

Abstract
Dalbavancin is indicated for the treatment of acute bacterial skin and skin structure infections caused by susceptible gram‐positive microorganisms. This analysis represents the update of the population pharmacokinetics (popPK) modeling and target attainment simulations performed with data from the single‐dose safety and efficacy study and an unrelated but substantial revision of the preclinical pharmacokinetic/pharmacodynamic target (fAUC/MIC, free area under concentration‐time curve/minimum inhibitory concentration ratio). A 3‐compartment distribution model with first‐order elimination provided an appropriate fit, with typical dalbavancin clearance of 0.05 L/h and total volume of distribution of ∼15 L. Impact of intrinsic factors was modest, although statistically significant (P < .05) relationships with total clearance were found for the following covariates: creatinine clearance, weight, and albumin — dose adjustment was only indicated for severe renal impairment. Under the new nonclinical target, simulations of the popPK model projected that >99% of subjects would achieve the nonclinical target at MIC values up to and including 2 mg/L.
Keywords: acute bacterial skin and skin structure infection, dalbavancin, long‐acting intravenous antibiotic, pharmacokinetics, target attainment
Acute bacterial skin and skin structure infections (ABSSSIs) are associated with high mortality and morbidity and a substantial financial burden on health‐care systems.1 The incidence and severity of disease has increased in recent years, in parallel with the emergence of community‐acquired methicillin‐resistant Staphylococcus aureus (MRSA) infections.2, 3 Community‐acquired MRSA is of global concern and has contributed to substantial health‐care resource utilization in the United States, where approximately half (46%) of all ABSSSIs caused by S. aureus were found to be the result of methicillin‐resistant isolates.4
Dalbavancin is a second‐generation lipoglycopeptide indicated for the treatment of ABSSSIs caused by susceptible isolates of the following gram‐positive microorganisms: methicillin‐susceptible and methicillin‐resistant strains of Staphylococcus aureus, Streptococcus pyogenes, Streptococcus agalactiae, Streptococcus dysgalactiae, Streptococcus anginosus group, and vancomycin‐susceptible strains of Enterococcus faecalis.5 Dalbavancin is given intravenously and has a terminal half‐life of >14 days,6 which allows administration either as a 2‐dose regimen (1000 mg on day 1 and 500 mg on day 8), as approved by the U.S. Food and Drug Administration (FDA) in 2014 or as the revised single‐dose regimen of 1500 mg approved in 2016.5 The pharmacokinetics (PK) of dalbavancin in humans is linear, dose‐proportional, and characterized by high protein binding7, 8; approximately 93% of an intravenous dose was reported to be bound to serum albumin, with the remaining 7% existing in unbound form, a proportion that is largely unchanged by drug concentration, renal impairment, or hepatic function.6, 9, 10
Initial distribution occurs in a volume of approximately 10 L, and drug levels in plasma decline rapidly over the first 48 hours as the drug distributes extensively into body tissues, including bone and articular tissue, with a total volume of distribution (Vd) of nearly 16 L.11, 12 In healthy volunteers, approximately one‐third of a 1000‐mg intravenous dose of dalbavancin is excreted in urine unmodified,8 with an additional one‐third of dalbavancin excreted in the feces and a further 12% excreted as a minor metabolite, hydroxyl‐dalbavancin, by 42 days postdose.10, 13 As a result, nonrenal methods play a major role in dalbavancin metabolism.13 Total drug clearance has been estimated to be approximately 0.04 L/h in healthy subjects7 and 0.058 L/h in patients.12 No dosage adjustment is necessary for patients with creatinine clearance (CLCR) > 30 mL/min, patients with renal impairment receiving hemodialysis, or those with mild hepatic impairment.6
Over the past 20 years, pharmacometric modeling techniques have been established as a key component of decision making for antimicrobial agents. The modeling is often performed in an iterative fashion as new data emerge, from initial dose selection to dose adjustment in important subpopulations through final dose justification and the establishment of in vitro susceptibility criteria (break points).14, 15, 16, 17, 18, 19 The overall methodological approach consists of 4 steps: (1) identification of appropriate pharmacokinetic/pharmacodynamic (PK/PD) targets from preclinical experiments (in vitro, in vivo)15; (2) population pharmacokinetic (popPK) analysis of clinical PK data to create a compartmental model of the concentration‐time relationship, and a quantitative understanding of the underlying intersubject variability, including identification of any significant intrinsic and/or extrinsic factors (covariates) influencing the relationship; (3) probability of target attainment (PTA) simulations, in which Monte Carlo simulation of the popPK model in the clinical population of interest is used to determine the percentage of the population that would be expected to meet the specified target(s); and (4) after results from the efficacy studies are available, attempts are made to establish a clinical PK/PD target via exposure‐response analysis of clinical data,20 and if one can be found, complementary target attainment simulations are performed with the clinical PK/PD target. Subsequent expert review for the establishment of an antimicrobial agent's susceptibility criteria for an agent nearly always includes PTA for preclinical targets in conjunction with review of microbiology surveillance data and clinical study results, but PTA using clinical targets is less common because of the computational challenges of determining a target from clinical data sets that often have high positive response rates and/or low variation in exposure.
For dalbavancin, earlier publications exist for popPK12 as well as for preclinical PTA.21 The prior popPK analysis used data from 3 safety and efficacy studies with PK sampling following different administration regimens in patients with catheter‐related bloodstream infections caused by gram‐positive bacterial pathogens and skin and soft‐tissue infections (Table 1). The preclinical PK/PD target was 24‐hour free drug area under the concentration‐time curve (fAUC)/minimum inhibitory concentration (MIC), based on a neutropenic murine thigh model.22
Table 1.
