

A potent drug delivery strategy promotes tumor infiltration of T cells and unleashes antitumor immunity.
Abstract
The response to programmed cell death protein-1 (PD-1)/programmed death ligand-1 (PD-L1) blockade in cancer immunotherapy is limited because of multiple immune evasion mechanisms. Here, a previously unknown strategy is proposed to synergize the nuclear factor κB (NF-κB) inhibition and PD-1 blockade for antitumor immunotherapy. A dual pH-sensitive nanocarrier loading curcumin (CUR) and anti–PD-1 monoclonal antibody (aPD-1) may bind to circulating PD-1+ T cells and then follow their infiltration into the tumor. Furthermore, the nanodrug bound to PD-1+ T cells may be released in the tumor microenvironment, leaving aPD-1 to block PD-1 on T cells and generating a CUR-encapsulated cationic nanodrug that can be easily taken up by tumor cells/tumor associated macrophages (TAMs). Thus, not only the antitumor T cells mediate efficient CUR delivery to tumor but also the efficient CUR delivery promotes the tumor infiltration of antitumor T cells, thereby resulting in effective activation of antitumor immunity.
INTRODUCTION
Nowadays, immune checkpoint blockade (ICB) is emerging as one of the most appealing and effective means in cancer immunotherapy (1). In particular, blockade of the programmed cell death protein-1 (PD-1)/programmed death ligand-1 (PD-L1) axis, which can reinvigorate exhausted T cells to inhibit tumor growth, has shown great promise in clinical therapy for patients with melanoma and other types of cancers (2). Yet, more than half of the patients still cannot benefit from the PD-1/PD-L1 blockade, as tumor may develop multiple immune evasion mechanism (3). To date, it has been found that the regulatory T cells (Treg cells) and some anti-inflammatory cytokines such as transforming growth factor β (TGF-β) and interleukin-10 (IL-10) perform indispensable roles in various tumor immune evasion mechanisms. Treg cells can suppress CD4+ T cells and CD8+ T cells, while TGF-β and IL-10 secreted by tumor-associated macrophages (TAMs; mainly the M2 phenotype) can inhibit cytotoxic T cell activation and suppress the function of antigen-presenting cells (4, 5). Thus, the depletion of Treg cells and the suppression of TGF-β/IL-10 may probably improve the therapeutic effect of PD-1/PD-L1 blockade (6). Meanwhile, the CCL-22, a CC-chemokine subfamily member secreted by tumor cells and TAMs, may recruit the Treg cells to the tumor (7), which makes the suppression of CCL-22 secretion a potential means to reduce Treg cells in the tumor. Studies have shown that the expression levels of CCL-22, TGF-β, and IL-10 are closely related to the nuclear factor κ light-chain enhancer of activated B cells (NF-κB) signaling pathway and the suppression of the NF-κB pathway in cancer cells and TAMs may substantially inhibit the expressions of all these cytokines (8). Therefore, an NF-κB pathway inhibitor is likely to reduce the tumor immune evasion, which can be expected to synergize with an immune checkpoint inhibitor such as the anti–PD-1 antibody (aPD-1) to improve antitumor immunotherapy.
A potent strategy to deliver the aPD-1 and NF-κB inhibitor to their target cells is critical for a combination cancer therapy. Over the past decades, drug delivery with nanocarriers has exhibited potential to improve therapeutic outcome and reduce side effects (9). The in vivo performances of nanomedicines highly depend on their morphological characteristics, e.g., worm-like or spherical, and physiochemical properties, including particle size, switchable surface charge, and stimuli sensitivity (10, 11). Moreover, tumor-targeted drug delivery with nanocarriers is based on an early finding that solid tumors may show an enhanced permeation and retention (EPR) effect to macromolecules and nanoscale objects (10). However, recent studies have found that passive tumor accumulation of nanomedicines is often much less effective than expected because the EPR effect is heterogeneous in humans and may not exist in some tumors (12). Therefore, research on different drug delivery strategies is of great significance and becoming a hotspot nowadays (13).
Here, in consideration that the circulating PD-1+ T cells can bind to aPD-1 in the bloodstream and then traffic actively down chemokine gradients to sites of inflammation or tumors, we propose an unusual strategy not only by applying a nanocarrier to deliver aPD-1 for ICB but also using T cells to deliver an NF-κB inhibitor curcumin (CUR) for antitumor T cell recruitment, which may result in a synergistic tumor immunotherapy (scheme S1). Because of the pH sensitivity, the nanodrug delivered by the tumor-infiltrating PD-1+ T cells may be released in the acidic tumor microenvironment (TME), leaving aPD-1 to block PD-1 on antitumor T cells and meanwhile generating a new CUR-encapsulated nanodrug to act on tumor cells/TAMs. On the basis of the strategy, CUR delivery to tumor cells and TAMs for the NF-κB pathway inhibition is expected to convert a “cold” tumor into a “hot” one by improving tumor infiltration of total T cells and cytotoxic T cells (14, 15). Consequently, an effective antitumor immunotherapy may be expected via simultaneously blocking the immune checkpoint to restore tumor killing of cytotoxic T cells and inhibiting the NF-κB pathway to recruit antitumor T cells to the tumor.
RESULTS
Polymer synthesis and nanodrug preparation
A diblock copolymer (CDM-PEG-PDPA) of 2-propionic-3-methylmaleic anhydride (CDM)–terminated polyethylene glycol (CDM-PEG) and poly(2-diisopropylaminoethyl methacrylate) (PDPA) was synthesized by multistep reactions, as outlined in fig. S1A. As shown in Fig. 1A and fig. S1 (B to D), we assigned the characteristic peaks of prepolymers and diblock copolymers in their 1H nuclear magnetic resonance (NMR) spectra. According to the gel permeation chromatography histograms of PEG and final diblock copolymer, the copolymer not only maintained a unimodal molecular weight distribution but also displayed an obvious increase in molecular weight as compared with PEG (fig. S2A and table S1). These results verified the successful syntheses of prepolymers and diblock copolymers. The CDM-capped diblock copolymer was then self-assembled in aqueous solution into the nanomicelle CUR@PDPA-PEG-CDM (CUR@PPC) with a PDPA core encapsulating CUR. Because of the pendant tertiary amino groups, PDPA showed a pKa value (where Ka is the acid dissociation constant) of around 6.54, as shown in fig. S2B. Thus, the tertiary amino groups of PDPA are deprotonated at neutral pH to make PDPA hydrophobic, which allows the formation of stable micelle encapsulating hydrophobic CUR. Last, the micelle surface was readily decorated with aPD-1 via ammonolysis reaction of the CDM groups with primary amino groups of antibody, resulting in the aPD-1–targeted and dual pH-sensitive nanodrug (CUR@PPC–aPD-1). The loading contents of CUR and aPD-1 of nanodrug were determined to be 5.0 and 3.1%, respectively. As shown in fig. S2C, no obvious change in particle size of nanodrug against incubation time was detected over 12 hours in phosphate-buffered saline (PBS) supplemented with 10% fetal bovine serum (FBS), suggesting a good stability of CUR@PPC–aPD-1 in serum-containing media.
Fig. 1. Characterizations of CUR@PPC–aPD-1.
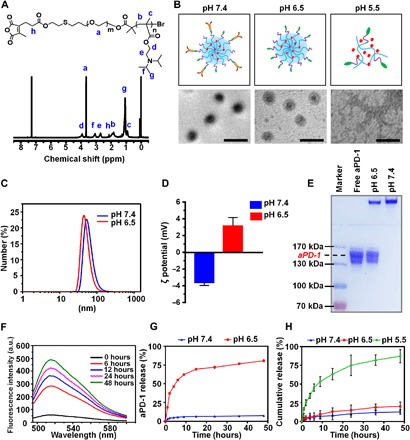
(A) 1H NMR spectrum of CDM-PEG-PDPA in CDCl3. ppm, parts per million. (B) Scheme and transmission electron microscopy (TEM) images of CUR@PPC–aPD-1 at various pH values. Scale bars, 100 nm. (C) The particle sizes of CUR@PPC–aPD-1 at pH values of 7.4 and 6.5 determined by dynamic light scattering (DLS). (D) The ζ potential of CUR@PPC–aPD-1 at pH values of 7.4 and 6.5 determined by DLS analysis (n = 3; means ± SD). (E) SDS–polyacrylamide gel electrophoresis (PAGE) picture of CUR@PPC–aPD-1 pretreated at pH values of 6.5 and 7.4 (5 μg of aPD-1 per sample). (F) Fluorescence spectra of Alexa Fluor 488–labeled nanoparticle (CUR@PPC–aPD-1/AF488) in PBS of pH 6.5 at different time points (concentration, 0.5 mg/ml). a.u., arbitrary units. (G) In vitro aPD-1 release from CUR@PPC–aPD-1 at pH values of 7.4 and 6.5 (n = 3; means ± SD). (H) In vitro CUR release from CUR@PPC–aPD-1 at pH values of 7.4, 6.5, and 5.5 (n = 3; means ± SD).
Dual pH sensitivity and drug release behaviors in vitro
As shown in fig. S2D, we measured the critical micellization concentrations (CMCs) of PPC at different pH values. According to the acid-base titration curve of HO-PEG-PDPA (fig. S2B), the pendant tertiary amino groups would be completely deprotonated at pH 7.4 to make PDPA highly hydrophobic, resulting in a CMC of PPC as low as 34 μg/ml. In contrast, the CMC of PPC at pH 6.5 was increased to 50 μg/ml, obviously due to a partial protonation of the tertiary amino groups according to fig. S2B. Moreover, the CMC of PPC was not detectable at pH 5.5 due to the protonation of all tertiary amino groups (fig. S2D), which made PDPA highly hydrophilic.
