

A TDNA mutant, in the single copy Blistering1 locus, was created in Penicillium expansum. The mutant showed reduced decay in apple fruit, undetectable levels of patulin, and blistered hyphae. Mass spectrometry of proteins in liquid medium and mycelia revealed that it failed to secrete degrative enzymes and ones involved in patulin synthesis. Quantitative mass spectrometry of mycelial proteins revealed altered cellular networks controlling protein processing. These data show that Blistering1 affects internal and external protein processing involving vesicle-mediated transport.
Keywords: Yeast, virulence, HPLC, electron microscopy, enzymes, exosomes, blue mold, fungal decay, patulin, protein secretion, T-DNA
Graphical Abstract

Highlights
The Blistering1 locus in Penicillium expansum encodes a novel protein with a DnaJ domain.
A single T-DNA insertion was generated in the blistering1 gene which manifested in reduced decay in apple and nearly undetectable levels of the mycotoxin patulin in the T625 mutant.
T625 failed to secrete a set of enzymes that degrade plant cell walls and the three final biosynthetic enzymes of patulin synthesis.
Quantitative mass spectrometry of mycelial proteins revealed that the mutant had altered cellular networks controlling protein processing in the endoplasmic reticulum, protein export, vesicle-mediated transport, and endocytosis.
Transmission electron microscopy of hyphal cross sections confirmed that the mutant formed abnormally enlarged endosomes or vacuoles.
These data reveal that Blistering1 affects internal and external protein processing involving vesicle-mediated transport in a family of fungi with medical, commercial, and agricultural importance.
Abstract
The blue mold fungus, Penicillium expansum, is a postharvest apple pathogen that contributes to food waste by rotting fruit and by producing harmful mycotoxins (e.g. patulin). To identify genes controlling pathogen virulence, a random T-DNA insertional library was created from wild-type P. expansum strain R19. One transformant, T625, had reduced virulence in apples, blistered mycelial hyphae, and a T-DNA insertion that abolished transcription of the single copy locus in which it was inserted. The gene, Blistering1, encodes a protein with a DnaJ domain, but otherwise has little homology outside the Aspergillaceae, a family of fungi known for producing antibiotics, mycotoxins, and cheese. Because protein secretion is critical for these processes and for host infection, mass spectrometry was used to monitor proteins secreted into liquid media during fungal growth. T625 failed to secrete a set of enzymes that degrade plant cell walls, along with ones that synthesize the three final biosynthetic steps of patulin. Consequently, the culture broth of T625 had significantly reduced capacity to degrade apple tissue and contained 30 times less patulin. Quantitative mass spectrometry of 3,282 mycelial proteins revealed that T625 had altered cellular networks controlling protein processing in the endoplasmic reticulum, protein export, vesicle-mediated transport, and endocytosis. T625 also had reduced proteins controlling mRNA surveillance and RNA processing. Transmission electron microscopy of hyphal cross sections confirmed that T625 formed abnormally enlarged endosomes or vacuoles. These data reveal that Blistering1 affects internal and external protein processing involving vesicle-mediated transport in a family of fungi with medical, commercial, and agricultural importance.
All commercial apple cultivars are susceptible to blue mold decay caused by Penicillium spp (1). Long term storage (from 6 to 12 months) coupled with a lack of host resistance leave producers few options other than fungicides to manage postharvest decay (2). As a result of their widespread use in the United States, most chemicals now have reduced efficacy to control blue mold of stored apple fruit (3, 4). Hence, there is a growing need to understand genes and regulatory networks in the fungus for the synthesis of new targeted controls.
P. expansum is the most common and aggressive species causing apple blue mold (5, 6). The disease is characterized by a soft, watery rot that is light brown accompanied by blue-green colored conidia that develop on the fruit surface. Blue mold lesions are symmetrical and round and have distinct margins, diagnostic features that distinguish P. expansum from other postharvest rots. An opportunistic fungus, P. expansum takes advantage of weakened and compromised cells arising from wounds caused by stem punctures and severe bruises that occur before, during, and after harvest. Infections through natural openings like lenticels, open calyx/sinus, and stem pull areas can occur but are far less common (7, 8). During infection, P. expansum secretes cell wall degrading enzymes (CWDE)1. Biochemical studies involving purified CWDEs including pectinases, cellulases, xylanses, and polygalacturonases (PG), provide foundational evidence for their involvement in decay development through tissue maceration, softening, and liquification of host tissues (5, 9). Some of these enzymes are adapted to function in different fruit hosts at varying pH and temperatures. For example, sensing its host, P. expansum will use one PG isozyme during postharvest decay of apple but deploy a biochemically different PG during pear fruit decay (5, 10). The diversity of CWDEs that enable the pathogenic success of P. expansum has been revealed in the genome sequences of several strains which contain hundreds of these genes (11, 12).
P. expansum also produces mycotoxins such as patulin, a polyketide beta unsaturated lactone that can withstand pasteurization (13, 14). The biochemical activity of patulin facilitates fruit cell death and pathogen colonization of the host (although some evidence disputes its role in pathogenicity) (11, 15, 16). Patulin is also toxic to humans and is a potential carcinogen. Consequently, the U.S. Food and Drug Administration has limited patulin content in fruit juices and processed pome fruit products for human consumption at 50 μg/L (17). Patulin also may have antimicrobial activity that favors P. expansum niche colonization. To this end, Penicillium spp. have a high genetic capacity to produce a variety of polyketides and secondary products including other mycotoxins like citrinin and penicillic acid and antibiotics like penicillin (12, 18–20).
Penicillium spp. belong to the same family of fungi as Aspergillus spp., some of which are well-studied for producing aflatoxin and causing disease in immuno-compromised humans (21, 22). Because of the industrial, medical, and agricultural importance of fungi in the Aspergillaceae family, the mechanisms by which these fungi incite disease, synthesize and secrete enzymes and secondary products warrant continued study. Thus, we created transfer-DNA (T-DNA) insertional mutants from the wild-type P. expansum isolate R19 to find genes that control fungal virulence during apple fruit decay. One transformant, T625, exhibited reduced decay in apple fruit had an insertion in a single gene, Blistering1. Our results show that this locus encodes a unique 35 kDa polypeptide with a DnaJ domain and that it influences multiple functions involved in proper CWDE secretion, patulin biosynthesis, protein processing and export, and endocytosis. Because this gene bears little homology to other fungi outside the Aspergillaceae, it is a likely that Blistering1 has a novel role that facilitates host adaptation in apple through proper regulation of protein secretion.
EXPERIMENTAL PROCEDURES
Fungal Transformation
Transformation of P. expansum R19 was performed following an Agrobacterium tumefaciens-mediated method developed by de Groot et al. (23) with minor modifications. The A. tumefaciens cells (EHA105) carrying the pPK2 binary vector are described by Chen et al. (24). R19 was cultured on Potato Dextrose Agar (PDA) at 25 °C for 3–5 days until prolific sporulation occurred. The conidia were collected, diluted to 107 spores per ml water, and mixed with transformation-primed A. tumefaciens (OD600 = 0.3) to obtain 200 μl of a 1:1 mixture. The mixture was spread onto the surface of a sterile nitrocellulose membrane placed on an IM agar plate supplemented with 200 μm acetosyringone. The plate was incubated for 54 h at 25 °C. After co-cultivation, the membrane was transferred to a 9 cm Petri plate containing PDA medium amended with 200 ng/μl cefotaxime and 100 ng/μl hygromycin B. The plate was placed in an incubator at 25 °C for 4 ∼ 6 days until individual colonies were visible. The colonies were individually streaked on the same selective medium and cultured at 25 °C for 3 days. Single colonies were selected and grown with 5 ml of liquid PDA medium containing 200 ng/μl cefotaxime and 100 ng/μl hygromycin B at 200 rpm for 3 days. Colonies positively verified for transformation by both PCR using primers specific for the hygromycin gene cassette via conventional PCR and standard cycling parameters. Sanger DNA sequencing was conducted at Eurofins, Inc. Transformants were stored at 4 °C for further study.
Virulence, Growth, and Germination Assays
Conidia were harvested from ∼7-day-old P. expansum R19 and transformant T625 growing on PDA and PDA containing 100 μg/ml hygromycin respectively. Conidia were removed from the plate surfaces by pipetting 1 ml of filter sterilized water until the solution was light green in color. Spore suspensions were quantified using a light microscope and hemocytometer and adjusted to achieve the desired spore concentration. To assess the virulence of T625 in apple fruit, conidial suspensions were harvested and adjusted to 1 × 105 per ml, and 10 μL of the defined conidial solution was pipetted into wounded apple fruit as described (5). Three apples with three wounds per isolate composed a replicate and the inoculated fruit were incubated at 25 °C for 5 days on cardboard trays in commercial apple boxes. Lesion sizes were measured using a digital micrometer. The mycelial growth rate of T625 was assessed by pipetting 10 μL of conidial suspension (1 × 105 conidia per ml) onto 3 locations on a 9 cm Petri plate containing PDA. Once the conidial suspension dried, the plates were sealed with parafilm and incubated at 25 °C in a temperature-controlled incubator with natural light. Colony diameters were measured using a digital micrometer 3 days later. To assess conidial germination, ∼1000 conidia were applied to the surface of 3 individual 4.5 cm Petri plates. Each plate was wrapped with parafilm and placed in a temperature-controlled incubator at 25 °C with natural light for 16 h. At least 100 spores for each plate were counted. A conidium was considered germinated when the germ tube was more than 2.5× the diameter of the spore. All experiments were conducted at least three times using R19 as a control.
Identification of T-DNA Insertional Location
Genomic DNA was extracted from mycelia using the Qiagen DNeasy Plant Mini Kit (Qiagen, Frederick, MD). The position of T-DNA in transformant T625 was identified by thermal asymmetric interlaced PCR (TAIL-PCR) (25). Thirteen primers including three T-DNA left border (Ppk2-LB1, 2, and 3), three right border (Ppk2-LB1, 2, and 3), and seven arbitrary degenerate ones (AD1–8) were used (supplemental Fig. S10). Three rounds of PCR were conducted on a Bio-Rad PCR machine (CFX96 Touch Real-Time PCR Detection System) with EX TaqDNA Polymerase (TaKaRa Bio, Mountain View, CA) using the PCR product of the previous reaction as a template. A common arbitrary primer and nested T-DNA specific primers were employed in a consecutive manner. Candidate bands from the second or third PCR were purified with the QIAquick PCR Purification Kit (Qiagen, Frederick, MD). The resulting DNA fragments were subcloned with the Invitrogen TOPO TA Cloning Kit (Invitrogen, Frederick, MD) and sequenced using the Sanger method at a commercial facility. The DNA sequences were compared with the R19 genome sequence (26), leading to the determination of the T-DNA location in gene R19_6445.
Southern Blot Analysis
Genomic DNA of P. expansum R19 and T625 was isolated from mycelia using the Qiagen DNeasy Plant Mini Kit (Qiagen, Frederick, MD), digested with restriction endonucleases BglII, separated by 1.3% agarose gel electrophoresis using Tris Acetate EDTA (TAE) buffer, and blotted overnight (12 h) onto an Amersham Biosciences Hybond-N+ nylon membrane at room temperature (GE Healthcare Life Sciences, Laurel, MD). A 283 base pair DNA fragment including a portion of the HYG gene and the Trp promoter was used as the probe. This fragment that carried a restriction site of BamHI was amplified from the pPK2 plasmid with the primer pair TTCGGGCGTACACAAATCGCCC/AGGCACTCTTTGCTGCTTGGACA. Hybridization and rinses were carried out as described in the manufacturer's instructions at 25 and 50 °C. The probes were labeled and detected using the DIG High Prime DNA Labeling and Detection Starter Kit II (Roche, Branchburg, NJ). Chemiluminescence was recorded on Kodak X-ray film.
