SEM (n = 3–5). *P < .05, **P < .005, ***P < .0005, ****P < .0001, Student’s t test was performed.
Administration of Tim-3 mAbs and PD-1 mAbs produced an antitumor effect in PD-1-resistant tumor models
PD-1 blockade has shown therapeutic efficacy in clinical trials. However, only a minority of patients respond to PD-1 blockade therapy.19 It is urgent to develop improved immunotherapies to further inhibit tumor progression. Combined PD-1 and Tim-3 mAbs20 or PD-L1 and Tim-3 mAbs11 have been shown to increase antitumor efficacy in some immunogenic tumor models. Consistently, we found that combining PD-1 and Tim-3 mAbs resulted in a slower tumor growth rate than PD-1 mAbs alone in an immunogenic MC38 tumor model (Figure 3a and Figure S2B). In contrast, the administration of Tim-3 mAbs alone had no effect on tumor growth.
Figure 3.
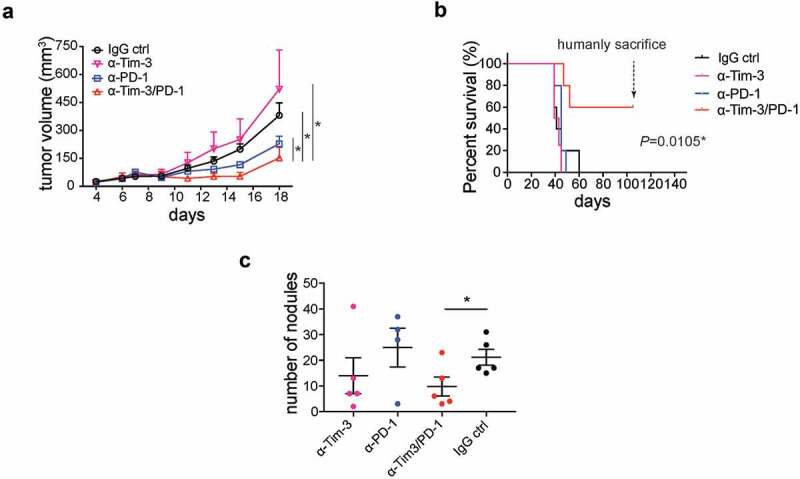
Administration of Tim-3 mAb PD-1 mAb synergistically inhibited tumor progression.
(a). Mean tumor volumes of MC38 tumor-bearing mice treated with indicated mAbs (n = 4 ~ 5). (b). Overall survival of ID8 tumor-bearing mice treated with indicated mAbs (n = 5). (c). Metastatic nodules in lung in 4T1 tumor-bearing mice treated with indicated mAbs (n = 4 ~ 5). *P < .05, one-way ANOVA test, Log-rank test, and student t test were performed.
We also found that Tim-3 mAbs synergized with PD-1 mAbs to prolong overall survival in mice inoculated intraperitoneally (i.p.) with ID8 cells, an ovarian tumor cell line (Figure 3b and Figure S2C). ID8 cell-injected mice that had been treated with control IgG antibodies had noticeable intra-abdominal tumor growth around 35 days after tumor cell inoculation and all died of tumor a few days later. Administration of neither Tim-3 mAbs nor PD-1 mAbs alone prolonged survival in this ovarian cancer model. In contrast, about 60% of the mice treated with Tim-3/PD-1 mAbs combination survived without any sign of tumor growth (Figure 3b). In another experiment, we injected ID8 cells again into tumor-free mice, which had been treated with Tim-3/PD-1 mAbs. No tumor formed in any of these mice, suggesting long-term T cell memory responses were generated in PD-1/Tim-3 mAbs treated mice (Figure S2C). To extend the finding to other tumor models, we examined the antitumor effects of these mAbs in mice inoculated with 4T1 breast tumor cells, which form tumors in the breast fat pad and can metastasize to the lung. Tim-3 mAbs, PD-1 mAbs, or Tim-3 and PD-1 mAbs in combination had no effect on the growth of primary tumors (Fig S2A). However, we noticed that the number of metastatic nodules in the lung was significantly reduced in the Tim-3 and PD-1 mAbs combination group compared to other groups (Figure 3c). Collectively, a combination of Tim-3 and PD-1 mAbs showed significant improved therapeutic effect in different cancer types.
Administration of Tim-3 and PD-1 mAbs enhanced CD8+ T cell function in the TME
Though Tim-3 and PD-1 mAbs combination produced a significant anti-tumor effect, the underlying mechanisms are still unclear. We next determined the cellular and molecular mechanisms mediating the effects of Tim-3 and PD-1 mAbs using the MC38 model (Figure 4a). We found that 24 h after antibody administration, Tim-3 and PD-1 mAbs in combination significantly enhanced the production of IFN-γ and Granzyme B by CD8+ T cells in the TME (Figure 4b,c). And IFN-γ and Granzyme B were increased in both Tim-3+ and Tim-3− subsets (Fig. S3A). In addition, the number of Ki67+ CD8+ T cells was modestly but statistically significantly increased in the group treated with Tim-3 and PD-1 mAbs in combination compared to other groups (Figure 4d,e). In contrast, no difference was observed in Foxp3−CD4+ T cells among the experimental groups (data not shown). Consistently, we observed that IFN-γ production was increased in CD8+ TIL upon Tim-3/PD-1 mAbs treatment in the mouse ovarian cancer ID8 model. Interestingly, in the ID8 model, Tim-3 mAbs treatment alone, when compared to control IgG treatment, also increased IFN-γ production by CD8+ T cells (Figure S3B). These data suggest an increase in CD8+ T cell immune responses immediately after treatment with Tim-3 and PD-1 mAbs.
