

LANA, the most abundantly expressed protein during latency, is a multifunctional protein which is absolutely required for the persistence of KSHV in the host cell. Even though the functions of LANA in aiding pathogenesis of the virus have been extensively studied, the mechanism of how LANA escapes host’s immune surveillance is not fully understood. This study sheds light on the autoregulatory role of LANA to modulate its expression and immune evasion through formation of G-quadruplexes in its mRNA. We used G-quadruplex-stabilizing ligand to define the inhibition in LANA expression and presentation on the cell surface through MHC class I. We defined the autoregulatory role of LANA and identified a cellular RNA helicase, hnRNP A1, regulating the translation of LANA mRNA. This interaction of hnRNP A1 with LANA mRNA could be exploited for controlling KSHV latency.
KEYWORDS: G-quadruplex, KSHV, LANA, latency, hRNP A1
ABSTRACT
During the latent phase, Kaposi’s sarcoma-associated herpes virus (KSHV) maintains itself inside the host by escaping the host immune surveillance mechanism through restricted protein expression. Latency-associated nuclear antigen (LANA), the most abundantly expressed protein, is essential for viral persistence, as it plays important roles in latent viral DNA replication and efficient segregation of the viral genome to the daughter cells following cell division. KSHV evades immune detection by maintaining the levels of LANA protein below a threshold required for detection by the host immune system but sufficient to maintain the viral genome. LANA achieves this by controlling its expression through regulation of its promoters and by inhibiting its presentation through interaction with the proteins of class I and class II major histocompatibility complex (MHC) pathways. In this study, we identified a mechanism of LANA expression and restricted immune recognition through formation of G-quadruplexes in LANA mRNA. We show that the formation of these stable structures in LANA mRNA inhibits its translation to control antigen presentation, which was supported by treatment of cells with TMPyP4, a G-quadruplex-stabilizing ligand. We identified heterogenous ribonucleoprotein A1 (hnRNP A1) as a G-quadruplex-unwinding helicase, which unfolds these stable secondary structures to regulate LANA translation.
IMPORTANCE LANA, the most abundantly expressed protein during latency, is a multifunctional protein which is absolutely required for the persistence of KSHV in the host cell. Even though the functions of LANA in aiding pathogenesis of the virus have been extensively studied, the mechanism of how LANA escapes host’s immune surveillance is not fully understood. This study sheds light on the autoregulatory role of LANA to modulate its expression and immune evasion through formation of G-quadruplexes in its mRNA. We used G-quadruplex-stabilizing ligand to define the inhibition in LANA expression and presentation on the cell surface through MHC class I. We defined the autoregulatory role of LANA and identified a cellular RNA helicase, hnRNP A1, regulating the translation of LANA mRNA. This interaction of hnRNP A1 with LANA mRNA could be exploited for controlling KSHV latency.
INTRODUCTION
Kaposi’s sarcoma-associated herpes virus (KSHV), the etiological agent of Kaposi’s sarcoma (KS), primary effusion lymphoma (PEL), and multicentric Castleman’s disease, displays two distinct phases in its life cycle: a predominant latent phase and a short productive lytic phase (1 – 4). Like other herpesviruses, KSHV establishes a persistent latent infection through the expression of a limited number of proteins, some of which play immunomodulatory roles to prevent recognition by the host immune surveillance system (5). Latency-associated nuclear antigen (LANA), encoded by ORF73, is the most predominantly expressed protein during latency and is essential for viral replication and maintenance of the viral genome through segregation of viral episomes to the daughter cells following mitosis (6 – 8). LANA achieves this by establishing simultaneous contacts with the host chromosome and the viral genome, with the involvement of many cellular proteins (9 – 11). One of the modes through which LANA aids in the pathogenesis is by helping the virus to evade host immune surveillance through inhibition of multiple pathways (12). LANA has been shown to avoid recognition by CD8+ T cells through inhibition of major histocompatibility complex class I (MHC-I) presentation by interfering with translocation of peptides generated by proteasome to the endoplasmic reticulum (ER) (13). Previous studies have demonstrated the role of LANA in inhibiting the CD4+ T cell responses by downregulating the major histocompatibility complex class II (MHC-II) genes through blocking of CIITA transcription and interaction with regulatory factor X (RFX) complex (14, 15).
G-quadruplexes are secondary DNA/RNA structures formed in nucleic acid sequences rich in guanine residues. G-quartets, which are formed by Hoogsteen hydrogen bonding between four guanine residues, stack on top of each other to form G-quadruplexes. These structures have been reported to play important roles in biological processes like DNA replication, transcription, alternative splicing, and translation (16 – 20). The formation of G-quadruplex structures is regulated by various proteins which either stabilize or unwind these structures (21). G-quadruplexes have recently gained importance as potential drug targets against pathogenic viruses because of their occurrence in the genomes of many viruses and their regulatory role in the viral life cycle. The presence of G-quadruplex structures has been reported for the central polypurine tract in the integrase gene of human immunodeficiency virus (HIV), which promotes recombination by facilitating the dimerization of RNA templates and template switching (22). In addition, the formation of these structures has been reported for the U3 region of the long terminal repeats (LTR) in the Sp1 and NF-κB binding sites, which negatively regulates the transcriptional activation of HIV (23). Also, the Nef region of HIV forms these secondary structures, which regulate the HIV infection by inhibiting the expression of the Nef gene (24). A recent study reported the presence of G-quadruplexes in the preS2/S gene promoter of hepatitis B virus, which positively regulated its transcription, and the disruption of these structures led to a reduction in virion production (25). Hepatitis C virus has also been shown to form G-quadruplexes, and stabilization of these structures led to an inhibition in viral replication and protein expression (26). Among herpesviruses, G-quadruplexes are present in the inverted repeat region of herpes simplex virus (HSV), and the stabilization of these structures led to a reduction in HSV-1 infection (27). These G-quadruplexes are enriched in the replication compartments following HSV-1 infection, which possibly regulate the viral life cycle by controlling viral replication (28). Epstein-Barr virus (EBV) nuclear antigen (EBNA1) has been reported to preferentially bind to the G-quadruplex-forming RNA and promotes EBNA1-dependent recruitment of origin recognition complex (ORC) to the origin in addition to attachment of EBNA1 to the metaphase chromosomes (29). A recent study also reported the formation of G-quadruplexes in EBNA1 mRNA, which serve as steric blocks for the movement of ribosomes to inhibit the translation of EBNA1 mRNA (30). Our lab has reported the formation of G-quadruplexes in the terminal repeat region of KSHV, and their stabilization led to a reduction in latent viral DNA replication (31). These studies substantiate the vital role of G-quadruplex regulatory structures in the pathogenesis of viruses.
G-quadruplexes are thermodynamically stable structures whose formation is tightly regulated by the proteins that promote stabilization or unwinding of these structures in order to stop their hindrance in RNA or DNA metabolism (32, 33). A number of such proteins are already identified, but the mechanism of how and when these proteins are recruited at definite sites is still unclear. A few helicases, including DHX9 (DEAH box helicase 9), RHAU (an RNA helicase of the DEAH box family), DDX1 (DEAD box RNA helicase 1), and DDX21, have been reported to unwind RNA G-quadruplexes (34 – 37). Another family of proteins which have been implicated in regulating G-quadruplex formation is the heterogenous nuclear ribonucleoproteins (hnRNPs). This is the RNA binding family of proteins that regulate gene expression by performing a variety of functions, including mRNA splicing, mRNA stability and trafficking, and modulation of protein translation (38). A member of the hnRNP family, hnRNP A1, has been reported to enhance translation of RON receptor tyrosine kinase by binding to the G-quadruplexes formed in the 5' untranslated region (UTR) (39). Another study has reported the interaction of hnRNP H/F with the G-quadruplex at the 3' end of p53 mRNA, which enhanced its expression during DNA damage (40). Similarly, hnRNP A2 positively regulated the translation of FMR1 (fragile X mental retardation) mRNA by destabilizing the G-quadruplex structure formed by the (CGG)n repeat, which is responsible for the silenced gene product of FMR1 patients with fragile X mental syndrome (41).
Here we report the formation of G-quadruplexes in the mRNA of LANA, through biophysical and biochemical methods. The stabilization of these structures with TMPyP4, a G-quadruplex-stabilizing ligand, inhibited the translation of LANA, which resulted in a reduced protein expression in KSHV-infected as well as LANA-expressing cells. We studied the regulatory role of G-quadruplexes through a G-quadruplex-containing LANA sequence (G4 wild type) along with a clone containing a codon-modified sequence of the G4 wild type that disrupts the ability to form G-quadruplexes. Furthermore, we also demonstrated the regulatory role of G-quadruplexes in inhibiting endogenous antigen presentation to CD8+ T cells owing to a reduced translation of LANA mRNA. Finally, we identified cellular and viral proteins involved in regulation of G-quadruplex formation. We provide evidence regarding the role of hnRNP A1 in enhancing the translation of LANA and also demonstrate the ability of LANA to regulate its translation.
RESULTS
LANA mRNA forms G-quadruplexes.
G-quadruplexes are formed by Hoogsteen hydrogen bonding between G-quartets and are stabilized by the presence of K+ ions. Due to their roles in modulating vital biological processes, these structures have recently gained importance as potential drug targets. EBV and KSHV, both members of the herpesvirus family, are known to share functional similarities. Of our particular interest is the expression of latent proteins and immune evasion strategies during latency. Expression of EBNA1, the functional homolog of KSHV LANA in EBV, is regulated by G-quadruplexes in its mRNA, which led us to speculate that the levels KSHV LANA are regulated by G-quadruplexes (30). We analyzed the LANA mRNA sequence for the presence of G-quadruplexes using a web-based tool, QGRS Mapper (http://bioinformatics.ramapo.edu/QGRS/index.php), that predicts the formation of these structures (42). The LANA mRNA was imported into the software, and the probability of G-quadruplex formation, represented as G-scores (a high value implies a higher likelihood of G-quadruplex formation), was determined (the yellow highlighted region in Fig. 1A is the region with high scores). Based on the G-scores, a region in the QE domain of LANA displayed a higher number of G-quadruplex-forming sites (see Fig. 3A, panel a). G-quartets, the building blocks of G-quadruplexes, are formed by Hoogsten hydrogen binding between four guanine residues as illustrated in Fig. 1B. These coplanar G-quartets stack upon each other to form a G-quadruplex, and this interaction is stabilized by the presence of N+ or K+ ions (Fig. 1B).
FIG 1.