Studies Included in the Population Pharmacokinetic Data Set
| Study | Title | Regimen | n (PK Sampling) | PK Sampling Schedule | Reference |
|---|---|---|---|---|---|
| VER001‐4 | Phase 2, Randomized, Open‐Label, Multi‐Center Study to Evaluate the Safety and Efficacy of Dalbavancin Versus Vancomycin in the Treatment of Catheter‐Related Bloodstream Infections With Suspected or Confirmed Gram‐Positive Bacterial Pathogens (2002) | 1000 mg on day 1 and 500 mg on day 8 | 30 | Days 1, 3‐8, EOT, TOC | Raad et al (2005)29 |
| VER001‐5 | Phase 2, Pilot, Randomized, Open‐Label, Multi‐Center Study to Evaluate the Safety and Efficacy of Dalbavancin Versus Investigator/Physician‐Designated Comparator in Skin and Soft Tissue Infection (2001) | 1100 mg on day 1; 1000 mg on day 1 and 500 mg on day 8 | 34 | Day 8, EOT, follow‐up | Seltzer et al (2003)30 |
| VER001‐9 | Phase 3, Randomized, Double‐Blind, Multi‐Center Study to Evaluate the Safety and Efficacy of Dalbavancin Versus Linezolid in the Treatment of Complicated Skin and Soft Tissue Infections with Suspected or Confirmed Gram‐Positive Bacterial Pathogens (2003) | 1000 mg on day 1 and 500 mg on day 8 | 468 | Days 1, 4, 7, 8, EOT, TOC | Jauregui et al (2005)31 |
| DUR001‐303 | A Phase 3b, Double‐Blind, Multi‐Center, Randomized Study to Compare the Efficacy and Safety of Single Dose Dalbavancin to a 2 Dose Regimen of Dalbavancin for the Treatment of Acute Bacterial Skin and Skin Structure Infections | Intravenous single‐dose dalbavancin: 1500 mg on day 1 (1000 mg for patients with CLCR < 30 mL/min not receiving dialysis), placebo intravenously on day 8 Intravenous 2‐dose dalbavancin: 1000 mg on day 1, 500 mg on day 8 (750 mg on day 1 and 375 mg on day 8 for patients with CLCR < 30 mL/min not receiving dialysis) | 171 | Day 1 (1 hour ± 30 minutes); 18 ± 2 hours, 23 ± 4 hours, and 36‐48 hours after day 1 dose | Dunne et al (2016)24 |
CLCR, creatinine clearance; EOT, end of therapy; TOC, test of cure.
Since the time of these publications, significant and relevant additional preclinical and clinical research has been performed. First, the initial in vivo PK/PD target using the neutropenic murine thigh infection model has been revised by the research group that performed the initial analysis.23 The new analysis improved on the prior analysis by using a more sensitive and accurate drug assay method (liquid chromatography‐tandem mass spectrometry vs the prior microbiologic assay), a more robust sampling scheme, and a set of organisms with higher MIC values. The derived bacterial stasis target for 24‐hour fAUC/MIC was estimated in the new analysis to be 27.1, in contrast to the value of 265 in the earlier publication, a change of approximately 10‐fold.23
In addition to the update of the preclinical target value, an additional pivotal phase 3 study (DUR001‐303) with PK sampling has since been performed, comparing a single 1500‐mg intravenous dose on day 1 with the prior standard 2‐dose regimen (1000 mg intravenously on day 1, 500 mg intravenously on day 8). Notably, this phase 3 study incorporated additional PK sampling times in the first 48 hours, in contrast to the earlier studies (Table 1), potentially allowing for a more precise characterization of the underlying structural PK model.
The primary objectives of the current modeling analysis were therefore to (1) reevaluate the dalbavancin popPK model through incorporation of the PK sampling data from DUR001‐303, and (2) conduct a new iteration of the preclinical target attainment simulation, using the updated murine thigh model preclinical target and the updated popPK model. As a secondary objective, the data from DUR001‐303 were explored for evidence of a clinical PK/PD target, that is, any potential relationship between a metric of exposure and efficacy end points in the study.
Material and Methods
Participants
The patient population of study DUR001‐303 consisted of adults (18–85 years old) who met the clinical definition for ABSSSIs as described previously.24 These data were combined with data from 3 earlier clinical studies, VER001‐4, VER001‐5, and VER001‐9 (Table 1). Appropriate participant informed consent and institutional review board (IRB) approvals had been obtained at the time of each study. Additional detailed information on each of the 4 studies, including study sites and IRBs, is provided as an online Supplemental Table.
Data Inclusion and Exclusion Criteria
Combination of data from studies VER001‐4, VER001‐5, and VER001‐9 used in the earlier popPK analysis12 with the new data from study DUR001‐303 resulted in an analysis data set of 2310 observations. No imputation of values was performed; any observations with missing values or below the quantification limit were omitted from the analysis. Overall, only 2 dalbavancin plasma concentrations were missing or reported below the limit of quantitation (0.5 µg/mL); both were from DUR001‐303 and were removed from the popPK analyses. One outlier data point (from VER001‐9) excluded in the previous model was also removed in this analysis.
Assessments and Analytical Methods
In DUR001‐303, blood samples were obtained at 1 hour ± 30 minutes, 18 ± 2 hours, 23 ± 4 hours, and between 36 and 48 hours after the end of the day 1 infusion. In DUR001‐303, subjects with CLCR < 30 mL/min and who were not receiving regular hemodialysis or peritoneal dialysis received a reduced dose: for the single‐dose group, dose was reduced from 1500 to 1000 mg on day 1; and for the 2‐dose group, dose was reduced from 1000 to 750 mg on day 1 and from 500 to 375 mg on day 8.
