

Abstract
Background:
Despite huge efforts, the underlying molecular mechanisms of diabetic nephropathy (DN) are yet elusive, and holistic views have rarely been generated. Considering the complexity of DN pathogenesis, the integration of datasets from different molecular types to construct a multilayer map of DN can provide a comprehensive insight toward the disease mechanisms and also can generate new knowledge. Here, we have re-analyzed two mRNA microarray datasets related to glomerular and tubulointerstitial compartments of human diabetic kidneys.
Materials and Methods:
The quality of the datasets was confirmed by unsupervised hierarchical clustering and principal component analysis. For each dataset, differentially expressed (DE) genes were identified, and transcription factors (TFs) regulating these genes and kinases phosphorylating the TFs were enriched. Furthermore, microRNAs (miRNAs) targeting the DE genes, TFs, and kinases were detected. Based on the harvested genes for glomeruli and tubulointerstitium, key signaling pathways and biological processes involved in diseases pathogenesis were recognized. In addition, the interaction of different elements in each kidney compartment was depicted in multilayer networks, and topology analysis was performed to identify key nodes. Central miRNAs whose target genes were most likely to be related to DN were selected, and their expressions were quantitatively measured in a streptozotocin-induced DN mouse model.
Results:
Among the examined miRNAs, miR-208a-3p and miR-496a-3p are, for the first time, found to be significantly overexpressed in the cortex of diabetic kidneys compared to controls.
Conclusion:
We predict that miR-208 is involved in oxygen metabolism and regulation of cellular energy balance. Furthermore, miR-496 potentially regulates protein metabolism and ion transport. However, their exact functions remain to be investigated in future studies. Taken together, starting from transcriptomics data, we have generated multilayer interaction networks and introduced novel players in DN.
Keywords: Diabetic nephropathy, gene expression profiling, gene regulatory networks, microRNAs, systems biology
INTRODUCTION
Diabetic nephropathy (DN) is the most common complication of diabetes mellitus and a leading cause of end-stage renal disease.[1,2] Although many studies have shown the role of individual genes in DN pathogenesis,[3] the molecular mechanisms of this divesting disorder are not fully understood. Systems biology provides an invaluable opportunity to process omics-scale data to obtain a holistic view of the complex interactions underlying chronic diseases. Although this approach has been employed in a few recent studies on DN,[4,5] the construction of multilayer networks has rarely been performed. Using multi-omics integration approaches, the flow of biological information can be explored and also novel interacting genes that drive DN disease can be identified.[6]
In this study, we reanalyzed two microarray datasets of kidney glomerular and tubulointerstitial compartments. The differentially expressed (DE) genes were identified and exploited to infer transcription factors (TFs), kinases, and microRNAs (miRNAs) related to this disorder. Finally, the multilayer interaction maps of these different elements were constructed and analyzed to identify the central nodes and predict the main signaling pathways and biological functions. Considering the key function of miRNAs in the pathogenesis of DN,[7,8,9] a systematic framework is followed to select important miRNAs in the integrative networks of kidney glomerule and tubulointerstitium and the expression alternation of two novel miRNAs is experimentally confirmed in an animal model of DN. This study suggests an innovative strategy for the investigation of complex disorders to identify novel players. The approach employed in this study is shown in Figure 1.
Figure 1.

The schematic representation of proposed approach. In this study, we employed a holistic integrative approach to identify novel player in diabetic nephropathy pathogenesis
MATERIALS AND METHODS
Microarray data analysis
GSE30528 and GSE30529 datasets deposited by Woroniecka et al.,[10] were obtained from the Gene Expression Omnibus (GEO) database.[11] GSE30528 includes the mRNA expression profiles of kidney glomeruli from 9 DN patients and 13 healthy individuals. GSE30529 contains the data of kidney tubulointerstitium from 10 DN and 12 controls. To evaluate microarray data quality, we performed unsupervised hierarchal clustering using ClusterMaker[12] application of Cytoscape 3.5.1[13] and principal component analysis (PCA) using ggplot2 package[14] of R software. GEO2R tool of GEO database was used for the identification of DE genes, and P value was corrected using Benjamini–Hochberg method. Volcano plots were prepared using the ggplot2 R package.
Enrichment analyses
To identify the regulators of the DE genes, TF enrichment analysis was performed using ChEA tool of Enrichr database.[15] Furthermore, kinase enrichment analysis was performed using KEA application of Enrichr to harvest kinases that regulate the identified TFs. In addition, gene ontology (GO) terms related to all gene sets were identified by GO term enrichment analysis using ClueGO v3.2.2 plugin of Cytoscape[16] and the parents of enriched GO terms were detected using REVIGO tool.[17] In addition, pathway enrichment analysis was performed by ClueGO, which retrieved data from KEGG and REACTOME databases with medium network specificity. In addition, the ggplot2 R package was used to visualize the enriched pathways as scatter plots. Rich factor was calculated as the number of genes enriched in a certain pathway relative to the total number of genes in the pathway. miRNA enrichment analysis was done using TargetScan 2017 application of Enrichr. For all these analyses, adjusted P ≤ 0.05 was considered as statistical significance threshold.
Molecular interaction network
Cytoscape CluePedia v1.3.3[18] application was used to construct protein–protein interaction networks encompassing DE genes, TFs, and kinases. For network construction, the interaction confidence cutoff was set at 0.6, and only edges with experimental validation evidence were retrieved from STRING v10.0.[19] Next, miRNA-target interactions were merged into the networks. Network topology analysis was performed using Network Analyser tool of Cytoscape.
MicroRNA-mRNA interaction
The predicted and validated targets of selected miRNAs were harvested using TargetScan[20] and miRTarBase,[21] respectively. The miRNA seed conservation was evaluated using TargetScan.
