

SUMMARY
The Ras GTPases are frequently mutated in human cancer, and, although the Raf kinases are essential effectors of Ras signaling, the tumorigenic properties of specific Ras-Raf complexes are not well characterized. Here, we examine the ability of individual Ras and Raf proteins to interact in live cells using bioluminescence resonance energy transfer (BRET) technology. We find that C-Raf binds all mutant Ras proteins with high affinity, whereas B-Raf exhibits a striking preference for mutant K-Ras. This selectivity is mediated by the acidic, N-terminal segment of B-Raf and requires the K-Ras polybasic region for high-affinity binding. In addition, we find that C-Raf is critical for mutant H-Ras-driven signaling and that events stabilizing B-Raf/C-Raf dimerization, such as Raf inhibitor treatment or certain B-Raf mutations, can allow mutant H-Ras to engage B-Raf with increased affinity to promote tumorigenesis, thus revealing a previously unappreciated role for C-Raf in potentiating B-Raf function.
In Brief
The Raf kinases bind to active Ras proteins and function to transmit signals that control cell growth and tumorigenesis. The study by Terrell et al. reveals distinct binding preferences between individual Ras and Raf family members and identifies events that can alter these interactions to upregulate Ras-driven cancer signaling.
Graphical Abstract
INTRODUCTION
Ras proteins are membrane-associated, small GTPases that function to transmit a multitude of cellular signals (Pylayeva-Gupta et al., 2011). All Ras family members, which include H-Ras, K-Ras4A/4B, and N-Ras, can relay signals received by cell surface receptors due to their ability to cycle between a GDP-bound “off” state and a GTP-bound “on” state (Cox and Der, 2010; Simanshu et al., 2017). Typically, receptor engagement results in the recruitment of guanine nucleotide exchange factors (GEFs) to the cell surface where they facilitate the GTP-loading of Ras and, in turn, the interaction of Ras with downstream effectors. Following signal transmission, Ras cycles back to its inactive state as a result of GTPase-activating proteins (GAPs) that stimulate the intrinsic GTPase activity of Ras (Bos et al., 2007).
Consistent with its central role in cell signaling, dysregulation of Ras cycling can promote human disease states, with somatic mutations in the Ras genes being prominent drivers of tumorigenesis and Ras germline mutations contributing to a group of related developmental disorders known as the RASopathies (Fernández-Medarde and Santos, 2011; Schubbert et al., 2007). Importantly, disease-associated mutations tend to render Ras insensitive to GAP stimulation and reduce its intrinsic GTPase activity, leaving Ras in a constitutively active state that promotes pathway activation in an unregulated manner (Prior et al., 2012).
One of the essential effector cascades required for Ras signaling is the ERK cascade, comprised of the Raf, MEK, and ERK protein kinases (Lavoie and Therrien, 2015). All Raf family members, which include A-Raf, B-Raf, and C-Raf, possess a conserved Ras-binding domain (RBD) that resides in the Raf N-terminal regulatory domain. In quiescent cells, the Rafs exist as autoinhibited monomers in the cytosol (Nan et al., 2013). However, when growth signals are received, the Raf kinases are recruited by Ras to the plasma membrane where they become activated through an allosteric mechanism that requires dimerization of the C-terminal Raf kinase domains (Hu et al., 2013). In normal Ras-dependent signaling, B-Raf/C-Raf heterodimers predominate (Freeman et al., 2013) and function to initiate the phosphorylation cascade that results in MEK and ERK activation. Once activated, ERK plays a critical role in the forward transmission of signals but also participates in the attenuation of Ras signaling through the phosphorylation of upstream pathway components, which, in the case of the Rafs, inhibit both Ras/Raf binding and Raf dimerization (Dougherty et al., 2005; Ritt et al., 2010).
Despite being one of the most frequently mutated signaling pathways in human cancer, various aspects of Ras biology are still poorly understood. For example, even though it is well known that the C-terminal hypervariable region (HVR) of the Ras proteins results in differential lipid processing and membrane localization (Prior and Hancock, 2012), the extent to which these differences influence Ras signaling and/or effector interactions is not clear. Moreover, a puzzling aspect of Ras-induced tumorigenesis is that, although the Ras proteins are highly conserved and rather ubiquitously expressed, their mutational frequency can vary significantly among cancer types, with K-Ras mutations being the predominant driver among all Ras-associated tumors but other family members being the primary driver in select tumor types. Therefore, given the central role of the Raf kinases in Ras signaling, studies examining the Ras/Raf interaction in live cells could reveal valuable information needed to tease apart unique tumorigenic properties of individual Ras members and may prove helpful in the pursuit of more effective therapeutic strategies.
Here, we examine the Ras/Raf interaction utilizing bioluminescence resonance energy transfer (BRET), a technique that allows quantitative measurements to be obtained under conditions that preserve crucial features of Ras and Raf regulation, including lipid processing, intracellular trafficking, membrane microdomain targeting, and protein phosphorylation. Strikingly, we find that different Ras and Raf family members exhibit distinct binding preferences and that these differences have important implications for disease-associated Ras signaling.
RESULTS
Live-Cell BRET Analysis of the Ras/Raf Interaction
Bioluminescence resonance energy transfer (BRET) was used to investigate the requirements for Ras/Raf binding in live cells (Pfleger and Eidne, 2006). In this system, a BRET signal is generated when a protein tagged with an energy donor comes in close proximity and can transfer energy to a protein tagged with an energy acceptor. Donor, acceptor, and BRET signals are each monitored individually, providing internal controls for protein expression and producing a sensitive ratiometric readout that is independent of cell number. Quantitative data regarding the interaction can also be obtained by generating a saturation curve in which expression of the energy donor remains constant, while expression of the energy acceptor increases. In this type of analysis, a specific interaction will generate a hyperbolic curve, with BRETmax being reflective of the total number of binding pairs that can form and BRET50 being a relative measure of binding affinity.
For our analysis, the Raf members functioned as the energy donor, tagged at a conserved C-terminal position with the Rluc8 enzyme, and the Ras proteins served as the energy acceptor, tagged at the N terminus with the Venus fluorophore. It should be noted that our initial studies were performed using proteins that encode the entire Raf regulatory domain (RafReg) but lack the kinase domain. This approach was taken in order to mitigate any indirect effects on Ras/Raf binding that might be caused by dimerization of the Raf kinase domains or due to inhibitory feedback loops generated by Raf catalytic activation. As shown in Figure 1A, a strong BRET signal was observed when wild-type (WT) C-RafReg was co-expressed with the Q61R mutant of K-Ras4B (hereon referred to as K-Ras). However, if the RBD of C-RafReg contained an arginine to leucine (R > L) mutation known to disrupt the Ras/Raf interaction (Fabian et al., 1994), the BRET signal was dramatically reduced. In addition, when the C-terminal CAAX motif of K-RasQ61R was mutated to prevent the lipid processing and membrane localization oi K-Ras (K-RasQ61R/C>A), the BRET signal was significantly compromised as was the GTP loading of Ras (Figures 1A and 1B). In co-immunoprecipitation assays, WT-C-RafReg, but not the R > L mutant, was strongly detected in K-RasQ61R complexes, and only faint levels of WT-C-RafReg were observed in K-RasQ61R/C>A complexes (Figure 1B). Moreover, live-cell imaging studies verified the cytosolic localization of K-RasQ61R/C>A and showed that K-RasQ61R could recruit WT-C-Raf to the cell surface but not the R > L mutant (Figure 1C). Thus, these findings confirm that the Ras and Raf proteins generated for use in the BRET assay exhibit their expected subcellular localization and protein binding properties.
Figure 1. Analysis of Raf-Binding Interactions with Activated K-Ras Mutants.

(A) BRET saturation curves are shown examining the interaction of WT or RBD mutant (R > L) C-RafReg-Rluc proteins with Venus-K-RasQ61R and the interaction of WT C-RafReg-Rluc with the CAAX mutant (C185A) Venus-K-RasQ61R/C>A. BRET50 values are listed.
(B) K-Ras and C-Raf proteins analyzed in (A) were examined in co-immunoprecipitation assays. Venus-K-Ras proteins were also evaluated for GTP loading in Raf-RBD pull-down assays.
(C) Live-cell imaging shows the intracellular localization of the indicated K-Ras and C-Raf proteins.
(D) BRET saturation curves are shown examining the interaction of WT or R > L RafReg-Rluc proteins with the indicated Venus-K-Ras mutants.
(E) BRET50 values from (D) are listed and the expression level of the K-Ras mutants is shown.
(F) WT and R > L RafReg-Rluc proteins were examined in co-immunoprecipitation assays for binding to Venus-K-RasQ61R. Lysates were also monitored for RafReg-Rluc expression, and all experiments were conducted in 293FT cells.