As shown in Fig. 1B, we investigated the morphologies of the CUR@PPC–aPD-1 nanodrug using transmission electron microscopy (TEM) at different pH values. At pH 7.4, the nanodrug showed highly uniform and spherical morphology revealing a core-shell structure, i.e., dark core of dense PDPA and gray shell of sparse PEG terminated by antibody. Although the spherical nanosphere was still observed at pH 6.5, its shell became less manifested as a result of antibody detachment via CDM cleavage. In contrast, the nanosphere completely dissembled at pH 5.5, and thus, only random aggregates were observed, which was formed most likely in the drying process of sample preparation. According to the dynamic light scattering (DLS) analyses, the hydrodynamic diameter of CUR@PPC–aPD-1 slightly decreased when the solution pH was adjusted to 6.5 from 7.4 (43 versus 50 nm), apparently owing to antibody release (Fig. 1C). Moreover, the ζ potentials of the nanodrug CUR@PPC–aPD-1 were −3.62 ± 0.35 and +3.15 ± 0.99 mV at pH values of 7.4 and 6.5, respectively (Fig. 1D). Considering that aPD-1 was negatively charged (fig. S2E) and PDPA was completely deprotonated at pH 7.4, it is reasonable that the aPD-1–decorated micelle should be negatively charged at this pH. In contrast, detachment of aPD-1 and partial protonation of PDPA would occur at pH 6.5 to result in nanoparticles with slight positive charge, which is a desirable feature because a negative surface is favorable for a long blood circulation, whereas a positive surface facilitates cell uptake of nanomedicines (16).
Then, the in vitro drug release profiles of the CUR@PPC–aPD-1 nanodrug were explored at pH values of 6.5 and 5.5, which mimic the tumor extracellular microenvironment and lysosomal environment, respectively. The pH-responsive CDM cleavage leading to aPD-1 release was further verified by gel electrophoresis assay [SDS–polyacrylamide gel electrophoresis (PAGE)]. As shown in Fig. 1E, no electrophoretic band for free aPD-1 (~150 kDa) was found at pH 7.4, whereas a clear band for free aPD-1 appeared at pH 6.5, indicating that the CDM linkage was cleaved to release negatively charged aPD-1 migratable in the electric field. The aPD-1 on the nanodrug was labeled with secondary antibody immunoglobulin G (IgG) Alexa Fluor 488, after the solution pH was adjusted to 6.5 from 7.4; the Alexa Fluor 488 fluorescence of CUR@PPC–aPD-1 solution at 525 nm exhibited a gradual increase against time, obviously owing to the CDM cleavage–induced release of aPD-1 causing fluorescence dequenching (Fig. 1F).
The in vitro release profiles of aPD-1 and CUR from CUR@PPC–aPD-1 were further quantified using enzyme-linked immunosorbent assay (ELISA) and measuring the absorbance of released CUR at 425 nm, respectively. As shown in Fig. 1G, aPD-1 was barely released at pH 7.4 due to the high stability of CDM linkage at neutral pH. On the contrary, the release of aPD-1 became very fast at pH 6.5, with 70% release in only 15 hours due to the cleavage of CDM linkage. The CUR release was very slow as well at pH 7.4, with 12.7% release over 48 hours, and it was only slightly accelerated at pH 6.5 (Fig. 1H). However, the CUR release became very fast at pH 5.5, which was attributed to the disassembly of micelle as revealed by TEM measurements. The above results evidenced the dual pH sensitivity of CUR@PPC–aPD-1 to enable a spatiotemporal release of aPD-1 and CUR inside the tumor, which allows aPD-1 and CUR to act on their target cells.
T cell binding and cellular uptake of nanodrug
We investigated the nanodrug binding to and release from PD-1+ T cells at pH values of 7.4 and 6.5. To show the potential of nanodrug to activate anticancer immunity via effectively binding to CD8+ T cells, we used CD8+ T cells as a target in the in vitro binding study and labeled with Alexa Fluor 647 (purple fluorescence). A dual fluorescence-visible nanomicelle NR@PPC–aPD-1 was prepared using the aPD-1 labeled with Alexa Fluor 488 (green fluorescence) and hydrophobic red fluorochrome Nile Red (NR) replacing CUR. As shown in fig. S3 (A and B) at pH 7.4, both the green fluorescence and red fluorescence increased against incubation time, implying gradual binding of nanodrug to CD8+ T cells owing to the PD-1/aPD-1 interaction. Moreover, the green fluorescence and red fluorescence were mainly on the surface of CD8+ T cells, indicating that the nanodrug was situated on the cell membrane rather than internalized into CD8+ T cells. This is vital for CD8+ T cells to mediate nanodrug delivery in consideration of an easy release. In contrast, the red fluorescence on cell membrane quickly attenuated against incubation time at pH 6.5, indicating detachment of the NR-labeled nanomicelle from CD8+ T cells via CDM linkage cleavage at pH 6.5 (fig. S3, A and D). All these results imply that the CD8+ T cell–bound nanodrug may be released when the CD8+ T cells infiltrate into the TME (pH ~6.5).
Then, the cell uptake potential of aPD-1–targeted nanodrug was evaluated in vitro by confocal laser scanning microscopy (CLSM) and flow cytometry analyses at pH 6.5, i.e., the TME acidity, in B16F10 cancer cells and RAW264.7 macrophages as the CUR target cells. To visualize the cell uptake and intracellular distribution, we used the nanodrug encapsulating NR as an analog of CUR and stained the nuclei blue. As shown under CLSM observation, cell incubation at pH 6.5 significantly increased the internalization of nanodrug into both the B16F10 and RAW264.7 cells (Fig. 2, A and C). After incubation for 4 hours at pH 6.5, the cells displayed intensive NR red fluorescence in the cytoplasm. In contrast, cells incubated at pH 7.4 showed almost no cell uptake. We obtained consistent results in quantitative analysis with flow cytometry (Fig. 2, B and D). That is, after nanodrug incubation for 4 hours at pH 7.4, the B16F10 and RAW264.7 cells only displayed 8.9% and 5.0% cell uptake, respectively. On the contrary, the B16F10 and RAW264.7 cells incubated at pH 6.5 showed much higher cell uptake levels up to 63.9% and 52.3%, respectively. Furthermore, after incubation with nanodrug for 10 hours at pH 6.5, only weak green fluorescence could be observed in both cells, implying that only a small amount of aPD-1 still conjugated on the nanodrug was internalized into the B16F10 and RAW264.7 cells (fig. S3C). As shown in fig. S3E, the small amount of aPD-1 internalized into B16F10 and RAW264.7 cells showed no cytotoxicity. Conceivably, the nanodrug accumulated in the tumor stroma (pH ~6.5) could release aPD-1 to enhance the internalization of CUR-loaded micelle into tumor cells and macrophages, which was in line with the DLS measurements that the negatively charged nanodrug at pH 6.5 was converted into a positively charged one facilitating cell uptake (16).
Fig. 2. T cell binding and cellular uptake of nanodrug.
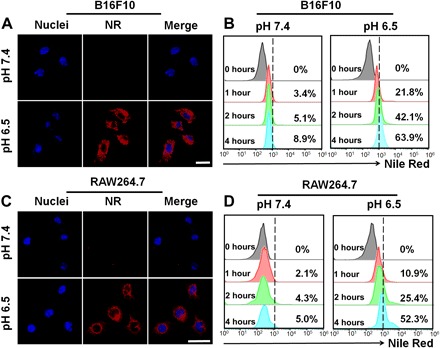
Cellular uptake of NR@PPC–aPD-1 in B16F10 tumor cells at pH values of 7.4 and 6.5 determined by CLSM imaging (A) and flow cytometry (B) analyses (concentration of NR@PPC–aPD-1, 1 μM). Scale bar, 25 μm. Cellular uptake of NR@PPC–aPD-1 in RAW264.7 macrophages at pH values of 7.4 and 6.5 determined by CLSM imaging (C) and flow cytometry (D) analyses (concentration of NR@PPC–aPD-1, 1 μM). Scale bar, 25 μm.
Together, the above results indicate that the nanodrug transported into the acidic TME could release the antibody to promote further CUR delivery into its target cells, no matter whether the nanodrug was delivered to the tumor by PD-1+ T cells or simply via a tumor EPR effect. The cytotoxicities of the empty PPC micelle and CUR-loaded CUR@PPC micelle in B16F10 cells were evaluated by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. As shown in fig. S4 (A and B), the empty PPC micelle almost showed no cytotoxicity, indicating a good compatibility. Meanwhile, the CUR@PPC micelle only displayed very weak cytotoxicity, suggesting a limited anticancer efficacy of CUR itself.