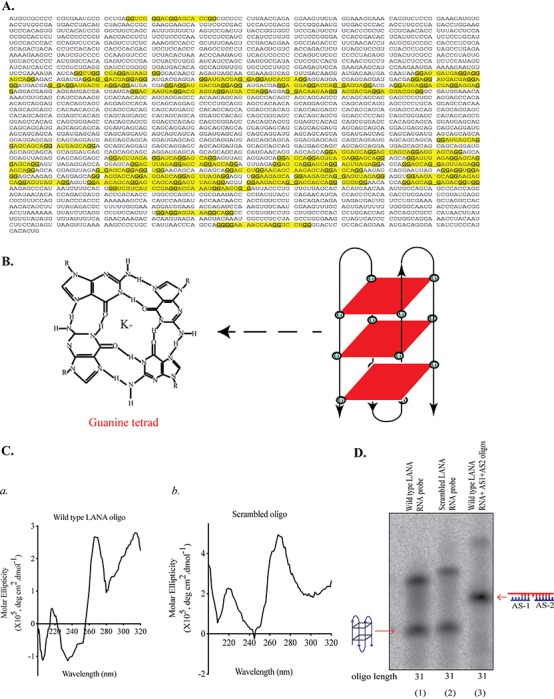
LANA mRNA formed stable G-quadruplex structures. (A) Sequence of LANA mRNA showing G-quadruplex-forming regions, highlighted in yellow. LANA mRNA sequence was imported into QGRS Mapper software, which predicts G-quadruplex formation as a function of G-score. The sequence highlighted in yellow are the region with high G-score. (B) A guanine tetrad showing Hoogsteen hydrogen bonding between four guanine residues stabilized by potassium ion in the center and schematic representation of a G-quadruplex structure formed by stacking of three guanine tetrads. (C) Circular-dichroism spectral analysis of LANA wild-type RNA oligonucleotide having a high G-score (a) and scrambled RNA oligonucleotide with no G-quadruplex formation (b), used as a negative control. The oligonucleotides were scanned for a wavelength range of 320 nm to 200 nm, and molar ellipticity was plotted on the y axis, with wavelength on the x axis. (D) Electrophoretic mobility shift assay (EMSA) performed in the presence of K+ ions on LANA wild-type and scrambled RNA oligonucleotides labeled with 32P and resolved on a native polyacrylamide gel. Antisense oligonucleotides (AS1 and AS2) complementary to LANA wild-type RNA oligonucleotide, added in molar excess, were used in the indicated lanes to confirm the specificity of the mobility shift by G-quadruplex-forming sequence.
FIG 3.

Destabilization of G-quadruplexes by codon modification-enhanced translation. (A) (a) Schematic of LANA showing various domains with potential G-quadruplex-forming sites. mRNA sequence with a high G-score in the DNA/chromatin binding region was chosen for our experiments. (b) Sequence of G4 wild-type clone representing the G-quadruplex-forming region from the QE-rich domain of LANA. (c) Sequence of G4 disrupted clone where G residues have been modified so it can no longer form a G-quadruplex structure. (B) (a) In vitro translation assay of G4 wild-type and G4 disrupted sequences, representing differences in the protein levels (marked by asterisk). Briefly, pA3F-G4 wild type and pA3F-G4 disrupted were translated using methionine and the TNT T7 translation system, and the resulting product was resolved by SDS-PAGE and detected using anti-Flag antibody. (b) mRNA levels of the in vitro-translated product of G4 wild-type and G4 disrupted clones. The in vitro translation products of pA3F-G4 wild type and G4 disrupted were incubated at 30°C for 1 h in separate reactions, RNA was extracted, and cDNA was synthesized and quantified using gene-specific primers. (C) (a) Translation efficiency of transiently expressed G4 wild-type and G4 disrupted clones in HEK293T cells. HEK293T cells were transfected with pA3F-G4 wild type and pA3F-G4 disrupted, harvested, and lysed 24 h posttransfection, and the lysates were resolved by SDS-PAGE and immunoblotted using anti-Flag antibody. Anti-GAPDH antibody was used to ensure equal loading of the proteins. (b) mRNA levels of transiently expressed G4 wild-type and G4 disrupted clones in HEK293T cells. RNA was extracted from cells transfected with pA3F-G4 wild type and pA3F-G4 disrupted, and cDNA was synthesized and quantified using gene-specific primers. (D) (a) In vitro translation assay of the G4 wild type with antisense oligonucleotide complementary to the G-rich region of the G4 wild type. pA3F-G4 wild type was translated using methionine and the TNT T7 translation system in the presence of 1,000 nM specific oligonucleotides, AS1 and AS2 (Sp-AS), and the resulting product was resolved by SDS-PAGE, followed by detection with anti-Flag antibody. An in vitro translation reaction for the G4 wild type with 1,000 nM nonspecific antisense oligonucleotides (nSp-AS) was used as a control. The numbers represent relative band densities determined by ImageJ software by taking the G4 wild-type clone translated alone as a reference. (b) Effects of antisense oligonucleotides on transcription of the G4 wild type. The in vitro translation products of pA3F-G4 wild type along with antisense oligonucleotides and nonspecific oligonucleotides were incubated at 30°C for 1 h in separate reactions, RNA was extracted, and cDNA was synthesized and quantified using gene-specific primers.
Due to their complex structure and folding, G-quadruplexes possess unique biophysical and biochemical properties. Circular-dichroism (CD) spectroscopy has been extensively used to study the conformational properties of DNA and RNA. Molar ellipticities of G-quadruplexes, analyzed by CD spectroscopy, have distinctive positive and negative peaks at specific wavelengths, which vary for duplex RNA or DNA (43). CD spectroscopy is used to analyze the effects of various factors affecting structural conformations, such as salt concentration, the presence of cations, differences in temperature, and ligand binding. To validate the presence of G-quadruplexes in LANA mRNA, we performed CD spectroscopy on an RNA oligonucleotide encompassing the G-quadruplex site of LANA mRNA (wild-type LANA oligonucleotide). We also used an oligonucleotide of the same length with scrambled nucleotides (scrambled oligonucleotide) as a control. We observed a CD spectral pattern with a maximum absorption at 260 nm and a negative minimum at 240 nm for wild-type LANA oligonucleotide, which is a characteristic pattern of RNA G-quadruplexes (Fig. 1C, panel a). In contrast, the scrambled RNA oligonucleotide did not show this specific absorption patterns (Fig. 1C, panel b), confirming the biophysical nature of G-quadruplexes in LANA mRNA. Furthermore, the formation of G-quadruplexes in LANA mRNA was confirmed by an electrophoretic mobility shift assay (EMSA). The wild-type and scrambled LANA RNA oligonucleotides were labeled with 32P and resolved in the presence of K+ ions on a 15% native gel. Autoradiography to determine the mobility of these oligonucleotides showed that the wild-type LANA mRNA sequence-containing RNA oligonucleotide migrated faster than the scrambled LANA RNA oligonucleotide due to the formation of a compact G-quadruplex structure in the wild-type LANA RNA oligonucleotide (Fig. 1D, compare lane 2 with lane 1). In order to further confirm that the slower mobility of the wild-type LANA RNA oligonucleotide was due to the formation of these secondary structures, we disrupted the formation of G-quadruplexes by incubating the wild-type RNA oligonucleotide with two antisense oligonucleotides (AS1, TTCCTGCTCTTCCAC, and AS2, CTCCTCTAACTCCTG), which were complementary to each arm of the wild-type LANA RNA oligonucleotide. These wild-type oligonucleotides along with the antisense oligonucleotides were resolved on a native gel, which showed a further shift in the mobility of the LANA RNA oligonucleotide (Fig. 1D, lane 3). This shift is attributed to the disruption of G-quadruplex structure formation by antisense oligonucleotides as they bind to the complementary G-rich region on LANA oligonucleotide, thereby making it unavailable for G-quadruplex formation. These results validated the presence of G-quadruplex structures in LANA mRNA.
G-quadruplex-stabilizing compound inhibited translation of LANA mRNA.
G-quadruplex formation has been shown to influence a number of biological processes, including the translation of mRNA. Since the translation of EBNA1, a homolog of LANA, was severely impaired due to the stabilization of G-quadruplexes, we asked whether the G-quadruplex in LANA mRNA affected its translation. To address this, we used TMPyP4, a widely used compound that stabilizes the formation of G-quadruplexes, to treat KSHV-positive BC3 and BCBL-1 cells to analyze the effects of G-quadruplex formation on LANA expression. Upon treatment with the compound for 24 h, we observed a reduction in the levels of LANA in both BC3 and BCBL-1 cells compared with that in control dimethyl sulfoxide (DMSO)-treated cells (Fig. 2A, panel a, compare lanes 1 and 3 with lanes 2 and 4, respectively). We analyzed the specificity of TMPyP4 treatment by detecting the expression of another viral protein, v-cyclin, which does not have a G-quadruplex-forming site in its mRNA. The levels of v-cyclin were similar in TMPyP4- and DMSO-treated cells, confirming that reduction in LANA expression was due to the stabilization of G-quadruplexes. Since the G-quadruplex-stabilizing compounds are known to act on G-quadruplexes formed on both DNA and RNA, we wanted to ensure that the transcription of LANA mRNA did not play a role in reducing LANA expression. Hence, we extracted RNA from TMPyP4- or DMSO-treated BC3 and BCBL-1 cells and quantified LANA mRNA by quantitative PCR (qPCR) using gene-specific primers. The relative mRNA levels of LANA, normalized to the mRNA levels of glyceraldehyde-3-phosphate dehydrogenase (GAPDH), were similar in both the control and TMPyP4-treated cells (Fig. 2A, panel b). Next, we wanted to confirm that the reduction in LANA expression in KSHV-infected cell lines was not because of any other viral latent proteins, so we determined the effect of TMPyP4 on cells exogenously expressing LANA. BJAB cells stably expressing LANA (LYFP-Flag) and HEK293L cells transfected with LANA-yellow fluorescent protein (YFP)-Flag were treated with DMSO or TMPyP4 for 24 h before assaying for LANA using anti-Flag antibody. Similar to the above-described observations, we detected a reduction in LANA expression in TMPyP4-treated cells compared to that in control DMSO-treated cells (Fig. 2A, panels c and e). We also ensured that TMPyP4 treatment did not lead to any disparity in LANA mRNA levels due to the compound treatment (Fig. 2A, panels d and f). We also analyzed the reduction in LANA expression quantitatively by detecting the levels of LANA-fused YFP signals through flow cytometry after treating the cells with TMPyP4 or DMSO. Cells transfected with YFP alone were used as a control in our experiments. Reduction in fluorescence intensity, representing the levels of YFP-fused LANA protein, confirmed TMPyP4-mediated reduction in LANA expression compared to that in the YFP-expressing cells, which did not show much reduction in fluorescence intensity following treatment (Fig. 2B). Furthermore, we determined the effects of TMPyP4 on LANA’s translation in a luciferase assay in which the luciferase gene was fused downstream, in frame with the LANA gene (Fig. 2C, panel a). HEK293L cells transfected with pGL3-LANA were treated with TMPyP4 for 24 h and lysed, and luminescence was quantified after incubation with specific substrate. We observed luminescence reduction in LANA-luciferase-expressing cells treated with TMPyP4 compared to that of the control DMSO-treated cells (Fig. 2C). As expected, levels of mRNA in TMPyP4- and DMSO-treated cells were similar (Fig. 2C, panel b). This supported our hypothesis that stable G-quadruplexes are formed on LANA mRNA and stabilization of these structures leads to a reduction in translation, possibly because of the steric roadblock for the movement of translation machinery.
