

Abstract
Long non-coding RNAs (lncRNAs) are a growing focus of cancer genomics studies, creating the need for a resource of lncRNAs with validated cancer roles. Furthermore, it remains debated whether mutated lncRNAs can drive tumorigenesis, and whether such functions could be conserved during evolution. Here, as part of the ICGC/TCGA Pan-Cancer Analysis of Whole Genomes (PCAWG) Consortium, we introduce the Cancer LncRNA Census (CLC), a compilation of 122 GENCODE lncRNAs with causal roles in cancer phenotypes. In contrast to existing databases, CLC requires strong functional or genetic evidence. CLC genes are enriched amongst driver genes predicted from somatic mutations, and display characteristic genomic features. Strikingly, CLC genes are enriched for driver mutations from unbiased, genome-wide transposon-mutagenesis screens in mice. We identified 10 tumour-causing mutations in orthologues of 8 lncRNAs, including LINC-PINT and NEAT1, but not MALAT1. Thus CLC represents a dataset of high-confidence cancer lncRNAs. Mutagenesis maps are a novel means for identifying deeply-conserved roles of lncRNAs in tumorigenesis.
Subject terms: Cancer genomics, Comparative genomics
Joana Carlevaro-Fita, Andrés Lanzós et al. present the Cancer LncRNA Census (CLC), a manually curated dataset of 122 long noncoding RNAs (lncRNAs) with experimentally-validated functions in cancer based on data from the ICGC/TCGA Pan-Cancer Analysis of Whole Genomes (PCAWG) Consortium. CLC lncRNAs have unique gene features, and a number display evidence for cancer-driving functions that are conserved from humans to mice.
Introduction
Tumorigenesis is driven by a series of genetic mutations that promote cancer phenotypes and consequently experience positive selection1. The systematic discovery of such driver mutations, and the genes whose functions they alter, has been made possible by tumour genome sequencing. By collecting the entirety of such genes for every cancer type, it should be possible to develop a comprehensive view of underlying processes and pathways, and thereby formulate effective, targeted therapeutic strategies.
The cast of genetic elements implicated in tumorigenesis has recently grown as diverse new classes of non-coding RNAs and regulatory features have been discovered. These include the long non-coding RNAs (lncRNAs), of which tens of thousands have been catalogued2–5. LncRNAs are >200 nt long transcripts with no protein-coding capacity. Their evolutionary conservation and regulated expression, combined with a number of well-characterised examples, have together led to the view that lncRNAs are bona fide functional genes6–9. Current thinking holds that lncRNAs function by forming complexes with proteins and RNA both inside and outside the nucleus10,11.
LncRNAs have been shown to play important roles in various cancers. For example, MALAT1, an oncogene across numerous cancers, is restricted to the nucleus and plays a housekeeping role in splicing12,13. MALAT1 is overexpressed in a variety of cancer types, and its knockdown potently reduces not only proliferation but also metastasis in vivo in mouse xenograft assays14. MALAT1 is subjected to elevated mutational rates in human tumours, although it has not yet been established whether these mutations drive tumorigenesis15,16. On the other hand, lncRNAs may also function as tumour suppressors. LincRNA-p21 acts as a downstream effector of p53 regulation through recruitment of the repressor hnRNP-K17.
Demonstrably conserved functions between human and mouse is potent evidence for gene’s importance, both in cancer and more generally. For well-known protein-coding genes with cancer roles in human, such as TP53 and MYC, mutations in mouse models can recapitulate the human disease18,19. For lncRNAs, evolutionary evidence has been mainly limited to discovery of sequence or positional orthologues, with no evidence for conserved functions20. Further doubt has been introduced by the fact that mouse knockouts of iconic cancer-related lncRNAs MALAT1 and NEAT1 display little to no aberrant phenotype21–24. However, a recent study of human and mouse orthologues of LINC-PINT showed that both have tumour-suppressor activity in cell lines, acting through a relatively short, conserved region25. Nevertheless, it remains unclear whether this generalises to other identified lncRNAs, and whether mutations in them can induce tumours.
These and other examples of lncRNAs linked to cancer, raise the question of how many more remain to be found amongst the ~99% of annotated lncRNAs that are presently uncharacterised5,26,27. Recent tumour genome sequencing studies, in step with advanced bioinformatic driver-gene prediction methods, have yielded hundreds of new candidate protein-coding driver genes28. For economic reasons, these studies initially restricted their attention to exomes or the ~2% of the genome covering protein-coding exons29. Unfortunately such a strategy ignores mutations in the remaining ~98% of genomic sequence, home to the majority of lncRNAs5,12. Driver-gene identification methods rely on statistical models that make a series of assumptions about and simplifications of complex tumour mutation patterns30. It is critical to test the performance of such methods using true-positive lists of known cancer driver genes. For protein-coding genes, this role has been fulfilled by the Cancer Gene Census (CGC)31, which is collected and regularly updated by manual annotators. Comparison of driver predictions to CGC genes facilitates further method refinement and comparison between methods32–35.
In addition to its benchmarking role, the CGC resource has also been useful in identifying unique biological features of cancer genes. For example, CGC genes tend to be more conserved and longer. Furthermore, they are enriched for genes with transcription regulator activity and nucleic acid binding functions36,37.
Until very recently, efforts to discover cancer lncRNAs have depended on classical functional genomics approaches of differential expression using microarrays or RNA sequencing17,27. While valuable, differential expression per se is not direct evidence for causative roles in tumour evolution. To more directly identify lncRNAs that drive cancer progression, a number of methods, including several within the Pan-Cancer Analysis of Whole Genomes (PCAWG) Network16, have recently been developed to search for signals of positive selection using mutation maps of tumour genomes. OncodriveFML utilises nucleotide-level functional impact scores like those inferred from predicted changes in RNA secondary structure together with an empirical significance estimate, to identify lncRNAs with an excess of high-impact mutations34. Another method, ExInAtor, identifies candidates with elevated mutational load, using trinucleotide-adjusted local background15. Furthermore, The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes (PCAWG) Consortium aggregated whole genome sequencing data from 2658 cancers across 38 tumour types generated by the ICGC and TCGA projects38, and applied diverse tools to identify cancer driver lncRNAs16. A clear impediment in such analyses has been the lack of true-positive set of known lncRNA driver genes, analogous to CGC. Valuable resources of cancer lncRNAs have been created, notably LncRNADisease39 and Lnc2Cancer40. These include minimally filtered data from numerous sources, which is beneficial in creating inclusive gene lists, but has drawbacks arising from permissive criteria for inclusion (including expression changes), and inconsistent gene identifiers.
To facilitate the future discovery of cancer lncRNAs, and gain insights into their biology, we have compiled a highly-curated set of cases with roles in cancer processes. Here we present the Cancer LncRNA Census (CLC), the first compendium of lncRNAs with direct functional or genetic evidence for cancer roles. We demonstrate the utility of CLC in assessing the performance of driver lncRNA predictions. Through analysis of this gene set, we demonstrate that cancer lncRNAs have a unique series of features that may in future be used to assist de novo predictions. Finally, we show that CLC genes have conserved cancer roles across the ~80 million years of evolution separating humans and rodents.
Results
Definition of cancer-related lncRNAs
As part of recent efforts to identify driver lncRNAs by the Drivers and Functional Interpretation Group (PCAWG-2-5-9-14) within the ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Network (henceforth PCAWG)16,38, we discovered the need for a high-confidence reference set of cancer-related lncRNA genes, which we henceforth refer to as cancer lncRNAs. We here present Version 1 of the Cancer LncRNA Census (CLC).
Cancer lncRNAs were identified from the literature using defined and consistent criteria, being direct experimental (in vitro or in vivo) or genetic (somatic or germline) evidence for roles in cancer progression or phenotypes (see Methods). Alterations in expression alone were not considered sufficient evidence. Importantly, only lncRNAs with GENCODE identifiers were included to allow direct integration and comparison between large-scale genomic projects41. For every cancer lncRNA, one or more associated cancer types were collected.
Attesting to the value of this approach, we identified several cases in semi-automatically annotated cancer lncRNA databases of lncRNAs that were misassigned GENCODE identifiers, usually with an overlapping protein-coding gene39. We also excluded a number of published lncRNAs for which we could not find evidence to meet our criteria, for example CONCR, SRA1 and KCNQ1OT142–44. We plan to collect these excluded lncRNAs in future versions of CLC.
Version 1 of CLC contains 122 lncRNA genes, however, eight of them are annotated as pseudogenes rather than lncRNAs by GENCODE. The remaining 114 CLC genes correspond to 0.72% of a total of 15,941 lncRNA gene loci annotated in GENCODE v245,45 (Fig. 1). For comparison, the Cancer Gene Census (CGC) (COSMIC v78, downloaded 3 October 2016) lists 561 or 2.8% of protein-coding genes31. The entire remaining set of 15,827 lncRNA loci is henceforth referred to as non-CLC (Fig. 1). The full CLC dataset is found in Supplementary Data 1.
Fig. 1. Overview of the Cancer LncRNA Census.

Rows represent the 122 CLC genes, columns represent 29 cancer types. Asterisks next to gene names indicate that they are predicted as drivers by PCAWG, based either on gene or promoter evidence (see Supplementary Data 1). Blue cells indicate evidence for the involvement of a given lncRNA in that cancer type. Left column indicates functional classification: tumour suppressor (TSG), oncogene (OG) or both (OG/TSG). Above and to the right, barplots indicate the total counts of each column/row. The piechart shows the fraction that CLC represents within GENCODE v24 lncRNAs. Note that 8 CLC genes are classified as “pseudogenes” by GENCODE. “nonCLC” refers to all other GENCODE-annotated lncRNAs, which are used as background in comparative analyses.
The cancer classification terminology used amongst the source literature for CLC was not uniform. Therefore, using the International Classification of Diseases for Oncology46, we reassigned the cancer types described in the original research articles to a reduced set of 29 (Fig. 1 and Supplementary Fig. 1).
Altogether, CLC contains 333 unique lncRNA-cancer type relationships. Out of 122 genes, 77 (63.1%) were shown to function as oncogenes, 35 (28.7%) as tumour suppressors, and 10 (8.2%) with evidence for both activities depending on the tumour type (Fig. 1 and Supplementary Fig. 1). It is unclear whether the difference in the frequencies of oncogenes and tumour suppressors has a biological explanation, or is simply the result of ascertainment bias. For protein-coding genes in the CGC (COSMIC v85, downloaded 25 May 2018), approximately equal numbers of oncogenes and tumour-suppressor genes are recorded (43% and 44%, respectively). It is important to take into account that the oncogene and tumour-suppressor classifications were deduced from the collected references. While a gene has shown oncogenic properties in a particular cancer type, future publications could show that it functions as tumour suppressor in a different tissue, for example, the most studied lncRNAs in CLC (top of Fig. 1) are enriched in dual functions.
The most prolific lncRNAs, with ≥16 recorded cancer types, are HOTAIR, MALAT1, MEG3 and H19 (Fig. 1 and Supplementary Fig. 1). It is not clear whether this reflects their unique pan-cancer functionality, or is simply a result of their being amongst the most early-discovered and widely-studied lncRNAs.
In vitro experiments were the most frequent evidence source, usually consisting of RNAi-mediated knockdown in cultured cell lines, coupled to phenotypic assays such as proliferation or migration (Supplementary Fig. 1). Far fewer have been studied in vivo, or have cancer-associated somatic or germline mutations. Nineteen lncRNAs had three or more independent evidence sources (Supplementary Fig. 1).
CLC and other databases
There are a number of relevant lncRNA databases presently available: the Lnc2Cancer database (n = 654)40, the LncRNADisease database (n = 121)39 and lncRNAdb (n = 191)26. CLC covers between 17% and 31% of these databases (Lnc2Cancer and LncRNADisease, respectively) but none of these resources contain the complete list of genes presented here (Fig. 2a). It is important to note that the other databases also include a minority of non-GENCODE genes, ranging from 40 to 316 (33 and 48%) (Fig. 2a). In addition, we intersected the four databases (Supplementary Fig. 2) using only GENCODE-annotated genes. It is clear that CLC has the greatest overlap with the other three, suggesting that it has the greatest specificity.
Fig. 2. Intersection of CLC with public databases.