Inhibited secretion of immunosuppressive cytokines in vitro
The NF-κB signaling pathway plays a key role in regulating tumor immune response. According to previous researches (17, 18), the NF-κB signaling pathway and the mechanism for CUR-mediated NF-κB inhibition could be illustrated as in Fig. 3A. Activation of the NF-κB signaling pathway by lipopolysaccharide (LPS) promotes inhibitor of NF-κBα (IκBα) degradation to allow nuclear translocation of the phosphorylated NF-κB p65 (pho-p65), resulting in up-regulation of the CCL-22 and PD-L1 genes in the B16F10 cells, as well as the CCL-22, PD-L1, TGF-β, and IL-10 genes in the RAW264.7 cells, which leads to immune evasion and tumor growth. However, CUR could block the NF-κB signaling pathway by inhibiting p65 translocation to the nucleus and IκBα degradation (19). Thus, CUR transported with the pH-sensitive nanodrug is expected to inhibit the expressions of these immunosuppressive cytokines. To verify this potential, we evaluated the secretion levels of these cytokines in B16F10 melanoma cells and RAW264.7 macrophages incubated with CUR@PPC by real-time reverse transcription polymerase chain reaction (PCR) assay. As shown in Fig. 3B, CUR@PPC treatment down-regulated the expression of the CCL-22 gene in B16F10 cells and the expressions of CCL-22, TGF-β, and IL-10 genes in RAW264.7 cells in a dose-dependent manner. CUR@PPC could effectively suppress the expressions of CCL-22 gene in B16F10 cells and CCL-22, TGF-β, and IL-10 genes in RAW264.7 cells at 10 μM CUR@PPC, which was therefore used in the following experiments. The p65 phosphorylation levels in cells incubated with PBS, LPS, CUR-S (the surfactant formulation of CUR; see experimental details) (10 μM CUR), and CUR@PPC (10 μM CUR) were further compared using immunofluorescence staining assay for pho-p65 (Fig. 3C). According to CLSM imaging, CUR@PPC treatment resulted in the weakest green fluorescence in the nucleus of B16F10 cells and the weakest purple fluorescence in the nucleus of RAW264.7 cells, indicating that CUR@PPC was more potent in inhibiting the NF-κB phosphorylation than the CUR-S likely due to an effective delivery. We also performed Western blot assays to analyze the NF-κB phosphorylation by analyzing the expression level of pho-p65 protein. As shown in Fig. 3 (D to F), CUR@PPC treatment significantly down-regulated the expression of pho-p65 protein in both the B16F10 and RAW264.7 cells regardless of prestimulation of LPS. Similarly, the expression of PD-L1 protein in B16F10 and RAW264.7 cells incubated with CUR@PPC was effectively suppressed as well. As CUR@PPC–aPD-1 could release CUR@PPC at the TME acidity (pH ~6.5) as already demonstrated (fig. S3, A and B), the above results imply that the dual pH-sensitive nanodrug (CUR@PPC–aPD-1) delivered to the tumor via either T cells mediation or EPR-driven passive accumulation will not only block the PD-1 immune check point but also effectively inhibit the NF-κB pathway in both tumor cells and TAMs to suppress the secretion of immunosuppressive cytokines (CCL-22, TGF-β, and IL-10) and the expression of PD-L1.
Fig. 3. CUR released from CUR@PPC suppressed cytokines (CCL-22, TGF-β, and IL-10) secretion and PD-L1 expression in tumor.
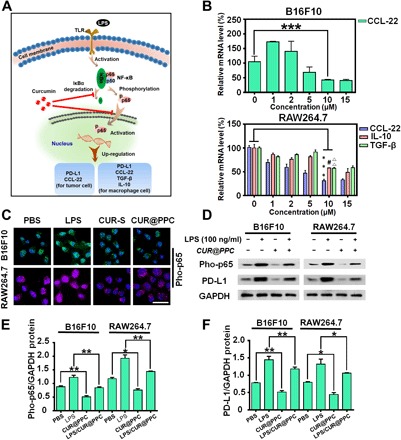
(A) Schematic illustration of CUR to suppress cytokines (CCL-22, TGF-β, and IL-10) secretion and PD-L1 expression via inhibiting the NF-κB pathway of tumor cells and macrophages. (B) Inhibited expressions of the CCL-22 gene in B16F10 cells and the CCL-22, IL-10, and TGF-β genes in RAW264.7 cells by CUR@PPC determined by real-time reverse transcription PCR (n = 3; means ± SD; ***P < 0.001, #P < 0.05, ΔΔP < 0.01). (C) CLSM images showed that CUR@PPC significantly inhibits the NF-κB pathway of B16F10 and RAW264.7 cells. Pho-p65 was labeled with Alexa Fluor 488 (green fluorescence) in B16F10 cells or Alexa Fluor 647 (purple fluorescence) in RAW264.7 cells (concentration of CUR@PPC, 10 μM). Scale bar, 25 μm. (D) Western blot assay showed that the NF-κB pathway and PD-L1 expression in B16F10 cells and RAW264.7 cells were inhibited by CUR@PPC (concentration of CUR@PPC, 10 μM). GAPDH, glyceraldehyde phosphate dehydrogenase. Protein expression levels of PD-L1 (E) and pho-p65 (F) quantified from Western blot. (n = 3; means ± SD; *P < 0.05, **P < 0.01). Statistical analyses were performed using analysis of variance (ANOVA) with Tukey’s test.
Drug delivery in vivo
As the B16F10 cells showed clear CCL-22 suppression at CUR concentrations above 10 μM in vitro (Fig. 3B), the intratumor CUR concentrations were determined using liquid chromatography–mass spectrometry after tail vein injection of CUR@PPC–aPD-1 into mice (fig. S4, C to E). CUR (molecular weight, 369) extracted from tumor tissue was determined on the basis of full-scan mass spectra. The quantitative analysis showed that the CUR levels in the tumor were above 10 μM from 12 to 48 hours after injection, implying that CUR can down-regulate the three immunosuppressive cytokines expression in the tumor after intravenous administration of CUR@PPC–aPD-1. Then, binding of the nanodrug to PD-1+ T cells was investigated in vivo. After NR@PPC–aPD-1 was injected into mice via tail vein, observation of blood lymphocytes under CLSM showed the binding of PD-1+ T cells with nanodrug. In addition to the binding to CD8−PD-1+ T cells (white arrow), NR@PPC–aPD-1 bound to the CD8+PD-1+ T cells effectively (red arrow), and most CD8+PD-1+ T cells were decorated with NR@PPC–aPD-1 at 12 hours after nanodrug injection (Fig. 4A and fig. S5). As the aPD-1 in nanodrug attached to tumor-infiltrating CD8+PD-1+ T cells was labeled with Alexa Fluor 555, the numbers of CD8+ T cells (gate on CD3+CD4−CD8+) blocked by aPD-1 in tumor site were measured by flow cytometry. As shown in Fig. 4B, only 19.7% of the CD8+ T cells inside the tumor were blocked by aPD-1 at 24 hours after intravenous administration of free aPD-1. However, 65.3% of CD8+ T cells inside the tumor could be blocked by aPD-1 when CUR@PPC–aPD-1 was injected, indicating effective blockade of the PD-1/PD-L1 axis via our drug delivery strategy.
Fig. 4. Evidence for T cell–mediated drug delivery in mice bearing subcutaneous B16F10 tumor.
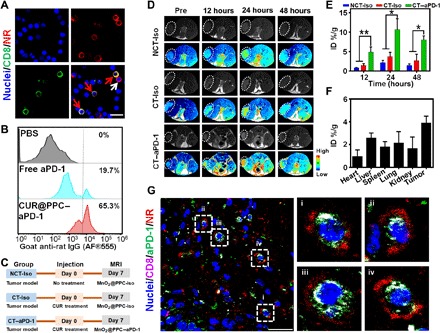
(A) CLSM images illustrate the binding of NR@PPC–aPD-1 to PD-1+ T cells at 12 hours after tail vein injection. All lymphocytes were separated from the mice blood. CD8+ T cells were labeled with Alexa Fluor 488 (green fluorescence), cell nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole) (blue fluorescence), and NR (red fluorescence) replacing CUR was encapsulated for fluorescence visualization. Scale bar, 30 μm. Red arrows indicate nanodrug-bound PD-1+CD8+ T cells, and white arrow illustrates a PD-1+CD8− T cell attached with nanodrug. See fig. S5 for images of low magnification acquired at three different time points (dose of NR@PPC–aPD-1 in vivo, 300 μg/kg body weight). (B) Flow cytometry analyses of CD8+ T cells (gated on CD3+CD4−CD8+ T cells) were blocked by Alexa Fluor 555–labled aPD-1 in tumor at 3 days after tail vein injection of PBS, free PD-1, and CUR@PPC–aPD-1 (dose of aPD-1 in vivo, 1.2 mg/kg body weight). (C) Illustration of in vivo magnetic resonance imaging (MRI) study schedule. (D) In vivo T1-weighted MRI (T1WI) of the B16F10 tumor-bearing mice after tail vein injection of MnO2@PPC–aPD-1 or MnO2@PPC-Iso at a dose of 13 mg/kg body weight. MnO2 replacing CUR was encapsulated for T1WI measurements. Black and white and pseudocolored images are shown for each group. (E) Quantitative analysis of Mn2+ contents in tumor site with inductively coupled plasma optical emission spectrometry (ICP-OES) at 12, 24, and 48 hours after tail vein injection of MnO2@PPC–aPD-1 (n = 3; means ± SD; *P < 0.05, **P < 0.01). Statistical analyses were performed using ANOVA with Tukey’s test. CT-Iso and CT–aPD-1 groups were pretreated with CUR. (F) Quantitative analysis of Mn2+ distribution in major organs and tumor tissue with ICP-OES at 24 hours after tail vein injection of MnO2@PPC–aPD-1 without CUR pretreatment (n = 3; means ± SD). (G) CLSM images of tumor section exhibiting distribution of aPD-1 (green fluorescence) and NR (red fluorescence) at 24 hours after tail vein injection of NR@PPC–aPD-1 at a dose of 300 μg/kg body weight. CD8+ T cells were labeled with Alexa Fluor 647 (purple fluorescence), cell nuclei were stained with DAPI (blue fluorescence), NR with red fluorescence replacing CUR was encapsulated for fluorescence visualization. See fig. S7C for nonoverlapped fluorescence images. Scale bar, 30 μm.
