

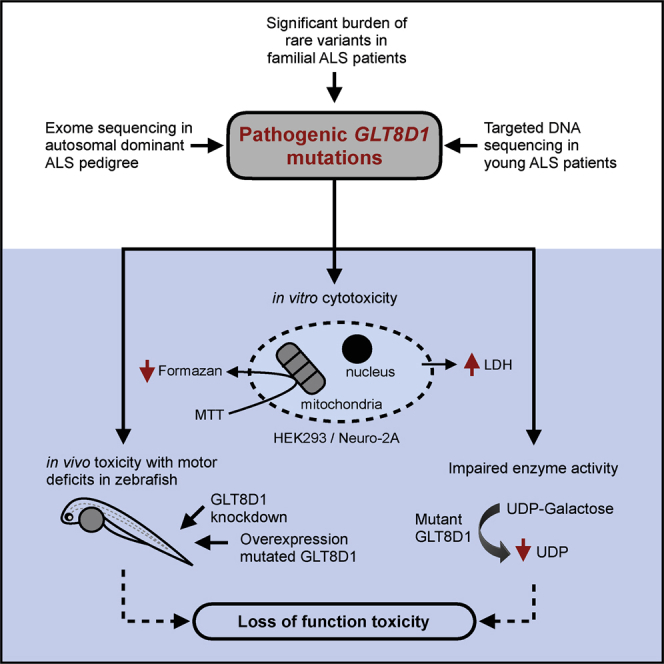
Summary
Amyotrophic lateral sclerosis (ALS) is a severe neurodegenerative disorder without effective neuroprotective therapy. Known genetic variants impair pathways, including RNA processing, axonal transport, and protein homeostasis. We report ALS-causing mutations within the gene encoding the glycosyltransferase GLT8D1. Exome sequencing in an autosomal-dominant ALS pedigree identified p.R92C mutations in GLT8D1, which co-segregate with disease. Sequencing of local and international cohorts demonstrated significant ALS association in the same exon, including additional rare deleterious mutations in conserved amino acids. Mutations are associated with the substrate binding site, and both R92C and G78W changes impair GLT8D1 enzyme activity. Mutated GLT8D1 exhibits in vitro cytotoxicity and induces motor deficits in zebrafish consistent with ALS. Relative toxicity of mutations in model systems mirrors clinical severity. In conclusion, we have linked ALS pathophysiology to inherited mutations that diminish the activity of a glycosyltransferase enzyme.
Keywords: amyotrophic lateral sclerosis, genetics, GLT8D1, glycosyltransferase, cell model, zebrafish
Graphical Abstract
Highlights
-
•
ALS-causing mutations found within the gene encoding the glycosyltransferase GLT8D1
-
•
Five ALS-associated GLT8D1 mutations proximate to the substrate binding site
-
•
GLT8D1 mutations exhibit in vitro cytotoxicity and impair enzyme activity
-
•
GLT8D1 mutations induce motor deficits in zebrafish consistent with ALS
Amyotrophic lateral sclerosis (ALS) is an incurable neurodegeneration. Cooper-Knock et al. report ALS-causing mutations within GLT8D1. Mutations are associated with the substrate binding site and impair enzyme activity. Mutated GLT8D1 is neurotoxic and induces an ALS-like zebrafish phenotype.
Introduction
Amyotrophic lateral sclerosis (ALS) is an aggressive and incurable neurodegenerative disorder. Progress in understanding of the pathogenesis of ALS has come primarily from the study of genetic variants. Approximately 10% of ALS is autosomal dominant but even in sporadic ALS estimates of heritability are as high as 61% (Al-Chalabi et al., 2010); indeed, genetic mutations identified in familial ALS patients are often also present in sporadic ALS patients. Therapeutic targets identified by characterization of genetic variants, therefore, have the potential to be broadly applicable. To date, the majority of described genetic variants are associated with RNA processing, axonal transport, or protein homeostasis (Kapeli et al., 2017, Chia et al., 2018). Discovery of an additional genetic variant that highlights upstream disease biology represents a significant advance.
Using exome sequencing in autosomal dominant familial ALS cases, we identified heterozygous p.R92C mutations in glycosyltransfersase 8 domain containing 1 (GLT8D1) that co-segregate with disease. Targeted sequencing in 103 familial and young sporadic ALS cases identified five additional patients carrying two missense mutations within GLT8D1: the p.R92C mutation identified in the index pedigree and p.G78W. Both mutations lie within exon 4, which encodes the substrate binding domain of GLT8D1. Sequencing of an international ALS cohort confirmed significant ALS-association with genetic variation in exon 4 of GLT8D1 in familial ALS patients, including three additional rare deleterious missense mutations.
GLT8D1 encodes a glycosyltransferase enzyme of unknown function, which is widely expressed (http://www.gtexportal.org/home/). Importantly, this class of proteins has not previously been associated with neurodegeneration, although GLT8D1 has been identified as a schizophrenia risk gene (Sasayama et al., 2014, Yang et al., 2018). It is noteworthy that ALS and schizophrenia share common genetic risk (McLaughlin et al., 2017).
We have localized wild-type and mutant GLT8D1 to the Golgi apparatus. We have shown that ALS-associated GLT8D1 variants are toxic in vivo and in vitro and, moreover, the relative toxicity of the mutations mirrors the clinical phenotype. Both knock down and expression of mutated GLT8D1 in zebrafish embryos produces a locomotor deficit consistent with a role in motor neuron function. Mutation of GLT8D1 negatively impacts upon measured glycosyltransferase activity. Taken together, our data would be consistent with a haploinsufficiency mechanism of toxicity.
Results
Exome Sequencing in an Autosomal Dominant ALS Pedigree Identifies Candidate Deleterious Variants
Exome sequencing in two related individuals with autosomal dominant ALS (Figure 1A) identified five potential causal variants. Candidate monogenic causal variants were shared by both affected individuals, rare, and predicted deleterious. Variants were selected for further analysis if the Exome Aggregation Consortium (ExAC) frequency was <1/10,000 controls (Lek et al., 2016) and the Phred-scaled Combined Annotation Dependent Depletion (CADD) score was >25 (Kircher et al., 2014). Heterozygous variants in five genes met the filtering criteria: p.R92C (GenBank: NM_018446: c.274C>T) in GLT8D1, p.P529L (GenBank: NM_001267619: c.1586C>T) in ARPP21, p.A266T (GenBank: NM_003848: c.796G>A) in SUCLG2, p.R1252H (GenBank: NM_173689: c.3755G>A) in CRB2, and p.C116R (GenBank: NM_052837: c.346T>C) in SCAMP3. Subsequently, another family member developed unilateral weakness and upper motor neuron dysfunction suggestive of ALS (Figure 1A). Detailed neurological investigation did not fulfil El-Escorial diagnostic criteria, but no alternative cause was identified. Screening this individual for the five candidate mutations revealed only p.R92C in GLT8D1 and p.P529L in ARPP21.
Figure 1.

Discovery of ALS-Associated Mutations within Exon 4 of GLT8D1 in Close Proximity to the Putative Substrate Binding Site
(A) Original pedigree in which p.R92C mutations were discovered. Exome sequencing was performed in two related individuals with ALS (∗). Sanger sequencing (of red shapes) confirmed the p.R92C mutation is carried by ALS patients (shaded gray) and absent from unaffected individuals.
(B) Rare deleterious mutations identified within exon 4 of GLT8D1.
(C) Phenotype information for patients carrying mutations in GLT8D1 and/or ARPP21.
(D) Identified structural and topological domains within GLT8D1, including the site of identified mutations within exon 4.
(E) Sequence homology analysis within exon 4 localizes ALS-associated mutations to close proximity with the substrate binding site of GLT8D1. ALS-associated amino acid changes (yellow highlight) affect evolutionary conserved bases (red text) with one exception. Amino acids which form the putative substrate binding site are indicated (blue highlight). Displayed sequence is glycosyltransferase domain encoded by exon 4 (amino acid [aa] 66–aa116).