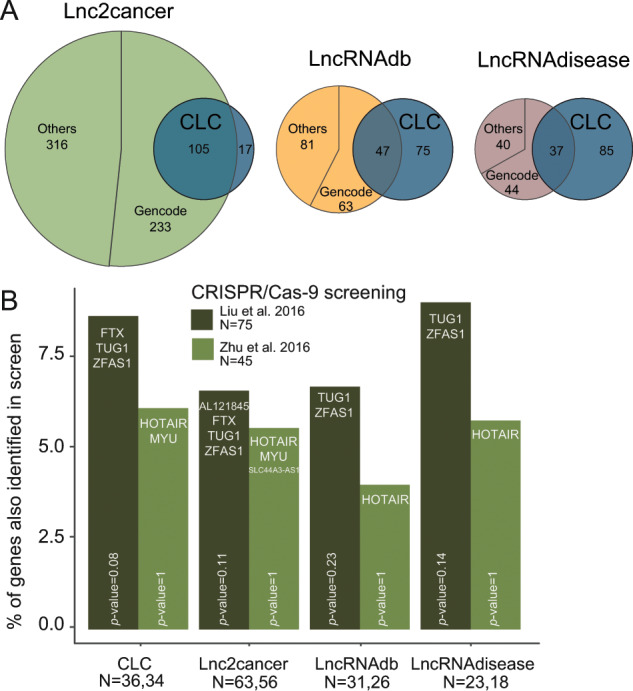
a Proportional Venn diagrams displaying the overlap between CLC set and the three indicated databases. Shown are the total numbers of unique human lncRNAs contained in each intersection (note that for LncRNADisease, numbers refer only to cancer-related genes). Databases are divided into genes that belong to GENCODE v24 annotation and others. b Barplot shows the percent of GENCODE v24 lncRNAs of each database that is present in the final list of cancer lncRNA candidates of two CRISPR/Cas-9 cancer screenings (Liu et al.9 and Zhu et al.47). N represents the number of GENCODE v24 lncRNAs from each database that were tested in each of the two CRISPR/Cas-9 screenings. Names of the genes that overlap between the databases and the screenings are shown in each bar. p-values were calculated using Fisher’s exact test.
We sought to use recent unbiased proliferation screen data to independently compare cancer lncRNA databases9,47. Using only GENCODE-annotated genes, CLC is the resource that overall has the most nearly-significant (p-value = 0.08, Fisher’s exact test) fraction of independently-identified proliferation lncRNAs, although the sparse nature of the data means that this conclusion is not definitive (Fig. 2b).
Finally, we downloaded and collected 8416 bioinformatically-predicted Gencode v24 lncRNAs from a recent TCGA publication48, but found no significant overlap with CLC (69 gene; p-value = 0.13, Fisher’s exact test).
CLC for benchmarking lncRNA driver prediction methods
One of the primary motivations for CLC is to develop a high-confidence functional set for benchmarking and comparing methods for identifying driver lncRNAs. In the domain of protein-coding driver-gene predictions, the Cancer Gene Census (CGC) has become such a gold standard training set31. Typically, the predicted driver genes belonging to CGC are judged to be true positives, and the fraction of these amongst predictions is used to estimate the positive predictive value (PPV), or precision. This measure can be calculated for increasing cutoff levels, to assess the optimal cutoff.
First, we used CLC to examine the performance of the lncRNA driver predictor ExInAtor15 in recalling CLC genes using PCAWG tumour mutation data16. A total of 2687 GENCODE lncRNAs were tested here, of which 82 (3.1%) belong to CLC. Driver predictions on several cancers at the standard false discovery rate (q-value) cutoff of 0.1 are shown for selected cancers in Fig. 3a. That panel shows the CLC-defined precision (y-axis) as a function of predicted driver genes ranked by q-value (x-axis). We observe rather heterogeneous performance across cancer cohorts. This may reflect a combination of intrinsic biological differences and differences in cohort sizes, which differs widely between the datasets shown. For the merged pan-cancer dataset, ExInAtor predicted three CLC genes amongst its top ten candidates (q-value < 0.1), a rate far in excess of the background expectation (baseline, fraction of all lncRNAs in CLC). Similar enrichments are observed for other cancer types. These results support both the predictive value of ExInAtor, and the usefulness of CLC in assessing lncRNA driver predictors. In addition, we repeated the same analysis for each of the three mentioned databases (lnc2cancer, lncRNAdb and lncRNAdisease) (q-value < 0.2) (Supplementary Fig. 3). The precision level of all databases is around 40%, except lncRNAdisease that shows the overall lowest precision. As deduced from Fig. 2, the low number of intersecting genes does not allow a definitive conclusion. However, it is interesting to notice that CLC shows a similar performance to the other databases in terms of sensitivity while increasing specificity. This is likely due to the stringent, function-based inclusion criteria of CLC.
Fig. 3. CLC as benchmark for cancer driver predictions.

a CLC benchmarking of ExInAtor driver lncRNA predictions using PCAWG whole genome tumours at q-value (false discovery rate) cutoff of 0.1. Genes sorted increasingly by q-value are ranked on x-axis. Percentage of CLC genes amongst cumulative set of predicted candidates at each step of the ranking (precision), are shown on the y-axis. Black line shows the baseline, being the percentage of CLC genes in the whole list of genes tested. Coloured dots represent the number of candidates predicted under the q-value cutoff of 0.1. “n” in the legend shows the number of CLC and total candidates for each cancer type. b Rate of driver-gene predictions amongst CLC and non-CLC genesets (q-value cutoff of 0.1) by all the individual methods and the combined list of drivers developed in PCAWG. p-value is calculated using Fisher’s exact test for the difference between CLC and non-CLC genesets. c Rate of driver-gene predictions amongst CGC and nonCGC genesets (q-value cutoff of 0.1) by all the individual methods and the combined list of drivers developed in PCAWG. p-value is calculated using Fisher’s exact test for the difference between CGC and nonCGC genesets.
Finally, we assessed the precision (i.e. positive predictive value) of PCAWG lncRNA and protein-coding driver predictions across all cancers and all prediction methods16. Using a q-value cutoff of 0.1, we found that across all cancer types and methods, a total of 8 (8.5%) of lncRNA predictions belong to CLC (Fig. 3b), while a total of 139 (23.1%) of protein-coding predictions belong to CGC (Fig. 3c). In terms of sensitivity, 9.8% and 25.1% of CLC and CGC genes are predicted as candidates, respectively. Despite the lower detection of CLC genes in comparison with CGC genes, both sensitivity rates significantly exceed the prediction rate of non-CLC and nonCGC genes (p-value = 0.007 and p-value < 0.001 Fisher’s exact tests, respectively), again highlighting the usefulness of the CLC gene set (Fig. 3c).
CLC genes are distinguished by function- and disease-related features
We recently found evidence, using a smaller set of cancer-related LncRNAs (CRLs), that cancer lncRNAs are distinguished by various genomic and expression features indicative of biological function15. We here extended these findings using a large series of potential gene features, to search for those features distinguishing CLC from non-CLC lncRNAs (Fig. 4a).
Fig. 4. Distinguishing features of CLC genes.

a Panel showing a hypothetic feature analysis example to illustrate the content of the following figures. All panels in this figure display features (dots), plotted by their log-fold difference (odds ratio in case of panel (b)) between CLC/non-CLC genesets (y-axis) and statistical significance (x-axis). In all plots dark and light green dashed lines indicate 0.05 and 0.01 significance thresholds, respectively. b Cancer and non-cancer disease-related data from indicated sources: y-axis shows the log2 of the odds ratio obtained by comparing CLC to non-CLC by Fisher’s exact test; x-axis displays the estimated p-value from the same test. “CGC 1 kb TSS” refers to the fraction of genes that have a nearby known CGC cancer protein-coding gene. This is explored in more detail in the next Figure. “Non-cancer SNPs” refers to GWAS SNPs associated with diseases/traits other than cancer. c Sequence and gene properties: y-axis shows the log2 fold difference of CLC/non-CLC means; x-axis represents the p-value obtained. d Evolutionary conservation: “Phastc mean” indicates average base-level PhastCons score; “Elements” indicates percent coverage by PhastCons conserved elements (see Methods). Colours distinguish exons (blue) and promoters (purple). e Tumour RNA-seq: expression levels of lncRNA genes in different cancer tissues obtained from RNA-seq expression data from PCAWG. For (b–d), statistical significance was calculated using Wilcoxon test.
First, associations with expected cancer-related features were tested (Fig. 4b). CLC genes are significantly more likely to have their transcription start site (TSS) within 100 kb of cancer-associated germline SNPs (cancer SNPs 100 kb TSS), and more likely to be either differentially expressed or epigenetically-silenced in tumours49 (Fig. 4b). Intriguingly, we observed a tendency for CLC lncRNAs to be more likely to lie within 1 kb of known cancer protein-coding genes (CGC 1 kb TSS). While searching for additional evidence of functionality for CLC genes, we found that they are significantly closer to non-cancer, phenotype-associated germline SNPs (non-cancer SNPs 100 kb TSS) in comparison with non-CLC genes (Fig. 4b). Proximity to cancer and non-cancer SNPs support the both cancer roles and general biological functionality of CLC genes.
We next investigated the properties of the genes themselves. As seen in Fig. 4c, and consistent with our previous findings15, CLC genes (gene length) and their spliced products (exonic length) are significantly longer than average. No difference was observed in the ratio of exonic to total length (exonic content), nor overall exon repetitive sequence coverage (repeats coverage), nor GC content.
CLC genes also tend to have greater evidence of function, as inferred from evolutionary conservation. Base-level conservation at various evolutionary depths was calculated for lncRNA exons and promoters (Fig. 4d). Across all measures tested, using either average base-level scores or percent coverage by conserved elements, we found that CLC genes’ exons are significantly more conserved than other lncRNAs (Fig. 4d). The same was observed for conservation of promoter regions.
High levels of gene expression in normal tissues are known to correlate with lncRNA conservation, and are hypothesized to be a reflection of functionality50. In addition, genes with oncogenic roles tend to be highly expressed in cancer samples36. We found that CLC has consistently higher steady-state expression levels compared with non-CLC genes across PCAWG tumours (Fig. 4e), as well as healthy organs and cultured cell lines (Supplementary Fig. 4). As deduced from proximity to cancer and non-cancer SNPs, high levels of expression in cancer and normal samples reflect important functionality for CLC genes.
Finally, we investigated whether CLC transcripts might be initiated by any types of Transposable Elements (TEs) (see Methods). We found that CLC TSSs are enriched for one category, “Simple repeats” (Supplementary Fig. 5).
Evidence for genomic clustering of non-coding and protein-coding cancer genes
In light of recent evidence for colocalisation and coexpression of disease-related lncRNAs and protein-coding genes51, we were curious whether such an effect holds for cancer-related lncRNAs and protein-coding genes. We asked, more specifically, whether CLC genes tend to be closer to CGC genes than expected by chance, and whether this is manifested in a more co-regulated expression.
To this aim, we computed TSS-TSS distances from lncRNAs to protein-coding genes and we found that CLC genes on average tend to lie moderately closer to protein-coding genes of all types, compared with non-CLC lncRNAs (Supplementary Fig. 6A, B). Since CLC genes are enriched for functional features (i.e. expression and conservation), we could not rule out the possibility that proximity to protein-coding genes is a feature of functional lncRNAs rather than cancer lncRNA genes. In order to further investigate this possibility, we repeated the analysis dividing the non-CLC set into potentially functional non-CLC genes (PF-non-CLC) (non-CLC genes sampled to match CLC expression and conservation, N = 149, Supplementary Fig. 7) and “other nonCLC” (the rest of non-CLC). Interestingly, when comparing distances to any type of protein-coding genes, both CLC and PF-non-CLC are significantly closer than the rest of lncRNA (Wilcoxon test, p-value = 0.03 and 0.007, respectively), being the PF-non-CLC genes the closest ones (median 21.9, 29 and 37.8 kb, for PF-non-CLC, CLC and other non-CLC, respectively) (Supplementary Fig. 6C). However, when assessing specifically for distance to CGC genes, only CLC set is significantly closer than the rest of lncRNAs (Wilcoxon test, p-value = 0.0008) and it represents the group with the lowest distance (median 1122, 1330 and 1607 kb for CLC, PF-non-CLC and other non-CLC, respectively) (Fig. 5a). Thus, although proximity to protein-coding genes seems to be a feature of potentially functional lncRNAs, CLC genes are closer to cancer genes compared with other lncRNAs with similar function-like properties.
Fig. 5. Evidence for genomic clustering of non-coding and protein-coding cancer genes.

a Cumulative distribution of the genomic distance of lncRNA transcription start site (TSS) to the closest Cancer Gene Census (CGC) (protein-coding) gene TSS. LncRNAs are divided into CLC (n = 122), potentially functional non-CLC genes (PF-non-CLC) (n = 149), and other non-CLC genes (n = 15,678). b Boxplot shows the distribution of the gene expression correlation between CLC and their closest CGC genes in 11 human cell lines, including two control analyses (distance-matched non-CLC-CGC pairs, and shuffled CLC-CGC pairs). Correlation was calculated for gene pairs within each cell type, using Pearson method. p-value for Kolmogorov–Smirnov test is shown. c Genomic classification of lncRNAs. Genes are classified according to distance and orientation to the closest protein-coding gene, and these are grouped into three categories: genes closer than 10 kb to closest protein-coding gene, genes overlapping a protein-coding gene and intergenic genes (>10 kb from closest protein-coding gene). p-values for Fisher’s exact tests are shown. d The percentage of divergent CLC (left bar) and non-CLC (right bar) genes divergent to a cancer protein-coding gene (CGC). Numbers represent numbers of genes with which the percentage is calculated. p-value for Fisher’s exact test is shown. e Functional annotations of the 20 protein-coding genes (pc-genes) divergent to CLC genes from panel (c). Bars indicate the –log10 (corrected) p-value (see Methods) and are coloured based on the “enrichment”: the number of genes that contain the functional term divided by the total number of queried genes. Numbers at the end of the bars correspond to the number of genes that fall into the category.