Next, each Raf family member was evaluated for binding interactions with a panel of K-Ras mutants. As shown in Figure 1D, a BRET signal was detected for all the RafReg/K-Ras pairings, with the highest binding affinity (represented by lower BRET50 values and highest BRETmax observed with C-Raf, followed by A-Raf and then B-Raf. For each individual RafReg protein, BRET50 values were similar for all the K-Ras mutants, indicating a comparable binding affinity (Figure 1E). Interestingly, the highest BRETmax signals were observed with K-Ras proteins containing mutations in the Q61 site, likely reflecting the reported increased GTP occupancy of Q61 mutants (Buhrman et al., 2011; Hunter et al., 2015) and an increase in the number of K-Ras proteins available for pairing with the Rafs. Finally, incorporation of the RBD R > L mutation into each Raf member disrupted the Ras/Raf interaction in both the BRET and co-immunoprecipitation assays (Figures 1D–1F).
BRET Analysis Reveals Binding Preferences between Ras and Raf Family Members
To determine whether any of the Raf or Ras family members display preferential binding to one another in live cells, the ability of each RafReg protein to interact with G12V or Q61R mutants of H-Ras, N-Ras, or K-Ras was monitored (Figures 2A and S1A). Surprisingly, differences in the BRETmax and BRET50 values were observed among the different pairings, revealing that the Rafs do not bind the Ras family members equivalently. For A-Raf and B-Raf, the highest BRETmax and lowest BRET50 values were observed when they were paired with mutant K-Ras. In contrast, when C-Raf was paired with mutant K-Ras the BRETmax signals were lower than those observed with mutant H-Ras or N-Ras; however, all the C-Raf/Ras pairings were of similar high affinity (BRET50 values ranging from 0.165–0.172). As expected, the RBD R > L mutation significantly disrupted all Ras/Raf interactions (Figure 2A).
Figure 2. Binding Preferences between Ras and Raf Family Members.

(A) BRET saturation curves examining the interaction of WT or R > L RafRe9-Rluc proteins with the Venus-RasG12V proteins are shown, and the BRET50 values are listed.
(B) WT RafReg-Rluc proteins were examined in co-immunoprecipitation assays for binding to the Venus-RasG12V proteins.
(C) Immunoprecipitated Venus-RasG12V complexes were probed for the presence of endogenous B-Raf, C-Raf, or A-Raf and Venus-Ras. Lysates were also examined for B-Raf, C-Raf, A-Raf, and pMEK levels (upper). Endogenous C-Raf complexes were isolated from cells expressing the indicated Venus-RasG12V proteins and examined for dimerization with B-Raf (lower).
(D) HeLa cells expressing WT Venus-Ras proteins were treated or not with EGF prior to lysis. Immunoprecipitated Venus-Ras complexes were probed for the presence of endogenous B-Raf, C-Raf, or A-Raf and Venus-Ras. Lysates were also examined for Raf levels.
(E) Ras complexes were immunoprecipitated from Ras-deficient MEFs re-expressing either K-RasQ61L or H-RasQ61L and probed for the presence of endogenous B-Raf, C-Raf, or A-Raf and Ras. Endogenous C-Raf was also isolated from the MEF lines and examined for dimerization with B-Raf. Lysates were examined for B-Raf, C-Raf, A-Raf, and pMEK levels.
(F) MCF10A cells stably expressing Halo-tagged K-RasG12V or H-RasG12V and B-Raf-Cherry or C-Raf-Cherry were examined by live-cell imaging to visualize recruitment of the Rafs to the plasma membrane. Experiments shown in (A)–(C) were conducted in 293FT cells.
See also Figure S1.
In co-immunoprecipitation assays (Figures 2B and S1B) C-RafReg was detected at nearly equivalent levels in all the mutant Ras complexes. A-RafReg was also observed in all Ras complexes, but binding to K-Ras was increased. Strikingly, B-RafReg was found to co-immunoprecipitate almost exclusively with activated K-Ras. Of note, the observed co-immunoprecipitation results appear to align more closely with the BRET50 values, which are reflective of the affinity of the interaction and are consistent with the fact that binding interactions detected in co-immunoprecipitation assays must be of sufficient strength to withstand detergent-based cell lysis and immunopurification.
Similar Ras binding preferences were observed when the endogenous Raf kinases were evaluated for their ability to co-immunoprecipitate with constitutively active Ras mutants or with growth-factor-activated WT Ras proteins (Figures 2C and 2D and S1C). Preferential binding of B-Raf to activated K-Ras was further confirmed in co-immunoprecipitation assays using Ras-deficient mouse embryonic fibroblasts (MEFs) (Drosten et al., 2010) reconstituted to express untagged H-RasQ61L or K-RasQ61L at endogenous levels (Figure 2E). Moreover, in live-cell imaging studies using MCF10A cell lines that stably express Halo-tagged H-RasG12V or K-RasG12V and Cherry-tagged B-Raf or C-Raf, the plasma membrane recruitment of B-Raf was significantly increased in cells expressing K-RasG12V, whereas strong membrane localization of C-Raf was observed in both cell lines (Figure 2F). Consistent with the preferred binding of B-Raf to K-Ras, mutant K-Ras was found to be the strongest driver of endogenous B-Raf/C-Raf dimer formation as well as downstream MEK activation (Figures 2C and 2E). Taken together, these findings indicate that C-Raf and K-Ras can bind with high affinity to all Ras or Raf family members respectively, whereas H-Ras displays preferential binding to C-Raf, and B-Raf exhibits a striking selectivity for K-Ras.
Role of the Raf N-Terminal Segment in Determining Ras Binding Selectivity
Given that B-Raf and C-Raf were found to exhibit the most divergent binding to Ras members, experiments were conducted to determine which regions of the Rafs might account for these differences. The Raf regulatory domain contains two conserved areas: the RBD and the membrane-binding cysteine-rich domain (CRD) (Hekman et al., 2002; Williams et al., 2000). However, preceding the RBD, there lies an N-terminal segment (N′-segment) that varies significantly among the Rafs (Figure 3A). In particular, the N′-segment of B-Raf is comprised of 154 amino acids and has an acidic isoelectric point (pi) of 4.6, whereas the C-Raf N′-segment contains 55 amino acids and has a more neutral pl of 6.6. Of note, the A-Raf N′-segment also has a neutral pl (6.9) and is 18 amino acids in length.
Figure 3. Raf N-Terminal Segment Determines the Ras Binding Selectivity.

(A) Schematic depiction of the B-Raf and C-Raf regulatory domains with the RBD, CRD, and N′-segment indicated.
(B) RafReg-Rluc proteins were generated in which the RBD/CRD or N′-segment of B-Raf and C-Raf were exchanged. BRET (upper) and co-immunoprecipitation assays (lower) were performed examining the interaction of WT or domain-exchanged RafReg-Rluc proteins with Venus-tagged H-RasQ61R or K-RasQ61R. BRET50 values are listed.
(C) WT full-length B-RafFL-Rluc or B-RafFL-Rluc proteins lacking the N′-segment (Δ-N′) or containing the N′-segment of C-Raf (C-N′) were examined for their ability to interact with the indicated Venus-RasQ61R proteins in co-immunoprecipitation assays. Lysates were also monitored for Raf-Rluc expression in (B and C), and all experiments were conducted in 293FT cells.
See also Figure S2.
Therefore, B-Raf and C-Raf constructs were generated in which the N′-segments were exchanged, and the resulting proteins were evaluated in BRET and co-immunoprecipitation assays for binding interactions with activated H-Ras or K-Ras. As shown in Figure 3B, when the B-Raf N′-segment was replaced with that of C-Raf (B-Raf(C-N′)), the B-Raf/H-Ras interaction was significantly increased as BRETmax signals were higher, BRET50 values were lower, and B-Raf(C-N′) could be detected in H-Ras immunoprecipitates. In contrast, replacing the C-Raf N′-segment with that of B-Raf (C-Raf(B-N′)) greatly reduced the C-Raf/H-Ras interaction. Consistent with the ability of K-Ras to engage all Raf kinases with high affinity, exchange of the Raf N′-segments had no significant effect on the affinity of K-Ras binding in either co-immunoprecipitation or BRET assays (Figures 3B and S2). Exchange of the conserved RBD-CRD domains was also evaluated and found to have little effect on Ras/Raf interactions.
The role of the N′-segment in determining the Ras binding selectivity of B-Raf was further confirmed in co-immunoprecipitation assays using full-length B-Raf proteins in which the N′-segment was either deleted (Δ-N′) or replaced with that of C-Raf (C-N′). As shown in Figure 3C, all B-Raf proteins were detected in K-RasQ61R complexes; however, only proteins lacking the B-Raf N′-segment were present in H-Ras or N-Ras complexes, suggesting that the B-Raf N′-segment may impede or obstruct high-affinity binding to H-Ras and N-Ras.
Contribution of the Ras Hypervariable Region to the Ras/Raf Interaction
Next, we sought to identify the region of the Ras proteins that likewise determines the Raf binding preferences. Members of the Ras family are highly conserved and diverge primarily at the C-terminal hypervariable region (HVR) (Figure 4A), which contains distinct signals for lipid processing and membrane attachment (Parker and Mattos, 2015; Prior and Hancock, 2012). All Ras proteins end with a CAAX motif, which is processed to yield a C-terminal farnesylated cysteine residue that is carboxymethylated. The H-Ras and N-Ras HVRs contain additional cysteine residues that are palmitoylated and function with the farneysl group to mediate plasma membrane attachment. In contrast, the K-Ras4B HVR uniquely contains a lysine-rich polybasic region (PBR) that aids in membrane binding.