Since the melanin produced in melanoma would disturb the in vivo fluorescence imaging, magnetic resonance imaging (MRI) was used to detect tumor accumulation of nanodrug after intravenous injection into C57BL/6 mice bearing subcutaneous xenograft of B16F10 tumor. To better understand the T cell–mediated tumor accumulation, we injected a nanodrug decorated with an isotype control antibody for the two control animal groups with and without CUR pretreatments to recruit antitumor T cells to the tumor, i.e., the CT-Iso and NCT-Iso groups. At 7 days before nanodrug injection, we carried out the CUR pretreatment by tail vein injection of CUR@PPC (Fig. 4C). To enable MRI detection, we loaded the small MnO2 nanoparticles (10.4 nm) instead of CUR into the antibody-decorated nanomicelle. As shown in Fig. 4D and fig. S6A, the tumors of the NT-Iso and NCT-Iso groups exhibited hardly detectable enhancement in the MRI signals on T1-weighted MRI (T1WI) after nanomicelle injection. In contrast, injection of MnO2@PPC–aPD-1 into animals with CUR pretreatment (the CT–aPD-1 group) significantly enhanced the T1WI signals of the tumor, reaching the highest at 24 hours and then attenuating. Nevertheless, an obviously enhanced T1WI signal intensity was still detected even at 48 hours after nanomicelle injection. MRI scans of animal preinjected with free aPD-1 further demonstrated that the binding to circulating PD-1+ T cells promoted the tumor accumulation of nanodrug. At 24 hours after tail vein administration of free aPD-1 to block PD-1+ T cells, MnO2@PPC–aPD-1 was injected via the tail vein, and then, MRI scans were performed. As shown in fig. S6 (B and C), similar to the NT-Iso and NCT-Iso groups, the animal group pretreated with aPD-1 exhibited hardly detectable enhancement in the MRI signals of tumor on the T1WI.
It is well known that MnO2 is stable in the bloodstream with low r1 and r2 values. However, it can react with TME-enriched acid and H2O2 to generate Mn2+, which is a potent MRI T1 contrast agent (20). The above results indicated that the aPD-1–decorated nanomicelle (MnO2@PPC–aPD-1) accumulated much more effectively in the tumor sites of animals with CUR pretreatment (the CT–aPD-1 group) than the antibody isotype-decorated nanomicelle (MnO2@PPC-Iso) in the tumor sites of animals with CUR pretreatment (the CT-Iso group) or without CUR pretreatment (the NCT-Iso group). Furthermore, determination of the Mn2+ content in tumors of different treatment groups with inductively coupled plasma optical emission spectrometry (ICP-OES) at 12, 24, and 48 hours after nanomicelle injection achieved data highly supportive of the in vivo MRI results (Fig. 4E). After intravenous injection, the animals receiving MnO2@PPC–aPD-1 after CUR pretreatment (the CT–aPD-1 group) showed a significantly higher intratumor Mn2+ content than the CT-Iso and NCT-Iso groups. Furthermore, the Mn2+ content in tumors was higher than all other organs including the liver, which is known to be a preferential organ for nanoparticle accumulation after tail vein injection of MnO2@PPC–aPD-1 without CUR pretreatment (Fig. 4F). According to Fig. 4 (E and F), CUR pretreatment resulted in an approximately 6% increase in the intratumor Mn2+ content, which revealed the potency of PD-1+ T cell–mediated tumor-targeting delivery via CUR-promoted T cell recruitment.
To further verify that the aPD-1–decorated nanodrug responsive to TME acidity (~6.5) could be effectively delivered to the tumor by PD-1+ T cells, we determined the colocalization of PD-1 with aPD-1 by CLSM analysis. As shown in fig. S7 (A and B), the PD-1 red fluorescence on the T cell membrane overlapped with the green aPD-1 fluorescence. The fluorescence intensity profiles across the cell along the selected line (lines 1 to 3) showed that the signal of PD-1 was colocalized with that of aPD-1. To study the tumor distribution of nanomicelles, we injected mice bearing subcutaneous B16F10 tumor in the tail vein with NR@PPC–aPD-1 and NR@PPC and then euthanized them at 24 hours after injection to prepare tumor sections. The CD8+ T cells were stained with Alexa Fluor 647–labeled anti-CD8 antibody (purple fluorescence), and the aPD-1 was labeled with secondary antibody IgG Alexa Fluor 488 (green fluorescence) for visualization under CLSM observation. As shown in fig. S7C, NR@PPC with red fluorescence of NR accumulated in the tumor mainly via tumor EPR effect, without a sign for T cell–mediated delivery. However, when NR@PPC–aPD-1 was injected, in addition to distribution of red and green fluorescence unbound to T cells due to EPR-driven tumor accumulation, it was observed that some tumor-infiltrated CD8+ T cells were labeled by the aPD-1 fluorescence (green fluorescence) and meanwhile surrounded by the NR fluorescence (red fluorescence). Moreover, the purple T cell membrane fluorescence overlapped with the green aPD-1 fluorescence, whereas the surrounding red fluorescence was separated from the green fluorescence and the CD8+ T cells (Fig. 4G). These results provided direct evidence that the aPD-1–decorated nanodrug could be delivered to the tumor and then released by the tumor-infiltrating CD8+PD-1+ T cells, which provided a potent addition to the EPR effect–driven passive tumor accumulation of the nanodrug. The nanodrug release from tumor-infiltrated CD8+ T cells inside the tumor was attributed to the cleavage of the pH-sensitive CDM linkage for aPD-1 in the TME (pH ~6.5), as already demonstrated in vitro. These results are of particular significance in consideration of the long-standing controversy on the EPR-driven tumor accumulation of nanodrugs (21). Via our strategy, the aPD-1–decorated nanodrug could be effectively delivered to the tumor by circulating PD-1+ T cells, provided that these cells could be effectively recruited to tumor.
Tumor infiltration of T cells regulated by nanodrug
The regulation of tumor immune microenvironment with nanodrug was evaluated in mice bearing subcutaneous xenograft of B16F10 tumor. First, we investigated the effect of CUR treatment. The ELISA showed that, although the expression levels of the immunosuppressive CCL-22, TGF-β, and IL-10 were all down-regulated via the CUR-S treatment as compared with PBS treatment, the CUR-loaded nanodrug (CUR@PPC) exhibited the best effect to down-regulate these cytokines (Fig. 5A). These results could be attributed to the TME acidity–increased positive surface charge via partial PDPA protonation to facilitate the cell uptake of nanodrug and the lysosomal acidity–triggered intracellular CUR release, which markedly increased the in vivo performance of CUR to inhibit the NF-κB signaling pathway in tumor cells and TAMs. As CCL-22 has proved to modulate the tumor trafficking of Treg cells (8, 22), the amounts of Treg cells (CD4+Foxp3+) in tumor tissue of animals receiving different treatments were measured by flow cytometry. The CUR@PPC treatment significantly decreased the percentage of Treg cells in the tumor, as compared to the CUR-S treatment (3.6% versus 8.9%) or the PBS treatment (3.6% versus 16.7%) (Fig. 5B and fig. S8A). Immunohistochemical analysis of the Treg cell marker Foxp3 obtained consistent results on tumor infiltration of Treg cells. Among the three animal groups receiving different treatments, the animals receiving CUR@PPC showed the lowest expression level of Foxp3 in the tumor, indicating the least Treg cells (Fig. 5C). It is known that Treg cells can impair the proliferation or even directly kill the tumor-infiltrated CD8+ T cells, thereby strengthening immune evasion response (23). Thus, we further determined the amount of CD8+ T cells in tumor tissues of animals receiving the CUR@PPC treatment by flow cytometry and immunofluorescence assay. As shown in Fig. 5D and fig. S8B, the CUR@PPC treatment markedly increased the tumor infiltration of CD8+ T cells (CD3+CD4−CD8+) as compared to the PBS treatment (37.4% versus 6.6%), which demonstrated that the CUR treatment converted the tumor into a “hotter” one being more responsive to ICB (14). The CLSM images of immunofluorescence staining in tumor center revealed consistent results (Fig. 5E). The animals treated with PBS showed hardly detectable tumor infiltration of CD8+ T cells, and the CUR-S treatment only slightly increased the tumor infiltration of CD8+ T cells. In contrast, the CUR@PPC treatment much more effectively increased the tumor infiltration of CD8+ T cells. Meanwhile, the CUR@PPC treatment resulted in the lowest expression level of PD-L1 in the tumor tissue, implying a greatly impaired immune checkpoint PD-1/PD-L1 axis. The increased CD8+ T cells in the tumor tissue via CUR@PPC treatment may partially be due to a reactivation upon the PD-L1 suppression (24). Overall, these results demonstrated that the NF-κB pathway inhibition by CUR@PPC in vivo lowered the tumor infiltration of Treg cells, which in turn promoted the recruitment of CD8+ T cells to tumor.
Fig. 5. CUR@PPC promoted infiltration of CD8+ T cells into tumor.
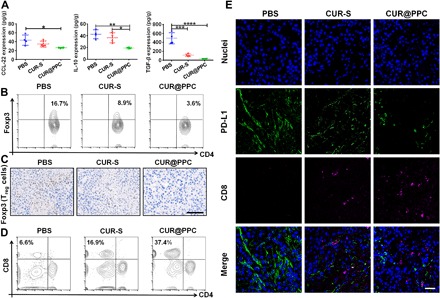
(A) Expression levels of immunosuppressive cytokines CCL-22, IL-10, and TGF-β analyzed by ELISA assay in B16F10 tumors of mice at 15 days of in vivo study schedule (see fig. S9A) (n = 4; means ± SD; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). Statistical analyses were performed using ANOVA with Tukey’s test. (B) Quantification of CD4+Foxp3+ T cells (i.e., Treg cells) in B16F10 tumors by flow cytometry (gated on CD3+CD4+ T cells). (C) Immunohistochemical staining of B16F10 tumors showed Treg cells (Foxp3+) (brown) at 15 days of in vivo study. Scale bar, 100 μm. (D) Quantification of CD8+ T cells in B16F10 tumors analyzed by flow cytometry (gated on CD3+ T cells). (E) Immunofluorescence of B16F10 tumor sections showed infiltration of CD8+ T cells and expression of PD-L1 at 15 days of in vivo study. CD8+ T cells were labeled with Alexa Fluor 647 (purple fluorescence), PD-L1 was labeled with Alexa Fluor 488 (green fluorescence), and cell nuclei were labeled with DAPI (blue fluorescence). Scale bar, 50 μm.