It has been widely proposed that proximal lncRNA/protein-coding gene pairs are involved in cis-regulatory relationships, which is reflected in expression correlation52. We next asked whether proximal CLC-CGC pairs exhibit this behaviour. An important potential confounding factor, is the known positive correlation between nearby gene pairs53, and this must be controlled for. Using gene expression data across 11 human cell lines, we observed a positive correlation between CLC-CGC gene pairs for each cell type (Fig. 5b). To control for the effect of proximity on correlation, we next randomly sampled a similar number of non-CLC lncRNAs with matched distances (TSS-TSS) from the same CGC genes, and found that this correlation was lost (Fig. 5b, “nonCLC-CGC”). To further control for a possible correlation arising from the simple fact that both CGC and CLC genes are involved in cancer, and CLC genes are in general enriched for conservation and expression, we next randomly shuffled the CLC-CGC pairs 1000 times, again observing no correlation (Fig. 5b, “Shuffled CLC-CGC”). Together these results show that genomically proximal protein-coding/non-coding gene pairs exhibit an expression correlation that exceeds that expected by chance, even when controlling for genomic distance.
These results prompted us to further explore the genomic localization of CLC genes relative to their proximal protein-coding gene and the nature of their neighbouring genes. Next, we observed an unexpected difference in the genomic organisation of CLC genes: when classified by orientation with respect to nearest protein-coding gene5, we found a significant enrichment of CLC genes immediately downstream and on the same strand as protein-coding genes (“Samestrand, pc up”, Fig. 5c). Moreover, CLC genes are approximately twice as likely to lie in an upstream, divergent orientation to a protein-coding gene (“Divergent”, Fig. 5c). Of these CLC genes, 20% are divergent to a CGC gene, compared with 5% for non-CLC genes (p-value = 0.018, Fisher’s exact test) (Fig. 5d), and several are divergent to protein-coding genes that have also been linked or defined to be involved in cancer, despite not being classified as CGCs (Supplementary Data 2).
Given this noteworthy enrichment of CGC genes among the divergent protein-coding genes of the CLC set, we next inspected the functional annotation of those protein-coding genes. Examining their Gene Ontology (GO) terms, molecular pathways and other gene function related terms, we found this group of genes to be enriched in GO terms for “sequence-specific DNA binding”, “DNA binding”, “tube development” and “transcriptional misregulation in cancer” (Fig. 5e and Supplementary Data 3), contrary to the GO terms of the divergent protein-coding genes of the non-CLC set (Supplementary Data 4). These results were confirmed by another, independent GO-analysis suite (see Methods). Interestingly, three out of the top four functional groups were observed previously in a study of protein-coding genes divergent to long upstream antisense transcripts in primary mouse tissues54.
Thus, CLC genes appear to be non-randomly distributed with respect to protein-coding genes, and particularly their CGC subset.
Evidence for anciently conserved cancer roles of lncRNAs
In mouse, numerous studies have employed unbiased forward genetic screens to identify genes that either inhibit or promote tumorigenesis55. These studies use engineered, randomly-integrating transposons carrying bidirectional polyadenylation sites as well as strong promoters. Insertions, or clusters of insertions, called “common insertion sites” (CIS) that are identified in sequenced tumour DNA, are assumed to act as driver mutations55, and thereby implicate the overlapping or neighbouring gene locus as either an oncogene or tumour-suppressor gene. Although these studies have traditionally been focused on identifying protein-coding driver genes, they can in principle also identify non-coding RNA driver loci55.
We thus reasoned that comparison of mouse CISs to orthologous human regions could yield independent evidence for the functionality of human cancer lncRNAs (Fig. 6a). To test this, we collected a comprehensive set of CISs in mouse56, consisting of 2906 loci from seven distinct cancer types (Supplementary Data 5). These sites were then mapped to orthologous regions in the human genome, resulting in 1301 non-overlapping human CISs, or hCISs. 6.9% (90) of these CISs lie outside of protein-coding gene boundaries.
Fig. 6. Evidence for ancient conserved cancer roles of lncRNAs.

a Functional conservation of human CLC genes was inferred by the presence of Common Insertion Sites (CIS), identified in transposon-mutagenesis screens, at orthologous regions in the mouse genome. Orthology was inferred from Chain alignments and identified using LiftOver utility. b Number of CLC and non-CLC genes that contain human orthologous common insertion sites (hCIS) (see Table 1). Significance was calculated using Fisher’s exact test. c UCSC browser screenshot of a CLC gene (SLNCR1, ENSG00000227036) intersecting a CIS (yellow arrow). d Number of basepairs and number of overlapping hCIS for cancer driver protein-coding genes (CGC), non-cancer driver protein-coding genes (nonCGC), cancer-related lncRNAs (CLC), rest of GENCODE lncRNAS (non-CLC) and the rest of the genome that do not overlap any of the previous element types (intergenic). Arrows indicate the number of hCIS and the percentage for each element type. e Number of overlapping hCIS per megabase of genomic span for each gene class.
Mapping hCISs to lncRNA annotations, we discovered altogether eight CLC genes (6.6%) carrying at least one insertion within their gene span: DLEU2, GAS5, MONC, NEAT1, PINT, PVT1, SLNCR1, XIST (Table 1). Two cases, DLEU2 and MONC, each have two independent hCIS sites. In contrast, just 64 (0.4%) non-CLC lncRNAs contained hCISs (Fig. 6b). A good example is SLNCR1, shown in Fig. 6c, which drives invasiveness of human melanoma cells57, and whose mouse orthologue contains a CIS discovered in pancreatic cancer. It is noteworthy that no hCIS was found to overlap MALAT1 despite its being amongst the most widely-studied cancer lncRNAs14. This agrees with the lack of strong phenotypic effects when deleting this gene in mouse models, as discussed in the Introduction21–23. We examined the possibility that hCIS insertions in these CLC genes could in fact be caused by nearby, protein-coding cancer genes. However, none of these eight CLC genes are within 100 kb of a CGC gene, with the exception of PVT1 lncRNA, lying 58 kb from c-MYC oncogene.
Table 1.
List of intergenic CIS human (GRCh38)/mouse (GRCm38) gene pairs.
| Human CLC name | Human CLC ID | Chr human | Start human | End human | Chr mouse | Start mouse | End mouse | PubMed ID | Cancer type mouse |
|---|---|---|---|---|---|---|---|---|---|
| DLEU2 | ENSG00000231607 | chr13 | 50,048,971 | 50,049,063 | chr14 | 61,631,880 | 61,631,972 | 24316982 | Liver |
| DLEU2 | ENSG00000231607 | chr13 | 50,049,117 | 50,049,206 | chr14 | 61,632,026 | 61,632,110 | 24316982 | Liver |
| GAS5 | ENSG00000234741 | chr1 | 173,864,370 | 173,864,435 | chr1 | 161,038,091 | 161,038,156 | 25961939 | Sarcoma |
| MONC | ENSG00000215386 | chr21 | 16,539,096 | 16,539,161 | chr16 | 77,598,935 | 77,599,000 | 23685747 | Nervous System |
| MONC | ENSG00000215386 | chr21 | 16,561,654 | 16,561,655 | chr16 | 77,616,439 | 77,616,440 | 24316982 | Liver |
| NEAT1 | ENSG00000245532 | chr11 | 65,444,511 | 65,444,512 | chr19 | 5,825,497 | 5,825,498 | 24316982 | Liver |
| PINT | ENSG00000231721 | chr7 | 131,049,455 | 131,049,456 | chr6 | 31,179,149 | 31,179,150 | 22699621 | Pancreatic |
| PVT1 | ENSG00000249859 | chr8 | 128,007,970 | 128,007,971 | chr15 | 62,186,646 | 62,186,647 | 22699621 | Pancreatic |
| SLNCR1 | ENSG00000227036 | chr17 | 72,507,275 | 72,507,276 | chr11 | 113,137,613 | 113,137,614 | 22699621 | Pancreatic |
| XIST | ENSG00000229807 | chrX | 73,841,539 | 73,841,540 | chrX | 103,473,862 | 103,473,863 | 24316982 | Liver |
This analysis would suggest that CLC genes are enriched for hCISs; however, there remains the possibility that this is confounded by their greater length and possible overlap with protein-coding genes. To account for this, we only selected hCIS elements that do not overlap protein-coding regions (90 hCIS) and we performed two separate validations using only regions that do not overlap protein-coding genes from the CLC and non-CLC genesets. First, groups of non-CLC genes with CLC-matched length were randomly sampled, and the number of intersecting hCISs per unit gene length (Mb) was counted (Supplementary Fig. 8A). Second, CLC genes were randomly relocated in the genome, and the number of genes intersecting at least one hCIS was counted (Supplementary Fig. 8B). Both analyses showed that the number of intersecting hCISs per Mb of CLC gene span is far greater than expected in comparison with both non-CLC genes (Supplementary Fig. 8A) and intergenic space (nucleotides that do not overlap neither lncRNAs neither protein-coding genes) (Supplementary Fig. 8B). Interestingly, non-CLC genes also show an enrichment for hCIS sites in comparison with intergenic regions (Supplementary Fig. 8C), suggesting that more cancer lncRNAs remain to be discovered.
We further compared the enrichment of hCIS in protein-coding genes, lncRNA genes and other intergenic space. Compared with the genomic space they occupy, there is a clear enrichment of hCIS elements in both protein-coding CGC genes, as well as CLC lncRNAs (Fig. 6d). Expressed as insertion rate per megabase of gene span, it is clear that CLC genes are targeted more frequently than background intergenic DNA and non-cancer-related lncRNA genes. Of note are the non-background insertion rates for non-cancer-related protein-coding (nonCGC) and lncRNA genes (non-CLC), suggesting that there remain substantial numbers of undiscovered cancer genes in both groups.
Together these analyses demonstrate that CLC genes are orthologous to mouse cancer-causing genomic loci at a rate greater than expected by random chance. These identified cases, and possibly other CLC genes, display cancer functions that have been conserved over tens of millions of years since human-rodent divergence.
Discussion
We have presented the Cancer LncRNA Census, the first controlled set of GENCODE-annotated lncRNAs with demonstrated roles in tumorigenesis or cancer phenotypes.
The present state of knowledge of lncRNAs in cancer, and indeed lncRNAs generally, remains incomplete. Consequently, our aim was to create a gene set with the greatest possible confidence, by eliminating the relatively large number of published cancer lncRNAs with as-yet unproven functional roles in disease processes. Thus, we defined cancer lncRNAs as those having direct experimental or genetic evidence supporting a causative role in cancer phenotypes. By this measure, gene expression changes alone do not suffice. By introducing these well-defined inclusion criteria, we hope to ensure that CLC contains the highest possible proportion of bona fide cancer genes, giving it maximum utility for de novo predictor benchmarking. In addition, its basis in GENCODE ensures portability across datasets and projects. Inevitably some well-known lncRNAs did not meet these criteria (including SRA1, CONCR, KCNQ1OT1)42–44; these may be included in future when more validation data becomes available. We believe that CLC will complement the established lncRNA databases such as lncRNAdb, LncRNADisease and Lnc2Cancer, which are more comprehensive, but are likely to have a higher false-positive rate due to their more relaxed inclusion criteria26,39,40.
De novo lncRNA driver-gene discovery is likely to become increasingly important as the number of sequenced tumours grow. The creation and refinement of statistical methods for driver-gene discovery will depend on the available of high-quality true-positive genesets such as CLC. It will be important to continue to maintain and improve the CLC in step with anticipated growth in publications on validated cancer lncRNAs. Very recently, CRISPR-based screens9,47 have catalogued large numbers of lncRNAs contributing to proliferation in cancer cell lines, which will be incorporated in future versions.
We used CLC to estimate the performance of de novo driver lncRNA predictions from the PCAWG project, made using the ExInAtor pipeline15. Supporting the usefulness of this approach, we found an enrichment for CLC genes amongst the top-ranked driver predictions. Extending this to the full set of PCAWG driver predictors, approximately ten percent of CLC genes (9.8%) are called as drivers by at least one method16, which is lower to the rate of CGC genes identified (25.1%).
The low rate of concordance between de novo predictions and CLC genes may be due to technical or biological factors. Indeed, it is important to state that we do not yet know whether CLC holds “cancer driver” lncRNAs, and indeed, how many such genes exist. In principle, lncRNAs may play two distinct roles in cancer: first, as driver genes, defined as those whose mutations are early and positively-selected events in tumorigenesis; or second, as “downstream genes”, which do make a genuine contribution to cancer phenotypes, but through non-genetic alterations in cellular networks resulting from changes in expression, localisation or molecular interactions. These downstream genes may not display positively-selected mutational patterns, but would be expected to display cancer-specific alterations in expression. A key question for the future is how lncRNAs break down between these two categories, and the utility of CLC in benchmarking de novo driver predictions will depend on this. However, the identification of lncRNAs whose silencing or overexpression is sufficient for tumour formation in mouse, would seem to suggest that they are true “driver genes”.
