

Abstract
Fms-like tyrosine kinase 3 ligand (FLT3L) stimulates the development of classical dendritic cells (DCs). Here we tested the hypothesis that classical DCs drive blood pressure elevation by promoting renal fluid retention. FLT3L deficient (FLT3L−/−) mice that lack classical DCs in the kidney had mean arterial pressures similar to wild-types (WTs) at baseline but had blunted hypertensive responses during 4 weeks of chronic angiotensin (Ang) II infusion. In FLT3L−/− mice, the proportions of effector memory T cells in the kidney were similar to WTs at baseline. However, after Ang II infusion, proportions of effector memory T cells were dramatically lower in the FLT3L−/− kidneys versus WTs, indicating that classical DCs augment the renal accumulation of effector T cells following renin angiotensin system (RAS) activation. Consistent with their lower blood pressures, the Ang II-infused FLT3L−/− mice had attenuated cardiac hypertrophy and lower renal mRNA expression for pro-hypertensive cytokines. Moreover, the Ang II-infused FLT3L−/− mice had lower urinary excretion of the oxidative stress marker 8-isoprostane and lower renal mRNA levels of NADPH oxidase 2. In an IP saline challenge test at day 7 of Ang II, FLT3L−/− mice excreted higher proportions of the injected volume and sodium than WTs. Consistent with this enhanced diuresis, mRNA expressions for the sodium-chloride cotransporter (NCC) and all 3 subunits of the epithelial sodium channel (ENaC) were diminished by more than 40% in FLT3L−/− kidneys compared to the WTs. Thus, classical FLT3L-dependent DCs promote renal T cell activation with consequent oxidative stress, fluid retention, and blood pressure elevation.
Keywords: Hypertension, Dendritic cells, T cells, oxidative stress, sodium transporters
Graphical Abstract
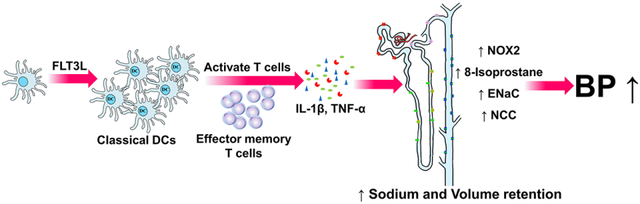
Summary
FLT3L stimulates the development of classical dendritic cells (cDCs), but the role of FLT3L-dependent dendritic cells in hypertension requires elucidation. In the current study, we find that FLT3L-dependent cDCs enhance the accumulation of effector memory T lymphocytes in the kidney and cause renal sodium and fluid retention during chronic Ang II infusion. These effects are associated with higher levels of oxidative stress in the kidney and induction of pro-hypertensive cytokines. Accordingly, FLT3L is required to sustain a full chronic hypertensive response following activation of the renin angiotensin system (RAS). This study thus identifies a key role for FLT3L-dependent cDCs in the pathogenesis of RAS-mediated hypertension. These findings should facilitate development of immune-directed therapies for patients with biologically resistant hypertension.
Introduction
Hypertension results from dysfunction in the vasculature, kidney, brain, and/or the immune system1, 2. To identify new targets for blood pressure control and target organ protection, recent studies have brought intense scrutiny to the immune system’s role in hypertension pathogenesis. Inappropriate immune activation contributes to hypertension in part via effects on kidney function3–6. A full hypertensive response requires T lymphocyte activation in multiple experimental models7, 8. In animal studies, T cells can raise blood pressure by promoting oxidative stress and sodium reabsorption in the kidney9–11. T cells mediate these effects through the elaboration of cytokines that influence renal epithelial function and even through direct contact with renal tubular cells6, 12–14. Dendritic cells (DCs) are the most potent antigen presenting cells that activate T cells and therefore play a fundamental role in immune-dependent blood pressure elevation15–17. Interactions between DCs and T cells to generate memory effector T cells marked by CD44hiCD62lo expression are therefore required to launch a full hypertensive response18, 19. Nevertheless, to interrupt the immune system’s contribution to hypertension, the subpopulations of DCs involved in promoting blood pressure elevation require elucidation.
Fms-like tyrosine kinase 3 ligand (FLT3L) is an endogenous factor that stimulates the differentiation of migratory and lymph node-resident classical DCs20. While these animals are healthy at baseline, their T cells exhibit impaired capacity for activation accruing from interactions with classical DCs. In the current studies, we examine the contribution of classical, Flt3L-dependent DCs to hypertension and volume retention by comparing levels of blood pressure elevation and renal T cell activation following activation of the renin angiotensin system (RAS) in mice with Flt3L deficiency and controls.
Methods
Animal experiments
C57BL/6 FLT3L−/− mice (Jackson Laboratories Stock# 37395-JAX) were backcrossed 6 generations onto the 129/SvEv strain and intercrossed to generate FLT3L+/+ (WT) and FLT3L−/− littermates for our experiments. Data, methods, and materials will be made available upon request per TOP Guidelines. All animal experiments were approved by the Durham Veterans’ Affairs Medical Center Institutional Animal Care and Use Committee. All mice were housed and bred in the animal facilities at the Durham Veterans’ Affairs Medical Center according to NIH guidelines. All animals were maintained on a 12:12-h light/dark cycle at temperatures ranging between 67–76 °F, 30–70 % relative humidity and free access to food and water.
Chronic Angiotensin II-induced Hypertension.