Dalbavancin plasma concentrations in DUR001‐303 were measured using a high‐performance liquid chromatography‐tandem mass spectrometry bioanalytical method,12 validated in the linear range from 0.5 to 500 µg/mL using 50 µL of K2‐ethylenediaminetetraacetic acid plasma.25 This was an updated version of the method details previously published,12 which were used in studies VER001‐4, VER001‐5, and VER001‐9.
Population Pharmacokinetics Analysis
Nonlinear mixed‐effects estimation for the population PK model was performed using NONMEM (version 7.3; ICON plc, Gaithersburg, Maryland). The first‐order conditional estimation method was used for structural pharmacokinetic modeling.26
The previous popPK model was thoroughly reevaluated, with attention paid to the appropriate number of compartments, covariate selection, model stability, and clinical relevance of the final model. One shortcoming of the prior model was the use of a 2‐compartment model, as more recent phase 1 data have shown that a 3‐compartment model is appropriate.11 In fact, the previous publication indicated that models with a third compartment successfully converged, but were not chosen because of the inability of the model to estimate interindividual variability (IIV) on all structural model parameters, a requirement that we would argue to be unnecessarily restrictive. As mentioned above, the additional PK sampling times of DUR001‐303 in the first 48 hours after the initial dose were believed to increase the stability for estimation of the third compartment.
Residual error, or within‐subject variability, was assumed to be a function of normally distributed random effects. IIV or random effects across individuals were incorporated using a log‐normal distribution. Following determination of the structural model, predictive value of individual patient characteristics was tested in a stepwise covariate search using the stepwise covariate modeling tool in Perl‐speaks‐NONMEM (PsN; version 4.2.0; Free Software Foundation Inc., Boston, Massachusetts).27 Stepwise forward inclusion was determined by P < .01 and P < .001 and was used for retention in the model during backward elimination. The covariates body weight, race, sex, age, and albumin were each tested on each parameter with intersubject variability in the base structural model. Based on biological plausibility, creatinine clearance was tested only on elimination clearance (CL).
Standard model diagnostic plots, visual predictive checks, and uncertainty of parameter estimates were used to assess model fit. Assessment of model fit was analyzed accounting for IIV using standard graphical diagnostic plots involving population predictions (PRED), individual predictions (IPRED), IIV, and visual predictive checks. A bootstrap procedure was used to obtain uncertainty estimates in model parameters.
Exposure‐Response Methods
Exposure‐response analysis for DUR001‐303 was performed using R version 3.1.1 (Comprehensive R Network; http://cran.rproject.org). Exploratory data analysis was undertaken to assess potential relationships between each of 4 efficacy end points: clinical response at 48‐72 hours, clinical status at end of treatment (14 days), clinical status at follow‐up (28 days), investigator assessed status at follow‐up (28 days), and predicted dalbavancin exposure, fAUC/MIC. Any potential relationships identified were to be quantified further using logistic regression analysis. AUC was simulated for each subject in DUR001‐303 in the popPK data set at 72 hours and 14 and 28 days. The exposure‐response analysis was restricted to 542 isolates in 420 subjects determined to be in the microbiologic intent‐to‐treat population.
Surveillance Data for MIC Distributions
MIC distributions for use in the probability of target attainment analyses were obtained from the 2017 surveillance program of isolates from 70 medical centers located across the 9 Census regions in the United States and 38 medical centers located in 18 countries in Europe. Susceptibility testing was conducted centrally at a reference laboratory (JMI Laboratories, North Liberty, Iowa) in accordance with Clinical and Laboratory Standards Institute (CLSI) reference methods (CLSI M07‐A10, 2015)28 for the following pathogens: Staphylococcus aureus (n = 8833), Streptococcus pyogenes (n = 941), Streptococcus dysgalactiae (n = 312), and vancomycin‐susceptible Enterococcus faecalis (n = 1060).
Target Attainment Simulation Methods
Preclinical target attainment using the established nonclinical PK/PD stasis target of 24‐hour fAUC/MIC = 27.123 was used to investigate the effect of dalbavancin concentration over time using the AUC‐time curve and allowed prediction of the efficacy of dalbavancin using the updated model. Patient‐level parameters (ie, CL, central volume of distribution [V1], etc.) were resampled (n = 1000) from the discrete individual‐level parameter predictions by the final population PK model. Calculation of AUC for the purposes of the fAUC/MIC index was aligned with the preclinical experiment that provided the index: mean average daily fAUC (to extrapolate from mouse to human; this was defined as fAUC0–120h/5 in humans) as the pharmacodynamic exposure metric. Simulations assumed plasma protein‐bound and free dalbavancin fractions to be approximately 93% and 7%, respectively, unaltered by drug concentration, renal impairment, or hepatic function.9
Results
Structural Model
Several candidate models were explored to find the best structural model. Because dalbavancin is given intravenously and the kinetics are linear, the model was evaluated only with respect to the number of distribution compartments and random‐effects structures. The residual error was assumed and verified to be proportional to the predicted concentration.
The distribution model was first explored using 1‐, 2‐, and 3‐compartment models with zero‐order input and first‐order elimination. The conditional weighted residuals for the 1‐ and 2‐compartment models showed positive bias (under prediction) with time, whereas the 3‐compartment model (see Supplementary Figure S1) reduced this bias and provided a significantly better fit than the 2‐compartment model. Concentrations inside the compartments (C1, C2, and C3) were a function of the parameters (CL, V1, Q2, V2, Q3, and V3), with drug entering compartment 1 via zero‐order infusion and moving between compartments and exiting the system via a first‐order process. The structural parameters incorporated IIV on CL, V1, V2, and V3, whereas the 2 distributional clearances, Q2 and Q3, had no IIV. A model progression table and further in‐depth details on the model‐building process are provided in the text and tables of the Supplemental Materials.