Analysis of the CLC gene set has broadened our understanding of the unique features of cancer lncRNAs, and generally supports the notion that lncRNAs have intrinsic biological functionality. Cancer lncRNAs are distinguished by a series of features that are consistent with both roles in cancer (e.g. tumour expression changes), and general biological functionality (e.g. high expression, evolutionary conservation). Elevated evolutionary conservation in the exons of CLC genes would appear to support their functionality as a mature RNA transcript, in contrast to the act of their transcription alone58. Another intriguing observation has been the colocalisation of cancer lncRNAs with known protein-coding cancer genes: these are genomically proximal and exhibit elevated expression correlation. This points to a regulatory link between cancer lncRNAs and protein-coding genes, perhaps through chromatin looping, as described in previous reports for CCAT1 and MYC, for example59.
One important caveat for all features discussed here is ascertainment bias: almost all lncRNAs discussed have been curated from published, single-gene studies. It is entirely possible that selection of genes for initial studies was highly non-random, and influenced by a number of factors—including high expression, evolutionary conservation and proximity to known cancer genes—that could bias our inference of lncRNA features. This may be the explanation for the observed excess of cancer lncRNAs in divergent configuration to protein-coding genes. However, the general validity of some of the CLC-specific features described here—including high expression and evolutionary conservation—were also observed in recent unbiased genome-wide screens9,15, suggesting that they are genuine.
Despite the relatively low concordance of CLC genes with PCAWG driver predictions, the results of this study strongly support the value and key cancer role of identified lncRNAs in cancer. Most notably, the existence of a core set of eight lncRNAs with independently-identified mouse orthologues with similar cancer functions, is a powerful evidence that these genes are bona fide cancer genes, whose overexpression or silencing can drive tumour formation. To our knowledge this is the most direct demonstration to date of anciently conserved functions and disease roles for lncRNAs. It will be intriguing to investigate in future whether more human-mouse orthologous lncRNAs have been identified in such screens.
Methods
Manual curation
All lncRNAs in lncRNAdb and those listed in Schmitt and Chang’s recent review article were collected26,60. To these were added all cases from LncRNADisease and Lnc2Cancer databases39,40. This primary list formed the basis for a manual literature search: all available publications for each gene were identified by keyword search in PubMed. If publications were found conforming to at least one of the inclusion criteria (below) and the gene has a GENCODE ID, then it was added to CLC, with appropriate information on the associated cancer, biological activity. For the numerous cases where no GENCODE ID was supplied in the original publication, any available ID, or primer or siRNA sequence was used to identify the gene using the UCSC Genome Browser Blat tool61.
Inclusion criteria sufficient to define a cancer lncRNA and link it to a cancer type were
Class t: In vitro demonstration that their knockdown and/or overexpression in cultured cancer cells results in changes to cancer-associated phenotypes. These typically include proliferation rates, migration, sensitivity to apoptosis, or anchorage-independent growth.
Class v: In vivo demonstration that their knockdown and/or overexpression in cancer cells alters their tumorigenicity when injected into animal models.
Class g: Germline mutations or variants that predispose humans to cancer.
Class s: Somatic mutations that show evidence for positive selection during tumour formation.
An additional criterion was allowed to link an lncRNA to a cancer type, only if at least one of the above criteria was already met for another cancer:
Class p: Prognosis, the lncRNAs expression is statistically linked to disease progression or response to treatment.
If an lncRNA was found to promote tumorigenesis or cancer phenotype, it was defined as “oncogene”. Conversely those found to inhibit such phenotypes were defined as “tumour suppressor”. Several lncRNAs were found to have both activities recorded in different cancer types, and were given both labels. For every lncRNA-cancer association, a single representative publication is recorded. Finally, it is important to note that no lncRNAs were included based on evidence from previous driver-gene discovery studies of the types represented by OncodriveFML, ExInAtor, ncdDetect or others described in PCAWG15,16,34,62.
CLC set at this stage relies on GENCODE v24 annotation, and therefore all CLC genes have a GENCODE v24 ID assigned. However, data relative to GENCODE v24 was not available for all types of data and analyses used in this study (i.e. all data relative to PCAWG is based on GENCODE v19). Thus, for some analyses only genes also present in GENCODE v19 could be used (specified in the corresponding methods sections) and the total number of genes analyzed in these cases is slightly lower (107 instead of 122 CLC genes and 13,503 instead of 15,827 non-CLC).
LncRNA and protein-coding driver prediction analysis
LncRNA and protein-coding predictions for ExInAtor and the rest of PCAWG methods, as well as the combined list of drivers, were extracted from the consortium database16. Parameters and details about each individual methods and the combined list of drivers can be found on the main PCAWG driver publication16 and false discovery rate correction was applied on each individual cancer type for each individual method in order to define candidates (q-value cutoffs of 0.1 and 0.2, specified in the corresponding sections). This way, we combined the predicted candidates of each individual method in each individual cancer type (including pan-cancer). To calculate sensitivity (percentage of true positives that are predicted as candidates) and precision (percentage of predicted candidates that are true positives) for lncRNA and protein-coding predictions we used the CLC and CGC (COSMIC v78, downloaded 3 October 2016) sets, respectively. To assess the statistical significance of sensitivity rates, we used Fisher’s exact test.
Feature identification
We compiled several quantitative and qualitative traits of GENCODE lncRNAs and used them to compare CLC genes to the rest of lncRNAs (referred to as “non-CLC”). Analysis of quantitative traits were performed using Wilcoxon test while qualitative traits were tested using Fisher' exact test. These methods principally refer to Figs. 4 and 5 as well as Supplementary Figs. 4, 5, 6 and 7.
Cancer SNPs: On 4 October 2016, we collected all 2192 SNPs related to “cancer”, “tumour” and “tumor” terms in the NHGRI-EBI Catalog of published genome-wide association studies63,64 (https://www.ebi.ac.uk/gwas/home). Then we calculated the closest SNP to each lncRNA TSS using closest function from Bedtools v2.1965 (GENCODE v24).
Non-cancer SNPs: On 31 July 2017, we collected all 29,813 SNPs not related to “cancer”, “tumour” and “tumor” terms in the NHGRI-EBI Catalog of published genome-wide association studies63,64 (https://www.ebi.ac.uk/gwas/home). Then we calculated the closest SNP to each lncRNA TSS using closest function from Bedtools v2.1965 (GENCODE v24).
Epigenetically-silenced lncRNAs: We obtained a published list of 203 cancer-associated epigenetically-silenced lncRNA genes present in GENCODE v2449. These candidates were identified due to DNA methylation alterations in their promoter regions affecting their expression in several cancer types.
Differentially expressed in cancer: We collected a list of 3533 differentially expressed lncRNAs in cancer compared with normal samples49 (GENCODE v24).
Sequence/gene properties: Exonic positions of each gene were defined as the the union of exons from all its transcripts. Introns were defined as all remaining non-exonic nucleotides within the gene span. Repeats coverage refers to the percent of exonic nucleotides of a given gene overlapping repeats and low complexity DNA sequence regions obtained from RepeatMasker data housed in the UCSC Genome Browser66. Exonic content refers to the fraction of total gene span covered by exons. For this section we used GENCODE v19.
Evolutionary conservation: Two types of PhastCons conservation data were used: base-level scores and conserved elements. These data for different multispecies alignments (GRCh38/hg38) were downloaded from UCSC genome browser66. Mean scores and percent overlap by elements were calculated for exons and promoter regions (GENCODE v24). Promoters were defined as the 200 nt region centred on the annotated gene start.
Expression: We used polyA+RNA-seq data from 10 human cell lines produced by ENCODE67,68, from various human tissues by the Illumina Human Body Map Project (HBM) (www.illumina.com; ArrayExpress ID: E-MTAB-513), and from cancer samples from PCAWG RNA-seq expression data16. In this last case, for each cancer type we computed the expression mean of genes across all RNA-seq samples belonging to that cancer type (GENCODE v19).
Transposable elements: We downloaded 5,520,016 transposable elements from the UCSC table browser69 on 3 August 2017. We separated them by element types and counted how many of them intersected or not with the transcription start sites of CLC and non-CLC genes, in order to detect any association with the Fisher' exact test.
Distance to protein-coding genes and CGC genes: For each lncRNA we calculated the TSS to TSS distance to the closest protein-coding gene (GENCODE v24) or CGC gene (downloaded on 3 October 2016 from Cosmic database)31 using closest function from Bedtools v2.1965. In order to divide non-CLC genes into potentially functional non-CLC (PF-non-CLC) and others, we sampled the list of all non-CLC genes to get a subsample that has a matched distribution to CLC genes in conservation (% of conserved elements, from Vertebrate Multiz Alignment 100 Species from UCSC genome browser data, in exonic regions). Then we sampled again the resulting subset to get a final subset that also matches CLC genes in terms of expression (median of expression across 16 human tissues, data from Illumina Human Body Map Project (HBM)). To create the non-CLC samples we used the matchDistribution script: https://github.com/julienlag/matchDistribution.
Coexpression with closest CGC gene: We took CLC-CGC gene pairs whose TSS-TSS distance was <200 kb. RNA-seq data from 11 human cell lines from ENCODE was used to assess expression levels67,68. ENCODE RNA-seq data were obtained from ENCODE Data Coordination Centre (DCC) in September 2016, https://www.encodeproject.org/matrix/?type=Experiment. All data is relative to GENCODE v24. We calculated the expression correlation of gene pairs within each of the 11 cell lines, using the Pearson measure. To control for the effect of proximity, we randomly sampled a subset of non-CLC-CGC pairs matching the same TSS-TSS distance distribution as above, and performed the same expression correlation analysis (“non-CLC-CGC”). Finally, to further control for the fact that CLC and CGC are both cancer genes, which may influence their expression correlation, we shuffled CLC-CGC pairs 1000 times, and tested expression correlation for each set (“Shuffled CLC-CGC”).
Genomic classification: We used an in-house script (https://github.com/gold-lab/shared_scripts/tree/master/lncRNA.annotator) to classify lncRNA transcripts into different genomic categories based on their orientation and proximity to the closest protein-coding gene (GENCODE v24): a 10 kb distance was used to distinguish “genic” from “intergenic” lncRNAs. When transcripts belonging to the same gene had different classifications, we used the category represented by the largest number of transcripts.
Functional enrichment analysis: The list of protein-coding genes (GENCODE v24) that are divergent and closer than 10 kb to CLC genes (or non-CLC) was used for a functional enrichment analysis (20 unique genes in the case of CLC analysis and 1202 in the case of non-CLC analysis). We show data obtained using g:Profiler web server70, g:GOSt, with default parameters for functional enrichment analysis of protein-coding genes divergent to CLC and using Bonferroni correction for protein-coding gene divergent to non-CLC. For CLC analysis we performed the same test with independent methods: Metascape (http://metascape.org)71 and GeneOntoloy (Panther classification system)72,73. In both cases similar results were found.
Mouse mutagenesis screen analysis
We extracted the genomic coordinates of transposon common insertion sites (CISs) in Mouse (GRCm38/mm10) http://ccgd-starrlab.oit.umn.edu/about.php56. This database contains target sites identified by transposon-based forward genetic screens in mice. LiftOver61 was used at default settings to obtain aligned human genome coordinates (hCISs) (GRCh38/hg38). We discarded hCIS regions longer than 1000 nucleotides for all the analyses; and also those that overlap protein-coding genes (except for Fig. 6b). The remainders (90 hCISs) were intersected with the genomic coordinates of CLC and non-CLC genes that do not overlap protein-coding genes.
To correctly assess the statistical enrichment of CLC in hCIS regions, we performed two control analyses:
Length-matched sampling: To calculate if the enrichment of hCIS intersecting genes in CLC set is higher and statistically different from non-CLC set, while controlling by gene length, we created 1000 samples of non-CLC genes with the same gene length distribution as CLC genes. Each sample was intersected with hCIS, and the number of intersecting hCISs per Mb of gene length was calculated. To create the non-CLC samples we used the matchDistribution script: https://github.com/julienlag/matchDistribution. Finally, we calculated an empirical p-value by counting how many of the simulated non-CLC enrichments were higher or equal than the real CLC value.
Randomly repositioning of CLC and non-CLC genes: We randomly relocated CLC/non-CLC genes 10,000 times within the non-protein-coding regions of the genome using the tool shuffle from BedTools v1965. In each iteration, we calculated the number of genes that intersected at least one hCIS, and created the distribution of these simulated values. Finally, we calculated an empirical p-value by counting how many of the simulated values were higher or equal than the real values. This analysis was performed separately for CLC and non-CLC genes.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Description of additional supplementary items
Acknowledgements
We wish to thanks Julien Lagarde (CRG) for help and advice in bioinformatic analysis. We acknowledge Romina Garrido (CRG), Deborah Re (DBMR), Silvia Roesselet (DBMR) and Marianne Zahn (Inselspital) for administrative support. We thank Ivo Buchhalter (DKFZ) and Sandra Koser (DKFZ) for preprocessing the SNV and expression data for the integrated analysis. Iñigo Martincorena (Sanger Institute) kindly provided the script for analysing driver prediction sensitivity. A.L. was supported by pre-doctoral fellowship FPU14/03371. This research was supported by the Swiss National Science Foundation through the National Centres for Competence in Research “RNA & Disease”, and by the Department of Medical Oncology of Inselspital. We acknowledge the contributions of the many clinical networks across ICGC and TCGA who provided samples and data to the PCAWG Consortium, and the contributions of the Technical Working Group and the Germline Working Group of the PCAWG Consortium for collation, realignment and harmonised variant calling of the cancer genomes used in this study. We thank the patients and their families for their participation in the individual ICGC and TCGA projects.