RNA extraction
The DN animal model was established using multiple low dose of streptozotocin injection, and the model was validated using biochemical and histopathological assessments in our previous study (unpublished). The kidney tissues of DN and normal mice were lysed with RNX (CinnaGen, Tehran, Iran) using the micro smash machine (TOMY Digital Biology, Tokyo, Japan). The homogenized tissues were transferred to new tubes, and 250 μl of chloroform (Merck, Darmstadt, Germany) was added and incubated at room temperature for 15 min. Then, samples were centrifuged at 4°C, 12,000 rpm for 20 min. The supernatant was transferred to new tubes, and 100% cold ethanol (Merck, Darmstadt, Germany) was added and gently mixed. The samples were stored overnight at −20°C. Next, samples were centrifuged at 4°C, 14,000 rpm for 45 min, and 1 ml 70% cold ethanol was added to the platelets. Samples were centrifuged at 4°C, 12,000 rpm for 10 min. Then, the supernatant was discarded, and 50 μl of distilled water was added to the platelets.
cDNA synthesis
For cDNA synthesis, 10 μl RNA and 1 μl of specific RT primer for each miRNA were mixed, and double-distilled water was added up to a total volume of 13.4 μl. The tubes were placed at 70°C for 5 min and then mixed with 4 μl first strand buffer, 1 μl dNTP, 0.5 μl RNase, and 1 μl reverse transcriptase M-MLV enzyme (YektaTajhiz, Tehran, Iran). The samples were incubated in polymerase chain reaction (PCR) machine (Eppendorf, Hamburg, Germany) at 37°C for 60 min, followed by 70°C for 5 min.
Quantitative polymerase chain reaction
Specific primers were designed [Supplementary Table 1] using AlleleID[22] and GeneRunner,[23] and their specificity was assessed using the NCBI-BLAST database sno202 and sno234 were chosen as references. For real-time PCR, 1 μl cDNA, 5 μl high ROX™ SYBR Green master mix (Ampliqon, Herlev, Denmark), 0.5 μl forward and 0.5 μl reverse primers, and 3 μl double distilled water were mixed and then the reaction was carried out using Applied Biosystems Real-Time machine (Carlsbad, USA). The temperature profile consisted of an initial step at 95°C for 15 min and then 40 cycles at 95°C for 15 s and 60°C for 1 min. REST software[24] was used to analyze the results.
Supplementary Table 1.
Specific primers were designed to evaluate the expression of candidate miRNAs
| miR name | Sequence |
|---|---|
| miR-921 | RT: GTCGTATGCACAGCAGGGTCCGAGGTATTCGCAGTGCATACGACGAATCC |
| F: CTAGTGAGGGACAGAACCA | |
| R: CAGCAGGGTCCGAGGT | |
| miR-505 | RT: GTCGTATG CACAGCAGGGTCCGAGGTATTCGCAGTGCATACGACAACATC |
| F: AGGGAGCCAGGAAGTATT | |
| R: CAGCAGGGTCCGAGGT | |
| miR-590-5p | RT: GTCGTATGCACAGCAGGGTCCGAGGTATTCGCAGTGCATACGACCTGCAC |
| F: GGTCCGAGCTTATTCATAAAA | |
| R: CAGCAGGGTCCGAGGT | |
| miR-383-5p | RT: GTCGTATGCACAGCAGGGTCCGAGGTATTCGCAGTGCATACGACAGCCAC |
| F: GGCGAGATCAGAAGGTGACT | |
| R: CAGCAGGGTCCGAGGT | |
| miR-208a-3p | RT: GTCGTATGCACAGCAGGGTCCGAGGTATTCGCAGTGCATACGACACAAGC |
| F: GCCGATAAGACGAGCAAAAA | |
| R: CAGCAGGGTCCGAGGT | |
| miR-496a-3p | RT: GTCGTATGCACAGCAGGGTCCGAGGTATTCGCAGTGCATACGACGAGATT |
| F: GCGTGAGTATTACATGGCC | |
| R: CAGCAGGGTCCGAGGT |
RESULTS
In order to explore DN transcriptomics profile, two microarray datasets related to human kidney glomerular (GSE30528) and tubulointerstitial (GSE30529) compartments were retrieved, and their quality was assessed using PCA and hierarchical clustering. Most samples were separated according to the study groups in an unsupervised manner, indicating the acceptable quality of both datasets. However, few samples not following the expected segregation were excluded to enhance the reliability of downstream analyses [Figure 2a and b]. Genes with adjusted P ≤ 0.05 and absolute logarithmic fold change (∣log2 FC∣) ≥1 were assumed as DE [Figure 2c and d].
Figure 2.
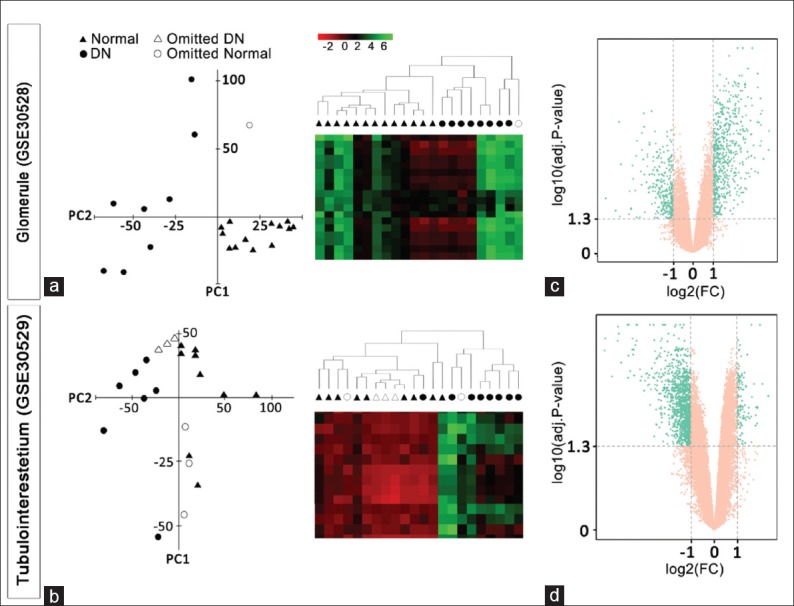
Datasets quality assessments. Principle component analysis and hierarchical clustering with all genes revealed an acceptable quality of both microarray datasets (a and b). The genes with adj. p-value ≤ 0.05 and │log FC│≥ 1 are considered as differentially expressed (DE) and depicted as green dots in the volcano graphs (c and d)
In the glomerule dataset, we identified 709 DE genes. TF enrichment analysis revealed that 61 TFs potentially regulating these genes. In turn, these TFs are proposed to be controlled by 91 kinases according to kinase enrichment analysis. Furthermore, 174 miRNAs regulating the DE genes, TFs, and kinases were identified by miRNA enrichment analysis [Supplementary Table 2 (529.3KB, pdf) ]. Similarly, 1372 DE genes, 91 TFs, 92 kinases, and 181 miRNAs were determined for the tubule dataset [Supplementary Table 3 (528.4KB, pdf) ]. In order to avoid the bias caused by the focus of previous studies on certain sets of miRNAs, the TargetScan algorithm was used for miRNA enrichment, which is based on bioinformatics predictions rather than experimental data for miRNA-mRNA interactions.