Covariate Summary by Study
Pharmacokinetic sampling came from 703 patients. Baseline demographics and clinical characteristics are summarized in Table 2. Patient demographics for DUR001‐303 were generally similar to the pooled data set of the earlier studies; the larger studies, VER001‐9 and DUR001‐303, were similar in terms of the distribution of covariates.
Table 2.
Patient Demographics and Baseline Characteristics
| Characteristic | VER001‐4 (n = 30) | VER001‐5 (n = 34) | VER001‐9 (n = 468) | DUR001‐303 (n = 171) | All (N = 703) |
|---|---|---|---|---|---|
| Age (y), median (range) | 58.5 (22‐78) | 51.5 (18‐86) | 44.0 (18‐93) | 50.0 (20‐84) | 47.0 (18‐93) |
| Male, n (%) | 13 (43.3) | 15 (44.1) | 287 (61.3) | 101 (59.1) | 416 (59.2) |
| Race | |||||
| Caucasian | 17 (56.7) | 33 (97.1) | 300 (64.1) | 154 (90.1) | 504 (71.7) |
| Hispanic | 1 (3.3) | 0 | 102 (21.8) | 0 | 103 (14.7) |
| Black | 12 (40.0) | 0 | 53 (11.3) | 13 (7.6) | 78 (11.1) |
| Asian | 0 | 0 | 5 (1.1) | 2 (1.2) | 7 (1.0) |
| Other | 0 | 1 (2.9) | 8 (1.7) | 2 (1.2) | 11 (1.6) |
| Weight, kg | |||||
| Mean ± SD | 81.2 ± 25 | 91.6 ± 19 | 92.3 ± 29 | 82.6 ± 21 | 89.5 ± 27 |
| Median (range) | 73.8 (43‐150) | 91.0 (46‐120) | 88.0 (44‐320) | 79.0 (50‐170) | 85.0 (43‐320) |
| BSA, m2 | |||||
| Mean ± SD | 1.9 ± 0.3 | 2.1 ± 0.2 | 2.1 ± 0.3 | 1.9 ± 0.2 | 2.0 ± 0.3 |
| Median (range) | 1.8 (1.4‐2.7) | 2.1 (1.4‐2.5) | 2.1 (1.4‐4.0) | 1.9 (1.5‐2.8) | 2.0 (1.4‐4.0) |
| CLCR, mg/mL | |||||
| Mean ± SD | 92.7 ± 31 | 119 ± 51 | 130 ± 55 | 91.8 ± 37 | 119 ± 53 |
| Median (range) | 82.6 (50‐180) | 120.0 (45‐240) | 121.0 (26‐440) | 90.8 (22‐190) | 113.0 (22‐440) |
| Albumin, g/dL | |||||
| Mean ± SD | 2.7 ± 0.8 | 3.9 ± 0.6 | 3.6 ± 0.6 | 4.0 ± 0.5 | 3.7 ± 0.7 |
| Median (range) | 2.8 (1.1‐4.3) | 3.7 (2.6‐4.9) | 3.6 (1.4‐5.1) | 4.1 (2.4‐5.1) | 3.7 (1.1‐5.1) |
BSA, body surface area; CLCR, creatinine clearance.
Covariate Model Building
The final model included relationships for CLCR, weight and albumin on CL, weight and albumin on V1, weight, albumin and age on V2, and weight and albumin on V3. The covariates age, body weight, race, sex, and albumin were considered for inclusion on all clearance and volume variables; CLCR was also considered for inclusion on CL. The covariate model was further optimized to ensure stable convergence by replacing an automatically selected covariate of age on V3 with weight and albumin and by reparametrizing the random‐effects covariance
The inclusion of these covariates reduced the level of intersubject variability estimated in the base structural model by 43% for clearance, by 63% for V1, by 24% for V2, and by 44% for V3. After inclusion of covariates, the final model estimated unexplained intersubject variability to be 22% for clearance and 24% for V1 (Table 3).
Table 3.
Fixed‐Effect Parameter Estimate Equations for Final Model
| Fixed‐Effect Parameter Estimate | Name | Estimate | RSE, % | 95%CI | Coefficient of Variation, % |
|---|---|---|---|---|---|
| θ1 | CL (L/h) | 0.0531 | 1.1 | 0.0519, 0.0543 | |
| θ2 | V1 (L) | 3.04 | 4.1 | 2.8, 3.28 | |
| θ3 | V2 (L) | 8.78 | 3.9 | 8.11, 9.44 | |
| θ4 | V3 (L) | 3.28 | 9.6 | 2.67, 3.9 | |
| θ5 | Q2 (L/h) | 0.288 | 13.2 | 0.213, 0.362 | |
| θ6 | Q3 (L/h) | 2.11 | 10.8 | 1.66, 2.55 | |
| θ7 | CL·ALB | −0.477 | 11.8 | −0.587, −0.367 | |
| θ8 | CL·CLCR | 0.273 | 12.2 | 0.208, 0.338 | |
| θ9 | CL·WT | 0.391 | 13.0 | 0.291, 0.491 | |
| θ10 | V1·ALB | −0.340 | 28.6 | −0.53, −0.149 | |
| θ11 | V1·WT | 0.683 | 11.0 | 0.537, 0.83 | |
| θ12 | V2·AGE | 0.486 | 12.1 | 0.371, 0.601 | |
| θ13 | V2·ALB | −0.413 | 27.2 | −0.633, −0.193 | |
| θ14 | V2·WT | 0.365 | 29.6 | 0.153, 0.577 | |
| θ16 | V3·ALB | −0.551 | 44.4 | −1.03, −0.0714 | |
| θ17 | V3·WT | 0.518 | 44.7 | 0.0644, 0.972 | |
| — | T1/2 (days)a | 8.77 | 1.47 | 8.51, 9.02 | |
| ω1.1 | ω2CL | 0.0489 | 15.5 | 0.034, 0.0638 | 22 |
| ω2.1 | ωCL,V2 | 0.0823 | 22.4 | 0.0462, 0.118 | |
| ω2.2 | ω2V2 | 0.153 | 29.7 | 0.064, 0.242 | 41 |
| ω3.3 | ω2V1 | 0.0566 | 26.6 | 0.0271, 0.0862 | 24 |
| ω4.3 | ω2V1,V3 | 0.111 | 37.5 | 0.0293, 0.192 | |
| ω4.4 | ω2v3 | 0.437 | 19.4 | 0.271, 0.602 | 74 |
| σ1.1 | σ2proportional | 0.0362 | 9.9 | 0.0292, 0.0432 |
ALB, albumin; CI, confidence interval; CL, clearance; CLCR, creatinine clearance; RSE, relative standard error; SE, standard error; V, volume; WT, weight; θ, parameter; ω, interindividual (co)variance estimate; σ, intraindividual variance estimate.