Author contributions
R.J. conceived the project, performed manual annotation of CLC, and supervised with advice and suggestions of J.S.P., L.F. and C.H. J.C.F. and A.L. performed the feature analysis and evolutionary analysis. D.M.-P. performed the intersection with public databases. A.L. performed mutation analysis. R.J., A.L. and J.C.F. drafted the manuscript and prepared the figures and supplementary material. All authors read and approved the final draft. The following are PCAWG Drivers and Functional Interpretation Group co-leaders or Project co-leaders: Mark Gerstein, Gad Getz, Michael S. Lawrence, Jakob Skou Pedersen, Benjamin J. Raphael, Joshua M. Stuart and David A. Wheeler.
Data availability
The data reported in this study are summarized in the manuscript and its Supporting Information files. The list of CLC genes are also available from the GOLD Lab website (https://www.gold-lab.org/clc). Somatic and germline variant calls, mutational signatures, subclonal reconstructions, transcript abundance, splice calls and other core data generated by the ICGC/TCGA Pan-cancer Analysis of Whole Genomes Consortium is described here38 and available for download at https://dcc.icgc.org/releases/PCAWG. Additional information on accessing the data, including raw read files, can be found at https://docs.icgc.org/pcawg/data/. In accordance with the data access policies of the ICGC and TCGA projects, most molecular, clinical and specimen data are in an open tier which does not require access approval. To access potentially identification information, such as germline alleles and underlying sequencing data, researchers will need to apply to the TCGA Data Access Committee (DAC) via dbGaP (https://dbgap.ncbi.nlm.nih.gov/aa/wga.cgi?page=login) for access to the TCGA portion of the dataset, and to the ICGC Data Access Compliance Office (DACO; http://icgc.org/daco) for the ICGC portion. In addition, to access somatic single nucleotide variants derived from TCGA donors, researchers will also need to obtain dbGaP authorisation.
Code availability
Custom code are available from the corresponding author upon request. The core computational pipelines used by the PCAWG Consortium for alignment, quality control and variant calling are available to the public at https://dockstore.org/search?search=pcawg under the GNU General Public License v3.0, which allows for reuse and distribution.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Joana Carlevaro-Fita, Andrés Lanzós
PCAWG Drivers and Functional Interpretation Working Group authors and their affiliations appear at the end of the paper
PCAWG Consortium members and their affiliations appear online
Change history
12/8/2022
A Correction to this paper has been published: 10.1038/s42003-022-03769-z
Contributor Information
Rory Johnson, Email: rory.johnson@dbmr.unibe.ch.
PCAWG Drivers and Functional Interpretation Group:
Federico Abascal, Samirkumar B. Amin, Gary D. Bader, Jonathan Barenboim, Rameen Beroukhim, Johanna Bertl, Keith A. Boroevich, Søren Brunak, Peter J. Campbell, Joana Carlevaro-Fita, Dimple Chakravarty, Calvin Wing Yiu Chan, Ken Chen, Jung Kyoon Choi, Jordi Deu-Pons, Priyanka Dhingra, Klev Diamanti, Lars Feuerbach, J. Lynn Fink, Nuno A. Fonseca, Joan Frigola, Carlo Gambacorti-Passerini, Dale W. Garsed, Mark Gerstein, Gad Getz, Abel Gonzalez-Perez, Qianyun Guo, Ivo G. Gut, David Haan, Mark P. Hamilton, Nicholas J. Haradhvala, Arif O. Harmanci, Mohamed Helmy, Carl Herrmann, Julian M. Hess, Asger Hobolth, Ermin Hodzic, Chen Hong, Henrik Hornshøj, Keren Isaev, Jose M. G. Izarzugaza, Rory Johnson, Todd A. Johnson, Malene Juul, Randi Istrup Juul, Andre Kahles, Abdullah Kahraman, Manolis Kellis, Ekta Khurana, Jaegil Kim, Jong K. Kim, Youngwook Kim, Jan Komorowski, Jan O. Korbel, Sushant Kumar, Andrés Lanzós, Erik Larsson, Michael S. Lawrence, Donghoon Lee, Kjong-Van Lehmann, Shantao Li, Xiaotong Li, Ziao Lin, Eric Minwei Liu, Lucas Lochovsky, Shaoke Lou, Tobias Madsen, Kathleen Marchal, Iñigo Martincorena, Alexander Martinez-Fundichely, Yosef E. Maruvka, Patrick D. McGillivray, William Meyerson, Ferran Muiños, Loris Mularoni, Hidewaki Nakagawa, Morten Muhlig Nielsen, Marta Paczkowska, Keunchil Park, Kiejung Park, Jakob Skou Pedersen, Oriol Pich, Tirso Pons, Sergio Pulido-Tamayo, Benjamin J Raphael, Jüri Reimand, Iker Reyes-Salazar, Matthew A. Reyna, Esther Rheinbay, Mark A. Rubin, Carlota Rubio-Perez, Radhakrishnan Sabarinathan, S. Cenk Sahinalp, Gordon Saksena, Leonidas Salichos, Chris Sander, Steven E. Schumacher, Mark Shackleton, Ofer Shapira, Ciyue Shen, Raunak Shrestha, Shimin Shuai, Nikos Sidiropoulos, Lina Sieverling, Nasa Sinnott-Armstrong, Lincoln D. Stein, Joshua M. Stuart, David Tamborero, Grace Tiao, Tatsuhiko Tsunoda, Husen M. Umer, Liis Uusküla-Reimand, Alfonso Valencia, Miguel Vazquez, Lieven P. C. Verbeke, Claes Wadelius, Lina Wadi, Jiayin Wang, Jonathan Warrell, Sebastian M. Waszak, Joachim Weischenfeldt, David A. Wheeler, Guanming Wu, Jun Yu, Jing Zhang, Xuanping Zhang, Yan Zhang, Zhongming Zhao, Lihua Zou, and Christian von Mering
PCAWG Consortium:
Lauri A. Aaltonen, Federico Abascal, Adam Abeshouse, Hiroyuki Aburatani, David J. Adams, Nishant Agrawal, Keun Soo Ahn, Sung-Min Ahn, Hiroshi Aikata, Rehan Akbani, Kadir C. Akdemir, Hikmat Al-Ahmadie, Sultan T. Al-Sedairy, Fatima Al-Shahrour, Malik Alawi, Monique Albert, Kenneth Aldape, Ludmil B. Alexandrov, Adrian Ally, Kathryn Alsop, Eva G. Alvarez, Fernanda Amary, Samirkumar B. Amin, Brice Aminou, Ole Ammerpohl, Matthew J. Anderson, Yeng Ang, Davide Antonello, Pavana Anur, Samuel Aparicio, Elizabeth L. Appelbaum, Yasuhito Arai, Axel Aretz, Koji Arihiro, Shun-ichi Ariizumi, Joshua Armenia, Laurent Arnould, Sylvia Asa, Yassen Assenov, Gurnit Atwal, Sietse Aukema, J. Todd Auman, Miriam R. R. Aure, Philip Awadalla, Marta Aymerich, Gary D. Bader, Adrian Baez-Ortega, Matthew H. Bailey, Peter J. Bailey, Miruna Balasundaram, Saianand Balu, Pratiti Bandopadhayay, Rosamonde E. Banks, Stefano Barbi, Andrew P. Barbour, Jonathan Barenboim, Jill Barnholtz-Sloan, Hugh Barr, Elisabet Barrera, John Bartlett, Javier Bartolome, Claudio Bassi, Oliver F. Bathe, Daniel Baumhoer, Prashant Bavi, Stephen B. Baylin, Wojciech Bazant, Duncan Beardsmore, Timothy A. Beck, Sam Behjati, Andreas Behren, Beifang Niu, Cindy Bell, Sergi Beltran, Christopher Benz, Andrew Berchuck, Anke K. Bergmann, Erik N. Bergstrom, Benjamin P. Berman, Daniel M. Berney, Stephan H. Bernhart, Rameen Beroukhim, Mario Berrios, Samantha Bersani, Johanna Bertl, Miguel Betancourt, Vinayak Bhandari, Shriram G. Bhosle, Andrew V. Biankin, Matthias Bieg, Darell Bigner, Hans Binder, Ewan Birney, Michael Birrer, Nidhan K. Biswas, Bodil Bjerkehagen, Tom Bodenheimer, Lori Boice, Giada Bonizzato, Johann S. De Bono, Arnoud Boot, Moiz S. Bootwalla, Ake Borg, Arndt Borkhardt, Keith A. Boroevich, Ivan Borozan, Christoph Borst, Marcus Bosenberg, Mattia Bosio, Jacqueline Boultwood, Guillaume Bourque, Paul C. Boutros, G. Steven Bova, David T. Bowen, Reanne Bowlby, David D. L. Bowtell, Sandrine Boyault, Rich Boyce, Jeffrey Boyd, Alvis Brazma, Paul Brennan, Daniel S. Brewer, Arie B. Brinkman, Robert G. Bristow, Russell R. Broaddus, Jane E. Brock, Malcolm Brock, Annegien Broeks, Angela N. Brooks, Denise Brooks, Benedikt Brors, Søren Brunak, Timothy J. C. Bruxner, Alicia L. Bruzos, Alex Buchanan, Ivo Buchhalter, Christiane Buchholz, Susan Bullman, Hazel Burke, Birgit Burkhardt, Kathleen H. Burns, John Busanovich, Carlos D. Bustamante, Adam P. Butler, Atul J. Butte, Niall J. Byrne, Anne-Lise Børresen-Dale, Samantha J. Caesar-Johnson, Andy Cafferkey, Declan Cahill, Claudia Calabrese, Carlos Caldas, Fabien Calvo, Niedzica Camacho, Peter J. Campbell, Elias Campo, Cinzia Cantù, Shaolong Cao, Thomas E. Carey, Joana Carlevaro-Fita, Rebecca Carlsen, Ivana Cataldo, Mario Cazzola, Jonathan Cebon, Robert Cerfolio, Dianne E. Chadwick, Dimple Chakravarty, Don Chalmers, Calvin Wing Yiu Chan, Kin Chan, Michelle Chan-Seng-Yue, Vishal S. Chandan, David K. Chang, Stephen J. Chanock, Lorraine A. Chantrill, Aurélien Chateigner, Nilanjan Chatterjee, Kazuaki Chayama, Hsiao-Wei Chen, Jieming Chen, Ken Chen, Yiwen Chen, Zhaohong Chen, Andrew D. Cherniack, Jeremy Chien, Yoke-Eng Chiew, Suet-Feung Chin, Juok Cho, Sunghoon Cho, Jung Kyoon Choi, Wan Choi, Christine Chomienne, Zechen Chong, Su Pin Choo, Angela Chou, Angelika N. Christ, Elizabeth L. Christie, Eric Chuah, Carrie Cibulskis, Kristian Cibulskis, Sara Cingarlini, Peter Clapham, Alexander Claviez, Sean Cleary, Nicole Cloonan, Marek Cmero, Colin C. Collins, Ashton A. Connor, Susanna L. Cooke, Colin S. Cooper, Leslie Cope, Vincenzo Corbo, Matthew G. Cordes, Stephen M. Cordner, Isidro Cortés-Ciriano, Kyle Covington, Prue A. Cowin, Brian Craft, David Craft, Chad J. Creighton, Yupeng Cun, Erin Curley, Ioana Cutcutache, Karolina Czajka, Bogdan Czerniak, Rebecca A. Dagg, Ludmila Danilova, Maria Vittoria Davi, Natalie R. Davidson, Helen Davies, Ian J. Davis, Brandi N. Davis-Dusenbery, Kevin J. Dawson, Francisco M. De La Vega, Ricardo De Paoli-Iseppi, Timothy