10–12 week old male mice were subjected to our hypertension protocol as previously described21. To render the animals more salt-sensitive, all mice underwent left nephrectomy followed one week later by implantation of a pressure-sensing radiotelemetry catheter. After 10 days of recovery, an osmotic mini-pump was implanted to infuse Ang II (500 ng/kg/min) continuously for 28 days. Blood pressure measurements were recorded for 21 days of Ang II, and during the 4th week of Ang II, mice were placed in metabolic cages as detailed below.
Cell preparations and flow cytometry
Mouse kidneys were harvested and digested into single cell suspensions. In the DC panel, cells were stained with fluorescently-labeled anti-CD45, anti-CD11c, anti-CD11b, and anti-MHCII prior to analysis. For the T cell panel, cells were stained with fluorescently-labeled anti-CD45, anti-CD3, anti-CD4, anti-CD8, anti-CD62L, and anti-CD44 as described22 and subjected to flow cytometric analysis.
RNA extraction and real-time quantitative PCR
Total RNA was extracted from samples according to the manufacturer’s instructions of the RNeasy Plus Mini kit (Cat#: 74136, Qiagen). RNA was reverse transcribed with a high capacity cDNA reverse transcriptase kit (Cat#: 4368813, Invitrogen), and real time quantitative PCR (RT-qPCR) was performed with SYBR (Cat#: 4367663) or Taqman assay (Cat#: 4370074) from Invitrogen. Taqman primers and probes were used for GAPDH, TNF-α, IL-1β, CCL2, CCL5 and IFN-γ. SYBR primers were used genes encoding NOX2, NOX4, α-ENaC, γ-ENaC, NCC, NKCC2, and NHE3.
Urinary 8-isoprostane measurement.
After 25 days of AngII infusion, mice were placed into metabolic cages for 24 hours with free access to food and water. Urine was collected and centrifuged at 3000 rpm for 3 minutes. The clear urine was used quantitate 8-isoprostane per instructions of the 8-Isoprostane ELISA kit (Cat#:516351, Cayman Chemical). The 8-isoprostane concentration was then multiplied by the 24-hour urine volume.
Saline challenge test.
In brief, following uni-nephrectomy and 7 days of Ang II infusion just as in the initial experiment, mice were anesthetized with isoflurane and then injected i.p. with a volume of warmed isotonic saline equivalent to 10% of their body weight23. Mice then awoke and were placed immediately in metabolic cages for urine collection over 4 hours. Urine volumes and sodium concentrations were quantitated, and results were tabulated as the percentage of the volume or sodium load originally injected.
Statistical analysis.
The values of each parameter within a group are expressed as the mean ± the standard error of the mean (SEM). For the blood pressure measurement experiment, comparisons between the groups were performed using the two-way ANOVA with repeated measures. For comparisons between two groups with normally distributed data, statistical significance was assessed using an unpaired student’ t-test. For comparisons between more than two groups with normally distributed variables, the one-way ANOVA was employed. p<0.05 was considered statistically significant. *, p<0.05. #, p<0.05. **, p<0.01. ***, p<0.005.
Results
Genetic deficiency of FLT3L limits numbers of classical DCs in the kidney.
Kidneys from Fms-like tyrosine kinase 3 ligand knockout (FLT3L−/−) mice and littermate controls (FLT3+/+= WT) were harvested and used for the flow cytometric analysis. As illustrated by our gating strategy (Figure 1A), cells were stained with fluorescently-labeled anti-CD45, anti-CD11c, anti-CD11b, and Anti-MHCII. Mature classical dendritic cells are marked by CD11chi expression plus MHCIIhi positivity in the kidney24. Kidneys from the FLT3L−/− animals in comparison to WT controls contained lower percentages of classical DCs as a proportion of CD11c- or CD11b-expressing myeloid cells (Figure 1C, 10.9 ± 0.6 vs. 2.8 ± 0.4 % of DCs, p<0.0001). Additionally, the absolute number of the CD11chiMHCIIhi cells were markedly reduced in the FLT3L−/− kidneys (Figure 1D, 2.2 ± 0.37 vs. 0.4 ± 0.04 ×104, p<0.005). By contrast, the numbers of CD3+ T cells, CD19+ B cells, and total CD11b+ and CD11c+ myeloid cell numbers were similar in the kidneys from the 2 groups. In the spleen, the numbers of each of these populations were reduced in the FLT3L−/− cohort, but only significantly so for the CD11c+ myeloid cells. Kidney, heart, and spleen weights were similar between the groups at baseline (Figure S1). These data confirm that FLT3L−/− mice have fewer classical DCs in the kidney.
Figure 1. Genetic deficiency of FLT3L reduces the classical DCs in the kidney.