Figure 4.
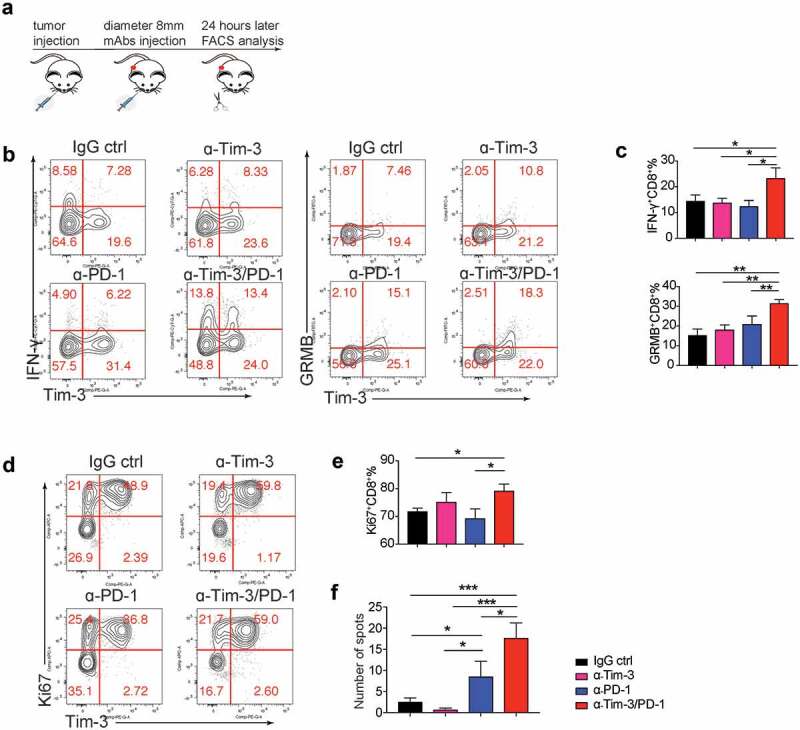
Acute effects of administration of Tim-3 and PD-1 mAbs.
(a-e). MC38 tumor-bearing mice were treated with Tim-3, PD-1, Tim3/PD-1 mAbs or IgG control, 24 h later, tumor-infiltrating lymphocytes were analyzed by flow cytometry. (a). Graphic schematics of the mouse experiment. (b-c). IFN-γ and granzyme B expressions of CD8+ T cells. (d-e). Proliferation of CD8+ T cells. (f). Splenocytes were isolated from mice treated with Tim-3, PD-1, Tim3/PD-1 mAbs or IgG control for 4 times, and applied to ELISpot assay, IFN-γ spots were counted and calculated. Data are presented as mean ± SEM (n = 4 ~ 7), *P < .05, **P < .005, ***P < .0005, one-way ANOVA test and Student’s t test were performed.
Kinetic changes of TIL upon Tim-3 and PD-1 mAbs treatment were also examined at 96 h after antibody administration (Fig S3C). Administration of either Tim-3 or PD-1 mAbs resulted in similar increases in the percentage of IFN-γ-producing CD8+ T cells but not CD4+ T cells in the TME (Fig S3C). Tim-3 and PD-1 mAbs in combination did not further increase IFN-γ+CD8+ T cells (Fig S3C). The cytolytic markers granzymes A and B were also examined. We found that granzyme A+ CD8+ T cells, but not granzyme B+ CD8+ T cells, were significantly increased after PD-1 mAbs treatment and trended even higher with Tim-3/PD-1 mAbs combined treatment (Fig S3C). We also found that proliferation and granzyme B production of CD4+ T cells were significantly increased after PD-1 mAbs treatment and were further increased with Tim-3/PD-1 mAbs combined treatment (Fig S3C). Besides changes in the TME, it was also found that tumor antigen-specific T cells in the spleen were increased upon treatment with Tim3/PD-1 mAbs (Figure 4f). Collectively these data demonstrated that Tim-3/PD-1 mAbs combination increased effector function of Th1 and CD8+ T cells in the TME.
Lag-3 conferred resistance to Tim-3/PD-1combination therapy
We have shown combination therapy with Tim-3 and PD-1 mAbs improved the efficacy of anti-PD-1 therapy. Nevertheless, the tumor growth was not stopped even with both Tim-3 and PD-1 mAbs. We found the expression of a checkpoint inhibitor, Lag-3, was increased on both Foxp3−CD4+ T cells and CD8+ T cells in the Tim-3 and PD-1 mAbs combination group compared to other groups after 24 h treatments (Figure 5a-b) and 96 h treatments (Figure S3D), suggesting Lag-3 might mediate resistance to Tim-3/PD-1 combo-immunotherapy. Besides Lag-3, GITR, a costimulatory receptor, was also induced upon combined treatment with Tim-3/PD-1 mAbs in CD4+ and CD8+ TIL (Figure S3E). We then determined whether the triple combination of Tim-3, PD-1, and Lag-3 mAbs could further improve the antitumor efficacy. Our results indicated that triple combination produced a much greater antitumor effect than Tim-3/PD-1 or PD-1/Lag-3 double combinations (Figure 5c), whereas Tim-3 and Lag-3 mAbs in combination didn’t affect tumor growth when compared to the IgG control group (Figure 5c).
Figure 5.