Defreitas, Angelo P. Dei Tos, Olivier Delaneau, John A. Demchok, Jonas Demeulemeester, German M. Demidov, Deniz Demircioğlu, Nening M. Dennis, Robert E. Denroche, Stefan C. Dentro, Nikita Desai, Vikram Deshpande, Amit G. Deshwar, Christine Desmedt, Jordi Deu-Pons, Noreen Dhalla, Neesha C. Dhani, Priyanka Dhingra, Rajiv Dhir, Anthony DiBiase, Klev Diamanti, Li Ding, Shuai Ding, Huy Q. Dinh, Luc Dirix, HarshaVardhan Doddapaneni, Nilgun Donmez, Michelle T. Dow, Ronny Drapkin, Oliver Drechsel, Ruben M. Drews, Serge Serge, Tim Dudderidge, Ana Dueso-Barroso, Andrew J. Dunford, Michael Dunn, Lewis Jonathan Dursi, Fraser R. Duthie, Ken Dutton-Regester, Jenna Eagles, Douglas F. Easton, Stuart Edmonds, Paul A. Edwards, Sandra E. Edwards, Rosalind A. Eeles, Anna Ehinger, Juergen Eils, Roland Eils, Adel El-Naggar, Matthew Eldridge, Kyle Ellrott, Serap Erkek, Georgia Escaramis, Shadrielle M. G. Espiritu, Xavier Estivill, Dariush Etemadmoghadam, Jorunn E. Eyfjord, Bishoy M. Faltas, Daiming Fan, Yu Fan, William C. Faquin, Claudiu Farcas, Matteo Fassan, Aquila Fatima, Francesco Favero, Nodirjon Fayzullaev, Ina Felau, Sian Fereday, Martin L. Ferguson, Vincent Ferretti, Lars Feuerbach, Matthew A. Field, J. Lynn Fink, Gaetano Finocchiaro, Cyril Fisher, Matthew W. Fittall, Anna Fitzgerald, Rebecca C. Fitzgerald, Adrienne M. Flanagan, Neil E. Fleshner, Paul Flicek, John A. Foekens, Kwun M. Fong, Nuno A. Fonseca, Christopher S. Foster, Natalie S. Fox, Michael Fraser, Scott Frazer, Milana Frenkel-Morgenstern, William Friedman, Joan Frigola, Catrina C. Fronick, Akihiro Fujimoto, Masashi Fujita, Masashi Fukayama, Lucinda A. Fulton, Robert S. Fulton, Mayuko Furuta, P. Andrew Futreal, Anja Füllgrabe, Stacey B. Gabriel, Steven Gallinger, Carlo Gambacorti-Passerini, Jianjiong Gao, Shengjie Gao, Levi Garraway, Øystein Garred, Erik Garrison, Dale W. Garsed, Nils Gehlenborg, Josep L. L. Gelpi, Joshy George, Daniela S. Gerhard, Clarissa Gerhauser, Jeffrey E. Gershenwald, Mark Gerstein, Moritz Gerstung, Gad Getz, Mohammed Ghori, Ronald Ghossein, Nasra H. Giama, Richard A. Gibbs, Bob Gibson, Anthony J. Gill, Pelvender Gill, Dilip D. Giri, Dominik Glodzik, Vincent J. Gnanapragasam, Maria Elisabeth Goebler, Mary J. Goldman, Carmen Gomez, Santiago Gonzalez, Abel Gonzalez-Perez, Dmitry A. Gordenin, James Gossage, Kunihito Gotoh, Ramaswamy Govindan, Dorthe Grabau, Janet S. Graham, Robert C. Grant, Anthony R. Green, Eric Green, Liliana Greger, Nicola Grehan, Sonia Grimaldi, Sean M. Grimmond, Robert L. Grossman, Adam Grundhoff, Gunes Gundem, Qianyun Guo, Manaswi Gupta, Shailja Gupta, Ivo G. Gut, Marta Gut, Jonathan Göke, Gavin Ha, Andrea Haake, David Haan, Siegfried Haas, Kerstin Haase, James E. Haber, Nina Habermann, Faraz Hach, Syed Haider, Natsuko Hama, Freddie C. Hamdy, Anne Hamilton, Mark P. Hamilton, Leng Han, George B. Hanna, Martin Hansmann, Nicholas J. Haradhvala, Olivier Harismendy, Ivon Harliwong, Arif O. Harmanci, Eoghan Harrington, Takanori Hasegawa, David Haussler, Steve Hawkins, Shinya Hayami, Shuto Hayashi, D. Neil Hayes, Stephen J. Hayes, Nicholas K. Hayward, Steven Hazell, Yao He, Allison P. Heath, Simon C. Heath, David Hedley, Apurva M. Hegde, David I. Heiman, Michael C. Heinold, Zachary Heins, Lawrence E. Heisler, Eva Hellstrom-Lindberg, Mohamed Helmy, Seong Gu Heo, Austin J. Hepperla, José María Heredia-Genestar, Carl Herrmann, Peter Hersey, Julian M. Hess, Holmfridur Hilmarsdottir, Jonathan Hinton, Satoshi Hirano, Nobuyoshi Hiraoka, Katherine A. Hoadley, Asger Hobolth, Ermin Hodzic, Jessica I. Hoell, Steve Hoffmann, Oliver Hofmann, Andrea Holbrook, Aliaksei Z. Holik, Michael A. Hollingsworth, Oliver Holmes, Robert A. Holt, Chen Hong, Eun Pyo Hong, Jongwhi H. Hong, Gerrit K. Hooijer, Henrik Hornshøj, Fumie Hosoda, Yong Hou, Volker Hovestadt, William Howat, Alan P. Hoyle, Ralph H. Hruban, Jianhong Hu, Taobo Hu, Xing Hua, Kuan-lin Huang, Mei Huang, Mi Ni Huang, Vincent Huang, Yi Huang, Wolfgang Huber, Thomas J. Hudson, Michael Hummel, Jillian A. Hung, David Huntsman, Ted R. Hupp, Jason Huse, Matthew R. Huska, Barbara Hutter, Carolyn M. Hutter, Daniel Hübschmann, Christine A. Iacobuzio-Donahue, Charles David Imbusch, Marcin Imielinski, Seiya Imoto, William B. Isaacs, Keren Isaev, Shumpei Ishikawa, Murat Iskar, S. M. Ashiqul Islam, Michael Ittmann, Sinisa Ivkovic, Jose M. G. Izarzugaza, Jocelyne Jacquemier, Valerie Jakrot, Nigel B. Jamieson, Gun Ho Jang, Se Jin Jang, Joy C. Jayaseelan, Reyka Jayasinghe, Stuart R. Jefferys, Karine Jegalian, Jennifer L. Jennings, Seung-Hyup Jeon, Lara Jerman, Yuan Ji, Wei Jiao, Peter A. Johansson, Amber L. Johns, Jeremy Johns, Rory Johnson, Todd A. Johnson, Clemency Jolly, Yann Joly, Jon G. Jonasson, Corbin D. Jones, David R. Jones, David T. W. Jones, Nic Jones, Steven J. M. Jones, Jos Jonkers, Young Seok Ju, Hartmut Juhl, Jongsun Jung, Malene Juul, Randi Istrup Juul, Sissel Juul, Natalie Jäger, Rolf Kabbe, Andre Kahles, Abdullah Kahraman, Vera B. Kaiser, Hojabr Kakavand, Sangeetha Kalimuthu, Christof von Kalle, Koo Jeong Kang, Katalin Karaszi, Beth Karlan, Rosa Karlić, Dennis Karsch, Katayoon Kasaian, Karin S. Kassahn, Hitoshi Katai, Mamoru Kato, Hiroto Katoh, Yoshiiku Kawakami, Jonathan D. Kay, Stephen H. Kazakoff, Marat D. Kazanov, Maria Keays, Electron Kebebew, Richard F. Kefford, Manolis Kellis, James G. Kench, Catherine J. Kennedy, Jules N. A. Kerssemakers, David Khoo, Vincent Khoo, Narong Khuntikeo, Ekta Khurana, Helena Kilpinen, Hark Kyun Kim, Hyung-Lae Kim, Hyung-Yong Kim, Hyunghwan Kim, Jaegil Kim, Jihoon Kim, Jong K. Kim, Youngwook Kim, Tari A. King, Wolfram Klapper, Kortine Kleinheinz, Leszek J. Klimczak, Stian Knappskog, Michael Kneba, Bartha M. Knoppers, Youngil Koh, Jan Komorowski, Daisuke Komura, Mitsuhiro Komura, Gu Kong, Marcel Kool, Jan O. Korbel, Viktoriya Korchina, Andrey Korshunov, Michael Koscher, Roelof Koster, Zsofia Kote-Jarai, Antonios Koures, Milena Kovacevic, Barbara Kremeyer, Helene Kretzmer, Markus Kreuz, Savitri Krishnamurthy, Dieter Kube, Kiran Kumar, Pardeep Kumar, Sushant Kumar, Yogesh Kumar, Ritika Kundra, Kirsten Kübler, Ralf Küppers, Jesper Lagergren, Phillip H. Lai, Peter W. Laird, Sunil R. Lakhani, Christopher M. Lalansingh, Emilie Lalonde, Fabien C. Lamaze, Adam Lambert, Eric Lander, Pablo Landgraf, Luca Landoni, Anita Langerød, Andrés Lanzós, Denis Larsimont, Erik Larsson, Mark Lathrop, Loretta M. S. Lau, Chris Lawerenz, Rita T. Lawlor, Michael S. Lawrence, Alexander J. Lazar, Ana Mijalkovic Lazic, Xuan Le, Darlene Lee, Donghoon Lee, Eunjung Alice Lee, Hee Jin Lee, Jake June-Koo Lee, Jeong-Yeon Lee, Juhee Lee, Ming Ta Michael Lee, Henry Lee-Six, Kjong-Van Lehmann, Hans Lehrach, Dido Lenze, Conrad R. Leonard, Daniel A. Leongamornlert, Ignaty Leshchiner, Louis Letourneau, Ivica Letunic, Douglas A. Levine, Lora Lewis, Tim Ley, Chang Li, Constance H. Li, Haiyan Irene Li, Jun Li, Lin Li, Shantao Li, Siliang Li, Xiaobo Li, Xiaotong Li, Xinyue Li, Yilong Li, Han Liang, Sheng-Ben Liang, Peter Lichter, Pei Lin, Ziao Lin, W. M. Linehan, Ole Christian Lingjærde, Dongbing Liu, Eric Minwei Liu, Fei-Fei Fei Liu, Fenglin Liu, Jia Liu, Xingmin Liu, Julie Livingstone, Dimitri Livitz, Naomi Livni, Lucas Lochovsky, Markus Loeffler, Georgina V. Long, Armando Lopez-Guillermo, Shaoke Lou, David N. Louis, Laurence B. Lovat, Yiling Lu, Yong-Jie Lu, Youyong Lu, Claudio Luchini, Ilinca Lungu, Xuemei Luo, Hayley J. Luxton, Andy G. Lynch, Lisa Lype, Cristina López, Carlos López-Otín, Eric Z. Ma, Yussanne Ma, Gaetan MacGrogan, Shona MacRae, Geoff Macintyre, Tobias Madsen, Kazuhiro Maejima, Andrea Mafficini, Dennis T. Maglinte, Arindam Maitra, Partha P. Majumder, Luca Malcovati, Salem Malikic, Giuseppe Malleo, Graham J. Mann, Luisa Mantovani-Löffler, Kathleen Marchal, Giovanni Marchegiani, Elaine R. Mardis, Adam A. Margolin, Maximillian G. Marin, Florian Markowetz, Julia Markowski, Jeffrey Marks, Tomas Marques-Bonet, Marco A. Marra, Luke Marsden, John W. M. Martens, Sancha Martin, Jose I. Martin-Subero, Iñigo Martincorena, Alexander Martinez-Fundichely, Yosef E. Maruvka, R. Jay Mashl, Charlie E. Massie, Thomas J. Matthew, Lucy Matthews, Erik Mayer, Simon Mayes, Michael Mayo, Faridah Mbabaali, Karen McCune, Ultan McDermott, Patrick D. McGillivray, Michael D. McLellan, John D. McPherson, John R. McPherson, Treasa A. McPherson, Samuel R. Meier, Alice Meng, Shaowu Meng, Andrew Menzies, Neil D. Merrett, Sue Merson, Matthew Meyerson, William Meyerson, Piotr A. Mieczkowski, George L. Mihaiescu, Sanja Mijalkovic, Tom Mikkelsen, Michele Milella, Linda Mileshkin, Christopher A. Miller, David K. Miller, Jessica K. Miller, Gordon B. Mills, Ana Milovanovic, Sarah Minner, Marco Miotto, Gisela Mir Arnau, Lisa Mirabello, Chris Mitchell, Thomas J. Mitchell, Satoru Miyano, Naoki Miyoshi, Shinichi Mizuno, Fruzsina Molnár-Gábor, Malcolm J. Moore, Richard A. Moore, Sandro Morganella, Quaid D. Morris, Carl Morrison, Lisle E. Mose, Catherine D. Moser, Ferran Muiños, Loris Mularoni, Andrew J. Mungall, Karen Mungall, Elizabeth A. Musgrove, Ville Mustonen, David Mutch, Francesc Muyas, Donna