Gene Expression Analysis
RNA was isolated from frozen mycelia from both R19 and transformant T625 using the RNeasy Plant Mini Kit (Qiagen, Frederick, MD). The mycelia were harvested with forceps from 10-day-old cultures growing on PDA and from infected apple fruit 10 days post inoculation. Genomic DNA contamination was removed from RNA with TURBO DNase (Invitrogen, Frederick, MD). First strand cDNA synthesis was conducted using quantified RNA and iScriptTM Transcription Supermix (Bio-RAD, Hercules, CA). The primer pairs, F1-R19_6445 CACGGTCTTGCCACTTGCTCGT/R1-R19_6445 TCGTTTCGAGTACGTCGCGCTG and, F1-ACT ACGTCGTCCCCATCTACGAG/R1-ACT GCTCAGCGAGGATCTTCATC were used for the expression analysis of the R19_6445 gene and the housekeeping gene actin, respectively (Genbank accession number XM_016739901). Quantitative PCR was performed in CFX96 Touch Real-Time PCR Detection System (Bio-RAD, Hercules, CA) using GoTaq qPCR Master Mix (Promega, Durham, NC). A melt curve analysis was performed to check the specificities of PCR products. Relative expression of the R19_6445 gene was normalized to the expression of actin and shown in fold changes (lowest value = 1) using the cycle threshold (Ct) 2−ΔΔCt method. All PCR experiments were conducted twice and performed with three technical replicates.
Phylogenetic Tree Construction
Evolutionary analyses were conducted in MEGA X (27). The evolutionary history of 18 select protein sequences with various levels of BLASTP homology to R19_6445 was inferred by using the Maximum Likelihood method based on the JTT matrix-based model (28). Bootstrapping was performed (29). Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using a JTT model and then selecting the topology with superior log likelihood value. There was a total of 493 positions in the final dataset.
Apple Broth Cultures
Freeze dried apple cortexes from organic 'Golden Delicious' apples were powdered and added to water at 10 g/L. The solution was stirred vigorously and filtered to clarify particulate materials. A salt solution (containing 10 g/L potassium nitrate, 5 g/L potassium monobasic phosphate, 2.5 g/L magnesium sulfate and 0.002 g/L ferric chloride) was added. After autoclaving, the pH of the medium was 4.3, which is physiologically representative of apple fruit tissue. Fifty ml of the apple broth was dispensed into 125 ml flasks and 50 μL of 1 × 106 conidial suspension of either R19 or T625 was added and placed in a temperature-controlled incubator at 25 °C with shaking at 150 rpm. Three flasks were inoculated with R19 and three for T625 for each experiment and was performed 3 times. After 3 days, the mycelial mats were harvested, filtered to remove residual broth, weighed, frozen in liquid nitrogen, and placed in a −80 °C freezer. Spent broth was collected and placed in labeled 50 ml conical tubes. All mycelial mats and broth samples were not pooled and kept separate. The pH was determined, and the broth was frozen in liquid nitrogen and stored at −80 °C.
Mass Spectrometry of Apple Broth Medium
Proteins were precipitated from three (biological replicate) 25 ml aliquots of post-culture apple broth with 2 volumes 75% acetone/25% trichloroacetic acid followed by centrifugation at 21,000 × g. Protein pellets were washed four times in acetone, dried, and resuspended in 100 mm Tris, pH 8.5/8 M urea. Protein concentration was determined by bicinchoninic assay (Pierce, Rockford, IL). Proteins (160 μg) were reduced in Tris(2-carboxyethyl)phosphine, carboxyamidomethylated with iodoacetamide, and digested overnight at 37 °C with Poroszyme immobilized trypsin (Thermo Fisher Scientific, Waltham, MA). The digested peptides were purified by reverse phase chromatography using Waters Oasis HLB 3 cc Vac Cartridges (Waters Corporation, Milford MA).
Peptides (500 ng) were separated on a 75 μm (inner diameter) fused silica capillary emitter packed with 20 cm of 2.5 micron Synergi Hydro-RP C18 (Phenomenex, Torrance, CA) coupled directly to a Dionex UltiMate 3000 RSLCnano System (Thermo Fisher Scientific, Waltham, MA) controlling a 180-min linear gradient from 3.2% to 40% acetonitrile and 0.1% formic acid at a flow rate of 300 nL per minute. Peptides were electrosprayed at 2.4 kV into an Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Fisher Scientific) operating in data-dependent mode with positive polarity and using m/z 445.12003 as an internal mass calibrant. Quadrupole isolation was enabled, and survey scans were recorded in the Orbitrap at 120,000 resolution over a mass range of 400–1600 m/z. The automatic gain control (AGC) target was 500,000 and the maximum injection time was 50 milliseconds (msec). The instrument was operated in Top Speed mode with a cycle time of 3 s. The most abundant ions (intensity threshold 5000) were fragmented by high-energy collision-induced dissociation (30% energy) and fragment ions were detected in the linear ion trap (AGC 10,000, 35 msec maximum injection). Analyzed precursors were dynamically excluded for 20 s.
MS2 spectrum data were extracted from the raw data with MSConvert (http://proteowizard.sourceforge.net/tools.shtml; ProteoWizard 3.0.10386) and searched with Mascot 2.6.1 (30) against the P. expansum R19 protein database (GenBank accession JHUC01000000 (26); 10,560 records provided in supplemental Table S1). Search parameters were for tryptic digests with one possible missed cleavage, fixed amino acid modification for carbamidomethylation (+57.021 Da, C), variable oxidized methionine (+15.995 Da, M), monoisotopic mass values, ±10 ppm parent ion mass tolerance, ±0.8 Da fragment ion mass tolerance, and #13C = 1.
Mascot output files were processed by PANORAMICS2, a probability-based program that determines the likelihood that peptides are correctly assigned to proteins (31–33). Analysis was limited to peptides having Mascot Ions score-Identity score differences not less than negative 5. Spectral counting was performed, and this procedure has been empirically validated for the relative quantitation of protein abundance (31). A count of 1 was assigned to each peptide for each top-ranked matched spectrum that received a positive peptide probability in PANORAMICS2. The total count for a distinct peptide was based on the total number of qualified spectra, but the total count for a shared peptide was divided by the number of protein groups with which it was shared. These numbers accounting for the identification of all shared and distinct peptides assigned to any one protein group were summed to create a spectral count. Proteins with probabilities > 95% comprising at least 1 peptide with at least 1 spectral count were further evaluated. A normalized spectral abundance factor (NSAF) was generated for each protein by dividing its summed spectral count by its molecular mass and then normalizing that value by the sum of all summed spectral counts divided by molecular mass for the set of proteins discovered (34). A t test (p < 0.01) was performed to identify significantly different natural log-transformed NSAF values for 156 proteins that appeared in all three replicates for both T625 and R19 spent growth media (supplemental Table S2). An additional cut-off was applied to identify proteins exhibiting 2-fold accumulation changes. Separately, there was a set of 38 proteins that was found across three replicates of T625 spent broth (present) but never observed (zero qualified spectral counts) in three replicates of R19 spent broth (absent), and vice-versa, for which no t test could be applied. In this case, a protein was present if the sum of its spectral counts across three replicates was greater than 50 (the median sum of spectral counts for R19 proteins that passed the t test). Signals targeting proteins for secretion were predicted with SignalP 4.1 (35).
Mass Spectrometry of Fungal Mycelial Proteins
Lyophilized mycelia mats collected from triplicate in vitro cultures were frozen in liquid nitrogen and triturated with a mortar and pestle in 100 mm Tris pH 8.5, 1 mm EDTA, 10 mm DTT, 0.5% n-dodecyl-beta-d-maltoside, and proteinase inhibitor mixture (Roche, Mannheim, Germany). The slurry was centrifuged at 22,000 × g, and proteins were precipitated from the supernatant using acetone/trichloroacetic acid. Protein was resuspended in 100 mm Tris, pH 8.5/8 M urea/0.5% n-dodecyl-beta-d-maltoside. Protein concentration was determined by bicinchoninic assay (Pierce, Rockford, IL). Proteins (600 μg) were reduced in Tris(2-carboxyethyl)phosphine, carboxyamidomethylated with iodoacetamide, and digested overnight at 37 °C with Poroszyme immobilized trypsin (Thermo Fisher Scientific, Waltham, MA). The digested peptides were purified by reverse phase chromatography using Waters Oasis HLB 3 cc Vac Cartridges (Waters Corporation, Milford MA). One-hundred micrograms of peptides from each sample were labeled with Tandem Mass Tag (TMT) 6-plex reagents per manufacturer instructions (Thermo Fisher Scientific). The samples were dried, resuspended in 0.1% trifluoroacetic acid, and peptides were C18 purified and their concentrations measured with the Pierce Quantitative Colorimetric Peptide Assay (Thermo Fisher Scientific). Equivalent quantities of the tagged samples were combined to yield a 500-ng aliquot which was analyzed by mass spectrometry (below) to determine label incorporation percentage (> 99%) and to estimate quantitative ratios between samples. On the basis of the quantitative ratios, labeled samples were mixed together and fractionated by high-pH reverse-phase HPLC through a Waters Xbridge 3.5 μm C18 column (4.6 × 15 cm) with a Dionyx UltiMate 3000 pump controlling a 38-min linear gradient from 4% to 28% acetonitrile and 0.5% triethylamine pH 10.7 (36). Seventy-four fractions were pooled by concatenation, dried, and resuspended in 5% acetonitrile and 0.1% formic acid, and the peptide concentrations were measured for 14 pools.
Peptides (∼500 ng per pool) were separated as before and analyzed on the Orbitrap Fusion Lumos Tribrid mass spectrometer. The instrument was operated in Top Speed mode using the multinotch MS3 method with a cycle time of 3 s (37, 38). The AGC target was set to 200,000 and the maximum injection time was set to 50 msec. The most abundant precursor ions (intensity threshold 5000) were fragmented by collision-induced dissociation (35% energy), and fragment ions were detected in the linear ion trap (AGC 10,000, 50 msec maximum injection). Analyzed precursors were dynamically excluded for 45 s. Multiple MS2 fragment ions were captured using isolation waveforms with multiple frequency notches and fragmented by high energy collision-induced dissociation (65% normalized collision energy). MS3 spectra were acquired in the Orbitrap (AGC 100,000; maximum injection time 120 msec, 60,000 resolution scanning from 100–500 m/z).
Mass spectrometry data files were processed with Proteome Discoverer 2.1 (Thermo Fisher Scientific) which extracted MS2 spectra for peptide identification and MS3 spectra for peptide quantitation. MS2 spectra were searched with Mascot against the P. expansum R19 protein database as before. Search parameters were for tryptic digests with two possible missed cleavages, fixed amino acid modification for chemically modified cysteine and labeled N-terminal and internal lysine (+57.021 Da, C; +229.163 Da, K), variable oxidized methionine (+15.995 Da, M), monoisotopic mass values, ±10 ppm parent ion mass tolerance, and ±0.6 Da fragment ion mass tolerance. Peptide spectrum matches (PSM) were processed by Percolator (39) using delta Cn (0.05), strict false discovery rate (FDR) (0.01), relaxed FDR (0.05) and PEP (0.05) settings. Additional filters limited Mascot Ions scores (greater than or equal to 13) and PSM and peptide PEPs (strict 0.01; relaxed 0.05). The estimated PSM strict and relaxed FDR was 0.003. Peptides were assigned to logical protein groups using parsimony. Proteins were quantified on summed signal-to-noise (S/N) ratios for each TMT channel for 43,638 qualified PSMs for unique peptides (isolation interference < 25%, S/N ratio average of all channels > 8; at least medium confidence). The most confident centroid within 0.003 Da of the expected mass of the reporter ions was used. TMT signals were also corrected for isotope impurities (lot specific data provided by the manufacturer). Missing TMT signal values were replaced with a minimum value of 1 for 2,588 qualified PSMs. Matches to contaminants and decoys were removed from the dataset as were proteins with quantitative signal sums across all 6 channels < 100 or less than 2 PSMs contributing to the qualified quantitative signal sum. Protein quantitative values for each channel were log2 transformed and median normalized. A t test was used to measure significant differences and the Benjamini and Hochberg correction was applied to limit false discovery to less than or equal to 0.01. Protein sequences were compared with the Swiss-Prot database (downloaded 11/14/2017) with BLASTP. Signals targeting proteins for secretion were determined with SignalP 4.1 (35). Subcellular localization was predicted with DeepLoc-1.0 (40). Kyoto Encyclopedia of Genes and Genomes (KEGG) identifiers were assigned with the P. digitatum reference and mapped to biochemical pathways (www.kegg.jp).