Next, we further analyzed the effect of aPD-1 delivery on tumor infiltration of PD-1+ T cells by immunofluorescence staining (Fig. 6A). Although the PPC–aPD-1 treatment promoted tumor infiltration of both CD8+ T cells and CD4+ T cells especially in the tumor boundary, the CUR@PPC–aPD-1 treatment showed an even better effect. That is, an effective tumor infiltration of CD8+ T cells was observed both in the boundary and center of tumors in animals receiving CUR@PPC–aPD-1. On the basis of these results, we hypothesized that an immunotherapy with aPD-1 increased the immunogenicity of tumor site, e.g., killing cancer cells to release tumor antigens, to promote recruitment of antitumor T cells (25). The CUR treatment, as already described in the previous section, is also essential for a highly effective tumor recruitment of cytotoxic T cells. Furthermore, the tumor infiltration of total T cells (CD3+) and Treg cells was analyzed by immunofluorescence staining and immunohistochemical assay, respectively. As shown in fig. S8 (C and D), among the four animal groups receiving different treatments, the animals receiving CUR@PPC–aPD-1 showed the highest expression level of CD3 but the lowest expression level of Foxp3 in the tumor, indicating the most T cells but the least Treg cells infiltrating into tumor. Obviously, the highly efficient delivery of dual pH-sensitive nanodrug to the tumor via both tumor EPR effect and PD-1+ T cells, as already shown in Fig. 4 and fig. S7C, is critical for the effective tumor infiltration of CD8+ T cells and CD4+ T cells in animals undergoing the CUR@PPC–aPD-1 treatment. Flow cytometry assay further revealed that the CUR@PPC–aPD-1 treatment most effectively enhanced infiltration of T helper 1 cells into the tumor (fig. S8, E and F), which obviously contributed significantly to the increased number of CD4+ T cells in tumor of animals subjected to the CUR@PPC–aPD-1 treatment.
Fig. 6. In vivo immune response regulated by CUR@PPC–aPD-1 and antitumor therapeutic effect of nanodrug.
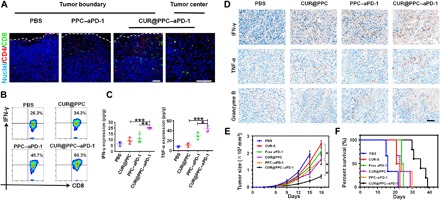
(A) Immunofluorescence of B16F10 tumor showed tumor infiltrations of CD8+ T cells and CD4+ T cells at 15 days of in vivo study schedule (see fig. S9A). CD8+ T cells were labeled with Alexa Fluor 488 (green fluorescence), CD4+ T cells were labeled with Alexa Fluor 555 (red fluorescence), and cell nuclei were labeled with DAPI (blue fluorescence). Scale bars, 100 μm. (B) Flow cytometry analysis of CD8+IFN-γ+ T cells in B16F10 tumors at 15 days of in vivo study (gated on CD3+CD4−CD8+ T cells). (C) The expression levels of interferon-γ (IFN-γ) and tumor necrosis factor–α (TNF-α) in B16F10 tumors of mice analyzed by ELISA assay at 15 days of in vivo study (n = 4; means ± SD; *P < 0.05, **P < 0.01, ***P < 0.001). Statistical analyses were performed using ANOVA with Tukey’s test. (D) Immunohistochemical staining of tumor-killing cytokines (IFN-γ, TNF-α, and granzyme B) (brown) in B16F10 tumor sections at 15 days of in vivo study. Scale bar, 50 μm. (E) Tumor growth and (F) cumulative survival of mice receiving different treatments (PBS, CUR-S, free aPD-1, CUR@PPC, PPC–aPD-1, and CUR@PPC–aPD-1) (n = 5; means ± SD; *P < 0.05, **P < 0.01). Statistical analyses in (E) were performed using ANOVA with Tukey’s test.
In vivo immune response regulated by nanodrug
It is known that CD8+ T cells secrete cytokines interferon-γ (IFN-γ) and tumor necrosis factor–α (TNF-α) to kill tumor cells (26, 27). However, this function was often impaired by the immune checkpoint PD-1/PD-L1 axis in the TME (28, 29), which makes the blockage of PD-1/PD-L1 axis with aPD-1 a promising strategy in tumor immunotherapy (30). Thus, flow cytometry assay was performed to evaluate the activation of CD8+ T cells in the tumor of animals receiving tail vein injections of various formulations via quantifying the CD8+ T cells expressing IFN-γ. As shown in Fig. 6B and fig. S8G, the ratio of CD8+ T cells with intracellular IFN-γ expression increased in the CUR@PPC and PPC–aPD-1 treatments as compared with the PBS treatment. Moreover, the CUR@PPC–aPD-1 treatment resulted in the highest ratio of IFN-γ–expressing cells, which indicated a combination effect of CUR and aPD-1 on the activation of CD8+ T cells. Then, the total expression levels of IFN-γ and TNF-α in the tumor tissue of various treatment groups were further evaluated by ELISA and immunohistochemical assays, respectively. As shown in Fig. 6 (C and D), in line with the flow cytometry results, although the CUR@PPC and PPC–aPD-1 treatments up-regulated the expression levels of IFN-γ and TNF-α, the CUR@PPC–aPD-1 treatment exhibited the best effect to restore the tumor immune microenvironment, namely, presenting the highest expression levels of IFN-γ and TNF-α. It is well known that the granzyme B as a cytotoxin for induction of DNA fragmentation and apoptosis in target cells was secreted specifically by activated cytotoxic T cells (31). Immunohistochemical assay (Fig. 6D) showed that the CUR@PPC–aPD-1 treatment resulted in the most up-regulated granzyme B expression among the four treatment groups, which further indicated that the restored immune responses in tumor tissues was closely related to the activation of cytotoxic T cells, as revealed by flow cytometry assay as well. Likely, the CUR@PPC–aPD-1 treatment regulated the tumor immune microenvironment most effectively via a synergistic mechanism in which CUR treatment promoted tumor infiltration of CD8+ T cells, while aPD-1 treatment reactivated CD8+ T cells, both of which depended on a highly efficient tumor-targeting delivery of the nanodrug via a combination delivery strategy proposed here.
In vivo anticancer effect
Last, the antitumor effect in vivo was explored in C57BL/6 mice bearing subcutaneous xenograft of B16F10 tumor. Beginning at day 6 after the construction of B16F10 tumor xenograft, the randomly grouped animals were subjected to a treatment schedule shown in fig. S9A. That is, animals in different groups were injected via the tail vein every 3 days with PBS, CUR-S, free PD-1, CUR@PPC, PPC–aPD-1, and CUR@PPC–aPD-1, respectively. As shown in Fig. 6 (E and F), compared to the PBS treatment, the CUR-S and free aPD-1 injections only showed minor effects on tumor growth and survival time, whereas obviously improved therapeutic effects that inhibit tumor growth and prolong survival were shown in animals receiving CUR@PPC or PPC–aPD-1. These results indicated that an efficient delivery with the pH-sensitive nanomicelle was vital for both drugs to exert their immunotherapeutic impacts. The CUR and aPD-1 combination therapy using CUR@PPC–aPD-1 most effectively inhibited tumor growth and prolonged animal survival among different treatment regimens, obviously owing to the notably promoted tumor infiltration and effective activation of cytotoxic T cells according to the aforementioned results on nanodrug-regulated tumor immune microenvironment. Moreover, nanodrug treatments caused no obvious damage to major organs and did not change the levels of the serum markers for liver and renal functions, and all mice receiving nanodrug treatments exhibited no body weight loss (fig. S9, B to G). These results indicated the low side effects of nanodrug in vivo, including CUR@PPC–aPD-1.
DISCUSSION
Compelling evidence in both preclinical and clinical researches is showing the great promise of immune checkpoint inhibition in tumor treatment (2). Suppressing the Treg cells recruitment while increasing the antitumor T cells recruitment to the tumor via down-regulating the expression levels of TGF-β, IL-10, and Treg cell–recruiting CCL-22 likely synergizes with PD-1/PD-L1 blockade to improve antitumor immunotherapy. The expression levels of CCL-22, TGF-β, and IL-10 in tumor can significantly be down-regulated by inhibiting the NF-κB signaling pathway in cancer cells and TAMs (29, 32). Leaving aside the great potential of combining PD-1/PD-L1 blockade with NF-κB pathway inhibition to overcome tumor immune evasion, a highly efficient delivery of aPD-1 and an NF-κB inhibitor to their target cells remains to be attained first. New studies are raising more and more questions nowadays about the mechanism based on an EPR effect widely adopted for passive tumor accumulation of nanodrugs (12). In this context, more workable strategies beyond the questionable tumor EPR effect should be developed to improve tumor-targeted delivery of nanodrugs.