In order to explore the key underlying molecular and cellular phenomena in DN, the signaling pathways associated to DE genes, TFs, and kinases were obtained by pathway enrichment analysis [Supplementary Tables 4 (461.1KB, pdf) and 5 (461.5KB, pdf) ]. A considerable fraction of the pathways with the highest rich factors is related to immune responses and tissue fibrosis [Figure 3]. In addition, some well-known pathways in DN, including hypoxia, FoxO, VEGF, and AGE-RAGE signaling pathways, as well as platelet aggregation[25,26,27,28] are identified. In addition, ErbB signaling pathway is enriched in the glomerule dataset, which is in line with the identification of EGF as a promising urinary DN biomarker.[29] As expected, the complement cascade,[30] which is underscored by the initial developers of the current datasets, is among the top pathways in the glomerule dataset. Interestingly, the Hedgehog signaling pathway whose role in DN is just recently shown is also enriched.[31] In addition to the previously recognized pathways, some novel interesting ones such as circadian clock and neurotrophin pathway are also enriched. Although the importance of the circadian rhythm in normal kidney function and nephrectomy-induced fibrosis is just studied,[32] its involvement in DN remains an interesting topic for future studies. Similarly, neurotrophin signaling is studied in diabetes, and its some other complications,[33] however, to the best of our knowledge, it is not yet investigated in DN. Moreover, the GO terms were identified and summarized as parent terms [Figure 4 and Supplementary Figures 1 (798KB, tif) and 2 (522.9KB, tif) ]. Interestingly, the majority of biological process terms for the glomerule and tubule genes are related to blood vessels and immune responses, respectively. This is in accordance with the histopathological features of DN, including glomerular capillary injuries and tubulointerstitial inflammation.
Figure 3.

Signaling pathways related to the glomerule and tubulointerstitium networks. Pathway enrichment analysis was performed with the differentially expressed genes, transcription factors, and kinases in each network. The horizontal axis is rich-factor, and pathways with adjusted P ≤ 0.05 are shown. The pathways with one star are known to be involved in the pathogenesis of diabetic nephropathy. Specifically, the pathways associated with immune response and inflammation are marked with double starts. The underlined pathways have not been previously described to be involved in diabetic nephropathy
Figure 4.

The ontology of the nodes in the glomerule and tubulointerstitium networks. Gene ontology enrichment analysis was performed with the differentially expressed genes, transcription factors and kinases in each network. Gene ontology biological process parents are illustrated. The horizontal axis is the numbers of children for each parent term. adjusted P ≤ 0.05 is considered as the threshold of statistical significance
The harvested DE genes, TFs, kinases, and miRNAs were employed to construct four-layer molecular interaction networks for the glomerule and tubulointerstitial sets. The connected networks were analyzed, and topology parameters were determined [Supplementary Tables 6 (554.5KB, pdf) and 7 (891KB, pdf) ]. It is supposed that the central genes in protein–protein interactions are critical for disease pathogenesis.[34,35] Hence, we identified the nodes with the highest degree and betweenness centralities in DE gene, TF, kinase, and miRNA layers of the glomerule and tubule networks [Figure 5].
Figure 5.

Central nodes in the glomerule and tubulointerstitium multilayer networks. The topology of the networks is analyzed, and top 5% differentially expressed genes as well as top 10% transcription factors, kinases, and microRNAs that are most central based on degree and betweenness are shown
Considering the critical role of miRNAs in DN pathogenesis,[7] we focused on the most central 16 and 17 miRNAs identified in the networks of glomerule and tubulointerstitium, respectively. Remarkably, among them is miR-21, a well-known player in DN.[36] To concentrate on miRNAs that are most likely involved in DN, the validated and predicted targets of each miRNA were determined and compared with the list of genes known to be involved in DN manually retrieved from literature [Supplementary Table 8]. The targets of miR-505-3p, miR-590-3p, miR-496a-3p, miR-208a-3p, miR-921, and miR-383-5p had the most overlap with DN-related genes. We hypothesized that these miRNAs are involved in DN albeit not described by previous investigators. Therefore, we planned to experimentally assess their expressions in diabetic kidneys. A mouse model of DN previously established in our laboratory was exploited, and after RNA extraction and miRNA-specific cDNA synthesis, the expression of the candidate miRNAs was quantified. Notably, for miR-921 no ortholog is reported in mouse, and quantitative PCR with human primers was not successful. Five miRNAs which have murine ortholog with conserved seeds were used for specific primer design. In the examined kidney tissues, miR-590-3p was undetectable. The expressions of miR-505-3p, miR-496a-3p, miR-208a-3p, and miR-383-5p were measured in the cortex and medulla compartments [Figure 6a]. Remarkably, miR-208a-3p and miR-496a-3p were almost three-fold overexpressed in the cortex of diabetic kidneys (P ≤ 0.05).