Apple Maceration Assay
Spent apple culture broth (15 ml) was filter sterilized using a 0.2 μm membrane. Three tubes for each sample (9 in total) from three liquid broth cultures of R19 and T625 and uninoculated broth were analyzed. Apple cortical tissue plugs were harvested aseptically using a #5 cork borer and cut into 1 cm pieces. Three pieces of tissue were weighed together, recorded, and placed in one of three 50 ml conical tubes for each sample. Tubes were placed in a temperature-controlled incubator at 25 °C with shaking at 65 rpm for 7 days. Apple plugs were then removed from each tube, blotted for 10 s each, and weighed collectively.
Patulin Extraction and HPLC Quantification
Liquid cultures were centrifuged for 30 min at 15,000 × g, and the supernatants were filter sterilized through 0.2 μm membranes. Three ml of the sample were treated with chloroform to remove proteins. After centrifugation at 10,000 × g for 10 min, the supernatant was collected and extracted with 3 ml of methanol-water (60:40, volume/volume). The mixture was vortexed for 2 min and mixed with ethyl acetate. The combined organic layer (upper layer) was evaporated to dryness under nitrogen gas. The residue was re-dissolved in 1 ml of methanol: water (80:20, volume/volume) and filtered with a 0.45 μm nylon membrane (Waters Associates, Milford, MA) before HPLC analysis. HPLC was performed using a liquid chromatography system consisting of an Agilent 1200 HPLC with a binary pump, diode array detector, vacuum degasser, column oven, and an autosampler (Agilent Technologies, Palo Alto, CA) (41). The HPLC conditions were as follows: a symmetry C18 column (4.6 mm × 250 mm, 5 μm) (Phenomenex Inc, CA) was used with a column temperature set to 30 °C. Mobile phase A consisted of methanol and mobile phase B consisted of 0.1% phosphoric acid in water. The isocratic elution program was applied for 15 mins: 80% A: 20% B. The flow rate was 0.5 ml/min and injection volume was 20 μl. The diode array detector wavelength was set to 222 nm because the highest absorbance for the reference peak was observed at this wavelength. Patulin was identified by its UV-visible spectrum, and the order of elution previously described for similar acquisition conditions. Patulin was quantified by using known concentrations of commercial standards and integrating the peak areas using Agilent ChemStation software (Agilent Technologies, Palo Alto, CA).
Electron Microscopy
Specimens were observed by Low Temperature-Scanning Electron Microscopy as described by Bolton et al. (42). Fungal cultures, grown on Petri plates containing PDA for 7 days with natural light in a temperature-controlled incubator at 25 °C, were used for analysis. Small blocks of fungal mycelia growing on PDA media were cut out of the growing cultures and secured to 15 cm x 30 cm copper plates or mycelia lifted from the cultures were lightly brushed across ultra-smooth, round (12 mm diameter), carbon adhesive tabs (Electron Microscopy Sciences, Inc., Hatfield, PA). The specimens were frozen conductively in a Styrofoam box by placing the plates on the surface of a pre-cooled (−196 °C) brass bar, the lower half of which was submerged in liquid nitrogen. After 20–30 s, the holders containing the frozen samples were transferred to a Quorum PP2000 cryo-prep chamber (Quorum Technologies, East Sussex, UK) attached to an S-4700 field emission scanning electron microscope (Hitachi High Technologies America, Inc., Dallas, TX). The specimens were etched inside the cryo-transfer system to remove any surface contamination (condensed water vapor) by raising the temperature of the stage to −90 °C for 10–15 min. Following etching, the temperature inside the chamber was lowered below −130 °C, and the specimens were coated with a 10 nm layer of platinum using a magnetron sputter head equipped with a platinum target. The specimens were transferred to a pre-cooled (−130 °C) cryostage in the scanning electron microscope for observation. An accelerating voltage of 5 kV was used to view the specimens. Images were captured using a 4pi Analysis System (Durham, NC). Individual images were re-sized in Adobe Photoshop to format them for publication.
For transmission electron microscopy, mycelia were excised from agar plates with a 1 mm biopsy punch and fixed in 2% paraformaldehyde, 2.5% glutaraldehyde, 0.05% Tween20, 0.05 m NaCacodylate, and 0.005 m CaCl2 at 4 °C overnight. Samples were further processed for TEM using the Pelco Biowave Pro+ Microwave (Ted Pella Inc, Redding, CA) as previously described (43). Ultrathin 70 nm sections were cut using a Reichert/AO Ultracut ultramicrotome with a Diatome diamond knife (Diatome US, Hatfield, PA), mounted onto 100 mesh carbon-formvar coated copper grids, stained for 5 min with 3% Reynolds lead citrate, and imaged at 80 kV with a Hitachi HT-7700 transmission electron microscope (Hitachi High Tech America, Inc., Dallas, TX).
Heterologous Yeast Expression
The plasmid backbone, I2, containing a segment of the yeast 2μ circle for replication, a gene for tryptophan biosynthesis (a selectable marker), genes for ampicillin selection and replication in bacteria, and the phosphoglycerate kinase promoter to drive yeast transcription of inserted DNA was enzymatically assembled in the Gibson Assembly Master Mix (New England BioLabs, Ipswich, MA) with DNA fragments of Blistering1, in forward and reverse directions and with 20 bp overlaps with I2, directly behind the promoter transcriptional start site (44). Saccharomyces cerevisiae strain 2966 [trp1 genotype] was transformed with assembled plasmids and cultured on SC-TRP medium (44).
Experimental Design and Statistical Rationale
Virulence, growth rate, and germination assays were performed at least three times. Quantitative RT-PCR was performed twice on biological replicates with three technical replicates. Apple broth culturing was performed multiple times to yield 3 biological replicates of broth for spectral counting, 3 biological replicates of mycelia for TMT analysis, 3 biological replicates of broth for patulin detection, and 3 biological replicates for maceration assays. Biological replicates consisted of T625 and R19 (control). Excel was used to create charts and to calculate means and standard deviations. Significant differences were determined using a one-way ANOVA followed by post hoc testing for means separation using Tukey's HSD (noted in the figure legends). Special statistical procedures for the mass spectrometry data is specified in the previous sections.
RESULTS
The blistering1 Mutant Has Reduced Virulence in Apple Fruit
Wild-type P. expansum R19 was transformed by Agrobacterium-mediated integration with a gene for hygromycin resistance, and 450 transformants were selected and analyzed for virulence in mechanically wounded apple fruit. One transformant, T625, produced lesions that were on average 6.6 mm smaller (18.3 mm versus 24.9 mm) in apple at 5 days post inoculation (Fig. 1A, 1C), and its mycelial growth rate was slightly reduced by 1.4 mm/day (4.95 mm/day versus 6.54 mm/day) in vitro (Fig. 1B, 1D). The observed defect in apple was not a function of selection pressure of T-DNA maintenance (Fig. 1D) and was not caused by reduced conidial germination (Fig. 1E). Scanning electron microscopy revealed that T625 had surface alterations including blisters and protrusions that were widespread across the surface of the hyphae and at the conidiophore base whereas the R19 surfaces were smooth (Fig 2A, 2B). No morphological defects in conidia, numbers of conidia, or germ tube malformations were observed.
Fig. 1.

Phenotypic evaluation of P. expansum wild-type R19 and transformant T625. A, Apples inoculated with equal amounts of spore suspensions of T625 (left) and R19 (right). B, Cultures of T625 (left) and R19 (right) on apple broth solid medium after 7 days of growth at 25 °C. C, Mean lesion diameter for apples inoculated with conidial suspensions of R19 and T625 incubated for 5 days at 25 °C. D, Mean mycelial growth rate of R19 and T625 on and off hygromycin selection. E, Mean conidial germination rate of R19 and T625 on and off hygromycin selection. Treatments with different letters are statistically significant following ANOVA and Tukey's HSD test and error bars represent standard deviation.
Fig. 2.
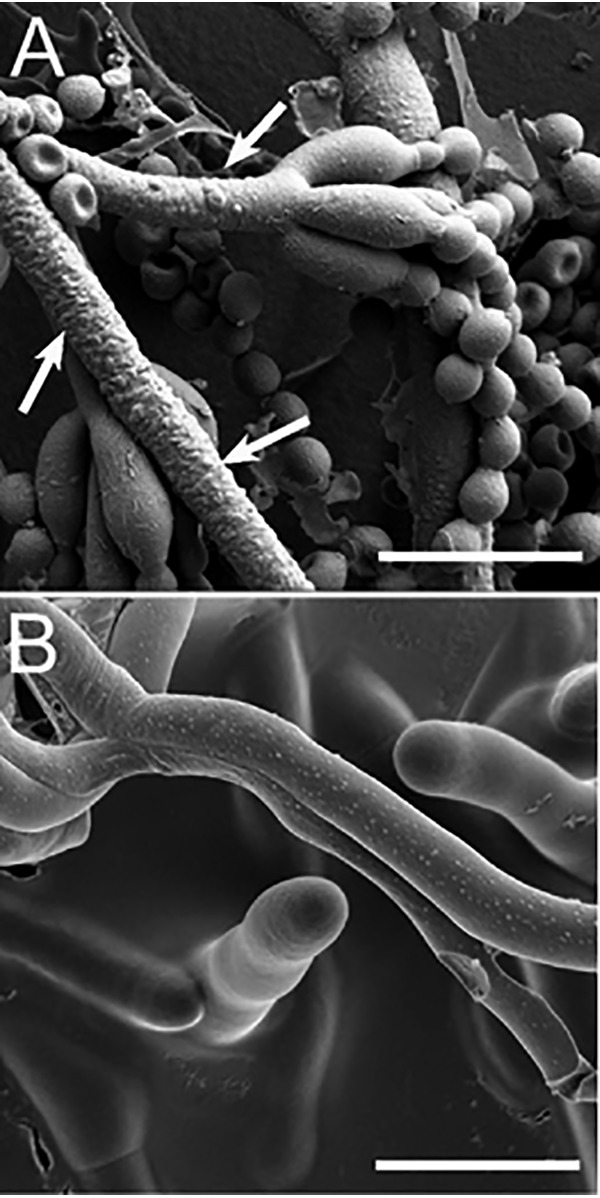
Scanning electron micrograph of Penicillium expansum. A, Transformant T625. White arrows point to bumps and blisters on the hyphae. B, R19 smooth hyphal surfaces. White bar in each panel is 10 microns long.
Southern blot hybridization, using the hygromycin resistance gene as a probe, confirmed single copy integration in the T625 genome (Fig. 3A). TAIL-PCR revealed T-DNA insertion 137 bp downstream from the predicted start codon in gene R19_6445 (contig JHUC01000202.1 of the R19 genome sequence assembly (26)) (Fig. 3B). Quantitative RT-PCR confirmed that the expression of R19_6445 was below our level of detection during apple fruit decay and during growth on PDA in vitro (Fig. 3C). R19_6445 was named Blistering1, and its mutation by T-DNA insertion (blistering1) is responsible for the growth and virulence defects in transformant T625.
Fig. 3.

Confirmation of T-DNA copy number, T-DNA site integration, and gene expression in transformant T625. A, Southern blot hybridized with a portion of the HYG gene, indicating a single T-DNA integration in the T-625 genome as shown by the single band. Lanes 3 and 4 contained 1 μg of genomic DNA digested with BglII. Lane 1 = high molecular weight marker, 2 = low molecular weight marker, 3 = R19, 4 = T625. B, Illustration showing location of the T-DNA in the coding sequence of 918 bp of Blistering1. Positions for the T-DNA and DnaJ domain are shown. C, Quantitative RT-PCR of Blistering1 during in vitro growth and apple fruit decay (10 days post-inoculation) for R19 and T625, error bars represent standard deviation. D, Phylogenetic analysis by Maximum Likelihood method for Blistering1 and top BLAST hits from 18 fungal, soybean, and human proteins. Evolutionary analyses were conducted in MEGA X. The evolutionary history was inferred by using the Maximum Likelihood method based on the JTT matrix-based model. The tree with the highest log likelihood (-8707.21) is shown. Bootstrap values are indicated on each branch. The percentage of trees in which the associated taxa clustered together is shown next to the branches. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site.