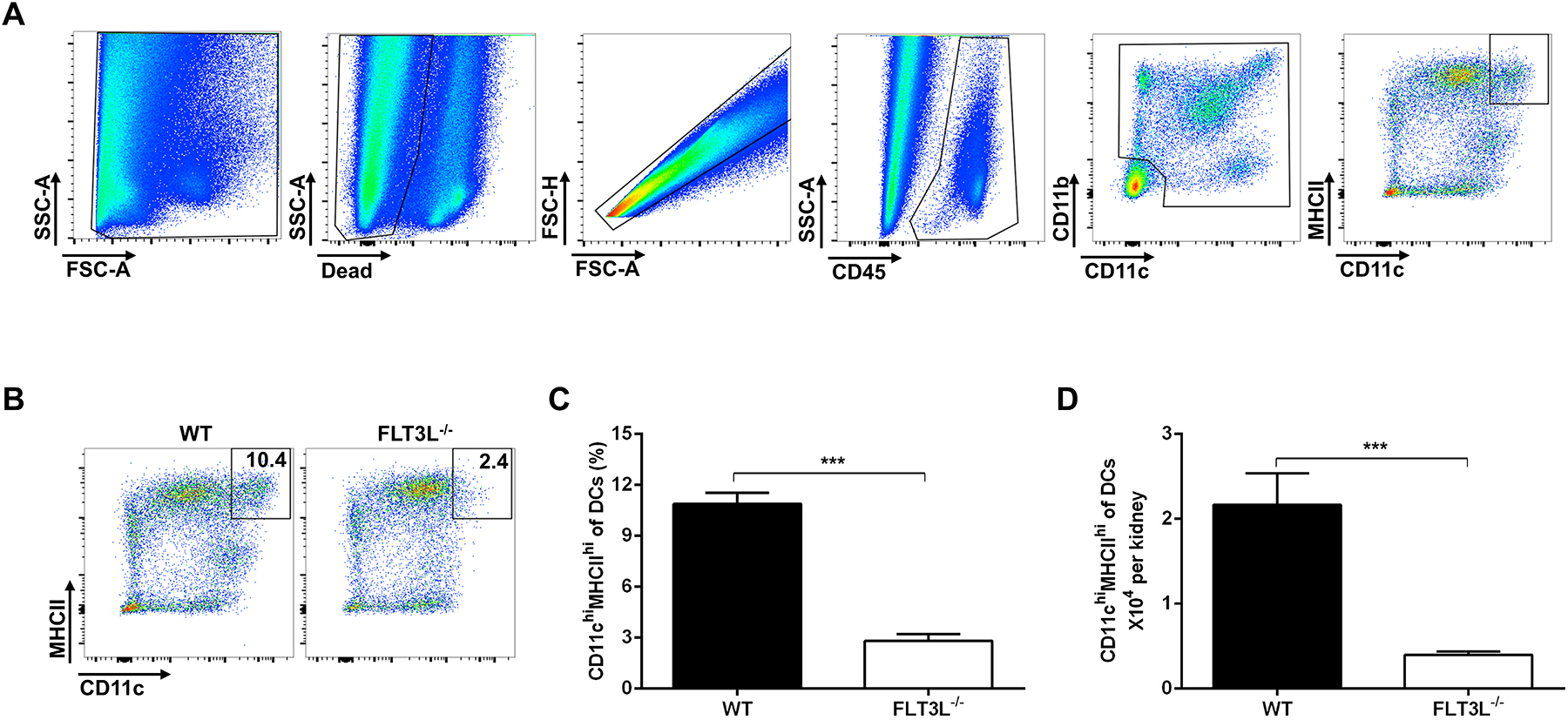
(A) In gating strategy, mature classical DCs were identified by high co-expression of CD11c and MHCII. (B) Representative flow plots of CD11chiMHCIIhi cells. (C) The proportions of classical DCs from wild-type (WT) and FLT3L−/− kidneys at baseline as a percent of CD45+/CD11c+ or CD11b+ myeloid cells. (D) The absolute number of classical DCs from WT and FLT3L−/− kidneys at baseline. N=4 mice/group. Data are mean ± SE.
Classical DCs mediate angiotensin (Ang) II-dependent hypertension.
FLT3L−/− mice and WT controls underwent unilateral nephrectomy to enhance salt sensitivity followed by implantation of pressure-sensing radiotelemetry catheters. Baseline blood pressures measured by telemetry were virtually identical in the WT and FLT3L−/− mice (Figure 2A, Figure S2). To induce hypertension, mice were infused subcutaneously with Ang II (500ng/kg/min) via osmotic mini-pump. During Ang II infusion, mean arterial blood pressures (MAPs) in the WT group rose approximately 30 mmHg and remained elevated throughout the infusion period. However, the elevation of blood pressure was blunted in the FLT3L−/− cohort compared to WT controls with significantly lower blood pressures covering the period from day 13 to day 21 of Ang II infusion (Figure 2A, 166 ± 2 vs.178 ± 4 mmHg at day 21, p<0.05). Consistent with these lower blood pressures, the FLT3L−/− mice had mitigated cardiac hypertrophy following 4 weeks of hypertension (Figure 2B, 6.67 ± 0.13 vs. 7.84 ± 0.36 mg heart weight/g body weight, p<0.05). Thus, FLT3-expressing classical DCs mediate Ang II-induced hypertension. Interestingly, circulating FLT3L levels were reduced in the WT group following chronic Ang II infusion (Figure S3).
Figure 2. Genetic deletion of classical DCs protects against the angiotensin (Ang) II-induced hypertension.

(A) Mean arterial pressures measured by radio-telemetry in WT mice (circles) and FLT3L−/− (squares) mice at baseline (“pre”) and during chronic Ang II infusion. ** p<0.005 for days 13–21 of Ang II. - (B) The ratio of heart weight/body weight (mg/g) in the WT and FLT3L−/− groups after 28 days of Ang II infusion. N=13–14 mice/group. Data are mean ± SE.
FLT3L deficiency decreases accumulation of activated T cells in the kidney during hypertension.
Naïve T cells are marked by CD62LhiCD44lo expression, whereas effector memory T cells exhibit a CD62LloCD44hi profile25. These effector memory T cells can have dramatic effects on blood pressure regulation19. Therefore, to quantify effector memory T cells in the kidney, we performed flow cytometric analysis on single cell suspensions from the kidney at the baseline and after Ang II infusion. As illustrated by our gating strategy (Figure 3A), cells were stained with fluorescently-labeled anti-CD45, anti-CD3, anti-CD4, Anti-CD8, Anti-CD62L, and Anti-CD44. We found that the proportions in the kidney of CD62LloCD44hi among all CD3+ T cells were similar in the groups at baseline (Figure 3B). By contrast, after Ang II infusion, this proportion was markedly increased in the WT cohorts and significant higher than in the FLT3L−/− kidneys (Figure 3B). Similarly, within both the CD4+ and CD8+ T cell subsets, the proportions of effector memory T cells, were not different between the WT and FLT3L−/− kidneys at baseline (Figure 3C–D), but were markedly increased after Ang II infusion only in the WT kidneys (Figure 3C–D). Co-culture studies confirmed the capacity of CD11c+ myeloid cells to drive the proliferation of effector memory T cells in vitro (Figure S4). Thus, classical DCs contribute to renal T cell activation during hypertension.