M. Muzny, Alfonso Muñoz, Jerome Myers, Ola Myklebost, Peter Möller, Genta Nagae, Adnan M. Nagrial, Hardeep K. Nahal-Bose, Hitoshi Nakagama, Hidewaki Nakagawa, Hiromi Nakamura, Toru Nakamura, Kaoru Nakano, Tannistha Nandi, Jyoti Nangalia, Mia Nastic, Arcadi Navarro, Fabio C. P. Navarro, David E. Neal, Gerd Nettekoven, Felicity Newell, Steven J. Newhouse, Yulia Newton, Alvin Wei Tian Ng, Anthony Ng, Jonathan Nicholson, David Nicol, Yongzhan Nie, G. Petur Nielsen, Morten Muhlig Nielsen, Serena Nik-Zainal, Michael S. Noble, Katia Nones, Paul A. Northcott, Faiyaz Notta, Brian D. O’Connor, Peter O’Donnell, Maria O’Donovan, Sarah O’Meara, Brian Patrick O’Neill, J. Robert O’Neill, David Ocana, Angelica Ochoa, Layla Oesper, Christopher Ogden, Hideki Ohdan, Kazuhiro Ohi, Lucila Ohno-Machado, Karin A. Oien, Akinyemi I. Ojesina, Hidenori Ojima, Takuji Okusaka, Larsson Omberg, Choon Kiat Ong, Stephan Ossowski, German Ott, B. F. Francis Ouellette, Christine P’ng, Marta Paczkowska, Salvatore Paiella, Chawalit Pairojkul, Marina Pajic, Qiang Pan-Hammarström, Elli Papaemmanuil, Irene Papatheodorou, Nagarajan Paramasivam, Ji Wan Park, Joong-Won Park, Keunchil Park, Kiejung Park, Peter J. Park, Joel S. Parker, Simon L. Parsons, Harvey Pass, Danielle Pasternack, Alessandro Pastore, Ann-Marie Patch, Iris Pauporté, Antonio Pea, John V. Pearson, Chandra Sekhar Pedamallu, Jakob Skou Pedersen, Paolo Pederzoli, Martin Peifer, Nathan A. Pennell, Charles M. Perou, Marc D. Perry, Gloria M. Petersen, Myron Peto, Nicholas Petrelli, Robert Petryszak, Stefan M. Pfister, Mark Phillips, Oriol Pich, Hilda A. Pickett, Todd D. Pihl, Nischalan Pillay, Sarah Pinder, Mark Pinese, Andreia V. Pinho, Esa Pitkänen, Xavier Pivot, Elena Piñeiro-Yáñez, Laura Planko, Christoph Plass, Paz Polak, Tirso Pons, Irinel Popescu, Olga Potapova, Aparna Prasad, Shaun R. Preston, Manuel Prinz, Antonia L. Pritchard, Stephenie D. Prokopec, Elena Provenzano, Xose S. Puente, Sonia Puig, Montserrat Puiggròs, Sergio Pulido-Tamayo, Gulietta M. Pupo, Colin A. Purdie, Michael C. Quinn, Raquel Rabionet, Janet S. Rader, Bernhard Radlwimmer, Petar Radovic, Benjamin Raeder, Keiran M. Raine, Manasa Ramakrishna, Kamna Ramakrishnan, Suresh Ramalingam, Benjamin J. Raphael, W. Kimryn Rathmell, Tobias Rausch, Guido Reifenberger, Jüri Reimand, Jorge Reis-Filho, Victor Reuter, Iker Reyes-Salazar, Matthew A. Reyna, Sheila M. Reynolds, Esther Rheinbay, Yasser Riazalhosseini, Andrea L. Richardson, Julia Richter, Matthew Ringel, Markus Ringnér, Yasushi Rino, Karsten Rippe, Jeffrey Roach, Lewis R. Roberts, Nicola D. Roberts, Steven A. Roberts, A. Gordon Robertson, Alan J. Robertson, Javier Bartolomé Rodriguez, Bernardo Rodriguez-Martin, F. Germán Rodríguez-González, Michael H. A. Roehrl, Marius Rohde, Hirofumi Rokutan, Gilles Romieu, Ilse Rooman, Tom Roques, Daniel Rosebrock, Mara Rosenberg, Philip C. Rosenstiel, Andreas Rosenwald, Edward W. Rowe, Romina Royo, Steven G. Rozen, Yulia Rubanova, Mark A. Rubin, Carlota Rubio-Perez, Vasilisa A. Rudneva, Borislav C. Rusev, Andrea Ruzzenente, Gunnar Rätsch, Radhakrishnan Sabarinathan, Veronica Y. Sabelnykova, Sara Sadeghi, S. Cenk Sahinalp, Natalie Saini, Mihoko Saito-Adachi, Gordon Saksena, Adriana Salcedo, Roberto Salgado, Leonidas Salichos, Richard Sallari, Charles Saller, Roberto Salvia, Michelle Sam, Jaswinder S. Samra, Francisco Sanchez-Vega, Chris Sander, Grant Sanders, Rajiv Sarin, Iman Sarrafi, Aya Sasaki-Oku, Torill Sauer, Guido Sauter, Robyn P. M. Saw, Maria Scardoni, Christopher J. Scarlett, Aldo Scarpa, Ghislaine Scelo, Dirk Schadendorf, Jacqueline E. Schein, Markus B. Schilhabel, Matthias Schlesner, Thorsten Schlomm, Heather K. Schmidt, Sarah-Jane Schramm, Stefan Schreiber, Nikolaus Schultz, Steven E. Schumacher, Roland F. Schwarz, Richard A. Scolyer, David Scott, Ralph Scully, Raja Seethala, Ayellet V. Segre, Iris Selander, Colin A. Semple, Yasin Senbabaoglu, Subhajit Sengupta, Elisabetta Sereni, Stefano Serra, Dennis C. Sgroi, Mark Shackleton, Nimish C. Shah, Sagedeh Shahabi, Catherine A. Shang, Ping Shang, Ofer Shapira, Troy Shelton, Ciyue Shen, Hui Shen, Rebecca Shepherd, Ruian Shi, Yan Shi, Yu-Jia Shiah, Tatsuhiro Shibata, Juliann Shih, Eigo Shimizu, Kiyo Shimizu, Seung Jun Shin, Yuichi Shiraishi, Tal Shmaya, Ilya Shmulevich, Solomon I. Shorser, Charles Short, Raunak Shrestha, Suyash S. Shringarpure, Craig Shriver, Shimin Shuai, Nikos Sidiropoulos, Reiner Siebert, Anieta M. Sieuwerts, Lina Sieverling, Sabina Signoretti, Katarzyna O. Sikora, Michele Simbolo, Ronald Simon, Janae V. Simons, Jared T. Simpson, Peter T. Simpson, Samuel Singer, Nasa Sinnott-Armstrong, Payal Sipahimalani, Tara J. Skelly, Marcel Smid, Jaclyn Smith, Karen Smith-McCune, Nicholas D. Socci, Heidi J. Sofia, Matthew G. Soloway, Lei Song, Anil K. Sood, Sharmila Sothi, Christos Sotiriou, Cameron M. Soulette, Paul N. Span, Paul T. Spellman, Nicola Sperandio, Andrew J. Spillane, Oliver Spiro, Jonathan Spring, Johan Staaf, Peter F. Stadler, Peter Staib, Stefan G. Stark, Lucy Stebbings, Ólafur Andri Stefánsson, Oliver Stegle, Lincoln D. Stein, Alasdair Stenhouse, Chip Stewart, Stephan Stilgenbauer, Miranda D. Stobbe, Michael R. Stratton, Jonathan R. Stretch, Adam J. Struck, Joshua M. Stuart, Henk G. Stunnenberg, Hong Su, Xiaoping Su, Ren X. Sun, Stephanie Sungalee, Hana Susak, Akihiro Suzuki, Fred Sweep, Monika Szczepanowski, Holger Sültmann, Takashi Yugawa, Angela Tam, David Tamborero, Benita Kiat Tee Tan, Donghui Tan, Patrick Tan, Hiroko Tanaka, Hirokazu Taniguchi, Tomas J. Tanskanen, Maxime Tarabichi, Roy Tarnuzzer, Patrick Tarpey, Morgan L. Taschuk, Kenji Tatsuno, Simon Tavaré, Darrin F. Taylor, Amaro Taylor-Weiner, Jon W. Teague, Bin Tean Teh, Varsha Tembe, Javier Temes, Kevin Thai, Sarah P. Thayer, Nina Thiessen, Gilles Thomas, Sarah Thomas, Alan Thompson, Alastair M. Thompson, John F. F. Thompson, R. Houston Thompson, Heather Thorne, Leigh B. Thorne, Adrian Thorogood, Grace Tiao, Nebojsa Tijanic, Lee E. Timms, Roberto Tirabosco, Marta Tojo, Stefania Tommasi, Christopher W. Toon, Umut H. Toprak, David Torrents, Giampaolo Tortora, Jörg Tost, Yasushi Totoki, David Townend, Nadia Traficante, Isabelle Treilleux, Jean-Rémi Trotta, Lorenz H. P. Trümper, Ming Tsao, Tatsuhiko Tsunoda, Jose M. C. Tubio, Olga Tucker, Richard Turkington, Daniel J. Turner, Andrew Tutt, Masaki Ueno, Naoto T. Ueno, Christopher Umbricht, Husen M. Umer, Timothy J. Underwood, Lara Urban, Tomoko Urushidate, Tetsuo Ushiku, Liis Uusküla-Reimand, Alfonso Valencia, David J. Van Den Berg, Steven Van Laere, Peter Van Loo, Erwin G. Van Meir, Gert G. Van den Eynden, Theodorus Van der Kwast, Naveen Vasudev, Miguel Vazquez, Ravikiran Vedururu, Umadevi Veluvolu, Shankar Vembu, Lieven P. C. Verbeke, Peter Vermeulen, Clare Verrill, Alain Viari, David Vicente, Caterina Vicentini, K. VijayRaghavan, Juris Viksna, Ricardo E. Vilain, Izar Villasante, Anne Vincent-Salomon, Tapio Visakorpi, Douglas Voet, Paresh Vyas, Ignacio Vázquez-García, Nick M. Waddell, Nicola Waddell, Claes Wadelius, Lina Wadi, Rabea Wagener, Jeremiah A. Wala, Jian Wang, Jiayin Wang, Linghua Wang, Qi Wang, Wenyi Wang, Yumeng Wang, Zhining Wang, Paul M. Waring, Hans-Jörg Warnatz, Jonathan Warrell, Anne Y. Warren, Sebastian M. Waszak, David C. Wedge, Dieter Weichenhan, Paul Weinberger, John N. Weinstein, Joachim Weischenfeldt, Daniel J. Weisenberger, Ian Welch, Michael C. Wendl, Johannes Werner, Justin P. Whalley, David A. Wheeler, Hayley C. Whitaker, Dennis Wigle, Matthew D. Wilkerson, Ashley Williams, James S. Wilmott, Gavin W. Wilson, Julie M. Wilson, Richard K. Wilson, Boris Winterhoff, Jeffrey A. Wintersinger, Maciej Wiznerowicz, Stephan Wolf, Bernice H. Wong, Tina Wong, Winghing Wong, Youngchoon Woo, Scott Wood, Bradly G. Wouters, Adam J. Wright, Derek W. Wright, Mark H. Wright, Chin-Lee Wu, Dai-Ying Wu, Guanming Wu, Jianmin Wu, Kui Wu, Yang Wu, Zhenggang Wu, Liu Xi, Tian Xia, Qian Xiang, Xiao Xiao, Rui Xing, Heng Xiong, Qinying Xu, Yanxun Xu, Hong Xue, Shinichi Yachida, Sergei Yakneen, Rui Yamaguchi, Takafumi N. Yamaguchi, Masakazu Yamamoto, Shogo Yamamoto, Hiroki Yamaue, Fan Yang, Huanming Yang, Jean Y. Yang, Liming Yang, Lixing Yang, Shanlin Yang, Tsun-Po Yang, Yang Yang, Xiaotong Yao, Marie-Laure Yaspo, Lucy Yates, Christina Yau, Chen Ye, Kai Ye, Venkata D. Yellapantula, Christopher J. Yoon, Sung-Soo Yoon, Fouad Yousif, Jun Yu, Kaixian Yu, Willie Yu, Yingyan Yu, Ke Yuan, Yuan Yuan, Denis Yuen, Christina K. Yung, Olga Zaikova, Jorge Zamora, Marc Zapatka, Jean C. Zenklusen, Thorsten Zenz, Nikolajs Zeps, Cheng-Zhong Zhang, Fan Zhang, Hailei Zhang, Hongwei Zhang, Hongxin Zhang, Jiashan Zhang, Jing Zhang, Junjun Zhang, Xiuqing Zhang, Xuanping Zhang, Yan Zhang, Zemin Zhang, Zhongming Zhao, Liangtao Zheng, Xiuqing Zheng, Wanding Zhou, Yong Zhou, Bin Zhu, Hongtu Zhu, Jingchun Zhu, Shida Zhu, Lihua Zou, Xueqing Zou, Anna deFazio, Nicholas van As, Carolien H. M. van Deurzen, Marc J. van de Vijver, L. van’t Veer, and Christian von Mering
Supplementary information
Supplementary information is available for this paper at 10.1038/s42003-019-0741-7.
