

Abstract
Yak (Bos grunniens) is an important and dominant livestock species in the challenging environment of the Qinghai–Tibetan Plateau. Rumen microbiota of the solid, liquid, and epithelium fractions play key roles in nutrient metabolism and contribute to host adaptation in ruminants. However, there is a little knowledge of the microbiota in these rumen fractions of yak. Therefore, we collected samples of solid, liquid, dorsal, and ventral epithelium fractions from five female yaks, then amplified bacterial 16S rRNA gene V4 regions and sequenced them using an Illumina MiSeq platform. Principal coordinates analysis detected significant differences in bacterial communities between the liquid, solid, and epithelium fractions, and between dorsal and ventral epithelium fractions. Rikenellaceae RC9, the families Lachnospiraceae and Ruminococcaceae, and Fibrobacter spp. were the abundant and enriched bacteria in solid fraction, while the genera Prevotella and Prevotellaceae UCG 003 were higher in the liquid fraction. Campylobacter spp., Comamonas spp., Desulfovibrio spp., and Solobacterium spp. were significantly higher in dorsal epithelium, while Howardella spp., Prevotellaceae UCG 001, Ruminococcaceae UCG 005, and Treponema 2 were enriched in the ventral epithelium. Comparison of predictive functional profiles among the solid, liquid, and dorsal, and ventral epithelium fractions also revealed significant differences. Microbiota in the ventral fraction of yak rumen also significantly differ from reported microbiota of cattle. In conclusion, our results improve our knowledge of the taxonomic composition and roles of yak rumen microbiota.
Keywords: Bos grunniens, Campylobacter spp., dorsal and ventral epithelium, ecology niches, Howardella spp, rumen, solid and liquid
In this present study, we explored microbiota in the solid, liquid, and epithelium (dorsal and ventral) fractions of yak rumen, and compared functional profiles of microbiota of the four fractions, and also compared microbiota of the ventral epithelium between yak and cattle. Our results indicated the significant differences in bacterial communities between the liquid, solid, and epithelium fractions, and between dorsal and ventral epithelium fractions. Moreover, comparison of functional profiles among the solid, liquid, and dorsal and ventral epithelium fractions also revealed significant differences. Finally, we found that the microbial composition and functional profile in the ventral epithelium between yak and cattle significantly differed, suggesting the specific role of microbiota in the ventral epithelium of yak rumen.
![]()
1. INTRODUCTION
The yak (Bos grunniens) is a remarkable domesticated ruminant species of the Qinghai–Tibetan Plateau (QTP), which provides basic necessities for Tibetans including meat, milk, and transportation (Wiener, Han, & Long, 2003). However, yak is seriously challenged by the harsh environmental conditions, such as high altitude (>3,000 m), low temperatures, and oxygen tension (Zhou, Zhong, et al., 2017a; Zhou, Fang, et al., 2017b), and limitations in both quality and availability of food associated with the harsh conditions and short growing season (mid‐May to mid‐September; Wang et al., 2011). Hence, yaks have evolved unique genomic features and associated traits that enable them to survive in this environment. For instance, significant enrichment or expansion of genes involved in sensory perception, hypoxia tolerance, and nutrient metabolism has been found in the yak genome, relative to the genome of closely related low‐altitude cattle (Qiu et al., 2012). Previous studies have also shown that yaks have lower daily fasting nitrogen excretion rates (Wang et al., 2011) and higher nitrogen retention than cattle (Wang et al., 2011), suggesting that they have higher dietary efficiency. Moreover, recent studies have demonstrated that methanogen communities in yak and cattle rumen significantly differ (Huang, Tan, Long, Liang, & Wright, 2012), and the yak rumen microbiome is significantly enriched with genes linked to pathways yielding volatile fatty acids (VFAs), and genes associated with VFA transport and absorption in the ruminal epithelium are significantly upregulated (Zhang et al., 2016). Together, these results indicate that the rumen microbiota are highly important in yak nutrient metabolism and adaptation.
The complex rumen microbial ecosystem can be divided into three: solid, liquid, and epithelial fractions (Cho et al., 2006; De Mulder et al., 2017; Liu, Zhang, Zhang, Zhu, & Mao, 2016; Sadet, Martin, Meunier, & Morgavi, 2007; Schären et al., 2017). The solid microbiota attached to ingested plant material play a key role in fiber digestion (McAllister, Bae, Jones, & Cheng, 1994), while the liquid phase contains bacteria that strongly participate in metabolism of soluble nutrients, and transmits components of the solid‐adherent biofilms to newly ingested feed (De Mulder et al., 2017). There are also significant differences between the microbial communities in the solid and liquid fractions (De Mulder et al., 2017; Larue, Yu, Parisi, Egan, & Morrison, 2005; McAllister et al., 1994). Microbiota of the epithelial fraction also have distinct functions, particularly oxygen scavenging (Cheng, Mccowan, & Costerton, 1979), urea hydrolysis (Cheng & Wallace, 1979), and recycling of epithelial tissue (Dinsdale, Cheng, Wallace, & Goodlad, 1980). These previous findings clearly suggest that fractions of rumen microbiota may generally have distinct compositional and functional signatures. However, although the microbiota in the yak rumen and other stomach components have been examined (Guo et al., 2015; Huang et al., 2012; Xue et al., 2016, 2018; Zhou, Zhong, et al., 2017a; Zhou, Fang, et al., 2017b), we have a little knowledge of the composition of microbiota of the three fractions in the yak rumen and differences between them.
The rumen epithelium can also be divided into dorsal and ventral fractions. Previous studies demonstrated that the dorsal epithelium faction faces an accumulation of gas dome, while the ventral epithelium fraction meets to a relatively fluid digest in the rumen (Tafaj et al., 2004). The degree of papillation also significantly differs between dorsal and ventral regions (Clauss, Hofmann, Fickel, Streich, & Hummel, 2009). Moreover, yak produces significantly lower levels of methane and higher levels of VFAs than cattle, and rumen microbiomes of yak and cattle differ (Zhang et al., 2016). Therefore, comparing the ventral epithelium microbiota in yak and cattle could enhance understanding of roles of microbiota in adaptations of yak and other ruminants.
Thus, aims of this study were to characterize and compare microbiota in the solid, liquid, and epithelium (dorsal and ventral) fractions of yak rumen; to compare functional profiles of microbiota of the four fractions; and to compare microbiota of the ventral epithelium in yak and cattle.
2. MATERIAL AND METHODS
2.1. Animals and sampling
In this study, five female 4‐year‐old yaks in Hezuo city, Gansu province, China (latitude >3,000 m) were used. The yaks were raised by the local herdsman, who traditionally grazes yaks on natural alpine meadow grassland (mainly consisted of Kobresia spp. and Cyperaceae spp.) without feed supplements, from September to November 2018. On the morning, the yaks were fed the collected pasture from the same grassland, which were then slaughtered after 3 hours of feeding. To minimize potential contamination from other gut regions, each yak carcass was placed in a natural position and the rumen chambers were tied off using cotton rope. Then, each animal's rumen content was carefully collected and homogenized. Approximately 200 g portions of whole contents were taken by hands with sterile gloves and squeezed through four layers of cheesecloth to separate them into solid and liquid fractions. Finally, the solid and liquid samples obtained were stored in DNase‐ and RNase‐free tubes. In addition, dorsal and ventral epithelium samples were collected by cutting ca. 3 cm2 pieces of epithelial tissue from dorsal and ventral rumen, then washing them with cold 0.01 M phosphate‐buffered saline three times. After that, rumen epithelium‐associated microbiota samples were scraped off using sterilized glass slides. All samples were immediately placed in liquid nitrogen and stored at −80°C for further analysis.
2.2. DNA extraction, library construction, and next‐generation sequencing
The microbial genomic DNA in each rumen sample was extracted following published methods (Yu & Morrison, 2004). Its integrity and quantity was evaluated by 1.0% agarose gel electrophoresis and spectroscopic analysis with a ND‐1000 spectrometer (NanoDrop). The V4 region of bacterial 16S rRNA genes in each sample was amplified in triplicate (Klindworth et al., 2012). Amplicons were purified using an AxyPrep DNA Gel Extraction Kit (Axygen Biosciences) according to the manufacturer's instructions. Purified PCR products were quantified using a Qubit®3.0 fluorometer (Invitrogen). A MiSeq Reagent Kit v2 was then used to construct an Illumina Pair‐End library according to the manufacturer's instructions. Finally, the obtained amplicons were sequenced using an Illumina MiSeq platform to generate paired 250‐bp reads.
2.3. Bioinformatics and statistical analysis
To compare the epithelial microbiota in yak and cattle, sequences from cattle obtained in a previous study, using the same primers (De Mulder et al., 2017), were downloaded. The paired end sequences were first assembled into contigs using FLASH (Magoč & Salzberg, 2011) with truncation of reads at any site receiving an average quality score <20 over a 50‐bp sliding window, removal of reads shorter than 50 bp after truncation, no mismatches in primers, and merger of sequences with >10 bp overlaps. Then, all sequences were analyzed using QIIME 1.9.1 (Caporaso et al., 2010). The operational taxonomy units (OTUs) were clustered using UPARSE based on 97% sequence similarity (Edgar, 2013). Chimeric sequences were identified and removed using UCHIME (Edgar, Haas, Clemente, Quince, & Knight, 2011). Representative sequences of the OTUs were used for taxonomic classification in conjunction with the SILVA database (version 123; Quast et al., 2013; Wang, Garrity, Tiedje, & Cole, 2007). Representative sequences in each OTU were also aligned using MUSCLE (Edgar, 2004), then used to construct a phylogenetic tree with FastTree (Price, Dehal, & Arkin, 2009). Singletons were removed, and the sequences from each sample were subsampled to the minimum number (36,574) in order to decrease the effects of sequencing depth. The alpha diversity indices including Chao1 and Shannon were also calculated using QIIME 1.9.1 (Caporaso et al., 2010).