Figure 3. FLT3L enhances accumulation of effector memory T cells in the kidney during hypertension.

Flow cytometric analysis of single cell suspensions from the kidney harvested at baseline or after 4 weeks of Ang II infusion. (A) Gating strategy for parsing T cell populations in the kidney. (A) Proportions of the CD44hiCD62Llo T cells among CD3+ T lymphocytes from WT and FLT3L−/− groups. (B) Proportions of the CD44hiCD62Llo T cells among CD4+ T lymphocytes from WT and FLT3L−/− cohort. (C) Proportions of the CD44hiCD62Llo T cells among CD8+ T lymphocytes from WT and FLT3L−/− mice. N=5 mice/group. Data are mean ± SE.
FLT3L augments the induction of inflammatory mediators in the hypertensive kidney.
As pro-inflammatory cytokines such as IL-1β and TNF-α can contribute to blood pressure elevation through diverse actions in the kidney13, 26–28, we measured the renal mRNA expression of these cytokines in the kidneys of the Ang II-infused WT and FLT3L−/− mice. Consistent with the reduction of the effector memory T cells in the FLT3L−/− hypertensive kidney, renal expression of the T cell chemokine CCL5 was significantly lower in the FLT3L−/− cohort (Figure 4A). Similarly, mRNA levels of IL-1β and TNF-α were significantly lower in the FLT3L−/− kidneys than in WT controls (Figure 4B–C), whereas renal expression of IFN-γ was similar in the experimental groups (Figure S5). Thus, classical DCs contribute to the induction of pro-hypertensive cytokines in the kidney.
Figure 4. FLT3L enhances the expression inflammatory mediators in the hypertensive kidney.

At the end of the experiment (28 days), kidneys from WT and FLT3L−/− mice were harvested for real-time QPCR analysis. (A-C) mRNA levels for (A) CCL5, (B) IL-1β, and (C) TNF-α. N=5 mice/group. Data are mean ± SE.
FLT3L deficiency reduces oxidative stress in the hypertensive kidney.
T cells augment hypertension in part by inducing oxidative stress via the activation of NADPH oxidase7, 29. We therefore measured urinary excretion of 8-isoprostane as marker of oxidative stress in the kidneys of the WT and FLT3L−/− mice during hypertension. We found that the 24-hour excretion of 8-isoprostane was decreased in the FLT3L−/− group (Figure 5A). As NOX2 and NOX4 are the major isoforms of NADPH oxidase in the kidney, we also determined the renal expression of those two isoforms during Ang II-induced hypertension. By real-time qPCR analysis, the mRNA levels of NOX2 but not NOX4 were significently lower in the FLT3L−/− cohort (Figure 5B–C). Immunostaining localized the NOX2 protein primarily to interstitial cells and, to a lesser extent, the renal epithlium (Figure S6). These data suggest that classical DCs can promote blood pressure elevation by activating NOX2-derived oxidative stress in the kidney.
Figure 5. FLT3L deficiency attenuates oxidative stress in the hypertensive kidney.
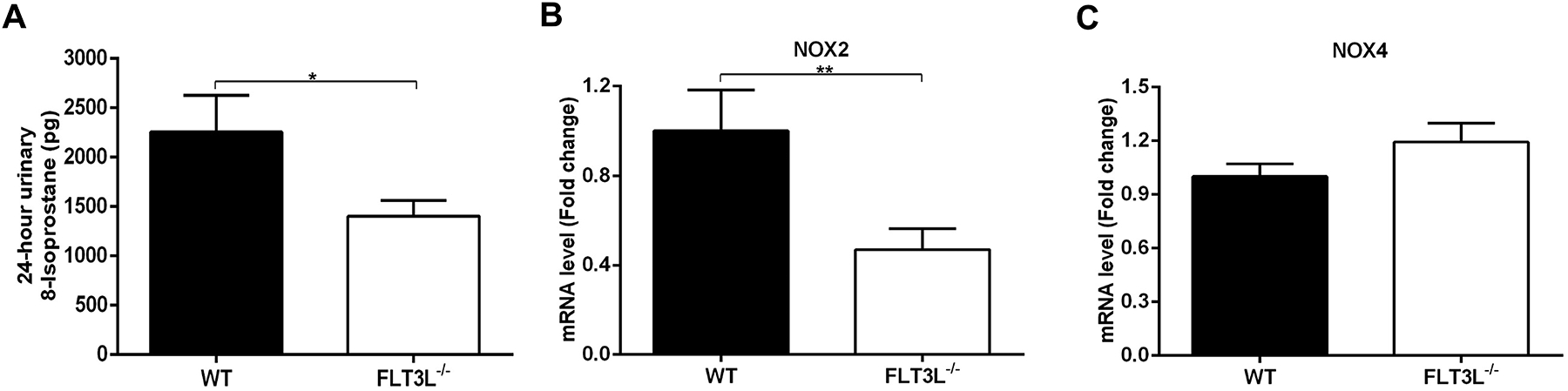
After 25 days of Ang II infusion, mice were placed in metabolic cages, and urine was collected over 24 hours for analysis. (A) 24-hour urinary 8-isoprostane. At the end of the experiment (28 days), kidneys from WT and FLT3L−/− mice were harvested for real-time QPCR analysis. (B-C) mRNA levels for (B) NOX2 and (C) NOX4. N≥5 mice/group. Data are mean ± SE.