In this study, we developed a core-shell structural nanodrug carrying the aPD-1 on its surface and encapsulating the NF-κB inhibitor CUR in its core based on a tailor-made block copolymer with dual pH sensitivity. In addition to a passive tumor accumulation driven by the tumor EPR effect, the nanodrug may bind to circulating PD-1+ T cells to allow a different tumor-targeted delivery pathway, i.e., a delivery via tumor recruitment of nanodrug-bound T cells. Owing to the dual pH-sensitive design, the nanodrug showed sequential drug release inside the tumor. First, the nanodrug delivered to the tumor tissue by either pathway may release aPD-1 by responding to the TME weak acidity (pH ~6.5), which also caused a surface charge conversion from negative to positive for an easy internalization of CUR-encapsulated nanodrug into cancer cells and macrophages. The aPD-1 released in the tumor extracellular matrix may block the PD-1/PD-1 axis to activate the antitumor function of cytotoxic T cells. Then, nanodrug rapidly released CUR in the lysosomal microenvironment (pH ~5.5) to inhibit the NF-κB pathway to down-regulate the expression levels of multiple cytokines (CCL-22, TGF-β, and IL-10), which inhibited the recruitment of Treg cells but boosted the recruitment of antitumor T cells. Thus, not only do PD-1+ T cells mediate efficient drug delivery to tumor but also the efficient drug delivery to tumor promotes tumor infiltration of antitumor T cells, thereby resulting in highly effective activation of antitumor immunity. Although engineering T cells as drug carriers may likely enhance the quantity of the drug being delivered to tumor (33, 34), the potential is greatly limited by the poor tumor infiltration of T cells due to the immunosuppressive TME. Obviously, our strategy may provide a solution to this challenge.
The combination therapy of aPD-1 and CUR demonstrated markedly improved antitumor immunotherapeutic effect both in vitro and in vivo. We applied the NF-κB inhibitor CUR to synergize with the immune checkpoint inhibitors such as aPD-1 for antitumor immunotherapy through nanotechnology. In addition, the nanodrug showed low side effects in vivo. Theoretically, the described strategy based on the TME-sensitive nanomedicines may be applied in other combination antitumor immunotherapy of aPD-1 with other immunomodulators requiring an efficient tumor-targeting delivery that is hard to achieve solely with the questionable tumor EPR effect.
MATERIALS AND METHODS
Preparation of aPD-1–decorated nanodrug CUR@PPC–aPD-1
The aPD-1 was conjugated to the surface of CUR@PPC micelle by a reaction between anhydride and primary amino. Typically, 10 ml of CUR@PPC micelle solution (1 mg/ml) and 1 ml of aPD-1 (1.1 mg/ml) were mixed for 4 hours at 4°C. Then, free PD-1 antibody was removed via dialysis against PBS [molecular weight cutoff (MWCO), 500 kDa]. After the solution was concentrated, the intensity of CUR absorbance was measured by ultraviolet-visible (UV-vis) analysis, and the intensity of aPD-1 absorbance was measured using ELISA kit (NEOBIOSCIENCE, China). The loading contents of CUR and aPD-1 as weight percentages in micelle were calculated on the basis of the calibration curves.
In vitro drugs release study
The in vitro release of aPD-1 and CUR was investigated at different pH values using dialysis diffusion method. Briefly, a dialysis bag (MWCO, 500 kDa) containing 4 ml of CUR@PPC–aPD-1 solution was immersed in 6 ml of release medium at different pH values and placed in a constant temperature shaker (37°C at 100 rpm). At predetermined time intervals, 1 ml of solution outside the dialysis bag was taken out and replaced with an equal volume of fresh medium. The content of aPD-1 and CUR was measured using ELISA kit (NEOBIOSCIENCE, China) and UV-vis on the basis of the absorbance intensity at 425 nm, respectively. The in vitro release experiments were performed in triplicate.
Serum stability assay
CUR@PPC–aPD-1 was incubated in PBS supplemented with 10% FBS at 37°C. Particles sizes were measured over time for 12 hours using Zetasizer Nano ZS instrument at 25°C.
Antibody labeling and release
CUR@PPC–aPD-1 was incubated with Alexa Fluor 488 IgG (goat anti-rat) at pH 7.4. The free secondary antibodies were removed by ultrafiltration. Then, the samples were adjusted to pH 6.5 and incubated for different times (0, 6, 12, 24, and 48 hours). The fluorescence intensity of Alexa Fluor 488 was determined at the emission wavelength of 525 nm. Furthermore, antibody conjugation and release studies were tested with SDS-PAGE as follows. Sample solution (20 μl; 5 μg of aPD-1) was mixed with 5 μl of loading buffer. Then, 25 μl of sample solution was loaded onto acrylamide gel. Among these samples, CUR@PPC–aPD-1 samples were pretreated at pH 7.4 or pH 6.5 and then incubated at 37°C for 4 hours before SDS-PAGE.
Cellular culture
B16F10 or RAW264.7 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, USA) supplemented with 10% FBS (Gibco, USA), penicillin (100 U/ml), and streptomycin (100 μg/ml; Gibco, USA). All cells were maintained at 37°C in a humidified chamber with 5% CO2.
In vitro T cell binding
Live T cells were purified by Ficoll gradient (MP Biomedicals, USA) from the spleen of healthy C57BL/6 mice, washed with PBS, and resuspended in RPMI 1640 (Gibco, USA) supplemented with 10% FBS at a density of 1 × 107 cells/ml. NR@PPC–aPD-1 was added to incubate the cells for different times (0, 6, 12, 24, and 48 hours) at pH 7.4 to allow binding. To further evaluate the detachment of nanodrug from T cells, the medium pH was adjusted to pH 6.5, and the cells were incubated for different times (6, 12, and 24 hours). For fluorescence visualization, cells were stained with Alexa Fluor 647 anti-mouse CD8 antibody and secondary antibody Alexa Fluor 488 for aPD-1 for 1 hour. The nuclei were stained with Hoechst 33342 for 15 min, and then, the cells were observed under CLSM (LSM710, Germany).
Cell uptake of nanodrug
B16F10 cells or RAW264.7 cells were seeded in 35-mm confocal dishes at a density of 1 × 104 cells per well and cultured overnight. Then, cells were incubated with NR@PPC–aPD-1 at pH value of 7.4 or 6.5 for 4 hours, washed with PBS, and fixed with 4% paraformaldehyde. After, DAPI (4′,6-diamidino-2-phenylindole) was added to stain nuclei for 5 min. The cells were observed under CLSM. For flow cytometric analysis [fluorescence-activated cell sorting (FACS) analysis], B16F10 or RAW264.7 cells were seeded in 12-well plates at a density of 1 × 106 cells per well and cultured for 24 hours. NR@PPC–aPD-1, which was pretreated at 37°C for 4 hours at pH value of 7.4 or 6.5, was added to incubate the B16F10 or RAW264.7 cells for 0, 1, 2, and 4 hours. Then, the cells were collected and resuspended in PBS for FACS analysis with an Attune NxT Acoustic Focusing Cytometer (Thermo Fisher Scientific, USA). The data were analyzed using FlowJo 10.0 (Treestar, USA).
In vitro cytotoxicity assay
B16F10 cells were seeded at a density of 8 × 103 cells per well in a 96-well plate and incubated at various micelle concentrations for 24 hours. The cell viability was determined using MTT kit (Invitrogen, Carlsbad, CA). Untreated cells as control were considered to be 100% viable.
Real-time reverse transcription PCR
Total RNA was extracted from the B16F10 or RAW264.7 cells using TRIzol reagent (Life Technologies, USA) according to the manufacturers’ instruction. Synthesis of complementary DNA (cDNA) was conducted according to the RNA PCR kit protocol (Takara Bio, China). Then, cDNAs were used for PCR (CCL-22, IL-10, TGF-β, and β-actin) with SYBR Green reagents under the reaction conditions. The primer sequences are shown in table S2. The relative value of mRNA expression was calculated by the comparative ΔΔCt method using β-actin as a reference gene.
Cell immunofluorescence assay
B16F10 or RAW264.7 cells were seeded in a confocal dish and incubated with PBS, CUR-S [10 μM CUR dissolved in 5% dimethyl sulfoxide (DMSO) + 30% PEG300 + 5% Tween 80 + double-distilled H2O (ddH2O)], and CUR@PPC (10 μM CUR) for 24 hours. The cells were permeated with 0.3% Triton X-100 for 5 min and incubated with 5% bovine serum albumin (BSA) for 30 min at room temperature. After the cells were incubated overnight at 4°C with the pho-p65 antibody, the corresponding secondary antibody IgG Alexa Fluor 488 or IgG Alexa Fluor 647 was added to incubate the cells for 1 hour at 37°C. After rinsing with PBS three times, cells were nucleus-stained with DAPI. Immunofluorescence imaging was performed using CLSM.
Western blot assay
Total proteins were extracted from B16F10 or RAW264.7 cells using radioimmunoprecipitation assay lysis buffer with protease inhibitors. The equivalent of protein was separated using a 10% gel and then transferred onto a polyvinylidene difluoride (PVDF) membrane (Bio-Rad, USA). Afterward, the PVDF membrane was blocked for 1 hour with 5% nonfat milk and then incubated overnight at 4°C with primary antibody, e.g., PD-L1, pho-p65, and glyceraldehyde phosphate dehydrogenase. Next, the membrane was washed five times with Tris-buffered saline with Tween 20 (TBST), incubated with secondary IgG–horseradish peroxidase (HRP) antibody for 1 hour, and washed with TBST. Last, the immunoreactive protein bands were visualized on a chemiluminescent detection system (ProteinSimple FR 0183, USA).