Blistering1 Encodes a Unique DnaJ-Containing Protein
Blistering1 is a single copy gene in the R19 genome, is 918 bp, and does not contain introns. Blistering1 is flanked upstream by gene R19_6429 (forkhead domain) which is transcribed from the opposite strand and downstream by R19_6449 (histidine kinase domain) which is transcribed from the same strand. The orthologous Blistering1 in P. expansum strain MD-8 (11) is gene Pex2_008940, also flanked by a histidine kinase domain gene (Pex2_008930) and a forkhead domain gene (Pex2_008950) in contig NW_015971442. The Blistering1 protein from R19 is predicted to have a mass of 35 kDa, is 305 amino acids long, and has a DnaJ domain proximal to the N terminus (Fig. 3B). Other motifs such as a nuclear localization signature and a signal domain targeting the protein for secretion were not identified. Instead, the protein is predicted by DeepLoc to target to the mitochondrion (40). BLASTP analysis showed the highest sequence identity to P. digitatum XP_014536403.1 (89%), which was annotated as a HSP40 co-chaperone, but our BLASTP tests revealed no similarities (except for a DnaJ domain) to canonical HSP40 proteins in P. expansum R19, yeast, or humans. Likewise, Blistering1 lacked broader sequence similarity to Sec63 or signal-recognition particles involved in protein translocation (45), or ERdj4 that interacts with IRE1 to repress the unfolded protein response in the endoplasmic reticulum (ER) (46). Phylogenetic analysis showed that Blistering1 forms a well-supported clade with proteins from other Penicillium spp. and Aspergillus spp. (Fig. 3D). Blistering1 diverges from proteins of fungal pathogens of humans such as Histoplasma capsulatum, Coccoides posadasii, Paracoccidoides lutzi. DnaJ-containing proteins from fission yeast (Schizosaccharomyces pombe), plants (Glycine max), or humans are distantly related and bear little sequence homology. Thus, Blistering1 encodes a specialized protein of Aspergillaceae.
The Blistering1 Mutant Has Altered Protein Secretion in Apple Broth Culture
P. expansum secretes CWDEs and other enzymes to degrade apple tissues (9, 10). Consequently, we hypothesized that T625 had altered protein secretions during apple fruit decay. We used mass spectrometry and spectral counting to analyze the proteins in the spent liquid apple broth after in vitro culture of R19 and T625. Overall, there were 37 proteins absent or with reduced amounts in T625 spent broth compared with R19 spent broth, and 33 of these were predicted to have in their coding genes sequence motifs targeting them for secretion (Table I). These proteins included CWDE-class proteins such as xylosidase, amylase, glycosyl hydrolase, pectin methylesterase, and PG. They also included three enzymes that catalyze the final 3 steps of patulin biosynthesis, PatB, PatO, and PatE (17). These results indicated that the loss of Blistering1 eliminated or reduced the secretion of two groups of enzymes important to P. expansum virulence.
Table I. Proteins absent in T625 spent broth but detected in R19 control spent broth or reduced more than one log2-fold compared to R19 control spent broth.
| Protein | Absent in T625 or Log2 fold decrease in T625 broth | Annotation | Secretion Signal |
|---|---|---|---|
| R19_4596 | Absent | Beta-xylosidase | Yes |
| R19_5734 | Absent | Glycosyl hydrolases family 11 | Yes |
| R19_6220 | Absent | Fungal alpha-l-arabinofuranosidase | Yes |
| R19_6244 | Absent | Starch binding domain-containing | Yes |
| R19_7091 | Absent | Alpha-amylase | Yes |
| R19_733 | Absent | Gmc oxidoreductase, PatE | Yes |
| R19_735 | Absent | Carboxylesterase family, PatB | Yes |
| R19_737 | Absent | FAD binding domain, PatO | Yes |
| R19_8562 | Absent | Unknown | Yes |
| R19_8648 | Absent | Pectin methylesterase | Yes |
| R19_8672 | Absent | Extracellular exo-polygalacturonase | Yes |
| R19_4239 | -3.3 | Glycoside hydrolase family 12 | Yes |
| R19_5814 | -3.0 | MFS sugar transporter | No |
| R19_10025 | -3.0 | Extracellular polygalacturonase | Yes |
| R19_5568 | -2.9 | Glucose oxidase | Yes |
| R19_9156 | -2.5 | Glucose oxidase | Yes |
| R19_8887 | -2.4 | Extracellular phytase | Yes |
| R19_110 | -2.3 | Endo-1,4-beta-xylanase | Yes |
| R19_7198 | -2.2 | Exoinulinase | Yes |
| R19_3017 | -2.0 | Unknown | Yes |
| R19_8291 | -1.9 | Beta-1,6-glucanase Neg1 | Yes |
| R19_2831 | -1.8 | Extracellular thaumatin domain | Yes |
| R19_1057 | -1.8 | Feruloyl esterase | No |
| R19_6005 | -1.6 | Extracellular rhamnogalacturonase | Yes |
| R19_5260 | -1.6 | FAD binding domain-containing | Yes |
| R19_6116 | -1.4 | Endoglucanase | Yes |
| R19_8601 | -1.4 | Extracellular cell wall glucanase Crf1 allergen Asp F9 | Yes |
| R19_4357 | -1.4 | Unknown | Yes |
| R19_10138 | -1.3 | Extracellular endoglucanase | Yes |
| R19_7121 | -1.3 | EPD1 precursor | Yes |
| R19_3964 | -1.2 | Triacylglycerol lipase | Yes |
| R19_5804 | -1.2 | GPI anchored dioxygenase | Yes |
| R19_1949 | -1.1 | Extracellular invertase | No |
| R19_1958 | -1.1 | Mycelial catalase Cat1 | Yes |
| R19_1479 | -1.1 | Triacylglycerol lipase | No |
| R19_5589 | -1.0 | Extracellular aldonolactonase | Yes |
| R19_885 | -1.0 | Carboxypeptidase S1 | Yes |
By contrast, there were 47 proteins absent in R19 spent broth or with reduced accumulation in R19 spent broth compared with T625 (Table II). In other words, these proteins were present in T625 broth. Forty-one of these were predicted to have in their coding genes sequence motifs targeting them for secretion. Some of the proteins were CWDEs, but they were different than those that were absent from T625 spent broth. Furthermore, a polyketide synthase, triacylglycerol lipase, and a glutaminase were present despite not having apparent secretion signals. Hence, the loss of Blistering1 prevented the secretion of some CWDEs and 3 specific patulin biosynthesis enzymes, but also resulted in the mis-secretion of other CWDEs and other enzymes.
Table II. Proteins absent in R19 spent broth but found in T625 spent broth or reduced more than one log2-fold in R19 compared to T625 control spent broth.
| Protein | Absent in R19 or Log2 fold decrease in R19 broth | Annotation | Secretion signal |
|---|---|---|---|
| R19_1444 | Absent | Extracellular | Yes |
| R19_1890 | Absent | SUN domain (Adg3) | Yes |
| R19_1967 | Absent | Unknown | Yes |
| R19_2538 | Absent | Exo-beta-1,3-glucanase (Exg1) | Yes |
| R19_2557 | Absent | Glycoside hydrolase family 2 | Yes |
| R19_2943 | Absent | Exo-beta-1,3-glucanase | Yes |
| R19_3280 | Absent | Bys1 family | Yes |
| R19_3591 | Absent | Beta-N-hexosaminidase | Yes |
| R19_3649 | Absent | Hypothetical protein PDIP_83530 | Yes |
| R19_4294 | Absent | Endo-1,3-beta-glucanase Engl1 | Yes |
| R19_4662 | Absent | Alpha-glucosidase | Yes |
| R19_5520 | Absent | Beta-d-glucoside glucohydrolase | No |
| R19_5656 | Absent | Extracellular matrix | Yes |
| R19_5771 | Absent | Endo-arabinase | Yes |
| R19_5874 | Absent | Glucose oxidase | Yes |
| R19_6082 | Absent | Unknown | Yes |
| R19_7076 | Absent | Unknown | Yes |
| R19_7318 | Absent | Class III chitinase | Yes |
| R19_7474 | Absent | Alpha-amylase | Yes |
| R19_766 | Absent | Polyketide synthase | No |
| R19_868 | Absent | Choline dehydrogenase | Yes |
| R19_8870 | Absent | Allergen Asp f 15 | Yes |
| R19_8906 | Absent | Endo-chitosanase, pseudogene | Yes |
| R19_8923 | Absent | Aspergillopepsin, putative | Yes |
| R19_9104 | Absent | Triacylglycerol lipase | No |
| R19_9217 | Absent | Unknown | Yes |
| R19_9976 | Absent | Pheromone processing carboxypeptidase (Sxa2) | Yes |
| R19_4666 | -1.1 | Extracellular cell wall glucanase | Yes |
| R19_8762 | -1.3 | Unknown | Yes |
| R19_3584 | -1.3 | Purine nucleoside permease | Yes |
| R19_3589 | -1.5 | Pectate lyase A | Yes |
| R19_6841 | -1.6 | Extracellular conserved serine-rich | Yes |
| R19_8185 | -1.7 | SUN domain (Uth1) | No |
| R19_4912 | -1.8 | FAD binding domain | Yes |
| R19_3052 | -1.9 | GPI anchored endo-1,3(4)-beta-glucanase, | Yes |
| R19_9201 | -1.9 | Amidase family | Yes |
| R19_9517 | -2.0 | Mono- and diacylglycerol lipase precursor | Yes |
| R19_4525 | -2.1 | Lipase, class 2 | Yes |
| R19_4345 | -2.2 | Unknown | Yes |
| R19_9832 | -2.3 | Thioredoxin reductase | Yes |
| R19_1565 | -2.3 | Unknown | Yes |
| R19_5059 | -2.3 | I-asparaginase | Yes |
| R19_2341 | -2.6 | Allergen Asp f 4 | Yes |
| R19_4911 | -3.0 | Glutaminase | No |
| R19_2474 | -3.1 | Aminopeptidase | No |
| R19_3269 | -3.5 | Glycosyl hydrolase family 43 | Yes |
| R19_10265 | -4.0 | Unknown | Yes |
The absence and reduction of CWDEs in T625 spent broth implied that the broth itself may have a reduced capacity to degrade apple tissue. Using apple cortical tissue plugs to test digestion, we found that the maceration capacity of filter-sterilized spent broth was ∼3-fold greater for R19 compared with T625, leading to a significant reduction in apple tissue mass in vitro (Fig. 4B). Morphological changes such as softening, loss of shape, discoloration were apparent in the plugs incubated with R19 spent broth compared with T625 spent broth and to unspent broth (Fig. 4A). The mass of the mycelial mat of T625 retrieved from the liquid culture was 0.19 g smaller than R19 (0.74 g versus 0.93 g) after the same period of growth (Fig. 4C). That slight growth reduction, however, does not alone account for the absence of CWDEs in T625 spent broth. Rather, secretion irregularity likely explains the loss of maceration capacity and, consequently, the reduction in virulence for T625.
Fig. 4.