Targeted Sequencing Supports Pathogenicity of Variants within Exon 4 of GLT8D1
Targeted DNA sequencing of the five candidate genes was performed in a cohort of 103 familial and young sporadic ALS cases from the North of England. The cohort included 34 familial ALS patients in whom a genetic cause had not been identified despite screening for ALS-associated mutations in SOD1, C9ORF72, TARDBP, and FUS; 61 young-onset sporadic ALS patients; and 13 C9ORF72-ALS patients. SUCLG2, CRB2, and SCAMP3 were not mutated in any additional cases and were excluded from further analysis. In addition to the two individuals from the index pedigree, four of the screened patients also carried p.R92C (GenBank: NM_018446: c.274C>T) within GLT8D1 exon 4 (Figure 1C). Another patient carried a rare deleterious p.G78W (GenBank: NM_018446: c.232C>A) change within the same exon of GLT8D1, suggesting a common pathogenic effect (Figure 1D). Mutations were confirmed by Sanger sequencing by using independent DNA samples. In addition to an ExAC frequency of <1/10,000 controls, similar mutations are absent from North of England controls (n = 220) and from unaffected family members within the index pedigree (Figure 1A). No patient with a mutation in GLT8D1 carried an additional ALS-associated mutation, as determined from the ALS Online Genetics Database (Abel et al., 2012).
Of the six cases carrying the p.R92C mutation in GLT8D1, five cases including the two index cases also carried a p.P529L change (GenBank: NM_001267619: c.1586C>T) within cyclic AMP (cAMP)-regulated phosphoprotein 21 (ARPP21). In support of pathogenicity of the p.R92C GLT8D1 mutation in the absence of the ARPP21 variant, one patient carried only the GLT8D1 mutation without an ARPP21 change. Subsequent analysis of Project MinE whole-genome sequencing data from 4,493 ALS patients (Project MinE ALS Sequencing Consortium, 2018), identified two additional patients carrying the p.R92C mutation without a variant in ARPP21. Comparison between the 4,493 Project MinE patients and 60,706 controls sequenced by the ExAC (http://exac.broadinstitute.org/) revealed that the p.R92C change is significantly associated with ALS (Fisher exact test, odds ratio [OR] = 54.1, p = 2.03E-08), and this association remains significant when cases carrying a variant in ARPP21 are excluded (Fisher exact test, OR = 20.3, p = 0.0029).
Exon 4 of GLT8D1 Is Significantly Enriched with ALS-Associated Rare Deleterious Variants Affecting Conserved Amino Acids
Burden analysis was performed using Sequence Kernal Association Test-Optimal Unified Test (SKAT-O) (Lee et al., 2012) to compare rate of genetic variation within exon 4 of GLT8D1 in a cohort of 1,138 familial ALS patients and 19,450 controls (as described in Kenna et al., 2016). Rare deleterious variants are significantly enriched in exon 4 in the familial ALS cohort (SKAT-O, p = 0.0025); there is no significant ALS-association within GLT8D1 when exon 4 is excluded (Table S1). Across all cohorts, we identified five distinct rare deleterious mutations of GLT8D1 exon 4 in fourteen ALS patients (Figures 1B–1E). Identification of additional familial ALS cases from an independent population with similar mutations in GLT8D1 strongly suggests that GLT8D1 is an ALS gene.
Four of five identified GLT8D1 mutations affect amino acids that show high evolutionary conservation, as determined by relative entropy using the NCBI “Conserved Domains Tool” (https://www.ncbi.nlm.nih.gov/Structure/cdd/cddsrv.cgi) (Figure 1E). All mutations are closely associated with the proposed GLT8D1 ligand-binding site involving amino acid residues 71–76 (Bourne and Henrissat, 2001), suggesting that the discovered mutations may modify this activity.
Clinical Characteristics of Patients with Mutated GLT8D1
Clinical characteristics of cases carrying a mutation in GLT8D1 are summarized in Figure 1C. Overall, disease characteristics are within the expected spectrum of ALS (Cooper-Knock et al., 2013). Phenotype data were not available from all cases; however eight patients with p.R92C mutations suffered a particularly aggressive disease course with average survival of 21 months, but patients with p.G78W or p.I70T mutations lived >5 years. Average age of onset across all patients was 49.3 years, and three patients developed symptoms in their fourth decade; early onset is consistent with monogenic disease (Cooper-Knock et al., 2013).
Synergy between p.P529L ARPP21 and p.R92C GLT8D1
We identified six patients carrying both the p.R92C change in GLT8D1 and the p.P529L change in ARPP21. Co-segregation of two rare deleterious mutations is unusual. Within our targeted sequencing experiment, no other rare SNPs (ExAC frequency, <0.0001) were shared between individuals who carried both GLT8D1 and ARPP21 mutations except within the index pedigree. We suggest that both GLT8D1 and ARPP21 changes are neurotoxic and synergy between them might account for their high concurrence in the ALS population. To support this, we have compiled phenotype data from ALS patients with either or both mutations (Figure 1C). ALS in the context of both mutations is significantly more severe than in the presence of either mutation in isolation. Patients carrying both changes lived <16 months compared to an average survival of 36 months in ALS patients carrying p.R92C-GLT8D1 in isolation and 55 months in the presence of p.P529L-ARPP21 in isolation (Wilcoxon rank-sum test, p = 0.001).
GLT8D1 Is Localized to the Golgi Network in Neuronal and Non-neuronal Cells
GLT8D1 is expressed in neurons (http://www.gtexportal.org/home/) and contains an arginine-lysine motif in its cytoplasmic domain (Figure 1D), which likely represents a Golgi localization signal (Uemura et al., 2015). To confirm this, non-neuronal HEK293 and neuronal N2A cells were transfected with pEGFP-N1 expression vectors containing the GLT8D1 nucleotide sequence. Cells were co-stained using anti-JL8 to label GFP-tagged proteins and anti-TGN46 that is normally localized to the Golgi network membrane. Confocal microscopy confirmed significant overlap between GLT8D1 and TGN-46 that supports localization of GLT8D1 to the Golgi apparatus (Figure 2A). Localization was not altered by expression of R92C or G78W mutations compared to the wild-type protein (data not shown).
Figure 2.

When Overexpressed in HEK293 and N2A Cells, GLT8D1 Is Localized to the Golgi Network and Produces Significant Toxicity
(A) The Golgi network was imaged in N2A (top) and HEK293 (bottom) cells using anti-TGN46 (red); GLT8D1-GFP fusion protein was imaged using anti-JL8 (green). Nuclear counterstain (Hoechst) is shown in blue. Scale bar, 50 μm.
(B–E) Overexpression of mutated GLT8D1 increases cytotoxicity (B and C) and reduces metabolic activity (D and E) in HEK293 (B and D) and N2A (C and E) cells compared to overexpression of wild-type protein; all experiments included 6 biological replicates and either 2 (MTT assays) or 4 (LDH assays) technical replicates per biological replicate. ∗p < 0.05.
Error bars represent mean and SD.
Effect of ALS-Associated Mutations of GLT8D1 on Cell Metabolism and Cell Viability
Mutant and wild-type GLT8D1 were overexpressed in non-neuronal HEK293 and neuronal N2A cells. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cell proliferation assays were used to assess metabolic activity; lactate dehydrogenase (LDH) quantification was used to assess cell death. Mutated GLT8D1 was toxic compared to wild-type protein as measured by both LDH (Figures 2B and 2C; HEK293: n = 6, ANOVA p = 0.01; N2A: n = 6, ANOVA p = 0.01) and MTT (Figures 2D and 2E; HEK293: n = 6, ANOVA p = 0.008; N2A: n = 6, ANOVA p = 0.01) assays. In all cases except the MTT assay in HEK293 cells, R92C overexpression produced relatively more toxicity than G78W overexpression, in line with observed relative clinical severity (Figure 1C). Immunoblotting confirmed equivalent expression of mutated and wild-type proteins (Figure S1).