FIG 2.

Stabilization of G-quadruplex formation by TMPyP4 inhibited LANA translation. (A) (a) Immunoblot (IB) depicting endogenous protein levels in BC3 and BCBL-1 cells following treatment with TMPyP4. BC3 and BCBL-1 cells were treated with 10 μM TMPyP4 for 24 h, following which cells were harvested and lysed in NP-40 lysis buffer and the lysates were resolved by SDS-PAGE for the detection of LANA using anti-LANA and anti-v-cyclin antibodies. Cells treated with DMSO were used as a negative control. Anti-GAPDH antibody was used to ensure equal loading of the proteins. (b) Relative mRNA levels of LANA in BC3 and BCBL-1 cells following treatment with 10 μM TMPyP4 for 24 h. RNA was extracted from compound-treated cells, and cDNA was synthesized and quantified using primers specific for LANA. The values were normalized against GAPDH. DMSO-treated cells were used as a negative control. (c) Immunoblot depicting endogenous protein levels in BJAB-LYFP cells following treatment with TMPyP4. BJAB-LYFP cells were treated with 10 μM TMPyP4 for 24 h, the cellular lysates were resolved by SDS-PAGE, and proteins were detected using anti-Flag antibody. Cells treated with DMSO were used as a negative control. Anti-GAPDH antibody was used to ensure equal loading of the proteins. (d) Relative mRNA levels of LANA in BJAB-LYFP cells following treatment with 10 μM TMPyP4 for 24 h. RNA was extracted from compound-treated cells, and cDNA was synthesized and quantified using primers specific for LANA. The values were normalized against GAPDH. DMSO-treated cells were used as a negative control. (e) Immunoblot depicting the levels of overexpressed protein LYFP following treatment with TMPyP4. HEK293L cells were transfected with LYFP, treated with 10 μM TMPyP4, and harvested 24 h posttreatment. The cellular lysates were resolved by SDS-PAGE, and proteins were detected using anti-Flag antibody. Cells treated with DMSO were used as a negative control. Anti-GAPDH antibody was used to ensure equal loading of the proteins. (f) Relative mRNA levels of LANA in HEK293L cells transfected with LYFP following treatment with 10 μM TMPyP4 for 24 h. RNA was extracted from compound-treated cells, and cDNA was synthesized and quantified using primers specific for LANA. The values were normalized against GAPDH. DMSO-treated cells were used as a negative control. ns, not significant. (B) Mean reduction in fluorescence intensity following TMPyP4 treatment. HEK-293L cells were transfected with YFP or LYFP, treated with 10 μM TMPyP4 or DMSO for 24 h, and fixed with paraformaldehyde. Flow cytometry was performed on the cells to quantify the number of GFP-positive cells. Reduction in fluorescence was determined by comparing the levels of fluorescence in TMPyP4-treated and DMSO-treated cells. LYFP-expressing cells showed a quantifiable reduction in fluorescence following TMPyP4 treatment. (C) (a) Schematic illustration of LANA luciferase plasmid. LANA was subcloned into pGL3 basic vector upstream of the luciferase gene in frame to make a LANA-luciferase fusion protein. (b) Luciferase assay showing reduction in luciferase levels following TMPyP4 treatment in HEK293L cells. Cells were transfected with LANA-pGL3 and treated with TMPyP4 or DMSO for 24 h. These cells were used for the extraction of RNA and luciferase assay. mRNA levels are expressed relative to the GAPDH. Luciferase levels were significantly reduced in TMPyP4-treated cells despite similar RNA levels.
Region of LANA mRNA with G-quadruplex sites negatively regulated translation.
After having established that G-quadruplex stabilization inhibited LANA translation, we wanted to selectively analyze the G-rich sequence of LANA mRNA for its translation by disrupting the G-quadruplex-forming sites. To achieve this, we generated a 250-nucleotide (nt)-long QE region of LANA (857 to 915 amino acids [aa]) in pA3F vector (G4 wild type) encompassing the G-rich residues that form G-quadruplexes (Fig. 3A, panels a and b). We also generated a G4 disrupted LANA construct in which the G residues were mutated (marked in red in Fig. 3A, panel c) so it could no longer form a G-quadruplex but still coded for the same amino acids as the G4 wild type. In order to assess the effect of G-quadruplex on translation of LANA mRNA, we first performed an in vitro translation assay using G4 wild-type and G4 disrupted constructs. The resulting in vitro-translated protein was resolved by SDS-PAGE and detected using anti-Flag antibody. Comparison of the protein bands showed significantly reduced levels in the G4 wild type compared to the G4 disrupted clone, suggesting that the presence of G-quadruplexes in LANA mRNA modulated its translation (Fig. 3B, panel a). It can be argued that the differences in the translation levels could have been due to a difference in the transcription from two different constructs. To address this, we isolated RNA from an in vitro transcription/translation reaction for G4 wild-type and G4 disrupted clones and quantified them through qPCR using gene-specific primers, which showed almost similar levels in G4 wild-type and G4 disrupted samples (Fig. 3B, panel b).
To analyze the effects of G-quadruplexes on the expression in human cells, we transfected these constructs in HEK293T cells and detected the expression of these proteins 24 h posttransfection following SDS-PAGE and anti-Flag detection. Interestingly, we did not detect any expression in cells transfected with the G4 wild type, compared to cells transfected with the G4 disrupted construct, which displayed a band specific for the G4 disrupted protein (Fig. 3C, panel a, lanes 2 and 3). This suggested that G4 wild-type mRNA formed stable G-quadruplex structures, which abrogated the translation completely, as the levels of transcribed mRNA were almost similar (Fig. 3C, panel b). To further confirm that the reduction in expression was specifically due to the formation of G-quadruplex structures, we disrupted these structures with antisense oligonucleotides, used in our gel shift assay. We used antisense nonspecific oligonucleotides, which were unable to disrupt the G4 structure, as a control. In vitro transcription/translation using the G4 wild type, the G4 wild type with specific antisense oligonucleotides (Sp-As; AS1, 5'-TTCCTGCTCTTCCAC-3', and AS2, 5'-CTCCTCTAACTCCTG-3'), and the G4 wild type with nonspecific oligonucleotides (nSp-As; NS1, 5'-GCCACCGAACAACCCC-3', and NS2, 5'-CACTAGCCCCCCCCC-3'), and resolution by SDS-PAGE followed by detection with Flag antibody showed a significant increase in the expression of the G4 wild type in the presence of specific antisense oligonucleotides, compared to an overall lower expression when the G4 wild type was translated alone or in the presence of nonspecific antisense oligonucleotides (Fig. 3D, panel a). The difference in the mRNA levels was statistically insignificant under these conditions (Fig. 3D, panel b). These observations confirmed that G-quadruplex formation inhibits the translation of G-rich LANA mRNA.
G-quadruplexes modulated antigen presentation.
LANA evades the host’s immune surveillance by inhibiting MHC class I and class II antigen presentation. In this study, we showed that G-quadruplexes in LANA mRNA regulate its expression; therefore, we wanted to investigate how these G-quadruplexes affect immunomodulation. To test this, we cloned the ovalbumin epitope (SIINFEKL) downstream of the G4 wild type, the G4 disrupted construct, and the full-length LANA coding sequence to generate ovalbumin fusion constructs designated G4 wild type-ova, G4 disrupted-ova, and LANA-ova, respectively. These constructs were transfected into HEK293Kbc2 antigen-presenting cells. Cells stably expressing G4 wild type-ova or G4 disrupted-ova were harvested at 24 h posttransfection and incubated with different ratios of T cell receptor cell line B3Z in a 96-well plate at 37°C for 18 h. A total of 150 μl of medium was replaced with solution containing 5 mM o-nitrophenyl-β-d-galactopyranoside (ONPG) and 0.5% NP-40 and incubated at 37°C for 4 h, following which the β-galactosidase activity was quantified by measuring the optical density (OD) at 450 nm. Upon comparing relative levels of β-galactosidase between G4 wild type-ova- and G4 disrupted-ova-transfected cells, we found a higher β-galactosidase activity, i.e., antigen presentation efficiency, in cells expression G4 disrupted-ova (Fig. 4B, panel a). This confirmed that G-quadruplex formation in the G4 wild-type mRNA inhibited its translation, leading to reduced expression and a lower surface antigen presentation on HEK293Kbc2 cells. Similarly, the β-galactosidase activity of B3Z cells incubated with TMPyP4-treated LANA-ova-transfected cells was lower than that of DMSO-treated cells (Fig. 4C, panel a). This confirmed that G-quadruplex formation in LANA mRNA leads to reduced translation and hence a lower surface expression and antigen presentation. We made sure to check the mRNA levels, which were uniform in all the samples, negating any effect on antigen presentation due to altered transcription (Fig. 4B, panel b, and C, panel b).
FIG 4.

G-quadruplex formation reduced antigen presentation of LANA. (A) (a) Schematic depicting antigen presentation assay. Vector encoding a sequence specific for ovalbumin epitope SIINFEKL were transfected in (target or antigen-presenting) HEK293Kbc2 cells and incubated with (effector) T cell hybridoma B3Z cells that recognize the epitope and activate the β-galactosidase gene. Upon incubation with a chromogenic substrate for β-galactosidase, the change in absorbance was recorded to quantitatively measure T cell activation. (B) (a) T cell activation following incubation with cells transiently expressing G4 wild-type and G4 disrupted clones. HEK293Kbc2 cells were transfected with pA3F-G4 wild type ova and pA3F-G4 disrupted ova clones. Twenty-four hours posttransfection, cells were incubated with B3Z cells at effector-to-target ratios of 1:2, 1:1, and 1:0.5. Eighteen hours postincubation, ONPG was added to the cells and β-galactosidase activity was measured at 450 nm using a plate reader. (b) Quantification of mRNA levels in antigen-presenting cells. HEK293Kbc2 cells were transfected with pA3F-G4 wild type ova and pA3F-G4 disrupted ova. Twenty-four hours posttransfection, RNA was extracted, and cDNA was synthesized and quantified using gene-specific primers. mRNA levels are expressed relative to the GAPDH. (C) (a) Effect of TMPyP4 treatment on endogenous antigen presentation. HEK293Kbc2 cells were transfected with pA3F-LANA ova and treated with TMPyP4. DMSO-treated cells were used as a control. Twenty-four hours posttransfection, HEK293Kbc2 cells were incubated with B3Z cells at effector-to-target ratios of 1:2, 1:1, and 1:0.5. Eighteen hours postincubation, ONPG was added to the cells, and β-galactosidase activity was measured at 450 nm using a plate reader. (b) Effect of TMPyP4 treatment on transcription of LANA in antigen-presenting cells. HEK293Kbc2 cells were transfected with pA3F-LANA ova and treated with DMSO or TMPyP4. Twenty-four hours posttransfection, RNA was extracted, and cDNA was synthesized and quantified using gene-specific primers. mRNA levels are expressed relative to the GAPDH.
RNA G-quadruplexes interacted with cellular proteins.