Classical DCs contribute to volume and sodium retention during hypertension.
To assess the effects of classical DCs on RAS-dependent volume expansion, uni-nephrectomized WT and FLT3L−/− mice were challenged intraperitoneally at baseline or day 7 of Ang II with a saline volume equivalent of 10% of their body weight and placed in metabolic cages for urine collection23. The ratio of urine volume to the injected volume at each time point was quantitated as a measure of diuresis while the ratio of sodium amount to that injected at each time point was quantitated as a measure of natriuresis. At baseline, WT and FLT3L−/− mice excreted similar proportions of the injected volumes and sodium (Figure 6A–B). Following one week of Ang II, the WT group excreted lower proportions of the injected volume and sodium compared to the baseline condition, consistent with Ang II-induced volume and sodium retention. However, in comparison to the Ang II-infused WT cohort, the Ang II-infused FLT3L−/− animals had blunted volume and sodium excretions, suggesting a reduced tendency for Ang II-mediated volume and sodium retention (Figure 6A–B). Consistent with this finding, mRNA levels of sodium transporters including the three subunits of the epithelial sodium channel (α-ENaC, β-ENaC, γ-ENaC) and the sodium-chloride cotransporter (NCC) were significantly decreased in the FLT3L−/− kidneys compared to WT controls (Figure 6C). By contrast, the levels of sodium-hydrogen antiporters 3 (NHE3), and sodium-potassium-chloride cotransporter 2 (NKCC2) were similar in the two groups (Figure 6C). Thus, following RAS activation, deficiency of FLT3L limits volume expansion and sodium transporter expression, which can protect against blood pressure elevation.
Figure 6. Mice lacking classical DCs have less volume and sodium retention during hypertension.
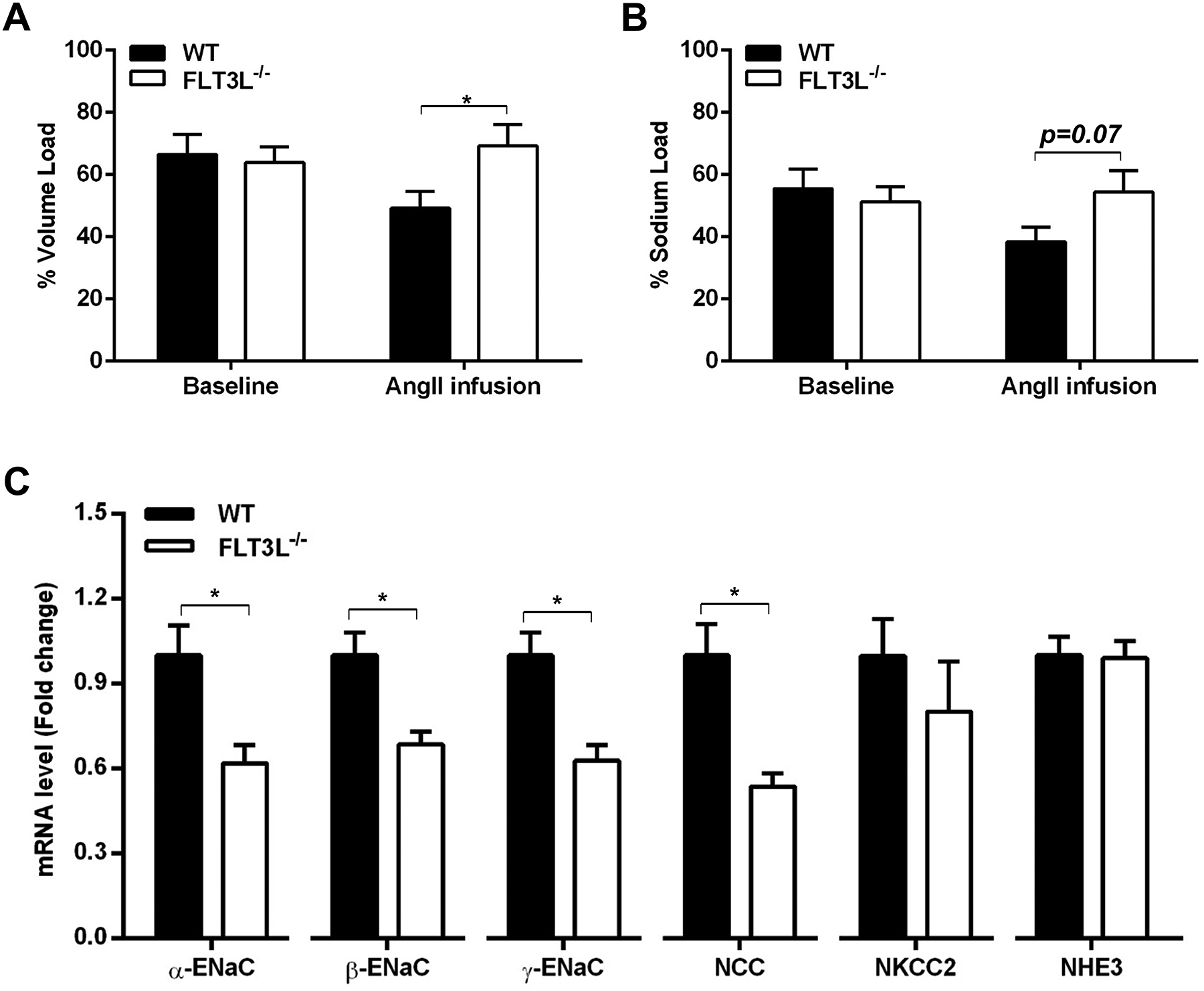
(A-B) At baseline and after 7 days of Ang II infusion, WT and FLT3L−/− mice were challenged with an i.p. bolus of saline equivalent to 10% of their body weights and placed in metabolic cages for 4 hours of urine collection. The ratios of urine volumes to injected volumes and the ratio of sodium to injected sodium are shown. N=9–10 mice/group. (C) After 4 weeks of Ang II infusion, renal mRNA levels of α-ENaC, β-ENaC, γ-ENaC, NCC, NKCC2, and NHE3 were determined by real-time QPCR. N=5 mice/group. Data are mean ± SE.