T1/2 calculated from 100 000 random samples drawn from mean and covariance of estimated model parameters and is consistent with values from multiple clinical trials.
The goodness of fit for the final model is shown in Figure 1. The final model appeared robust and fit the observed data accurately. The visual predictive check showed that model predictions were in good agreement with the observed data, individual‐level and population‐level predictions were unbiased versus observed data, and the model had good predictive performance over the range of the observed data (Figure 2).
Figure 1.
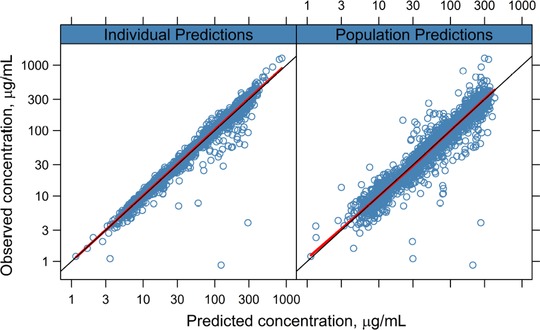
Goodness of fit for the final model. Observed values plotted against predicted values overlaid with smoothing function (solid red line) and unity line (solid black line).
Figure 2.
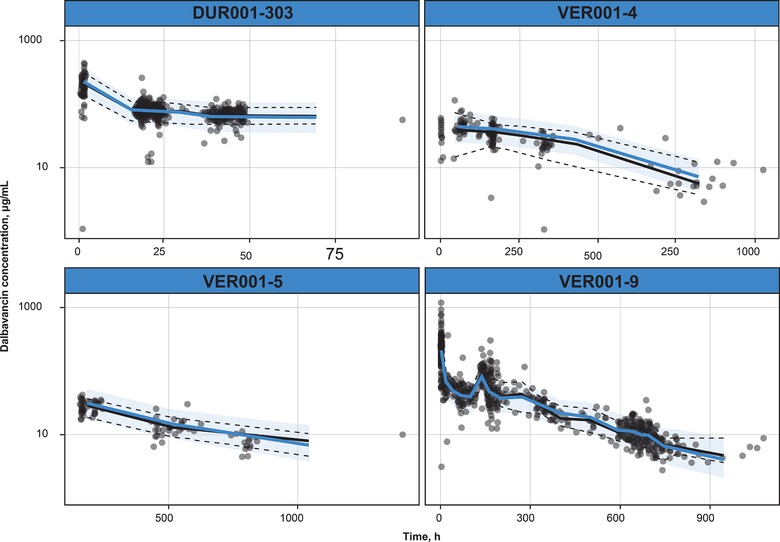
Visual predictive check of final PK model by study. Dots are prediction‐correction data. Black lines indicate observed median (solid line) and 2.5th and 97.5th percentiles (dashed lines) of the observed data. Shaded area indicates the 95% prediction interval around the prediction‐corrected median (blue line).
Exposure Response
The exploratory data analysis of the 4 efficacy end points in DUR001‐303 did not reveal any relationship between drug exposure and clinical outcome. Logistic regression models of the 4 end points against their respective AUC/MIC metrics showed no statically significant trends (Figure 3; P = .33‐1.00). All gradients were very small, with large coefficients of variation and P > .05, indicating a lack of correlation between end points and AUC. Given the high positive response rate observed in the study, this result was unsurprising.
Figure 3.
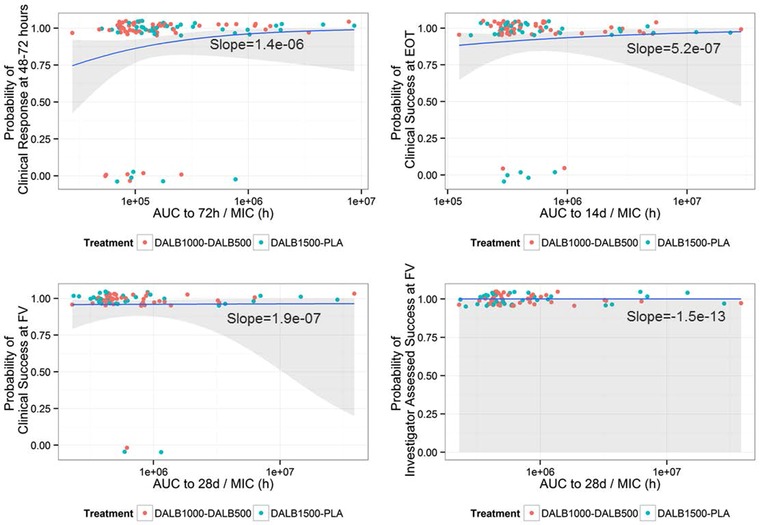
Logistic regression models of end points against AUC/MIC. X axis is logarithmic scale. AUC, area under the curve; MIC, minimum inhibitory concentration. Responses (points) jittered at 0 (failure) or 1 (success) overlaid with logistic regression (blue line) and 95%CI (shaded area).