GLT8D1 Knock Down Leads to Impaired Mobility of Zebrafish Embryos
Zebrafish larvae behavior was analyzed at 5 days post-fertilization following knock down of endogenous glt8d1 by using splice-blocking antisense morpholino oligonucleotides. Mean distance moved by 160 zebrafish embryos from three separate clutch mates was tracked using Viewpoint Zebralab software (Scott et al., 2016). Knock down of glt8d1 was associated with a reduction in distance moved by zebrafish (Mann-Whitney U-test, p < 0.0001) (Figure 3A). No significant non-motor phenotype was observed (Video S1). Knock down was confirmed by RT-PCR (Figure S2).
Figure 3.

Knock Down of GLT8D1 and Overexpression of Mutant GLT8D1 Produce Toxicity and Motor Impairment in Zebrafish Embryos
(A, B, and D) Mean distance moved (mm) during three light-dark cycles at 5 days post-fertilization is reduced following injection with 1.5 ng GLT8D1 splice blocking morpholino (A) compared to 1.5-ng control morpholino (n = 160 from 3 clutches); and following injection with 1-nl mature mRNA encoding human GLT8D1-R92C (B) (n = 96 from 2 clutches) or GLT8D1-G78W (D) (n = 48 from one clutch) compared to 1-nl mature mRNA encoding wild-type human GLT8D1.
(C) Injection of 1-nl mature mRNA encoding human GLT8D1-R92C significantly reduces survival of zebrafish embryos at 24 hours and 48 hours compared to mature mRNA encoding wild-type human GLT8D1. WT, wild-type; SB, splice blocking morpholino. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗∗p < 0.0001.
Error bars represent SEM (A, B, and D) or SD (C).
Overexpression of Mutant GLT8D1 Produces Toxicity and Impairs Mobility of Zebrafish Embryos Compared to Wild-Type GLT8D1
Zebrafish embryos were injected at the one-cell stage with mRNA-encoding human wild-type or mutant forms of GLT8D1 to investigate the effects of transient gene expression on motor activity and viability. Expression of wild-type GLT8D1, GLT8D1-R92C, and GLT8D1-G78W protein was confirmed by immunoblotting (Figure S2). Zebrafish embryo survival was reduced in fish exposed to GLT8D1-R92C compared to wild-type GLT8D1 at a 1-nl dose (∼700 pg) (Figure 3C, t test, p = 0.02). Survival was not significantly impaired with injection of a 0.5-nl (∼350 pg) dose (data not shown), but mean distance moved was reduced at 5 days post-fertilization (Figure 3B, Mann-Whitney U-test, p = 0.03); to measure movement, ninety-six zebrafish embryos from two separate clutch mates were tracked. A similar dose (0.5 nl, ∼350 pg) of GLT8D1-G78W RNA also reduced mean distance moved at 5 days post-fertilization (Figure 3D, t test, p = 0.001); forty-eight zebrafish embryos from a single clutch mate were tracked. No significant non-motor phenotype was observed in any of the tracked fish (Videos S2 and S3).
The Glycosyltransferase Activity of GLT8D1 Is Impaired by ALS-Linked Mutations
Based on sequence homology, GLT8D1 is a member of glycosyltransferase family 8 and exon 4, which contains ALS-associated mutations and encodes the substrate binding site within the glycosyltransferase domain (Figure 1D). To assess the impact of discovered mutations on glycosyltransferase activity, we used a uridine diphosphate (UDP)-Glo glycosyltransferase assay kit (Promega) with UDP-galactose as substrate.
FLAG-tagged wild-type and mutated GLT8D1-R92C and GLT8D1-G78W proteins were overexpressed in HEK293 cells and then immunopurified onto anti-FLAG-coated beads prior to competitive elution with a 3× FLAG peptide. All three proteins were purified to a high degree and in equivalent amounts (Figure 4A). To validate the specificity of the signal, we used miRNA knock down of expressed GLT8D1 (Figure S1).
Figure 4.

ALS-Linked GLT8D1-R92C Mutation Impairs Glycosyltransferase Activity
(A) FLAG-tagged WT, GLT8D1-G78W, and GLT8D1-R92C proteins were immunopurified in 1M NaCl-containing buffer to maximize dissociation of interacting partners and eluted in native conditions prior to analysis by SDS-PAGE and Coomassie staining. Input represents ∼0.1% of whole-cell protein extracts loaded onto anti-FLAG coated beads. Arrow indicates FLAG-GLT8D1 proteins.
(B) Standard curve for UDP-Glo assay illustrates linear correlation for <500 μmol/min UDP.
(C) Initial reaction velocity at fixed enzyme concentration with variable substrate concentration; Michaelis-Menten curves were fitted with nonlinear regression. WT, wild-type; FT, flow-through.
In order to measure glycosyltransferase activity, we first determined the linear range for measurement of UDP concentration by the UDP-Glo assay (Figure 4B). Initial reaction velocity was measured at a fixed enzyme concentration with increasing substrate concentrations. Data were fitted to a standard Michaelis-Menten equation by nonlinear regression to enable determination of maximal enzyme velocity (Vmax) and the Michaelis constant (Km) (Figure 4C). Purified GLT8D1 proteins behaved as expected for enzymes of this class and were purified in their active forms. Vmax was reduced ∼30%–40% (WT: 67.5 μmol/min, G78W: 58.6 μmol/min, R92C: 51.4 μmol/min), and Km was reduced ∼50% (WT: 3.5 mmol/l, G78W 2.3 mmol/l, R92C 2.5 mmol/l) in GLT8D1-R92C and GLT8D1-G78W compared to wild-type enzyme. Similar to our cell-based assays, the relative effect of the two tested mutations mirrored observed clinical severity, with R92C producing a more severe phenotype. Next, we fixed substrate concentration to ∼5-fold higher than Km in order to measure near maximal enzymatic velocity and plotted enzyme activity as a function of enzyme concentration. At all enzyme concentrations, the activity of GLT8D1-R92C and GLT8D1-G78W proteins was reduced compared to wild-type enzyme (Figure S3).
Discussion
To date, understanding and translational approaches to ALS have been underpinned by described genetic variants. In recent years, these genetic variants have focused onto a small number of pathways, including RNA processing, protein homeostasis, and axonal function (Kapeli et al., 2017, Chia et al., 2018). This convergence has limited the new information from each discovery. In light of this, the discovery of a genetic variant within a novel functional category is a major step forward for ALS research.
We identified mutations within exon 4 of GLT8D1 that segregated with disease in an autosomal dominant pedigree. Moreover, we discovered that there is significant enrichment of GLT8D1 exon 4 mutations in an international cohort of familial ALS cases compared to matched controls; this is consistent with a monogenic disease. Overall, analysis of sequencing data from local and international cohorts revealed five rare deleterious mutations within GLT8D1 exon 4 in ALS patients.
