

Abstract
Deficiency of the adaptor protein complex 4 (AP-4) leads to childhood-onset hereditary spastic paraplegia (AP-4-HSP): SPG47 (AP4B1), SPG50 (AP4M1), SPG51 (AP4E1) and SPG52 (AP4S1). This study aims to evaluate the impact of loss-of-function variants in AP-4 subunits on intracellular protein trafficking using patient-derived cells. We investigated 15 patient-derived fibroblast lines and generated six lines of induced pluripotent stem cell (iPSC)-derived neurons covering a wide range of AP-4 variants. All patient-derived fibroblasts showed reduced levels of the AP4E1 subunit, a surrogate for levels of the AP-4 complex. The autophagy protein ATG9A accumulated in the trans-Golgi network and was depleted from peripheral compartments. Western blot analysis demonstrated a 3–5-fold increase in ATG9A expression in patient lines. ATG9A was redistributed upon re-expression of AP4B1 arguing that mistrafficking of ATG9A is AP-4-dependent. Examining the downstream effects of ATG9A mislocalization, we found that autophagic flux was intact in patient-derived fibroblasts both under nutrient-rich conditions and when autophagy is stimulated. Mitochondrial metabolism and intracellular iron content remained unchanged. In iPSC-derived cortical neurons from patients with AP4B1-associated SPG47, AP-4 subunit levels were reduced while ATG9A accumulated in the trans-Golgi network. Levels of the autophagy marker LC3-II were reduced, suggesting a neuron-specific alteration in autophagosome turnover. Neurite outgrowth and branching were reduced in AP-4-HSP neurons pointing to a role of AP-4-mediated protein trafficking in neuronal development. Collectively, our results establish ATG9A mislocalization as a key marker of AP-4 deficiency in patient-derived cells, including the first human neuron model of AP-4-HSP, which will aid diagnostic and therapeutic studies.
Introduction
The hereditary spastic paraplegias (HSP) are a group of more than 80 neurodegenerative diseases with diverse molecular defects (1). Bi-allelic variants in genes that encode subunits of the adaptor protein complex 4 (AP-4) lead to prototypical yet poorly understood forms of complex HSP in children, called AP-4-associated HSP (AP-4-HSP) (2–5). This includes four different conditions: SPG47 (AP4B1, OMIM #614066); SPG50 (AP4M1, OMIM #612936), SPG51 (AP4E1, OMIM #613744) and SPG52 (AP4S1, OMIM #614067). The molecular mechanism in all four diseases is a loss-of-function of the AP-4 complex; hence, they share the same clinical phenotype (2). AP-4 belongs to a family of adaptor proteins (AP-1 through AP-5), which are evolutionarily conserved heterotetrameric protein complexes. The adaptor protein complexes function by selectively incorporating transmembrane cargo proteins into vesicles and by recruiting the machinery necessary for vesicle budding and transport (6). AP-4 is composed of four subunits (β4, ε, μ4 and σ4) forming an obligate complex (7,8) and has been implicated in trafficking of transmembrane proteins from the trans-Golgi network (TGN) to early (9,10) and late endosomes (11). Recent studies in cultured cells have demonstrated that the autophagy protein ATG9A is a cargo of AP-4 and that loss of AP-4 leads to a mislocalization of ATG9A, potentially impacting the transport and function of ATG9A in axons (12–14). The recent characterization of an Ap4e1 knockout mouse has revealed widespread axonal pathology that includes reduced axonal development and prominent axonal swellings (14). ATG9A is important for autophagic vesicle formation (15,16) and is essential for axonal function as demonstrated in CNS-specific Atg9a knockout mice (Atg9aF/F: Nes-Cre, CKO) (17). Insights into the molecular mechanisms of AP-4-HSP and the pathogenic variants in the AP-4 subunit genes promise to not only lead to opportunities for targeted treatment, but broaden our understanding of AP-4 related protein trafficking, ATG9A biology and autophagy in neurons. To this end, the consequences of a loss of function of AP-4 remain to be assessed systematically in patient-derived cells.
Here we report a detailed investigation of cellular phenotypes in 15 patient-derived fibroblast lines covering a wide range of frameshift, non-sense and missense variants in all four subunits of the AP-4 complex. We also detail the generation of the first induced pluripotent stem cell (iPSC)-derived neurons from patients with AP-4-HSP. We establish mislocalization and accumulation of the AP-4 cargo protein, ATG9A, as a marker of AP-4 deficiency in patient-derived cells, including cortical neurons, and define the impact on autophagy, mitochondrial function, iron homeostasis, neurite outgrowth and branching. Collectively, our results define AP-4-HSP as a paradigm of childhood-onset HSP caused by defective protein trafficking.
Results
Trafficking of the AP-4 cargo protein ATG9A is impaired in AP-4-deficient patient-derived fibroblasts
Using unbiased proteomic approaches, ATG9A was recently identified as the major cargo of AP-4 (12,13). In AP-4-depleted HeLa cells, SH-SY5Y cells, fibroblasts (12,13) and neurons of Ap4e1 knockout mice (14), ATG9A was found to be mostly retained in the TGN. This provides strong evidence that AP-4 is required for the export of ATG9A from the TGN to peripheral compartments. To assess whether this cellular phenotype was uniformly present in patient-derived cells, we generated fibroblast lines from 15 well-characterized patients carrying homozygous or compound-heterozygous variants in AP4B1 (7 lines), AP4M1 (3 lines), AP4E1 (1 line) and AP4S1 (4 lines) (Table 1 and Supplementary Material, Table S1). Most variants were frameshifting, truncating variants but a few patients also carried missense changes and, although heterogeneous in terms of age and stage of disease progression, all patients showed core clinical and radiographic features of AP-4-HSP (2,18). We found that fibroblasts from three patients with compound-heterozygous variants in AP4B1 [Lof/LoF] showed reduced levels of AP4E1 (Fig. 1A and B), a surrogate for levels of the AP-4 complex. This supports the notion that the stability of all AP-4 subunits depends on the presence of each subunit and the formation of a stable complex. Confirming the absence of AP-4 assembly, we found no co-immunoprecipitation of AP4B1 and AP4E1 in lysates from patient fibroblasts but did detect such binding in lysates from heterozygous controls (Fig. 1C). We found that whole cell levels of ATG9A were robustly increased in all patient fibroblasts (Fig. 1A and D). Relative mRNA levels of ATG9A, however, were unchanged, suggesting that a post-translational mechanism accounts for the increase in ATG9A protein levels (Supplementary Material, Fig. S1A). To characterize the subcellular localization of ATG9A in the context of AP-4 deficiency, we examined endogenous ATG9A and its colocalization with the trans-Golgi marker TGN46. In all three patient-derived fibroblasts, ATG9A was retained in a juxtanuclear region, colocalizing with TGN46, and depleted from the cell periphery (Fig. 1E). In contrast, fibroblasts with heterozygous variants in AP4 subunits [WT/LoF] displayed ATG9A staining in the TGN and the cell periphery (Fig. 1E), consistent with the known distribution of ATG9A in various cell lines and similar to the pattern in fibroblasts from healthy donors [WT/WT] (Fig. 2B). We performed automated quantification of the area of ATG9A signal overlapping with the TGN in relation to the total area covered by ATG9A staining (a localization index). This revealed a significant increase of ATG9A overlapping with the TGN in patient fibroblast lines compared to controls (Fig. 1E). Extending these findings to AP-4-HSP patients with loss-of-function variants in the other AP-4 subunits, we discovered that ATG9A robustly increased in the area of the TGN in all patient fibroblast lines with some variation but no exception (Fig. 2A–C). Increased whole-cell levels of ATG9A were also found in all patient-derived lines to between 3-5x the level of heterozygous controls and of wild-type controls (Fig. 2D). Again, levels of AP4E1, a surrogate for the formation and stability of the AP-4 complexes and thus the other three subunits, were robustly decreased (Fig. 2D). As expected, patient #66, who carries a homozygous variant in AP4E1, showed no residual levels of AP4E1, indicating that truncating loss-of-function variants lead to no detectable residual protein. To assess whether ATG9 mislocalization and accumulation in the TGN was indeed a result of loss of AP-4, we used a lentiviral vector to express AP4B1 in fibroblasts with bi-allelic loss-of-function variants in AP4B1 (Fig. 3A–E). We found that restoring expression of AP4B1 led to a decrease in ATG9A (Fig. 3C and D) and a redistribution to areas outside the TGN to levels similar to controls (Fig. 3E).
Table 1.