Since the G-quadruplexes are known to interact with a number of proteins that help to open or stabilize the secondary structures, we wanted to identify proteins that interacted with LANA G4 mRNA. To achieve this, we performed a pulldown assay with biotinylated RNA, in which LANA RNA oligonucleotide with wild-type G4 sites and a scrambled oligonucleotide with disrupted G4 were first biotinylated at the 3' end. The biotinylated RNA was purified and incubated with cellular extract from KSHV-positive BCBL-1 cells for 3 h, following which streptavidin beads were added for 2 h to capture the proteins associated with the RNA. The beads were washed stringently to remove any nonspecific proteins before they were subjected to mass spectrometry analysis. Our assay identified a large number of proteins enriched in wild-type LANA RNA pulldown samples, in contrast to the scrambled RNA sample (Table 1). Among these, we found the hnRNP family of proteins specifically associating with the wild-type RNA oligonucleotide rather than the scrambled oligonucleotide. We confirmed this interaction through immunoblotting after resolving the proteins pulled down with biotinylated RNA oligonucleotide, which specifically came down with oligonucleotide having wild-type G4 sites (Fig. 5A). Further, we wanted to determine whether hnRNP A1 binds to the RNA with G4 sites within the cells. To this end, we performed a cross-linking and immunoprecipitation (CLIP) assay on HEK239T cells transfected with G4 wild-type or G4 disrupted constructs. Cells were harvested 24 h posttransfection, and the cell lysates were incubated with hnRNP A1 antibody to capture RNA associated with hnRNP A1. The RNA-protein immune complexes were reverse cross-linked, the bound RNA was quantified using primers specific for G4 wild-type or G4 disrupted mRNA, and the bindings were calculated relative to those of the input samples. Our data showed that hnRNP A1 specifically binds to G4 wild-type mRNA rather than the G4 disrupted mRNA, thus confirming our previous observation by biotinylated RNA pulldown that hnRNP A1 binds to G-quadruplex-forming mRNA (Fig. 5B, panel a). To ensure the specificity of hnRNP A1 binding, we quantified its binding on a known, 7sk mRNA, which showed similar degrees of binding in both the samples (Fig. 5B, panel b). This confirmed that the difference in hnRNP A1 binding to mRNA is dependent on G4 sites. The next step was to analyze whether hnRNP A1 bound LANA mRNA in KSHV-infected cells. To this end, we performed a cross-linking and immunoprecipitation assay with hnRNP A1 on BCBL-1 cells. We compared the degrees of binding of hnRNP A1 on LANA mRNA with and without stabilizing the G-quadruplexes by treatment with TMPyP4 and DMSO, respectively. Comparison of degrees of hnRNP A1 binding to the G-rich region in BCBL-1 cells treated with DMSO and TMPyP4 showed higher affinity of hnRNP A1 in the DMSO-treated cells than in TMPyP4-treated cells (Fig. 5C, panel a). This suggested that hnRNP A1 selectively binds to the G-quadruplexes and that stabilization with ligands, e.g., TMPyP4, reduces its binding. We also analyzed the binding of hnRNP A1 to a region on LANA mRNA (aa 1 to 32), which does not form G-quadruplex structures, that showed significantly reduced binding confirming specificity of hnRNP A1 binding to the G-quadruplexes (Fig. 5C, panel b).
TABLE 1.
List of proteins bound to the LANA RNA with G-quadruplexes
| Name of protein | UniProt accession no. |
|---|---|
| Heterogeneous nuclear ribonucleoproteins A2/B1 | ROA2_HUMAN |
| Epididymis secretory sperm binding protein | A0A024RB53_HUMAN |
| Heterogeneous nuclear ribonucleoprotein A3 | P51991 |
| Heterogeneous nuclear ribonucleoprotein A/B | D6R9P3_HUMAN |
| Heterogeneous nuclear ribonucleoprotein A1 | P04256 |
| Histone H2A | A0A024R017_HUMAN |
| Cell growth-inhibiting protein 34 | Q08ES8_HUMAN |
| 40S ribosomal protein S16 | RS16_HUMAN |
| ATP synthase subunit gamma, mitochondrial | ATPG_HUMAN |
| Nucleoside diphosphate kinase A | E7ERL0_HUMAN |
| Heterogeneous nuclear ribonucleoprotein H3 | P31942 |
| Propionyl-CoAa carboxylase beta chain, mitochondria | P05166 |
| Coiled-coil domain-containing protein | A2RUR9 |
| Four and a half LIM domains protein 3 | Q13643 |
| Elongation factor 1-beta | P24534 |
| Serine/arginine-rich splicing factor 1 | Q07955 |
| Poly(rC)-binding protein 1 | O19048 |
| Calcium-activated chloride channel regulator 2 | Q8BG22 |
| 60S ribosomal protein L11-1 | A8WQ43 |
| Heterogeneous nuclear ribonucleoprotein C | G3V9R8 |
| RNA-binding motif protein, X chromosome | Q9WV02 |
| TAR DNA-binding protein 43 | TARDBP |
| Heterogeneous nuclear ribonucleoprotein K | O19049 |
| Elongation factor 1-beta | P24534 |
| Serine/arginine-rich splicing factor 3 | P84104 |
| Succinate-CoA ligase (ADP-forming) subunit beta | Q482S1 |
| Small nuclear ribonucleoprotein Sm D1 | Q4R5F6 |
| THAP domain-containing protein 10 | Q5NVM3 |
| Interleukin enhancer-binding factor 2 | Q5RFJ1 |
| 30S ribosomal protein S8 | Q06FM9 |
| Guanine deaminase | Q9WTT6 |
| Glycine-tRNA ligase | Q8L785 |
| Adenosine deaminase | C6DH28 |
| Quinolinate synthase A | A5VZW1 |
| 5-hydroxytryptamine receptor 3B | Q9JJ16 |
| Nucleophosmin | P06748 |
| Signal peptidase complex subunit 2 | Q15005 |
| Myb-related protein A | P10243 |
| Kinesin-like protein KIF14 | Q15058 |
| E3 UFM1-protein ligase 1 | A8WN14 |
| Signal peptidase complex subunit 2 | SPCS2 |
| Vitamin D-binding protein | P02774 |
| Solute carrier organic anion transporter family member 6A1 | Q86UG4 |
| Bifunctional purine biosynthesis protein PurH | A4QCK |
| Glutamate-cysteine ligase | Q72RD4 |
| Pentatricopeptide repeat-containing protein | Q9SQU6 |
| Eukaryotic translation initiation factor 3 subunit G | Q6BT10 |
CoA, coenzyme A.
FIG 5.

hnRNP A1 interacted directly with G-rich RNA of LANA and enhanced translation. (A) Affinity pulldown assay of wild-type (WT) LANA RNA. Wild-type LANA RNA oligonucleotide was biotinylated and incubated with lysate from KSHV-positive BCBL-1 cells, and streptavidin beads were added to pull down RNA-bound proteins, followed by resolution of the samples on an SDS-PAGE gel and detection by hnRNP A1. Scrambled (Scrmb) RNA oligonucleotide was used as a negative control. WT LANA and Scrmb LANA RNA lanes were imaged at high exposure to detect binding. WT LANA RNA showed relatively higher binding with LANA than with the scrmb LANA RNA. The input lane was imaged at a lower exposure for comparable band intensities. (B) RNA CLIP assay confirming the direct interaction between hnRNP A1 and G-rich RNA of LANA. HEK293T cells transfected with pA3F-G4 wild type and pA3F-G4 disrupted plasmids were harvested and fixed 24 h posttransfection. The cells were lysed and then incubated with hnRNP A1 antibody and protein A/G beads. Bead-bound RNA was purified and quantified using an iTaq Universal SYBR green one-step kit with G4 wild-type and G4 disrupted clone-specific primers (a) and 7sk gene-specific primers (b), which served as a positive control for hnRNP A1 RNA CLIP. (C) RNA CLIP assay confirming the direct interaction between endogenous hnRNP A1 and G-rich RNA of LANA in KSHV-positive cells. BCBL-1 cells were treated with DMSO and TMPyP4 for 24 h, following which the cells were lysed and incubated with hnRNP A1 antibody and protein A/G beads. Bead-bound RNA was purified and quantified using an iTaq Universal SYBR green one-step kit with primers specific to the G-rich region (amino acids 857 to 916) (a) and specific to the region from amino acids 1 to 32, which does not form any G-quadruplexes. (D) Luciferase assay showing an increase in translation of LANA with increasing concentrations of hnRNP A1. HEK293L cells were transfected with pGL3-LANA and pA3F-hnRNA1 (at various concentrations). Cell lysates were used for the luciferase levels using a dual-luciferase assay. (E) Effects of hnRNP A1 on translation of the G4 wild type. HEK293T cells were transfected with pA3F-G4 wild type and pA3F-G4 disrupted plasmids along with pA3F-hnRNP A1 at increasing concentrations. Twenty-four hours posttransfection, the cell lysates were resolved by SDS-PAGE and proteins were detected using anti-Flag antibody. Anti-GAPDH antibody was used to ensure equal loading of the proteins.
hnRNP A1 has been shown to regulate a number of biological processes by unfolding the G-quadruplex structures through selectively binding to the G-rich region of the mRNA (44, 45). Since LANA mRNA with G-quadruplex sites showed reduced translation, we wanted to analyze the role of hnRNP A1-mediated unfolding of G-quadruplexes in LANA mRNA. We addressed this by analyzing the effects of hnRNP A1 expression on full-length LANA through a luciferase assay. For this, we transfected HEK293L cells with LANA luciferase constructs in the presence of different concentrations of hnRNP A1. The cells were harvested 24 h posttransfection and lysed, and the lysates were incubated with luciferase substrate for determining the levels of LANA-fused luciferase. Our data showed a gradual increase in the luciferase, indicating a higher expression of LANA-fused proteins with increasing concentrations of hnRNP A1 (Fig. 5D). This suggested that hnRNP A1 unwinds the G-quadruplexes of LANA mRNA, thereby increasing the levels of translated LANA. Furthermore, we confirmed the role of hnRNP A1 in enhancing the expression of G-quadruplex-containing mRNA by using G4 wild-type and G4 disrupted clones used in earlier experiments. HEK293T cells were transfected with either pA3F-G4 wild type or pA3F-G4 disrupted, along with increasing concentrations of hnRNP A1, and the lysates were resolved by SDS-PAGE 24 h posttransfection. Detection of LANA G4 wild-type protein with anti-Flag antibody was progressively enhanced from no detectable expression to a significant level with an increasing concentrations of hnRNP A1 (Fig. 5E, IB:Flag). In contrast, hnRNP A1 expression had a limited effect on the G4 disrupted protein expression, which was evident by the almost similar level of G4 disrupted protein expression even with increasing amounts of hnRNP A1 (Fig. 5E, panel b). This corroborated our previous results and led us to conclude that hnRNP A1 binds selectively to G-quadruplex-forming mRNA of LANA, unfolds these secondary structures, and facilitates the translation of LANA mRNA.
LANA inhibited the nuclear export of G-rich mRNA by selectively binding.