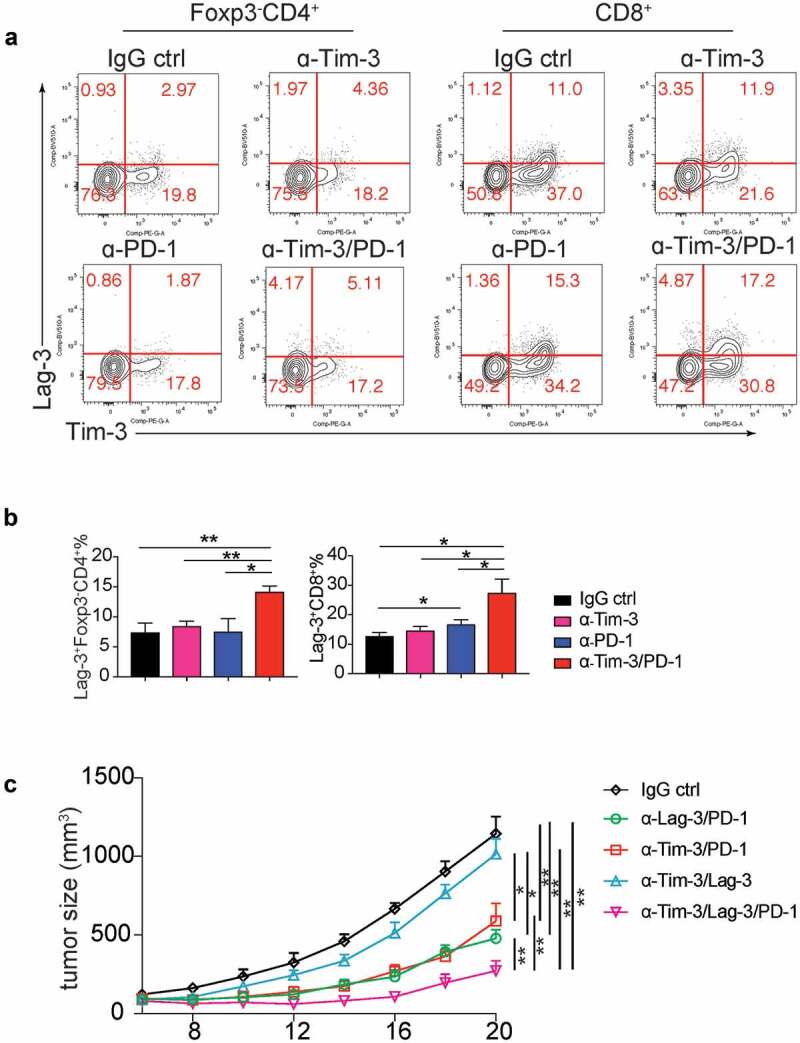
Triple therapy with Tim-3, PD-1 and Lag-3 mAbs synergistically increased the antitumor activity.
a-b. MC38 tumor-bearing mice were treated with Tim-3, PD-1, Tim-3/PD-1 mAbs or IgG control, 24 h later, tumor-infiltrating lymphocytes were analyzed for Lag3 expression by flow cytometry. (a). Representative flow plots of Lag3 expression on tumor-infiltrating CD4+ and CD8 T+ cells. (b). Statistical analysis of Lag3 expression depicted in A (n = 4). (c). Mean tumor volumes of MC38 tumor-bearing mice treated with indicated mAbs (n = 5). Data are presented as mean ± SEM, *P < .05, **P < .005, one-way ANOVA test and Student’s t test were performed.
To further study the molecular mechanisms of the improved anti-tumor effect mediated by the triple therapy, we analyzed TIL at 24 h after treatments. We found that the triple treatment further enhanced Granzyme B production compared to IgG control, Tim-3/PD-1, Tim-3/Lag-3, and PD-1/Lag-3 groups (Figure 6a-b). In contrast, IFN-γ was increased by Tim-3/PD-1, and triple treatment did not result in greater increases compared to Tim-3/PD-1 double treatment. To further determine that the improved anti-tumor efficacy of the triple combination was due to the enhanced cytotoxicity of tumor cell-specific T cells, we performed the ex vivo cytolytic assay using the CD8+ TIL isolated from tumors. Consistently, CD8+ TIL from the triple combination group had much higher specific killing activities (Figure 6c). Collectively, these data established that triple treatment with PD-1, Tim-3, and Lag-3 mAbs synergistically increased the responses of tumor immunotherapy.
Figure 6.
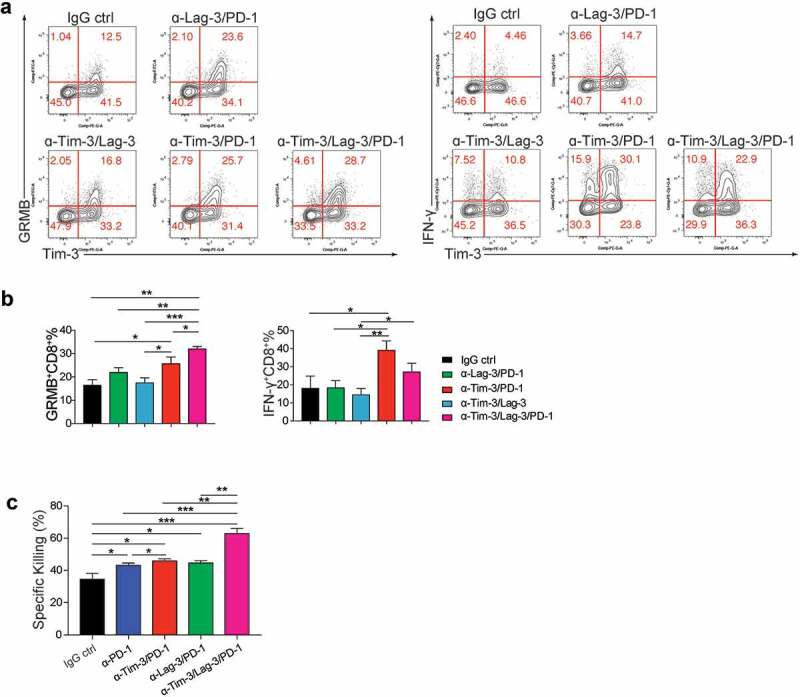
Triple administration with Tim-3, PD-1 and Lag-3 mAbs greatly increased the effector function of tumor-infiltrating CD8+ T cells.