Based on sequence homology, GLT8D1 is a member of glycosyltransferase family 8 and is expected to catalyze the transfer of a glycosyl group from a donor to acceptor via a “retaining” mechanism involving a glycosyl-enzyme intermediate (http://www.cazy.org/GT8_structure.html). Although the crystal structure of GLT8D1 is unknown, it contains a conserved “DXD” metal ion binding site that indicates that it is a Letoir glycosyltransferase where donor and acceptor binding sites are divided by a single “GT-A” fold (Bourne and Henrissat, 2001). Discovered ALS-associated mutations in GLT8D1 cluster together in a short sequence of ∼20 amino acids in close proximity to the substrate binding site, suggesting a common effect on this function. Consistent with this, we observed a significant reduction in enzyme activity in mutated GLT8D1-R92C and GLT8D1-G78W compared to wild-type protein. Measured Km was reduced in the mutated proteins commensurate with an increase in substrate affinity, which could impair cycling of substrate through the enzyme and, thus, reduce overall velocity. Moreover, higher substrate affinity could result in a dominant-negative effect with a competitive antagonism of wild-type enzyme function. Precedence exists; for example dominant-negative mutations in the GTPase Ras increase its substrate affinity and, thus, allow it to act as a competitive antagonist of the wild-type protein (Nassar et al., 2010). We cautiously note the example of SOD1 mutations that were originally thought to cause ALS through loss of function but were subsequently shown to cause gain of function toxicity (Boillée et al., 2006); our work awaits validation in a higher organism. Of note, measured Km values were relatively high compared to reports of other members of the glycosyltransferase 8 family (Persson et al., 2001), suggesting low substrate affinity. It is possible that absolute substrate affinity would be higher under physiological conditions, particularly as the identity of the normal donor and acceptor for GLT8D1 are unknown and glycosyltransferase enzymes with a GT-A fold structure have been observed to show cooperativity between donor and acceptor binding (Shoemaker et al., 2008). Our reaction conditions utilized UDP-galactose and occurred without an acceptor.
Our functional data confirm the molecular toxicity of the discovered mutations in GLT8D1 in vitro and in vivo. In neuronal and non-neuronal cells, overexpression of mutated GLT8D1 is toxic compared to equivalent expression of the wild-type protein. To confirm that GLT8D1 mutations are toxic within a whole organism, we expressed human mutated and wild-type GLT8D1 in zebrafish embryos. At higher doses, the expression of mutated GLT8D1 produced increased rates of embryo death compared to wild-type; at lower doses, motor function was specifically impaired in fish expressing either the GLT8D1-R92C or GLT8D1-G78W mutated protein. This is consistent with a specific effect on the motor system. Further supporting a role for GLT8D1 in motor function, knock down of the endogenous glt8d1 protein in zebrafish embryos produced a specific deficit of motor function without observable off-target toxicity. Overlap between the effect of glt8d1 knock down and overexpression of mutated GLT8D1 is consistent with a dominant-negative mechanism.
Clinically, patients carrying mutations in GLT8D1 are within the spectrum of sporadic ALS, suggesting that they may share a common disease mechanism. Unfortunately, pathological material was not available from these cases and so we were not able to ascertain the presence of TDP-43-positive neuronal inclusions, which are the hallmark pathology in most cases of ALS. All patients suffered relatively young-onset disease, which is consistent with a monogenic disorder. In measures of toxicity and enzyme activity, the relative severity of the studied mutations mirrored the relative clinical severity—R92C produced a more severe effect than G78W.
In a number of patients, the p.R92C-GLT8D1 mutation occurred in the presence of a rare deleterious p.P529L-ARPP21 mutation. Despite this, our data clearly suggest that GLT8D1 mutations are toxic in isolation: the p.R92C mutation is significantly enriched in ALS patients even when patients also carrying an ARPP21 change are excluded. Moreover, we observed a significant clustering of five different rare deleterious mutations within GLT8D1 exon 4 in familial ALS patients, but only the R92C change occurred in the presence of p.P529L-ARPP21. Finally, all of our functional data are consistent with a common effect for two different ALS-associated GLT8D1 mutations: p.R92C and p.G78W. It is possible that an ARPP21 mutation is an independent toxic factor that acts synergistically with GLT8D1 mutation; this might explain their high concurrence in the ALS population. Consistent with this hypothesis, we have identified a significant increase in disease severity in ALS patients carrying both p.R92C-GLT8D1 and p.P529L-ARPP21 compared to either mutation in isolation.
Gangliosides are sialic acid-containing glycosphingolipids that are particularly abundant within the CNS (Vajn et al., 2013). Gangliosides within the CNS are typically synthesized in the endoplasmic reticulum (ER) from a lactosylceramide precursor and are remodeled during transit from the cis-Golgi to the trans-Golgi network by a series of glycosyltransferase enzymes that incorporate galactose and GalNAc groups. Consistent with a role in this process, GLT8D1 shows prominent CNS expression; moreover, we have shown that GLT8D1 carries a Golgi localization signal, is prominently expressed within the Golgi, and is able to accept UDP-galactose as a substrate. Mature gangliosides are carried to the cell surface where they function prominently in cell signaling (Yu et al., 2012). Interestingly, autoantibodies against specific gangliosides produce an inflammatory disease of spinal motor neurons known as multifocal motor neuropathy with conduction block (Harschnitz et al., 2014), which is a common differential diagnosis of ALS. Altered levels of gangliosides have been reported in animal models of ALS and in post-mortem CNS tissue from ALS patients (Dodge et al., 2015, Ariga, 2014). We have shown that ALS-linked mutations in GLT8D1 impair its glycosyltransferase activity that we predict will negatively impact on ganglioside signaling.
In conclusion, we have discovered that mutations in exon 4 of GLT8D1 are a cause of ALS. GLT8D1 is a glycosyltransferase, a class of proteins that has not previously been associated with neurodegeneration. Several lines of our evidence suggest that ALS-associated mutations are associated with haploinsufficiency or even a dominant-negative effect: (1) mutations cluster in proximity to the substrate binding domain, (2) mutated protein exhibits reduced enzyme activity compared to wild-type, and (3) overexpression of mutant GLT8D1 and knock down of endogenous glt8d1 in zebrafish both produce a locomotor deficit consistent with ALS. Classically, pathogenic enzyme mutations are thought to be recessive, but it is estimated that as many as 25% of enzyme mutations are autosomal dominant (Veitia, 2002) as in our patients. Our discovery places GLT8D1 glycosyltransferase activity firmly upstream in the pathogenesis of ALS, making it an attractive therapeutic target. Currently, we do not know the extent to which GLT8D1 activity is impaired in ALS patients with and without GLT8D1 mutations or whether augmenting GLT8D1 activity could be beneficial irrespective of background activity.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-GLT8D1 | GeneTex | Cat.# GTX123636; RRID: AB_11169529 |