A number of studies have reported the autoregulatory mechanisms through which LANA controls its expression and antigen presentation. Here we show that the expression of LANA is controlled by G-quadruplexes through the involvement of cellular proteins. We speculated that LANA regulates its expression by associating with G4 sites. We confirmed this by testing the binding of LANA to G4 wild-type mRNA and G4 disrupted mRNA through RNA a cross-linking and immunoprecipitation assay. HEK293L cells were transfected with pA3F-G4 wild type or pA3F-G4 disrupted, along with full-length LANA, and these cells were harvested 24 h posttransfection for chromatin isolation (Fig. 6A, panel a). The LANA-bound chromatin was immunoprecipitated using anti-LANA antibody, and the RNA associated with the chromatin was purified and quantified using gene-specific primers. Our data normalized to respective input samples demonstrated enrichment of G4 wild-type mRNA compared to G4 disrupted mRNA with chromatin bound to LANA, thus confirming the binding of LANA to the G-quadruplex-forming RNA (Fig. 6A, panel b). Next, we wanted to confirm our findings that LANA binds to the G-rich region of LANA mRNA in KSHV-positive cells. We achieved this by treating the cells with G4-stabilizing compound TMPyP4. Twenty-four hours posttreatment, the cells were fixed with formaldehyde and the chromatin was sheared to immunoprecipitate with anti-LANA antibody. LANA-bound RNA in the immune complex was purified for quantitation using a one-step iTaq SYBR kit with primers specific to the G-rich region. Our data showed that LANA has more affinity for the G-rich mRNA from the cells treated with TMPyP4 than from the control DMSO-treated cells, suggesting that LANA preferentially binds to stabilized G-quadruplexes (Fig. 6B, panel a). The quantification of the region of LANA mRNA from positions 1 to 32, which does not form G-quadruplexes, was used as a control to confirm the specificity of LANA’s binding to the G-quadruplex region of LANA mRNA (Fig. 6B, panel b).
FIG 6.

LANA bound to the G-rich of its mRNA and inhibited its translation at higher concentrations through inhibiting mRNA export to the cytoplasm. (A) RNA CLIP assay confirming direct interaction between LANA and the G-rich RNA of LANA. HEK293T cells transfected with pA3F-G4 wild type and pA3F-G4 disrupted plasmids along with LANA were harvested and fixed 24 hposttransfection. The cells were harvested, and a sample of cells was lysed to detect proteins using anti-Flag antibody (a) or lysed and incubated with anti-LANA antibody and protein A/G beads (b). Bead-bound RNA was purified and quantified using an iTaq Universal SYBR green one-step kit with gene-specific (G4 wild type and G4 disrupted) primers. (B) RNA CLIP assay confirming the direct interaction between endogenous LANA and G-rich RNA of LANA in KSHV-positive cells. BCBL-1 cells were treated with DMSO and TMPyP4 for 24 h, following which the cells were lysed and incubated with anti-LANA antibody and protein A/G beads. Bead-bound RNA was purified and quantified using an iTaq Universal SYBR green one-step kit with G-rich region-specific primers (amino acids 857 to 916) (a) and primers specific to the region from amino acids 1 to 32, which does not form any G-quadruplexes (b). (C) Luciferase assay showing a decrease in the translation of LANA at higher concentrations. HEK293L cells were transfected with pGL3-LANA and pA3F-LANA (at various concentrations). Cell lysates were used for quantifying luciferase levels in a dual-luciferase assay. (D and E) Effect of LANA on nuclear export of G4 wild-type (D) and G4 disrupted (E) mRNAs. HEK293L cells were transfected with pA3F-G4 wild type (D) and pA3F-G4 disrupted (E) along with pA3F-LANA at increasing concentrations. Nuclear/cytoplasmic fractionations were performed using the Active Motif kit. (a) Efficacy of the fractionation was analyzed using anti-lamin B1 antibody, which serves as a nuclear protein marker, and GAPDH, which serves as a cytoplasmic protein marker. (b) RNA was extracted from the cytoplasmic fractions, and cDNA was synthesized and quantified using gene-specific primers. Cytoplasmic mRNA levels were determined with reference to GAPDH. Cytoplasmic RNA levels were calculated relative to total mRNA levels.
We further wanted to explore how G-quadruplexes maintain the minimal levels of LANA expression. To do this, we overexpressed LANA at various concentrations along with LANA clones in frame with luciferase (pGL3-LANA). The cells were harvested 24 h posttransfection and lysed, and the luciferase levels were measured. We found an interesting pattern in luciferase expression with different LANA concentrations. Luciferase units, indirectly representing the levels of LANA expression through G-quadruplexes, gradually increased with increasing LANA concentration; however, they started to decrease at larger amounts of LANA (Fig. 6C). This led us to conclude that LANA has a self-regulatory role in controlling its expression, and our findings prove that hnRNP A1 and LANA modulate LANA’s expression through interaction with the G-quadruplexes in LANA mRNA.
Since our results showed that transfection of a pA3F-G4 wild-type clone in HEK293T cells did not show any protein expression despite levels of mRNA similar to those of the pA3F-G4 disrupted clone, we hypothesized that LANA could block the export of mRNA for translation through interacting with the G-quadruplexes. To prove this, we transfected HEK293L cells with G4 wild-type or G4 disrupted clones along with LANA (at different concentrations), following which cells were harvested and the cytoplasmic RNA was extracted. We also extracted total RNA from a portion of cells used for the cytoplasmic RNA extraction. We calculated the amounts of mRNA exported to the cytoplasm (to determine the amounts available for translation) relative to the total mRNA following normalization with the GAPDH gene, a housekeeping gene. Comparison of the cytoplasmic mRNA levels of the G4 wild type in cells transfected with different concentrations of LANA showed a slight increase in the cytoplasmic G4 wild-type mRNA levels at lower LANA concentrations (Fig. 6D, panel b). However, at higher LANA concentrations, we observed a decline in the cytoplasmic levels of G4 wild-type mRNA available for translation (Fig. 6D, panel b). In contrast, there was no significant difference in the cytoplasmic RNA levels of G4 disrupted mRNA in cells transfected with different amounts of LANA (Fig. 6E, panel b) This confirmed that LANA retained G4 wild-type mRNA, possibly through binding with the G-quadruplexes in the mRNA.
LANA inhibited the interaction of hnRNP A1 with G-rich RNA.
Through the above-described experiments, we were able to confirm the interaction between hnRNP A1 and G-rich mRNA of LANA, which led to unfolding of G-quadruplexes and enhanced translation. Moreover, we also showed that LANA can also bind to G-quadruplex-containing mRNA, thereby inhibiting the cytoplasmic export of the mRNA. We next wanted to analyze if LANA interferes with the binding of hnRNP A1 to the G-rich mRNA. For this, we performed an RNA CLIP assay on HEK293L cells transfected with pA3F-G4 wild type or pA3F-G4 disrupted along with full-length LANA at two different concentrations 24 h posttransfection (Fig. 7A). The hnRNP A1-bound chromatin was immunoprecipitated using anti-hnRNP A1 antibody, and the RNA associated with the chromatin was purified and quantified using gene-specific primers. Our data, normalized to respective input samples, demonstrated that increasing concentrations of LANA reduced the binding of hnRNP A1 to the G-rich mRNA of LANA (Fig. 7A). This effect was not observed in cells transfected with the G4 disrupted clone, as the relative hnRNP A1 binding to the mRNA was mostly unaltered with LANA (Fig. 7B). This confirmed that hnRNP A1 binding to LANA mRNA is reduced in cells with excess LANA levels. Reduction in hnRNP A1 binding to LANA mRNA may lead to lower unwinding of G-quadruplexes and thus reduced expression of LANA protein.
FIG 7.

LANA inhibits the binding of hnRNP A1 to G-rich mRNA at higher concentrations. RNA CLIP assay confirming the reduction in the interaction of hnRNP A1 with G-rich RNA at higher LANA concentrations. HEK293T cells transfected with pA3F-G4 wild type (A) and pA3F-G4 disrupted (B) plasmids, along with various concentrations of LANA, were harvested and fixed 24 h posttransfection. The cells were harvested and lysed, following by incubation with anti-hnRNP A1 antibody and protein A/G beads. Bead-bound RNA was purified and quantified using an iTaq Universal SYBR green one-step kit with gene-specific (G4 wild type and G4 disrupted) primers.
DISCUSSION
KSHV establishes persistent latent infection in host cells and employs various mechanisms to avoid immune recognition. During latency, the virus expresses only a limited number of proteins, of which LANA is the most abundantly expressed, which ensures efficient viral replication and distribution of viral genome to the daughter cells (46). LANA has been shown to autoregulate its expression by regulating its promoter as well as inhibition of peptide synthesis and proteasomal degradation through two central region domains (CR2 and CR3; aa 442 to 768 and aa 769 to 920, respectively) (47 – 50). Additionally, the CR1 (aa 330 to 442) domain of LANA has been shown to play a role in restricted MHC class I antigen presentation, through reduced translocation of peptides to the endoplasmic reticulum (ER) (13, 51). We have previously shown that LANA interacts with regulatory factor X proteins and disrupts the association of CIITA for MHC-II gene expression (14). These studies substantiate the role of LANA in regulating its expression and restricting recognition by the host’s immune system; however, there has been no direct evidence demonstrating a link between these two processes. Here we report the formation of G-quadruplexes in the G-rich regions of LANA mRNA and the immunological role of these structures in restricted immune recognition.
G-quadruplexes have gained recognition as regulatory nucleic acid structures that influence a number of biological processes. Many members of the Herpesviridae family have been shown to form G-quadruplexes that regulate viral gene expression, viral DNA replication, and virion production (52, 53). Moreover, G-quadruplexes have gained recognition as immunomodulatory structures in a few gammaherpesviruses, such as EBV and alcelaphine herpesvirus 1 (AIHV), in which these structures abrogate protein synthesis and restrict antigen presentation, thereby helping in immune evasion (30, 54). Upon analysis of the LANA mRNA sequence through QGRS Mapper, G-quadruplex motif-predicting software, we found regions of LANA to form potentially stable G-quadruplexes. CD spectroscopy and electrophoretic mobility assay further confirmed the presence of these secondary structures. We demonstrated the functional significance of G-quadruplex formation through treatment with G-quadruplex-stabilizing ligand TMPyP4, which resulted in reduction of LANA mRNA translation despite similar levels of mRNA synthesis. This was also confirmed by treating the HEK293T cells expressing LANA with G-quadruplex-stabilizing ligand and determining the levels of translated LANA through Western blotting. Stabilizing the G-quadruplexes though TMPyP4 of LANA mRNA fused to YFP showed a quantifiable reduction in LANA-YFP expression by flow cytometry. Additionally, fusion of the LANA-coding sequence upstream of the luciferase reporter gene demonstrated that stabilizing the G-quadruplexes of LANA mRNA reduces the expression of LANA-luciferase gene, possibly by blocking the movement of translational machinery through these secondary structures. All these assays convincingly demonstrated the role of G-quadruplexes in controlling LANA expression. Further, we tested the specific role of these G-quadruplexes by generating a small clone of LANA peptide from the region of the LANA gene containing G-quadruplexes. We also generated G-quadruplex disrupted clones but with the same amino acid sequence, through codon optimization, to ensure that the peptide sequence did not affect translation efficiency. The presence of almost no to very little expression in G-quadruplex clone, despite equal mRNA levels, confirmed the regulatory role of these secondary structures. Disruption of these secondary structures with antisense oligonucleotides in an in vitro translation assay with enhanced expression further substantiated the role of G-quadruplexes in regulating mRNA translation. Antisense oligonucleotides to the target mRNA could potentially degrade the mRNA through a microRNA (miRNA)-induced silencing complex, but the antisense RNA was smaller (15 bp) than required for miRNA. Moreover, the TNT T7 in vitro reaction mix lacks RNA-induced silencing complex (RISC), and the levels of mRNA were comparable in samples with antisense oligonucleotides.