(a-b). MC38 tumor-bearing mice were treated with indicated combinations of mAbs or IgG isotype control, 24 h later, tumor-infiltrating lymphocytes were analyzed by flow cytometry. Representative flow plots and Statistical analysis of IFN-γ and Granzyme B expression on tumor-infiltrating CD8+ T cells (n = 4). (c). Statistical analysis of specific cytolytic activities of CD8+ T cells sorted from tumors treated with indicated groups of mAbs (n = 3–4). Data were presented as mean ± SEM, *P < .05, **P < .005, ***P < .0005, one-way ANOVA test and Student’s t test were performed.
Discussion
In this study, we found that Tim-3 expression in the TME was upregulated in large fractions of Treg, CD4, CD8 T cells, M1, DC1 and small percentages of B cells, NK cells, γδT cells, M2, and MDSC. We also demonstrated that Tim3+ TIL had an effector T cell phenotype, indicating Tim-3 helps regulate effector T cell function. Surprisingly, we found that Tim-3+PD-1+ CD8+ TIL produced similar amounts of effector molecules, such as IFN-γ and granzyme B and had higher cytolytic activities compared to other TIL subsets, suggesting PD-1 and Tim-3 play a role in limiting the function of effector cells rather than maintaining exhaustion state in our model. In addition, we found Tim-3/PD-1 mAbs combination treatment overcame PD-1 resistance in an ovarian cancer model. Interestingly, the combined Tim-3/PD1 mAbs treatment resulted in the upregulation of other immune regulatory receptors such as Lag-3 and GITR on TIL. Triple treatment with PD-1, Tim-3 and Lag-3 mAbs further increased antitumor efficacy compared to Tim3/PD-1, and PD-1/Lag-3 mAbs through increasing granzyme B+ CD8+ TIL and cytolytic activities of CD8+ TIL. Our studies demonstrated cross-regulation between different immune checkpoint molecules in TIL and established that the combination of multiple checkpoint inhibitors is necessary to further improve current tumor immunotherapy.
CD4+ and CD8+ TIL co-expressing multiple immune regulatory receptors such as PD-1, Tim-3, Lag-3, OX-40, GITR, and TIGIT have been reported.9–11,13 Whether, and how, these molecular pathways cross-regulate each other is not fully understood. We found that PD-1 mAbs increase the levels of Tim-3 on the TIL (data not shown), consistent with previous reports.21–23 Our data demonstrate that Tim-3/PD-1 mAbs combination overrode resistance to therapeutic PD-1 blockade.21 In addition, we found that Tim-3/PD-1 mAbs treatment further increased Lag-3 and GITR expression on CD8+ and CD4+ TIL, demonstrating cross-regulation between these immune receptors. Upregulation of Lag-3 and GITR in response to PD-1 and Tim-3 blockade suggests these immune regulatory receptors might play redundant roles. Indeed, we showed that Tim-3/PD-1/Lag-3 mAbs triple combination can further increase antitumor efficacy, supporting such hypothesis. Our finding that Lag3 is upregulated right after PD-1/Tim-3 mAbs treatment supports the practice that all three antibodies should be used together for cancer treatment.
Tim-3+PD-1+ TIL have been reported to be functionally exhausted T cells.9–11,24-26 In addition, TIL expressing multiple checkpoint inhibitors have been shown to be more functionally exhausted.14 However, our data indicated that Tim-3+PD-1+ TIL in the MC38 model were effector T cells. These differences might be due to the models that are used. MC38 is a very immunogenic tumor model and is known to respond well to PD-1 blockade immunotherapy. In addition, the time point we studied might reflect the effector stage of antitumor immune responses. As a result, the effect of PD-1 and Tim-3 mAbs observed in our study was in line with the enhancement of the effector function rather than a reinvigoration of exhausted T cells.
Although we focus on studying Tim-3 expression on effector Th1 and CD8+ T cells, we also found Tim-3 expression on myeloid cells, particularly CD103+ DC1 and M1 cells. Many studies have demonstrated that Tim-3 expression on DC and macrophages might be involved in regulating the function of these myeloid cells during antitumor immunity.27–33 It is foreseeable that expression of Tim-3 on DC1 and M1 might regulate cross-presentation as well as IL-12 production, and thereby regulate antitumor Th1 and CD8 T cell function.28,29,34 The role of Tim-3 on these cells during checkpoint inhibition needs to be further determined. Equally, important, we have also confirmed our previous finding that Tim-3 was specifically upregulated in the Treg in the TME of human lung cancer.9 We have recently found that Tim-3+ Treg represents an effector Treg.35 The role of Tim-3 on tumoral Treg is also elusive at this stage and should be further investigated. Despite strong antitumor activities of PD-1 in combination with Lag3 and Tim-3 mAbs, we did not see the total regression of tumors in the MC38 model or the ID8 model. There are many reasons for this. It is possible that additional immune checkpoint molecules. It is also possible that the MHC-I is down-regulated through epigenetic mechanisms. Future studies on the adaptive immune resistance mechanisms should provide further insights.