| Rabbit anti-TGN46 | Abcam | Cat.# ab50595; RRID: AB_2203289 |
| Mouse anti-Tubulin | Sigma-Aldrich | Cat.# T6199; RRID: AB_477583 |
| Mouse anti-JL8 | Takara Bio | Cat.# 632380; RRID: AB_10013427 |
| Mouse anti-FLAG-M2 | Sigma | Cat.# F3165; RRID: AB_259529 |
| Donkey anti-mouse Alexa568 secondary | Life Technologies | Cat.# A-10037; RRID: AB_2534013 |
| Donkey anti-mouse Alexa488 secondary | Life Technologies | Cat.# A-21202; RRID: AB_141607 |
| Goat anti-rabbit Alexa568 secondary | Life Technologies | Cat.# A-11011; RRID: AB_143157 |
| Donkey anti-rabbit Alexa488 secondary | Life Technologies | Cat.# A-21206: RRID: AB_141708 |
| Chemicals, Peptides and Recombinant Proteins | ||
| Normal goat serum | Vector | Cat.# S-1000 |
| Fluorescent mounting medium | Dako | Cat.# GM304 |
| Hoechst 33342 | ThermoFisher Scientific | Cat.# 62249 |
| Pre-stained protein ladder | Cleaver Scientific | Cat.# CSL-PPL |
| Unstained SDS-PAGE Standard | Bio-Rad | Cat.# 1610317 |
| Bradford reagent | Bio-Rad | Cat.# 5000001 |
| Clarity Western ECL blotting substrate | Bio-Rad | Cat.# 1705060S |
| 10x FastDigest Green Buffer | ThermoFisher Scientific | Cat.# B72 |
| Trypsin | Sigma-Aldrich | Cat.# 59427C |
| Thiazolyl Blue Tetrazolium Bromide (MTT) | Sigma-Aldrich | Cat.# M2128 |
| opti-MEM | ThermoFisher Scientific | Cat.# 11058021 |
| Laemmli buffer | Bio-Rad | Cat.# 1610747 |
| SigmaFAST™ Protease Inhibitor Cocktail tablets | Sigma-Aldrich | Cat.# S8820 |
| Lysogeny broth | ThermoFisher Scientific | Cat.# 10855021 |
| LB agar | ThermoFisher Scientific | Cat.# 22700025 |
| β-mercaptoethanol | Sigma-Aldrich | Cat.# M6250 |
| Triton X-100 | Sigma-Aldrich | Cat.# X100 |
| Hygromycin B | Invitrogen | Cat.# 10687010 |
| Blasticidin | ThermoFisher Scientific | Cat.# A1113902 |
| Penicillin-Streptomycin | Sigma-Aldrich | Cat.# P4333 |
| DMF | Fisher Scientific | Cat.# D/3840/17 |
| SDS | Fisher Scientific | Cat.# S/5200/53 |
| SDS - Lauryl Sulfate | Melford | Cat.# L22010-1000 |
| Sodium chloride 99.5% | Fisher Scientific | Cat.# S/3161/65 |
| EDTA | Sigma-Aldrich | Cat.# E5134 |
| DTT | Sigma-Aldrich | Cat.# D9779 |
| HEPES | Sigma-Aldrich | Cat.# H3375 |
| Glycerol | Sigma-Aldrich | Cat.# G9012 |
| Manganese (II) chloride | Fisher Scientific | Cat.# M/1850/53 |
| 1M Tris buffer | Vivantis | Cat.# PB0855-1L |
| Dulbecco’s Modified Eagle’s Medium (DMEM) | Corning | Cat.# 10-017-CV |
| Tetracycline-free FBS | Biosera | Cat.# FB-1001T |
| FBS | ThermoFisher Scientific | Cat.# 10270106 |
| BSA | Sigma-Aldrich | Cat.# A2058 |
| Agarose | Melford | Cat.# MB1200 |
| 3x FLAG® peptide | Sigma-Aldrich | Cat.# F4799 |
| Anti-FLAG® M2 magnetic beads | Sigma-Aldrich | Cat.# M8823 |
| Critical Commercial Assays | ||
| mMESSAGE mMACHINE-SP6 transcription kit | Fisher Scientific | Cat.# AM1340 |
| LDH Cytotoxicity Assay Kit | Pierce | Cat.# 88953 |
| UDP-glycosyltransferase assay | Promega | Cat.# V6961 |
| UDP-galactose 100mM | Promega | Cat.# V7071 |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | Cat.# ACS-4500 |
| HEK293 FlpIN | Invitrogen | Cat.# R75007 |
| N2A | ATCC | Cat.# CCL-131 |
| HEK293 | ATCC | Cat.# CRL-1573 |
| Experimental Models: Organisms/Strains | ||
| Zebrafish | AB zebrafish | zfin.org/ZDB-GENO-960809-7 |
| Oligonucleotides | ||
| See Table S2 for oligo information | ||
| Recombinant DNA | ||
| pcDNA5/FRT/TO_GFP vector | Addgene | Cat.# 19444; RRID: Addgene_19444 |
| pcDNA6.2-GW/EmGFP vector | ThermoFisher Scientific | Cat.# K493600 |
| Software and Algorithms | ||
| SKAT-O | Lee et al., 2012 | http://cran.r-project.org/web/packages/SKAT/index.html |
| R | The R Project for Statistical Computing | http://cran.r-project.org/mirrors.html |
| Galaxy | The Galaxy Project | https://usegalaxy.org/ |
| snpStats | University of Cambridge | http://www.bioconductor.org/packages/release/bioc/html/snpStats.html |
| VariantAnnotation | Obenchain et al., 2014 | http://www.bioconductor.org/packages/release/bioc/html/VariantAnnotation.html |
| Plink | Purcell et al., 2007 | http://zzz.bwh.harvard.edu/plink/download.shtml |
| PRISM 7 | GraphPad | https://www.graphpad.com/ |
| Zebralab | Viewpoint | http://www.viewpoint.fr/en/p/software/zebralab |
| Fiji (Fiji Is Just ImageJ) | NIH | https://imagej.net/Fiji |
| wANNOVAR | Yang and Wang, 2015 | http://wannovar.wglab.org/ |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Johnathan Cooper-Knock (j.cooper-knock@sheffield.ac.uk).
Experimental Model and Subject Details
Selection of patients and controls for genetic sequencing
The study was approved by the South Sheffield Research Ethics Committee and informed consent was obtained for all samples. Genomic DNA was extracted from 103 ALS patients (33 female, 65 male, 5 unavailable) from the North of England. In this cohort mean age of onset was 49 years and mean survival (censored) was 67 months. The cohort included 34 familial ALS patients in whom a genetic cause had not been identified despite screening for ALS associated mutations in SOD1, C9ORF72, TARDBP, and FUS; and 61 young-onset sporadic ALS patients. Two additional cohorts of patients were utilized for genetic screening: the Project MinE latest data freeze including 4493 ALS patients and 1924 controls (3764 males and 2632 females, 21 unavailable); and the ALS Variant Server familial ALS cohort (http://als.umassmed.edu/index.php#FALSbrowser, no phenotype information available) including 1138 familial ALS patients.
Immortalized cell lines
Human embryonic kidney-293 (HEK293) cells and mouse neuro-2A (N2A) cells were cultured in DMEM medium supplemented with 10% (v/v) fetal bovine serum (Thermo-Fisher Scientific), 50 units/mL of penicillin and 50 μg/mL of streptomycin. N2A cell media was supplemented with an additional 500 mM sodium pyruvate (Thermo-Fisher Scientific). Cell lines were maintained at 5% CO2 in a 37°C incubator and split when ∼80% confluent. All experimental work was performed using HEK293T and N2A cells within the range of 7-32 passages.
Zebrafish models
All applicable international, national and/or institutional guidelines for the care and use of animals were followed. This includes review by the local ethical review panel at the University of Sheffield, UK. Zebrafish of the AB strain were raised in recirculating systems provided by Techniplast, at a density of 1 zebrafish per 0.25 l. Water was maintained at pH 7.4, a temperature of 28°C, and electrolytic conductivity of 500 μS/cm. Zebrafish were exposed to 14 hour light and 10 hour dark cycles. The night before breeding, adult male and female zebrafish were transferred to breeding tanks, separated by a clear plastic divider. At the start of the light cycle the following day, the dividers were removed and eggs were collected. Embryos were transferred to Petri dishes containing fresh E3 media and incubated at 28°C.
Method Details
High Throughput DNA sequencing
Exome sequencing was performed by Oxford Gene Technology (https://www.ogt.com/). Genomic DNA was enriched by SureSelect All Exon V4 capture in prepare libraries. Sequencing was performed using an Illumina HiSeq 2000 with 100bp paired end reads and minimum 50X coverage.
For targeted validation of candidate genes genomic DNA was enriched for selected genes using a custom designed Agilent SureSelect in solution kit. Sequencing was performed using an Illumina HiScan platform according to manufacturer’s instructions.
All sequencing data was aligned using Galaxy (http://usegalaxy.org). Reads were mapped to the human genome assembly GRCh37 (hg19) reference genome using BWA (Li and Durbin, 2009). GATK (McKenna et al., 2010) was used to produce vcf. files which were annotated using wANNOVAR (Yang and Wang, 2015).