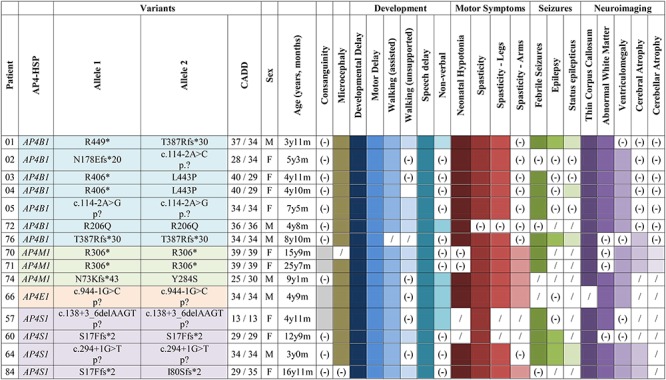
Summary of AP-4-HSP patients included in this study.
|
|
Reference sequences: AP4B1: NM_001253852.1; AP4M1: NM_004722.3; AP4E1: NM_007347.4; AP4S1: NM_007077.4. (−), not present; /, data not available.
Figure 1.
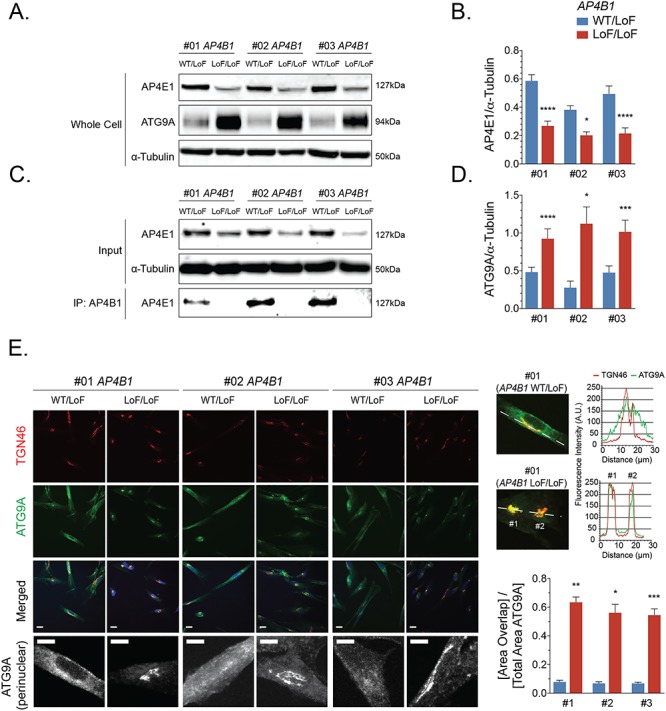
The AP-4 cargo protein ATG9A accumulates in the trans-Golgi network of fibroblasts from patients with AP4B1-associated HSP. (A, B) Western blotting of whole cell lysates of fibroblasts from three patients with bi-allelic variants in AP4B1 reveals a reduction in the levels of AP4E1, a subunit and surrogate for levels of the AP-4 complex. (C) Co-immunoprecipitation of AP4B1 and AP4E1 demonstrates AP-4 complex formation in lysates from heterozygous controls but not in fibroblasts from patients with bi-allelic variants in AP4B1. (D) Whole cell levels of ATG9A by western blotting are significantly increased in patient-derived fibroblasts. (E) Immunocytochemistry for ATG9A (green), trans-Golgi marker TGN46 (red) and nuclear marker DAPI (blue) demonstrates an accumulation of ATG9A in the perinuclear trans-Golgi network in AP-4-deficient patient fibroblasts. Line blots confirm that the ATG9A signal in patient-derived fibroblasts largely overlaps with TGN46 while the signal outside the trans-Golgi network is diminished. Quantification of the area of ATG9A staining overlapping with TGN46 as a ratio to the total area of ATG9A staining confirms the accumulation of ATG9A in the trans-Golgi network of AP-4-deficient fibroblasts. Scale bar: 10 μm (merged); 5 μm (ATG9A perinuclear). A.U., arbitrary units; LoF, loss of function; WT, wild type.
Figure 2.
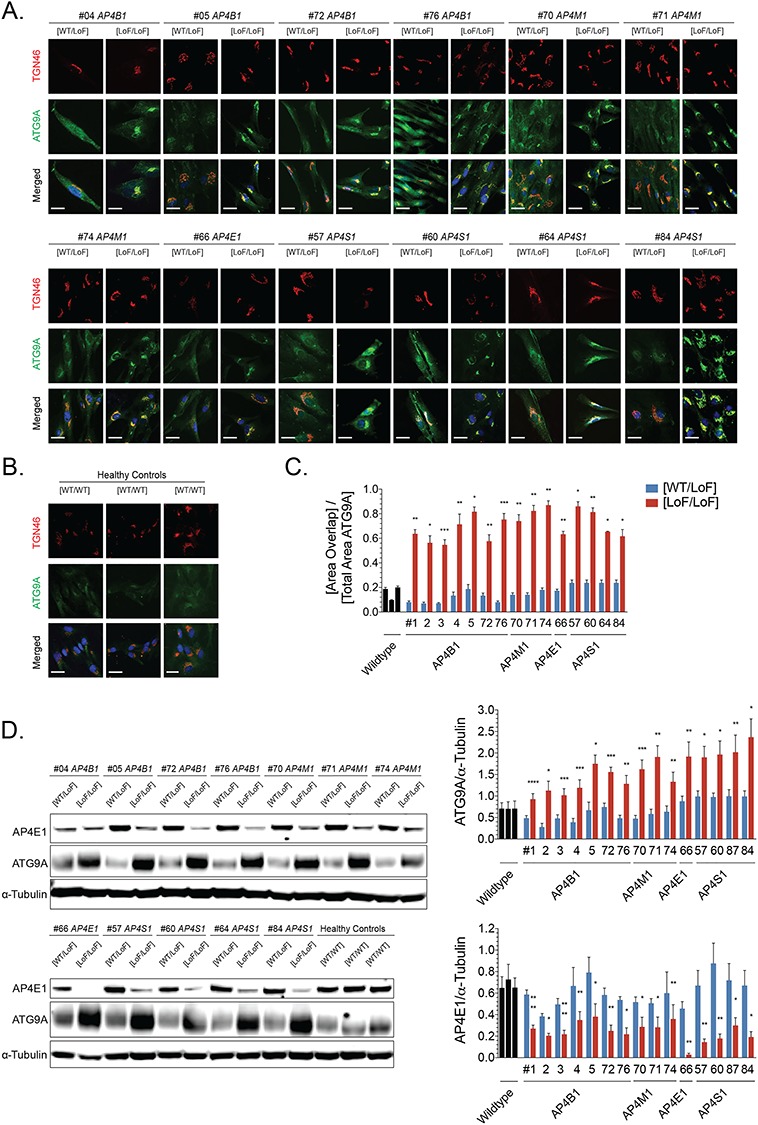
Trafficking of ATG9A is impaired in AP-4-deficient fibroblasts from patients with bi-allelic variants in different AP-4 subunits. (A) Immunocytochemistry for ATG9A (green), trans-Golgi marker TGN46 (red) and nuclear marker DAPI (blue) demonstrates an accumulation of ATG9A in the perinuclear trans-Golgi network in fibroblasts from patients with bi-allelic variants in AP4B1, AP4M1, AP4E1 or AP4S1. (B) In fibroblasts from healthy controls, ATG9A shows an even cytoplasmic distribution. (C) Quantification of the area of ATG9A staining overlapping with TGN46 as a ratio to the total area of ATG9A staining confirms the accumulation of ATG9A in the trans-Golgi network of AP-4-deficient fibroblasts. (D) Western blotting of whole cell lysates from patient fibroblasts with a variety of bi-allelic variants in different AP-4 subunits shows a significant increase in levels of ATG9A and a reduction in levels of AP4E1. The latter serves as a surrogate for levels of the AP-4 complex. Scale bar: 10 μm. LoF, loss of function; WT, wild type.
Figure 3.