Figure 4. The Ras HVRs Contribute to the Ras/Raf Binding Preferences.

(A) Shown are the HVR sequences of the various Ras proteins.
(B) Venus-RasQ61R proteins were generated in which the HVRs of H-Ras and K-Ras4B were exchanged. BRET (upper) and co-immunoprecipitation assays (lower) were performed examining the interaction of B-RafReg or C-RafReg-Rluc with WT or HVR-exchanged Venus-RasQ61R proteins. BRET50 values are listed.
(C) Co-immunoprecipitation assays were performed examining the interaction of B-RafReg or C-RafReg-Rluc with Venus-tagged K-Ras4AQ61R or K-Ras4BQ61R.
(D) Cells co-expressing the indicated GFP-RasG12V proteins and B-RafReg or C-RafReg-Rluc were lysed, and GFP-Ras complexes were immunoprecipitated from the cell lysates and examined for RafReg-Rluc binding.
(E) WT or N′-segment mutant B-RafReg-Rluc proteins were examined in co-immunoprecipitation assays for binding to Venus-tagged K-RasQ61R or H-RasQ61R. Lysates were monitored for the indicated proteins in (B–E), and all experiments were conducted in 293FT cells.
See also Figure S3.
To investigate whether the Ras HVRs might also contribute to the Raf binding preferences, Ras proteins were analyzed in which the HVRs of mutant K-Ras4B (amino acids 165–188) and H-Ras (amino acids 165–189) were exchanged. As shown in Figure 4B, placing the K-Ras4B HVR sequences onto H-RasQ61R increased the binding of B-RafReg such that the BRETmax signals and BRET50 values were similar to those observed with K-RasQ61R. Conversely, exchanging the K-Ras4B HVR with that of H-Ras reduced the K-RasQ61R/B-RafReg interaction to levels observed with H-RasQ61R. Moreover, B-RafReg and endogenous B-Raf were only able to co-immunoprecipitate with Ras proteins that contained the K-Ras4B HVR (Figures 4B and S3A).
Consistent with the high-affinity binding of C-Raf to all Ras members, C-RafReg and endogenous C-Raf were detected in all of the mutant Ras complexes, and all C-RafReg pairings exhibited high-affinity BRET50 values (Figures 4B and S3A). However, the highest BRETmax signals were observed with proteins that contained the H-Ras HVR, suggesting that the H-Ras HVR sequence itself or the localization of these Ras proteins allows more C-Raf binding pairs to form. In addition, the K-Ras4A splice variant, whose HVR contains a palmitoylated cysteine residue instead of the PBR, interacted with the Rafs in a manner similar to H-Ras and N-Ras, as only C-Raf proteins could bind with sufficient affinity to co-immunoprecipitate with K-Ras4AQ61R (Figures 4C and S3B).
The above findings suggest that the PBR-containing HVR of K-Ras4B also contributes to the B-Raf selectivity, and by utilizing a panel of previously characterized K-RasG12V PBR mutants (Zhou et al., 2017), we further found that the positive charge of the PBR was critical for high-affinity B-Raf binding. As shown in Figure 4D, substitution of each individual PBR lysine residue to an uncharged glutamine reduced binding of B-RafReg but had little effect on C-RafReg binding. Moreover, the reduction in B-RafReg binding was equivalent for all the PBR mutants, correlating with an equivalent reduction in the net basic charge of the PBR. In addition, replacing all six lysine residues with similarly charged arginine residues (6R) had minimal effect on B-RafReg binding; however, mutation of the serine phosphorylation site adjacent to the PBR to a phosphomimetic acidic residue (S181D) reduced the B-RafReg interaction, whereas mutation of the site to a neutral alanine residue had little effect (Figure 4D). Similar results were obtained when a subset of these mutants was evaluated for binding to endogenous B-Raf or C-Raf or when they were assessed in BRET assays (Figures S3D and S3E).
Finally, to investigate whether the positively charged K-Ras PBR might interact with the negatively charged B-Raf N′-segment, several cancer-associated mutations that alter acidic residues in the B-Raf N′-segment were analyzed in co-immunoprecipitation assays (Figure 4E). Strikingly, the E46K mutation resulted in reduced binding of B-RafReg to mutant K-Ras but increased binding to mutant H-Ras. Collectively, these findings support a model whereby the PBR contributes to the B-Raf/K-Ras interaction by engaging the B-Raf N′-segment, thus disrupting its inhibitory effect to facilitate high-affinity RBD contact.
Dimerization with C-Raf Can Influence the Affinity of the B-Raf/H-RasQ61R Interaction
B-Raf and C-Raf are known to form heterodimers, and, given that C-Raf exhibits high-affinity binding to all Ras proteins, experiments were initiated to determine whether B-Raf/C-Raf dimerization might alter the ability of B-Raf to interact with Ras members that lack the PBR. For these studies, full-length B-Raf proteins containing well-characterized mutations in the Raf dimer interface were utilized: dimerization-deficient R509H-B-Raf and dimerization-enhanced E586K-B-Raf. These mutants and WT-B-Raf were then evaluated in BRET and co-immunoprecipitation assays for binding to activated H-Ras or K-Ras. As indicated by the BRET50 values, full-length WT-B-Raf (B-RafFL) exhibited a similar binding affinity to mutant H-Ras, as did the B-RafReg protein, and showed little ability to co-immunoprecipitate with H-Ras (Figures 5A and 2A). The R509H mutant also displayed low-affinity binding to H-Ras, whereas E586K-B-Raf exhibited an increased binding affinity and co-immunoprecipitated with mutant H-Ras in a manner that correlated with its increased ability to dimerize with C-Raf (Figure 5A). As expected, all of the B-RafFL proteins bound mutant K-Ras with a similar high affinity (Figure S4A).
Figure 5. B-Raf/C-Raf Dimerization Can Modulate the B-Raf/H-RasQ61R Interaction.

(A) BRET (left) and co-immunoprecipitation assays (right) are shown examining the interaction of WT, R509H (dimer-defective), or E586K (dimer-enhanced) B-RafFL-Rluc proteins with Venus-H-RasQ61R. The B-RafFL-Rluc proteins were also monitored for dimerization with C-Raf.
(B) BRET saturation curves were performed examining the effect of 1 h DMSO or Raf inhibitor SB590885 (SB) treatment on the interaction of B-RafFL-Rluc with Venus-tagged H-RasQ61R or K-RasQ61R. BRET50 values are listed.
(C) MCF10A cells stably expressing Halo-H-RasG12V and B-RafFL-Cherry were treated for 1 h with DMSO or SB590885 prior to live-cell imaging. Recruitment of B-Raf to the plasma membrane in SB590885-treated cells is indicated by white arrows.
(D) 293FT cells expressing B-RafFL-Rluc with Venus-H-RasQ61R or Venus-K-RasQ61R or expressing B-RafReg-Rluc with Venus-H-RasQ61R were treated for 1 h with DMSO or SB590885 prior to lysis. Immunoprecipitated Venus-Ras complexes were probed for B-Raf-Rluc and Venus-Ras.
(E) BRET (left) and co-immunoprecipitation (right) assays were performed examining the effect of various Raf inhibitors on the interaction of B-RafFL-Rluc with Venus-H-RasQ61R. BRET50 values are listed. B-RafFL-Rluc proteins were also examined for dimerization with C-Raf.
(F) BRET (left) and co-immunoprecipitation (right) assays were performed examining the effect of SB590885 treatment on the interaction of B-RafFL-Rluc and Venus-H-RasQ61R in control (NT) or C-Raf-depleted (C-Raf-T) 293FT cells. BRET50 values are listed. Co-immunoprecipitation of endogenous C-Raf with Venus-H-RasQ61R is also shown.
(G) BRET (left) and co-immunoprecipitation (right) assays were performed examining the effect of SB590885 treatment on the interaction of WT or R > L B-RafFL-Rluc with Venus-H-RasQ61R. BRET50 values are listed. Co-immunoprecipitation of endogenous C-Raf with Venus-H-RasQ61R is also shown. Lysates were monitored for the indicated protein levels in (A, D, and E–G).
See also Figures S4 and S5.
Further supporting the model that dimerization with C-Raf can facilitate the interaction between B-Raf and H-Ras, stabilizing B-Raf/C-Raf dimers by mutation of the ERK-mediated feedback phosphorylation sites (which function to disrupt Raf dimerization) also resulted in increased B-Raf/H-Ras binding (Figure S4B). Of note, Ras proteins have also been proposed to dimerize; however, a mutation (D154Q) reported to impair Ras dimer formation (Ambrogio et al., 2018) was found to have little effect on Ras/Raf binding in either the BRET or co-immunoprecipitation assays (Figure S4C).