One of the most striking findings in this study is the that Lag3, Tim-3 and PD-1 mAbs triple treatment synergistically increased the cytolytic activity of tumor-infiltrating CD8+ T cells. Previous studies showed that PD-1/Tim-3 mAbs treatment increased T cell proliferation and cytokine production. However, the cytolytic activities were not studied. We showed that the increased cytolytic activities were associated with increased granzyme B protein expression. Other molecular mechanisms can also be involved and warrant future studies.
Material and method
Cell lines
Mouse colorectal cancer cell line MC38 and mouse ovarian cancer cell line ID8 were cultured in complete DMEM medium supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin. Mouse breast cancer cell line 4T1 and mouse melanoma B16 cells were cultured in complete RPMI-1640 medium supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin.
Reagents and antibodies
Anti-mouse Tim-3 (clone: RMT3-23), anti-mouse PD-1 (clone: J43), anti-mouse Lag-3 (clone: C9B7W), Rat IgG isotype control (clone: 2A3) and polyclonal American hamster IgG were purchased from Bioxcell company for tumor therapy. For Flow cytometry, anti-mouse CD45 (clone: 30-F11), anti-mouse CD4 (clone: GK1.5), anti-mouse CD8a (clone: 53–6.7), anti-mouse Foxp3 (MF23), anti-mouse γδTCR (clone: GL3), anti-mouse CD103 (clone: M290), anti-mouse Tim-3 (clone: 5D12), anti-mouse Lag-3 (clone: C9B7W), anti-mouse Lag-3 (clone: C9B7W), anti-mouse CD206 (clone: MR5D3), anti-mouse CD62L (clone: MEL-14), anti-mouse CD44(clone: IM7), anti-mouse OX40 (OX-86), anti-mouse IL7R (clone: SB/199), anti-mouse GITR (clone:DTA-1) and anti-mouse CD11b (clone: M1/70) were purchased from BD Bioscience. anti-mouse Ki67(clone: SolA15), IFN-γ (clone: XMG1.2), CD11b (M1/70) were purchased from ebioscience. anti-mouse B220 (clone: RA3-6B2), anti-mouse CD8a (clone: 53–6.7), anti-human/mouse Granzyme B (clone: GB11), anti-mouse Granzyme A (clone: 3G8.5), anti-mouse MHC II (clone: M5/114.15.2), anti-mouse Gr-1 (clone: RB6-8C5), anti-mouse CD24 (clone: M1/69), anti-mouse F4/80 (clone: BM8) and anti-mouse NK1.1 (clone: PK136) were purchased from Biolegend. Mitotracker Deep Red FM labeling kit was purchased from Invitrogen. Ghost 510 and Zombie NIR dye was purchased from Tonbo Biosciences and Biolegend.
Mouse tumor model
6 ~ 8 weeks old C57BL/6 and BALB/c mice were purchased from The Jackson Laboratory and maintained at the specific pathogen-free animal facility of University of Pittsburgh. For MC38 model, C57BL/6 mice were injected with 1 million cells intradermally (i.d.). On day 4 or 8, mice were randomly divided into 4 groups. Anti-Tim-3, anti-PD-1, anti-Tim-3 plus anti-PD-1 or IgG isotype, anti-Tim-3/Lag-3, anti-Tim-3/PD-1, anti-Lag-3/PD-1 or anti-Tim-3/PD-1/Lag-3 groups, mAbs were injected intraperitoneally (i.p.) 200 μg per mouse. Mice were treated with mAbs every 4 days for a total of 4 times. Tumor sizes were measured every 2–3 days by caliper, and the tumor volume was calculated as L× W2/2. Mice were humanely euthanized when tumor diameter reached 20 mm. For ID8 model, C57BL/6 mice were injected i.p. with 2 million ID8 cells. On day 5 or 10, mice were randomly divided into 4 groups, mAbs mentioned above were injected i.p.every 5 days for a total of 5 times. Mice were monitored every 2 days. For 4T1 model, BABL/c mice were injected with 0.05 million 4T1 cells into the mammary fat pad. On day 5, mice were randomly divided into 4 groups. mAbs were injected i.p.every 4 days for a total of 3 times. Tumor sizes were measured every 2–3 days. When mice had apparent expiratory dyspnea, they were sacrificed and lungs were isolated and stained with 15% Indian ink solution and then washed with wash buffer (70% alcohol, 10% formalin and 5% acetic acid). Metastatic nodules were counted. All mouse experiments have been approved by the Institute of Animal Care and Use Committee of the University of Pittsburgh.
Tissue processing and flow cytometry
In MC38 model, C57BL/6 mice were injected with 1 million tumor cells i.d., when tumor sizes reached about 8 mm in diameter, mice were injected i.p. with Anti-Tim-3, anti-PD-1, anti-Tim-3 plus anti-PD-1, anti-Tim-3/Lag-3, anti-Lag-3/PD-1, anti-Tim-3/PD-1/Lag-3 or IgG isotype mAbs. Twenty-four hours or 96 h later, mice were sacrificed and tumors were isolated. In ID8 model, C57BL/6 mice were injected with 2 million tumor cells i.p. and when mice had obvious ascites, mice were injected i.p. with anti-Tim-3, anti-PD-1, anti-Tim-3 plus anti-PD-1 or IgG isotype mAbs. Twenty-four hours later, mice were sacrificed and tumors were isolated. Isolated tumors were cut into small pieces and digested with 0.25mg/ml Liberase TL (Roche) and 0.33mg/ml Dnase (Sigma) in 37 degrees for 30 min. Single-cell suspensions were obtained. For Mitochondrial staining, cells were stained with Mitotracker Deep Red along with surface markers followed by fixation and permeabilization as described before.14 For IFN-γ, Granzyme B or Granzyme A staining, cells were stimulated for 4 h with 50ng/ml phorbol 12-myristate 13-acetate (PMA, Sigma) and 1μg/ml ionomycin (Sigma) in the presence of 10μg/ml Brefeldin A. After stimulation, cells were stained for antibodies to surface marker, followed by fixation permeabilization with Fixation and Permeabilization buffer (ebioscience) according to the manufacturer’s instructions. Then, cells were stained with antibodies to intracellular markers. All the samples were applied to LSRII or Fortessa FACS (BD Biosciences) or Aurora (Cytek Biosciences) and analyzed by using Flowjo software (Tree star).