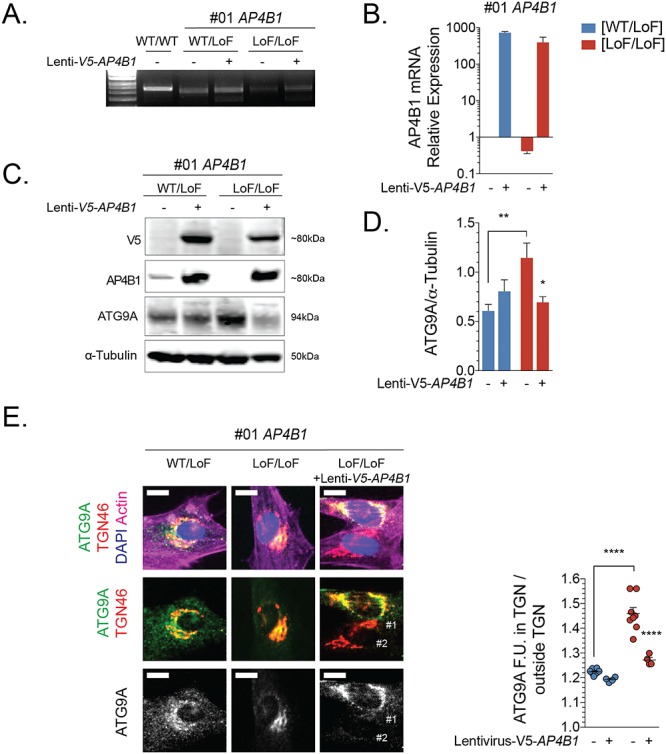
Re-expression of AP4B1 in fibroblasts from patients with AP4B1-associated HSP leads to a reduction of total ATG9A levels and redistribution from the trans-Golgi network to the cell periphery. (A, B) mRNA expression levels of AP4B1 in wild-type fibroblasts and fibroblasts with heterozygous or bi-allelic loss-of-function variants in AP4B1 with and without treatment with lentivirus to express human AP4B1. (C, D) Re-expression of AP4B1 in AP4B1-deficient patient-derived fibroblasts lowers ATG9A protein levels to levels that are not different from controls. (E) Re-expression of AP4B1 in AP4B1-deficient patient-derived fibroblasts leads to re-distribution of ATG9A from the trans-Golgi network to the cell periphery. Quantification of the area of ATG9A staining overlapping with TGN46 as a ratio to the total area of ATG9A staining demonstrates that the pattern after re-expression of AP4B1 is not significantly different from the pattern in heterozygous controls. Scale bar: 5 μm. Lenti, lentivirus; LoF, loss of function; TGN, trans-Golgi network; WT, wild type.
Attempts to determine changes in the subcellular localization of two other known cargo proteins of AP-4, SERINC1 and SERINC3 (Serine incorporator 1 and 3) (13) were limited, since there are no commercial antibodies that allow a reliable detection of endogenous levels of these low-abundance proteins. APP (amyloid precursor protein) has also been proposed as a cargo of AP-4 in cultured cells (9); however, we did not detect a difference in the distribution of APP in fibroblasts with bi-allelic loss-of-function variants in AP4B1 (Supplementary Material, Fig. S1B).
We conclude that bi-allelic loss-of-function variants in any of the AP-4 subunit genes lead to loss of AP-4 function and subsequently to accumulation of ATG9A in the TGN area and depletion from peripheral compartments. The robust nature of this cellular phenotype makes it suitable as a supporting diagnostic marker of AP-4-HSP in patient-derived fibroblasts.
Autophagic flux is intact in AP-4-deficient patient-derived fibroblasts
Because of the known role of ATG9A in the early steps of the autophagy (19,20), we investigated AP-4-HSP patient-derived fibroblast lines for any changes in autophagic vesicle formation. In a nutrient-rich environment, levels of the autophagosome marker LC3-II (as a ratio to LC3-I) and the autophagy substrate p62 were unchanged (Fig. 4A). To assess autophagic flux, we employed a commonly used paradigm of starvation-mediated autophagy induction and blockage of autophagosome–lysosome fusion with bafilomycin A1. We found that the combination of autophagy induction and blockage of the late stages of the pathway led to a significant increase in the LC3II/I ratio in both patient-derived fibroblasts with bi-allelic variants in AP4B1 and heterozygous controls (Fig. 4B and C). Starvation-mediated induction of autophagy and/or blocking autophagy with bafilomycin did not significantly alter levels of AP4E1 or ATG9A (Fig. 4D and E). This indicates that increased levels of ATG9A in patient-derived fibroblasts are not the result of decreased turnover of ATG9A through autophagy.
Figure 4.
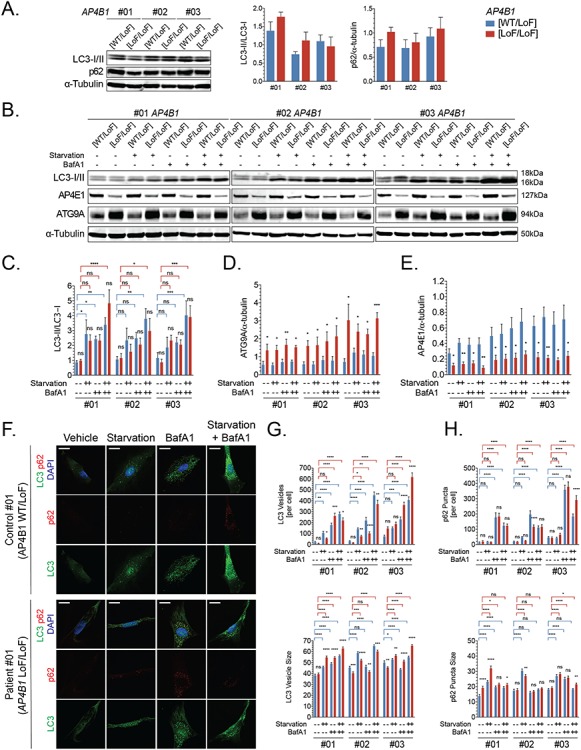
Autophagic flux is intact in AP-4-deficient patient-derived fibroblasts. (A) Whole cell levels of autophagosome marker LC-3 (as a ratio of LC3-II/LC3-I) and autophagy substrate p62 are similar in fibroblasts with bi-allelic loss-of-function variants in AP4B1 and heterozygous controls under nutrient-rich conditions. (B) When challenged in a paradigm of autophagy induction through starvation and autophagy blockade with bafilomycin A1, AP-4-deficient patient fibroblast shows (C) an increase in LC3II/I indicative of preserved autophagic flux. (D, E) Levels of ATG9A and AP4E1 remain unchanged by autophagy induction or inhibition. (F-H) Immunocytochemistry for LC3-positive autophagosomes and p62 demonstrates a significant increase in vesicle/punctae number and size following autophagy induction and blockade, again arguing that autophagic flux is maintained in AP-4-deficient fibroblasts. Scale bar: 20 μm. BafA1, bafilomycin A1; LoF, loss of function; WT, wild type.
To ensure that the lack of difference in autophagic flux was not simply due to a restoration of autophagy by other mechanisms after prolonged induction or blockage, we tested several time points including shorter incubation (1 and 2 h) as a well as longer incubation (8 h) (Supplementary Material, Fig. S2). We found that AP-4-deficient fibroblasts are capable of mounting an autophagy induction in response to starvation or mTOR-inhibition with a trajectory that is similar to that of control cell lines. Levels of LC3-II as a ratio to LC3-I were significantly increased (Supplementary Material, Fig. S2A and B). ATG9A levels again did not change with autophagy induction or blockage and remained significantly elevated in AP-4-deficient fibroblasts (Supplementary Material, Fig. S2A and C). The localization of ATG9A, as assessed by immunofluorescence, also remained unchanged in the setting of autophagy induction, blockage or both, though the fluorescence signal in the TGN intensified under all conditions in AP-4-deficient fibroblasts (Supplementary Material, Fig. S2E). Levels of phosphorylated S6 as a marker of mTORC1 activity did not differ between patient-derived fibroblasts and controls and showed the expected reduction in response to starvation or pharmacological mTORC1 blockade (Supplementary Material, Fig. S2A and D).
To detect more subtle changes in autophagosome formation, we employed quantitative immunocytochemistry to assess the number and size of LC3-positive autophagic vesicles (Fig. 4F). We found that at baseline, the number of LC3 vesicles per cell was not significantly different in AP-4-deficient fibroblasts. Autophagosomes were, however, smaller in two out of the three lines tested (Fig. 4G). Autophagic flux, defined as the formation of new vesicles over time, and an increase in autophagosome size were evident when a combined induction and downstream block of the pathway was applied (Fig. 4G). In addition, p62 aggregates increase in number and size in AP-4-deficient cells and controls with a block in autophagy flux through treatment with bafilomycin A1 (Fig. 4H).
From this set of experiments, we conclude that AP-4-HSP patient-derived fibroblasts show intact autophagic flux at baseline and when challenged, with no detectable difference in the turnover of autophagosomes.