Stable B-Raf/C-Raf dimer formation can also be driven by treatment of cells with ATP-competitive Raf inhibitors (Durrant and Morrison, 2018). Therefore, we next used a B-Raf inhibitor known to strongly promote Raf dimerization, SB590885, to determine whether inhibitor treatment would alter B-Raf interactions (Figures 5B–5D). In the BRET system, SB590885 treatment resulted in a dramatic increase in binding of B-RafFL to mutant H-Ras, as evidenced by increased BRETmax signals and reduced BRET50 values (Figure 5B). SB590885 treatment also allowed B-RafFL to stably co-immunoprecipitate with mutant H-Ras (Figure 5D) and resulted in a significant increase in the membrane localization of B-Raf-Cherry in MCF10A cells expressing Halo-H-RasG12V (Figure 5C). Binding between B-RafFL and mutant K-Ras was also enhanced in SB590885-treated cells; however, the increases were not as pronounced (Figures 5B, 5D, and S5A). Importantly, the enhancing effect of SB590885 treatment required binding of the inhibitor to the B-Raf kinase domain as SB590885 treatment had no effect on the interaction of H-Ras and the B-RafReg protein, which lacks the kinase domain that mediates Raf dimerization (Figures 5D and S5B).
When a panel of Raf inhibitors was evaluated, we found that all of the inhibitors tested, with the exception of the second-generation “paradox-breaker” inhibitor PLX7904 (Zhang et al., 2015), increased the level and affinity of the B-RafFL/H-Ras interaction and that the increased affinity correlated with the degree to which the inhibitors promoted B-Raf/C-Raf dimerization (Figure 5E). Further establishing C-Raf as a mediator of the upregulated interaction between B-Raf and H-Ras, depletion of endogenous C-Raf prevented SB590885 from increasing the B-Raf/H-Ras interaction in BRET or co-immunoprecipitation assays (Figure 5F). Finally, our findings suggest that inhibitor-stabilized B-Raf/C-Raf dimerization impacts the ability of B-Raf to directly contact H-Ras, as no increased binding to H-Ras was observed in SB590885-treated cells if the B-RafFL protein contained the RBD R > L mutation (Figure 5G).
Co-occurrence of B-Raf and H-Ras Mutations
Although H-Ras is not a prevalent driver of human cancer, 85% of Ras mutations in bladder cancer occur in H-Ras, and genomic analysis of metadata from cBioPortal and COSMIC databases indicates that mutations in H-Ras co-occur with B-Raf mutations at a statistically significant level (p value = 0.003). Strikingly, the majority of the co-occurring B-Raf mutations cause alterations in the B-Raf kinase domain that are known to promote increased dimerization with C-Raf (Yao et al., 2017). When a panel of these mutants was compared against WT-B-RafFL in the BRET and co-immunoprecipitation assays, all of the kinase domain mutants exhibited an increased affinity for H-RasQ61R that correlated with the extent to which the mutations augmented B-Raf/C-Raf dimerization (Figures 6A and S6A). In these cells, MEK activation was also increased, indicating enhanced H-Ras-driven signaling (Figures 6A and S6A). As was observed for Raf inhibitor treatment, the increased B-Raf/H-Ras interaction was dependent on C-Raf in that co-immunoprecipitation of the G466V- and D594G-B-Raf mutants with H-RasQ61R was reduced to background levels in C-Raf-depleted cells (Figure 6B). Moreover, the interaction with C-Raf again appeared to promote direct binding of G466V-B-Raf to H-Ras, as no increase in B-Raf/H-Ras co-immunoprecipitation was observed if G466V-B-Raf contained the RBD R > L mutation (Figure S6B).
Figure 6. Co-occurring B-Raf and H-Ras Mutations in Cancer.

(A) BRET (left) and co-immunoprecipitation assays (right) were performed comparing the interaction of WT and mutant B-RafFL-Rluc proteins with Venus- H-RasQ61R. BRET50 values are listed. Endogenous C-Raf was also examined for dimerization with the B-RafFL-Rluc mutants. Lysates were monitored for pMEK and B-Raf-Rluc levels.
(B) Control (NT) or C-Raf-depleted (C-Raf-T) 293FT cells expressing WT, G466V, or D594G B-RafFL-Rluc with Venus-H-RasQ61R were examined in co-immunoprecipitation assays for binding of B-RafFL-Rluc to Venus-H-RasQ61R.
(C) Control (sh-Neg) or C-Raf-depleted (sh-C-Raf) NIH 3T3 cells were infected with retroviruses expressing the indicated Ras proteins. After two weeks of culture, focus formation was visualized by methylene blue staining. Shown are focus plates from a representative experiment.
(D) T24 and RL95–2 cells were infected with lentiviruses expressing Cas9 and either a non-targeting sgRNA (NT) or sgRNAs targeting A-Raf, B-Raf, C-Raf, or H-Ras. Cells were assessed for 2D proliferation (left), 3D growth (middle), and expression of A-Raf, B-Raf, C-Raf, or H-Ras proteins (right). Data are represented as mean ± SD. ***p < 0.001.
(E) Control (NT) or C-Raf-depleted (C-Raf-T) lines were serum-starved for 18 h and then treated for 1 h with DMSO or SB590885 prior to lysis. Endogenous mutant H-Ras proteins from T24 and RL95–2 cells and endogenous mutant K-Ras proteins from H358 and SW480 were immunoprecipitated and examined for the presence of endogenous B-Raf. Lysates were monitored for the indicated protein levels in (A–E).
See also Figure S6.
Importance of C-Raf in H-Ras-Driven Signaling
Given that H-Ras binds C-Raf with the highest affinity and that C-Raf can promote increased B-Raf/H-Ras binding through B-Raf/C-Raf dimer formation, it is possible that C-Raf may be required for efficient transmission of H-Ras-mediated signals. To test this hypothesis, we first monitored the transformation potential of mutant H-Ras in focus-forming assays using NIH 3T3 cells that were depleted or not of endogenous C-Raf. As shown in Figure 6C, the number of foci induced by H-RasG12V expression was dramatically reduced (~80%) in cells lacking C-Raf, suggesting a dependence on C-Raf. In comparison, K-RasG12V-induced focus formation was only modestly affected by C-Raf loss (15%–20% reduction), and the effect of C-Raf depletion on H-Ras- and K-Ras-mediated transformation could be reversed by exchanging the C′-terminal HVR sequences (Figure 6C), further demonstrating the role of the Ras HVR in determining Raf engagement.
Next, we examined the effect of C-Raf depletion on the transformation potential and proliferative growth of two human cancer cell lines expressing mutant H-Ras proteins: T24 bladder carcinoma cells and the RL95–2 endometrial carcinoma line. Using the CRISPR/Cas9 system to individually deplete each of the Raf kinases or H-Ras, loss of C-Raf was found to reduce the 2D proliferative and 3D spheroid growth of T24 and RL95–2 cells to a similar extent as did H-Ras depletion, whereas loss of A-Raf or B-Raf had minimal effect (Figure 6D). When a similar analysis was performed on cancer lines expressing mutant K-Ras proteins, H358 lung carcinoma cells and the SW480 colorectal line, individually depleting each Raf member was found to have little effect on 2D proliferation. Spheroid growth could be reduced by depletion of either B-Raf or C-Raf; however, the effect was not as great as that observed for K-Ras depletion (Figure S6C). Taken together, the above depletion experiments demonstrate that C-Raf is critical for H-Ras-mediated transformation.
Finally, the cancer cell lines were utilized to further validate the effects of Raf inhibitor treatment on Ras/Raf binding. For these studies, previously characterized Ras antibodies (Waters et al., 2017) were used to selectively immunoprecipitate the endogenous mutant Ras proteins from cells that had been depleted or not of C-Raf (Figures 6E and S6D and S6E). In the mutant K-Ras lines, H358 and SW480, co-immunoprecipitation of B-Raf and mutant K-Ras was observed in the presence or absence of Raf inhibitor treatment, and depletion of C-Raf had no significant effect on the B-Raf/K-Ras interaction. However, for the mutant H-Ras lines, T24 and RL95–2, B-Raf was only detected in H-Ras immunoprecipitates from cells that had been treated with Raf inhibitor, and this interaction was reduced to background levels by C-Raf depletion (Figure 6E). These findings further support the model that B-Raf/C-Raf dimerization can allow mutant H-Ras to engage B-Raf with increased affinity and may provide an explanation for why melanoma patients treated with Raf inhibitors often developed secondary cancers driven by activating H-Ras mutations.
DISCUSSION
The Raf kinases are essential effectors of Ras signaling, and, although it has been over 20 years since they were first shown to possess a Ras-binding domain, whether these kinases differ in their ability to interact with an individual Ras family member in live cells has been unclear. In this study, we have utilized BRET technologies to further investigate the interactions of the Raf kinases with Ras members. In contrast to in vitro Ras/Raf binding studies, the BRET system allows for this important interaction to be monitored in the context of the plasma membrane and under conditions where post-translational modifications and lipid processing still occur, events that can strongly influence protein binding as well as signal progression. Despite the highly conserved nature of the Ras effector domains and the Raf RBDs, our findings reveal pronounced binding preferences between the Ras and Raf family members.