Principal coordinate analysis (PCoA) was applied to compare the bacterial communities in the solid, liquid, dorsal, and ventral fractions based on Bray–Curtis, unweighted UniFrac and weighted UniFrac distances. Analysis of similarities (ANOSIM) was applied to test whether the microbial communities among these fractions are significantly different, and Adonis was employed to describe the strength and significance between them. In addition, the linear discriminant analysis (LDA) effect size (LEfSe) was used to identify the enriched bacterial taxonomy from each fraction (Segata et al., 2011). LEfSe first applies the nonparametric factorial Kruskal–Wallis rank‐sum test to detect significantly differing features (here, taxa), and then LDA to estimate the effect size of each feature. A significance level of p < .05 and effect‐size threshold of 3 were applied in this study to identify significant taxa. Finally, phylogenetic investigation of communities by reconstruction of unobserved states (PICRUSt) was used to predict functional profiles of the four fractions resulting from reference‐based OTU picking against the Greengenes database (Langille et al., 2013). The predicted genes were then clustered and categorized according to Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways.
Differences in alpha diversity indices and functional category abundances among the four fractions were analyzed using one way ANOVA with the post hoc Tukey‐HSD test. All p‐values were corrected to account for the false discovery rate (FDR) by the Benjamini–Hochberg method, and FDR‐corrected p‐values below .05 were considered as significance. All values reported here are expressed as mean ± standard deviation (SD) unless otherwise stated.
2.4. Quantitative real‐time PCR for bacterial community
The bacterial 16S rRNA gene copy number was determined by quantitative PCR on a ABI 7300 real‐time PCR System (Life Technologies) suing SYBR Premix Ex Taq dye (TaKaRa Biotechnology). The primers reported in previous study were used for the quantitative PCR (Maeda et al., 2003). Each 20 µl reaction mixture contained 10 µl SYBR Premix Ex Taq™ (TaKaRa Biotechnology), 0.4 µl of each primer (10 µM), 0.4 µl ROX Reference Dye (TaKaRa Biotechnology), 6.8 µl of nuclease‐free water, and 2 μl of the template. The PCR was performed in a two‐step thermal cycling process that consisted of hot start activation at 95°C for 30 s, followed by 40 cycles at 95°C for 5 s, and 60°C for 1 min. The quantification of bacterial 16S rRNA gene copies in each sample was performed in triplicate, and the mean value was calculated. A standard curve was prepared by using a 10‐fold serial dilution of purified plasmid DNA containing the 16S rRNA gene sequence. The total number of gene copies was expressed as log10 numbers of marker loci gene copies per ng DNA.
3. RESULTS
3.1. 16S rRNA gene sequencing and bacterial diversity in the rumen of yak
From the sequencing of bacterial 16S rRNA genes in the solid, liquid, dorsal, and ventral epithelial fractions of yak rumen, we obtained 1,009,806 raw sequence reads in total, with 50,490 ± 6,620 from each sample on average. After quality control, a total of 957,559 high‐quality sequences were retained, with 47,877 ± 6,443 on average (range: 36,574–60,816) per sample. As already mentioned, to minimize the sequencing depth effect, we subsampled sequences from each sample to the minimum number (36,574) and identified 4,200 OTUs in total in all samples based on 97% similarity.
All three indices (number of OTUs, Shannon and Chao 1) indicated that bacterial diversity was highest and lowest in the liquid and dorsal epithelium fractions, respectively (Figure 1). The Chao 1 index did not significantly differ among the four fractions (p > .05, Figure 1). However, the number of OTUs and Shannon diversity did significantly differ between them (p < .05, Figure 1).
Figure 1.
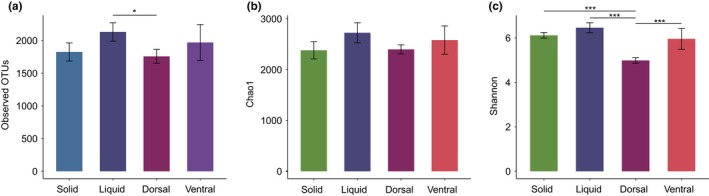
Alpha diversity indices of the solid, liquid, and epithelial fractions of yak rumen. *and ***indicate p < .05 and p < .001, respectively
3.2. Bacterial community composition in the rumen solid, liquid, and epithelial fractions of yak
Overall, a total of 19 phyla, 37 classes, 75 orders, 121 families, and 287 genera were identified in the four yak rumen fractions (Figure 2 and Figure A1 in Appendix). At the phylum level, the most abundant bacteria, accounting for more than 76% of the bacterial community in total were Bacteroidetes (57.6 ± 2.9, 52.2 ± 3.7 and 42.2 ± 3.9% in solid, liquid, and ventral epithelium, respectively) and Firmicutes (27.9 ± 2.1, 27.3 ± 4.1 and 34.6 ± 5.7% in solid, liquid, and ventral epithelium, respectively). However, in the dorsal epithelium fraction, the most abundant phylum was Bacteroidetes (28.3 ± 1.5%), followed by Firmicutes (27.4 ± 5.7%) and Epsilonbacteraeota (26.9 ± 4.5%).
Figure 2.
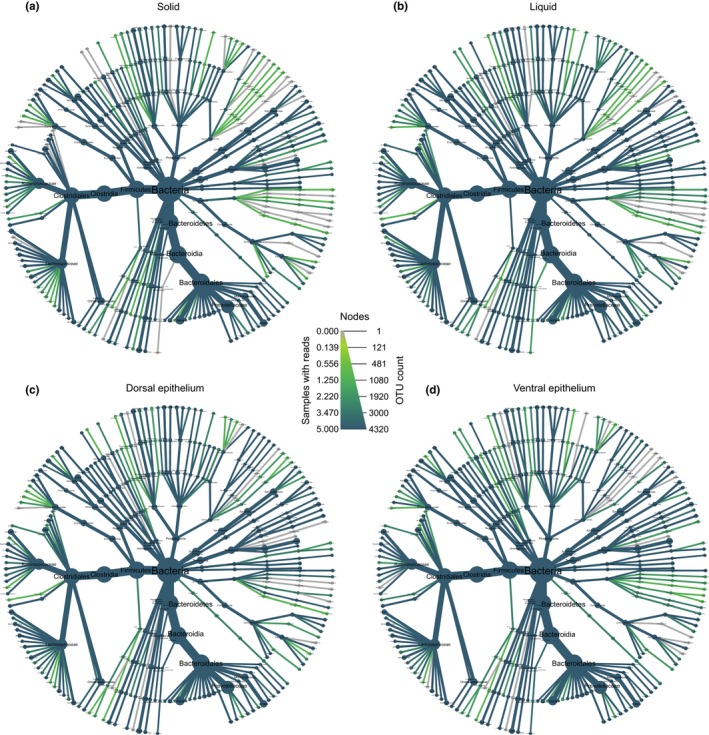
Heat trees showing the bacterial community composition in the (a) solid, (b) liquid, (c) dorsal epithelium, and (d) ventral epithelium fractions of yak rumen. Sizes of nodes and edges indicate OTU richness, and color indicates abundance
At genus level, the most abundant bacteria in the solid, liquid, and ventral epithelium fractions were members of the Rikenellaceae RC9 gut group (22.5 ± 3.5, 14.5 ± 4.6, and 8.3 ± 2.7%, respectively) and Prevotella (12.8 ± 2.2%, 14.6 ± 2.3, and 8.6 ± 3.0%, respectively; Figure 2 and Figure A1 in Appendix). However, the most abundant genera in the dorsal epithelium fraction were Campylobacter (26.9 ± 4.6%), followed by Rikenellaceae RC9 (6.5 ± 1.9%) and Prevotella (5.3 ± 1.6%).
3.3. Comparison of microbiota in the four yak rumen fractions
To compare the microbial community among the different rumen fractions, the PCoA based on the Bray–Curtis distance, unweighted UniFrac distance, and weighted UniFrac distance was employed (Figure 3). The results showed that the bacterial communities clearly separated according to the rumen fractions. Moreover, the microbiota in the dorsal epithelium also distinguished from that in the ventral epithelium. Adonis analysis and ANOSIM based on the three distance matrices also revealed significant differences in microbial communities of the four fractions (p = .001, Figure 3).
Figure 3.
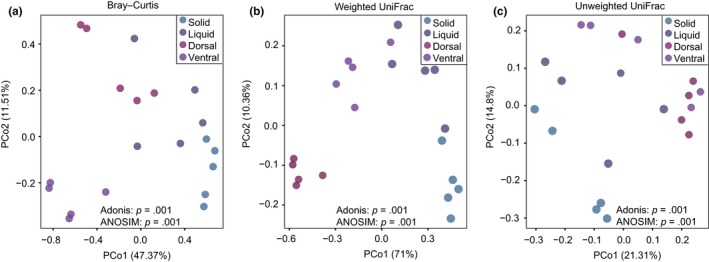
Principal coordinate analysis (PCoA) of microbial communities in the solid, liquid, dorsal epithelium, and ventral epithelium fractions of yak rumen based on (a) Bray–Curtis distance, (b) weighted uniFrac distance, and (c) unweighted uniFrac distance
Application of LEfSe identified 120 bacterial taxa that were significantly enriched in the fractions (Figure 4): 27, 49, 16, and 28 in samples of the solid, liquid, dorsal epithelium, and ventral epithelium fractions, respectively. In the solid fraction, 13 genera were identified with relative abundance greater than 0.1%: Rikenellaceae RC9, Fibrobacter, Anaerovorax, Lachnoclostridium 10, Lachnospiraceae FCS020, Lachnospiraceae NK4A136, Lachnospiraceae UCG 006, Mailhella, Papillibacter, Pelobacter, Ruminiclostridium 6, Ruminococcus 1, and Succiniclasticum. In the liquid fraction, 11 genera met this criterion: Bacteroidales RF16, Prevotella, Prevotellaceae UCG 003, Roseburia, Ruminococcus gauvreauii, Ruminococcaceae UCG 010, Erysipelotrichaceae UCG 004, Quinella, Selenomonas 1, Veillonellaceae UCG 001, and Fretibacterium. In the dorsal epithelium fraction, four enriched genera were identified: Comamonas, Campylobacter, Desulfovibrio, and Solobacterium. Finally, in the ventral epithelium fraction, 13 genera with relative abundance >0.1% were identified: Treponema 2, Howardella, Lachnospiraceae UCG 010, Lachnospiraceae UCG 008, Lachnospiraceae NK3A20, Acetitomaculum, Mogibacterium, Alloprevotella, Syntrophococcus, Ruminococcaceae UCG 005, Ruminococcaceae UCG 009, Prevotellaceae UCG 001, and Eubacterium nodatum (Table 1).