Mitochondrial function and iron content are preserved in AP-4-deficient patient-derived fibroblasts
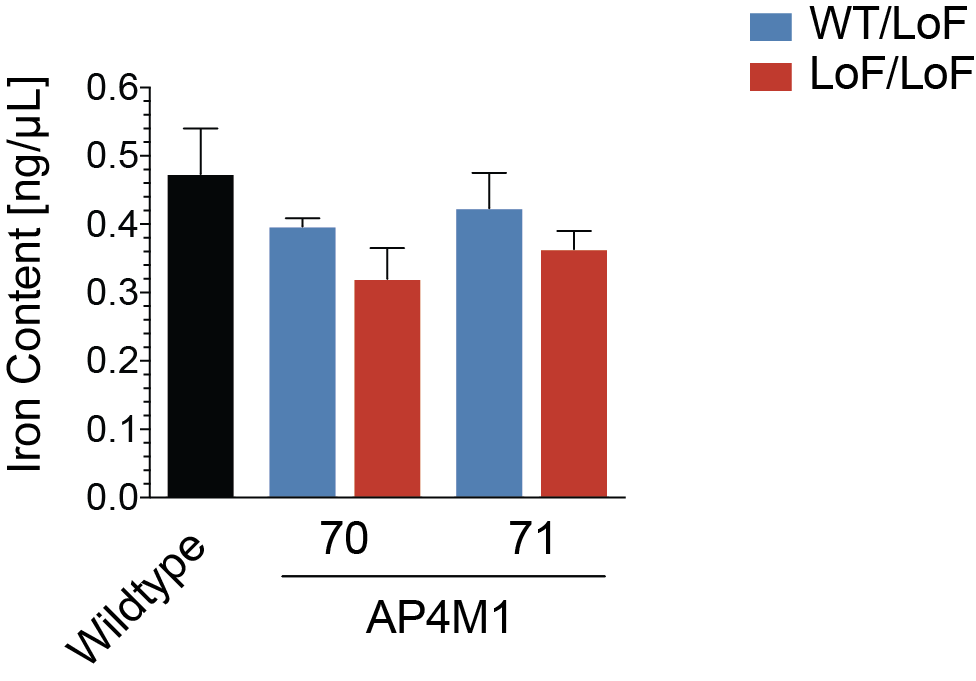
Mitochondrial dysfunction as a result of altered autophagy is a common finding in congenital disorders of autophagy (21). To explore any changes in mitochondrial homeostasis, we measured the mitochondrial membrane potential using TMRE and JC-1, two fluorescent indicators. We found that the membrane potential was not different between fibroblasts from patients with homozygous loss-of-function variants and heterozygous controls (Supplementary Material, Fig. S3A and B). Similarly, the mitochondrial mass was not significantly different (Supplementary Material, Fig. S3A and B) and although not assessed quantitatively, the shape and distribution of mitochondria seemed unaltered. In four AP-4-HSP patients from two families (22,23), iron accumulation in the basal ganglia has been reported, raising the question of altered iron metabolism in AP-4-HSP. To explore iron homeostasis, we derived fibroblasts from two patients with AP4M1-associated HSP and known brain iron accumulation (patients #70 and #71). No difference in cellular iron concentration was appreciated between patient-derived fibroblasts and controls (Supplementary Material, Fig. S4).
ATG9A is mislocalized in human iPSC-derived neurons from AP-4-HSP patients
The promise of using human iPSC-derived neuron disease models for identification of disease-relevant cellular phenotypes is starting to bear fruit across a variety of neurological diseases (24). iPSCs from three patients with AP4B1-associated SPG47 and same-sex parents (heterozygous carriers) were generated and underwent differentiation to excitatory cortical neurons using overexpression of neurogenin-2 (25) (Fig. 5A). To explore the ATG9A retention phenotype in this first human neuron model of AP-4-HSP, we plated neurons in a 96-well format and subjected them to automated high-content confocal microscopy (Fig. 5A). This allowed us to investigate > 30 000 neurons per round of differentiation. Patient and control lines from each family were differentiated in parallel, and three independent differentiations per line were studied. In patient-derived neurons, ATG9A was mainly concentrated to a high-intensity juxtanuclear area that overlapped with GM130, a Golgi marker, with lower fluorescence intensity in the remainder of the soma (Fig. 5A and B). In neurons from controls, ATG9A was distributed more evenly in the soma, and the juxtanuclear high-intensity area, if present, was often smaller (Fig. 5B). A significant increase in the juxtanuclear ATG9A area occurred in patient-derived neurons (Fig. 5B–D). This phenotype was robustly present in independent differentiations of all three lines (Fig. 5C and D). This indicates that in human neurons derived from patients with AP4B1-associated SPG47, similar to our findings in fibroblasts, ATG9A accumulates in the TGN and is depleted from other compartments. Western blotting confirmed a decrease in AP4E1 in patient-derived neurons while ATG9A showed a robust increase to about 2–3-fold the levels of heterozygous controls (Fig. 5E–G). Whole cell levels of autophagy markers at baseline, in a nutrient enriched environment, showed a reduced LC3II/I ratio in patient-derived neurons (Fig. 5E and H) but no change in p62 levels (Fig. 5E and I). This contrasts our findings in fibroblasts where we observed no change in LC3 levels. Thus, the reduction in levels of lipidated LC3 may point to subtle and neuron-specific changes in autophagosome formation.
Figure 5.
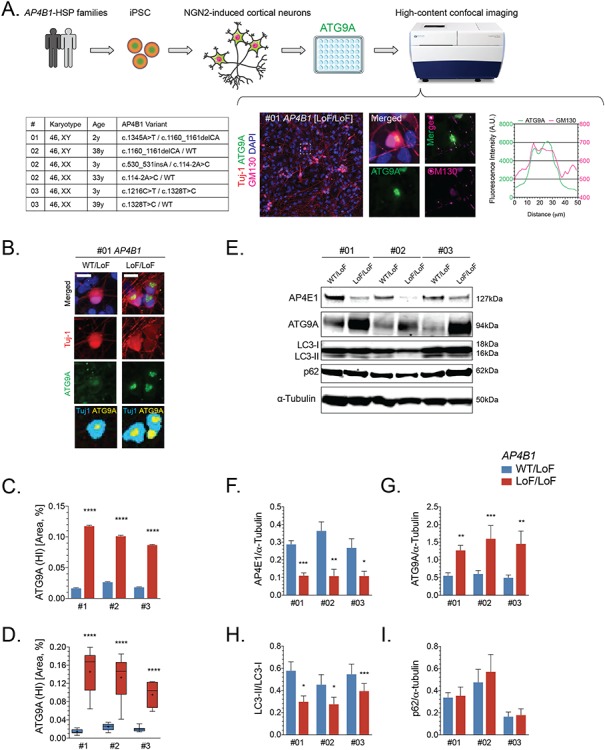
ATG9A is mislocalized in iPSC-derived cortical neurons from AP-4-HSP patients. (A) Fibroblasts from three families with AP4B1-associated HSP were reprogrammed into iPSCs and subsequently into excitatory cortical neurons using overexpression of neurogenin 2. iPSC-derived neurons were grown in 96 well plates and subjected to high-content confocal imaging to assess the localization of ATG9A. Immunocytochemistry was performed for the neuronal marker Tuj-1 (red), ATG9A (green), Golgi marker GM130 (pink) and nuclear marker DAPI (blue). ATG9A concentrated to a high-intensity juxtanuclear area that overlapped with GM130 with lower fluorescence intensity in the remainder of the soma. (B) In neurons from heterozygous controls, ATG9A was distributed in the soma, and the juxtanuclear high-intensity area (marked in yellow), if present, was often small. A significant increase in the high intensity juxtanuclear ATG9A area occurred in patient-derived neurons. The ratio between soma size by Tuj-1 staining (marked in cyan) and the ATG9A high intensity area (marked yellow) was significantly increased in cortical neurons from AP-4-HSP patients compared to controls. This was quantified on (C) a per cell basis with over 90x103 neurons analyzed per condition or (D) per differentiation with three independent rounds of differentiation per pair of cell lines. (E–I) Western blotting of whole cell lysates of iPSC-derived neurons from three patients with bi-allelic variants in AP4B1 reveals a reduction in the levels of AP4E1, a subunit and surrogate for levels of the AP-4 complex, increased levels of ATG9A and a decrease in the ratio of LC3-II to LC3-I. Scale bar: 20 μm. HI, high intensity; iPSC, induced pluripotent stem cells; LoF, loss of function; NGN2, neurogenin 2; WT, wild type.
Collectively, our experiments establish mislocalization and increased levels of ATG9A as cellular phenotypes in the first human neuronal model of AP-4 deficiency.
Neurite outgrowth is impaired in iPSC-derived cortical neurons from AP-4-HSP patients
To explore neuron-specific phenotypes due to a loss of AP-4 function, we evaluated our mutant AP4B1 patient-derived cortical neurons during the initial steps of neurite initiation and outgrowth using live cell imaging (Supplementary Material, Videos S1 and S2). We found that at this early stage of neuronal development, AP-4-deficient neurons showed reduced neurite outgrowth with a shorter average neurite length and a reduced number of branches per cell (Fig. 6A and B; Supplementary Material, Videos S1 and S2). Despite the significant reduction compared to controls, neurites continued to grow at a steady rate throughout the first 24 h after plating (Fig. 6B). Of note, even at this early developmental stage, we found a robust accumulation of ATG9A in the juxtanuclear area that corresponds to the TGN (Fig. 6C).
Figure 6.