Discussion
As a key modifiable risk factor for cardiovascular and renal disease, hypertension causes one-third of deaths worldwide30. Inappropriate immune responses promote hypertension by acting on cardiovascular control centers including the heart, kidney, vasculature, and nervous system31. Activated dendritic cells (DCs) augment the hypertensive response by stimulating T lymphocytes to alter vascular and renal epithelial cell functions15, 16. Fms-like tyrosine kinase 3 ligand (FLT3L) is a growth factor that drives the development of classical DCs. Accordingly, in the current study, we find that mice lacking FLT3L have markedly fewer classical DCs in the kidney. In turn, these animals have blunted T cell activation in the kidney and an attenuated hypertensive response.
As the most potent antigen presenting cells in the body, DCs can invite the participation of the adaptive immune system by stimulating T lymphocytes. Interactions between DCs and T cells resulting in the generation of effector memory T cells, underpin a full hypertensive response18, 19. DCs are generated from common DC precursors that are then restricted to plasmacytoid DCs and classical DCs32. Classical DCs are identified by expression of FLT3 and high co-expression of CD11c and MHCII. Culture of bone marrow cells with the growth factor FLT3L can generate DCs subtypes that are functionally and phenotypically equivalent to mature DCs in vivo. FLT3L−/− mice therefore have significantly fewer classical DCs in the kidney than WTs, allowing us to use this murine mutant line to study the function of classical DCs in the development of RAS-dependent hypertension.
In our hypertension model, WT and FLT3L−/− mice have similar blood pressures at baseline indicating that classical DCs do not control normal blood pressure homeostasis. Nevertheless, during chronic Ang II infusion, FLT3L−/− mice have partially attenuated blood pressure elevation evident by the 2nd week of AngII infusion with correspondingly less cardiac hypertrophy. Thus, classical DCs are required to maintain the full hypertensive response following RAS activation rather than initiate blood pressure elevation. By contrast, Hevia et al. found that ablation of all CD11c-expressing myeloid cells prevents the onset of hypertension in response to Ang II infusion plus a high-salt diet17. Thus, our study elucidates a distinct role for FLT3-responsive classical DCs to sustain RAS-dependent hypertension. While we have not yet examined the actions of classical DCs in other models of hypertension, others have demonstrated the importance of DCs in multiple models of experimental hypertension15, 16.
Following their activation by DCs, T lymphocytes exacerbate the hypertensive response as seen in multiple experimental models7, 33. Upon activation, naïve T cells can downregulate their surface expression of CD62L and upregulate CD4425 to become effector memory T cells During hypertension, interactions between DCs and T cells lead to the accumulation of effector memory T cells in the vascular wall and in the kidney18, 19. Consistent with the result the similar blood pressures in the WT and FLT3L−/− groups at baseline, we found no baseline differences between the groups in the proportions of effector memory T cells in the kidney. Whereas the proportion of effector memory T cells in the WT kidney was dramatically increased following Ang II infusion, FLT3L deficiency mice prevented the increase in effector memory T cells. congruent with attenuated level of hypertension in this group. Thus, following RAS activation, classical DCs are required for the induction of renal effector memory T cells that play a key role in sustaining chronic hypertension.
Following activation by DCs, T cells can modulate blood pressure through the elaboration of inflammatory cytokines and oxidative stress, which then influence renal epithelial cell function and systemic vascular responses6, 7, 34–36. Among these cytokines, IL-1β and TNF-α, and other Interleukins have all been shown to promote hypertension through actions in the kidney. We found that RAS-dependent induction of mRNAs for IL-1β and TNF-α was diminished with FLT3L deficiency, consistent with a role for classical DCs to promote blood pressure elevation through the downstream release of pro-hypertensive cytokines. T cell activation can also augment hypertension by triggering oxidative stress in the kidney10, and our data indicate that FLT3L-dependent classical DCs promote oxidative stress in the kidney as measured by urinary excretion of 8-isoprostane. Whereas NADPH oxidase has seven isoforms, NOX2 and NOX4 are the dominant isoforms detected in the kidney. In the current study, we find that RAS-mediated upregulation of NOX2 but not NOX4 requires classical DCs. By contrast, the broader set of CD11c+ myeloid cells regulate blood pressure via the activation of both NOX2 and NOX417, suggesting that CD11c-expressing cells other than those induced by FLT3L may contribute to hypertension by activating the NOX4-derived oxidative stress pathway.