Figure 4.
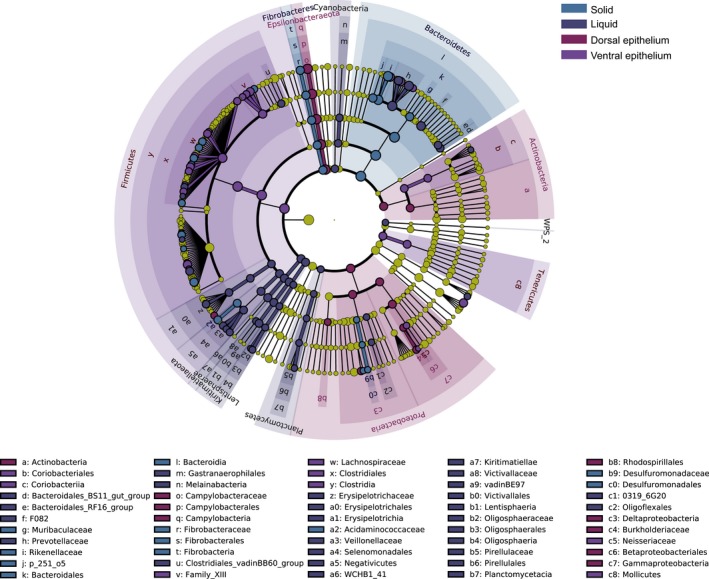
LEfSe cladogram showing the taxonomic differences among the four fractions in the rumen of yak. All identified taxonomy was significantly different based on Kruskal–Wallis test (p < .05) and a LDA score larger than 3.0
Table 1.
Relative abundances of bacterial taxa identified by LEfSe in microbiota of the solid, liquid, dorsal, and ventral epithelium fractions of yak rumen
| Fraction | Enriched taxa | Relative abundance (Mean ± SD) | |||
|---|---|---|---|---|---|
| Solid | Liquid | Dorsal Ep | Ventral Ep | ||
| Solid | Anaerovorax | 0.51 ± 0.09 | 0.44 ± 0.20 | 0.32 ± 0.11 | 0.19 ± 0.04 |
| Fibrobacter | 3.48 ± 0.62 | 1.14 ± 0.60 | 2.60 ± 1.33 | 1.92 ± 0.82 | |
| Lachnoclostridium 10 | 0.80 ± 0.35 | 0.29 ± 0.22 | 0.23 ± 0.19 | 0.22 ± 0.12 | |
| Lachnospiraceae FCS020 | 0.63 ± 0.09 | 0.21 ± 0.06 | 0.22 ± 0.13 | 0.20 ± 0.11 | |
| Lachnospiraceae NK4A136 | 0.83 ± 0.26 | 0.64 ± 0.20 | 0.48 ± 0.26 | 0.31 ± 0.14 | |
| Lachnospiraceae UCG 006 | 0.30 ± 0.03 | 0.16 ± 0.10 | 0.14 ± 0.10 | 0.07 ± 0.04 | |
| Mailhella | 0.10 ± 0.07 | 0.06 ± 0.02 | 0.05 ± 0.02 | 0.03 ± 0.02 | |
| Papillibacter | 1.15 ± 0.43 | 0.64 ± 0.20 | 0.46 ± 0.25 | 0.41 ± 0.09 | |
| Pelobacter | 0.11 ± 0.02 | 0.03 ± 0.01 | 0.04 ± 0.02 | 0.02 ± 0.01 | |
| probable genus 10 | 0.40 ± 0.13 | 0.22 ± 0.07 | 0.19 ± 0.12 | 0.21 ± 0.06 | |
| Rikenellaceae RC9 | 22.55 ± 3.52 | 14.52 ± 4.64 | 8.26 ± 2.74 | 6.47 ± 1.91 | |
| Ruminiclostridium 6 | 0.05 ± 0.04 | 0.04 ± 0.01 | 0.02 ± 0.02 | 0.01 ± 0.01 | |
| Ruminococcus 1 | 3.57 ± 0.70 | 1.31 ± 0.62 | 1.00 ± 0.40 | 1.08 ± 0.73 | |
| Succiniclasticum | 1.57 ± 0.77 | 0.73 ± 0.30 | 0.41 ± 0.17 | 0.19 ± 0.08 | |
| Liquid | Eubacterium oxidoreducens | 0.02 ± 0.01 | 0.01 ± 0.01 | 0.02 ± 0.01 | 0.03 ± 0.02 |
| Ruminococcus gauvreauii | 0.05 ± 0.04 | 0.35 ± 0.21 | 0.09 ± 0.02 | 0.07 ± 0.03 | |
| Bacteroidales RF16 | 0.23 ± 0.03 | 1.09 ± 0.51 | 0.98 ± 0.37 | 0.65 ± 0.34 | |
| Enterorhabdus | 0.01 ± 0.00 | 0.05 ± 0.03 | 0.01 ± 0.01 | 0.01 ± 0.01 | |
| Erysipelotrichaceae UCG 004 | 0.25 ± 0.08 | 0.91 ± 0.50 | 0.76 ± 0.24 | 0.44 ± 0.15 | |
| Fretibacterium | 0.04 ± 0.01 | 0.17 ± 0.07 | 0.14 ± 0.07 | 0.11 ± 0.08 | |
| horsej‐a03 | 0.14 ± 0.06 | 0.25 ± 0.03 | 0.14 ± 0.04 | 0.10 ± 0.05 | |
| Oscillospira | 0.01 ± 0.01 | 0.04 ± 0.03 | 0.02 ± 0.02 | 0.00 ± 0.00 | |
| p−1088‐a5 gut group | 0.09 ± 0.05 | 0.15 ± 0.08 | 0.03 ± 0.02 | 0.01 ± 0.01 | |
| Prevotella | 12.82 ± 2.23 | 14.59 ± 2.35 | 8.62 ± 3.03 | 5.26 ± 1.59 | |
| Prevotellaceae UCG 003 | 2.61 ± 0.47 | 3.82 ± 0.47 | 1.61 ± 0.43 | 1.08 ± 0.22 | |
| Pseudobacteroides | 0.00 ± 0.00 | 0.03 ± 0.02 | 0.02 ± 0.01 | 0.01 ± 0.01 | |
| Quinella | 0.07 ± 0.06 | 0.68 ± 0.41 | 0.13 ± 0.09 | 0.04 ± 0.01 | |
| Roseburia | 0.02 ± 0.01 | 0.1 ± 0.04 | 0.09 ± 0.03 | 0.09 ± 0.04 | |
| Ruminococcaceae UCG 010 | 1.15 ± 0.27 | 1.98 ± 0.53 | 1.06 ± 0.37 | 0.85 ± 0.30 | |
| Selenomonas 1 | 0.06 ± 0.02 | 0.21 ± 0.09 | 0.03 ± 0.02 | 0.04 ± 0.02 | |
| Tyzzerella 3 | 0.01 ± 0.01 | 0.02 ± 0.01 | 0.01 ± 0.01 | 0.01 ± 0.01 | |
| Veillonellaceae UCG 001 | 0.32 ± 0.08 | 0.37 ± 0.15 | 0.08 ± 0.04 | 0.08 ± 0.03 | |
| Dorsal epithelium | Campylobacter | 0.01 ± 0.01 | 3.13 ± 5.64 | 4.95 ± 2.4 | 26.91 ± 4.55 |
| Comamonas | 0.00 ± 0.00 | 0.16 ± 0.22 | 0.31 ± 0.35 | 0.50 ± 0.29 | |
| Desulfovibrio | 0.08 ± 0.03 | 0.22 ± 0.03 | 0.38 ± 0.17 | 0.62 ± 0.65 | |
| Solobacterium | 0.01 ± 0.01 | 0.06 ± 0.09 | 0.08 ± 0.08 | 0.26 ± 0.18 | |
| Ventral epithelium | Eubacterium nodatum | 0.02 ± 0.01 | 0.25 ± 0.23 | 2.26 ± 1.24 | 1.92 ± 0.92 |
| Acetitomaculum | 0.05 ± 0.02 | 0.16 ± 0.06 | 0.57 ± 0.62 | 0.31 ± 0.29 | |
| Alloprevotella | 0.00 ± 0.01 | 0.08 ± 0.13 | 0.35 ± 0.36 | 0.27 ± 0.31 | |
| Blvii28 wastewater‐sludge group | 0.00 ± 0.00 | 0.51 ± 1.08 | 5.50 ± 4.82 | 1.90 ± 1.05 | |
| Family XIII AD3011 | 0.07 ± 0.03 | 0.18 ± 0.11 | 0.48 ± 0.25 | 0.38 ± 0.17 | |
| GCA−900066575 | 0.00 ± 0.00 | 0.02 ± 0.04 | 0.14 ± 0.23 | 0.02 ± 0.02 | |
| Howardella | 0.00 ± 0.00 | 0.08 ± 0.11 | 0.99 ± 0.86 | 0.79 ± 0.82 | |
| Lachnospiraceae NK3A20 | 0.15 ± 0.06 | 0.21 ± 0.10 | 0.6 ± 0.32 | 0.34 ± 0.14 | |
| Lachnospiraceae UCG008 | 0.50 ± 0.17 | 0.34 ± 0.13 | 1.47 ± 0.97 | 0.81 ± 0.18 | |
| Lachnospiraceae UCG 010 | 0.01 ± 0.01 | 0.09 ± 0.18 | 0.4 ± 0.47 | 0.16 ± 0.09 | |
| Mogibacterium | 0.01 ± 0.00 | 0.03 ± 0.02 | 0.21 ± 0.17 | 0.11 ± 0.04 | |
| Prevotellaceae UCG 001 | 2.65 ± 0.63 | 3.46 ± 1.15 | 5.58 ± 1.63 | 4.05 ± 1.16 | |
| Ruminococcaceae UCG 004 | 0.01 ± 0.01 | 0.02 ± 0.01 | 0.04 ± 0.02 | 0.04 ± 0.03 | |
| Ruminococcaceae UCG 005 | 0.42 ± 0.23 | 0.77 ± 0.63 | 3.85 ± 2.5 | 2.65 ± 1.21 | |
| Ruminococcaceae UCG 009 | 0.02 ± 0.01 | 0.03 ± 0.02 | 0.1 ± 0.15 | 0.09 ± 0.04 | |
| Sutterella | 0.01 ± 0.01 | 0.02 ± 0.01 | 0.02 ± 0.01 | 0.00 ± 0.00 | |
| Syntrophococcus | 0.02 ± 0.01 | 0.05 ± 0.03 | 0.41 ± 0.41 | 0.16 ± 0.11 | |
| Treponema 2 | 3.62 ± 1.09 | 1.13 ± 1.42 | 3.72 ± 1.92 | 2.32 ± 1.51 | |
Abbreviation: Ep, epithelium.