For all Ras proteins, C-Raf was found to exhibit the highest level and affinity of binding, followed by A-Raf, and then B-Raf, which surprisingly demonstrated a strong selectivity for K-Ras. These findings were further supported in co-immunoprecipitation studies, where the ability of the Ras/Raf interaction to withstand detergent cell lysis and immunopurification was found to correlate with lower BRET50 values, which are indicative of higher binding affinities. The preferential binding of B-Raf to activated K-Ras was also observed in live-cell imaging experiments as well as in co-immunoprecipitation assays examining the ability of endogenous B-Raf to bind Ras members in cells overexpressing Venus-tagged Ras proteins, in Ras-deficient MEFs reconstituted to express untagged mutant H-Ras or K-Ras proteins at endogenous levels, and in human cancer cell lines harboring H-Ras or K-Ras mutant alleles.
Through the generation of various chimeric Ras and Raf proteins, we found that the B-Raf N′-segment and polybasic residues (PBR) in the K-Ras HVR account for the K-Ras binding selectivity of B-Raf. With regard to Ras members that lack the PBR, the B-Raf N′-segment, which carries an acidic charge and is 100–150 amino acids larger than the N′-segment of C-Raf or A-Raf, appears to act in an inhibitory manner as removal of the N′-segment allowed B-Raf to bind all Ras members with high affinity. It should be noted that our findings differ from a previous study where B-Raf was reported to bind with high affinity to farnesylated, GTP-bound H-Ras in surface plasmon resonance (SPR) assays (Fischer et al., 2007). However, in the SPR studies, B-Raf was coupled to the biosensor chip via a GST tag that was fused to the N′-segment, likely causing conformation changes or steric constraints that may have abrogated the inhibitory effect of the B-Raf N′-segment. In addition, the absence of crucial cellular components, including 14–3-3 dimers that stabilize the Raf autoinhibited state, and the lack of an authentic membrane environment, features which are preserved in the BRET system, may also contribute to the observed differences.
Nevertheless, through BRET, co-immunoprecipitation, and live-cell imaging experiments, all of our results indicate that the B-Raf N′-segment results in reduced binding to Ras proteins that lack the PBR. For these Ras members (H-Ras, N-Ras, and K-Ras4A), it is possible that the B-Raf N′-segment, with its increased size and acidic charge, might occlude the RBD or act to repel B-Raf from the negatively charged plasma membrane such that contact with the RBD cannot be established. However, for K-Ras, our findings suggest that basic residues in the PBR may engage acidic residues in the B-Raf N′-segment to disrupt its inhibitory effect and facilitate high-affinity RBD binding (model depicted in Figure 7). Support for this model comes from the observations that reducing the basic charge of the PBR as well as reversing the charge of an acidic residue in the B-Raf N′-segment could reduce the affinity of the B-Raf/K-Ras interaction. Although further studies are needed to fully define the points of contact between B-Raf and K-Ras, these findings indicate the existence of other interactions, in addition to RBD binding, that uniquely contribute to the B-Raf/K-Ras interaction.
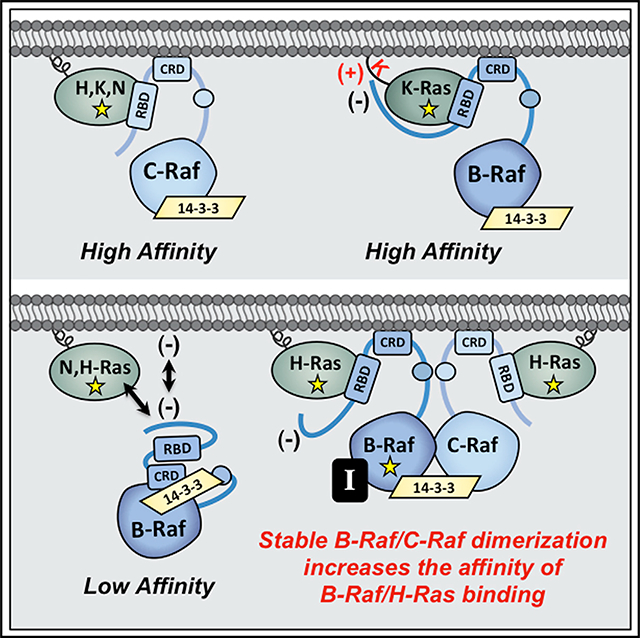
Figure 7. Model for Ras/Raf Binding Preferences.

The C-Raf kinase exhibits high-affinity binding to all Ras family members. In contrast, B-Raf, whose N-terminal segment is larger and possesses an overall acidic charge, only binds with high affinity to mutant K-Ras, whose HVR contains a series of polybasic lysine residues (upper). In the context of H-Ras or N-Ras, the B-Raf N′-segment might occlude the RBD or act to repel B-Raf from the negatively charged plasma membrane. However, events that promote stable B-Raf/C-Raf dimer formation, such as B-Raf mutations (depicted as yellow star) or treatment with B-Raf inhibitors (black box containing the letter I), allow mutant H-Ras to engage B-Raf with increased affinity to upregulate ERK cascade signaling (lower).
The distinct binding properties of the various Ras and Raf proteins also suggest that certain Raf kinases may play a more important role in cancers driven by a specific Ras family member. For example, our results implicate C-Raf as being required for H-Ras-driven transformation in that depletion of C-Raf, but not B-Raf or A-Raf, could suppress cell proliferation and the spheroid growth of two human cancer cell lines expressing mutant H-Ras alleles, T24 and RL95–2. Moreover, in NIH 3T3 focus-forming assays, C-Raf depletion severely reduced the transformation potential of H-RasG12V, whereas it had only a modest effect on K-RasG12V-mediated transformation. Notably, C-Raf was also found to impact the H-Ras/B-Raf interaction as B-Raf mutations or drug treatments stabilizing B-Raf/C-Raf dimerization significantly increased the affinity of B-Raf/H-Ras binding in a manner that required C-Raf (Figure 7). It is unclear whether dimerization with C-Raf alters the conformation of the B-Raf N-terminal domain or facilitates B-Raf localization at the membrane such that binding of H-Ras to the B-Raf RBD can occur. Nevertheless, augmented dimer formation with C-Raf appears to promote direct contact between B-Raf and mutant H-Ras, as no increase in H-Ras binding was observed if B-Raf contained the RBD R > L mutation.
Finally, our results indicate that the ability of C-Raf to facilitate the binding of B-Raf to non-PBR-containing Ras proteins may have important biological consequences. In particular, these findings likely explain why melanoma patients treated with the B-Raf inhibitors vemurafenib and dabrafenib often developed secondary cancers driven by H-Ras mutations (Boussemart et al., 2016; Oberholzer et al., 2012; Su et al., 2012). In this case, inhibitor-stabilized B-Raf/C-Raf dimerization would allow mutant H-Ras to engage B-Raf with increased affinity, thus upregulating ERK cascade signaling to levels that promote tumorigenesis. Likewise, B-Raf mutations that increase B-Raf/C-Raf dimerization and co-occur with oncogenic mutations in non-PBR-containing Ras members may be functionally relevant, acting to augment the signaling potential of these Ras mutants in human cancer. In conclusion, our study highlights the importance of elucidating the distinct roles of individual Ras and Raf family members in cell signaling and tumorigenesis and may aid in the design of new therapeutic strategies.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Deborah Morrison (morrisod@mail.nih.gov). Plasmids and cell lines are available for use upon request to the Lead Contact.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines and Culture Conditions
293FT, NIH 3T3, Phoenix-Eco, RL95–2, and RAS-deficient MEFs were cultured in DMEM. H358 cells were cultured in RPMI, T24 cells in McCoy’s 5a, and SW480 in L-15. All media was supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, and 1% penicillin/streptomycin. MCF10A cells were cultured in DMEM/F12 supplemented with 5% horse serum, 0.5 μg/mL hydrocortisone, 20 ng/mL EGF, 100 ng/mL cholera toxin, 10 μg/mL insulin, and 1% penicillin/streptomycin. All cell lines were cultured at 37°C under 5% CO2 except for SW480 cells, which were cultured at 37°C under atmospheric conditions. Ras-deficient MEFs were sequenced by the provider (NCI-Ras Initiative) to confirm loss of endogenous Ras and integration of the transgene.