Renal generation of cytokines and oxidative stress can instigate blood pressure elevation by promoting renal fluid retention with consequent volume expansion per Ohm’s law6, 12, 13, 37–39. Consistent with that notion, we detected preserved diuresis and natriuresis in response to an IP saline challenge in our Ang II-infused FLT3L-deficient animals’ cohort compared to WT controls. Moreover, the FLT3L−/− hypertensive kidneys showed downregulated mRNA levels for multiple sodium channels, including the ENaC subunits and NCC, which should diminish the capacity for tubular sodium reabsorption. These findings are in agreement with previous studies that showing that induction of cytokines and oxidative stress in the kidney can regulate blood pressure via effects on the expression of renal sodium transporters. Nevertheless, in this study, we did not detect the differences between groups in NHE3 or NKCC2 mRNA expression, which suggest that classical DCs regulate sodium reabsorption in the distal nephron rather than the proximal tubule and thick ascending limb. While we speculate that sodium transport is reduced due to blunted T cell activation in the FLT3L−/− cohort, we acknowledge that our experiments do not explicitly demonstrate the mechanism through which FLT3L influences renal solute handling.
In sum, we find that FLT3L-dependent DCs mediate hypertension and enhance renal T cell activation and accumulation. With the accumulation of effector memory T cells in the kidney, local induction of pro-hypertensive cytokines and oxidative stress favor volume and sodium retention during RAS-mediated hypertension. These studies identify classical DCs as a specific DC subtype critical for sustained elevations in blood pressure elevation.
Perspectives
In the Ang II-induced hypertension model, we find that classical DCs mediate sustained blood pressure elevation and promote renal inflammation and oxidative stress with consequent fluid retention. FLT3L-deficient mice have less accumulation of effector memory T cells in the hypertensive kidney and reduced sodium retention during hypertension. Exploring the roles of specific populations of DCs involved in sustained hypertension should help us identify more effective treatment strategies for immune-mediated blood pressure elevation.
Supplementary Material
Novelty and Significance.
What is New?
FLT3L is required for a full chronic hypertensive response to Angiotensin (Ang) II
FLT3L facilitates the accumulation of effector memory T cells in the hypertensive kidney.
FLT3L-dependent dendritic cells can promote renal oxidative stress and salt and water retention.
What Is Relevant?
Dendritic cells are potent activators of T lymphocytes in the setting of hypertension.
Fms-like tyrosine kinase 3 ligand (FLT3L) stimulates the development of classical dendritic cells.
Stimulation of T cells by FLT3-expressing dendritic cells sustains hypertension following activation of the renin angiotensin system.
Acknowledgments
We thank Taylor Robinette for her technical and administrative assistance.
Sources of Funding
This work was supported by NIH grants DK118019, HL128355; Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development Grant BX000893; American Heart Association Grants 19POST34380480 and 18TPA34170047.
Footnotes
Disclosures
None
References
- 1.Harrison DG, Vinh A, Lob H, Madhur MS. Role of the adaptive immune system in hypertension. Curr Opin Pharmacol. 2010;10:203–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lu X, Crowley SD. Inflammation in salt-sensitive hypertension and renal damage. Curr Hypertens Rep. 2018;20:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rodriguez-Iturbe B, Pons H, Quiroz Y, Gordon K, Rincon J, Chavez M, Parra G, Herrera-Acosta J, Gomez-Garre D, Largo R, Egido J, Johnson RJ. Mycophenolate mofetil prevents salt-sensitive hypertension resulting from angiotensin ii exposure. Kidney Int. 2001;59:2222–2232 [DOI] [PubMed] [Google Scholar]
- 4.Mattson DL, James L, Berdan EA, Meister CJ. Immune suppression attenuates hypertension and renal disease in the dahl salt-sensitive rat. Hypertension. 2006;48:149–156 [DOI] [PubMed] [Google Scholar]
- 5.Xiao L, Kirabo A, Wu J, Saleh MA, Zhu L, Wang F, Takahashi T, Loperena R, Foss JD, Mernaugh RL, Chen W, Roberts J 2nd, Osborn JW, Itani HA, Harrison DG. Renal denervation prevents immune cell activation and renal inflammation in angiotensin ii-induced hypertension. Circulation research. 2015;117:547–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Norlander AE, Saleh MA, Kamat NV, Ko B, Gnecco J, Zhu L, Dale BL, Iwakura Y, Hoover RS, McDonough AA, Madhur MS. Interleukin-17a regulates renal sodium transporters and renal injury in angiotensin ii-induced hypertension. Hypertension. 2016;68:167–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the t cell in the genesis of angiotensin ii induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–2460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rudemiller N, Lund H, Jacob HJ, Geurts AM, Mattson DL, PhysGen Knockout P. Cd247 modulates blood pressure by altering t-lymphocyte infiltration in the kidney. Hypertension. 2014;63:559–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crowley SD, Song Y-S, Lin EE, Griffiths R, Kim H-S, Ruiz P. Lymphocyte responses exacerbate angiotensin ii-dependent hypertension. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1089–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Miguel C, Guo C, Lund H, Feng D, Mattson DL. Infiltrating t lymphocytes in the kidney increase oxidative stress and participate in the development of hypertension and renal disease. Am J Physiol Renal Physiol. 