Target Attainment
Simulations of the concentration profile over time for both approved regimens (1500 mg once weekly; 1000 and 500 mg on days 1 and 8; infusion time, 30 minutes for all) are shown in Figure 4. Target attainment simulations were run for the 1500‐mg single‐dose regimen using the revised nonclinical pharmacodynamic stasis target of 27.1. These simulations projected more than 90% of simulated subjects to achieve the nonclinical target at MIC ≤ 2 mg/L; target attainment of 99% at MIC = 2.0 and 87.7% at MIC = 4.0 was achieved (Figure 5). Figure 5 also displays the histograms of MIC distributions for the 4 most relevant species of pathogens obtained in the JMI‐analyzed microbial surveillance data from 2017. The separation of the rightmost MIC at which 90% target attainment is gained (ie, MIC = 2) from the rightmost histogram bar (ie, MIC = 0.25) indicates that the current dose would continue to provide attainment of the preclinical PK/PD target for several additional MIC dilutions of pathogen potency beyond what is currently observed in the United States and Europe.
Figure 4.
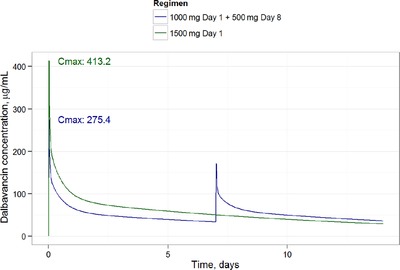
Simulation of the population mean PK profile by dose used in the DUR001‐303 study. Cmax, peak concentration.
Figure 5.
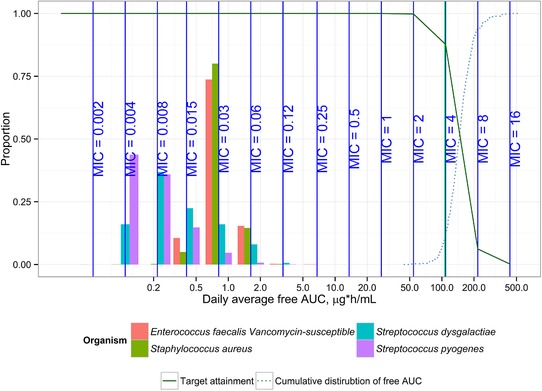
Target attainment for 1500‐mg single‐dose regimen. The projected target MIC attainment for 1500‐mg single‐dose dalbavancin regimen is indicated as a solid line. X axis is logarithmic scale. Histograms represent MIC distributions from 2017 surveillance data for the 4 most relevant species of pathogens. AUC, area under the curve; MIC, minimum inhibitory concentration. Target attainment simulations were run for the 1500‐mg single‐dose regimen using the revised nonclinical pharmacodynamic stasis target of 27.1. Daily average free AUC was calculated as AUC0‐120/5.
Target attainment was also calculated using a 1‐log kill target of 53.3 and a 2‐log kill target of 111.1, resulting in target attainment of 99% at MIC = 1.0 mg/L and MIC = 0.5 mg/L, respectively, for 1‐log and 2‐log kill.23 Simulations using the updated popPK model but the older stasis target showed >99% for the 1500‐mg dose at MIC = 0.25 mg/L but only 65% PTA at MIC = 0.50 mg/L.
Discussion
The objectives of the present study were to update the popPK model and related nonclinical target attainment simulations for dalbavancin. This update was driven primarily by the updated nonclinical PK/PD targets published by Lepak et al (2015) and Bowker et al (2006),9, 23 but also took advantage of the additional PK information provided by a new pivotal safety and efficacy study of dalbavancin in ABSSSI patients assessing the noninferiority of a single‐dose regimen to the previously approved 2‐dose regimen.
Graphical analysis of the PK data in the updated popPK data set showed a rapid increase in plasma concentration to the end of infusion, followed by a rapid distribution period and a slow extended elimination phase, in keeping with prior descriptions. In this iteration of dalbavancin popPK modeling, a 3‐compartment distribution model (1 central and 2 peripheral compartments) was found to best describe the multiphasic elimination profile of dalbavancin. The addition of a third compartment reduced visual bias in the fit and was statistically superior to the 2‐compartment model for the updated data set. This finding is in agreement with analyses of intensive PK sampling data from phase 1 studies in healthy volunteers.11 However, this contrasts with the prior publication of a 2‐compartment model; here, the incorporation of the additional PK sampling data within the first 24 hours postinfusion provided by the new clinical study is believed to have allowed identification of a clear 3‐compartment structure.
The effect of the covariates was modest and consistent with previous understanding of the intrinsic factors of the popPK of dalbavancin. The covariate of body surface area from the prior model was replaced by body weight in this model, providing easier clinical use and interpretation without any loss in model performance. As with the earlier model,12 renal function, as measured by creatinine clearance, was the most important predictor of the exposure‐response of patients to dalbavancin, with no other covariate showing a large enough influence to necessitate dosage adjustment. Despite this, because nonrenal methods play a significant role in dalbavancin clearance, patients with CLCR ≥ 30 mg/mL and patients with severe renal impairment receiving dialysis do not require dalbavancin dose adjustments.