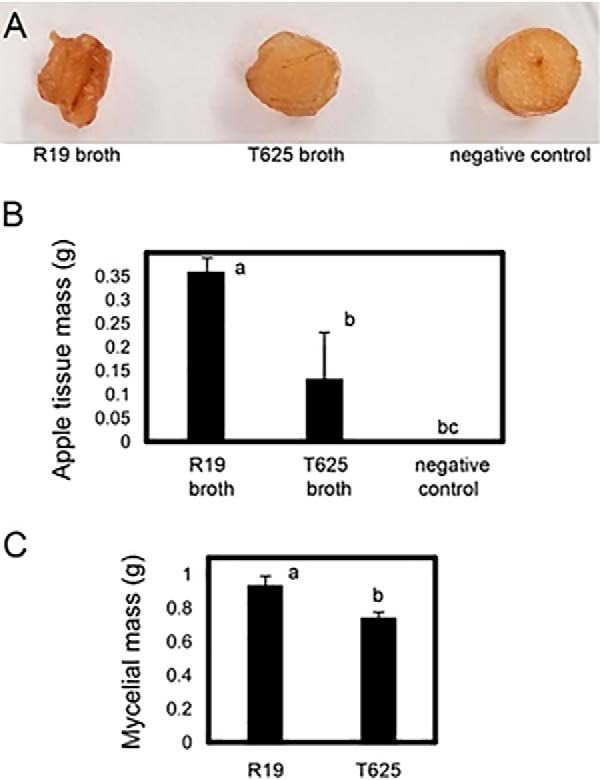
Maceration capacity of sterile, spent broth from R19 and transformant T625 liquid cultures. A, Apple tissue plugs 7 days after incubation with sterilized spent growth broth of R19, T625, and sterile unspent broth (negative control). B, Mean apple tissue mass (g) after incubation with sterilized spent growth broth as in Fig. 4A. C, Mean mycelial mass after growth in liquid broth. Treatments with different letters are statistically different following ANOVA and Tukey's HSD test and error bars represent standard deviation.
The loss of 3 patulin biosynthetic enzymes in spent broth for T625 also implied that the broth may have reduced amounts of patulin. Indeed, patulin was reduced by more than 30-fold in T625 spent broth compared with R19 (Fig. 5A). The PatN gene encodes the isoepoxydon dehydrogenase that produces phyllostine, the substrate of PatB (17). PatN does not encode a secretion signal, and PatN was not detected in the spent broth for T625 or R19. PatN, however, is optimally transcribed at pH 5.0 and decreases at higher pH (17). The final pH of T625 spent broth was higher at 5.9 compared with 5.3 for R19 (Fig. 5B). Although somewhat higher pH may have partially suppressed PatN and the production of phyllostine in T625, it also is reasonable to hypothesize that the loss of secretion of PatB, PatO, and PatE were major factors contributing to the 30-fold decrease in patulin production.
Fig. 5.
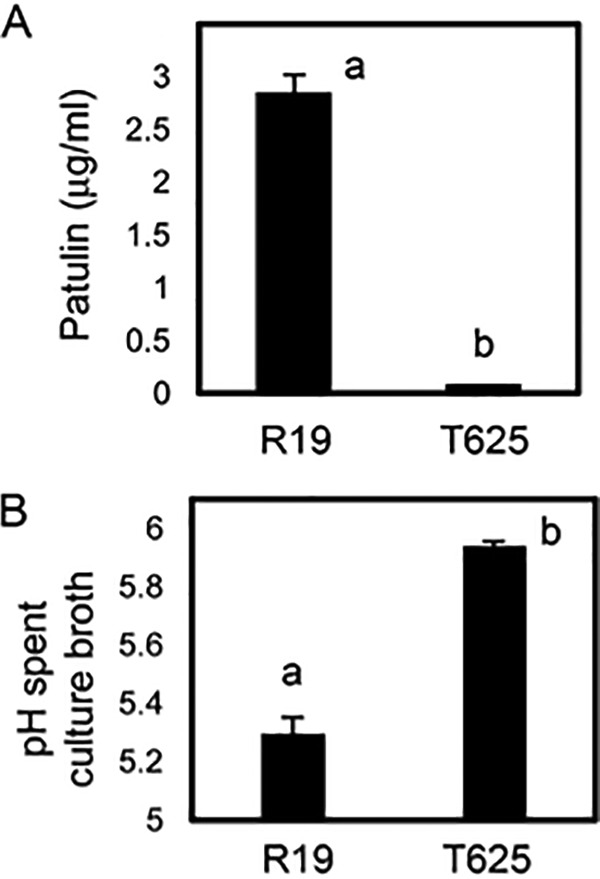
Amount of patulin and pH of spent growth broth for R19 and transformant T625. A, Mean patulin amounts as determined by HPLC. B, Mean spent broth pH. Treatments with different letters are statistically different following ANOVA and Tukey's HSD test and error bars represent standard deviation.
Alterations in the Accumulation of T625 Mycelial Proteins
Because of the drastic alteration in protein secretion in T625, we suspected that Blistering1 had widespread effects on intercellular protein processing and sorting machinery. Therefore, we purified proteins from R19 and T625 mycelial mats harvested from apple broth cultures grown in triplicate. Peptides were prepared, labeled with TMTs, and analyzed by mass spectrometry (Isasa et al., 2015). There were 4589 proteins detected, 3282 of which were quantified by their TMT signals, constituting 31% of the predicted genes in the R19 genome (26) (supplemental Tables S3 and S4). Pearson correlation clustering of the TMT quantitative values revealed the continuity between the biological replicates (Fig. 6A). The log2 fold changes for all proteins exhibited a normal distribution, with 806 proteins exhibiting significant increases and 798 proteins exhibiting significant decreases in T625 (FDR < 1%; Fig. 6B). Because Blistering1 gene expression was detectable in R19, we expected to find the Blistering1 protein. Blistering1, however, was not detected in any sample, although peptides were found for the genes immediately upstream, R19_6429 (-1.5 log2 fold change in T625), and downstream, R19_6449 (no significant change). This indicated that this genomic region was transcriptionally active in both R19 and T625 during liquid culture growth.
Fig. 6.

Hierarchical clustering and statistical representation of quantitative data for mycelial proteins from R19 and transformant T625. A, Pearson correlation clustering of the normalized and scaled tandem mass tag (TMT) reporter ion S/N values for three biological replicates of R19 and T625. The scale bar shows that blue is the highest amount of relative accumulation (>30) and white is the lowest (<5). Clustering was performed in Treeview 3.0. B, Volcano distribution of log2 average of normalized TMT reporter ion S/N values for T625 proteins divided by R19 controls versus the -log (t test p value) for each set of reporter ion S/N values for each protein. Proteins with p values with < 1% false discovery rate (FDR) are in black and those with >1% FDR are in gray. C, Distribution of predicted localizations for sets of significantly increased and decreased proteins in T625.
The cellular localizations of the quantified proteins were predicted (40), and their distributions revealed that the set of proteins with increased accumulation in T625 comprised more proteins with extracellular targeting motifs (Fig. 6C). By contrast, the group of proteins with decreased accumulation comprised more polypeptides with nuclear targeting motifs (Fig. 6C). In other words, proteins predicted to be extracellular had increased accumulation in T625 mycelia. For example, of the top 20 proteins with the greatest increased accumulation in the T625 mycelia (Table III), 16 were predicted to localize to the extracellular domain. Tellingly, 6 of these 16 proteins also were mis-secreted into the T625 culture broth (absent in R19) (Table II). These results reveal that the loss of Blistering1 led to the irregular, increased accumulation and increased secretion of extracellular proteins that might not normally be produced during lesion development in apple fruit. Meanwhile, none of the top 20 proteins with the greatest decreases in accumulation were predicted to be extracellular (they had other destinations) (Table III), and none were secreted into the T625 culture broth (Table I). These results show that decreased accumulation of some proteins correlates with their concomitant decrease in secretion.
Table III. Greatest protein abundance changes in T625 mycelia compared to R19 control.
| Protein | log2 fold change T625 over R19 | Annotation | Predicted localization |
|---|---|---|---|
| R19_4337 | 7.7 | Antigenic thaumatin-like protein | Extracellular |
| R19_3358 | 7.4 | unknown | Extracellular |
| R19_7793 | 6.9 | Conidial hydrophobin Hyp1 | Extracellular |
| R19_34 | 6.4 | Cell wall protein phiA | Extracellular |
| R19_820 | 6.3 | Unknown | Extracellular |
| R19_9242 | 6.2 | Probable pectin lyase A | Extracellular |
| R19_10303 | 6.1 | Agglutinin-like protein | Cell membrane |
| R19_5771 | 6.0 | Endo-arabinase | Extracellular |
| R19_7318 | 5.9 | Class III chitinase | Extracellular |
| R19_7076 | 5.6 | Unknown | Extracellular |
| R19_6024 | 5.6 | Probable feruloyl esterase A | Extracellular |
| R19_2325 | 5.5 | FAD-linked oxidoreductase azaL | Extracellular |
| R19_8870 | 5.5 | Allergen Asp f 15 | Extracellular |
| R19_3280 | 5.5 | Bys1 family | Extracellular |
| R19_4912 | 5.4 | FAD binding domain | Extracellular |
| R19_782 | 5.4 | Putative nitroreductase HBN1 | Cytoplasm |
| R19_3453 | 5.4 | Dimethylglycine oxidase | Peroxisome |
| R19_7079 | 5.3 | NADP-dependent alcohol dehydrogenase 6 | Cytoplasm |
| R19_3031 | 5.2 | Glycosyl hydrolase | Extracellular |
| R19_4294 | 5.2 | Endo-1,3-beta-glucanase Engl1 | Extracellular |
| R19_357 | -3.7 | Cell wall integrity and stress response component 3 | Cell membrane |
| R19_10321 | -3.7 | Casein kinase II subunit beta-1 | Nucleus |
| R19_6292 | -3.8 | Unknown | Mitochondrion |
| R19_6032 | -3.8 | Unknown | Cell membrane |
| R19_6241 | -3.8 | Uncharacterized peptidase YuxL | Cytoplasm |
| R19_7431 | -3.9 | Uncharacterized protein YbiU | Mitochondrion |
| R19_6774 | -3.9 | Unknown | Cell membrane |
| R19_151 | -3.9 | 37S ribosomal protein S17, mitochondrial | Plastid |
| R19_2677 | -3.9 | Unknown | Cell membrane |
| R19_9782 | -4.0 | Esterase | Cytoplasm |
| R19_3425 | -4.0 | Unknown | Cytoplasm |
| R19_1067 | -4.0 | Unknown | Nucleus |
| R19_9595 | -4.0 | Bouquet formation protein 4 | Nucleus |
| R19_6884 | -4.1 | SUR7 family protein pun1 | Cell membrane |
| R19_8934 | -4.1 | Protein SSO2, SNARE domain, STX1B | Golgi apparatus |
| R19_3505 | -4.1 | UBX domain-containing protein 2 | Cytoplasm |
| R19_6888 | -4.2 | PAB1-binding protein 1 | Cytoplasm |
| R19_10358 | -4.7 | Unknown | Cell membrane |
| R19_5407 | -4.7 | 28 kDa heat- and acid-stable phosphoprotein | Nucleus |
| R19_10370 | -4.7 | Peroxiredoxin | Nucleus |
The patulin biosynthetic enzymes PatB, PatE, and PatO that were not secreted into the T625 spent broth (Table I) were among those that exhibited decreased accumulation in T625 mycelia (Table IV). Other enzymes important for patulin biosynthesis also were found to be reduced. PatK, which catalyzes the first step of patulin biosynthesis, the ER-targeted cytochrome P450 PatH that catalyzes the third step, phyllostine producing PatN, and membrane transporters PatC and PatM all exhibited 4-fold or greater reductions in T625 mycelia (Table IV). These findings reveal two things. First, the overall decrease in accumulation of these enzymes, not just the secreted ones or those regulated by culture pH, likely resulted in the reduction of patulin biosynthesis. Second, not all the proteins involved in patulin biosynthesis were secreted. This observation, coupled with the prior results showing a greater proportion of nuclear-localized proteins with decreased accumulation in T625 (Fig. 6C), and the dramatic 14 to 22-fold decrease of proteins with various internal cellular destinations (Table III) together reveal that Blistering1 affects more than just the secretion of proteins during apple fruit decay.