Supplementry Table 8.
DN-associated genes are manually retrived from literature. For each miRNA, validated and predicted tragets known to be DN-associated are listed.
| Glomerule Compartment | Tubulointerstitium Compartment | |||||
|---|---|---|---|---|---|---|
| DN assosiated genes | microRNA | Common genes between validated targets and DN gene list | Common genes between predicted targets and DN gene list | microRNA | Common genes between validated targets and DN gene list | Common genes between predicted targets and DN gene list |
| Vegf | hsa-miR-590-5p | TGFBR2,SMAD3,SMAD7,FOXN2,FOXO3,PDCD4,TGFB1 | PPP3CA,SERP1 | hsa-miR-208a | LEP,CYP1B1,FOXP1,MAK16,MAP3K5,MAPK10, | TGFBR1,COL4A3,FNIP1,FOXG1,FOXP2,MAP3K2,MMP16,PPP3CB,PRKAR1A,ZEB2 |
| Fox | hsa-miR-921 | ANGPTL1,FOXN3,TNFAIP8L1 | MAP2K6,MAPK1 | hsa-miR-921 | ANGPTL1,FOXN3,PRKG1,TNFAIP8L1 | MAPK1 |
| Hif | hsa-miR-505 | PRKCA,ACER2,COL4A1,FOXE1 | MAPK1IP1L,PTEN,TNFSF11 | hsa-miR-496 | AKT1,COL19A1,FOXA1,FOXN2,LEPROTL1,MAPK8,PPP6C | TGFBR2,FOXN2,PPP6C,TNFRSF10D |
| Cyp | hsa-miR-383 | ADIPOQ,AGTRAP,ANGEL2,ANGPT4,COL8A1,CYP20A1,CYP51A1 | PRKAG1,VEGFA | hsa-miR-590-5p | TGFBR2,SMAD3,SMAD7,FOXN2,FOXO3,TGFB1 | PPP3CA,SERP1 |
| Ace2 | hsa-miR-3152-3p | PPP2CA,MAPK10,PPP1R16B | SMAD2,TGFBR1,ADI1,MMP16,TGFBR1,TNFSF14 | hsa-miR-3146 | IGF2R,PPP1R15B | COL4A4,SMAD9 |
| Adipoq | hsa-miR-4259 | COL18A1 | CYP20A1,HIP1,TNFRSF14 | hsa-miR-331-5p | SOD2,PPP1R1A | MAP2K6,MAP3K1,PDGFD,PRKAB2,SMAD2 |
| Agt | hsa-miR-4327 | HIF1AN,LEPROT,MAPK1IP1L | PTEN | hsa-miR-4484 | MAPKAPK5 | SOD3,FOXE1,SMAD4,SOD3, |
| Akr1b3 | hsa-miR-4445 | SOD2 | PPP2CA | hsa-miR-4637 | SOD2,FOXN3,PPP1R3G | TMEM236,PPP1R2 |
| Akt1 | hsa-miR-1284 | FGF2 | AKTIP | hsa-miR-4684-3p | PPP3R1 | PPP1R15B,PRKAA2,ZEB2 |
| Bdkrb1 | hsa-miR-4718 | IGF2BP1,PRKCB | ACER3,MAP2K6 | hsa-miR-4704-5p | MAP10,TNFSF15 | MAP2K4,SMAD9 |
| Col1a1 | hsa-miR-4423-3p | IGF1R,PPP1R2 | AGTRAP,PPP4R1L | hsa-miR-550b | PPP2CA,FOXA1,IGFBP5,MAPK1 | PPP2CA |
| Col2a1 | hsa-miR-501-3p | SOD2,COL23A1,CYP4F11 | COL10A1,PPP2R2C,PPP2R5E,PPP4R2 | hsa-miR-584 | LEP,CYP1B1,FOXP1,MAP3K5,MAPK10 | COLQ,FNDC3A,PPP6C |
| Col3a1 | hsa-miR-502-3p | SOD2 | hsa-miR-208b | SOD2,COL23A1,HIF1AN | COLEC10,SMAD4 | |
| Col4a1 | hsa-miR-508-3p | FLOT2,PPP1R15B | hsa-miR-4474-5p | MAP9 | PPP1R12B | |
| Ctgf | hsa-miR-3682-3p | COL4A4,MAP2K6,PPP1R12B | hsa-miR-770-5p | COL19A1,TNFAIP1, | COL4A4,MAP2K6,PPP1R12B | |
| Fn1 | hsa-miR-4694-5p | FOXN3,PRKCA,PTEN | hsa-miR-3689a-5p | SOD2 | ||
| Icam1 | hsa-miR-4445 | |||||
| Jun | ||||||
| Lep | ||||||
| Lepr | ||||||
| Mapk14 | ||||||
| Mmp9 | ||||||
| Nos3 | ||||||
| Pdgfb | ||||||
| Pdgfc | ||||||
| Pdgfd | ||||||
| Ppara | ||||||
| Ppp2ca | ||||||
| Prkca | ||||||
| Pten | ||||||
| Serpine1 | ||||||
| Smad2 | ||||||
| Smad3 | ||||||
| Smad7 | ||||||
| Sod2 | ||||||
| Sod3 | ||||||
| Spp1 | ||||||
| Srebf1 | ||||||
| Tgfb2 | ||||||
| Tgfbr1 | ||||||
| Tgfbr2 | ||||||
| Tgfbr3 | ||||||
| Tnf | ||||||
| Zeb1 | ||||||
| Yap | ||||||
| Foxn | ||||||
| Pdcd4 | ||||||
| Mmp9 | ||||||
| Ogg | ||||||
| Ros | ||||||
Figure 6.