LANA has been shown to modulate antigen presentation by interacting with the components of the antigen presentation pathway to limit its surface presentation (13, 14). Interestingly, we found a link between the antigen presentation and G-quadruplex formation in LANA mRNA, as the antigen presentation of LANA was downregulated upon stabilization of the G-quadruplex structures in KSHV-positive cells along with highly inhibited antigen presentation in cells expressing the G-rich region of LANA. This led us to conclude that G-quadruplex formation inhibits antigen presentation through a reduction in mRNA translation. G-quadruplex formation is shown to be regulated (stabilized/destabilized) through the involvement of a number of cellular proteins (21, 32). Our mass spectrometry results revealed hnRNP A1 as a highly specific interacting partner of the G-quadruplex-forming mRNA. Previous studies have found hnRNP A1 to be a G-quadruplex-destabilizing protein, which was similar to its effect on increasing the expression of G-quadruplex wild-type clones (44, 45). This may suggest that hnRNP A1 increased the expression by selectively binding and potentially destabilizing the G-quadruplexes. Interestingly, we also discovered LANA to be binding at the G-quadruplex region of its mRNA and modulating its translation. Importantly, binding of LANA to the G-quadruplex region on its RNA inhibited the export of those mRNA to the cytoplasm, as LANA primarily localizes in the nucleus. Based on these observations, we propose a model in which the G-quadruplex formation on various regions of LANA, including the region from aa 857 to 917 (a subregion of the CR3 domain), controls LANA mRNA translation and expression of LANA protein (Fig. 8). Expression of LANA is regulated by the relative amounts of LANA, and in cells in which LANA levels are lower, relative binding of hnRNP A1 to LANA mRNA is higher, which leads to its export to the cytoplasm and destabilization of the G-quadruplexes. The translation machinery of the cytoplasm translates LANA mRNA, leading to formation of mature protein or defective ribosomal proteins (DRiPs). The mature LANA proteins translocate to the nucleus, thus increasing the overall levels of LANA in the nucleus. When the concentration of LANA in the nucleus is sufficient, LANA binds to the G-quadruplex-forming region of its mRNA on relatively more copies than the hnRNP A1, thereby inhibiting mRNA export into the cytoplasm and thus making it unavailable for translation and hence lowering levels of proteins available for antigen presentation.
FIG 8.

Schematic model showing regulation of LANA expression through G-quadruplex formation and antigen presentation. LANA is required for maintaining the viral genome in latent cells, and its expression is modulated by the level of LANA in the cells. At lower levels of LANA, hnRNP A1 binds to the G-quadruplex region of LANA mRNA and exports it to the cytoplasm following destabilization of the G-quadruplex structures for translation. Once a sufficient or higher level of LANA is synthesized and translocated to the nucleus, it binds to the G-quadruplex region of the LANA mRNA and retains the G-quadruplex-forming mRNAs in the nucleus to control an excess synthesis of LANA protein. G-quadruplex-mediated control of LANA mRNA translation ensures optimal expression without overly producing mature or defective ribosomal proteins (DriPs), which are presented through MHC class I molecules to escape the host’s immune surveillance system.
MATERIALS AND METHODS
Cell lines, plasmids, and reagents.
The KSHV-positive BCBL-1 cell line was grown in RPMI 1640 supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine, 5 U/ml of penicillin, and 5 μg/ml of streptomycin. The BJAB-LYFP cell line was grown in RPMI 1640 supplemented with 10% FBS, 2 mM glutamine, 5 U/ml of penicillin, 5 μg/ml of streptomycin, and 0.5 μg/ml of puromycin. HEK293T and HEK293L cells were grown in Dulbecco modified Eagle medium (DMEM) supplemented with 8% bovine growth serum, 2 mM glutamine, 5 U/ml of penicillin, and 5 μg/ml of streptomycin (55). B3Z cells, a T cell hybridoma cell line and a kind gift from Nilabh Shastri (John Hopkins University) and Charles L. Stentman (Dartmouth University), were grown in RPMI 1640 supplemented with 10% FBS, 2 mM glutamine, 1 mM pyruvate, 50 μM 2-mercaptoethanol, 100 U/ml of penicillin, and 100 μg/ml of streptomycin. H-2Kb-expressing HEK293 (HEK293KbC2) cells, a gift from Jonathan Yewdell (NIH), were cultured in DMEM supplemented with 8% bovine growth serum, 2 mM glutamine, 5 U/ml of penicillin, and 5 μg/ml of streptomycin.
pA3F-LANA, pLVXYFP-Flag, and pLVX-LYFP-Flag have been described previously (56, 57). hnRNP A1 was generated by PCR amplification and cloning into a Flag-tagged vector, pA3F. LANA luciferase was generated by subcloning LANA in pGL3 basic vector. The constructs pA3F-G4 wild type (containing the G-quadruplex-forming sequence of LANA) and pA3F-G4 disrupted (codon optimized to disrupt the formation of G-quadruplexes but coding for the same amino acids) were commercially synthesized by Genscript, Inc. The constructs pA3F-G4 wild type ova, pA3F-G4 disrupted ova, and pA3F-LANA ova were generated by subcloning the sequence coding for the ovalbumin SIINFEKL peptide into pA3F-G4 wild type, pA3F-G4 disrupted, and pA3F-LANA, respectively. The integrity of the clones was confirmed by DNA sequencing performed at the Nevada Genomics Center, University of Nevada, Reno.
Antibodies.
The following commercial antibodies were used for this study: mouse anti-GAPDH (U.S. Biological), mouse anti-Flag M2 (Sigma-Aldrich, St. Louis, MO), hnRNP A1 (Santa Cruz Biotechnology), lamin B1 (Santa Cruz Biotechnology), and v-cyclin (Abcam). Mouse monoclonal anti-LANA hybridoma was generated at Genscript (Genscript, Inc.).
CD spectroscopy.
CD spectroscopy was performed on an Aviv Biomedical spectrometer, for which circularly polarized UV light was used to record CD spectra at 25°C in a series of progressive scans from 320 nm to 200 nm in a quartz cuvette with a path length of 1 mm. The RNA oligonucleotides used to record CD spectra were resuspended at a final concentration of 5 μM in sodium cacodylate buffer (10 mM; pH 7.4) with 100 mM KCl.
Electrophoretic mobility shift assay.
Electrophoretic mobility shift assay (EMSA) was performed as described before (31). Briefly, wild-type LANA (UGGAAGAGCAGGAAGAGCAGGAGUUAGAGGA) and scrambled LANA (UAACCGAUGAUAUGAGUCAGAUAUAUAAGCA) RNA oligonucleotides were labeled with radioactive 32P using T4 polynucleotide kinase (New England BioLabs). The oligonucleotides were resuspended at a final concentration of 2 μM in 10 mM sodium cacodylate buffer with 100 mM KCl and resolved on a 15% nondenaturing polyacrylamide gel. The oligonucleotides were resolved in 1× Tris-borate-EDTA (TBE) buffer with 100 mM KCl in either the presence or absence of antisense oligonucleotides. The gels were dried using Gelair gel dryer (Bio-Rad Inc.), and autoradiography was performed using a phosphorimager (GE Healthcare Life Sciences).
In vitro translation and RNA quantification.
In vitro translation of pA3F-G4 wild type and G4 disrupted was performed using the Promega TNT T7 quick-coupled transcription/translation system, in which 2 μg of the plasmid was translated in a 50-μl reaction mixture containing 1 mM methionine at 30°C for 2 h. The translated product was resolved using SDS-PAGE, and proteins were detected using anti-Flag antibody. RNA levels in the in vitro-translated samples were determined by extracting the RNA from the translation mix using TRIzol reagent (Thermo Fisher Scientific) as per the manufacturer’s protocol. cDNA was synthesized using a high-capacity RNA-to-cDNA kit (Applied Biosystems Inc.) as per the manufacturer’s protocol and quantified using gene-specific primers.
RNA quantification and protein expression following compound treatment.
Cells were treated with 10 μM TMPyp4 for 24 h, harvested, and lysed in NP-40 cell lysis buffer (1% Nonidet P-40, 50 mM Tris-HCl [pH 7.5], 150 mM NaCl, and 1 mM EDTA) supplemented with protease inhibitors (1 mM phenylmethylsulfonyl fluoride, 10 μg/ml of pepstatin, 10 μg/ml of leupeptin, and 10 μg/ml of aprotinin). Cells treated with DMSO were used as a control, the cellular lysates were resolved by SDS-PAGE, and proteins were detected using specific antibodies. To analyze the effect of TMPyP4 treatment on transcription, cells were treated with compound for 24 h, following which total RNA was extracted using an Illustra RNAspin minikit (GE Healthcare) according to the manufacturer’s protocol. cDNAs were synthesized using a high-capacity RNA-to-cDNA kit (Applied Biosystems Inc.) as per the manufacturer’s protocol and quantified using gene-specific primers. The threshold cycle (CT) values were normalized to the GAPDH gene, a housekeeping gene. All the reactions were run in triplicates.
RNA cross-linking immunoprecipitation assay.
RNA cross-linking immunoprecipitation assay was performed as described before (58). Briefly, 10 million cells were harvested, fixed in 1% formaldehyde, and quenched by the addition of 125 mM glycine, following which the cells were washed and the cell pellet was resuspended in cell lysis buffer [5 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES; KOH; pH 8.0), 85 mM KCl, 0.5% NP-40, fresh protease inhibitors, and RNAseOUT]. After incubation for 10 min on ice, the nuclei were isolated following centrifugation and resuspended in Diagenode chromatin shearing buffer D (with protease inhibitors and RNAseOUT). The nuclei were sheared to an average chromatin size of 200 to 400 bp using BioRuptor (Diagenode, Inc.), following which the cell debris was pelleted and the sheared chromatin was diluted with 3 volumes of dilution buffer (1.2 mM EDTA, 16.7 mM Tris [pH 8.0], protease inhibitors, and RNAseOUT). The diluted chromatin was incubated with specific antibodies overnight, and protein A/G magnetic Sepharose beads were added for 2 h to capture the immune complexes. The beads were washed twice with low-salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris [pH 8.0], 150 mM NaCl) and once with high-salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris [pH 8.0], 500 mM NaCl). The beads were finally washed with 1× TE and resuspended in 150 μl of elution buffer (1% SDS, 100 mM NaHCO3), with gentle vortexing for 15 min to elute the complex. The samples were reverse cross-linked overnight at 65°C using 0.3 M NaCl. The next day, the samples were treated with proteinase K at 45°C for 2 h, followed by DNase treatment at 37°C for 30 min. TRIzol was added to the samples and incubated for 5 min, followed by addition of chloroform and incubation at room temperature for 15 min. The samples were centrifuged and RNA in the aqueous phase was precipitated by the addition of 1× isopropanol and ammonium acetate. The samples were allowed to precipitate at –80°C for at least 30 min, following which the samples were centrifuged and the pellets were washed twice with 75% ethanol. Finally, the pellet was resuspended in 30 μl of nuclease-free water.