3.4. qPCR quantification of bacterial density
The real‐time PCR results showed that the bacterial 16S rRNA gene copy numbers in the solid and liquid fractions were significantly higher than that in the dorsal and ventral epithelium fractions (p < .001, Figure 7). However, the differences between the solid and liquid fractions, and between the dorsal and ventral epithelium fractions were not significant (p > .05). In addition, the bacterial count in the ventral epithelium increased in comparison with that in the dorsal epithelium.
Figure 7.
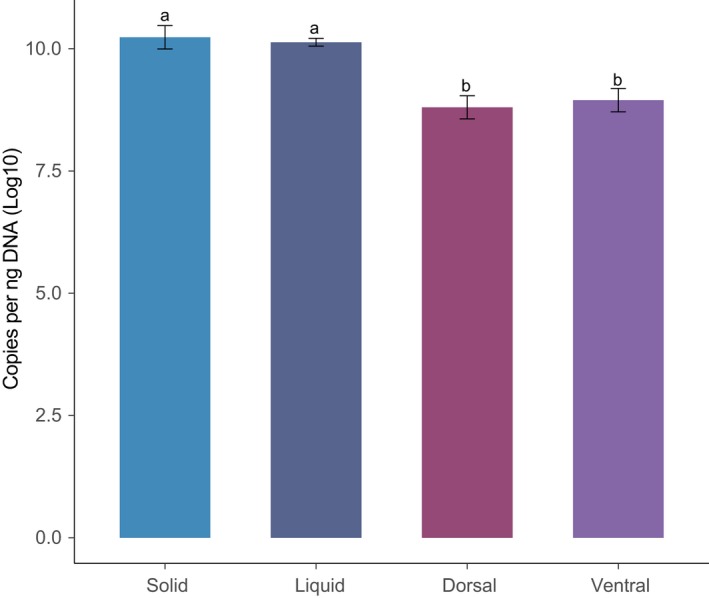
Density of bacteria in the solid, liquid, and epithelial fractions of yak rumen based on 16S rRNA gene. The results were expressed as log10 gene copies per ng of DNA. The different letters denote significant differences
3.5. Potential functional profiles of yak rumen fractions
To assess metabolic profiles of the ruminal microbiota, PICRUSt was applied to predict their functions (Figure 5). PCoA results showed that the functional profiles of the four fractions significantly differed (Figure 5a). Comparison of enriched KEGG pathways between the solid and liquid fractions indicated that amino acid metabolism, enzyme families, and energy metabolism pathways were enriched in the solid fraction, relative to the liquid fraction, while xenobiotic biodegradation and metabolism pathways were enhanced in the liquid fraction (Figure 5b). Further comparison revealed that energy metabolism, cell motility, and membrane transport were enriched in the dorsal epithelium fraction relative to the ventral epithelium (Figure 5c), while carbohydrate metabolism, enzyme families, metabolism of terpenoids and polyketides, and biodegradation of xenobiotics were stronger in the dorsal fraction than the ventral epithelium fraction (Figure 5c).
Figure 5.
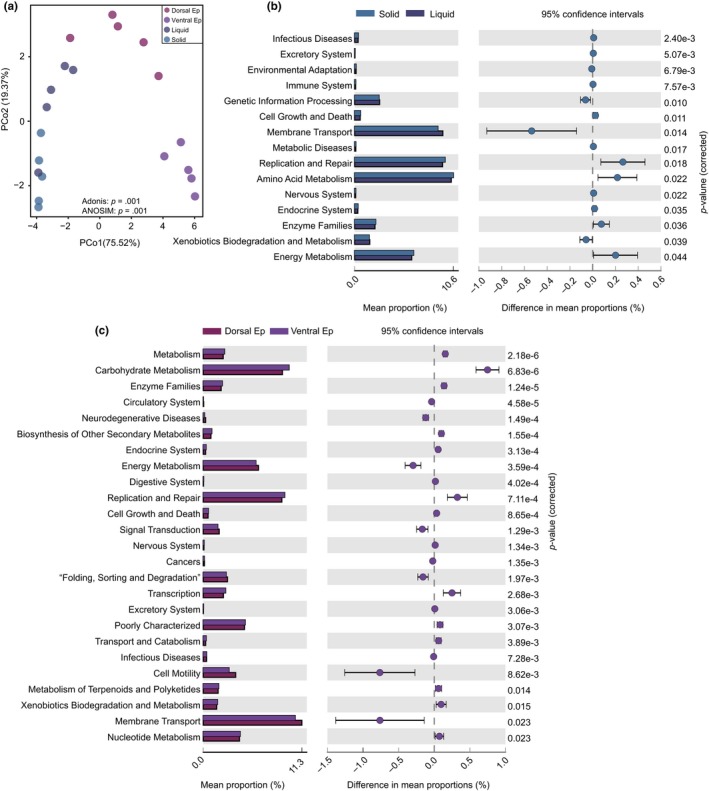
Functional profiles of microbial communities in the solid, liquid, and epithelial fractions. (a) PCoA plot revealing differences in predicted microbial functions based on Bray–Curtis distances. (b) Relative abundance of metabolic pathways in solid (green) and liquid (blue) fractions. The extended error bars show significantly different KEGG pathways between the fractions. (c) Relative abundances of pathways in the dorsal epithelium (orange) and ventral epithelium (maroon) fractions of yak. The extended error bars show significantly different KEGG pathways between the fractions. Ep, epithelium
3.6. Comparison of ventral epithelium microbiota in yak and cattle
PCoA based on Bray–Curtis, unweighted uniFrac, and weighted uniFrac distances, to further elucidate roles of microbiota in the ventral epithelium of yak rumen, showed that they significantly differed from corresponding microbiota in cattle (Figure 6a‐c). These results were corroborated by ANOSIM (p < .05) and Adonis analysis (p < .01).
Figure 6.
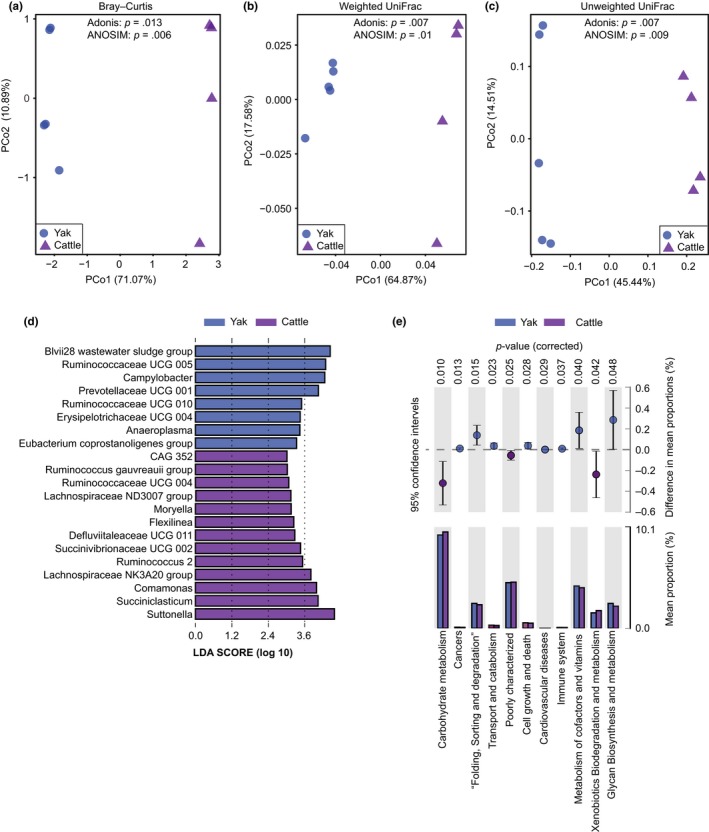
Differences in bacterial communities associated with the ventral epithelium between yak and cattle. Results of PCoA showing differences in the communities based on: (a) Bray–Curtis distance, (b) weighted uniFrac distance, and (c) unweighted uniFrac distance. (d) Results of LEfSe analysis showing taxa that significantly differed in the ventral epithelium of cattle and yak. (e) Differences in predicted functions between the ventral epithelium fraction of yak (orange) and cattle (purple). The extended error bars show significantly different KEGG pathways between the two ruminants’ ventral epithelium fractions
Application of LEfSe to explore the differences in microbial composition identified 20 genera as biomarkers distinguishing yak and cattle microbiota (Figure 6d). In the ventral epithelium fraction of yak, a total of seven taxa with relative abundance greater than 0.1% were identified (Table A1 in Appendix): Campylobacter, Ruminococcaceae UCG 005, Ruminococcaceae UCG 010, Prevotellaceae UCG 001, Erysipelotrichaceae UCG 004, Anaeroplasma, and Eubacterium coprostanoligenes group. In the ventral fraction of cattle, 13 genera were found to be enriched with relative abundance >0.1%: CAG 352, Ruminococcus gauvreauii group, Ruminococcaceae UCG 004, Lachnospiraceae ND3007 group, Moryella, Flexiinea, Defluviitaleaceae UCG 002, Succinivibrionaceae UCG 002, Ruminococcus 2, Lachnospiraceae NK3A30 group, Comamonas, Succiniclasticum, and Suttonella.