Expression assessments and functional analysis. The expressions of selected microRNAs with the greatest centrality values were assessed by quantitative polymerase chain reaction. Asterisks indicate P ≤ 0.05 (a). The Gene ontology biological process terms enriched with the validated targets of miR-208a-3p and miR-496b-3p are demonstrated. The terms that are most related to diabetic nephropathy are underlined (b)
The role of miR-208a-3p and miR-496a-3p in DN has not been investigated so far. In order to predict the biological processes in which these two novel miRNAs are involved, the ontology of their validated targets was determined [Figure 6b]. Among the GO terms enriched for miR-208a-3p are well-known DN-associated cellular functions including oxidative stress,[37] response to oxygen levels,[37] carbohydrate and protein metabolism,[38] response to nutrient levels,[39] regulation of transforming growth factor-beta signaling pathway,[40] apoptosis,[37] and tissue remodeling.[41] Furthermore, miR-496a-3p is mainly related to transcriptional, translational, and posttranslational gene expression control and anion transmembrane transport.
DISCUSSION
Chronic noncommunicable diseases are the main challenge of current medicine. Systems biology with its holistic view may assist to reveal the complex pathogenesis of these disorders. In order to provide an inclusive map of DN molecular pathogenesis, we have here re-analyzed two microarray datasets initially generated by Woroniecka et al.[10] These datasets have prominent advantages such as being derived from human subjects and separate profiling of glomerular and tubulointerstitial sections. Indeed, the diverse expression profiles of different anatomical kidney compartments are shown in previous studies.[42] Moreover, we have shown that these datasets fulfill the quality control criteria.
Biomedical phenomena are the result of complex interactions between thousands of molecules from different entities. Although data integration approach is underscored as a critical step to generate inclusive maps of complex biomedical phenomena,[43] it is commonly ignored in omics data analysis. In this study, the primary data were at the transcriptome level, and other potential interacting levels such as TFs, kinases, and miRNAs were predicted to generate multilayer networks. These integrated elements were found to be potentially involved in well-known DN-associated pathogenic processes such as Wnt, VEGF, FoxO, hypoxia, P53, AGE-RAGE signaling pathways, angiogenesis, and immunological reaction.[44,45,46,47,48,49,50,51] Pathway enrichment analysis also suggests that circadian clock is important in this disorder. This idea is not yet comprehensively investigated and can be an interesting subject for future studies.
Considering the pivotal role of miRNAs in the regulation of DN pathogenesis, we focused on the miRNA layer of the networks. miRNAs with the greatest network centrality whose target genes were most likely to be involved in the disease were selected for the experimental study. Gene expression quantification in tissue samples of an experimental animal model revealed that miR-208a-3p and miR-496a-3p are significantly overexpressed in the kidney cortex in DN. To the best of our knowledge, this is the first time that the involvement of these two miRNAs in DN is reported.
GO term enrichment analysis revealed that miR-208a-3p could be involved in response to oxygen levels and metabolic pathways, indicating the plausible key function of this miRNA in cellular energetics. In agreement with this assumption, miR-208 is extensively shown to be involved in myocardial ischemia.[44,45,46,47,48] In addition, circulating miR-208a is a candidate biomarker of coronary artery diseases.[44,45] Considering the fact that chronic kidney disease significantly increases the risk of cardiovascular disorders,[49] it is interesting to investigate if miR-208a secreted from injured kidneys can partly mediate this association. Remarkably, it is shown that miR-208 is associated with angiotensin-mediated blood pressure control in the heart.[46] Based on GO results, miR-496 is related to protein metabolism. In addition, this miRNA is reported to be involved in cell proliferation,[50] aging,[51] apoptosis,[52] and response to vasopressin in kidney collecting ducts.[53] However, it is a new identified miRNA, and further studies are definitely required to disclose its functions.
Taken together, based on a system approach, we have explored the underlying molecular mechanisms of DN and proposed two novel miRNAs. The top-down framework exploited in this study is of potential value for the investigation of other complex disorders.
Financial support and sponsorship
This work was supported by Isfahan University of Medical Sciences (grant numbers: 396377 and 195173) and Iran National Science Foundation (grant number: 96006608).
Conflicts of interest
There are no conflicts of interest.
SUPPLEMENTARY TABLES
Supplementary Table 1: Specific primers were designed to evaluate the expression of candidate microRNAs
Supplementary Table 2 (529.3KB, pdf) : Gene set enrichment analysis of the glomerule dataset (GSE30528). The genes with the adjusted P = 0.05 and │logFC│ =1 are considered as DE. TFs regulating the DE genes and kinases targeting TFs are harvested by enrichment analysis. In addition, microRNAs targeting either DE genes, TFs, or kinases are enriched. For enrichment analyses, adjusted P = 0.05 was considered as the statistical significance threshold. DE = Differentially expressed; TFs = Transcription factors
Gene set enrichment analysis of the glomerule dataset (GSE30528). The genes with the adj. P-value≤0.05 and │logFC│≥1 are considered as differentially expressed (DE). Transcription factors (TFs) regulating the DE genes and kinases trageting TFs are harvested by enrichment analysis. Also, miRNAs targeting either DE genes, TFs, or kinases are enriched. For enrichment analyses, adj. P-value≤0.05 was considered as statistical significance threshhold.
Supplementary Table 3 (528.4KB, pdf) : Gene set enrichment analysis of the tubulointerstitium dataset (GSE30529). The genes with the adjusted P= 0.05 and │logFC│ =1 are considered as DE. TFs regulating the DE genes and kinases targeting TFs are harvested by enrichment analysis. In addition, microRNAs targeting either DE genes, TFs, or kinases are enriched. For enrichment analyses, adjusted P = 0.05 was considered as the statistical significance threshold. DE = Differentially expressed; TFs = Transcription factors
Gene set enrichment analysis of the tubulointerstitium dataset (GSE30529). The genes with the adj. P-value≤0.05 and │logFC│≥1 are considered as differentially expressed (DE). Transcription factors (TFs) regulating the DE genes and kinases trageting TFs are harvested by enrichment analysis. Also, miRNAs targeting either DE genes, TFs, or kinases are enriched. For enrichment analyses, adj. P-value≤0.05 was considered as statistical significance threshhold.