Generation of GFP and FLAG-tagged GLT8D1 plasmids
For GFP-tagged fusions, the human GLT8D1 open reading frame (IMAGE:40116197) was amplified using oligonucleotides G8-1-koz-NheI5 and G8-371-XhoI3 (Table S2) and cloned as a NheI/XhoI PCR fragment into the NheI/XhoI restriction sites of the pEGFP-N1 vector (Clontech) to generate pEGFP-N1 _GLT8D1-eGFP.
For FLAG-tagged fusions, BamHI/HindIII restricted oligonucleotides (Table S2) bearing Kozac and 3xFLAG-M2 tag sequences were annealed and phosphorylated prior to cloning into the HindIII/BamHI sites of the backbone pcDNA5/FRT/TO_GFP vector (Addgene 19444) to generate a pcDNA5/FRT/TO_3xFLAG vector. The HindIII site of pcDNA5/FRT/TO_3xFLAG was further destroyed by site-directed mutagenesis. The human GLT8D1 open reading frame (IMAGE:40116197) was amplified using oligonucleotides G8-1-BclI5 and G8-371stp-XhoI3 (Table S2) and cloned as a BclI/XhoI PCR fragment into the BamHI/XhoI restriction sites of the pcDNA5/FRT/TO_3xFLAG vector to generate pcDNA5/FRT/TO_3xFLAG-GLT8D1.
G78W and R92C ALS-linked mutations were introduced on the pEGFP-N1 _GLT8D1-eGFP and pcDNA5/FRT/TO_3xFLAG-GLT8D1 plasmids by site-directed mutagenesis using the following oligonucleotides: G8-G78W_QC_fwd and G8-G78W_QC_rev; or G8-R92C_QC_fwd and G8-R92C_QC_rev (Table S2). All plasmids were validated by Sanger-sequencing and sequences are available upon request.
MTT assays
HEK293 and N2A cells were transfected with pEGFP-N1 vectors expressing wild-type and mutant GLT8D1 sequences. Control groups were transfected with empty pEGFP-N1 vectors. A colorimetric assay using 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) dye was used to assess N2A and HEK293 cellular metabolic activity at 2 and 3 days post transfection (dpt), respectively. 55 μL of 5mg/mL of MTT reagent in PBS was added per well of a 24-well culture plate and incubated at 37°C for 1 hour. 550 μL of un-precipitated 20% SDS in 50% di-methyl formamide (DMF) + dH2O (pH 7.4) was added per well and mixed thoroughly to lyse the cells. Cells were incubated in a dark environment on an orbital shaker for 1 hour. The colorimetric change was measured using a PHERAstar FS spectrophotometer (BMG Biotech), and absorbance readings taken at 590nm were normalized to media-only wells. Mean absorbance readings were calculated for each biological repeat and expressed as a percentage of controls.
LDH assay
HEK293 and N2A cells were transfected with pEGFP-N1 vectors expressing wild-type and mutant GLT8D1 sequences. Control groups were transfected with empty pEGFP-N1 vectors. The lactate dehydrogenase (LDH) assay was used to quantify the amount of LDH released into the culture media from HEK293 and N2A cells at 2 and 3 dpt, respectively. A Pierce™ LDH Cytotoxicity Assay Kit (ThermoFisher Scientific) was used according to the manufacturer’s instructions. Sterile, ultra-pure H2O was added to one set of triplicate cell-containing wells (10% v/v) as a measure of spontaneous LDH activity. 10x Lysis buffer was added to another set of triplicate cell-containing wells (10% v/v) to determine the maximum LDH activity. Cells were incubated at 37°C in 5% CO2 for 45 minutes. 50 μL of each sample medium was transferred to an optically clear 96-well plate in triplicate wells. 50 μL of reaction mixture (lyophilizate, 11.4mL ultra-pure H2O, 0.6mL assay buffer) was added to each sample well and plates were incubated in a dark environment for 30 minutes. 50 μL of stop solution was added to each sample well and the absorbance was measured at 490nm to 680nm using a PHERAstar FS spectrophotometer (BMG Biotech). The 680nm absorbance value was subtracted from the 490nm absorbance value to determine LDH activity. Mean absorbance readings were calculated for each biological repeat and expressed as a percentage of controls.
Zebrafish
Splice-blocking morpholino and control morpholino oligonucleotides (Table S2) were designed and provided by Gene Tools, LLC. The GLT8D1 start codon lies within exon 2, therefore skipping this exon was considered likely to change expression of the gene. The splice blocking morpholino sequence complementary to the splice junction target from 5′ - 3′ is shown in bold; brackets have been inserted either side of the pre-mRNA (sense strand) target to illustrate its position in the sequence:
ACATCAGAGTCTGCATACCTGAAGT[ACGCAGAGgtaactatattttacac]gactttca.
Morpholino powder was reconstituted in sterile Milli-Q water and 1% phenol red to create a 1mM stock. Reduction in expression of the zebrafish glt8d1 mRNA was confirmed by RT-PCR.
For overexpression studies, the human GLT8D1 sequences were cloned into PCS2+ downstream of an SP6 promotor and contained a 3′ polyA signal. Plasmids were amplified by transformation of DH5-α competent Escherichia coli cells before overexpression in zebrafish embryos. mRNA was synthesized using mMessage machine (Fisher scientific) as per manufacturer’s protocol, purified and resuspended in RNase free water.
Fertilized embryos were injected into the yolk at the one-cell stage using a pressure injector with either 0.5nL, 1.0nL or 2.0nL of injection material. Embryos were transferred to Petri dishes containing E3 media. Any unfertilized embryos, or embryos damaged during the injection process, were discarded. To increase the reliability of data, zebrafish embryos from separate clutch mates underwent the same injection process for each biological repeat. Embryos were raised to 2 days for western blotting for detecting reduction in expression levels and to 5 dpf for behavioral analysis.
Viewpoint Zebralab software was used to record the activity of individual larva in a 96-well plate at 5 days post-fertilization. Zebrafish embryos were washed in fresh E3 media (approximately 50 embryos per group) and transferred to an optically clear 96-well plate, one fish per well in 200 μL E3 medium. 100 μL of E3 media was removed from one column and replaced with 100 μL of Tricaine to create a non-motile control group. Fish were left to acclimatise to the 96-well plate for 30 minutes. Zebrafish were then placed inside a Zebrabox observation chamber (Viewpoint) and were habituated at 10% light intensity for 30 minutes prior to behavioral testing. Zebrafish were exposed to a 10 minute light (10% intensity) cycle followed by a 10 minute dark (0% intensity) cycle. These cycles were then repeated to equal a total of 40 minutes of video recording. Motor function was analyzed according to the mean distance traveled.
For immunoblotting: Dechorionated zebrafish embryos (2dpf) were transferred to eppendorf tubes (20 embryos per tube) containing 1mL E3 media (34.8g NaCl; 1.6g KCl; 5.8g CaCl2·2H2O; 9.78 g MgCl2·6H2O) and immersed in Tricaine (MS222) (400mg tricaine powder; 97.9mL dH2O; 2.1mL 1 M Tris; pH 7) (Sigma-Aldrich) (164 mg/L) for 5 minutes. E3 media was removed and replaced with 1 μL/embryo 2x Laemmli buffer (65.8 mM Tris HCl; 26.3% (w/v) glycerol; 2.1% SDS; 0.01% bromophenol blue; 355 mM 2-mercaptoethanol; pH 6.8). Zebrafish embryos were lysed by sonication (Soniprep 150, MSE) for 10 s at 25% amplitude followed by a 30 s incubation on ice; this process was repeated 3 times for each group. Embryos were subsequently boiled at 95°C for 10 minutes, then centrifuged at 10,000 xg for 3 minutes. 10 μL of lysate was added per lane and fractionated on a 12% SDS-PAGE gel.