In addition, comparison of KEGG pathways enriched in the ventral epithelium fractions of yak and cattle revealed that cell growth and death, immune system, metabolism of cofactors and vitamins, and glycan biosynthesis and metabolism pathways were stronger in the yak fraction than the cattle fraction, while carbohydrate metabolism and xenobiotic biodegradation pathways were enhanced in the cattle fraction.
4. DISCUSSION
Our examination of microbiota in the yak rumen revealed clear differences in microbial composition and functional profiles among the solid, liquid, and dorsal and ventral epithelium fractions. The results also revealed clear differences in microbiota in the ventral epithelium fractions of yak and cattle. Our results extend understanding of the composition and roles of rumen microbiota in yak living on the QTP, as summarized in the following sections.
Our results demonstrated that the bacteria density (Figure 7) and Shannon index of bacterial diversity were lower in the ventral epithelium fraction than in the solid and liquid fractions (Figure 1), in accordance with patterns previously detected in dairy cattle (De Mulder et al., 2017; Schären et al., 2017). Interestingly, we found that ventral epithelium had a much higher Shannon index and bacteria count than the dorsal epithelium, providing the first indications that the microbial community associated with the ventral epithelium is much more diverse.
We also found that members of the phyla Bacteroidetes and Firmicutes were the dominant bacteria in the solid, liquid, and ventral epithelium fractions of yak rumen, in accordance with previous findings in ruminants generally (Henderson et al., 2015) and yaks specifically (Hu et al., 2019; Xue et al., 2018; Zhang et al., 2016; Zhou, Zhong, et al., 2017a; Zhou, Fang, et al., 2017b). These results indicate that the two phyla have adapted to wide ranges of gastrointestinal tract environments and play important roles in rumen ecology. However, Epsilonbacteraeota was the most abundant bacterial phylum in the yak dorsal epithelium fraction, which has not been recorded in previous studies (An, Dong, & Dong, 2005; Guo et al., 2015; Hu et al., 2019; Xue et al., 2018; Zhang et al., 2016; Zhou, Zhong, et al., 2017a; Zhou, Fang, et al., 2017b). The comparative genomic analysis revealed that the host‐associated Epsilonbacteraeota lacked genes involved in carbon fixation, but possessed genes involved in osmoprotection, and transport of heme, lipopolysaccharide, and capsular polysaccharides (Waite et al., 2017), in accordance with presumed requirements for their epithelium‐associated lifestyle. These findings confirm, inter alia, the strength of effects of ecological niches on microbial communities.
We also found that Rikenellaceae RC9 and Prevotella were abundant genera in all four fractions, in accordance with previous findings regarding ruminants generally (Henderson et al., 2015) and yaks (Hu et al., 2019; Xue et al., 2018; Zhang et al., 2016). Rikenellaceae RC9 can reportedly degrade structural carbohydrates and starch in the rumen of cows (Asma et al., 2013), while Prevotella spp. have high genetic and metabolic diversity (Bekele, Koike, & Kobayashi, 2010; Purushe et al., 2010), and play key roles in metabolism of carbohydrates, such as hemicellulose, starch, xylan, and pectin (Cotta, 1992; Dehority, 1966; Gardner, Wells, Russell, & Wilson, 1995; Kabel et al., 2011), and nitrogen (Kim et al., 2017; Stevenson & Weimer, 2007). Thus, these results indicate that Rikenellaceae RC9 and Prevotella play crucial roles in basic metabolic processes in the yak rumen.
Our study also revealed significant differences among the microbial communities of the four fractions (Figure 4 and Table 1). The families Lachnospiraceae and Ruminococcaceae, Succiniclasticum spp., and Fibrobacter spp. were enriched in the solid fractions. Previous studies have shown that Lachnospiraceae and Ruminococcaceae are also relatively abundant in the solid phase of cattle rumen (De Mulder et al., 2017; Schären et al., 2017) and play important roles in degradation of cellulose and fermentation of plant fibers (Biddle, Stewart, Blanchard, & Leschine, 2013; Flint & Louis, 2009; Schwarz, 2001). Moreover, Ruminococcaceae are reportedly associated with feed efficiency of dairy cattle (Myer, Wells, Smith, Kuehn, & Freetly, 2015), Fibrobacter spp. are reportedly major degraders of cellulosic plant biomass in the herbivore gut (Ransom‐Jones, Jones, McCarthy, & McDonald, 2012), and Succiniclasticum spp. specialize in fermentation of succinate, yielding propionate as a major product (van Gylswyk, 1995). Therefore, these bacteria likely contribute strongly to digestion of fibers in the yak rumen, a hypothesis supported by our predictions of microbial functions (Figure 5).
The liquid fraction was enriched with Roseburia spp., Quinella spp., Fretibacterium spp., Ruminococcus gauvreauii, Erysipelotrichaceae UCG 004, and Selenomonas 1 (Figure 4 and Table 1). Roseburia spp. are important butyrate‐producing bacteria, which widely degrade starch through production of extracellular amylase (Duncan et al., 2006). Ruminococcus gauvreauii is also a glucose‐fermenting bacterium, mainly producing acetate (Domingo et al., 2008). Quinella spp. are reportedly associated with low methane production rates in sheep, and ferment sugars equimolarly to acetate and propionate (Krumholz et al., 1993). Thus, their abundance in the yak rumen is consistent with yaks’ low methane emissions (Zhang et al., 2016). Selenomonas spp. are obligately saccharolytic bacteria that participate in fermentation of soluble sugars and lactate in the rumen (Hespell, Paster, & Dewhirst, 2006). Interestingly, two species of the genus were previously isolated from yak rumen and shown to generate acetate and propionate through glucose fermentation (Zhang & Dong, 2009). Moreover, amino acid metabolism is enhanced in the liquid fraction relative to the solid fraction (Figure 5). Taken together, these results suggest that the enriched bacteria in the liquid fraction are important for the metabolism of soluble nutrients in the yak rumen.
Our results also revealed differences in the microbial composition and predicted functions between the dorsal and ventral epithelium (Figure 4 and Table 1). Campylobacter spp., Desulfovibrio spp., and Comamonas spp. were enriched in the dorsal epithelium, while Treponema 2, Howardella spp., members of the families Lachnospiraceae and Ruminococcaceae, Acetitomaculum spp., Alloprevotella spp., and Syntrophococcus spp. were identified in the ventral epithelium fraction. These findings are consistent with previous reports regarding microbiota in the ventral epithelium of cows (De Mulder et al., 2017; Liu et al., 2016; Mann, Wetzels, Wagner, Zebeli, & Schmitz‐Esser, 2018). Campylobacter spp. are microaerophilic bacteria that can consume oxygen, are positively correlated with the weight and papilla length of goat rumen (Jiao, Huang, Zhou, & Tan, 2015), and reportedly associated with nitrogen metabolism (Mann et al., 2018). Comamonas spp. are aerobic proteobacteria (Willems & De Vos, 2006), and thus are likely involved in oxygen scavenging. Desulfovibrio spp. can use oxygen as an electron acceptor under microaerophilic conditions and are involved in hydrogen metabolism (Voordouw, 1995). These results suggest that major activities of the microbiota in the dorsal epithelium likely involved in hydrogen metabolism and oxygen scavenging, in accordance with the predicted microbial functions (Figure 5).
Finally, we found significant differences between the ventral epithelium microbiota of yak and cattle, including enrichment of Ruminococcaceae UCG 005, Anaeroplasma, and Erysipelotrichaceae UCG 004 in yak, and enrichment of Succiniclasticum, Ruminococcus 2, and Lachnospiraceae NK3A20 in cattle (Figure 6 and Table A1 in Appendix). The representative OTU sequences of Ruminococcaceae UCG 005 are similar to those of Sporobacter termitidis, a hydrogen‐consuming acetogen isolated from the termite gut (91%–92% sequence similarity, according to BLAST analysis; Grech‐Mora et al., 1996). Members of Anaeroplasma reportedly induce increases in levels of mucosal IgA (Beller et al., 2019). Erysipelotrichaceae UCG 004 belongs to the family Erysipelotrichaceae, which is reportedly associated with inflammation‐related gastrointestinal diseases (Chen, Liu, Ling, Tong, & Xiang, 2012). Succiniclasticum reportedly plays an important role in fermentation of succinate to propionate (van Gylswyk, 1995). Ruminococcus 2 has amylolytic activity (Ferrario et al., 2017), and Lachnospiraceae NK3A20 can potentially biohydrogenate fatty acids (Wang et al., 2017). These findings are consistent with the functional predictions (Figure 6e) and indicate that microbiota associated with yak ventral epithelium may contribute more strongly to immune responses than the corresponding microbiota in cattle, which may have mainly metabolic roles. However, rumen microbiota are also significantly affected by dietary composition (Henderson et al., 2015). Therefore, the significant difference in the ventral epithelium microbiota between cattle and yak is likely to result from the dietary difference. In further study, it is necessary to compare rumen microbiota between cattle and yak fed the same diet.