Supplementary Table 4 (461.1KB, pdf) : Pathway enrichment analysis of the glomerule dataset. Signaling pathways related to the DE gene, TFs, and kinase of the glomerule network were determined. Adjusted P= 0.05 was considered as statistical significance threshold. DE = Differentially expressed; TFs = Transcription factors
Pathway enrichment analysis of the glomerule dataset. Signaling pathways related to the DE gene, TFs, and kinase of the glomerule network were detremined. Adj. P-value≤0.05 was considered as statistical significance threshhold.
Supplementary Table 5 (461.5KB, pdf) : Pathway enrichment analysis of the tubulointerstitium dataset. Signaling pathway related to the DE gene, TFs, and kinase of the tubulointerstitium network were determined. Adjusted P= 0.05 was considered as statistical significance threshold. DE = Differentially expressed; TFs = Transcription factors
Pathway enrichment analysis of the tubulointerstitium dataset. Signaling pathway related to the DE gene, TFs, and kinase of the tubulointerstitium network were detremined. Adj. P-value≤0.05 was considered as statistical significance threshhold.
Supplementary Table 6 (554.5KB, pdf) : Topology analysis of the glomerule multilayer network
Topology analysis of the glomerule multi-layer network.
Supplementary Table 7 (891KB, pdf) : Topology analysis of the multi-layer tubulointerstitium network
Topology analysis of the multi-layer Tubulointerstitium network.
Supplementary Table 8: Diabetic nephropathy -associated genes are manually retrieved from literature. For each microRNAs, validated and predicted targets known to be diabetic nephropathy-associated are listed.
The ontology of the nodes in the glomerule and tubulointerstitium networks. Gene ontology enrichment analysis was performed with the differentially expressed genes, transcription factors and kinases in each network. Gene ontology molecular function parents are illustrated. Horizontal axis is the numbers of children for each parent term. adjusted P ≤ 0.05 is considered as threshold of statistical significance
The ontology of the nodes in the glomerule and tubulointerstitium networks. Gene ontology enrichment analysis was performed with the differentially expressed genes, transcription factors and kinases in each network. Gene ontology cellular component parents are illustrated. Horizontal axis is the numbers of children for each parent term. adjusted P ≤ 0.05 is considered as threshold of statistical significance
REFERENCES
- 1.Ghaderian SB, Hayati F, Shayanpour S, Beladi Mousavi SS. Diabetes and end-stage renal disease; a review article on new concepts. J Renal Inj Prev. 2015;4:28–33. doi: 10.12861/jrip.2015.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lim AK. Diabetic nephropathy – Complications and treatment. Int J Nephrol Renovasc Dis. 2014;7:361–81. doi: 10.2147/IJNRD.S40172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wei L, Xiao Y, Li L, Xiong X, Han Y, Zhu X, et al. The susceptibility genes in diabetic nephropathy. Kidney Dis (Basel) 2018;4:226–37. doi: 10.1159/000492633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang WN, Zhang WL, Zhou GY, Ma FZ, Sun T, Su SS, et al. Prediction of the molecular mechanisms and potential therapeutic targets for diabetic nephropathy by bioinformatics methods. Int J Mol Med. 2016;37:1181–8. doi: 10.3892/ijmm.2016.2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van JA, Scholey JW, Konvalinka A. Insights into diabetic kidney disease using urinary proteomics and bioinformatics. J Am Soc Nephrol. 2017;28:1050–61. doi: 10.1681/ASN.2016091018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Civelek M, Lusis AJ. Systems genetics approaches to understand complex traits. Nat Rev Genet. 2014;15:34–48. doi: 10.1038/nrg3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kato M, Natarajan R. MicroRNAs in diabetic nephropathy: Functions, biomarkers, and therapeutic targets. Ann N Y Acad Sci. 2015;1353:72–88. doi: 10.1111/nyas.12758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cardenas-Gonzalez M, Srivastava A, Pavkovic M, Bijol V, Rennke HG, Stillman IE, et al. Identification, confirmation, and replication of novel urinary microRNA biomarkers in lupus nephritis and diabetic nephropathy. Clin Chem. 2017;63:1515–26. doi: 10.1373/clinchem.2017.274175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kantharidis P, Hagiwara S, Brennan E, McClelland AD. Study of microRNA in diabetic nephropathy: Isolation, quantification and biological function. Nephrology (Carlton) 2015;20:132–9. doi: 10.1111/nep.12374. [DOI] [PubMed] [Google Scholar]
- 10.Woroniecka KI, Park AS, Mohtat D, Thomas DB, Pullman JM, Susztak K. Transcriptome analysis of human diabetic kidney disease. Diabetes. 2011;60:2354–69. doi: 10.2337/db10-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, et al. NCBI GEO: Archive for functional genomics data sets – Update. Nucleic Acids Res. 2013;41:D991–5. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morris JH, Apeltsin L, Newman AM, Baumbach J, Wittkop T, Su G, et al. ClusterMaker: A multi-algorithm clustering plugin for cytoscape. BMC Bioinformatics. 2011;12:436. doi: 10.1186/1471-2105-12-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wickham H. Ggplot2: Elegant Graphics for Data Analysis. 2nd ed. Springer International Publishing; 2016. [Last accessed on 2018 Jul 22]. Available from: http://www.springer.com/gp/book/9783319242750 . [Google Scholar]