Table IV. Patulin operon protein changes in T625 mycelia compared to R19 control.
| Protein | log2 fold change T625 over R19 | Annotation | Predicted localization |
|---|---|---|---|
| R19_756 | Not found | PatA | Cell membrane |
| R19_735 | -2.4 | PatB | Extracellular |
| R19_747 | -2.8 | PatC | Cell membrane |
| R19_753 | No significant change | PatD | Cytoplasm |
| R19_733 | -1.9 | PatE | Extracellular |
| R19_742 | -3.9* | PatG | Peroxisome |
| R19_750 | -2.8 | PatH | Endoplasmic reticulum |
| R19_754 | No significant change | PatI | Endoplasmic reticulum |
| R19_749 | -3.3* | PatJ | Peroxisome |
| R19_725 | -3.2 | PatK | Cytoplasm |
| R19_740 | Not found | PatL | Cytoplasm |
| R19_746 | -2.6 | PatM | Cell membrane |
| R19_736 | -2.8 | PatN | Cytoplasm |
| R19_737 | -2.1 | PatO | Extracellular |
*FDR < 3%.
Because Blistering1 was predicted to localize to the mitochondrion, all 217 proteins with significant changes in accumulation and predicted to localize to mitochondria were analyzed. We hypothesized that the loss of Blistering1 could have altered the accumulation of proteins with which it associated. Among the 104 mitochondrion proteins with decreased accumulation, two (R19_7431 and R19_6292) were among the top 20 proteins with decreased accumulation in T625 (Table III). Their functions based on homology to other proteins, however, are unknown.
To gain insight into the broader cellular functions of Blistering1, the proteins identified with significant changes in accumulation were mapped to enzymatic, biochemical, and cellular pathways in the KEGG database. The mapped proteins revealed major alterations in T625 pathways for protein processing in the ER (Fig. 7). In the Sec-dependent pathway for protein translocation, there were decreased amounts of SEC61 and SEC63 translocase complex proteins and signal-recognition particles that selectively load proteins destined for secretion (Fig. 7; supplemental Fig. S1) (45). Protein decreases likely negatively affected exocytosis at the Golgi bodies (COPII coatomer Sec13/31 and Sec23/24) (47), and the unfolded protein response (IRE1) (46), the ER-associated degradation pathway (Hsp90, Hsp40, Ubx, Npl4) (48), and the ubiquitin ligase complex (Hsp40, Skp1, Cul1, Hrd1) used for proteolysis of mis-secreted or misfolded proteins (49, 50) (Fig. 7).
Fig. 7.

Changes in the accumulation of T625 proteins associated with protein processing in the endoplasmic reticulum. Yellow indicates that the protein decreased in abundance. Blue indicates that the protein increased in abundance. Mapping was generated at www. kegg. jp. KEGG numbers for each significant protein are: Sec61, K07342; Sec62, K09540; OSTs, K12668; GlcI, K01228; NEF, K09486; GlcII, K05546; UGGT, K11718; ERManI, K01230; VIP36, K10082; ERGIC53, K10080; Sec13/31, K14004; Sec23/24, K14007; IRE1, K08852; PDIs, K09580; OS9, K10088; Ubx, K14012; Hsp40, K09518; Hsp90, K04079; Np14, K14015; Hrd1, K10601; HRD1; K10601; UbcH5, K06689; Cul1, K03347; Skp1, K03094.
We also observed decreased amounts of Rab5, a Ras superfamily GTPase that regulates vesicle fusion and maturation of late endosomes (51), vacuolar protein sorting (VPS) proteins (VPS45, VPS35, VPS4) (47), sorting nexins (SNX1/2, Snx3/12) (51), and actin-filament capping proteins (CAPZA, CAPZB) (52) involved in endocytosis and the degradation of transmembrane proteins at the vacuole (Fig. 8). Thus, the processes of endocytosis and endosome maturation may have been negatively affected. There also were decreases in proteins that effect autophagy and mitophagy processes (supplemental Figs. S2 and S3). Decreases in SCF complex proteins that function to ubiquitinate proteins destined for degradation (50) and decreases in proteasome regulatory particles likely impacted the processing of proteins and vesicle content turnover (supplemental Figs. S4 and S5).
Fig. 8.

Changes in the accumulation of T625 proteins associated with endocytosis. Yellow indicates that the protein decreased in abundance. Blue indicates that the protein increased in abundance. Mapping was generated at www. kegg. jp. KEGG numbers for each significant protein are: PLD, K01115; Epsin, K12471; AMPH, K12562; AP-2, K11824; N-WASP, K05747; Arp2/3, K05758; ArfGAP, K12488; ArfGEF, K18442; CAPZB, K10365; CAPZA, K10364; Rab11, K07904; KIF5, K10396; Rab5, K07889; VPS45, K12479; Rab8, K07901; Rab11, K07904; VPS35, K18468; Snx3/12, K17918; IST1, K19476; VPS4, K12196; SNX1/2, K17917.
Hypothesizing that these global proteomic changes affected vesicle-mediated exocytosis and endocytosis processes, we examined transverse and longitudinal cross sections of T625 and R19 mycelia by transmission electron microscopy. Alterations in morphology for the T625 included the pronounced appearance of hollow domains resembling abnormally enlarged endosomes/vacuoles inside hyphae (Fig. 9E, 9F). Furthermore, the cell walls of T625 were markedly thicker (205 ± 48 nm for T625 versus116 ± 32 nm for R19), and the cross sections revealed evidence of round orbs of little electron density appearing to push toward the blisters at the surfaces on the cell walls (Fig. 9G). Cross sections of hyphal tips, the forefront of cellular growth and the most active secretion zone, revealed the accumulation of hundreds of small vesicles that were distinct from the hollow domains (Fig. 9D, 9H), but there appeared to be no overt difference in the numbers or the sizes of these small vesicles between R19 and T625 at the apex of fungal cellular growth. These observations imply that the loss of Blistering1 altered the loading of protein content in vesicles, disrupted the fusion of vesicles to the plasma membrane, inhibited the recycling of vesicles with misloaded contents, and/or impaired endosome turnover rather than affect the formation of small vesicles, per se.
Fig. 9.
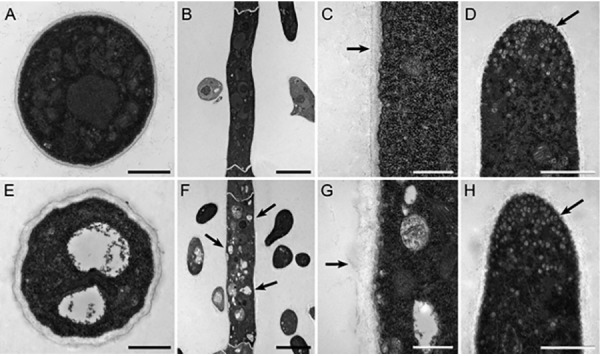
Transmission electron microscopy of thin sections of mycelia from R19 and transformant T625. A, Transverse cross section of R19 hypha. Bar is 1 μm. B, Longitudinal cross section of R19 hypha. Bar is 5 μm. C, Magnified longitudinal cross section of R19 hypha. Arrow points to cell wall boundary. Bar is 500 nm. D, R19 hyphal tip. Arrow points to small vesicles. Bar is 1 μm. E, Transverse cross section of T625 hypha. Note the two central hollow bodies compared with Fig. 9A. Bar is 1 μm. F, Longitudinal cross section of T625 hypha. Compared with Fig. 9B, arrows point to white enlarged endosomes. Bar is 5 μm. G, Magnified longitudinal cross section of T625 hypha. Arrow points to cell wall boundary. Note the thicker and bulbous cell wall compared with Fig. 9C. Bar is 500 nm. H, T625 hyphal tip. Arrow points to small vesicles as in Fig. 9D. Bar is 1 μm.
The preceding results reveal that Blistering1 has wide ranging effects on protein translocation and extracellular secretions that depend on efficient protein sorting, intercellular localization, degradation, and turnover. Such negative effects appeared to extend further to other cellular activities. For example, in T625 there were decreased amounts of proteins involved in mRNA surveillance, RNA splicing, RNA transport, and RNA degradation (Fig. 10; supplemental Figs. S6–S8). Many of these proteins have nuclear localization signals and account for the set of proteins with decreased accumulation in T625 (Fig. 6C). Because these proteins are subjected to internal transport from the ER across the nuclear membrane, it likely that the disruption of the same processing machinery involved in the transport of CWDEs and patulin biosynthetic enzymes affected these proteins that function in the nucleus. Overall, the results show that Blistering1 has pronounced effects on the accumulation of intercellular and extracellular proteins in P. expansum, especially those influencing the secretion of patulin and CWDEs needed for full virulence in apple fruit.
Fig. 10.

Changes in the accumulation of T625 proteins associated with mRNA surveillance. Yellow indicates that the protein decreased in abundance. Blue indicates that the protein increased in abundance. Mapping was generated at www. kegg. jp. KEGG numbers for each significant protein are: RNMT, K00565; CBP20, K12883; EIF4A3, K13025; Ref/Aly, K12881; UAP56, K12812; p15, K14285; PABP2, K14396; PAP, K14376; CPSF1, K14401; CPSF2, K14402; SWD2, K14962; GLC7, K06269; CSTF3, K14408; SYMPK, K06100; Musashi, K14411; PABP1, K13126; eRF1, K03265; eRF3, K03267; PP2A, K04354; Hbs1, K14416; Dom34, K06965.
On the Generation of Mutant Alleles and Heterologous Yeast Expression
Over the course of a year, we attempted many times to create a blistering1 knock-out through homologous recombination. A vector containing a hygromycin resistance gene flanked with 1,000 bp of DNA upstream and downstream of Blistering1 was used to transform P. expansum strain Pe21 ΔKu through an established transformation procedure (53). Pe21 ΔKu has a high rate of homologous recombination, and its Blistering1 flanking regions are 99–100% identical to R19. No transformants were recovered, but transformants of other independent gene knock-outs were recovered simultaneously. We suspect that Blistering1 resides in a genomic region that is inaccessible to the resident fungal homologous recombination machinery.
Taking another approach, we sought to express Blistering1 in yeast which has no ortholog. Using a Gibson cloning strategy, Blistering1 was engineered, in the sense and antisense directions, behind the phosphoglycerate kinase promoter in an I2 plasmid backbone containing the 2μ circle for yeast replication, and tryptophan biosynthesis and ampicillin resistance selectable markers (44). Oddly, we could only recover plasmids from Escherichia coli with Blistering1 in the antisense direction. It is possible that leaky expression was detrimental to the cells. Bypassing E. coli, we directly transformed yeast and recovered transformants in both the forward and antisense directions. Scanning electron microscopy of two independent transformants expressing Blistering1 mRNA revealed an altered, rough surface morphology compared with controls (supplemental Fig. S9). Although a proteomic investigation of Blistering1 in yeast will be a forthcoming subject of investigation, we hypothesize that overexpression of Blistering1 altered the yeast secretion system.
DISCUSSION
Blistering1 is Highly Conserved in Penicillium and Aspergillus spp
The Blistering1 gene was named for the appearance of the hyphae of P. expansum transformant T625 with a single-copy T-DNA insertion that eliminated the expression of a gene formerly designated by its model name, R19_6445 (Fig. 2, 3). A DnaJ domain is the only conserved recognizable feature. The DnaJ superfamily of proteins includes those involved in posttranslational modification, folding, and turnover, and chaperones which are localized to various cellular compartments including the ER and mitochondria (54). The J domain, glycine/phenylalanine rich region, and zinc binding motifs (CXXCXGXG) are conserved components with functions that involve protein-protein interactions, ATPase activity, and thiol-disulfide oxidoreductase activity (54). Blistering1 is phylogenetically restricted to the order Eurotiales, specifically the family Aspergillaceae (Fig. 3D) which comprises the Penicillium spp. and Aspergillus spp. that have diverse lifestyles as plant pathogens, saprophytes, human pathogens and which have industrial (cheese making) applications. Thus, Blistering1 is not only interesting because it is a novel gene that exerts broad proteomic influence in P. expansum, but also because it does not appear to have been duplicated or horizontally transferred to other fungal plant pathogens that occupy similar ecological niches, i.e. other fruit decay pathogens like Monilinia fructicola (brown rot) or Botrytis cinerea (gray mold). We speculate that the Blistering1 orthologs in other Penicillium spp. and Aspergillus spp. perform similar functions shown in this study.