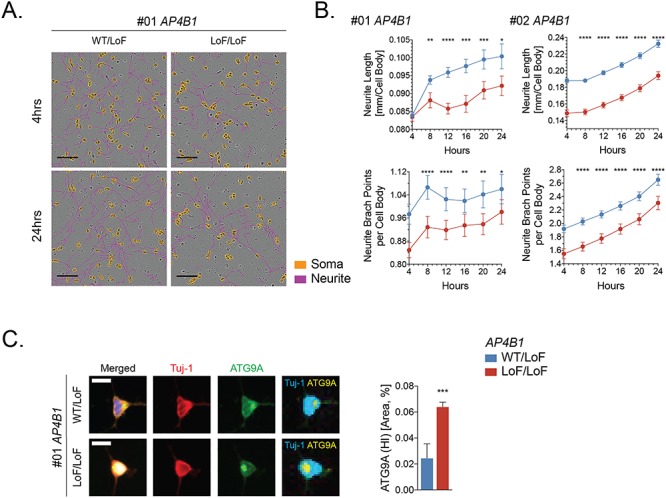
Neurite outgrowth is impaired in iPSC-derived cortical neurons from AP-4-HSP patients. (A, B) iPSC-derived cortical neurons from patients with AP4B1-associated HSP and their heterozygous same sex parent were analyzed using automated live cell imaging to assess neurite outgrowth and branching. Neurons were monitored from 4 h post plating until fixation at 24 h. Automated image analysis revealed reduced neurite outgrowth (neurites are pseudo-colored in violet) with a shorter average neurite length and a reduced number of branches per cell (cell bodies are pseudo-colored in orange) in AP4B1-deficient neurons. (C) At 24 h post plating, AP-4-deficient neurons show a robust increase in high-intensity juxtanuclear ATG9A signal (marked in yellow) compared to control, similar to more mature neurons at day 7 post plating (Fig. 5). Scale bar: 100 μm (A); 20 μm (C). HI, high intensity; LoF, loss of function; WT, wild type.
Taken together, these data show that loss of AP-4 in patient-derived human cortical neurons leads to neurite outgrowth impairment, which may represent the in vitro correlate of brain malformations, such as thin corpus callosum, seen in the majority of AP-4-HSP patients (Ebrahimi-Fakhari et al., submitted).
Discussion
Adaptor protein complexes are crucial for the selective transport of transmembrane proteins to different intracellular compartments. Efficient protein trafficking is particularly important to neurons given their complex architecture and long neurites. To better understand disorders that arise from genetic defects in the adaptor protein complexes, it is imperative to identify and characterize the missorting of their cargo proteins. To explore the effects of variants in the AP-4 subunit genes on AP-4 function and downstream cargo, we generated patient-derived fibroblast lines, iPSCs and iPSC-derived neurons. First, we confirmed that variants found in AP-4-HSP patients led to a reduction in AP4E1 levels indicating reduced stability of this subunit in the absence of a functional complex (6,26). To probe the functional consequences of AP-4 deficiency, we investigated the location and the levels of the AP-4 cargo ATG9A. ATG9A was recently identified as being missorted in AP-4-deficient HeLa and SH-SY5Y cells (12,13), as well as in neurons from Ap4e1 knockout mice (14,27) and fibroblasts (13,14). In keeping with these findings, we found that ATG9A accumulates in the TGN in AP-4-deficient fibroblasts and levels are significantly increased. This strong cellular phenotype was found to be AP-4-dependent and was present in all patient-derived fibroblast lines tested covering all four subunits and a great range of different variants including patients with missense changes. Thus, this assay appears to be a reliable and quantitative indicator of AP-4 assembly and function and may serve as an in vitro disease marker that can help establish a diagnosis in cases of variants of unclear significance. This has enabled us to support a diagnosis of AP-4-HSP in several patients in our cohort.
The cell periphery and other cellular compartments are depleted of ATG9A providing evidence for a model by which loss of AP-4 function leads to a loss of peripheral ATG9A with possible consequences to axonal homeostasis and autophagy. This loss-of-function model is supported by the fact that CNS-specific Atg9a knockout mice (Atg9aF/F: Nes-Cre, CKO) (17) resemble the phenotype of Ap4e1 knockout mice (14,27), with prominent dysgenesis of the corpus callosum and reduced neurite outgrowth.
ATG9A is the only known transmembrane protein required for autophagosome formation (16,28,29) and localizes to the endoplasmic reticulum, TGN, late endosomes/lysosomes and axons (20,28,30). ATG9A is sorted from one or more of these compartments to form cytoplasmic Atg9-containing membranes, which form vesicular structures (31,32). ATG9A is subject to phosphorylation by ULK1 (19) and phosphorylated ATG9A recruits ATG8 and ATG18 to the site of autophagosome formation to promote expansion of the isolation membrane (16). This process localizes to the distal axon in C. elegans neurons arguing that there is spatial regulation of ATG9-mediated autophagy (33). Clinically, there are parallels between AP-4-HSP and single gene disorders of the autophagy pathway known as congenital diseases of autophagy (34).
To investigate the impact of AP-4 variants on autophagy, we measured levels of autophagy markers in patient-derived fibroblasts under baseline conditions and in a paradigm of starvation- or mTORC1-inhibition-induced autophagy and pharmacological autophagy inhibition. To our surprise, we found that fibroblast lines derived from AP-4-HSP patients showed intact autophagic flux despite the changes in ATG9A distribution.
To search for more subtle changes in autophagy that might slowly occur over time, we investigated mitochondria since defective mitochondria are an important target of autophagy. We did not detect any changes in mitochondrial parameters in AP-4-deficient fibroblasts. Similarly, iron deposition in the basal ganglia has been reported in two families with AP-4-HSP (22,23) and cellular iron homeostasis is known to be at least in part subject to regulation by ATG9A (35) or autophagy (36). The latter concept is exemplified by one of the congenital disorders of autophagy, beta-propeller protein-associated neurodegeneration, where defective autophagy leads to a buildup of iron in the CNS (37) and in patient-derived cells (38). In fibroblasts from patients with AP-4-HSP and brain iron accumulation, we did not detect any changes in iron content, arguing against altered iron storage at least in peripheral cells. Taken together, we provide multiple lines of evidence that fibroblasts from AP-4-HSP patients do not show any significant impairment of autophagosome formation or autophagic turnover.
Human neurons differentiated from iPSCs have arisen as an important translational in vitro disease model that enables therapeutic drug discovery (24). iPSC-derived models of several HSPs have enabled the study of disease-relevant pathways in a cell type immediately relevant to patients (39–43). To shed light into the effects of AP-4 deficiency on human neurons, we generated iPSC lines from three families with AP4B1-associated SPG47 carrying different compound-heterozygous variants in AP4B1 and differentiated excitatory cortical neurons. We developed these neurons in a format that allows for high-throughput confocal imaging. We find that iPSC-derived cortical neurons from individuals with bi-allelic AP4B1 variants show reduced levels of AP4E1 demonstrating that the patient’s genetic variants impaired AP-4 complex formation and thus reduce AP-4 subunit levels. We next investigated these neurons for ATG9A localization and levels using automated high-content confocal imaging. We find that AP-4-HSP patient-derived neurons show a juxtanuclear accumulation of ATG9A in an area that overlaps with the Golgi marker GM130. At the same time, the peripheral soma shows a lower ATG9A signal arguing of a depletion of ATG9A in compartments outside of the Golgi apparatus. Whole cell lysates from patient iPSC neurons also showed a significant increase in ATG9A levels. These findings corroborate our findings in patient fibroblasts and establish ATG9A retention in the Golgi network as a strong cellular phenotype that is present in disease-relevant patient-derived neurons.
Contrasting our findings in patient-derived fibroblasts, we found significantly reduced baseline levels of the autophagosome marker LC3-II, as a ratio to unlipidated LC3-I, in neurons from patients with AP4B1-associated HSP. This could point to neuron-specific changes in autophagosome turnover. However, no changes in the autophagy cargo receptor and autophagy substrate p62 were observed. We speculate that the increased total ATG9A protein levels might present a compensatory mechanism that allows for some ATG9A to exit the Golgi network, thus preventing any severe impairment in autophagy that would impact substrate turnover or survival. Subtle changes in the delivery of ATG9A vesicle to axons and subsequently to autophagy initiation in the axonal compartment could, however, over time impact axonal homeostasis and neuronal function. This is corroborated by findings in cultured neurons from Ap4e1 knockout mice that show an ATG9A-dependent impairment in the ability to clear aggregated proteins from axons (14). Since autophagy in neurons is known to be regulated differently compared to peripheral cells (44), our human neuronal model of AP-4-HSP opens novel and unique avenues to investigate autophagy in the context of neuronal AP-4 deficiency in future studies.
Exploring neuronal phenotypes of AP-4-HSP, we find that neurite development and branching is impaired in iPSC-derived cortical neurons from patients with homozygous variants in AP4B1. This indicates that loss of AP-4 or subsequent ATG9A depletion from the periphery has a detrimental effect on neurite outgrowth. This is supported by circumstantial evidence in that neuron-specific Atg9 knockout mice show a strong axonopathy (17). The possibility that increased levels of ATG9A in the Golgi network are pathogenic should also be considered, as well as the possible involvement of other AP-4 cargo proteins. Our iPSC-derived neurons will serve as an important tool to explore these and other cellular phenotypes for phenotypic screens and will allow us to further delineate the molecular mechanisms at play.