METHOD DETAILS
DNA Constructs
The full-length Raf kinases and the Raf regulatory domain proteins were tagged at the C terminus with the Rluc8 enzyme and cloned into the pLHCX-CMV vector. The Raf regulatory domain constructs encode amino acids 1–288 of A-Raf, amino acids 1–435 of B-Raf, and amino acids 1–327 of C-Raf. Chimeric Raf proteins with various regions in the Raf regulatory domain exchanged were constructed using the GeneArt Seamless Cloning and Assembly Kit from Life Technologies. The Raf regions exchanged are based on the following amino acid designations: B-Raf N’-segment: amino acids 1–154, B-Raf RBD/CRD: amino acids 155–280, C-Raf N’-segment: amino acids 1–55, C-Raf RBD/CRD: amino acids 56–184. The Ras family members were tagged at the N terminus with the Venus fluorophore and cloned into the pCMV5 vector. For Ras HVR-exchanged constructs, the HVR of K-Ras4B was defined as amino acids 165–188, and the HVR of H-Ras as amino acids 165–189. Point mutations were generated by site-directed mutagenesis using the QuickChange II Kit from Agilent.
BRET Assay
293FT cells were seeded into 12-well dishes at a concentration of 1×105 cells/well. 16 h after plating, Venus-tagged and Rluc8- tagged constructs were transfected into cells using a calcium phosphate protocol. A 12-point saturation curve was generated in which the concentration of the energy donor construct (Rluc8) was held constant (62.5 ng) as the concentration of the energy acceptor plasmid (Venus) increased (0–1.0 μg). Live cells were collected 48 h after transfection, washed, and plated in PBS. The Rluc8 cofactor coelenterazine-h was added to a final concentration of 3.375 μM, and the BRET signal read 2 min after addition. The BRET signal was measured at 535 nm (bandwidth 30 nm) on the PHERAstar Plus plate reader (BMG Labtech) and the Rluc8 signal was simultaneously measured at 475 nm (bandwidth 30 nm). Venus fluorescence was measured independently using an excitation wavelength of 485 nm (5 nm bandwidth), and the emission spectra measured at 530 nm (5 nm bandwidth) on the Tecan Infinite M1000 plate reader. The BRET value for each data point was calculated by dividing the BRET ratio (BRET/Rluc8) by the background signal. The acceptor/donor ratio was equalized against a control where equal quantities of Venus and Rluc8 constructs were transfected. Data was analyzed using GraphPad Prism. Non-linear regression was used to plot the best fit hyperbolic curve and values for BRETmax and BRET50 were obtained from the calculated best fit curves.
Transfection, Lysis, and Co-immunoprecipitation
The indicated cell lines were plated at ~70% confluency 18–24 h prior to transfection. Cells were then transfected using the XtremeGENE9 transfection reagent per the manufacturer’s instructions, using a 2:1 ratio of XtremeGENE9 to DNA. For cell lysis, cells were washed twice with ice cold PBS and lysed for 15 min at 4°C in 1% NP-40 buffer (20mM Tris [pH 8.0], 137 mM NaCl, 10% glycerol, 1% NP-40 alternative, 0.15 U/mL aprotinin, 1 mM phenylmethylsulfonyl fluoride, 0.5 mM sodium vanadate, 20 μM leupeptin). Lysates were clarified by centrifugation at 14,000 rpm for 10 min at 4°C, following which the protein content was determined by Bradford assays. Lysates containing equivalent amounts of protein were incubated with the appropriate antibody and protein G Sepharose beads for 2 h at 4°C on a rocking platform. Complexes were washed extensively with 1% NP-40 buffer and then examined by immunoblot analysis along with aliquots of equalized lysate.
Live-cell Imaging
293FT or MCF10A cells expressing the indicated Halo- and mCherry-tagged proteins were plated onto collagen-coated glass surfaces (10 μg/mL human placenta type IV collagen). On the day of live cell imaging experiments, cells were washed with media lacking phenol red and incubated with the Halo Oregon green ligand for 15–30 min at 37°C. Cells were then washed in phenol red-free media and maintained in growth media lacking phenol red for the duration of image acquisition using either Zeiss Axiovert Z1 and LSM710.
Raf-RBD Pull-down Assays
To monitor the GTP-bound state of Ras, equalized cell lysates containing 5 μM MgCl2 were incubated with GST-tagged Raf-RBD bound to glutathione-Sepharose beads (Millipore) for 1 h at 4°C on a rocking platform. Complexes were washed extensively with 1% NP-40 buffer and then examined by immunoblot analysis.
shRNA and CRISPR/Cas9 Vectors
For depletion of C-Raf protein levels in NIH 3T3 cells, pLKO.1 lentiviral vectors expressing shC-Raf (TRCN0000001066) sequences were obtained from Open Biosystems. For CRISPR/Cas9 studies, a non-targeting (NT), single guide RNA (sgRNA) or sgRNAs targeting the A-Raf, B-Raf, C-Raf, H-Ras, or K-Ras gene were each cloned into pLentiCRISPRv2 (Sanjana et al., 2014).
Recombinant Lentiviruses and Cell Infection
For protein depletion experiments, lentiviral particles expressing the desired targeting constructs were generated by co-transfecting the pLKO.1 or pLentiCRISPRv2 constructs with the MISSION lentiviral packaging mix (Sigma) into 293T cells using the Mirus Trans-IT lenti transfection kit. 48 h post-transfection, viral supernatants were collected, centrifuged twice at 1500 rpm for 7 min, and either stored at −80°C or used directly. Cells were infected with viral supernatants containing 8 μg/mL polybrene. 48 h post-infection, cells were placed into selection media containing 6 μg/mL puromycin for 4 days and then shifted into media containing 3 μg/mL puromycin for an additional 6 days, prior to analysis. For protein expression studies, lentiviral particles were generated by co-transfecting the pUBC-Raf-mCherry or pCMV-Halo-RasG12V constructs with packaging plasmids pMD2.G and psPAX2 (3:1:2 ratio) into 293T cells using XtremeGENE9. 48 h post-transfection, viral supernatants were collected, centrifuged twice at 1500 rpm for 7 min, and either stored at −80°C or used directly. MCF10A cells were infected with lentivirus supernatants containing 8 μg/mL polybrene for 24 h, following which growth media supplemented with the appropriate antibiotic selection was added (Puromycin: 1 μg/mL, Hygromycin: 40 μg/mL).
NIH 3T3 Focus Formation Assay
Recombinant retroviruses expressing Halo-H-RasG12V or Halo-K-RasG12V constructs were generated by transfecting the pBabe-Halo-Ras constructs into Phoenix-Eco cells using the X-tremeGENE9 protocol described above. Viral supernatants were collected 3 days post-transfection, centrifuged twice at 1500 rpm for 7 min, and either stored at −80°C or used directly. Control (shNeg) or C-Raf-depleted (shC-Raf) NIH 3T3 cells were plated into 60 mm dishes at a concentration of 2 × 105/dish. After 18 h, cells were infected with the indicated recombinant retrovirus in media containing 4% FBS and 8 μg/mL polybrene for 24 h. Cells were trypsinized and plated into two 100 mm dishes, one of which contained 5 μg/mL puromycin. After two weeks of culture, cells were fixed with 3.7% formaldehyde and stained with 1% methylene blue.
Cell Proliferation Assay
Cell proliferation was assessed using the CellTiter-Glo 2.0 Reagent (Promega) with luminescence determined using a GloMax Discover Plate Reader (Promega). 1 × 103 cells were seeded into white-walled cell culture-coated 96-well plates (Promega). Cell number was assessed 24 h after plating (day 1) and then every 48 h for 7 days. Data were analyzed as an increase in luminescence over day 1.
Transformation and Spheroid Growth Assays
Transformation of T24 cells was assessed by CSC/spheroid frequency as previously described (Inouye et al., 2000). Briefly, serially diluted T24 cells were seeded into ultra-low attachment 96-well, flat-bottomed plates (1 cell /well – 1000 cells/well, Corning Corstar #3474), with 24 wells per condition. Cells were cultured for 7–10 days, and wells with spheroids > 100 μm were scored as spheroid positive. CSC/CIC frequency was calculated by ELDA website (http://bioinf.wehi.edu.au/software/elda/) (Hu and Smyth, 2009). Spheroid growth of R95–2, H358 and SW480 cells were conducted as described in (Sheffels et al., 2019). RL95–2, H358 or SW480 cells were seeded at 500–1000 cells/well in ultra-low attachment 96-well round bottomed plates (Corning Costar #7007). Cell number was assessed 18 h after plating to allow spheroids to form (day 0), and then at day 7 using CellTiter-Glo 2.0 reagent (Promega), which measures ATP content as a surrogate of overall cell number. Spheroid growth for each cell line was normalized to the CellTiter Glo signal at day 0, and the results are expressed as a fold-increase over day 0.
QUANTIFICATION AND STATISTICAL ANALYSIS
BRET data were transferred to GraphPad Prism for statistical analysis and curve fitting. Data was plotted as the acceptor to donor ratios versus mBRET values. Non-linear regression was used to fit a hyperbolic curve to the dataset and determine R-squared values. The BRETmax and BRET50 values as well as the corresponding 95% confidence intervals were calculated using GraphPad Prism8 software. For cell proliferation and spheroid growth assays, replicate wells (n = 6 per experiment) were seeded into 96-well plates and cell number was quantified at the indicated times. Data represent 3 independent experiments and are presented as mean ± SD. Significance was determined by ANOVA using the Tukey’s multiple comparisons test and calculatedusing GraphPad Prism8 software. The co-immunoprecipitation, live cell imaging, and BRET experiments shown are representative and reflect at least 3 independent experiments.