Animal model and antitumor activity
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Sun Yat-sen University (SYSU) and approved by the Animal Ethics Committee of SYSU. All experiments involving animals strictly followed the Animal Management Regulations of China (1988, revised 2017) and the Guideline on the Humane Treatment of Laboratory Animals of China (MOST 2006). After cleaning the hair, 1 × 106 B16F10 cells in 100 μl of PBS were subcutaneously injected into the right leg of C57BL/6 mice to establish the animal tumor model. Mice were then injected with PBS, CUR-S (CUR dissolved in 5% DMSO + 30% PEG300 + 5% Tween 80 + ddH2O), free PD-1, CUR@PPC, PPC–aPD-1, and CUR@PPC–aPD-1 at days 6, 9, 12, and 15. The aPD-1 and CUR doses per injection, if applied, were 1.2 and 2 mg/kg body mass, respectively. Tumor volume was measured using a caliper and calculated as follows: Tumor volume = 0.5 × length × width2.
In vivo T cell binding
Tumor-bearing mice were injected via tail vein with NR@PPC–aPD-1 (dose, 300 μg/kg body weight) and euthanized at different time points (0, 12, and 24 hours) to collect blood for further experiments. Live T cells were purified from blood using Ficoll-Hypaque solution (MP Biomedicals, USA). After being stained with CD8 antibody and Hoechst 33342, respectively, T cells were observed under CLSM. To analyze the aPD-1–blocked CD8+ T cells in tumor site, the tumor-infiltrated T cells were obtained by fully digesting total tumor in DMEM supplemented with collagenase (1 mg/ml; Sigma-Aldrich, USA) for 40 min at 37°C. After acquiring the tumor-infiltrated T cells, they were stained with antibodies (e.g., anti-CD3, anti-CD4, and anti-CD8) and secondary antibody IgG Alexa Fluor 555 (aPD-1) for 30 min. Last, the cells were analyzed using Attune NxT Acoustic Focusing Cytometer (Thermo Fisher Scientific, USA), and the data were analyzed using FlowJo 10.0 (Treestar, USA).
In vivo MRI and biodistribution
MnO2 nanoparticles were purchased from Aladdin (Shanghai, China) and loaded into the PPC micelle to get the MnO2@PPC–aPD-1 nanoparticles. After injecting the nanoparticles via tail vein (dose, 13 mg/kg body weight), the B16F10 tumor-bearing mice were anesthetized by chloral hydrate and then scanned with a 3-T clinical MR scanner (Philips, Achieva, Philips Medical Systems, Best, The Netherlands) equipped with a 50-mm small animal coil (Zhongzhi Medical Technologies, Suzhou, China) at different time points after injection (0, 12, 24, and 48 hours). For the biodistribution of nanomicelle, the mice were euthanized to obtain the major organs and tumors, and then, the tissues were solubilized in strong acid solution (HNO3:HCl:HClO4 = 3:1:2 by volume). After diluting to 10 ml with deionized water, Mn2+ concentration of all samples was determined via ICP-OES (CIROS VISION FVE12, Germany). Formula for biodistribution data: %ID/g = Corgan × Vorgan/(Ctotal × Vtotal × Morgan) × 100% (35). Further, B16F10 tumor-bearing mice were injected via tail vein with NR@PPC–aPD-1 (dose, 300 μg/kg body weight) and euthanized at 24 hours to collect the tumors, which were then frozen and sectioned in 8 μm for CLSM observation. The CD8+ T cells were stained purple with Alexa Fluor 647 anti-mouse CD8 antibodies, the aPD-1 were stained green with secondary antibody IgG Alexa Fluor 488, and the nuclei were stained blue with DAPI. Last, the sections were observed using CLSM.
Flow cytometry analysis
The staining for surface markers and intracellular markers of T cells was performed according to the standard protocols. Briefly, the tumor-infiltrated T cells were obtained as described above and then stained with surface antibodies (CD3, CD4, and CD8) for 30 min. The intracellular Foxp3 and IFN-γ were stained after fixing and perforating the T cells. The stained cells were detected using Attune NxT Acoustic Focusing Cytometer (Thermo Fisher Scientific, USA), and the data were analyzed using FlowJo 10.0 (Treestar, USA).
Histological analysis
Tumor tissues and major organs of B16F10 tumor-bearing mice were fixed for 48 hours with 4% paraformaldehyde, dehydrated using vacuum infiltration processor (Leica ASP200S, Germany), and embedded in paraffin. Two-micrometer sections of samples were obtained using a microtome (Leica RM2245, Germany) and stained with hematoxylin and eosin according to standard protocol. Photographs were taken with Pathology Imaging System (PerkinElmer Vectra, USA).
Enzyme-linked immunosorbent assay
The expressions of cytokines in the tumor sample including TGF-β, CCL-22, IL-10, IFN-γ, and TNF-α were detected by commercially available ELISA kits. The ELISA measurements were carried out strictly according to standard protocol provided by the manufacturer. All the ELISA kits were purchased from Westang Biotechnology (Shanghai, China).
Immunohistochemical staining
For immunohistochemical staining, the sections were put in xylene and rehydrated in graded ethanol solution (100 to 80%). The sections were pretreated with heated sodium citrate buffer (pH 6.0) for enzymatic antigen retrieval and steeped in hydrogen peroxide (5%) for 15 min at room temperature. After incubating with 5% BSA for 30 min, the samples were incubated with anti-Foxp3, anti–IFN-γ, anti–TNF-α, and anti–granzyme B antibodies overnight at 4°C. Then, the samples stained with HRP-conjugated secondary antibodies for 60 min at 37°C. The acquired sections were stained with diaminobenzidine (DAB) kit (ZSGB-BIO, China) and hematoxylin, respectively. After dehydration and mounting, the tissue sections were observed with Pathology Imaging System (PerkinElmer Vectra, USA).
Immunofluorescence staining
Immunofluorescence images of PD-L1/CD8– and CD8/CD4-stained tumor sections were acquired using the standard protocol. Tumor tissue sections were incubated with PD-L1/CD8, CD8/CD4, and CD3 antibodies overnight at 4°C. After washing with PBS, the samples were incubated at room temperature for 60 min with the secondary antibodies of goat anti-rabbit IgG Alexa Fluor 488/goat anti-rat IgG Alexa Fluor 647, goat anti-rabbit IgG Alexa Fluor 488/goat anti-rat IgG Alexa Fluor 555, and goat anti-rabbit IgG Alexa Fluor 555 diluted with PBS. Last, the sections were counterstained with DAPI and observed under CLSM.
Biochemistry index and histology analysis
Mice blood were collected at the last day of treatment to test the biomedical indexes including alkaline phosphatase (ALP), total bilirubin (TBIL), blood urea nitrogen (BUN), and creatinine (Cr) using an Auto Biochemical analyzer (Rayto Chemray 240, China).
Statistics
All data are expressed as means ± SD. Statistical differences were performed using one-way analysis of variance (ANOVA), followed by Tukey’s test with GraphPad Prism 6.0 software (GraphPad Software Inc., USA). Tukey’s post hoc tests and one-way ANOVA were performed for multiple comparisons. P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank the members of the Shuai laboratories for helpful discussions and technical assistance. We thank K. Zhu and Y. An for assistance in MRI scans. Funding: This research was supported by the National Basic Research Program of China (2015CB755500), the National Natural Science Foundation of China (51933011), the Natural Science Foundation of Guangdong Province (2014A030312018), and the Guangdong Innovative and Entrepreneurial Research Team Program (2013S086). Author contributions: X.S. conceived and designed the nanodrug and experimental program. Z.X., Z.S., J.H., L.L., and S.H. performed the experiments and data analysis. X.S. wrote the manuscript with the assistance of S.H., Z.X., and Z.S. All authors contributed feedback on the final manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: Correspondence and requests for materials should be addressed to X.S. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/6/eaay7785/DC1
Supplementary Materials and Methods
Scheme S1. Preparation and tumor-targeted delivery of dual pH-sensitive nanodrug.
Fig. S1. The synthetic route and 1H NMR spectra of copolymers.
Fig. S2. Characterizations of copolymers, antibody, and nanomicelle.
Fig. S3. T cell binding and cellular uptake of nanodrug at pH 6.5.
Fig. S4. Cytotoxicities of nanomicelles and concentrations of CUR in tumor tissue.
Fig. S5. PD-1+ T cell binding of NR@PPC–aPD-1 in vivo.
Fig. S6. T1WI signal intensities of tumors and T1WI of the B16F10 tumor-bearing mice.
Fig. S7. CLSM images showing the colocalization of aPD-1 and PD-1 and intratumor distribution of nanodrug.
Fig. S8. In vivo immune response regulated by nanodrug.
Fig. S9. Schedule of in vivo study and biosafety of nanodrug.
Table S1. Molecular weight of the synthesized polymers.
Table S2. Sequences for forward and reverse specific primers for real-time reverse transcription PCR amplification.