Immunocytochemistry
HEK293 and N2A cells were transfected with pEGFP-N1 vectors expressing wild-type and mutant GLT8D1 sequences. HEK293 and N2A cells were cultured on sterile coverslips in 24-well plates for 2/3 days prior to staining, respectively. Cells were blocked using 5% normal goat serum (NGS) (v/v%) in PBS with 0.01% Triton™ X-100 (v/v%), and were incubated at room temperature for 45 minutes. Blocking buffer was removed and cells were incubated in primary antibody, diluted in 5% NGS (v/v %), at room temperature for 1 hour or overnight at 4°C. Cells were washed 3x in PBS (5 minutes per wash), then incubated in a fluorescent secondary antibody, diluted in 5% NGS (v/v %), in a dark environment at room temperature for 1 hour. Cells were incubated with a nuclear counterstain (Hoechst 33342) in the dark for 5 minutes at room temperature and washed 3x with PBS (5 minutes per wash). Coverslips were mounted onto microscope slides using fluorescence mounting medium (DAKO) and stored in a dark environment at 4°C overnight to dry. Cells were imaged the following day using a using a Leica SP5 confocal microscope system with a × 63/1.4 oil immersion objective lens.
Immunoprecipitation and assessment of glycosyltransferase activity
HEK293 and N2A cells were transfected with pcDNA5/FRT/TO_3xFLAG vectors expressing mutant and wild-type GLT8D1. Cells were lysed 2 dpt in lysis buffer (150mM NaCl, 1mM EDTA, 1mM DTT, 0.5% Triton X-100, 50mM HEPES, 10% glycerol, pH 7.5) and a Bradford assay was used to determine overall protein concentration. Protein lysates were incubated with anti-FLAG® M2 magnetic beads (Sigma-Aldrich) for 4 hours on a rotating mixer at 4°C. FLAG-tagged GLT8D1 was eluted overnight in lysis buffer using a 3xFLAG® peptide (Sigma-Aldrich). Protein purification was confirmed via a combination of Coomassie Blue staining and immunoblotting. To validate the specificity of GLT8D1 immunoblot bands we performed miRNA knockdown of expressed GLT8D1. miRNA sequences against human GLT8D1 (NCBI Reference Sequence: NM_152932.2, transcript variant 1) were designed using the ‘miR RNAi’ Block-IT RNAi designer tool (ThermoFisher): GLT8D1_miR1 target: ATTGTAGGGCCTCAACCTATA starts at 241nt, GLT8D1_miR2 target: GAGCAGGAAACCAGTACAATT starts at 749nt, GLT8D1_miR3 target: TTTGTAAAGGCTGCCAAGTTA starts at 1075nt. Synthesized oligonucleotides (Table S2) were annealed and ligated into pcDNA6.2-GW/EmGFP vectors. HEK293 cells were co-transfected with pcDNA5/FRT/TO_3xFLAG vectors expressing wild-type GLT8D1 with and without pcDNA6.2-GW/EmGFP vectors expressing miRNA sequences. Knockdown was confirmed at two days post transfection via immunoblotting.
Glycosyltransferase activity was measured using a UDP-Glo Glycosyltransferase Assay kit (Promega) according to the manufacturer’s instructions. 25 μL reactions were initiated by adding 28mg/mL of purified protein in lysis buffer to a UDP-galactose substrate (Promega) in 3 μL 1x glycosyltransferase reaction buffer (50mM Tris pH 7.5 + 5mM MnCl2) + dH2O. Reactions were assembled in a solid white 96-well plate at room temperature and incubated at 37°C for 60 minutes. To determine kinetic parameters of the glycosyltransferase enzyme, multiple reactions with varying concentrations of either the enzyme or substrate were carried out simultaneously in the presence of fixed volumes of all other components. Glycosyltransferase reactions were terminated by the addition of 25 μL UDP detection reagent to each well of the assay plate. Plates were mixed for 30 s on a shaker and incubated in a dark environment at room temperature for 60 minutes. Luminescence was recorded using a Pherastar FS system (BMG Labtech). A standard curve of 0-25 μM UDP-standard was prepared in 1x glycosyltransferase reaction buffer to determine the relationship between [UDP] and luminescence measurement.
Quantification and Statistical Analysis
Statistical analysis was conducted in GraphPad Prism 7 (La Jolla, CA). All bar graphs show the mean ± SEM. Data comparing two variables was transformed to identify outliers and analyzed to identify statistical differences between the treatment groups. Statistical test used was a non-parametric analysis of variance (ANOVA) except zebrafish data which were analyzed using either a Mann-Whitney test or, in the case of survival data, a paired t test to account for continuity between time points.
Genetic burden analysis to determine the relative frequency of rare deleterious variants within exon 4 of GLT8D1 was performed using SKAT-O (Lee et al., 2012). Rare deleterious mutations were defined by frequency within the ExAC dataset of < 1/10,000 control alleles (Lek et al., 2016), and a Phred-scaled Combined Annotation Dependent Depletion (CADD) score > 25 (Kircher et al., 2014) based on the minimum score seen in ALS patients. For burden analysis, threshold CADD score was determined by the minimum seen in patients. Comparison of various pathogenicity prediction tools recently supported the sensitivity and specificity of CADD (Salgado et al., 2016). Given that we were focused on exonic changes with an effect on protein function, synonymous changes were excluded. We also excluded short indels and changes with a read depth < 10 due to the possibility that these represent sequencing errors; we validated GLT8D1 mutations by Sanger sequencing.
Acknowledgments
The authors would like to thank the ALS Variant Server (http://als.umassmed.edu), which is supported by funds from NIH (NINDS) (1R01NS065847), AriSLA (EXOMEFALS, NOVALS), the ALS Association, and the MND Association (MNDA); and the Project MinE data browser (http://databrowser.projectmine.com/). We acknowledge grants from EU Framework 7 (Euro-Motor), JPND and MRC (SOPHIA, STRENGTH, and ALS-CarE), IWT, Belgian ALS Liga, Opening the Future Fund, MNDA (Hautbergue/Apr16/846-791 and Ramesh/Apr17/854-791), IUAP (program P7/16), and FWO-Vlaanderen (E-RARE-2). T.M. is supported by the University of Sheffield Lee Newton PhD studentship. J.C.-K. holds a NIHR Clinical Lectureship. G.M.H. is supported by a MRC New Investigator Research Grant (MR/R024162/1). P.J.S. is supported as an NIHR Senior Investigator. A.A.C. receives salary support from the NIHR Dementia Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London. P.V.D. holds a senior clinical investigatorship from FWO-Vlaanderen. W.R. is supported through the E. von Behring Chair for Neuromuscular and Neurodegenerative Disorders. This work was also supported by the NIHR Sheffield Biomedical Research Centre for Translational Neuroscience and the Sheffield NIHR Clinical Research Facility. Biosample collection was supported by the MND Association and the Wellcome Trust (P.J.S.). We are grateful to patients and control subjects who generously donated biosamples.
Author Contributions
J.C.-K., A.H., G.M.H., T.R., J.K., and P.J.S. were responsible for the conception and design of the study. J.C.-K., T.M., A.H., T.R., L.C., I.F., A.B., P.V.D., M.M., W.R., O.H., M.P.P., A.A., J.S.M., A.N.B., K.E.M., C.E.S., A.A.C., J.E.L., H.R., I.N., P.H., M.W., T.W., M.K., C.M., G.M.H., J.V., J.K., and P.J.S. were responsible for generating reagents and acquiring data. J.C.-K., T.M., A.B., L.C., A.H., T.R., H.R., and I.N. were responsible for analysis of data. J.C.-K., T.M., T.R., A.H., G.M.H., L.C., J.K., and P.J.S. were responsible for interpretation of data. All authors were responsible for revising the manuscript and approving the final version for publication.
Declaration of Interests
The authors declare no competing interests.
Published February 26, 2019
Footnotes
Supplemental Information can be found with this article online at https://doi.org/10.1016/j.celrep.2019.02.006.
Contributor Information
Johnathan Cooper-Knock, Email: j.cooper-knock@sheffield.ac.uk.