DATA CODE AND AVAILABILITY
The datasets analyzed during this study are available at cBioPortal [http://www.cbioportal.org] and COSMIC [https://cancer.sanger.ac.uk/cosmic]. Raw data of immunoblots and live cell imaging are available through Mendeley Data (https://doi.org/10.17632/r6vvxjpskf.1). This study did not generate any code.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| B-Raf (H-145) rabbit polyclonal | Santa Cruz Biotechnology | cat# sc-9002; RRID:AB_2067494 |
| B-Raf (F-7) mouse monoclonal | Santa Cruz Biotechnology | cat# sc-5284; RRID:AB_2721130 |
| C-Raf (C-12) rabbit polyclonal | Santa Cruz Biotechnology | cat# sc-133; RRID:AB_632305 |
| C-Raf mouse monoclonal | BD Pharmagen | cat# 610152; RRID:AB_397553 |
| A-Raf (C-20) rabbit polyclonal | Santa Cruz Biotechnology | cat# sc-408; RRID:AB_630882 |
| H-Ras (C-20) rabbit polyclonal | Santa Cruz Biotechnology | cat# sc-520; RRID:AB_631670 |
| N-Ras (F155) mouse monoclonal | Santa Cruz Biotechnology | cat# sc-31; RRID:AB_628041 |
| K-Ras mouse monoclonal | Sigma | cat# WH0003845M1; RRID:AB_1842235 |
| pS217/221-MEK rabbit polyclonal | Cell Signaling Technology | cat# 9121; RRID:AB_331648 |
| Rluc rabbit polyclonal | MBL International | cat# PM047; RRID:AB_1520866 |
| GFP mouse monoclonal | Roche | cat# 11814460001; RRID:AB_390913 |
| GFP rat monoclonal | MBL International | cat# D153–3; RRID:AB_591817 |
| Pan-Ras [EPR3255] rabbit monoclonal | Abcam | cat# 108602; RRID:AB_10891004 |
| Pan-Ras [Ras10] mouse monoclonal | EMD Millipore | cat# 05–516; RRID:AB_11211664 |
| Actin (I-19) goat polyclonal | Santa Cruz Biotechnology | cat# sc-1616; RRID:AB_630836 |
| Donkey anti-goat IgG-HRP | Santa Cruz Biotechnology | cat# sc-2020; RRID:AB_631728 |
| Donkey anti-rabbit IgG-HRP | GE Healthcare | cat# NA934; RRID:AB_772206 |
| Sheep anti-mouse IgG-HRP | GE Healthcare | cat# NA931; RRID:AB_772210 |
| Goat anti-rat IgG-HRP | Cell Signaling Technology | cat# 7077; RRID:AB_10694715 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Vemurafenib (PLX4032) | SelleckChem | Cat# S1267 |
| Dabrafenib (GSK2118436) | SelleckChem | Cat# S2807 |
| LY3009120 | SelleckChem | Cat# S7842 |
| PLX7904 | SelleckChem | Cat# S7964 |
| SB-590885 | SelleckChem | Cat# S2220 |
| Coelenterazine-h | Promega | Cat# S2011 |
| Halo Oregon Green ligand | Promega | Cat# G2802 |
| Epidermal Growth Factor (EGF) | ThermoFisher | cat# PHG0311 |
| GST-RBD | Millipore | cat# 14–863 |
| X-tremeGENE 9 | Roche/Sigma | cat# 06365809001 |
| Collagen, Human Placenta Type IV | Millipore/Sigma | cat# C7521 |
| Critical Commercial Assays | ||
| CellTiter-Glo 2.0 | Promega | cat# G9241 |
| TransIT®-Lenti Transfection Reagent | Mirus | cat# MIR 6603 |
| Mission Lentiviral Packaging Mix | Sigma | cat# SHP001 |
| GeneArt Seamless Cloning and Assembly Kit | Life Technologies | cat# A13288 |
| QuickChange II Kit | Agilent | cat# 200523 |
| Deposited Data | ||
| Raw data of immunoblots and live cell imaging | Mendeley Data | https://doi.org/10.17632/r6vvxjpskf.1 |
| Experimental Models: Cell Lines | ||
| 293FT (human) | Invitrogen | cat# R70007 |
| Phoenix-Eco (human) | ATCC | cat# CRL-3214; RRID:CVCL_H717 |
| NIH 3T3 (mouse) | ATCC | cat# CRL-1658; RRID:CVCL_0594 |
| HeLa (human, female) | ATCC | cat# CCL-2; RRID:CVCL_0030 |
| SW480 (human, male) | ATCC | cat# CCL-228; RRID:CVCL_0546 |
| RL95–2 (human, female) | ATCC | cat# CRL-1671; RRID:CVCL_0505 |
| H358 (human, male) | ATCC | cat# CRL-5807; RRID:CVCL_1559 |
| T24 (human, female) | ATCC | cat# HTB-4; RRID:CVCL_0554 |
| MCF10A (human, female) | ATCC | cat# CRL-10317; RRID:CVCL_0598 |
| Ras−/− MEF + KRas Q61L (mouse) | NCI-Ras Initiative | N/A |
| Ras−/− MEF + HRas Q61L (mouse) | NCI-Ras Initiative | N/A |
| Oligonucleotides | ||
| CRISPR A-Raf sgRNA (TGGTCTACCGACTCATCAAG) | This paper | N/A |
| CRISPR B-Raf sgRNA (GGGCCAGGCTCTGTTCAACG) | This paper | N/A |
| CRISPR C-Raf sgRNA (GCCGAACAAGCAAAGAACAG) | This paper | N/A |
| CRISPR H-Ras sgRNA (ACGGAATATAAGCTGGTGG) | Sheffels et al., 2019 | N/A |
| CRISPR K-Ras sgRNA (TCATTGCACTGTACTCCTCT) | Sheffels et al., 2019 | N/A |
| CRISPR NT sgRNA (CCATATCGGGGCGAGACATG) | Sheffels et al., 2019 | N/A |
| shCRaf (CGGAGATGTTGCAGTAAAGAT) | Open Biosystems | TRCN0000001066 |
| Recombinant DNA | ||
| pLHCX-WT-RafReg-Rluc8 (A-, B-, C-Raf) | This paper | N/A |
| pLHCX-R > L-RafReg-Rluc8 (A-, B-, C-Raf) | This paper | N/A |
| pLHCX-RafFL-Rluc8 (A-, B-, C-Raf) | This paper | N/A |
| pLHCX-C-RafReg/B-Raf N’-segment-Rluc8 | This paper | N/A |
| pLHCX-C-RafReg/B-Raf RBD/CRD-Rluc8 | This paper | N/A |
| pLHCX-B-RafReg/C-Raf N’-segment-Rluc8 | This paper | N/A |
| pLHCX-B-RafReg/C-Raf RBD/CRD-Rluc8 | This paper | N/A |
| pLHCX-B-RafReg N′segment mutants-Rluc8 | This paper | N/A |
| pLHCX-B-RafFL kinase domain mutants-Rluc8 | This paper | N/A |
| pLHCX-R188L-B-RafFL-Rluc8 | This paper | N/A |
| pUBC-Raf-mCherry (B-, C-Raf) | NCI-Ras Initiative | N/A |
| pCMV5-Venus-RasQ61R (H-, N-, K-Ras4A, 4B) | NCI-Ras Initiative | N/A |
| pCMV5-Venus-RasG12V(H-, N-, K-Ras4B) | NCI-Ras Initiative | N/A |
| pCMV5-Venus-RasG12D (H-, N-, K-Ras4B) | NCI-Ras Initiative | N/A |
| pCMV5-Venus-RasG13D (H-, N-, K-Ras4B) | NCI-Ras Initiative | N/A |
| pCMV5-Venus-RasQ61L (H-, N-, K-Ras4B) | NCI-Ras Initiative | N/A |
| pCMV5-Venus-WT Ras (H-, N-, K-Ras4B) | NCI-Ras Initiative | N/A |
| pCMV5-Halo-RasG12V (H-, K-Ras4B) | NCI-Ras Initiative | N/A |
| EGFP-K-Ras4BG12V PBR mutants | Zhou et al., 2017 | N/A |
| pCMV-Venus-H-RasQ61R/K-Ras4B HVR | NCI-Ras Initiative | N/A |
| pCMV-Venus-K-RasQ61R/H-Ras HVR | NCI-Ras Initiative | N/A |
| pLKO.1 lentiviral vector | Open Biosystems/Dharmacon | cat# RHS4080 |
| pLKO.1 shCRaf lentiviral construct | Open Biosystems/Dharmacon | cat# RHS3979–201733340; TRCN0000001066 |
| pLentiCRISPRv2 | Addgene | cat# 52961 |
| Software and Algorithms | ||
| GraphPad Prism | GraphPad | https://www.graphpad.com |
| ImageJ | ImageJ | https://imagej.nih.gov/ij/ |
Highlights.