In summary, we describe the first human neuronal model of AP-4-HSP using iPSC-derived cortical neurons from patients with AP4B1-associated SPG47. The use of a large cohort of patient-derived cells has allowed us to establish ATG9A mislocalization as a highly penetrant cellular phenotype and biomarker of AP-4 deficiency in fibroblasts and neurons. This will serve as a platform for future investigations into AP-4 biology in neurons and screens for therapeutic targets.
Materials and Methods
Clinical and genetic data
Clinical information provided in Table 1 was collected from individuals consented under protocols approved by Boston Children’s Hospital (Protocol #10-02-0053) or the IRCCS Stella Maris, Pisa, Italy (Protocol #134/16) (Table 1). Combined Annotation-Dependent Depletion (CADD) scores were calculated (45). Written informed consent for donation of fibroblasts was obtained. The cellular studies were approved by the Institutional Review Board at Boston Children’s Hospital (Protocol #P00016119).
Patient fibroblasts
A total of 15 fibroblast lines from patients with AP-4-HSP carrying bi-allelic variants in AP4B1 (7 lines), AP4M1 (3 lines), AP4E1 (1 line) and AP4S1 (4 lines) and a same-sex parent (heterozygous carriers, clinically unaffected) were analyzed (Table 1 and Supplementary Material, Table S1). For fibroblasts obtained from patients #57, #60, #70, #71, #76 and #84, a heterozygous carrier of a variant in the same AP-4 subunit was used for comparisons since no sex-matched parent sample was available. For fibroblasts obtained from patient #66, a cell line established from his grandmother was used since samples from a parent were unavailable (grandmother is obligate heterozygous carrier due to first degree consanguinity of the parents). For further comparisons, three fibroblast lines from healthy donors were included. Fibroblasts were passaged and maintained in culture, as previously described (46).
Generation of human iPSC lines and iPSC-derived cortical neurons
The protocol for experiments involving human iPSCs was approved by the Institutional Review Board at Boston Children’s Hospital (IRB#: P00016119). In brief, fibroblast lines of three patients with AP4B1-associated SPG47 and sex-matched parents (heterozygous carriers, clinically unaffected) were reprogramed using Sendai virus to introduce reprogramming factors (Oct4, Sox2, Klf4, and L-Myc) (Supplementary Material, Table S1). A total of six independent iPSC lines were established. Karyotypes were normal; immunocytochemistry and RT-PCR analysis showed high expression of pluripotency markers in all iPSC lines and STR profiling matched respective fibroblast lines (47). iPSCs were maintained in StemFlex medium on Geltrex (Thermo Fisher Scientific), and passaged weekly with Gentle Cell Dissociation Reagent (STEMCELL Technologies). Cortical neurons were differentiated according to protocols published by Zhang et al. (25). For neurite outgrowth experiments, neurons were plated at a density of 1x104 cells per well in 96 well plates (Greiner Bio-One) and maintained in human astrocyte conditioned media. For protein extraction, neurons were cultured in human astrocyte conditioned media (Astro.4 U, Ncardia) after day 6 and collected on day 14. For high-content imaging, neurons were plated at 2 × 104 cells per well in 96 well plates and co-cultured with human iPSC-derived astrocytes (Astro.4 U, Ncardia) at 3 × 103 per well. Plates were fixed on day 14 for subsequent immunocytochemistry.
Antibodies and reagents
Antibodies and reagents are listed in Supplementary Material, Table S2. Ngn2 lentivirus was a kind gift from T. Südhof, Stanford University, Palo Alto, CA. A lentiviral vector carrying human AP4B1 with a V5 tag under a PGK promoter was generated using standard protocols (48), and fibroblasts were transduced at an MOI of 10 for 24 h.
Polymerase chain reaction
PCR was performed according to the standard protocol (49) with primer list in Supplementary Material, Table S2.
Immunocytochemistry
Immunocytochemistry in fibroblast lines was done as described previously (13,46). In brief, fibroblasts were taken into culture, expanded and grown onto cover slips. Cells were fixed using 3% PFA (staining with anti-ATG9A, anti-TGN46, anti-APP antibodies) or ice-cold methanol (staining with anti-LC3 and anti-p62 antibodies). For immunocytochemistry, iPSC-derived cortical neurons cells were grown in 96-well plates as described above. Neurons were fixed using 4% PFA and stained with antibodies against ATG9A, GM130, and Tuj-1. DAPI was used as a nuclear marker. Standard secondary antibodies were used for detection.
Immunoprecipitation
For immunoprecipitation, fibroblasts were grown in 100 mm plates. Cell were washed and scraped in ice-cold PBS with Triton X supplemented with protease inhibitor (AEBSF, Thermo Scientific). Following incubation on ice for 10 min, lysates were cleared by centrifugation at 21 000g for 20 min at 4°C. A protein assay was performed and, if required, lysates were adjusted to equal protein concentrations with lysis buffer. A portion of each lysate was retained as input and the remainder was incubated with an antibody against AP4B1 and PA sepharose beads. Beads were washed five times with ice-cold PBS-T and PBS and then boiled in NuPAGE LDS Sample Buffer to prepare for western blot analysis.
Western blotting
Western blotting was done as previously described (49,50). In brief, total protein concentration was determined using the BCA Protein Assay Kit (Thermo Fisher Scientific). Equal amounts of protein were solubilized in LDS buffer under reducing conditions, separated by gel electrophoresis, using 4–12% Bis-Tris gels and MOPS or MES buffer (Thermo Fisher Scientific) and transferred to a PVDF membrane (EMD Millipore). Following blocking with blocking buffer (LI-COR Biosciences), membranes were incubated overnight with the respective primary antibody. Near-infrared fluorescent-labeled secondary antibodies (IR800CW, IR680LT; LI-COR Biosciences) were used and quantification was done using the Odyssey infrared imaging system and Image Studio Software (LI-COR Biosciences).
IncuCyte live imaging and neurite outgrowth
Automated, non-invasive imaging and measurement of neurite outgrowth in iPSC-derived neurons was performed using the IncuCyte® S3 Live-Cell Analysis System (Essen BioScience). Four hours after replating, 96-well plates were moved to the IncuCyte® incubator, and phase-contrast images were acquired at 20X magnification at a rate of every 4 h.
Confocal microscopy
Confocal microscopy was performed using a spinning disk confocal microscope (Perkin Elmer) with the Nikon Ti-Eclipse imaging system at 20X or 60X lens magnification and an automated stage function to cover a large area and high number of cells (2–3 × 103 per fibroblast line) (50). Images were analyzed with CellProfiler (51).
High-throughput confocal imaging
High-throughput confocal imaging was performed on the ImageXpress Confocal High Content Screening System (Molecular Devices). iPSC-derived cortical neurons were grown onto 96-well plates at a density of 20 × 103 per well co-cultured with 3 × 103 iPSC-derived human astrocytes. PBS-only wells around the outside of the plate were included to prevent any edge effects. Plates were fixed and stained on day 14. Images were acquired using a 20X objective. Per well, 25 fields were acquired in a 5x5 format covering ~ 1000–1500 neurons per well on average. Analysis was done using a customized image analysis pipeline in MetaXpress (Molecular Devices).
Autophagy assays
To assess autophagic flux, fibroblasts were subjected to a paradigm of autophagy induction through starvation or Torin 1 treatment and/or inhibition of autophagosome-lysosome fusion using bafilomycin A1 according to well-established protocols (50,52–54). For autophagy induction, fibroblasts were placed in HBSS or treated with Torin at a concentration of 250 nM. For blocking autophagy, fibroblasts were exposed to bafilomycin A1 at a concentration of 200 nM. For an assessment of autophagic flux, the two treatments were combined. All treatments were carried out on the same plate and at the same time to avoid variability. In accordance with guidelines on assays monitoring autophagy (52), two independent methods (western blotting and immunocytochemistry) were used to detect changes in autophagy markers.
Mitochondrial assays
To approximate the mitochondrial mass in cells, we labeled mitochondria with the mitochondrial membrane potential-independent dye MitoTracker® Deep Red as reported previously (50). To determine the mitochondrial membrane potential (ΔΨm), we employed the potentiometric, cell-permeable fluorescent probe TMRE (tetramethylrhodamine, ethyl ester) and the mitochondrial membrane potential indicator dye JC1 (tetraethylbenzimidazolylcarbocyanine iodide) (50).
Iron assay
Cellular iron content was measured in lysates from 10x106 fibroblasts using the Iron Assay Kit (#MAK025-1KT, Sigma-Aldrich) according to the manufacturer’s instructions.