Blistering1 Influences Fungal Virulence
Two studies involving the entomopathogenic fungus Beauveria bassiana and the corn smut pathogen Ustilago maydis have explored the roles of small molecular weight proteins containing DnaJ domains (55, 56). Deletion of these single copy genes impacted virulence, conidial formation, osmotic sensitivity, blastospore development, and gall formation. Blistering1, however, does not resemble those proteins, and conidial germination rates of T625 were indistinguishable from R19 (Fig. 1E).
The blistering1 mutation in T625 reduced virulence on apple, but it also reduced T625 growth in culture, somewhat. For a strict necrotrophic fungus like P. expansum, virulence and growth are inherently intertwined, but it appears that the mutation has a more pronounced effect on virulence processes than growth. This is evident because T625 does not secrete several CWDEs that are critical for apple tissue maceration (Table I; Fig. 4A). Some of the reduced virulence also may be attributed to decreased amounts of patulin production caused by decreased amounts of patulin biosynthesis enzymes (Table I; Fig. 5A). Hence, it can be concluded that a functional Blistering1 locus is required for the secretion of CWDEs and to produce patulin needed for apple infection.
As an aside, the absence of PatB, PatO, and PatE in the T625 spent broth imply that the final 3 steps of patulin biosynthesis occur outside of the P. expansum cell. A recent study evaluating the patulin operon through gene mutation also suggests that the final step of patulin biosynthesis occurs outside of P. expansum (57). These findings prompt us to hypothesize that patulin formation outside the cell is a means for autoimmunity in which Blistering1 plays a part. In A. parasiticus, autoimmunity is conferred by confining the late stages of aflatoxin polyketide biosynthesis to vesicles for exocytosis (58). A role for a Blistering1 ortholog in A. parasiticus has not been elucidated, but the parallel of exocytosis for aflatoxin production and the secretion of the enzymes for patulin production is striking. Therefore, we hypothesize that a blistering1 mutant in A. parasiticus may also impact aflatoxin production and virulence in the host.
The blistering1 Mutation has Broad Pleiotropic Effects on the P. expansum Proteome
Quantitative analysis of 3,282 mycelia proteins in R19 and T625 revealed drastic changes that arose from the blistering1 mutation. There were 798 proteins with decreased accumulation, and many of these proteins were found to be associated with ones involved in protein translocation, cargo loading, exocytosis, endocytosis, protein degradation and turnover. The precise functional role of Blistering1 in this biological context is unknown especially given its predicted localization to the mitochondrion. Nevertheless, the mis-secretion of proteins in T625 imply that Blistering1 may regulate translocation, although Blistering1 is not like Sec proteins or signal-recognition particles involved in protein translocation. Meanwhile, the microscopy data show that Blistering1 does not appear to affect the formation of vesicles at hyphal tips (Fig. 9D, 9H). Rather, the mis-secretion results and the microscopic evidence of large hollow bodies in T625 cells (Fig. 9E, 9F, 9G) implicate Blistering1 in proper cargo loading of proteins destined for secretion, fusion of vesicles with the plasma membrane, recycling of mis-folded or misdirected proteins, and endosome turnover of recycled vesicles. Consequently, alterations in these processes likely led to the reduced formation and secretion of CWDEs and patulin biosynthetic enzymes, and the increased mis-secretion of other CWDEs not needed for apple virulence. Therefore, we surmise that Blistering1 has a specific role in controlling the formation, degradation, or secretion of a class of enzymes specific to Aspergillaceae that when disrupted leads to broader pleiotropic effects as these enzymes fail to reach their proper destinations. Hence, T625 may struggle at many levels as it fails to secrete the proper CWDEs, absorb nutrients through endocytosis, or reconcile the disturbances within its protein sorting and degradation systems.
The blistering1 mutation altered the ability of T625 to modulate its ambient pH in vitro (Fig. 5B). Ambient pH modulation by fungal pathogens is an important aspect of pathogen ingress, host colonization, and virulence. This has been demonstrated in a variety of phytopathogens including Colletotrichum acutatum, Sclerotinia sclerotiorum, and P. expansum (59–61). Modulation of host pH ensures the proper milieu for secreted enzymes to function during the degradation of host tissue and nutrient acquisition (9). Factors that explain the increased pH of liquid culture during growth for T625 include altered organic acid secretion or aberrations in the pH-signaling pathway. The PacC pH-regulated transcription factor (62, 63), however, did not have changes in protein accumulation (supplemental Table S3), and R19_6449, the next positional gene downstream from Blistering1 and having the most similarity (e−15) to ExoS of the two- component acid sensor ExoS/ChvI (64), also had no change in accumulation in T625 (supplemental Table S3). On the other hand, PalF ortholog R19_2375 exhibited a 6-fold decrease in T625 (supplemental Table S3). In the P. expansum-related A. nidulans, ubiquitylated-PalF interacts with Vps23 to recruit the endosomal sorting complex required for transport (ESCRT) to regulate pH-sensing at the plasma membrane (65). Given the potential perturbance to endosomal processes that utilize ESCRT complexes (Fig. 8) and the disruption of the ubiquitin ligase complex (Fig. 7), the ability of T625 to sense or regulate its environmental pH may have been weakened. In Candida albicans, the transcription for PRR1, a PalF ortholog, decreases as pH becomes alkaline, and a prr1 null mutant has impaired hyphal development (66). Hence, defects in pH regulation or sensing and subsequent retarded growth go together, but they may be smaller parts of a larger network disturbance that occurs in absence of Blistering1.
DATA AVAILABILITY
The mass spectrometry data associated with this project can be found at http://massive.ucsd.edu under accession number MSV000082957.
Supplementary Material
Footnotes
* This work was supported in part by base funds provided to USDA-ARS project plan #8042–42430–002-00D entitled “Development of Novel Tools to Manage Fungal Plant Pathogens That Cause Postharvest Decay of Pome Fruit to Reduce Food Waste” under USDA-ARS National Program 303 Plant Diseases. The authors declare that they have no conflicts of interest with the contents of this article.
 This article contains supplemental Figures and Tables.
This article contains supplemental Figures and Tables.
1 The abbreviations used are:
- CWDE
- cell wall degrading enzymes
- PG
- polygalacturonase
- T-DNA
- transfer DNA
- PDA
- potato dextrose agar
- TAIL-PCR
- thermal asymmetric interlaced PCR
- AGC
- automatic gain control
- msec
- milliseconds
- NSAF
- normalized spectral abundance factor
- TMT
- tandem mass tag
- PSM
- peptide spectrum matches
- FDR
- false discovery rate
- S/N
- signal-to-noise
- KEGG
- Kyoto Encyclopedia of Genes and Genomes
- ER
- endoplasmic reticulum
- VPS
- vacuolar protein sorting
- ESCRT
- endosomal sorting complex required for transport.
REFERENCES
- 1. Spotts R. A., Cervantes L. A., and Mielke E. A. (1999) Variability in postharvest decay among apple cultivars. Plant Dis. 83, 1051–1054 [DOI] [PubMed] [Google Scholar]
- 2. Rosenberger D. (2012) Sanitize apple storage rooms to minimize postharvest decays. Scaffolds Fruit J. 21, 4–5 [Google Scholar]
- 3. Yan H. J., Gaskins V. L., Vico I., Luo Y. G., and Jurick W. M. 2nd (2014) First report of Penicillium expansum isolates resistant to pyrimethanil from stored apple fruit in Pennsylvania. Plant Dis. 98, 1004–1004 [DOI] [PubMed] [Google Scholar]
- 4. Gaskins V. L., Vico I., Yu J., and Jurick W. M. (2015) First report of Penicillium expansum isolates with reduced sensitivity to fludioxonil from a commercial packinghouse in Pennsylvania. Plant Dis. 99, 1182–1182 [Google Scholar]
- 5. Jurick W. M. II., Vico I., Gaskins V. L., Garrett W. M., Whitaker B. D., Janisiewicz W. J., and Conway W. S. (2010) Purification and biochemical characterization of polygalacturonase produced by Penicillium expansum during postharvest decay of 'Anjou' pear. Phytopathology 100, 42–48 [DOI] [PubMed] [Google Scholar]
- 6. Frisvad J. C., and Samson R. A. (2004), Polyphasic taxonomy of Penicillium subgenus Penicillium: A guide to identification of food and air-borne terverticillate Penicillia and their mycotoxins. Studies Mycol. 49, 1–174 [Google Scholar]
- 7. Errampalli D. (2004) Effect of fludioxonil on germination and growth of Penicillium expansum and decay in apple cvs. Empire and Gala. Crop Prot. 23, 811–817 [Google Scholar]
- 8. Rosenberger D. A., Engle C. A., Meyer F. W., and Watkins C. B. (2006) Penicillium expansum invades apples through stems during controlled atmosphere storage. Plant Health Progress doi: 10.1094/PHP-2006-1213-01-RS [DOI] [Google Scholar]
- 9. Jurick W. M., Vico I., McEvoy J. L., Whitaker B. D., Janisiewicz W., and Conway W. S. (2009) Isolation, purification, and characterization of a polygalacturonase produced in Penicillium solitum-decayed 'golden delicious' apple fruit. Phytopathology 99, 636–641 [DOI] [PubMed] [Google Scholar]
- 10. Yao C. L., Conway W. S., and Sams C. E. (1996), Purification and characterization of a polygalacturonase produced by Penicillium expansum in apple fruit. Phytopathology 86, 1160–1166 [Google Scholar]
- 11. Ballester A. R., Marcet-Houben M., Levin E., Sela N., Selma-Lázaro C., Carmona L., Wisniewski M., Droby S., González-Candelas L., and Gabaldón T. (2015) Genome, transcriptome, and functional analyses of Penicillium expansum provide new insights into secondary metabolism and pathogenicity. Mol. Plant Microbe 28, 232–248 [DOI] [PubMed] [Google Scholar]
- 12. Li B., Zong Y., Du Z., Chen Y., Zhang Z., Qin G., Zhao W., and Tian S. (2015) Genomic characterization reveals insights into patulin biosynthesis and pathogenicity in penicillium species. Mol. Plant-Microbe Interactions 28, 635–647 [DOI] [PubMed] [Google Scholar]
- 13. McKinley E. R., and Carlton W. W. P. (1991) in Mycotoxins and Phytoalexins (Sharma R. P. and Salunkhe D. K., ed.) pp. 191–236, CRC Press, Boca Raton, FL [Google Scholar]
- 14. Andersen B., Smedsgaard J., and Frisvad J. C. (2004) Penicillium expansum: Consistent production of patulin, chaetoglobosins, and other secondary metabolites in culture and their natural occurrence in fruit products. J. Agr. Food Chem. 52, 2421–2428 [DOI] [PubMed] [Google Scholar]
- 15. Sanzani S. M., Reverberi M., Punelli M., Ippolito A., and Fanelli C. (2012) Study on the role of patulin on pathogenicity and virulence of Penicillium expansum. Int. J. Food Microbiol. 153, 323–331 [DOI] [PubMed] [Google Scholar]