5. CONCLUSION
In this study, we characterized and compared the microbiota in the solid, liquid, and epithelial (dorsal and ventral) fractions of yak rumen. The results show that the rumen microbiota significantly differ in the four ecological niches, in both composition and functions. They also provide the first information on microbial community in the dorsal epithelial fraction, which clearly differed from the community in the ventral epithelium. The predicted functional profiles showed that amino acid metabolism, enzyme families, and energy metabolism pathways were enriched in the solid fraction, while xenobiotic biodegradation and metabolic pathways were enriched in the liquid fraction. The results also indicate that microbiota of the ventral epithelium of yak and cattle substantially differ. Overall, the results extend knowledge of microbiota in the yak rumen and their roles in yaks’ adaptation to their harsh environments.
ETHICS STATEMENT
All procedures applied were approved by the Institutional Animal Care and Use Committee of Lanzhou University and were in accordance with the university's guidelines for animal research.
CONFLICT OF INTEREST
None declared.
AUTHOR CONTRIBUTIONS
QMR and HZS collected the samples and analyzed the data; QMR, HZS, XTY, and CL analyzed the data; QMR, ZPL, and QQ wrote the manuscript; LMD and RJL participated in methodology and investigation; ZPL and QQ designed the study and reviewed the manuscript. All authors approved the final manuscript.
ACKNOWLEDGMENTS
This study was supported by the National Natural Science Foundation of China (grant nos. 31661143020, 41620104007, 31801089), the National Program for Support of Top‐notch Young Professionals, and the Fok Ying Tung Education Foundation (151105).
Appendix 1.
Figure A1.
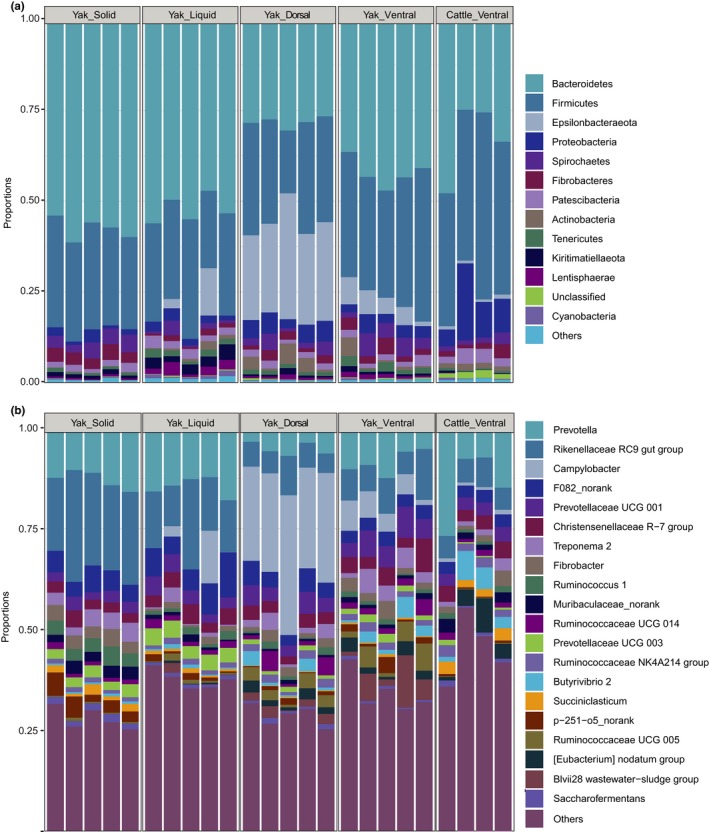
Stacked bar plot presenting the phylum composition in solid, liquid, dorsal epithelium and ventral epithelium fractions of yak and ventral epithelium fraction of cattle [Correction added on 6 January 2020 after first online publication: Figure A1 from Supporting Information has been moved to Appendix section]
Table A1.
Relative abundances of the taxa identified by LEfSe that significantly differed in abundance between ventral epithelium fractions of yak and cattle rumen [Correction added on 6 January 2020 after first online publication: Table A1 from Supporting Information has been moved to Appendix section]
| Fraction | Taxa | Relative abundance (Mean ± SD) | |
|---|---|---|---|
| Yak | Cattle | ||
| Yak | Blvii28 wastewater‐sludge | 5.50±4.82 | 0.01±0.01 |
| RuminococcaceaeUCG005 | 3.85±2.50 | 0.17±0.04 | |
| Campylobacter | 4.95±2.40 | 0.87±0.21 | |
| Prevotellaceae UCG 001 | 5.58±1.63 | 3.21±0.28 | |
| RuminococcaceaeUCG010 | 1.06±0.37 | 0.48±0.08 | |
| ErysipelotrichaceaeUCG004 | 0.76±0.24 | 0.18±0.07 | |
| Anaeroplasma | 0.72±0.54 | 0.16±0.08 | |
| Eubacterium coprostanoligenes | 0.75±0.10 | 0.32±0.15 | |
| Cattle | CAG‐352 | 0.0±0.0 | 0.21±0.23 |
| Ruminococcus gauvreauii | 0.09±0.02 | 0.32±0.26 | |
| Ruminococcaceae UCG 004 | 0.04±0.02 | 0.28±0.14 | |
| Lachnospiraceae ND3007 | 0.10±0.06 | 0.39±0.23 | |
| Moryella | 0.01±0.01 | 0.29±0.14 | |
| Flexilinea | 0.05±0.02 | 0.38±0.07 | |
| Defluviitaleaceae UCG 011 | 0.07±0.05 | 0.42±0.20 | |
| Succinivibrionaceae UCG 002 | 0.01±0.01 | 0.63±0.76 | |
| Ruminococcus 2 | 0.03±0.03 | 0.73±0.42 | |
| Lachnospiraceae NK3A20 | 0.60±0.32 | 1.86±0.71 | |
| Comamonas | 0.31±0.35 | 1.96±1.14 | |
| Succiniclasticum | 0.41±0.17 | 2.46±0.72 | |
| Suttonella | 0.08±0.11 | 6.15±6.24 | |
Ren Q, Si H, Yan X, et al. Bacterial communities in the solid, liquid, dorsal, and ventral epithelium fractions of yak (Bos grunniens) rumen. MicrobiologyOpen. 2020;9:e963 10.1002/mbo3.963
Ren and Si contribute equally to this work.
Contributor Information
Zhipeng Li, Email: lizhipeng01@caas.cn.
Qiang Qiu, Email: qiuqiang@lzu.edu.cn.