Statistical analysis
Statistical analysis was performed with Prism version 6.0 (GraphPad Software, Inc.). Data are shown as mean ± standard error of the mean. For experiments shown in Fig. 5D, the distribution of data points is expressed in box blots covering the 5th to 95th percentile. Unpaired t-test (Figs 5C and D and 6; Supplementary Material, Figs S1–S4), paired ratio t-test (Figs 1–3, 4A and 5F–I) or one-way ANOVA with Fisher’s LSD test (Fig. 4C–H) was used to determine the significance of differences between conditions. P < 0.05 was considered significant. P-values are denoted as follows: P < 0.05 (*), P < 0.01 (**), P < 0.001 (***) and P < 0.0001 (****).
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
The authors thank the patients and families who participated in this study and the CureSPG47 advisory board members for feedback and support of this study. The authors are also grateful to A. Athanasiou Fragkouli (London, UK), G. Berry (Boston, MA), E. Carmody (Boston, MA), A. Diplock (Boston, MA), C. Genetti (Boston, MA), S. Kölker (Heidelberg, Germany), S. Lakhani (New York City, NY), T. Südhof (Palo Alto, CA), J. Volkmann (Würzburg, Germany) and A. Williams (Northampton, UK) for supply of reagents or resources, the Harvard Stem Cell Institute for help with generating induced pluripotent stem cells and the Gene Discovery Core of The Manton Center for Orphan Disease Research for help with consenting patients.
Funding
CureSPG47 Inc., the Spastic Paraplegia Foundation (SPF) Inc., the Thrasher Research Fund and the Lovejoy Award (all to D.E.-F.); the University of Würzburg Graduate School of Life Sciences Scholarship (to R.B.); the German National Academic Foundation (to J.T.); the University of Siena ‘Pegaso Scholarship’ (to a.d.); National Institute for Health Research (to A.K.D.); German Research Foundation (DFG/Gottfried Wilhelm Leibniz Prize MA 1764/2–1 to G.H.H.B.); Max Planck Society for the Advancement of Science; Wellcome Trust Principal Research Fellowship (086598 to M.R.). The Translational Neuroscience Center at Boston Children’s Hospital is supported by the Massachusetts Life Sciences Center (MLSC). The BCH Intellectual and Developmental Disabilities Research Center Clinical/Translational Core and Cellular Imaging Core are supported by the National Institutes of Health (BCH IDDRC, 1U54HD090255).
Conflict of Interest statement
There are no conflicts of interest related to this study.
References
- 1. Blackstone C. (2018) Hereditary spastic paraplegia. Handb. Clin. Neurol., 148, 633–652. [DOI] [PubMed] [Google Scholar]
- 2. Ebrahimi-Fakhari D., Behne R., Davies A.K. and Hirst J. (2018) AP-4-Associated Hereditary Spastic Paraplegia. In Adam M.P., Ardinger H.H., Pagon R.A., Wallace S.E., Bean L.J.H., Stephens K. and Amemiya A. (eds), GeneReviews((R)). Seattle (WA): University of Washington; in press. [PubMed] [Google Scholar]
- 3. Abou Jamra R., Philippe O., Raas-Rothschild A., Eck S.H., Graf E., Buchert R., Borck G., Ekici A., Brockschmidt F.F., Nothen M.M. et al. (2011) Adaptor protein complex 4 deficiency causes severe autosomal-recessive intellectual disability, progressive spastic paraplegia, shy character, and short stature. Am. J. Hum. Genet., 88, 788–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Moreno-De-Luca A., Helmers S.L., Mao H., Burns T.G., Melton A.M., Schmidt K.R., Fernhoff P.M., Ledbetter D.H. and Martin C.L. (2011) Adaptor protein complex-4 (AP-4) deficiency causes a novel autosomal recessive cerebral palsy syndrome with microcephaly and intellectual disability. J. Med. Genet., 48, 141–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Verkerk A.J., Schot R., Dumee B., Schellekens K., Swagemakers S., Bertoli-Avella A.M., Lequin M.H., Dudink J., Govaert P., van Zwol A.L. et al. (2009) Mutation in the AP4M1 gene provides a model for neuroaxonal injury in cerebral palsy. Am. J. Hum. Genet., 85, 40–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hirst J., Irving C. and Borner G.H. (2013) Adaptor protein complexes AP-4 and AP-5: new players in endosomal trafficking and progressive spastic paraplegia. Traffic, 14, 153–164. [DOI] [PubMed] [Google Scholar]
- 7. Hirst J., Bright N.A., Rous B. and Robinson M.S. (1999) Characterization of a fourth adaptor-related protein complex. Mol. Biol. Cell, 10, 2787–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dell'Angelica E.C., Mullins C. and Bonifacino J.S. (1999) AP-4, a novel protein complex related to clathrin adaptors. J. Biol. Chem., 274, 7278–7285. [DOI] [PubMed] [Google Scholar]
- 9. Burgos P.V., Mardones G.A., Rojas A.L., daSilva L.L., Prabhu Y., Hurley J.H. and Bonifacino J.S. (2010) Sorting of the Alzheimer’s disease amyloid precursor protein mediated by the AP-4 complex. Dev. Cell, 18, 425–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Toh W.H., Tan J.Z., Zulkefli K.L., Houghton F.J. and Gleeson P.A. (2017) Amyloid precursor protein traffics from the Golgi directly to early endosomes in an Arl5b- and AP4-dependent pathway. Traffic, 18, 159–175. [DOI] [PubMed] [Google Scholar]
- 11. Aguilar R.C., Boehm M., Gorshkova I., Crouch R.J., Tomita K., Saito T., Ohno H. and Bonifacino J.S. (2001) Signal-binding specificity of the mu4 subunit of the adaptor protein complex AP-4. J. Biol. Chem., 276, 13145–13152. [DOI] [PubMed] [Google Scholar]
- 12. Mattera R., Park S.Y., De Pace R., Guardia C.M. and Bonifacino J.S. (2017) AP-4 mediates export of ATG9A from the trans-Golgi network to promote autophagosome formation. Proc. Nat. Acad. Sci. U.S.A., 114, E10697–E10706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Davies A.K., Itzhak D.N., Edgar J.R., Archuleta T.L., Hirst J., Jackson L.P., Robinson M.S. and Borner G.H.H. (2018) AP-4 vesicles contribute to spatial control of autophagy via RUSC-dependent peripheral delivery of ATG9A. Nat Commun, 9, 3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. De Pace R., Skirzewski M., Damme M., Mattera R., Mercurio J., Foster A.M., Cuitino L., Jarnik M., Hoffmann V., Morris H.D. et al. (2018) Altered distribution of ATG9A and accumulation of axonal aggregates in neurons from a mouse model of AP-4 deficiency syndrome. PLoS Genetics, 14, e1007363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Noda T., Kim J., Huang W.P., Baba M., Tokunaga C., Ohsumi Y. and Klionsky D.J. (2000) Apg9p/Cvt7p is an integral membrane protein required for transport vesicle formation in the Cvt and autophagy pathways. J. Cell Biol., 148, 465–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Papinski D., Schuschnig M., Reiter W., Wilhelm L., Barnes C.A., Maiolica A., Hansmann I., Pfaffenwimmer T., Kijanska M., Stoffel I. et al. (2014) Early steps in autophagy depend on direct phosphorylation of Atg9 by the Atg1 kinase. Mol. Cell, 53, 471–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yamaguchi J., Suzuki C., Nanao T., Kakuta S., Ozawa K., Tanida I., Saitoh T., Sunabori T., Komatsu M., Tanaka K. et al. (2017, in press) Atg9a deficiency causes axon-specific lesions including neuronal circuit dysgenesis. Autophagy, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ebrahimi-Fakhari D., Cheng C., Dies K., Diplock A., Pier D.B., Ryan C.S., Lanpher B.C., Hirst J., Chung W.K., Sahin M. et al. (2018) Clinical and genetic characterization of AP4B1-associated SPG47. Am. J. Med. Genet. A, 176, 311–318. [DOI] [PubMed] [Google Scholar]
- 19. Zhou C., Ma K., Gao R., Mu C., Chen L., Liu Q., Luo Q., Feng D., Zhu Y. and Chen Q. (2017) Regulation of mATG9 trafficking by Src- and ULK1-mediated phosphorylation in basal and starvation-induced autophagy. Cell Res., 27, 184–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Young A.R., Chan E.Y., Hu X.W., Kochl R., Crawshaw S.G., High S., Hailey D.W., Lippincott-Schwartz J. and Tooze S.A. (2006) Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J. Cell Sci., 119, 3888–3900. [DOI] [PubMed] [Google Scholar]
- 21. Teinert J., Behne R., Wimmer M. and Ebrahimi-Fakhari D. (2019) Novel insights into the clinical and molecular spectrum of congenital disorders of autophagy. J. Inherit. Metab. Dis., doi: 10.1002/jimd.12084 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 22. Roubertie A., Hieu N., Roux C.J., Leboucq N., Manes G., Charif M., Echenne B., Goizet C., Guissart C., Meyer P. et al. (2018) AP4 deficiency: a novel form of neurodegeneration with brain iron accumulation? Neurol Genet, 4, e217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vill K., Muller-Felber W., Alhaddad B., Strom T.M., Teusch V., Weigand H., Blaschek A., Meitinger T. and Haack T.B. (2017) A homozygous splice variant in AP4S1 mimicking neurodegeneration with brain iron accumulation. Mov. Disord., 32, 797–799. [DOI] [PubMed] [Google Scholar]