Seahorse assay
Tim-3+PD-1+ and Tim-3−PD-1− CD8+ T cells were first enriched from MC38 tumors using CD8 TIL beads (Miltenyi) and then stained for surface markers and further purified by FACS. T cells were plated on Cell-Tak coated Seahorse culture plate (100,000 T cells/well) in assay medium containing unbuffered DMEM supplemented with 1%BSA and 25mM glucose, 1mM pyruvate, and 2mM glutamine and analyzed using a Seahorse XFe96 (Agilent). Basal extracellular acidification and oxygen consumption rates were taken for 30 min. Cells were stimulated with oligomycin (2mM), FCCP (0.5mM), 2-deoxyglucose (100mM) and retenone/antimycin A (100mM) to obtain maximal reparatory and control values.
The enzyme-linked immunospot (ELISpot) assay
15 μg/ml anti-IFN-γ (clone:AN18, MabTech) was coated and incubated overnight at 4 degrees. On the other day, mice were sacrificed and spleens were isolated. 5*105 splenocytes were stimulated with 5*104, 200Gy irradiated MC38 cells for 48 h in complete medium at 37 degrees incubator. Forty-eight hours later, the plate was washed and incubated with 1.5 μg/ml biotinylated secondary antibody (clone: R4-6A2-biotin, MabTech) and then incubated and developed with VECTASTAIN Elite ABC HRP Kit (Peroxidase, standard) and AEC Peroxidase (HRP) Substrate Kit. The plate was scanned and counted using the ImmunoSpot Analyzer (Cellular Technology).
Ex vivo micro-cytolytic assay
C57BL/6 mice were inoculated with MC38 cells and when tumor sizes reached about 6 ~ 7 mm in diameter, mice were randomly divided into five groups. Anti-PD-1, anti-Tim-3/PD-1, anti-Lag-3/PD-1, anti-Tim-3/PD-1/Lag-3 or IgG isotype mAbs were injected 200 μg per mouse every 4 days for a total of 3 times. Forty-eight hours after the last treatment, mice were sacrificed and tumors were isolated. For untreated mice, when tumor sizes reached about 10 mm in diameter, mice were sacrificed and tumors were isolated. Single-cell suspensions were first enriched using CD8 TIL beads (Miltenyi) and then stained for surface markers and further purified by FACS to obtain CD8 TIL or Tim-3+PD-1+/Tim-3−PD-1− CD8 subsets. MC38 cells were pre-stimulated with 10ng/ml IFN-γ (Biolegend) overnight and then stained with CellTrace Violet (invitrogen). CD8 TIL and MC38 cells were co-cultured in the presence of PD-1 mAbs (10μg/ml) with the ratio of 10:1 (cell number 30000:3000). Twenty-four hours later, the remain MC38 cells along with reference cells stained with CellTrace Far-red (invitrogen) were analyzed with FACS. Specific killing of CD8 TIL was calculated as 100%* (1-RFeff/RFctrl). RF represents the relative frequency of remaining MC38 cells to reference cells, RFctrl represents the RF value in groups without CD8 TIL, RFeff represents the RF value in groups with CD8 TIL.
Statistical analysis
Statistical analyses, such as one-way ANOVA test, Log-rank test, student t test, were performed with Graphpad Prism.