DATA AVAILABILITY STATEMENT
The datasets generated in this study are available in the NCBI Sequence Read Archive database (SRP212418) at https://www.ncbi.nlm.nih.gov/sra/?term=SRP212418
REFERENCES
- An, D. , Dong, X. , & Dong, Z. (2005). Prokaryote diversity in the rumen of yak (Bos grunniens) and Jinnan cattle (Bos taurus) estimated by 16S rDNA homology analyses. Anaerobe, 11(4), 207–215. 10.1016/j.anaerobe.2005.02.001 [DOI] [PubMed] [Google Scholar]
- Asma, Z. , Sylvie, C. , Laurent, C. , Jérôme, M. , Christophe, K. , Olivier, B. , … Francis, E. (2013). Microbial ecology of the rumen evaluated by 454 GS FLX pyrosequencing is affected by starch and oil supplementation of diets. FEMS Microbiology Ecology, 83(2), 504–514. 10.1111/1574-6941.12011 [DOI] [PubMed] [Google Scholar]
- Bekele, A. Z. , Koike, S. , & Kobayashi, Y. (2010). Genetic diversity and diet specificity of ruminal Prevotella revealed by 16S rRNA gene‐based analysis. FEMS Microbiology Letters, 305(1), 49–57. 10.1111/j.1574-6968.2010.01911.x [DOI] [PubMed] [Google Scholar]
- Beller, A. , Kruglov, A. , Durek, P. , von Goetze, V. , Hoffmann, U. , Maier, R. , … Chang, H.‐D. (2019). P104 Anaeroplasma, a potential anti‐inflammatory probiotic for the treatment of chronic intestinal inflammation. Annals of the Rheumatic Diseases, 78, A45–A46. 10.1136/annrheumdis-2018-EWRR2019.92 [DOI] [Google Scholar]
- Biddle, A. , Stewart, L. , Blanchard, J. , & Leschine, S. (2013). Untangling the genetic basis of fibrolytic specialization by Lachnospiraceae and Ruminococcaceae in diverse gut communities. Diversity, 5(3), 627 10.3390/d5030627 [DOI] [Google Scholar]
- Caporaso, J. G. , Kuczynski, J. , Stombaugh, J. , Bittinger, K. , Bushman, F. D. , Costello, E. K. , … Knight, R. (2010). QIIME allows analysis of high‐throughput community sequencing data. Nature Methods, 7, 335 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, W. , Liu, F. , Ling, Z. , Tong, X. , & Xiang, C. (2012). Human intestinal lumen and mucosa‐associated microbiota in patients with colorectal cancer. PLoS ONE, 7(6), e39743 10.1371/journal.pone.0039743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, K. J. , Mccowan, R. P. , & Costerton, J. W. (1979). Adherent epithelial bacteria in ruminants and their roles in digestive tract function. The American Journal of Clinical Nutrition, 32(1), 139–148. 10.1093/ajcn/32.1.139 [DOI] [PubMed] [Google Scholar]
- Cheng, K. J. , & Wallace, R. J. (1979). The mechanism of passage of endogenous urea through the rumen wall and the role of ureolytic epithelial bacteria in the urea flux. British Journal of Nutrition, 42(3), 553–557. 10.1079/BJN19790147 [DOI] [PubMed] [Google Scholar]
- Cho, S. J. , Cho, K. M. , Shin, E. C. , Lim, W. J. , Hong, S. Y. , Choi, B. R. , … Yun, H. D. (2006). 16S rDNA analysis of bacterial diversity in three fractions of cow rumen. Journal of Microbiology and Biotechnology, 16(1), 92–101. [Google Scholar]
- Clauss, M. , Hofmann, R. R. , Fickel, J. , Streich, W. J. , & Hummel, J. (2009). The intraruminal papillation gradient in wild ruminants of different feeding types: Implications for rumen physiology. Journal of Morphology, 270(8), 929–942. 10.1002/jmor.10729 [DOI] [PubMed] [Google Scholar]
- Cotta, M. (1992). Interaction of ruminal bacteria in the production and utilization of maltooligosaccharides from starch. Applied and Environmental Microbiology, 58(1), 48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Mulder, T. , Goossens, K. , Peiren, N. , Vandaele, L. , Haegeman, A. , De Tender, C. , … De Campeneere, S. (2017). Exploring the methanogen and bacterial communities of rumen environments: Solid adherent, fluid and epimural. FEMS Microbiology Ecology, 93(3), fiw251 10.1093/femsec/fiw251 [DOI] [PubMed] [Google Scholar]
- Dehority, B. A. (1966). Characterization of several bovine rumen bacteria isolated with a xylan medium. Journal of Bacteriology, 91(5), 1724–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinsdale, D. , Cheng, K. J. , Wallace, R. J. , & Goodlad, R. A. (1980). Digestion of epithelial tissue of the rumen wall by adherent bacteria in infused and conventionally fed sheep. Applied and Environmental Microbiology, 39(5), 1059–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingo, M.‐C. , Huletsky, A. , Boissinot, M. , Bernard, K. A. , Picard, F. J. , & Bergeron, M. G. (2008). Ruminococcus gauvreauii sp. nov., a glycopeptide‐resistant species isolated from a human faecal specimen. International Journal of Systematic and Evolutionary Microbiology, 58(6), 1393–1397. 10.1099/ijs.0.65259-0 [DOI] [PubMed] [Google Scholar]
- Duncan, S. H. , Aminov, R. I. , Scott, K. P. , Louis, P. , Stanton, T. B. , & Flint, H. J. (2006). Proposal of Roseburia faecis sp. nov., Roseburia hominis sp. nov. and Roseburia inulinivorans sp. nov., based on isolates from human faeces. International Journal of Systematic and Evolutionary Microbiology, 56(10), 2437–2441. 10.1099/ijs.0.64098-0 [DOI] [PubMed] [Google Scholar]
- Edgar, R. C. (2004). MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research, 32(5), 1792–1797. 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R. C. (2013). UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nature Methods, 10(10), 996–998. 10.1038/nmeth.2604 [DOI] [PubMed] [Google Scholar]
- Edgar, R. C. , Haas, B. J. , Clemente, J. C. , Quince, C. , & Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics, 27(16), 2194–2200. 10.1093/bioinformatics/btr381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrario, C. , Statello, R. , Carnevali, L. , Mancabelli, L. , Milani, C. , Mangifesta, M. , … Turroni, F. (2017). How to feed the mammalian gut microbiota: Bacterial and metabolic modulation by dietary fibers. Frontiers in Microbiology, 8(1749), 10.3389/fmicb.2017.01749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint, H. J. , & Louis, P. (2009). Diversity, metabolism and microbial ecology of butyrate‐producing bacteria from the human large intestine. FEMS Microbiology Letters, 294(1), 1–8. 10.1111/j.1574-6968.2009.01514.x [DOI] [PubMed] [Google Scholar]
- Gardner, R. G. , Wells, J. E. , Russell, J. B. , & Wilson, D. B. (1995). The cellular location of Prevotella ruminicola beta‐1,4‐D‐endoglucanase and its occurrence in other strains of ruminal bacteria. Applied and Environmental Microbiology, 61(9), 3288–3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grech‐Mora, I. , Fardeau, M.‐L. , Patel, B. K. C. , Ollivier, B. , Rimbault, A. , Prensier, G. , … Garnier‐Sillam, E. (1996). Isolation and characterization of Sporobacter termitidis gen. nov., sp. nov., from the digestive tract of the wood‐feeding termite Nasutitermes lujae. International Journal of Systematic and Evolutionary Microbiology, 46(2), 512–518. 10.1099/00207713-46-2-512 [DOI] [Google Scholar]
- Guo, W. , Li, Y. , Wang, L. , Wang, J. , Xu, Q. , Yan, T. , & Xue, B. (2015). Evaluation of composition and individual variability of rumen microbiota in yaks by 16S rRNA high‐throughput sequencing technology. Anaerobe, 34, 74–79. 10.1016/j.anaerobe.2015.04.010 [DOI] [PubMed] [Google Scholar]
- Henderson, G. , Cox, F. , Ganesh, S. , Jonker, A. , Young, W. , & Janssen, P. H. (2015). Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Scientific Reports, 5, 14567 10.1038/srep14567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hespell, R. B. , Paster, B. J. , & Dewhirst, F. E. (2006). The genus Selenomonas In Dworkin M., Falkow S., Rosenberg E., Schleifer K.‐H., & Stackebrandt E. (Eds.), The prokaryotes: Volume 4: Bacteria: Firmicutes, cyanobacteria (pp. 982–990), New York, NY: Springer. [Google Scholar]
- Hu, R. , Zou, H. , Wang, Z. , Cao, B. , Peng, Q. , Jing, X. , … Kong, X. (2019). Nutritional interventions improved rumen functions and promoted compensatory growth of growth‐retarded yaks as revealed by integrated transcripts and microbiome analyses. Frontiers in Microbiology, 10(318), 10.3389/fmicb.2019.00318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, X. D. , Tan, H. Y. , Long, R. , Liang, J. B. , & Wright, A.‐D.‐G. (2012). Comparison of methanogen diversity of yak (Bos grunniens) and cattle (Bos taurus) from the Qinghai‐Tibetan plateau, China. BMC Microbiology, 12(1), 237 10.1186/1471-2180-12-237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao, J. , Huang, J. , Zhou, C. , & Tan, Z. (2015). taxonomic identification of ruminal epithelial bacterial diversity during rumen development in goats. Applied and Environmental Microbiology, 81(10), 3502–3509. 10.1128/aem.00203-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabel, M. A. , Yeoman, C. J. , Han, Y. , Dodd, D. , Abbas, C. A. , de Bont, J. A. , … Mackie, R. I. (2011). Biochemical characterization and relative expression levels of multiple carbohydrate esterases of the xylanolytic rumen bacterium Prevotella ruminicola 23 grown on an ester‐enriched substrate. Applied and Environmental Microbiology, 77(16), 5671–5681. 10.1128/AEM.05321-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. N. , Méndez‐García, C. , Geier, R. R. , Iakiviak, M. , Chang, J. , Cann, I. , & Mackie, R. I. (2017). Metabolic networks for nitrogen utilization in Prevotella ruminicola 23. Scientific Reports, 7(1), 7851 10.1038/s41598-017-08463-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klindworth, A. , Pruesse, E. , Schweer, T. , Peplies, J. , Quast, C. , Horn, M. , & Glöckner, F. O. (2012). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next‐generation sequencing‐based diversity studies. Nucleic Acids Research, 41(1), e1 10.1093/nar/gks808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumholz, L. R. , Bryant, M. P. , Brulla, W. J. , Vicini, J. L. , Clark, J. H. , & Stahl, D. A. (1993). Proposal of Quinella ovalis gen. nov., sp. nov., based on phylogenetic analysis. International Journal of Systematic Bacteriology, 43(2), 293–296. 10.1099/00207713-43-2-293 [DOI] [PubMed] [Google Scholar]