Pamela J. Shaw, Email: pamela.shaw@sheffield.ac.uk.
Supplemental Information
References
- Abel O., Powell J.F., Andersen P.M., Al-Chalabi A. ALSoD: a user-friendly online bioinformatics tool for amyotrophic lateral sclerosis genetics. Hum. Mutat. 2012;33:1345–1351. doi: 10.1002/humu.22157. [DOI] [PubMed] [Google Scholar]
- Al-Chalabi A., Fang F., Hanby M.F., Leigh P.N., Shaw C.E., Ye W., Rijsdijk F. An estimate of amyotrophic lateral sclerosis heritability using twin data. J. Neurol. Neurosurg. Psychiatry. 2010;81:1324–1326. doi: 10.1136/jnnp.2010.207464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariga T. Pathogenic role of ganglioside metabolism in neurodegenerative diseases. J. Neurosci. Res. 2014;92:1227–1242. doi: 10.1002/jnr.23411. [DOI] [PubMed] [Google Scholar]
- Boillée S., Vande Velde C., Cleveland D.W. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52:39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Bourne Y., Henrissat B. Glycoside hydrolases and glycosyltransferases: families and functional modules. Curr. Opin. Struct. Biol. 2001;11:593–600. doi: 10.1016/s0959-440x(00)00253-0. [DOI] [PubMed] [Google Scholar]
- Chia R., Chiò A., Traynor B.J. Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. Lancet Neurol. 2018;17:94–102. doi: 10.1016/S1474-4422(17)30401-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper-Knock J., Jenkins T., Shaw P.J. clinical and molecular aspects of motor neuron disease. Colloquium Series on Genomic and Molecular Medicine. 2013;2:1–60. [Google Scholar]
- Dodge J.C., Treleaven C.M., Pacheco J., Cooper S., Bao C., Abraham M., Cromwell M., Sardi S.P., Chuang W.L., Sidman R.L. Glycosphingolipids are modulators of disease pathogenesis in amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA. 2015;112:8100–8105. doi: 10.1073/pnas.1508767112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harschnitz O., Jongbloed B.A., Franssen H., Straver D.C., van der Pol W.L., van den Berg L.H. MMN: from immunological cross-talk to conduction block. J. Clin. Immunol. 2014;34(Suppl 1):S112–S119. doi: 10.1007/s10875-014-0026-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapeli K., Martinez F.J., Yeo G.W. Genetic mutations in RNA-binding proteins and their roles in ALS. Hum. Genet. 2017;136:1193–1214. doi: 10.1007/s00439-017-1830-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenna K.P., van Doormaal P.T., Dekker A.M., Ticozzi N., Kenna B.J., Diekstra F.P., van Rheenen W., van Eijk K.R., Jones A.R., Keagle P., SLAGEN Consortium NEK1 variants confer susceptibility to amyotrophic lateral sclerosis. Nat. Genet. 2016;48:1037–1042. doi: 10.1038/ng.3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M., Witten D.M., Jain P., O’Roak B.J., Cooper G.M., Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S., Emond M.J., Bamshad M.J., Barnes K.C., Rieder M.J., Nickerson D.A., Christiani D.C., Wurfel M.M., Lin X., NHLBI GO Exome Sequencing Project—ESP Lung Project Team Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am. J. Hum. Genet. 2012;91:224–237. doi: 10.1016/j.ajhg.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O’Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B., Exome Aggregation Consortium Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin R.L., Schijven D., van Rheenen W., van Eijk K.R., O’Brien M., Kahn R.S., Ophoff R.A., Goris A., Bradley D.G., Al-Chalabi A., Project MinE GWAS Consortium. Schizophrenia Working Group of the Psychiatric Genomics Consortium Genetic correlation between amyotrophic lateral sclerosis and schizophrenia. Nat. Commun. 2017;8:14774. doi: 10.1038/ncomms14774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassar N., Singh K., Garcia-Diaz M. Structure of the dominant negative S17N mutant of Ras. Biochemistry. 2010;49:1970–1974. doi: 10.1021/bi9020742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obenchain V., Lawrence M., Carey V., Gogarten S., Shannon P., Morgan M. VariantAnnotation: a Bioconductor package for exploration and annotation of genetic variants. Bioinformatics. 2014;30:2076–2078. doi: 10.1093/bioinformatics/btu168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson K., Ly H.D., Dieckelmann M., Wakarchuk W.W., Withers S.G., Strynadka N.C. Crystal structure of the retaining galactosyltransferase LgtC from Neisseria meningitidis in complex with donor and acceptor sugar analogs. Nat. Struct. Biol. 2001;8:166–175. doi: 10.1038/84168. [DOI] [PubMed] [Google Scholar]
- Project MinE ALS Sequencing Consortium Project MinE: study design and pilot analyses of a large-scale whole-genome sequencing study in amyotrophic lateral sclerosis. Eur. J. Hum. Genet. 2018;26:1537–1546. doi: 10.1038/s41431-018-0177-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salgado D., Desvignes J.P., Rai G., Blanchard A., Miltgen M., Pinard A., Levy N., Collod-Beroud G., Beroud C. UMD-Predictor: A High-Throughput Sequencing Compliant System for Pathogenicity Prediction for any Human cDNA Substitution. Hum. Mutat. 2016;37:439–446. doi: 10.1002/humu.22965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasayama D., Hori H., Yamamoto N., Nakamura S., Teraishi T., Tatsumi M., Hattori K., Ota M., Higuchi T., Kunugi H. ITIH3 polymorphism may confer susceptibility to psychiatric disorders by altering the expression levels of GLT8D1. J. Psychiatr. Res. 2014;50:79–83. doi: 10.1016/j.jpsychires.2013.12.002. [DOI] [PubMed] [Google Scholar]
- Scott C.A., Marsden A.N., Slusarski D.C. Automated, high-throughput, in vivo analysis of visual function using the zebrafish. Dev. Dyn. 2016;245:605–613. doi: 10.1002/dvdy.24398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker G.K., Soya N., Palcic M.M., Klassen J.S. Temperature-dependent cooperativity in donor-acceptor substrate binding to the human blood group glycosyltransferases. Glycobiology. 2008;18:587–592. doi: 10.1093/glycob/cwn043. [DOI] [PubMed] [Google Scholar]
- Uemura S., Shishido F., Kashimura M., Inokuchi J. The regulation of ER export and Golgi retention of ST3Gal5 (GM3/GM4 synthase) and B4GalNAcT1 (GM2/GD2/GA2 synthase) by arginine/lysine-based motif adjacent to the transmembrane domain. Glycobiology. 2015;25:1410–1422. doi: 10.1093/glycob/cwv071. [DOI] [PubMed] [Google Scholar]
- Vajn K., Viljetić B., Degmečić I.V., Schnaar R.L., Heffer M. Differential distribution of major brain gangliosides in the adult mouse central nervous system. PLoS ONE. 2013;8:e75720. doi: 10.1371/journal.pone.0075720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veitia R.A. Exploring the etiology of haploinsufficiency. BioEssays. 2002;24:175–184. doi: 10.1002/bies.10023. [DOI] [PubMed] [Google Scholar]
- Yang H., Wang K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat. Protoc. 2015;10:1556–1566. doi: 10.1038/nprot.2015.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C.P., Li X., Wu Y., Shen Q., Zeng Y., Xiong Q., Wei M., Chen C., Liu J., Huo Y. Comprehensive integrative analyses identify GLT8D1 and CSNK2B as schizophrenia risk genes. Nat. Commun. 2018;9:838. doi: 10.1038/s41467-018-03247-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu R.K., Tsai Y.T., Ariga T. Functional roles of gangliosides in neurodevelopment: an overview of recent advances. Neurochem. Res. 2012;37:1230–1244. doi: 10.1007/s11064-012-0744-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.