Funding Statement
This work was partly supported by National Natural Science Foundation of China International Collaboration Grant 31729001 (to J.J. and B.L.). This work was partly supported by startup funding to B.L. Min Yang was supported by China Scholarship Council (File No. 201706920070) and International Exchange Scholarship of Soochow University. Wenwen Du was supported by International Exchange Scholarship of Soochow University.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary Material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Dunn GP, Old LJ, Schreiber RD.. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329–11. doi: 10.1146/annurev.immunol.22.012703.104803. Epub 2004/03/23. PubMed PMID: 15032581. [DOI] [PubMed] [Google Scholar]
- 2.Spranger S, Gajewski TF. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat Rev Cancer. 2018; 18(3):139–147. doi: 10.1038/nrc.2017.117. Epub 2018/01/13. PubMed PMID: 29326431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. 2017; 17(9):559–572. doi: 10.1038/nri.2017.49. Epub 2017/05/31. PubMed PMID: 28555670; PubMed Central PMCID: PMCPMC5731833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu B, Yang M, Wang Q. Interleukin-33 in tumorigenesis, tumor immune evasion, and cancer immunotherapy. J Mol Med (Berl). 2016; 94(5):535–543. doi: 10.1007/s00109-016-1397-0. Epub 2016/ 02/29. PubMed PMID: 26922618. [DOI] [PubMed] [Google Scholar]
- 5.Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ. The immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology. 2013; 138(2):105–115. doi: 10.1111/imm.12036. Epub 2012/ 12/12. PubMed PMID: 23216602; PubMed Central PMCID: PMCPMC3575763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–296. doi: 10.1146/annurev.immunol.25.022106.141609. Epub 2006/ 12/01. PubMed PMID: 17134371; PubMed Central PMCID: PMCPMC2895922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frankel T, Lanfranca MP, Zou W. The Role of Tumor Microenvironment in Cancer Immunotherapy. Adv Exp Med Biol. 2017;1036:51–64. doi: 10.1007/978-3-319-67577-0_4. Epub 2017/ 12/25. PubMed PMID: 29275464. [DOI] [PubMed] [Google Scholar]
- 8.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015; 15(8):486–499. doi: 10.1038/nri3862. Epub 2015/ 07/25. PubMed PMID: 26205583; PubMed Central PMCID: PMCPMC4889009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao X, Zhu Y, Li G, Huang H, Zhang G, Wang F, Sun J, Yang Q, Zhang X, Lu B. TIM-3 expression characterizes regulatory T cells in tumor tissues and is associated with lung cancer progression. PLoS One. 2012;7(2):e30676. doi: 10.1371/journal.pone.0030676. Epub 2012/03/01. PubMed PMID: 22363469; PubMed Central PMCID: PMC3281852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, Kirkwood JM, Kuchroo V, Zarour HM. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. 2010;207(10):2175–2186. doi: 10.1084/jem.20100637. Epub 2010/ 09/08. PubMed PMID: 20819923; PubMed Central PMCID: PMC2947081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010; 207(10):2187–2194. doi: 10.1084/jem.20100643. Epub 2010/09/08. PubMed PMID: 20819927; PubMed Central PMCID: PMC2947065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, Betts MR, Freeman GJ, Vignali DAA, Wherry EJ. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. 2009;10(1):29–37. doi: 10.1038/ni.1679. Epub 2008/ 12/02. PubMed PMID: 19043418; PubMed Central PMCID: PMCPMC2605166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chihara N, Madi A, Kondo T, Zhang H, Acharya N, Singer M, Nyman J, Marjanovic ND, Kowalczyk MS, Wang C, et al. Induction and transcriptional regulation of the co-inhibitory gene module in T cells. Nature. 2018;558(7710):454–459. doi: 10.1038/s41586-018-0206-z. Epub 2018/06/15. PubMed PMID: 29899446; PubMed Central PMCID: PMCPMC6130914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, Ferris RL, Delgoffe GM. The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity. 2016;45(2):374–388. doi: 10.1016/j.immuni.2016.07.009. Epub 2016/08/09. PubMed PMID: 27496732; PubMed Central PMCID: PMCPMC5207350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vilgelm AE, Johnson DB, Richmond A. Combinatorial approach to cancer immunotherapy: strength in numbers. J Leukoc Biol. 2016; 100(2):275–290. doi: 10.1189/jlb.5RI0116-013RR. Epub 2016/ 06/04. PubMed PMID: 27256570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matsuzaki J, Gnjatic S, Mhawech-Fauceglia P, Beck A, Miller A, Tsuji T, Eppolito C, Qian F, Lele S, Shrikant P, et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc Natl Acad Sci U S A. 2010;107(17):7875–7880. doi: 10.1073/pnas.1003345107. Epub 2010/ 04/14. PubMed PMID: 20385810; PubMed Central PMCID: PMCPMC2867907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, Bettini ML, Gravano DM, Vogel P, Liu CL, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012;72(4):917–927. doi: 10.1158/0008-5472.CAN-11-1620. Epub 2011/ 12/22. PubMed PMID: 22186141; PubMed Central PMCID: PMCPMC3288154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, Barczak A, Rosenblum M, Daud A, Barber DL, et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell. 2014;26(5):638–652. doi: 10.1016/j.ccell.2014.09.007. Epub 2014/12/03. PubMed PMID: 25446897; PubMed Central PMCID: PMCPMC4254577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. doi: 10.1056/NEJMoa1200690. Epub 2012/06/05. PubMed PMID: 22658127; PubMed Central PMCID: PMC3544539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ngiow SF, von Scheidt B, Akiba H, Yagita H, Teng MW, Smyth MJ. Anti-TIM3 antibody promotes T cell IFN-gamma-mediated antitumor immunity and suppresses established tumors. Cancer Res. 2011; 71(10):3540–3551. doi: 10.1158/0008-5472.CAN-11-0096. Epub 2011/03/25. PubMed PMID: 21430066. [DOI] [PubMed] [Google Scholar]