C-Raf binds all Ras proteins equivalently, but B-Raf exhibits selectivity for K-Ras
Raf N-terminal segments and Ras HVR sequences determine binding preferences
C-Raf is critical for downstream transmission of H-Ras-driven signaling
Events that increase B-Raf/C-Raf dimerization augment the B-Raf/H-Ras interaction
ACKNOWLEDGMENTS
We thank Linda Miller (LCDS) and Vanessa Wall (FNLCR Protein Expression Laboratory) for technical support. This project has been funded by federal funds from the National Cancer Institute, United States under project number ZIA BC 010329 (D.K.M.) and contract number HHSN261200800001E (D.E.), by federal funds from the National Institutes of Health, United States under grant number R01 GM124233 (J.F.H.), and from the Congressionally Directed Medical Research Program, United States LC160222 (R.L.K.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.molcel.2019.09.004.
REFERENCES
- Ambrogio C, Köhler J, Zhou ZW, Wang H, Paranal R, Li J, Capelletti M, Caffarra C, Li S, Lv Q, et al. (2018). KRAS Dimerization Impacts MEK Inhibitor Sensitivity and Oncogenic Activity of Mutant KRAS. Cell 772, 857–868.e15. [DOI] [PubMed] [Google Scholar]
- Bos JL, Rehmann H, and Wittinghofer A (2007). GEFs and GAPs: critical elements in the control of small G proteins. Cell 129, 865–877. [DOI] [PubMed] [Google Scholar]
- Boussemart L, Girault I, Malka-Mahieu H, Mateus C, Routier E, Rubington M, Kamsu-Kom N, Thomas M, Tomasic G, Agoussi S, et al. (2016). Secondary Tumors Arising in Patients Undergoing BRAF Inhibitor Therapy Exhibit Increased BRAF-CRAF Heterodimerization. Cancer Res. 76, 1476–1484. [DOI] [PubMed] [Google Scholar]
- Buhrman G, Kumar VS, Cirit M, Haugh JM, and Mattos C (2011). Allosteric modulation of Ras-GTP is linked to signal transduction through RAF kinase. J. Biol. Chem 286, 3323–3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox AD, and Der CJ (2010). Ras history: The saga continues. Small GTPases 1, 2–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty MK, Müller J, Ritt DA, Zhou M, Zhou XZ, Copeland TD, Conrads TP, Veenstra TD, Lu KP, and Morrison DK (2005). Regulation of Raf-1 by direct feedback phosphorylation. Mol. Cell 17, 215–224. [DOI] [PubMed] [Google Scholar]
- Drosten M, Dhawahir A, Sum EY, Urosevic J, Lechuga CG, Esteban LM, Castellano E, Guerra C, Santos E, and Barbacid M (2010). Genetic analysis of Ras signalling pathways in cell proliferation, migration and survival. EMBO J. 29, 1091–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrant DE, and Morrison DK (2018). Targeting the Raf kinases in human cancer: the Raf dimer dilemma. Br. J. Cancer 118, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian JR, Vojtek AB, Cooper JA, and Morrison DK (1994). A single amino acid change in Raf-1 inhibits Ras binding and alters Raf-1 function. Proc. Natl. Acad. Sci. USA 91, 5982–5986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Medarde A, and Santos E (2011). Ras in cancer and developmental diseases. Genes Cancer 2, 344–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A, Hekman M, Kuhlmann J, Rubio I, Wiese S, and Rapp UR (2007). B- and C-RAF display essential differences in their binding to Ras: the isotype-specific N terminus of B-RAF facilitates Ras binding. J. Biol. Chem 282, 26503–26516. [DOI] [PubMed] [Google Scholar]
- Freeman AK, Ritt DA, and Morrison DK (2013). Effects of Raf dimerization and its inhibition on normal and disease-associated Raf signaling. Mol. Cell 49, 751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekman M, Hamm H, Villar AV, Bader B, Kuhlmann J, Nickel J, and Rapp UR (2002). Associations of B- and C-Raf with cholesterol, phosphatidylserine, and lipid second messengers: preferential binding of Raf to artificial lipid rafts. J. Biol. Chem 277, 24090–24102. [DOI] [PubMed] [Google Scholar]
- Hu Y, and Smyth GK (2009). ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J. Immunol. Methods 347, 70–78. [DOI] [PubMed] [Google Scholar]
- Hu J, Stites EC, Yu H, Germino EA, Meharena HS, Stork PJS, Kornev AP, Taylor SS, and Shaw AS (2013). Allosteric activation of functionally asymmetric RAF kinase dimers. Cell 154, 1036–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, and Westover KD (2015). Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol. Cancer Res. 13, 1325–1335. [DOI] [PubMed] [Google Scholar]
- Inouye K, Mizutani S, Koide H, and Kaziro Y (2000). Formation of the Ras dimer is essential for Raf-1 activation. J. Biol. Chem 275, 3737–3740. [DOI] [PubMed] [Google Scholar]
- Lavoie H, and Therrien M (2015). Regulation of RAF protein kinases in ERK signalling. Nat. Rev. Mol. Cell Biol 16, 281–298. [DOI] [PubMed] [Google Scholar]
- Nan X, Collisson EA, Lewis S, Huang J, Tamgüney TM, Liphardt JT, McCormick F, Gray JW, and Chu S (2013). Single-molecule superresolution imaging allows quantitative analysis of RAF multimer formation and signaling. Proc. Natl. Acad. Sci. USA 110, 18519–18524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberholzer PA, Kee D, Dziunycz P, Sucker A, Kamsukom N, Jones R, Roden C, Chalk CJ, Ardlie K, Palescandolo E, et al. (2012). RAS mutations are associated with the development of cutaneous squamous cell tumors in patients treated with RAF inhibitors. J. Clin. Oncol 30, 316–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker JA, and Mattos C (2015). The Ras-Membrane Interface: Isoform-specific Differences in The Catalytic Domain. Mol. Cancer Res 13, 595–603. [DOI] [PubMed] [Google Scholar]
- Pfleger KD, and Eidne KA (2006). Illuminating insights into protein-protein interactions using bioluminescence resonance energy transfer (BRET). Nat. Methods 3, 165–174. [DOI] [PubMed] [Google Scholar]
- Prior IA, and Hancock JF (2012). Ras trafficking, localization and compartmentalized signalling. Semin. Cell Dev. Biol 23, 145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior IA, Lewis PD, and Mattos C (2012). A comprehensive survey of Ras mutations in cancer. Cancer Res. 72, 2457–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pylayeva-Gupta Y, Grabocka E, and Bar-Sagi D (2011). RAS oncogenes: weaving a tumorigenic web. Nat. Rev. Cancer 11, 761–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritt DA, Monson DM, Specht SI, and Morrison DK (2010). Impact of feedback phosphorylation and Raf heterodimerization on normal and mutant B-Raf signaling. Mol. Cell. Biol 30, 806–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubbert S, Shannon K, and Bollag G (2007). Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer 7, 295–308. [DOI] [PubMed] [Google Scholar]
- Sheffels E, Sealover NE, Theard PL, and Kortum RL (2019). Anchorage-independent growth conditions reveal a differential SOS2 dependence for transformation and survival in RAS-mutant cancer cells. Small GTPases, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simanshu DK, Nissley DV, and McCormick F (2017). RAS Proteins and Their Regulators in Human Disease. Cell 170, 17–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su F, Viros A, Milagre C, Trunzer K, Bollag G, Spleiss O, Reis-Filho JS, Kong X, Koya RC, Flaherty KT, et al. (2012). RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N. Engl. J. Med 366, 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters AM, Ozkan-Dagliyan I, Vaseva AV, Fer N, Strathern LA, Hobbs GA, Tessier-Cloutier B, Gillette WK, Bagni R, Whiteley GR, et al. (2017). Evaluation of the selectivity and sensitivity of isoform- and mutation-specific RAS antibodies. Sci. Signal 10, eaao3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JG, Drugan JK, Yi GS, Clark GJ, Der CJ, and Campbell SL (2000). Elucidation of binding determinants and functional consequences of Ras/Raf-cysteine-rich domain interactions. J. Biol. Chem 275, 22172–22179. [DOI] [PubMed] [Google Scholar]
- Yao Z, Yaeger R, Rodrik-Outmezguine VS, Tao A, Torres NM, Chang MT, Drosten M, Zhao H, Cecchi F, Hembrough T, et al. (2017). Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 548, 234–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Spevak W, Zhang Y, Burton EA, Ma Y, Habets G, Zhang J, Lin J, Ewing T, Matusow B, et al. (2015). RAF inhibitors that evade paradoxical MAPK pathway activation. Nature 526, 583–586. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Prakash P, Liang H, Cho KJ, Gorfe AA, and Hancock JF (2017). Lipid-Sorting Specificity Encoded in K-Ras Membrane Anchor Regulates Signal Output. Cell 168, 239–251.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.