- 15.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–7. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, et al. ClueGO: A cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25:1091–3. doi: 10.1093/bioinformatics/btp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Supek F, Bošnjak M, Škunca N, Šmuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One. 2011;6:e21800. doi: 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bindea G, Galon J, Mlecnik B. CluePedia cytoscape plugin: Pathway insights using integrated experimental and in silico data. Bioinformatics. 2013;29:661–3. doi: 10.1093/bioinformatics/btt019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45:D362–8. doi: 10.1093/nar/gkw937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Agarwal V, Bell GW, Nam J, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. ELife. 2015;4:e05005. doi: 10.7554/eLife.05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chou CH, Chang NW, Shrestha S, Hsu SD, Lin YL, Lee WH, et al. MiRTarBase 2016: Updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res. 2016;44:D239–47. doi: 10.1093/nar/gkv1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Apte A, Singh S. AlleleID: A pathogen detection and identification system. Methods Mol Biol. 2007;402:329–46. doi: 10.1007/978-1-59745-528-2_17. [DOI] [PubMed] [Google Scholar]
- 23.Kumar A, Chordia N. In silico PCR primer designing and validation. Methods Mol Biol. 2015;1275:143–51. doi: 10.1007/978-1-4939-2365-6_10. [DOI] [PubMed] [Google Scholar]
- 24.Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002;30:e36. doi: 10.1093/nar/30.9.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kato M, Yuan H, Xu ZG, Lanting L, Li SL, Wang M, et al. Role of the Akt/FoxO3a pathway in TGF-beta1-mediated mesangial cell dysfunction: A novel mechanism related to diabetic kidney disease. J Am Soc Nephrol. 2006;17:3325–35. doi: 10.1681/ASN.2006070754. [DOI] [PubMed] [Google Scholar]
- 26.Tufro A, Veron D. VEGF and podocytes in diabetic nephropathy. Semin Nephrol. 2012;32:385–93. doi: 10.1016/j.semnephrol.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanajou D, Ghorbani Haghjo A, Argani H, Aslani S. AGE-RAGE axis blockade in diabetic nephropathy: Current status and future directions. Eur J Pharmacol. 2018;833:158–64. doi: 10.1016/j.ejphar.2018.06.001. [DOI] [PubMed] [Google Scholar]
- 28.Buch A, Kaur S, Nair R, Jain A. Platelet volume indices as predictive biomarkers for diabetic complications in type 2 diabetic patients. J Lab Physicians. 2017;9:84–8. doi: 10.4103/0974-2727.199625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Betz BB, Jenks SJ, Cronshaw AD, Lamont DJ, Cairns C, Manning JR, et al. Urinary peptidomics in a rodent model of diabetic nephropathy highlights epidermal growth factor as a biomarker for renal deterioration in patients with type 2 diabetes. Kidney Int. 2016;89:1125–35. doi: 10.1016/j.kint.2016.01.015. [DOI] [PubMed] [Google Scholar]
- 30.Kościelska-Kasprzak K, Bartoszek D, Myszka M, Zabińska M, Klinger M. The complement cascade and renal disease. Arch Immunol Ther Exp (Warsz) 2014;62:47–57. doi: 10.1007/s00005-013-0254-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao XP, Chang SY, Liao MC, Lo CS, Chenier I, Luo H, et al. Hedgehog interacting protein promotes fibrosis and apoptosis in glomerular endothelial cells in murine diabetes. Sci Rep. 2018;8:5958. doi: 10.1038/s41598-018-24220-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bonny O, Vinciguerra M, Gumz ML, Mazzoccoli G. Molecular bases of circadian rhythmicity in renal physiology and pathology. Nephrol Dial Transplant. 2013;28:2421–31. doi: 10.1093/ndt/gft319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Verge VM, Andreassen CS, Arnason TG, Andersen H. Mechanisms of disease: Role of neurotrophins in diabetes and diabetic neuropathy. Handb Clin Neurol. 2014;126:443–60. doi: 10.1016/B978-0-444-53480-4.00032-1. [DOI] [PubMed] [Google Scholar]
- 34.Abedi M, Gheisari Y. Nodes with high centrality in protein interaction networks are responsible for driving signaling pathways in diabetic nephropathy. PeerJ. 2015;3:e1284. doi: 10.7717/peerj.1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rabieian R, Abedi M, Gheisari Y. Central nodes in protein interaction networks drive critical functions in transforming growth factor beta-1 stimulated kidney cells. Cell J. 2017;18:514–31. doi: 10.22074/cellj.2016.4718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McClelland AD, Herman-Edelstein M, Komers R, Jha JC, Winbanks CE, Hagiwara S, et al. MiR-21 promotes renal fibrosis in diabetic nephropathy by targeting PTEN and SMAD7. Clin Sci (Lond) 2015;129:1237–49. doi: 10.1042/CS20150427. [DOI] [PubMed] [Google Scholar]
- 37.Sifuentes-Franco S, Padilla-Tejeda DE, Carrillo-Ibarra S, Miranda-Díaz AG. Oxidative stress, apoptosis, and mitochondrial function in diabetic nephropathy. Int J Endocrinol. 2018;2018:1875870. doi: 10.1155/2018/1875870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kikkawa R, Koya D, Haneda M. Progression of diabetic nephropathy. Am J Kidney Dis. 2003;41:S19–21. doi: 10.1053/ajkd.2003.50077. [DOI] [PubMed] [Google Scholar]