REFERENCES AND NOTES
- 1.Pardoll D. M., The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12, 252–264 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sharma P., Allison J. P., The future of immune checkpoint therapy. Science 348, 56–61 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Sharma P., Hu-Lieskovan S., Wargo J. A., Ribas A., Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168, 707–723 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.von Herrath M. G., Harrison L. C., Antigen-induced regulatory T cells in autoimmunity. Nat. Rev. Immunol. 3, 223–232 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Zou W., Regulatory T cells, tumour immunity and immunotherapy. Nat. Rev. Immunol. 6, 295–307 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Hoos A., Development of immuno-oncology drugs—From CTLA4 to PD1 to the next generations. Nat. Rev. Drug Discov. 15, 235–247 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Curiel T. J., Coukos G., Zou L., Alvarez X., Cheng P., Mottram P., Evdemon-Hogan M., Conejo-Garcia J. R., Zhang L., Burow M., Zhu Y., Wei S., Kryczek I., Daniel B., Gordon A., Myers L., Lackner A., Disis M. L., Knutson K. L., Chen L., Zou W., Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 10, 942–949 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Sun J., Sun J., Song B., Zhang L., Shao Q., Liu Y., Yuan D., Zhang Y., Qu X., Fucoidan inhibits CCL22 production through NF-kappaB pathway in M2 macrophages: A potential therapeutic strategy for cancer. Sci. Rep. 6, 35855 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goldman A., Kulkarni A., Kohandel M., Pandey P., Rao P., Natarajan S. K., Sabbisetti V., Sengupta S., Rationally designed 2-in-1 nanoparticles can overcome adaptive resistance in cancer. ACS Nano 10, 5823–5834 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Hubbell J. A., Chilkoti A., Chemistry. Nanomaterials for drug delivery. Science 337, 303–305 (2012). [DOI] [PubMed] [Google Scholar]
- 11.Petros R. A., DeSimone J. M., Strategies in the design of nanoparticles for therapeutic applications. Nat. Rev. Drug Discov. 9, 615–627 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Huang B., Abraham W. D., Zheng Y., Bustamante Lopez S. C., Luo S. S., Irvine D. J., Active targeting of chemotherapy to disseminated tumors using nanoparticle-carrying T cells. Sci. Transl. Med. 7, 291ra94 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Riley R. S., June C. H., Langer R., Mitchell M. J., Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 18, 175–196 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galon J., Bruni D., Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 18, 197–218 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Demaria S., Coleman C. N., Formenti S. C., Radiotherapy: Changing the game in immunotherapy. Trends Cancer 2, 286–294 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Du J.-Z., Du X.-J., Mao C.-Q., Wang J., Tailor-made dual pH-sensitive polymer-doxorubicin nanoparticles for efficient anticancer drug delivery. J. Am. Chem. Soc. 133, 17560–17563 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Ben-Neriah Y., Karin M., Inflammation meets cancer, with NF-κB as the matchmaker. Nat. Immunol. 12, 715–723 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Taniguchi K., Karin M., NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 18, 309–324 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Singh S., Aggarwal B. B., Activation of transcription factor NF-κB is suppressed by curcumin (diferuloylmethane). J. Biol. Chem. 270, 24995–25000 (1995). [DOI] [PubMed] [Google Scholar]
- 20.Lin L.-S., Song J., Song L., Ke K., Liu Y., Zhou Z., Shen Z., Li J., Yang Z., Tang W., Niu G., Yang H.-H., Chen X., Simultaneous fenton-like ion delivery and glutathione depletion by MnO2-based nanoagent to enhance chemodynamic therapy. Angew. Chem. Int. Ed. 57, 4902–4906 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Rosenblum D., Joshi N., Tao W., Karp J. M., Peer D., Progress and challenges towards targeted delivery of cancer therapeutics. Nat. Commun. 9, 1410 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Furudate S., Fujimura T., Kambayashi Y., Kakizaki A., Hidaka T., Aiba S., Immunomodulatory effect of imiquimod through CCL22 produced by tumor-associated macrophages in B16F10 melanomas. Anticancer Res. 37, 3461–3471 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Serrels A., Lund T., Serrels B., Byron A., McPherson R. C., von Kriegsheim A., Gomez-Cuadrado L., Canel M., Muir M., Ring J. E., Maniati E., Sims A. H., Pachter J. A., Brunton V. G., Gilbert N., Anderton S. M., Nibbs R. J., Frame M. C., Nuclear FAK controls chemokine transcription, Tregs, and evasion of anti-tumor immunity. Cell 163, 160–173 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang D., Wang T., Liu J., Yu H., Jiao S., Feng B., Zhou F., Fu Y., Yin Q., Zhang P., Zhang Z., Zhou Z., Li Y., Acid-Activatable versatile micelleplexes for PD-L1 blockade-enhanced cancer photodynamic immunotherapy. Nano Lett. 16, 5503–5513 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Huang A. C., Postow M. A., Orlowski R. J., Mick R., Bengsch B., Manne S., Xu W., Harmon S., Giles J. R., Wenz B., Adamow M., Kuk D., Panageas K. S., Carrera C., Wong P., Quagliarello F., Wubbenhorst B., D’Andrea K., Pauken K. E., Herati R. S., Staupe R. P., Schenkel J. M., McGettigan S., Kothari S., George S. M., Vonderheide R. H., Amaravadi R. K., Karakousis G. C., Schuchter L. M., Xu X., Nathanson K. L., Wolchok J. D., Gangadhar T. C., Wherry E. J., T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 545, 60–65 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen S., Lee L. F., Fisher T. S., Jessen B., Elliott M., Evering W., Logronio K., Tu G. H., Tsaparikos K., Li X., Wang H., Ying C., Xiong M., VanArsdale T., Lin J. C., Combination of 4-1BB agonist and PD-1 antagonist promotes antitumor effector/memory CD8 T cells in a poorly immunogenic tumor model. Cancer Immunol. Res. 3, 149–160 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Grinberg-Bleyer Y., Oh H., Desrichard A., Bhatt D. M., Caron R., Chan T. A., Schmid R. M., Klein U., Hayden M. S., Ghosh S., NF-κB c-Rel Is crucial for the regulatory T cell immune checkpoint in cancer. Cell 170, 1096–1108.e13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Errico A., Immunotherapy: PD-1-PD-L1 axis: Efficient checkpoint blockade against cancer. Nat. Rev. Clin. Oncol. 12, 63 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Huang Y., Kim B. Y. S., Chan C. K., Hahn S. M., Weissman I. L., Jiang W., Improving immune-vascular crosstalk for cancer immunotherapy. Nat. Rev. Immunol. 18, 195–203 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Balar A. V., Weber J. S., PD-1 and PD-L1 antibodies in cancer: Current status and future directions. Cancer Immunol. Immunother. 66, 551–564 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Darmon A. J., Nicholson D. W., Bleackley R. C., Activation of the apoptotic protease CPP32 by cytotoxic T-cell-derived granzyme B. Nature 377, 446–448 (1995). [DOI] [PubMed] [Google Scholar]
- 32.Klages K., Mayer C. T., Lahl K., Loddenkemper C., Teng M. W., Ngiow S. F., Smyth M. J., Hamann A., Huehn J., Sparwasser T., Selective depletion of Foxp3+ regulatory T cells improves effective therapeutic vaccination against established melanoma. Cancer Res. 70, 7788–7799 (2010). [DOI] [PubMed] [Google Scholar]
- 33.Stephan M. T., Moon J. J., Um S. H., Bershteyn A., Irvine D. J., Therapeutic cell engineering with surface-conjugated synthetic nanoparticles. Nat. Med. 16, 1035–1041 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmid D., Park C. G., Hartl C. A., Subedi N., Cartwright A. N., Puerto R. B., Zheng Y., Maiarana J., Freeman G. J., Wucherpfennig K. W., Irvine D. J., Goldberg M. S., T cell-targeting nanoparticles focus delivery of immunotherapy to improve antitumor immunity. Nat. Commun. 8, 1747 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu J., Chen Q., Zhu W., Yi X., Yang Y., Dong Z., Liu Z., Nanoscale-coordination-polymer-shelled manganese dioxide composite nanoparticles: A multistage Redox/pH/H2O2-responsive cancer theranostic nanoplatform. Adv. Funct. Mater. 27, 1605926 (2017). [Google Scholar]
- 36.Ma X., Wang Y., Zhao T., Li Y., Su L.-C., Wang Z., Huang G., Sumer B. D., Gao J., Ultra-pH-sensitive nanoprobe library with broad pH tunability and fluorescence emissions. J. Am. Chem. Soc. 136, 11085–11092 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Singha N. K., Gibson M. I., Koiry B. P., Danial M., Klok H.-A., Side-chain peptide-synthetic polymer conjugates via tandem “ester-amide/thiol-ene” post-polymerization modification of poly(pentafluorophenyl methacrylate) obtained using ATRP. Biomacromolecules 12, 2908–2913 (2011). [DOI] [PubMed] [Google Scholar]
- 38.Luo M., Wang H., Wang Z., Cai H., Lu Z., Li Y., Du M., Huang G., Wang C., Chen X., Porembka M. R., Lea J., Frankel A. E., Fu Y. X., Chen Z. J., Gao J., A STING-activating nanovaccine for cancer immunotherapy. Nat. Nanotechnol. 12, 648–654 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun C.-Y., Liu Y., Du J.-Z., Cao Z.-T., Xu C.-F., Wang J., Facile generation of tumor-pH-labile linkage-bridged block copolymers for chemotherapeutic delivery. Angew. Chem. Int. Ed. 55, 1010–1014 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Wenger M. D., Bowman A. M., Thorsteinsson M. V., Little K. K., Wang L., Zhong J., Lee A. L., DePhillips P., An automated homogeneous method for quantifying polysorbate using fluorescence polarization. Anal. Biochem. 337, 48–54 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/6/eaay7785/DC1
Supplementary Materials and Methods
Scheme S1. Preparation and tumor-targeted delivery of dual pH-sensitive nanodrug.
Fig. S1. The synthetic route and 1H NMR spectra of copolymers.
Fig. S2. Characterizations of copolymers, antibody, and nanomicelle.
Fig. S3. T cell binding and cellular uptake of nanodrug at pH 6.5.
Fig. S4. Cytotoxicities of nanomicelles and concentrations of CUR in tumor tissue.
Fig. S5. PD-1+ T cell binding of NR@PPC–aPD-1 in vivo.
Fig. S6. T1WI signal intensities of tumors and T1WI of the B16F10 tumor-bearing mice.
Fig. S7. CLSM images showing the colocalization of aPD-1 and PD-1 and intratumor distribution of nanodrug.
Fig. S8. In vivo immune response regulated by nanodrug.
Fig. S9. Schedule of in vivo study and biosafety of nanodrug.
Table S1. Molecular weight of the synthesized polymers.
Table S2. Sequences for forward and reverse specific primers for real-time reverse transcription PCR amplification.