- 16. Snini S. P., Tannous J., Heuillard P., Bailly S., Lippi Y., Zehraoui E., Barreau C., Oswald I. P., and Puel O. (2016) Patulin is a cultivar-dependent aggressiveness factor favouring the colonization of apples by Penicillium expansum. Mol. Plant Pathol. 17, 920–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Puel O., Galtier P., and Oswald I. P. (2010) Biosynthesis and toxicological effects of patulin. Toxins 2, 613–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang F. Q., Zhong J., Zhao Y., Xiao J., Liu J., Dai M., Zheng G., Zhang L., Yu J., Wu J., and Duan B. (2014) Genome sequencing of high-penicillin producing industrial strain of Penicillium chrysogenum. BMC genomics 15, S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Specht T., Dahlmann T. A., Zadra I., Kurnsteiner H., and Kück U. (2014) Complete sequencing and chromosome-scale genome assembly of the industrial progenitor strain P2niaD18 from the penicillin producer Penicillium chrysogenum. Genome Announc. 2, pii: e00577–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. van den Berg M. A., Albang R., Albermann K., Badger J. H., Daran J. M., Driessen A. J., Garcia-Estrada C., Fedorova N. D., Harris D. M., Heijne W. H., Joardar V., Kiel J. A., Kovalchuk A., Martín J. F., Nierman W. C., Nijland J. G., Pronk J. T., Roubos J. A., van der Klei I. J., van Peij N. N., Veenhuis M., von Döhren H., Wagner C., Wortman J., and Bovenberg R. A. (2008) Genome sequencing and analysis of the filamentous fungus Penicillium chrysogenum. Nat. Biotechnol. 26, 1161–1168 [DOI] [PubMed] [Google Scholar]
- 21. Yu J., Jurick W. M., and Bennett J. W., (2015) Current status of genomics research on mycotoxigenic fungi Int. J. Plant Biol. Res. 3, 1035 [Google Scholar]
- 22. van der Does H. C., and Rep M. (2017) Adaptation to the host environment by plant-pathogenic fungi. Ann. Rev. Phytopathol. 55, 427–450 [DOI] [PubMed] [Google Scholar]
- 23. de Groot M. J., Bundock P., Hooykaas P. J., and Beijersbergen A. G. (1998), Agrobacterium tumefaciens-mediated transformation of filamentous fungi. Nat. Biotechnol. 16, 839–842 [DOI] [PubMed] [Google Scholar]
- 24. Chen H., Nelson R. S., and Sherwood J. L., (1994) Enhanced recovery of transformants of Agrobacterium tumefaciens after freeze-thaw transformation and drug selection. BioTechniques 16, 664–668, 670 [PubMed] [Google Scholar]
- 25. Singer T., and Burke E. (2003) High-throughput TAIL-PCR as a tool to identify DNA flanking insertions. Methods Mol. Biol. 236, 241–272 [DOI] [PubMed] [Google Scholar]
- 26. Yu J., Jurick W. M. 2nd, Cao H., Yin Y., Gaskins V. L., Losada L., Zafar N., Kim M., Bennett J. W., and Nierman W. C. (2014) Draft genome sequence of Penicillium expansum strain R19, which causes postharvest decay of apple fruit. Genome Announc. 2, pii: e00635–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kumar S., Stecher G., Li M., Knyaz C., and Tamura K. (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evolution 35, 1547–1549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jones D. T., Taylor W. R., and Thornton J. M. (1992) The rapid generation of mutation data matrices from protein sequences. Computer Appl. Biosci. 8, 275–282 [DOI] [PubMed] [Google Scholar]
- 29. Felsenstein J. (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39, 783–791 [DOI] [PubMed] [Google Scholar]
- 30. Perkins D. N., Pappin D. J., Creasy D. M., and Cottrell J. S. (1999), Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 31. Cooper B., Feng J., and Garrett W. M. (2010), Relative, label-free protein quantitation: spectral counting error statistics from nine replicate MudPIT samples. J. Am. Soc. Mass Spectrom. 21, 1534–1546 [DOI] [PubMed] [Google Scholar]
- 32. Feng J., Garrett W. M., Naiman D. Q., and Cooper B. (2009) Correlation of multiple peptide mass spectra for phosphoprotein identification. J. Proteome Res. 8, 5396–5405 [DOI] [PubMed] [Google Scholar]
- 33. Feng J., Naiman D. Q., and Cooper B. (2007) Probability model for assessing proteins assembled from peptide sequences inferred from tandem mass spectrometry data. Anal. Chem. 79, 3901–3911 [DOI] [PubMed] [Google Scholar]
- 34. Zybailov B., Mosley A. L., Sardiu M. E., Coleman M. K., Florens L., and Washburn M. P. (2006) Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J. Proteome Res. 5, 2339–2347 [DOI] [PubMed] [Google Scholar]
- 35. Petersen T. N., Brunak S., von Heijne G., and Nielsen H. (2011), SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786 [DOI] [PubMed] [Google Scholar]
- 36. Wang Y., Yang F., Gritsenko M. A., Clauss T., Liu T., Shen Y., Monroe M. E., Lopez-Ferrer D., Reno T., Moore R. J., Klemke R. L., Camp D. G. 2nd, and Smith R. D. (2011) Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics 11, 2019–2026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Isasa M., Rose C. M., Elsasser S., Navarrete-Perea J., Paulo J. A., Finley D. J., and Gygi S. P. (2015) Multiplexed, proteome-wide protein expression profiling: yeast deubiquitylating enzyme knockout strains. J. Proteome Res. 14, 5306–5317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McAlister G. C., Nusinow D. P., Jedrychowski M. P., Wuhr M., Huttlin EL, Erickson BK, Rad R, Haas W, and Gygi SP. (2014) MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem. 86, 7150–7158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kall L., Canterbury J. D., Weston J., Noble W. S., and MacCoss M. J. (2007) Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 4, 923–925 [DOI] [PubMed] [Google Scholar]
- 40. Armenteros J. J. A., Sonderby C. K., Sonderby S. K., Nielsen H., and Winther O. (2017) DeepLoc: prediction of protein subcellular localization using deep learning. Bioinformatics 33, 4049–4049 [DOI] [PubMed] [Google Scholar]
- 41. Sun J., Kou L., Geng P., Huang H., Yang T. Luo Y., and Chen P. (2015) Metabolomic assessment reveals an elevated level of glucosinolate content in CaCl(2) treated broccoli microgreens. J. Agric. Food Chem. 63, 1863–1868 [DOI] [PubMed] [Google Scholar]
- 42. Bolton S. J., Klompen H., Bauchan G. R., and Ochoa R. (2014) A new genus and species of Nematalycidae (Acari: Endeostigmata). J. Nat. Hist. 48, 1359–1373 [Google Scholar]
- 43. Mowery J., and Bauchan G. (2018) Optimization of rapid microwave processing of botanical samples for transmission electron microscopy. Microsc. Microanal. 24, 1202–1203 [Google Scholar]
- 44. Cooper B., Campbell K. B., and Garrett W. M. (2016) Expression of a synthetic rust fungal virus cDNA in yeast. Arch. Virol. 161, 111–123 [DOI] [PubMed] [Google Scholar]
- 45. Linxweiler M., Schick B., and Zimmermann R. (2017) Let's talk about Secs: Sec61, Sec62 and Sec63 in signal transduction, oncology and personalized medicine. Signal Transduction Targeted Therapy 2, 17002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Amin-Wetzel N., Saunders R. A., Kamphuis M. J., Rato C., Preissler S., Harding H. P., and Ron D. (2017) A J-protein co-chaperone recruits BiP to monomerize IRE1 and repress the unfolded protein response. Cell 171, 1625–1637.e1613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Feyder S., De Craene J. O., Bar S., Bertazzi D. L., and Friant S. (2015) Membrane trafficking in the yeast Saccharomyces cerevisiae model. Int. J. Mol. Sci. 16, 1509–1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hassdenteufel S., Klein M. C., Melnyk A., and Zimmermann R. (2014), Protein transport into the human ER and related diseases, Sec61-channelopathies. Biochem. Cell Biol. 92, 499–509 [DOI] [PubMed] [Google Scholar]
- 49. Wu X., and Rapoport T. A. (2018) Mechanistic insights into ER-associated protein degradation. Curr. Opinion Cell Biol. 53, 22–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nakatsukasa K., Okumura F., and Kamura T. (2015) Proteolytic regulation of metabolic enzymes by E3 ubiquitin ligase complexes: lessons from yeast. Critical Rev. Biochem. Mol. Biol. 50, 489–502 [DOI] [PubMed] [Google Scholar]
- 51. Langemeyer L., Frohlich F., and Ungermann C., (2018) Rab GTPase function in endosome and lysosome biogenesis. Trends Cell Biol. 28, 57–970 [DOI] [PubMed] [Google Scholar]
- 52. Derivery E., Sousa C., Gautier J. J., Lombard B., Loew D., and Gautreau A. (2009) The Arp2/3 activator WASH controls the fission of endosomes through a large multiprotein complex. Developmental Cell 17, 712–723 [DOI] [PubMed] [Google Scholar]
- 53. Tannous J., Kumar D., Sela N., Sionov E., Prusky D., and Keller N. P. (2018) Fungal attack and host defence pathways unveiled in near-avirulent interactions of Penicillium expansum creA mutants on apples. Mol. Plant Pathol. 19, 2635–2650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Walsh P., Bursac D., Law Y. C., Cyr D., and Lithgow T. (2004) The J-protein family: modulating protein assembly, disassembly and translocation. EMBO Reports 5, 567–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang J., Ying S. H., Hu Y., and Feng M. G. (2017) Vital role for the J-domain protein Mdj1 in asexual development, multiple stress tolerance, and virulence of Beauveria bassiana. Appl. Microbiol. Biotechnol. 101, 185–195 [DOI] [PubMed] [Google Scholar]
- 56. Lo Presti L., Diaz C. L., Turra D., Di Pietro A., Hampel M., Heimel K., and Kahmann R. (2016) A conserved co-chaperone is required for virulence in fungal plant pathogens. New Phytol. 209, 1135–1148 [DOI] [PubMed] [Google Scholar]
- 57. Li B., Chen Y., Zong Y., Shang Y., Zhang Z., Xu X., Wang X., Long M., and Tian S. (2019) Dissection of patulin biosynthesis, spatial control and regulation mechanism in Penicillium expansum. Environmental Microbiol. 21, 1124–1139 [DOI] [PubMed] [Google Scholar]
- 58. Chanda A., Roze L. V., Kang S., Artymovich K. A., Hicks G. R., Raikhel N. V., Calvo A. M., and Linz J. E. (2009) A key role for vesicles in fungal secondary metabolism. Proc. Natl. Acad. Sci. U.S.A. 106, 19533–19538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Prusky D., McEvoy J. L., Leverentz B., and Conway W. S. (2001), Local modulation of host pH by Colletotrichum species as a mechanism to increase virulence. Mol. Plant-Microbe Interactions 14, 1105–1113 [DOI] [PubMed] [Google Scholar]
- 60. Prusky D., McEvoy J. L., Saftner R., Conway W. S., and Jones R. (2004) Relationship between host acidification and virulence of Penicillium spp. on apple and citrus fruit. Phytopathology 94, 44–51 [DOI] [PubMed] [Google Scholar]
- 61. Jurick W. M. (2007) 2nd and Rollins, J. A., Deletion of the adenylate cyclase (sac1) gene affects multiple developmental pathways and pathogenicity in Sclerotinia sclerotiorum. Fungal Gen. Biol. 44, 521–530 [DOI] [PubMed] [Google Scholar]
- 62. Rollins J. A. (2003) The Sclerotinia sclerotiorum pac1 gene is required for sclerotial development and virulence. Mol. Plant-Microbe Interactions 16, 785–795 [DOI] [PubMed] [Google Scholar]
- 63. Prusky D., and Yakoby N. (2003) Pathogenic fungi: leading or led by ambient pH? Mol Plant Pathol 4, 509–516 [DOI] [PubMed] [Google Scholar]
- 64. Chen E. J., Fisher R. F., Perovich V. M., Sabio E. A., and Long S. R. (2009) Identification of direct transcriptional target genes of ExoS/ChvI two-component signaling in Sinorhizobium meliloti. J. Bacteriol. 191, 6833–6842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Galindo A., Calcagno-Pizarelli A. M., and Arst H. N. (2012) Jr., and Penalva, M. A., An ordered pathway for the assembly of fungal ESCRT-containing ambient pH signalling complexes at the plasma membrane. J. Cell Sci. 125, 1784–1795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Porta A., Ramon A. M., and Fonzi W. A. (1999) PRR1, a homolog of Aspergillus nidulans palF, controls pH-dependent gene expression and filamentation in Candida albicans. J. Bacteriol. 181, 7516–7523 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry data associated with this project can be found at http://massive.ucsd.edu under accession number MSV000082957.