In vitro RNA pulldown assay.
Wild-type LANA RNA oligonucleotide (5'-UGGAAGAGCAGGAAGAGCAGGAGUUAGAGGA-3') and scrambled LANA RNA oligonucleotide (5'-UAACCGAUGAUAUGAGUCAGAUAUAUAAGCA-3') were biotinylated at the 3' end with a Pierce 3' RNA biotinylation kit according to the instruction manual. To prepare the cellular lysate, approximately 20 million KSHV-positive BCBL-1 cells were harvested and lysed in 1% NP-40 lysis buffer with protease inhibitors and RNaseOUT. The samples were sonicated and centrifuged to remove cellular debris. The purified biotinylated RNA oligonucleotides were incubated with the lysate for 3 h at 4°C. Pierce streptavidin-agarose beads (Thermo Fisher Scientific) were then added to the samples to pull down the proteins bound to the biotinylated RNA oligonucleotides following incubation with the beads for 2 h. The beads were washed thrice with 1% NP-40 lysis buffer, following which they were loaded onto a 9% SDS-PAGE gel, transferred onto a nitrocellulose membrane, and probed with specific antibodies. The immunoprecipitation samples were also subjected to liquid chromatography-mass spectrometry (LC-MS) analysis at Mitch Hitchcock Nevada Proteomics Center, University of Nevada, Reno.
Dual-luciferase assay.
LANA was subcloned into pGL3 basic vector with the luciferase gene downstream of the LANA coding region. HEK293T cells were transfected with 250 ng of pGL3-LANA and 10 ng of Renilla luciferase-expressing plasmid pRRLSV40. Twenty-four hours posttransfection, the cells were harvested, lysed in 1% NP-40 lysis buffer, and centrifuged to remove cell debris. The firefly reporter was measured by adding luciferase assay reagent II (LARII; Promega, Inc.) to a 96-well plate containing supernatant from the transfected cells and quantified with a luminometer. The reaction was stopped and Renilla luciferase activity was measured by addition of Stop and Glo reagent and quantified using a luminometer. The relative luciferase levels were calculated after normalization of firefly luciferase with Renilla luciferase to account for the transfection discrepancies.
Flow cytometry.
YFP and LYFP plasmids were transfected into HEK293T cells as described previously. Four hours posttransfection, the cells were treated with DMSO or TMPyP4. Twenty-four hours posttreatment, the cells were trypsinized, washed with phosphate-buffered saline (PBS), and then fixed in PBS supplemented with 2% paraformaldehyde for 15 min at room temperature. Finally, the cells were washed once with PBS and the cell pellet was resuspended in PBS. Green fluorescent protein (GFP)-positive cells were detected using a FACSCalibur flow cytometer equipped with CellQuest Pro software and analyzed using FlowJo software. The fluorescence intensity was expressed as a percentage of total number of cells, and the reduction in fluorescence intensity due to TMPyP4 treatment was calculated for both YFP- and LYFP-transfected cells.
Antigen presentation assay.
HEK293Kbc2 cells were transfected with LANA-ova, G4 wild type-ova, and G4 disrupted-ova plasmids and harvested 24 h posttransfection. The cells were incubated with T cell hybridoma B3Z cells for 18 h at effector-to-target ratios of 1:2, 1:1, and 1:0.5. The cells were incubated with buffer containing 0.125% NP-40, 9 mM MgCl2, 100 mM β-mercaptoethanol, and 5 mM ONPG for 4 h at 37°C. The absorbance was measured at 450 nm using an EMax absorbance microplate reader (Molecular Devices, USA) and read using SoftMax Pro v. 6.4 software.
RNA subcellular isolation.
293L cells were transfected with the G4 wild type or G4 disrupted clones along with different concentrations of LANA. Cells were harvested 24 h posttransfection; total and cytoplasmic RNA extraction was performed using an Active Motif RNA subcellular isolation kit according to the manufacturer’s instructions. cDNA was synthesized using an ABI cDNA synthesis kit, and G4 wild-type/G4 disrupted RNA was quantified using gene-specific primers. Normalization of cytoplasmic RNA was performed using total RNA content, and the RNA contents of the cytoplasmic fractions were compared between the samples.
Statistical analysis.
P values were calculated by two-tailed t test using GraphPad Inc. (Prism 8) software for statistical significance. In figures, asterisks represent P values as follows: *, P value < 0.05; **, P value < 0.01; and ***, P value < 0.001.
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (CA174459 and AI105000). P.D. was partially supported by the Reno Cancer Foundation.
We thank Matthew Tucker and Yftah Tal-Gan, Department of Chemistry, University of Nevada, Reno, for helping with CD spectroscopy.
REFERENCES
- 1.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 2.Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. 1995. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med 332:1186–1191. doi: 10.1056/NEJM199505043321802. [DOI] [PubMed] [Google Scholar]
- 3.Renne R, Zhong W, Herndier B, McGrath M, Abbey N, Kedes D, Ganem D. 1996. Lytic growth of Kaposi’s sarcoma–associated herpesvirus (human herpesvirus 8) in culture. Nat Med 2:342–346. doi: 10.1038/nm0396-342. [DOI] [PubMed] [Google Scholar]
- 4.Decker LL, Shankar P, Khan G, Freeman RB, Dezube BJ, Lieberman J, Thorley-Lawson DA. 1996. The Kaposi sarcoma-associated herpesvirus (KSHV) is present as an intact latent genome in KS tissue but replicates in the peripheral blood mononuclear cells of KS patients. J Exp Med 184:283–288. doi: 10.1084/jem.184.1.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rezaee SA, Cunningham C, Davison AJ, Blackbourn DJ. 2006. Kaposi’s sarcoma-associated herpesvirus immune modulation: an overview. J Gen Virol 87:1781–1804. doi: 10.1099/vir.0.81919-0. [DOI] [PubMed] [Google Scholar]
- 6.Rainbow L, Platt GM, Simpson GR, Sarid R, Gao SJ, Stoiber H, Herrington CS, Moore PS, Schulz TF. 1997. The 222- to 234-kilodalton latent nuclear protein (LNA) of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) is encoded by orf73 and is a component of the latency-associated nuclear antigen. J Virol 71:5915–5921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kedes DH, Lagunoff M, Renne R, Ganem D. 1997. Identification of the gene encoding the major latency-associated nuclear antigen of the Kaposi’s sarcoma-associated herpesvirus. J Clin Invest 100:2606–2610. doi: 10.1172/JCI119804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kellam P, Boshoff C, Whitby D, Matthews S, Weiss RA, Talbot SJ. 1997. Identification of a major latent nuclear antigen, LNA-1, in the human herpesvirus 8 genome. J Hum Virol 1:19–29. [PubMed] [Google Scholar]
- 9.Ballestas ME, Kaye KM. 2011. The latency-associated nuclear antigen, a multifunctional protein central to Kaposi’s sarcoma-associated herpesvirus latency. Future Microbiol 6:1399. doi: 10.2217/fmb.11.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barbera AJ, Chodaparambil JV, Kelley-Clarke B, Joukov V, Walter JC, Luger K, Kaye KM. 2006. The nucleosomal surface as a docking station for Kaposi’s sarcoma herpesvirus LANA. Science 311:856–861. doi: 10.1126/science.1120541. [DOI] [PubMed] [Google Scholar]
- 11.Matsumura S, Persson LM, Wong L, Wilson AC. 2010. The latency-associated nuclear antigen interacts with MeCP2 and nucleosomes through separate domains. J Virol 84:2318–2330. doi: 10.1128/JVI.01097-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee H-R, Lee S, Chaudhary PM, Gill P, Jung JU. 2010. Immune evasion by Kaposi’s sarcoma-associated herpesvirus. Future Microbiol 5:1349–1365. doi: 10.2217/fmb.10.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kwun HJ, da Silva SR, Qin H, Ferris RL, Tan R, Chang Y, Moore PS. 2011. The central repeat domain 1 of Kaposi’s sarcoma-associated herpesvirus (KSHV) latency associated-nuclear antigen 1 (LANA1) prevents cis MHC class I peptide presentation. Virology 412:357–365. doi: 10.1016/j.virol.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thakker S, Purushothaman P, Gupta N, Challa S, Cai Q, Verma SC. 2015. Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen inhibits major histocompatibility complex class II expression by disrupting enhanceosome assembly through binding with the regulatory factor X complex. J Virol 89:5536–5556. doi: 10.1128/JVI.03713-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cai Q, Banerjee S, Cervini A, Lu J, Hislop AD, Dzeng R, Robertson ES. 2013. IRF-4-mediated CIITA transcription is blocked by KSHV encoded LANA to inhibit MHC II presentation. PLoS Pathog 9:e1003751. doi: 10.1371/journal.ppat.1003751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beaudoin J-D, Perreault J-P. 2010. 5′-UTR G-quadruplex structures acting as translational repressors. Nucleic Acids Res 38:7022–7036. doi: 10.1093/nar/gkq557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Didiot M-C, Tian Z, Schaeffer C, Subramanian M, Mandel J-L, Moine H. 2008. The G-quartet containing FMRP binding site in FMR1 mRNA is a potent exonic splicing enhancer. Nucleic Acids Res 36:4902–4912. doi: 10.1093/nar/gkn472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gomez D, Lemarteleur T, Lacroix L, Mailliet P, Mergny J-L, Riou J-F. 2004. Telomerase downregulation induced by the G-quadruplex ligand 12459 in A549 cells is mediated by hTERT RNA alternative splicing. Nucleic Acids Res 32:371–379. doi: 10.1093/nar/gkh181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marcel V, Tran PL, Sagne C, Martel-Planche G, Vaslin L, Teulade-Fichou MP, Hall J, Mergny JL, Hainaut P, Van Dyck E. 2011. G-quadruplex structures in TP53 intron 3: role in alternative splicing and in production of p53 mRNA isoforms. Carcinogenesis 32:271–278. doi: 10.1093/carcin/bgq253. [DOI] [PubMed] [Google Scholar]