- 24. Engle S.J., Blaha L. and Kleiman R.J. (2018) Best practices for translational disease modeling using human iPSC-derived neurons. Neuron, 100, 783–797. [DOI] [PubMed] [Google Scholar]
- 25. Zhang Y., Pak C., Han Y., Ahlenius H., Zhang Z., Chanda S., Marro S., Patzke C., Acuna C., Covy J. et al. (2013) Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron, 78, 785–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Borner G.H., Antrobus R., Hirst J., Bhumbra G.S., Kozik P., Jackson L.P., Sahlender D.A. and Robinson M.S. (2012) Multivariate proteomic profiling identifies novel accessory proteins of coated vesicles. J. Cell Biol., 197, 141–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ivankovic D., Drew J., Lesept F., White I.J., Lopez Domenech G., Tooze S.A. and Kittler J.T. (2019) Axonal autophagosome maturation defect through failure of ATG9A sorting underpins pathology in AP-4 deficiency syndrome. Autophagy, 1–17. 10.1080/15548627.2019.1615302 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Orsi A., Razi M., Dooley H.C., Robinson D., Weston A.E., Collinson L.M. and Tooze S.A. (2012) Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol. Biol. Cell, 23, 1860–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Karanasios E., Walker S.A., Okkenhaug H., Manifava M., Hummel E., Zimmermann H., Ahmed Q., Domart M.C., Collinson L. and Ktistakis N.T. (2016) Autophagy initiation by ULK complex assembly on ER tubulovesicular regions marked by ATG9 vesicles. Nat Commun, 7, 12420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tamura H., Shibata M., Koike M., Sasaki M. and Uchiyama Y. (2010) Atg9A protein, an autophagy-related membrane protein, is localized in the neurons of mouse brains. J. Histochem. Cytochem., 58, 443–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yamamoto H., Kakuta S., Watanabe T.M., Kitamura A., Sekito T., Kondo-Kakuta C., Ichikawa R., Kinjo M. and Ohsumi Y. (2012) Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J. Cell Biol., 198, 219–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mari M., Griffith J., Rieter E., Krishnappa L., Klionsky D.J. and Reggiori F. (2010) An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J. Cell Biol., 190, 1005–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stavoe A.K., Hill S.E., Hall D.H. and Colon-Ramos D.A. (2016) KIF1A/UNC-104 transports ATG-9 to regulate neurodevelopment and autophagy at synapses. Dev. Cell, 38, 171–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ebrahimi-Fakhari D., Saffari A., Wahlster L., Lu J., Byrne S., Hoffmann G.F., Jungbluth H. and Sahin M. (2016) Congenital disorders of autophagy: an emerging novel class of inborn errors of neuro-metabolism. Brain, 139, 317–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Goodwin J.M., Dowdle W.E., DeJesus R., Wang Z., Bergman P., Kobylarz M., Lindeman A., Xavier R.J., McAllister G., Nyfeler B. et al. (2017) Autophagy-independent lysosomal targeting regulated by ULK1/2-FIP200 and ATG9. Cell Rep, 20, 2341–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dowdle W.E., Nyfeler B., Nagel J., Elling R.A., Liu S., Triantafellow E., Menon S., Wang Z., Honda A., Pardee G. et al. (2014) Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat. Cell Biol., 16, 1069–1079. [DOI] [PubMed] [Google Scholar]
- 37. Hayflick S.J., Kruer M.C., Gregory A., Haack T.B., Kurian M.A., Houlden H.H., Anderson J., Boddaert N., Sanford L., Harik S.I. et al. (2013) Beta-propeller protein-associated neurodegeneration: a new X-linked dominant disorder with brain iron accumulation. Brain, 136, 1708–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Seibler P., Burbulla L.F., Dulovic M., Zittel S., Heine J., Schmidt T., Rudolph F., Westenberger A., Rakovic A., Munchau A. et al. (2018) Iron overload is accompanied by mitochondrial and lysosomal dysfunction in WDR45 mutant cells. Brain, 141, 3052–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhu P.P., Denton K.R., Pierson T.M., Li X.J. and Blackstone C. (2014) Pharmacologic rescue of axon growth defects in a human iPSC model of hereditary spastic paraplegia SPG3A. Hum. Mol. Genet., 23, 5638–5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Denton K.R., Lei L., Grenier J., Rodionov V., Blackstone C. and Li X.J. (2014) Loss of spastin function results in disease-specific axonal defects in human pluripotent stem cell-based models of hereditary spastic paraplegia. Stem Cells, 32, 414–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mishra H.K., Prots I., Havlicek S., Kohl Z., Perez-Branguli F., Boerstler T., Anneser L., Minakaki G., Wend H., Hampl M. et al. (2016) GSK3ss-dependent dysregulation of neurodevelopment in SPG11-patient iPSC model. Ann. Neurol., 79(5), 826–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Perez-Branguli F., Mishra H.K., Prots I., Havlicek S., Kohl Z., Saul D., Rummel C., Dorca-Arevalo J., Regensburger M., Graef D. et al. (2014) Dysfunction of spatacsin leads to axonal pathology in SPG11-linked hereditary spastic paraplegia. Hum. Mol. Genet., 23, 4859–4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Denton K., Mou Y., Xu C.C., Shah D., Chang J., Blackstone C. and Li X.J. (2018) Impaired mitochondrial dynamics underlie axonal defects in hereditary spastic paraplegias. Hum. Mol. Genet., 27, 2517–2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Maday S. and Holzbaur E.L. (2016) Compartment-specific regulation of autophagy in primary neurons. J. Neurosci., 36, 5933–5945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kircher M., Witten D.M., Jain P., O'Roak B.J., Cooper G.M. and Shendure J. (2014) A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet., 46, 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ebrahimi-Fakhari D., Wahlster L., Bartz F., Werenbeck-Ueding J., Praggastis M., Zhang J., Joggerst-Thomalla B., Theiss S., Grimm D., Ory D.S. et al. (2016) Reduction of TMEM97 increases NPC1 protein levels and restores cholesterol trafficking in Niemann-pick type C1 disease cells. Hum. Mol. Genet., 25, 3588–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Teinert J., Behne R., D'Amore A., Wimmer M., Dwyer S., Chen T., Buttermore E.D., Chen I.P.-F., Sahin M. and Ebrahimi-Fakhari D. (2019) Generation and characterization of six human induced pluripotent stem cell lines (iPSC) from three families with AP4B1-associated hereditary spastic paraplegia (SPG47). Stem Cell Res., 40, 101575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kirby J., Ning K., Ferraiuolo L., Heath P.R., Ismail A., Kuo S.W., Valori C.F., Cox L., Sharrack B., Wharton S.B. et al. (2011) Phosphatase and tensin homologue/protein kinase B pathway linked to motor neuron survival in human superoxide dismutase 1-related amyotrophic lateral sclerosis. Brain, 134, 506–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ebrahimi-Fakhari D., Cantuti-Castelvetri I., Fan Z., Rockenstein E., Masliah E., Hyman B.T., McLean P.J. and Unni V.K. (2011) Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of alpha-synuclein. J. Neurosci., 31, 14508–14520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ebrahimi-Fakhari D., Saffari A., Wahlster L., Di Nardo A., Turner D., Lewis T.L. Jr., Conrad C., Rothberg J.M., Lipton J.O., Kolker S. et al. (2016) Impaired mitochondrial dynamics and mitophagy in neuronal models of tuberous sclerosis complex. Cell Rep, 17, 1053–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Carpenter A.E., Jones T.R., Lamprecht M.R., Clarke C., Kang I.H., Friman O., Guertin D.A., Chang J.H., Lindquist R.A., Moffat J. et al. (2006) CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol., 7, R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Klionsky D.J., Abdelmohsen K., Abe A., Abedin M.J., Abeliovich H., Acevedo Arozena A., Adachi H., Adams C.M., Adams P.D., Adeli K. et al. (2016) Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy, 12, 1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mizushima N. and Yoshimori T. (2007) How to interpret LC3 immunoblotting. Autophagy, 3, 542–545. [DOI] [PubMed] [Google Scholar]
- 54. Sahani M.H., Itakura E. and Mizushima N. (2014) Expression of the autophagy substrate SQSTM1/p62 is restored during prolonged starvation depending on transcriptional upregulation and autophagy-derived amino acids. Autophagy, 10, 431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.