Overall, the PK profile of dalbavancin was well characterized by the final popPK model, and we believe that this model will be sufficiently robust to simulate dalbavancin PK in all adult patients except those with severe renal impairment, as the data set did not include enough subjects in this category to allow for simulation under CLCR of 30 mL/min. Future extensions of this work could involve the addition of the data from phase 1 studies in renal impairment, as well as the addition of data from studies in pediatric subjects, to better enable simulations in these subpopulations.12
Nonclinical target attainment simulations were conducted based on the updated popPK model and the updated murine thigh model‐derived fAUC/MIC stasis target of 27.1 for S. aureus. The results of these simulations projected a nonclinical S. aureus target attainment of greater than 90% at MIC ≤ 2 mg/L for both approved dosing regimens, in contrast to the value of 0.25 mg/L achieved by simulations of the older nonclinical target. As clinical isolates with dalbavancin MICs greater than 0.12 mg/L are exceedingly rare, the nonclinical target attainment results, in theory, provide some evidence for potential efficacy for isolates with MICs higher than those typically currently encountered. These results were submitted to the FDA for the supplemental approval application with the single‐dose study DUR001‐303. Although many factors are taken into account for the setting of break points, during this approval, the CLSI updated the break point from 0.12 to 0.25 mg/L.
Conclusions
This analysis updated prior work on population PK and nonclinical target attainment analysis of dalbavancin, incorporating data from the pivotal single‐dose efficacy and safety study as well as using the substantially revised nonclinical PK/PD target. Results from the popPK modeling showed that the pharmacokinetic profile of dalbavancin is well characterized, with low intersubject variation and a limited effect of intrinsic factors. A 3‐compartment model with first‐order elimination provided a robust and accurate fit to the clinical PK data from the 4 dalbavancin safety and efficacy studies. The additional PK sampling times provided by the most recent study allowed for more robust identification of the third compartment, compared with a prior analysis. Renal function, weight, albumin, and age were found to be statistically significant, but not necessarily clinically significant covariates, as only severe renal impairment (CLCR < 30 mL/min) requires a dose adjustment. When target attainment was simulated using the updated nonclinical 24‐hour fAUC/MIC stasis target of 27.1, more than 99% of simulated subjects were predicted to achieve this nonclinical PK/PD target at MICs up to and including 2 mg/L, a value several dilutions higher than the MICs currently seen in microbiologic surveillance studies.
Declaration of Conflicting Interests
Two of the authors were employees of Allergan at the time of study conduct and analysis. Therefore, the sponsor of the study played a role in study design; in the collection, analysis, and interpretation of data; in the writing of the report; and in the decision to submit the article for publication.
Funding
This work was supported by Allergan plc (Dublin, Ireland).
Ethical Approval
Not required.
Author Contributions
T.J.C. was involved in the conception and design of the study. T.J.C. and I.C. were involved in acquisition of the data. All authors contributed to data analysis/interpretation and to each stage of drafting/critical review of the manuscript and approved the final version for submission.
Competing Interests
Timothy J. Carrothers is an employee of Allergan. Jason Chittenden is an employee of qPharmetra, LLC, and was contracted to undertake the analysis described here. Ian Critchley was an employee of Allergan at the time of study conduct and analysis and is a current employee of Spero Therapeutics.
Authors, either individually or collectively, have signed an agreement with the sponsor of the research reported in the Contribution, Allergan, that did not place any requirements on their publication of the research findings, such as preventing them from publishing both positive and negative results or that forbids them from publishing the research without the prior approval of the sponsor.
Supporting information
Supporting Information
Acknowledgments
Writing and editorial assistance was provided to the authors by Jennifer Venzie, PhD, and John E. Fincke, PhD, of Complete Healthcare Communications, LLC (North Wales, Pennsylvania), a CHC Group company, and funded by Allergan. All authors met the ICMJE authorship criteria. Neither honoraria nor payments were made for authorship.
References
- 1. Pollack CV Jr, Amin A, Ford WT Jr, et al. Acute bacterial skin and skin structure infections (ABSSSI): practice guidelines for management and care transitions in the emergency department and hospital. J Emerg Med. 2015;48(4):508‐519. [DOI] [PubMed] [Google Scholar]
- 2. Ramdeen S, Boucher HW. Dalbavancin for the treatment of acute bacterial skin and skin structure infections. Expert Opin Pharmacother. 2015;16(13):2073‐2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hersh AL, Chambers HF, Maselli JH, Gonzales R. National trends in ambulatory visits and antibiotic prescribing for skin and soft‐tissue infections. Arch Intern Med. 2008;168(14):1585‐1591. [DOI] [PubMed] [Google Scholar]
- 4. Ray GT, Suaya JA, Baxter R. Incidence, microbiology, and patient characteristics of skin and soft‐tissue infections in a U.S. population: a retrospective population‐based study. BMC Infect Dis. 2013;13:252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dalbavancin (Xydalba) . Severe bacterial infections of the skin: a teicoplanin me‐too. Prescrire Int. 2016;25(171):123‐125. [Google Scholar]