- 20.Siddiqui-Jain A, Grand CL, Bearss DJ, Hurley LH. 2002. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc Natl Acad Sci U S A 99:11593–11598. doi: 10.1073/pnas.182256799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rhodes D, Lipps HJ. 2015. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res 43:8627–8637. doi: 10.1093/nar/gkv862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Piekna-Przybylska D, Sharma G, Bambara RA. 2013. Mechanism of HIV-1 RNA dimerization in the central region of the genome and significance for viral evolution. J Biol Chem 288:24140–24150. doi: 10.1074/jbc.M113.477265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perrone R, Nadai M, Frasson I, Poe JA, Butovskaya E, Smithgall TE, Palumbo M, Palù G, Richter SN. 2013. A dynamic G-quadruplex region regulates the HIV-1 long terminal repeat promoter. J Med Chem 56:6521–6530. doi: 10.1021/jm400914r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perrone R, Nadai M, Poe JA, Frasson I, Palumbo M, Palù G, Smithgall TE, Richter SN. 2013. Formation of a unique cluster of G-quadruplex structures in the HIV-1 nef coding region: implications for antiviral activity. PLoS One 8:e73121. doi: 10.1371/journal.pone.0073121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Biswas B, Kandpal M, Vivekanandan P. 2017. A G-quadruplex motif in an envelope gene promoter regulates transcription and virion secretion in HBV genotype B. Nucleic Acids Res 45:11268–11280. doi: 10.1093/nar/gkx823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang S-R, Min Y-Q, Wang J-Q, Liu C-X, Fu B-S, Wu F, Wu L-Y, Qiao Z-X, Song Y-Y, Xu G-H, Wu Z-G, Huang G, Peng N-F, Huang R, Mao W-X, Peng S, Chen Y-Q, Zhu Y, Tian T, Zhang X-L, Zhou X. 2016. A highly conserved G-rich consensus sequence in hepatitis C virus core gene represents a new anti–hepatitis C target. Sci Adv 2:e1501535. doi: 10.1126/sciadv.1501535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Artusi S, Nadai M, Perrone R, Biasolo MA, Palu G, Flamand L, Calistri A, Richter SN. 2015. The herpes simplex virus-1 genome contains multiple clusters of repeated G-quadruplex: implications for the antiviral activity of a G-quadruplex ligand. Antiviral Res 118:123–131. doi: 10.1016/j.antiviral.2015.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Artusi S, Perrone R, Lago S, Raffa P, Di Iorio E, Palu G, Richter SN. 2016. Visualization of DNA G-quadruplexes in herpes simplex virus 1-infected cells. Nucleic Acids Res 44:10343–10353. doi: 10.1093/nar/gkw968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Norseen J, Johnson FB, Lieberman PM. 2009. Role for G-quadruplex RNA binding by Epstein-Barr virus nuclear antigen 1 in DNA replication and metaphase chromosome attachment. J Virol 83:10336–10346. doi: 10.1128/JVI.00747-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murat P, Zhong J, Lekieffre L, Cowieson NP, Clancy JL, Preiss T, Balasubramanian S, Khanna R, Tellam J. 2014. G-quadruplexes regulate Epstein-Barr virus–encoded nuclear antigen 1 mRNA translation. Nat Chem Biol 10:358–364. doi: 10.1038/nchembio.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Madireddy A, Purushothaman P, Loosbroock CP, Robertson ES, Schildkraut CL, Verma SC. 2016. G-quadruplex-interacting compounds alter latent DNA replication and episomal persistence of KSHV. Nucleic Acids Res 44:3675–3694. doi: 10.1093/nar/gkw038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brázda V, Hároníková L, Liao JCC, Fojta M. 2014. DNA and RNA quadruplex-binding proteins. Int J Mol Sci 15:17493–17517. doi: 10.3390/ijms151017493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lane AN, Chaires JB, Gray RD, Trent JO. 2008. Stability and kinetics of G-quadruplex structures. Nucleic Acids Res 36:5482–5515. doi: 10.1093/nar/gkn517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen MC, Murat P, Abecassis K, Ferre-D’Amare AR, Balasubramanian S. 2015. Insights into the mechanism of a G-quadruplex-unwinding DEAH-box helicase. Nucleic Acids Res 43:2223–2231. doi: 10.1093/nar/gkv051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chakraborty P, Grosse F. 2011. Human DHX9 helicase preferentially unwinds RNA-containing displacement loops (R-loops) and G-quadruplexes. DNA Repair (Amst) 10:654–665. doi: 10.1016/j.dnarep.2011.04.013. [DOI] [PubMed] [Google Scholar]
- 36.Ribeiro de Almeida C, Dhir S, Dhir A, Moghaddam AE, Sattentau Q, Meinhart A, Proudfoot NJ. 2018. RNA helicase DDX1 converts RNA G-quadruplex structures into R-loops to promote IgH class switch recombination. Mol Cell 70:650–662.e658. doi: 10.1016/j.molcel.2018.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McRae EKS, Booy EP, Moya-Torres A, Ezzati P, Stetefeld J, McKenna SA. 2017. Human DDX21 binds and unwinds RNA guanine quadruplexes. Nucleic Acids Res 45:6656–6668. doi: 10.1093/nar/gkx380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Geuens T, Bouhy D, Timmerman V. 2016. The hnRNP family: insights into their role in health and disease. Hum Genet 135:851–867. doi: 10.1007/s00439-016-1683-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cammas A, Lacroix-Triki M, Pierredon S, Le Bras M, Iacovoni JS, Teulade-Fichou M-P, Favre G, Roché H, Filleron T, Millevoi S, Vagner S. 2016. hnRNP A1-mediated translational regulation of the G quadruplex-containing RON receptor tyrosine kinase mRNA linked to tumor progression. Oncotarget 7:16793–16805. doi: 10.18632/oncotarget.7589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Decorsiere A, Cayrel A, Vagner S, Millevoi S. 2011. Essential role for the interaction between hnRNP H/F and a G quadruplex in maintaining p53 pre-mRNA 3′-end processing and function during DNA damage. Genes Dev 25:220–225. doi: 10.1101/gad.607011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khateb S, Weisman-Shomer P, Hershco I, Loeb LA, Fry M. 2004. Destabilization of tetraplex structures of the fragile X repeat sequence (CGG)(n) is mediated by homolog-conserved domains in three members of the hnRNP family. Nucleic Acids Res 32:4145–4154. doi: 10.1093/nar/gkh745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kikin O, D’Antonio L, Bagga PS. 2006. QGRS Mapper: a web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res 34:W676–W682. doi: 10.1093/nar/gkl253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paramasivan S, Rujan I, Bolton PH. 2007. Circular dichroism of quadruplex DNAs: applications to structure, cation effects and ligand binding. Methods 43:324–331. doi: 10.1016/j.ymeth.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 44.Xodo L, Paramasivam M, Membrino A, Cogoi S. 2008. Protein hnRNPA1 binds to a critical G-rich element of KRAS and unwinds G-quadruplex structures: implications in transcription. Nucleic Acids Symp Ser (Oxf) 2008(52):159–160. doi: 10.1093/nass/nrn081. [DOI] [PubMed] [Google Scholar]
- 45.Kruger AC, Raarup MK, Nielsen MM, Kristensen M, Besenbacher F, Kjems J, Birkedal V. 2010. Interaction of hnRNP A1 with telomere DNA G-quadruplex structures studied at the single molecule level. Eur Biophys J 39:1343–1350. doi: 10.1007/s00249-010-0587-x. [DOI] [PubMed] [Google Scholar]
- 46.Purushothaman P, Dabral P, Gupta N, Sarkar R, Verma SC. 2016. KSHV genome replication and maintenance. Front Microbiol 7:54. doi: 10.3389/fmicb.2016.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.De Leo A, Deng Z, Vladimirova O, Chen H-S, Dheekollu J, Calderon A, Myers KA, Hayden J, Keeney F, Kaufer BB, Yuan Y, Robertson E, Lieberman PM. 2019. LANA oligomeric architecture is essential for KSHV nuclear body formation and viral genome maintenance during latency. PLoS Pathog 15:e1007489. doi: 10.1371/journal.ppat.1007489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Renne R, Barry C, Dittmer D, Compitello N, Brown PO, Ganem D. 2001. Modulation of cellular and viral gene expression by the latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus. J Virol 75:458–468. doi: 10.1128/JVI.75.1.458-468.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jeong JH, Orvis J, Kim JW, McMurtrey CP, Renne R, Dittmer DP. 2004. Regulation and autoregulation of the promoter for the latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus. J Biol Chem 279:16822–16831. doi: 10.1074/jbc.M312801200. [DOI] [PubMed] [Google Scholar]
- 50.Kwun HJ, da Silva SR, Shah IM, Blake N, Moore PS, Chang Y. 2007. Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen 1 mimics Epstein-Barr virus EBNA1 immune evasion through central repeat domain effects on protein processing. J Virol 81:8225–8235. doi: 10.1128/JVI.00411-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zaldumbide A, Ossevoort M, Wiertz EJ, Hoeben RC. 2007. In cis inhibition of antigen processing by the latency-associated nuclear antigen I of Kaposi sarcoma herpes virus. Mol Immunol 44:1352–1360. doi: 10.1016/j.molimm.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 52.Ruggiero E, Richter SN. 2018. G-quadruplexes and G-quadruplex ligands: targets and tools in antiviral therapy. Nucleic Acids Res 46:3270–3283. doi: 10.1093/nar/gky187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ravichandran S, Kim Y-E, Bansal V, Ghosh A, Hur J, Subramani VK, Pradhan S, Lee MK, Kim KK, Ahn J-H. 2018. Genome-wide analysis of regulatory G-quadruplexes affecting gene expression in human cytomegalovirus. PLoS Pathog 14:e1007334. doi: 10.1371/journal.ppat.1007334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sorel O, Chen T, Myster F, Javaux J, Vanderplasschen A, Dewals BG. 2017. Macavirus latency-associated protein evades immune detection through regulation of protein synthesis in cis depending upon its glycin/glutamate-rich domain. PLoS Pathog 13:e1006691. doi: 10.1371/journal.ppat.1006691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thakker S, Strahan RC, Scurry AN, Uppal T, Verma SC. 2018. KSHV LANA upregulates the expression of epidermal growth factor like domain 7 to promote angiogenesis. Oncotarget 9:1210–1228. doi: 10.18632/oncotarget.23456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Verma SC, Cai Q, Kreider E, Lu J, Robertson ES. 2013. Comprehensive analysis of LANA interacting proteins essential for viral genome tethering and persistence. PLoS One 8:e74662. doi: 10.1371/journal.pone.0074662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Purushothaman P, McDowell ME, McGuinness J, Salas R, Rumjahn SM, Verma SC. 2012. Kaposi’s sarcoma-associated herpesvirus-encoded LANA recruits topoisomerase IIbeta for latent DNA replication of the terminal repeats. J Virol 86:9983–9994. doi: 10.1128/JVI.00839-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rossetto CC, Pari G. 2012. KSHV PAN RNA associates with demethylases UTX and JMJD3 to activate lytic replication through a physical interaction with the virus genome. PLoS Pathog 8:e1002680. doi: 10.1371/journal.ppat.1002680. [DOI] [PMC free article] [PubMed] [Google Scholar]