- Langille, M. G. I. , Zaneveld, J. , Caporaso, J. G. , McDonald, D. , Knights, D. , Reyes, J. A. , … Huttenhower, C. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nature Biotechnology, 31, 814 10.1038/nbt.2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larue, R. , Yu, Z. , Parisi, V. A. , Egan, A. R. , & Morrison, M. (2005). Novel microbial diversity adherent to plant biomass in the herbivore gastrointestinal tract, as revealed by ribosomal intergenic spacer analysis and rrs gene sequencing. Environmental Microbiology, 7(4), 530–543. 10.1111/j.1462-2920.2005.00721.x [DOI] [PubMed] [Google Scholar]
- Liu, J.‐H. , Zhang, M.‐L. , Zhang, R.‐Y. , Zhu, W.‐Y. , & Mao, S.‐Y. (2016). Comparative studies of the composition of bacterial microbiota associated with the ruminal content, ruminal epithelium and in the faeces of lactating dairy cows. Microbial Biotechnology, 9(2), 257–268. 10.1111/1751-7915.12345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda, H. , Fujimoto, C. , Haruki, Y. , Maeda, T. , Kokeguchi, S. , Petelin, M. , … Takashiba, S. (2003). Quantitative real‐time PCR using TaqMan and SYBR Green for Actinobacillus actinomycetemcomitans, Porphyromonas gingivalis, Prevotella intermedia, tetQ gene and total bacteria. FEMS Immunology & Medical Microbiology, 39(1), 81–86. 10.1016/s0928-8244(03)00224-4 [DOI] [PubMed] [Google Scholar]
- Magoč, T. , & Salzberg, S. L. (2011). FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics, 27(21), 2957–2963. 10.1093/bioinformatics/btr507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann, E. , Wetzels, S. U. , Wagner, M. , Zebeli, Q. , & Schmitz‐Esser, S. (2018). Metatranscriptome sequencing reveals insights into the gene expression and functional potential of rumen wall bacteria. Frontiers in Microbiology, 9(43), 10.3389/fmicb.2018.00043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister, T. A. , Bae, H. D. , Jones, G. A. , & Cheng, K. J. (1994). Microbial attachment and feed digestion in the rumen. Journal of Animal Science, 72(11), 3004–3018. 10.2527/1994.72113004x [DOI] [PubMed] [Google Scholar]
- Myer, P. R. , Wells, J. E. , Smith, T. P. L. , Kuehn, L. A. , & Freetly, H. C. (2015). Cecum microbial communities from steers differing in feed efficiency. Journal of Animal Science, 93(11), 5327–5340. 10.2527/jas.2015-9415 [DOI] [PubMed] [Google Scholar]
- Price, M. N. , Dehal, P. S. , & Arkin, A. P. (2009). FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Molecular Biology and Evolution, 26(7), 1641–1650. 10.1093/molbev/msp077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purushe, J. , Fouts, D. , Morrison, M. , White, B. , Mackie, R. , Coutinho, P. , … Nelson, K. (2010). Comparative genome analysis of Prevotella ruminicola and Prevotella bryantii: Insights into their environmental niche. Microbial Ecology, 60(4), 721–729. 10.1007/s00248-010-9692-8 [DOI] [PubMed] [Google Scholar]
- Qiu, Q. , Zhang, G. , Ma, T. , Qian, W. , Wang, J. , Ye, Z. , … Liu, J. (2012). The yak genome and adaptation to life at high altitude. Nature Genetics, 44, 946 10.1038/ng.2343 [DOI] [PubMed] [Google Scholar]
- Quast, C. , Pruesse, E. , Yilmaz, P. , Gerken, J. , Schweer, T. , Yarza, P. , … Glockner, F. O. (2013). The SILVA ribosomal RNA gene database project: Improved data processing and web‐based tools. Nucleic Acids Research, 41(D1), D590–D596. 10.1093/nar/gks1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransom‐Jones, E. , Jones, D. L. , McCarthy, A. J. , & McDonald, J. E. (2012). The fibrobacteres: An important phylum of cellulose‐degrading bacteria. Microbial Ecology, 63(2), 267–281. 10.1007/s00248-011-9998-1 [DOI] [PubMed] [Google Scholar]
- Sadet, S. , Martin, C. , Meunier, B. , & Morgavi, D. P. (2007). PCR‐DGGE analysis reveals a distinct diversity in the bacterial population attached to the rumen epithelium. Animal, 1(7), 939–944. 10.1017/S1751731107000304 [DOI] [PubMed] [Google Scholar]
- Schären, M. , Kiri, K. , Riede, S. , Gardener, M. , Meyer, U. , Hummel, J. , … Dänicke, S. (2017). Alterations in the rumen liquid‐, particle‐ and epithelium‐associated microbiota of dairy cows during the transition from a silage‐ and concentrate‐based ration to pasture in spring. Frontiers in Microbiology, 8(744), 10.3389/fmicb.2017.00744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz, W. (2001). The cellulosome and cellulose degradation by anaerobic bacteria. Applied Microbiology and Biotechnology, 56(5), 634–649. 10.1007/s002530100710 [DOI] [PubMed] [Google Scholar]
- Segata, N. , Izard, J. , Waldron, L. , Gevers, D. , Miropolsky, L. , Garrett, W. S. , & Huttenhower, C. (2011). Metagenomic biomarker discovery and explanation. Genome Biology, 12(6), R60 10.1186/gb-2011-12-6-r60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson, D. M. , & Weimer, P. J. (2007). Dominance of Prevotella and low abundance of classical ruminal bacterial species in the bovine rumen revealed by relative quantification real‐time PCR. Applied Microbiology and Biotechnology, 75(1), 165–174. 10.1007/s00253-006-0802-y [DOI] [PubMed] [Google Scholar]
- Tafaj, M. , Junck, B. , Maulbetsch, A. , Steingass, H. , Piepho, H. P. , & Drochner, W. (2004). Digesta characteristics of dorsal, middle and ventral rumen of cows fed with different hay qualities and concentrate levels. Archives of Animal Nutrition, 58(4), 325–342. 10.1080/00039420412331273259 [DOI] [PubMed] [Google Scholar]
- van Gylswyk, N. O. (1995). Succiniclasticum ruminis gen. nov., sp. nov., a ruminal bacterium converting succinate to propionate as the sole energy‐yielding mechanism. International Journal of Systematic and Evolutionary Microbiology, 45(2), 297–300. 10.1099/00207713-45-2-297 [DOI] [PubMed] [Google Scholar]
- Voordouw, G. (1995). The genus desulfovibrio: The centennial. Applied and Environmental Microbiology, 61(8), 2813–2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waite, D. W. , Vanwonterghem, I. , Rinke, C. , Parks, D. H. , Zhang, Y. , Takai, K. , … Hugenholtz, P. (2017). Comparative genomic analysis of the class Epsilonproteobacteria and proposed reclassification to Epsilonbacteraeota (phyl. nov.). Frontiers in Microbiology, 8(682), 10.3389/fmicb.2017.00682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H. , Long, R. , Liang, J. B. , Guo, X. , Ding, L. , & Shang, Z. (2011). Comparison of nitrogen metabolism in yak (Bos grunniens) and indigenous cattle (Bos taurus) on the Qinghai‐Tibetan Plateau. Asian Australasian Journal of Animal Sciences, 24(6), 766–773. 10.5713/ajas.2011.10350 [DOI] [Google Scholar]
- Wang, Q. , Garrity, G. M. , Tiedje, J. M. , & Cole, J. R. (2007). Naïve bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and Environmental Microbiology, 73(16), 5261–5267. 10.1128/aem.00062-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Z. , Elekwachi, C. , Jiao, J. Z. , Wang, M. , Tang, S. X. , Zhou, C. S. , … Forster, R. J. (2017). Changes in metabolically active bacterial community during rumen development, and their alteration by rhubarb root powder revealed by 16s rrna amplicon sequencing. Frontiers in Microbiology, 8, 22 10.3389/fmicb.2017.00159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiener, G. , Han, J. L. , & Long, R. J. (2003). Origins, domestication and distribution of yak. Yak. [Google Scholar]
- Willems, A. , & De Vos, P. (2006). Comamonas In Dworkin M., Falkow S., Rosenberg E., Schleifer K.‐H., & Stackebrandt E. (Eds.), The prokaryotes: Volume 5: Proteobacteria: Alpha and beta subclasses (pp. 723–736), New York, NY: Springer. [Google Scholar]
- Xue, D. , Chen, H. , Chen, F. , He, Y. , Zhao, C. , Zhu, D. , … Li, W. (2016). Analysis of the rumen bacteria and methanogenic archaea of yak (Bos grunniens) steers grazing on the Qinghai‐Tibetan Plateau. Livestock Science, 188, 61–71. 10.1016/j.livsci.2016.04.009 [DOI] [Google Scholar]
- Xue, D. , Chen, H. , Luo, X. , Guan, J. , He, Y. , & Zhao, X. (2018). Microbial diversity in the rumen, reticulum, omasum, and abomasum of yak on a rapid fattening regime in an agro‐pastoral transition zone. Journal of Microbiology, 56(10), 734–743. 10.1007/s12275-018-8133-0 [DOI] [PubMed] [Google Scholar]
- Yu, Z. , & Morrison, M. (2004). Improved extraction of PCR‐quality community DNA from digesta and fecal samples. BioTechniques, 36(5), 808–812. 10.2144/04365st04 [DOI] [PubMed] [Google Scholar]
- Zhang, K. , & Dong, X. (2009). Selenomonas bovis sp. nov., isolated from yak rumen contents. International Journal of Systematic and Evolutionary Microbiology, 59(8), 2080–2083. 10.1099/ijs.0.007641-0 [DOI] [PubMed] [Google Scholar]
- Zhang, Z. , Xu, D. , Wang, L. , Hao, J. , Wang, J. , Zhou, X. , … Shi, P. (2016). Convergent evolution of rumen microbiomes in high‐altitude mammals. Current Biology, 26(14), 1873–1879. 10.1016/j.cub.2016.05.012 [DOI] [PubMed] [Google Scholar]
- Zhou, J. W. , Zhong, C. L. , Liu, H. , Degen, A. A. , Titgemeyer, E. C. , Ding, L. M. , … Long, R. J. (2017a). Comparison of nitrogen utilization and urea kinetics between yaks (Bos grunniens) and indigenous cattle (Bos taurus). Journal of Animal Science, 95(10), 4600–4612. 10.2527/jas2017.1428 [DOI] [PubMed] [Google Scholar]
- Zhou, Z. , Fang, L. , Meng, Q. , Li, S. , Chai, S. , Liu, S. , & Schonewille, J. T. (2017b). Assessment of ruminal bacterial and archaeal community structure in yak (Bos grunniens). Frontiers in Microbiology, 8(179), 10.3389/fmicb.2017.00179 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated in this study are available in the NCBI Sequence Read Archive database (SRP212418) at https://www.ncbi.nlm.nih.gov/sra/?term=SRP212418