- 39.Sas KM, Kayampilly P, Byun J, Nair V, Hinder LM, Hur J, et al. Tissue-specific metabolic reprogramming drives nutrient flux in diabetic complications. JCI Insight. 2016;1:e86976. doi: 10.1172/jci.insight.86976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McKnight AJ, Savage DA, Patterson CC, Sadlier D, Maxwell AP. Resequencing of genes for transforming growth factor beta1 (TGFB1) type 1 and 2 receptors (TGFBR1, TGFBR2), and association analysis of variants with diabetic nephropathy. BMC Med Genet. 2007;8:5. doi: 10.1186/1471-2350-8-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kundu S, Pushpakumar S, Sen U. MMP-9- and NMDA receptor-mediated mechanism of diabetic renovascular remodeling and kidney dysfunction: Hydrogen sulfide is a key modulator. Nitric Oxide. 2015;46:172–85. doi: 10.1016/j.niox.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Higgins JP, Wang L, Kambham N, Montgomery K, Mason V, Vogelmann SU, et al. Gene expression in the normal adult human kidney assessed by complementary DNA microarray. Mol Biol Cell. 2004;15:649–56. doi: 10.1091/mbc.E03-06-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gligorijević V, Pržulj N. Methods for biological data integration: Perspectives and challenges. J R Soc Interface. 2015:12. doi: 10.1098/rsif.2015.0571. pii: 20150571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nabiałek E, Wańha W, Kula D, Jadczyk T, Krajewska M, Kowalówka A, et al. Circulating microRNAs (miR-423-5p, miR-208a and miR-1) in acute myocardial infarction and stable coronary heart disease. Minerva Cardioangiol. 2013;61:627–37. [PubMed] [Google Scholar]
- 45.Linhares-Lacerda L, Granato A, Gomes-Neto JF, Conde L, Freire-de-Lima L, de Freitas EO, et al. Circulating plasma microRNA-208a as potential biomarker of chronic indeterminate phase of chagas disease. Front Microbiol. 2018;9:269. doi: 10.3389/fmicb.2018.00269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang Y, Yang Y, He Y, Huang C, Meng X, Li J. MicroRNA-208a potentiates angiotensin II-triggered cardiac myoblasts apoptosis via inhibiting nemo-like kinase (NLK) Curr Pharm Des. 2016;22:4868–75. doi: 10.2174/1381612822666160210143047. [DOI] [PubMed] [Google Scholar]
- 47.Diniz GP, Takano AP, Barreto-Chaves ML. MiRNA-208a and miRNA-208b are triggered in thyroid hormone-induced cardiac hypertrophy-role of type 1 angiotensin II receptor (AT1R) on miRNA-208a/α-MHC modulation. Mol Cell Endocrinol. 2013;374:117–24. doi: 10.1016/j.mce.2013.04.010. [DOI] [PubMed] [Google Scholar]
- 48.Białek S, Górko D, Zajkowska A, Kołtowski Ł, Grabowski M, Stachurska A, et al. Release kinetics of circulating miRNA-208a in the early phase of myocardial infarction. Kardiol Pol. 2015;73:613–9. [PubMed] [Google Scholar]
- 49.Whitman IR, Feldman HI, Deo R. CKD and sudden cardiac death: Epidemiology, mechanisms, and therapeutic approaches. J Am Soc Nephrol. 2012;23:1929–39. doi: 10.1681/ASN.2012010037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu DM, Wen X, Han XR, Wang S, Wang YJ, Shen M, et al. Role of circular RNA DLEU2 in human acute myeloid leukemia. Mol Cell Biol. 2018:38. doi: 10.1128/MCB.00259-18. pii: e00259-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rubie C, Kölsch K, Halajda B, Eichler H, Wagenpfeil S, Roemer K, et al. MicroRNA-496 – A new, potentially aging-relevant regulator of mTOR. Cell Cycle. 2016;15:1108–16. doi: 10.1080/15384101.2016.1158360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yao X, Yao R, Yi J, Huang F. Upregulation of miR-496 decreases cerebral ischemia/reperfusion injury by negatively regulating BCL2L14. Neurosci Lett. 2019;696:197–205. doi: 10.1016/j.neulet.2018.12.039. [DOI] [PubMed] [Google Scholar]
- 53.Kim JE, Jung HJ, Lee YJ, Kwon TH. Vasopressin-regulated miRNAs and AQP2-targeting miRNAs in kidney collecting duct cells. Am J Physiol Renal Physiol. 2015;308:F749–64. doi: 10.1152/ajprenal.00334.2014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Gene set enrichment analysis of the glomerule dataset (GSE30528). The genes with the adj. P-value≤0.05 and │logFC│≥1 are considered as differentially expressed (DE). Transcription factors (TFs) regulating the DE genes and kinases trageting TFs are harvested by enrichment analysis. Also, miRNAs targeting either DE genes, TFs, or kinases are enriched. For enrichment analyses, adj. P-value≤0.05 was considered as statistical significance threshhold.
Gene set enrichment analysis of the tubulointerstitium dataset (GSE30529). The genes with the adj. P-value≤0.05 and │logFC│≥1 are considered as differentially expressed (DE). Transcription factors (TFs) regulating the DE genes and kinases trageting TFs are harvested by enrichment analysis. Also, miRNAs targeting either DE genes, TFs, or kinases are enriched. For enrichment analyses, adj. P-value≤0.05 was considered as statistical significance threshhold.
Pathway enrichment analysis of the glomerule dataset. Signaling pathways related to the DE gene, TFs, and kinase of the glomerule network were detremined. Adj. P-value≤0.05 was considered as statistical significance threshhold.
Pathway enrichment analysis of the tubulointerstitium dataset. Signaling pathway related to the DE gene, TFs, and kinase of the tubulointerstitium network were detremined. Adj. P-value≤0.05 was considered as statistical significance threshhold.
Topology analysis of the glomerule multi-layer network.
Topology analysis of the multi-layer Tubulointerstitium network.
The ontology of the nodes in the glomerule and tubulointerstitium networks. Gene ontology enrichment analysis was performed with the differentially expressed genes, transcription factors and kinases in each network. Gene ontology molecular function parents are illustrated. Horizontal axis is the numbers of children for each parent term. adjusted P ≤ 0.05 is considered as threshold of statistical significance
The ontology of the nodes in the glomerule and tubulointerstitium networks. Gene ontology enrichment analysis was performed with the differentially expressed genes, transcription factors and kinases in each network. Gene ontology cellular component parents are illustrated. Horizontal axis is the numbers of children for each parent term. adjusted P ≤ 0.05 is considered as threshold of statistical significance