References
- 1.Yates LR, Campbell PJ. Evolution of the cancer genome. Nat. Rev. Genet. 2012;13:795–806. doi: 10.1038/nrg3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guttman M, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–7. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jia H, et al. Genome-wide computational identification and manual annotation of human long noncoding RNA genes. RNA. 2010;16:1478–87. doi: 10.1261/rna.1951310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cabili MN, et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011;25:1915–27. doi: 10.1101/gad.17446611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Derrien, T. et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 22, 1775–89 (2012). [DOI] [PMC free article] [PubMed]
- 6.Grote, P. et al. The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev. Cell24, 206–214 (2013). [DOI] [PMC free article] [PubMed]
- 7.Sauvageau M, et al. Multiple knockout mouse models reveal lincRNAs are required for life and brain development. Elife. 2013;2:e01749. doi: 10.7554/eLife.01749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ulitsky I, Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell. 2013;154:26–46. doi: 10.1016/j.cell.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu SJ, et al. CRISPRi-based genome-scale identification of functional long noncoding RNA loci in human cells. Science. 2017;355:eaah7111. doi: 10.1126/science.aah7111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guttman M, Rinn JL. Modular regulatory principles of large non-coding RNAs. Nature. 2012;482:339–46. doi: 10.1038/nature10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson R, Guigó R. The RIDL hypothesis: transposable elements as functional domains of long noncoding RNAs. RNA. 2014;20:959–76. doi: 10.1261/rna.044560.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gutschner T, Diederichs S. The hallmarks of cancer: a long non-coding RNA point of view. RNA Biol. 2012;9:703–19. doi: 10.4161/rna.20481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engreitz JM, et al. RNA-RNA interactions enable specific targeting of noncoding RNAs to nascent Pre-mRNAs and chromatin sites. Cell. 2014;159:188–99. doi: 10.1016/j.cell.2014.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gutschner T, et al. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013;73:1180–9. doi: 10.1158/0008-5472.CAN-12-2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lanzós A, et al. Discovery of cancer driver long noncoding RNAs across 1112 tumour genomes: new candidates and distinguishing features. Sci. Rep. 2017;7:41544. doi: 10.1038/srep41544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rheinbay, E. et al. Analyses of non-coding somatic drivers in 2,658 cancer whole genomes. Nature. 10.1038/s41586-020-1965-x (2020). [DOI] [PMC free article] [PubMed]
- 17.Huarte M, et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell. 2010;142:409–19. doi: 10.1016/j.cell.2010.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Symonds H, et al. p53-Dependent apoptosis suppresses tumor growth and progression in vivo. Cell. 1994;78:703–711. doi: 10.1016/0092-8674(94)90534-7. [DOI] [PubMed] [Google Scholar]
- 19.Corcoran LM, Adams JM, Dunn AR, Cory S. Murine T lymphomas in which the cellular myc oncogene has been activated by retroviral insertion. Cell. 1984;37:113–122. doi: 10.1016/0092-8674(84)90306-4. [DOI] [PubMed] [Google Scholar]
- 20.Hezroni H, et al. Principles of long noncoding RNA evolution derived from direct comparison of transcriptomes in 17 species. Cell Rep. 2015;11:1110–1122. doi: 10.1016/j.celrep.2015.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakagawa S, et al. Malat1 is not an essential component of nuclear speckles in mice. RNA. 2012;18:1487–1499. doi: 10.1261/rna.033217.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang B, et al. The lncRNA Malat1 is dispensable for mouse development but its transcription plays a cis-regulatory role in the adult. Cell Rep. 2012;2:111–23. doi: 10.1016/j.celrep.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eißmann M, et al. Loss of the abundant nuclear non-coding RNA MALAT1 is compatible with life and development. RNA Biol. 2012;9:1076–87. doi: 10.4161/rna.21089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakagawa S, Naganuma T, Shioi G, Hirose T. Paraspeckles are subpopulation-specific nuclear bodies that are not essential in mice. J. Cell Biol. 2011;193:31–9. doi: 10.1083/jcb.201011110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marín-Béjar O, et al. The human lncRNA LINC-PINT inhibits tumor cell invasion through a highly conserved sequence element. Genome Biol. 2017;18:202. doi: 10.1186/s13059-017-1331-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quek XC, et al. lncRNAdb v2.0: expanding the reference database for functional long noncoding RNAs. Nucleic Acids Res. 2015;43:D168–73. doi: 10.1093/nar/gku988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iyer, M. K. et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet.47, 199 (2015). [DOI] [PMC free article] [PubMed]
- 28.Tamborero D, et al. Comprehensive identification of mutational cancer driver genes across 12 tumor types. Sci. Rep. 2013;3:2650. doi: 10.1038/srep02650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chang K, et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013;45:1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lawrence MS, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Futreal P, et al. A census of human cancer genes. Nat. Rev. Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sjoblom T, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 33.Redon R, et al. Global variation in copy number in the human genome. Nature. 2006;444:444. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mularoni L, Sabarinathan R, Deu-Pons J, Gonzalez-Perez A, López-Bigas N. OncodriveFML: a general framework to identify coding and non-coding regions with cancer driver mutations. Genome Biol. 2016;17:128. doi: 10.1186/s13059-016-0994-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tokheim CJ, Papadopoulos N, Kinzler KW, Vogelstein B, Karchin R. Evaluating the evaluation of cancer driver genes. Proc. Natl Acad. Sci. USA. 2016;113:14330–14335. doi: 10.1073/pnas.1616440113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Furney S, Higgins D, Ouzounis C, López-Bigas N. Structural and functional properties of genes involved in human cancer. BMC Genomics. 2006;7:3. doi: 10.1186/1471-2164-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Furney SJ, Madden SF, Kisiel TA, Higgins DG, Lopez-Bigas N. Distinct patterns in the regulation and evolution of human cancer genes. Silico Biol. 2008;8:33–46. [PubMed] [Google Scholar]
- 38.The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature. 10.1038/s41586-020-1969-6 (2020).
- 39.Chen G, et al. LncRNADisease: a database for long-non-coding RNA-associated diseases. Nucleic Acids Res. 2013;41:D983–6. doi: 10.1093/nar/gks1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ning S, et al. Lnc2Cancer: a manually curated database of experimentally supported lncRNAs associated with various human cancers. Nucleic Acids Res. 2016;44:D980–5. doi: 10.1093/nar/gkv1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Uszczynska-Ratajczak, B., Lagarde, J., Frankish, A., Guigó, R. & Johnson, R. Towards a complete map of the human long non-coding RNA transcriptome. Nat. Rev. Genet.110.1038/s41576-018-0017-y (2018). [DOI] [PMC free article] [PubMed]
- 42.Marchese FP, et al. A long noncoding RNA regulates sister chromatid cohesion. Mol. Cell. 2016;63:397–407. doi: 10.1016/j.molcel.2016.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lanz RB, et al. A steroid receptor coactivator, SRA, functions as an RNA and is present in an SRC-1 complex. Cell. 1999;97:17–27. doi: 10.1016/S0092-8674(00)80711-4. [DOI] [PubMed] [Google Scholar]
- 44.Higashimoto K, Soejima H, Saito T, Okumura K, Mukai T. Imprinting disruption of the CDKN1C/KCNQ1OT1 domain: the molecular mechanisms causing Beckwith-Wiedemann syndrome and cancer. Cytogenet. Genome Res. 2006;113:306–12. doi: 10.1159/000090846. [DOI] [PubMed] [Google Scholar]
- 45.Harrow J, et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012;22:1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.World Health Organization. International Classification of Diseases for Oncology (ICD-O). 3rd edn, 1st revision (2013).
- 47.Zhu S, et al. Genome-scale deletion screening of human long non-coding RNAs using a paired-guide RNA CRISPR–Cas9 library. Nat. Biotechnol. 2016;34:1279–1286. doi: 10.1038/nbt.3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chiu H-S, et al. Pan-cancer analysis of lncRNA regulation supports their targeting of cancer genes in each tumor context. Cell Rep. 2018;23:297–312.e12. doi: 10.1016/j.celrep.2018.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yan, X. et al. Comprehensive genomic characterization of long non-coding rnas across human cancers. Cancer Cell10.1016/j.ccell.2015.09.006 (2015). [DOI] [PMC free article] [PubMed]
- 50.Managadze D, Rogozin IB, Chernikova D, Shabalina SA, Koonin EV. Negative correlation between expression level and evolutionary rate of long intergenic noncoding RNAs. Genome Biol. Evol. 2011;3:1390–1404. doi: 10.1093/gbe/evr116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tan JY, et al. cis-acting complex-trait-associated lincRNA expression correlates with modulation of chromosomal architecture. Cell Rep. 2017;18:2280–2288. doi: 10.1016/j.celrep.2017.02.009. [DOI] [PubMed] [Google Scholar]
- 52.Ponjavic J, Oliver PL, Lunter G, Ponting CP. Genomic and transcriptional co-localization of protein-coding and long non-coding RNA pairs in the developing brain. PLoS Genet. 2009;5:e1000617. doi: 10.1371/journal.pgen.1000617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marques AC, et al. Chromatin signatures at transcriptional start sites separate two equally populated yet distinct classes of intergenic long noncoding RNAs. Genome Biol. 2013;14:R131. doi: 10.1186/gb-2013-14-11-r131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lepoivre C, et al. Divergent transcription is associated with promoters of transcriptional regulators. BMC Genomics. 2013;14:914. doi: 10.1186/1471-2164-14-914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Copeland NG, Jenkins NA. Harnessing transposons for cancer gene discovery. Nat. Rev. Cancer. 2010;10:696–706. doi: 10.1038/nrc2916. [DOI] [PubMed] [Google Scholar]
- 56.Abbott KL, et al. The candidate cancer gene database: a database of cancer driver genes from forward genetic screens in mice. Nucleic Acids Res. 2015;43:D844–D848. doi: 10.1093/nar/gku770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schmidt K, et al. The lncRNA SLNCR1 mediates melanoma invasion through a conserved SRA1-like region. Cell Rep. 2016;15:2025–37. doi: 10.1016/j.celrep.2016.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Latos PA, et al. Airn transcriptional overlap, but not its lncRNA products, induces imprinted Igf2r silencing. Science. 2012;338(80):1469–1472. doi: 10.1126/science.1228110. [DOI] [PubMed] [Google Scholar]
- 59.Xiang JF, et al. Human colorectal cancer-specific CCAT1-L lncRNA regulates long-range chromatin interactions at the MYC locus. Cell Res. 2014;24:513–531. doi: 10.1038/cr.2014.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schmitt AM, et al. Long noncoding RNAs in cancer pathways. Cancer Cell. 2016;29:452–463. doi: 10.1016/j.ccell.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kent WJ, et al. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Juul, M. et al. Non-coding cancer driver candidates identified with a sample- and position-specific model of the somatic mutation rate. Elife6, e21778 (2017). [DOI] [PMC free article] [PubMed]
- 63.Hindorff La, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl Acad. Sci. USA. 2009;106:9362–7. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Welter D, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42:1001–1006. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tyner C, et al. The UCSC Genome Browser database: 2017 update. Nucleic Acids Res. 2017;45:D626–D634. doi: 10.1093/nar/gkw1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Djebali S, et al. Landscape of transcription in human cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.ENCODE Project Consortium, T. et al. An integrated encyclopedia of DNA elements in the human genome. Nature489, 57 (2012). [DOI] [PMC free article] [PubMed]
- 69.Karolchik D, et al. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004;32:D493–6. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Reimand, U. et al. g:Profiler––a web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res. 10.1093/nar/gkw199 (2016). [DOI] [PMC free article] [PubMed]
- 71.Tripathi S, et al. Meta- and orthogonal integration of influenza “OMICs” data defines a role for UBR4 in virus budding. Cell Host Microbe. 2015;18:723–35. doi: 10.1016/j.chom.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mi H, Muruganujan A, Casagrande JT, Thomas PD. Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 2013;8:1551–1566. doi: 10.1038/nprot.2013.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mi H, et al. PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017;45:D183–D189. doi: 10.1093/nar/gkw1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of additional supplementary items
Data Availability Statement
The data reported in this study are summarized in the manuscript and its Supporting Information files. The list of CLC genes are also available from the GOLD Lab website (https://www.gold-lab.org/clc). Somatic and germline variant calls, mutational signatures, subclonal reconstructions, transcript abundance, splice calls and other core data generated by the ICGC/TCGA Pan-cancer Analysis of Whole Genomes Consortium is described here38 and available for download at https://dcc.icgc.org/releases/PCAWG. Additional information on accessing the data, including raw read files, can be found at https://docs.icgc.org/pcawg/data/. In accordance with the data access policies of the ICGC and TCGA projects, most molecular, clinical and specimen data are in an open tier which does not require access approval. To access potentially identification information, such as germline alleles and underlying sequencing data, researchers will need to apply to the TCGA Data Access Committee (DAC) via dbGaP (https://dbgap.ncbi.nlm.nih.gov/aa/wga.cgi?page=login) for access to the TCGA portion of the dataset, and to the ICGC Data Access Compliance Office (DACO; http://icgc.org/daco) for the ICGC portion. In addition, to access somatic single nucleotide variants derived from TCGA donors, researchers will also need to obtain dbGaP authorisation.
Custom code are available from the corresponding author upon request. The core computational pipelines used by the PCAWG Consortium for alignment, quality control and variant calling are available to the public at https://dockstore.org/search?search=pcawg under the GNU General Public License v3.0, which allows for reuse and distribution.