- 6. Marbury T, Dowell JA, Seltzer E, Buckwalter M. Pharmacokinetics of dalbavancin in patients with renal or hepatic impairment. J Clin Pharmacol. 2009;49(4):465‐476. [DOI] [PubMed] [Google Scholar]
- 7. Dorr MB, Jabes D, Cavaleri M, et al. Human pharmacokinetics and rationale for once‐weekly dosing of dalbavancin, a semi‐synthetic glycopeptide. J Antimicrob Chemother. 2005;55(suppl 2):ii25‐ii30. [DOI] [PubMed] [Google Scholar]
- 8. Leighton A, Gottlieb AB, Dorr MB, et al. Tolerability, pharmacokinetics, and serum bactericidal activity of intravenous dalbavancin in healthy volunteers. Antimicrob Agents Chemother. 2004;48(3):940‐945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bowker KE, Noel AR, MacGowan AP. Pharmacodynamics of dalbavancin studied in an in vitro pharmacokinetic system. J Antimicrob Chemother. 2006;58(4):802‐805. [DOI] [PubMed] [Google Scholar]
- 10. Leuthner KD, Buechler KA, Kogan D, Saguros A, Lee HS. Clinical efficacy of dalbavancin for the treatment of acute bacterial skin and skin structure infections (ABSSSI). Ther Clin Risk Manag. 2016;12:931‐940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dunne MW, Puttagunta S, Sprenger CR, et al. Extended‐duration dosing and distribution of dalbavancin into bone and articular tissue. Antimicrob Agents Chemother. 2015;59(4):1849‐1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Buckwalter M, Dowell JA. Population pharmacokinetic analysis of dalbavancin, a novel lipoglycopeptide. J Clin Pharmacol. 2005;45(11):1279‐1287. [DOI] [PubMed] [Google Scholar]
- 13. Galluzzo M, D'Adamio S, Bianchi L, Talamonti M. Pharmacokinetic drug evaluation of dalbavancin for the treatment of skin infections. Expert Opin Drug Metab Toxicol. 2018;14(2):197‐206. [DOI] [PubMed] [Google Scholar]
- 14. Louie A, Kaw P, Liu W, et al. Pharmacodynamics of daptomycin in a murine thigh model of Staphylococcus aureus infection. Antimicrob Agents Chemother. 2001;45(3):845‐851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Craig WA. Pharmacokinetic/pharmacodynamic parameters: rationale for antibacterial dosing of mice and men. Clin Infect Dis. 1998;26(1):1‐10; quiz 11‐12. [DOI] [PubMed] [Google Scholar]
- 16. Li J, Lovern M, Green ML, et al. Ceftazidime‐avibactam population pharmacokinetic modeling and pharmacodynamic target attainment across adult indications and patient subgroups. Clin Transl Sci. 2019;12(2):151‐163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Drusano GL. Antimicrobial pharmacodynamics: critical interactions of 'bug and drug'. Nat Rev Microbiol. 2004;2(4):289‐300. [DOI] [PubMed] [Google Scholar]
- 18. Van Wart SA, Ambrose PG, Rubino CM, et al. Pharmacokinetic‐pharmacodynamic target attainment analyses to evaluate in vitro susceptibility test interpretive criteria for ceftaroline against Staphylococcus aureus and Streptococcus pneumoniae. Antimicrob Agents Chemother. 2014;58(2):885‐891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dudley MN, Ambrose PG. Pharmacodynamics in the study of drug resistance and establishing in vitro susceptibility breakpoints: ready for prime time. Curr Opin Microbiol. 2000;3(5):515‐521. [DOI] [PubMed] [Google Scholar]
- 20. Ambrose PG, Bhavnani SM, Rubino CM, et al. Pharmacokinetics‐pharmacodynamics of antimicrobial therapy: it's not just for mice anymore. Clin Infect Dis. 2007;44(1):79‐86. [DOI] [PubMed] [Google Scholar]
- 21. Dowell JA, Goldstein BP, Buckwalter M, Stogniew M, Damle B. Pharmacokinetic‐pharmacodynamic modeling of dalbavancin, a novel glycopeptide antibiotic. J Clin Pharmacol. 2008;48(9):1063‐1068. [DOI] [PubMed] [Google Scholar]
- 22. Andes D, Craig WA. In vivo pharmacodynamic activity of the glycopeptide dalbavancin. Antimicrob Agents Chemother. 2007;51(5):1633‐1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lepak A, Marchillo K, VanHecker J, Andes D. Impact of glycopeptide resistance in Staphylococcus aureus on the dalbavancin in vivo pharmacodynamic target. Antimicrob Agents Chemother. 2015;59(12):7833‐7836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dunne MW, Puttagunta S, Giordano P, et al. A randomized clinical trial of single‐dose versus weekly dalbavancin for treatment of acute bacterial skin and skin structure infection. Clin Infect Dis. 2016;62(5):545‐551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Alebic‐Kolbah T, Demers R, Cojocaru L. Dalbavancin: quantification in human plasma and urine by a new improved high performance liquid chromatography‐tandem mass spectrometry method. J Chromatogr B Analyt Technol Biomed Life Sci. 2011;879(25):2632‐2641. [DOI] [PubMed] [Google Scholar]
- 26. Wang Y. Derivation of various NONMEM estimation methods. J Pharmacokinet Pharmacodyn. 2007;34(5):575‐593. [DOI] [PubMed] [Google Scholar]
- 27. Lindbom L, Ribbing J, Jonsson EN. Perl‐speaks‐NONMEM (PsN)–a Perl module for NONMEM related programming. Comput Methods Programs Biomed. 2004;75(2):85‐94. [DOI] [PubMed] [Google Scholar]
- 28. Clinical and Laboratory Standards Institute . Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard ‐ Tenth Edition. CLSI document M07‐A10. Wayne, PA: Clinical and Laboratory Standards Institute; 2015. [Google Scholar]
- 29. Raad I, Darouiche R, Vazquez J, et al. Efficacy and safety of weekly dalbavancin therapy for catheter‐related bloodstream infection caused by gram‐positive pathogens. Clin Infect Dis. 2005;40(3):374‐380. [DOI] [PubMed] [Google Scholar]
- 30. Seltzer E, Dorr MB, Goldstein BP, et al. Once‐weekly dalbavancin versus standard‐of‐care antimicrobial regimens for treatment of skin and soft‐tissue infections. Clin Infect Dis. 2003;37(10):1298‐1303. [DOI] [PubMed] [Google Scholar]
- 31. Jauregui LE, Babazadeh S, Seltzer E, et al. Randomized, double‐blind comparison of once‐weekly dalbavancin versus twice‐daily linezolid therapy for the treatment of complicated skin and skin structure infections. Clin Infect Dis. 2005;41(10):1407‐1415. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information