- 21.Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, Gandhi L, Redig AJ, Rodig SJ, Asahina H, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:10501. doi: 10.1038/ncomms10501. PubMed PMID: 26883990; PubMed Central PMCID: PMC4757784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kato R, Yamasaki M, Urakawa S, Nishida K, Makino T, Morimoto-Okazawa A, Kawashima A, Iwahori K, Suzuki S, Ueda R, et al. Increased Tim-3+ T cells in PBMCs during nivolumab therapy correlate with responses and prognosis of advanced esophageal squamous cell carcinoma patients. Cancer Immunol Immunother. 2018;67(11):1673–1683. doi: 10.1007/s00262-018-2225-x. Epub 2018/ 08/22. PubMed PMID: 30128737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi L, Chen L, Wu C, Zhu Y, Xu B, Zheng X, Sun M, Wen W, Dai X, Yang M, et al. PD-1 Blockade Boosts Radiofrequency Ablation-Elicited Adaptive Immune Responses against Tumor. Clin Cancer Res. 2016;22(5):1173–1184. doi: 10.1158/1078-0432.CCR-15-1352. PubMed PMID: 26933175; PubMed Central PMCID: PMC4780056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li J, Shayan G, Avery L, Jie HB, Gildener-Leapman N, Schmitt N, Lu BF, Kane LP, Ferris RL. Tumor-infiltrating Tim-3(+) T cells proliferate avidly except when PD-1 is co-expressed: evidence for intracellular cross talk. Oncoimmunology. 2016;5(10):e1200778. doi: 10.1080/2162402X.2016.1200778. Epub 2016/ 11/18. PubMed PMID: 27853635; PubMed Central PMCID: PMCPMC5087305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Granier C, Dariane C, Combe P, Verkarre V, Urien S, Badoual C, Roussel H, Mandavit M, Ravel P, Sibony M, et al. Tim-3 Expression on Tumor-Infiltrating PD-1(+)CD8(+) T Cells Correlates with Poor Clinical Outcome in Renal Cell Carcinoma. Cancer Res. 2017;77(5):1075–1082. doi: 10.1158/0008-5472.CAN-16-0274. Epub 2016/ 11/23. PubMed PMID: 27872087. [DOI] [PubMed] [Google Scholar]
- 26.Mohme M, Schliffke S, Maire CL, Runger A, Glau L, Mende KC, Matschke J, Gehbauer C, Akyüz N, Zapf S, et al. Immunophenotyping of Newly Diagnosed and Recurrent Glioblastoma Defines Distinct Immune Exhaustion Profiles in Peripheral and Tumor-infiltrating Lymphocytes. Clin Cancer Res. 2018;24(17):4187–4200. doi: 10.1158/1078-0432.CCR-17-2617. Epub 2018/02/16. PubMed PMID: 29444930. [DOI] [PubMed] [Google Scholar]
- 27.Anderson AC, Anderson DE, Bregoli L, Hastings WD, Kassam N, Lei C, Chandwaskar R, Karman J, Su EW, Hirashima M, et al. Promotion of tissue inflammation by the immune receptor Tim-3 expressed on innate immune cells. Science. 2007;318(5853):1141–1143. doi: 10.1126/science.1148536. PubMed PMID: 18006747. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Y, Ma CJ, Wang JM, Ji XJ, Wu XY, Moorman JP, Yao ZQ. Tim-3 regulates pro- and anti-inflammatory cytokine expression in human CD14+ monocytes. J Leukoc Biol. 2012;91(2):189–196. doi: 10.1189/jlb.1010591. Epub 2011/ 08/17. PubMed PMID: 21844165; PubMed Central PMCID: PMCPMC3290426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Mingo Pulido A, Gardner A, Hiebler S, Soliman H, Rugo HS, Krummel MF, Coussens LM, Ruffell B. TIM-3 Regulates CD103(+) Dendritic Cell Function and Response to Chemotherapy in Breast Cancer. Cancer Cell. 2018;33(1):60–74 e6. doi: 10.1016/j.ccell.2017.11.019. Epub 2018/01/10. PubMed PMID: 29316433; PubMed Central PMCID: PMCPMC5764109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ocana-Guzman R, Torre-Bouscoulet L, Sada-Ovalle I. TIM-3 Regulates Distinct Functions in Macrophages. Front Immunol. 2016;7:229. doi: 10.3389/fimmu.2016.00229. Epub 2016/07/06. PubMed PMID: 27379093; PubMed Central PMCID: PMCPMC4904032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma CJ, Ren JP, Li GY, Wu XY, Brockstedt DG, Lauer P, Moorman JP, Yao ZQ. Enhanced virus-specific CD8+ T cell responses by Listeria monocytogenes-infected dendritic cells in the context of Tim-3 blockade. PLoS One. 2014;9(1):e87821. doi: 10.1371/journal.pone.0087821. Epub 2014/ 02/06. PubMed PMID: 24498204; PubMed Central PMCID: PMCPMC3909257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, Fujioka Y, Ohba Y, Gorman JV, Colgan JD, et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol. 2012;13(9):832–842. doi: 10.1038/ni.2376. Epub 2012/07/31. PubMed PMID: 22842346; PubMed Central PMCID: PMCPMC3622453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nagahara K, Arikawa T, Oomizu S, Kontani K, Nobumoto A, Tateno H, Watanabe K, Niki T, Katoh S, Miyake M, et al. Galectin-9 increases Tim-3+ dendritic cells and CD8+ T cells and enhances antitumor immunity via galectin-9-Tim-3 interactions. J Immunol. 2008;181(11):7660–7669. doi: 10.4049/jimmunol.181.11.7660. Epub 2008/ 11/20.PubMed PMID: 19017954; PubMed Central PMCID: PMCPMC5886706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moller-Hackbarth K, Dewitz C, Schweigert O, Trad A, Garbers C, Rose-John S, Scheller J. A disintegrin and metalloprotease (ADAM) 10 and ADAM17 are major sheddases of T cell immunoglobulin and mucin domain 3 (Tim-3). J Biol Chem. 2013;288(48):34529–34544. doi: 10.1074/jbc.M113.488478. Epub 2013/ 10/15. PubMed PMID: 24121505; PubMed Central PMCID: PMCPMC3843067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Z, McMichael EL, Shayan G, Li J, Chen K, Srivastava R, Kane LP, Lu B, Ferris RL. Novel Effector Phenotype of Tim-3(+) Regulatory T Cells Leads to Enhanced Suppressive Function in Head and Neck Cancer Patients. Clin Cancer Res. 2018;24(18):4529–4538. doi: 10.1158/1078-0432.CCR-17-1350. Epub 2018/ 05/02. PubMed PMID: 29712685; PubMed Central PMCID: PMCPMC6139056. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.