2011;300:F734–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mathis KW, Venegas-Pont M, Masterson CW, Stewart NJ, Wasson KL, Ryan MJ. Oxidative stress promotes hypertension and albuminuria during the autoimmune disease systemic lupus erythematosus. Hypertension. 2012;59:673–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li K, Guo D, Zhu H, Hering-Smith KS, Hamm LL, Ouyang J, Dong Y. Interleukin-6 stimulates epithelial sodium channels in mouse cortical collecting duct cells. Am J Physiol Regul Integr Comp Physiol. 2010;299:R590–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kamat NV, Thabet SR, Xiao L, Saleh MA, Kirabo A, Madhur MS, Delpire E, Harrison DG, McDonough AA. Renal transporter activation during angiotensin-ii hypertension is blunted in interferon-gamma−/− and interleukin-17a−/− mice. Hypertension. 2015;65:569–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hashmat S, Rudemiller N, Lund H, Abais-Battad JM, Van Why S, Mattson DL. Interleukin-6 inhibition attenuates hypertension and associated renal damage in dahl salt-sensitive rats. Am J Physiol Renal Physiol. 2016;311:F555–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kirabo A, Fontana V, de Faria APC, Loperena R, Galindo CL, Wu J, Bikineyeva AT, Dikalov S, Xiao L, Chen W, Saleh MA, Trott DW, Itani HA, Vinh A, Amarnath V, Amarnath K, Guzik TJ, Bernstein KE, Shen XZ, Shyr Y, Chen S-c, Mernaugh RL, Laffer CL, Elijovich F, Davies SS, Moreno H, Madhur MS, Roberts J 2nd, Harrison DG. Dc isoketal-modified proteins activate t cells and promote hypertension. The Journal of clinical investigation. 2014;124:4642–4656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barbaro NR, Foss JD, Kryshtal DO, Tsyba N, Kumaresan S, Xiao L, Mernaugh RL, Itani HA, Loperena R, Chen W, Dikalov S, Titze JM, Knollmann BC, Harrison DG, Kirabo A. Dendritic cell amiloride-sensitive channels mediate sodium-induced inflammation and hypertension. Cell Rep. 2017;21:1009–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hevia D, Araos P, Prado C, Fuentes Luppichini E, Rojas M, Alzamora R, Cifuentes-Araneda F, Gonzalez AA, Amador CA, Pacheco R, Michea L. Myeloid cd11c+ antigen-presenting cells ablation prevents hypertension in response to angiotensin ii plus high-salt diet. Hypertension. 2018;71:709–718 [DOI] [PubMed] [Google Scholar]
- 18.Vinh A, Chen W, Blinder Y, Weiss D, Taylor WR, Goronzy JJ, Weyand CM, Harrison DG, Guzik TJ. Inhibition and genetic ablation of the b7/cd28 t-cell costimulation axis prevents experimental hypertension. Circulation. 2010;122:2529–2537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Itani HA, Xiao L, Saleh MA, Wu J, Pilkinton MA, Dale BL, Barbaro NR, Foss JD, Kirabo A, Montaniel KR, Norlander AE, Chen W, Sato R, Navar LG, Mallal SA, Madhur MS, Bernstein KE, Harrison DG. Cd70 exacerbates blood pressure elevation and renal damage in response to repeated hypertensive stimuli. Circ Res. 2016;118:1233–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anandasabapathy N, Feder R, Mollah S, Tse S-W, Longhi MP, Mehandru S, Matos I, Cheong C, Ruane D, Brane L, Teixeira A, Dobrin J, Mizenina O, Park CG, Meredith M, Clausen BE, Nussenzweig MC, Steinman RM. Classical flt3l-dependent dendritic cells control immunity to protein vaccine. The Journal of experimental medicine. 2014;211:1875–1891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang JD, Patel MB, Song YS, Griffiths R, Burchette J, Ruiz P, Sparks MA, Yan M, Howell DN, Gomez JA, Spurney RF, Coffman TM, Crowley SD. A novel role for type 1 angiotensin receptors on t lymphocytes to limit target organ damage in hypertension. Circ Res. 2012;110:1604–1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang JD, Patel MB, Griffiths R, Dolber PC, Ruiz P, Sparks MA, Stegbauer J, Jin H, Gomez JA, Buckley AF, Lefler WS, Chen D, Crowley SD. Type 1 angiotensin receptors on macrophages ameliorate il-1 receptor-mediated kidney fibrosis. J Clin Invest. 2014;124:2198–2203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trott DW, Thabet SR, Kirabo A, Saleh MA, Itani H, Norlander AE, Wu J, Goldstein A, Arendshorst WJ, Madhur MS, Chen W, Li CI, Shyr Y, Harrison DG. Oligoclonal cd8+ t cells play a critical role in the development of hypertension. Hypertension. 2014;64:1108–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kawakami T, Lichtnekert J, Thompson LJ, Karna P, Bouabe H, Hohl TM, Heinecke JW, Ziegler SF, Nelson PJ, Duffield JS. Resident renal mononuclear phagocytes comprise five discrete populations with distinct phenotypes and functions. J Immunol. 2013;191:3358–3372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gerberick GF, Cruse LW, Miller CM, Sikorski EE, Ridder GM. Selective modulation of t cell memory markers cd62l and cd44 on murine draining lymph node cells following allergen and irritant treatment. Toxicol Appl Pharmacol. 1997;146:1–10 [DOI] [PubMed] [Google Scholar]
- 26.Dalekos GN, Elisaf M, Bairaktari E, Tsolas O, Siamopoulos KC. Increased serum levels of interleukin-1beta in the systemic circulation of patients with essential hypertension: Additional risk factor for atherogenesis in hypertensive patients? J Lab Clin Med. 1997;129:300–308 [DOI] [PubMed] [Google Scholar]
- 27.Bautista LE, Vera LM, Arenas IA, Gamarra G. Independent association between inflammatory markers (c-reactive protein, interleukin-6, and tnf-alpha) and essential hypertension. J Hum Hypertens. 2005;19:149–154 [DOI] [PubMed] [Google Scholar]
- 28.Sriramula S, Haque M, Majid DS, Francis J. Involvement of tumor necrosis factor-alpha in angiotensin ii-mediated effects on salt appetite, hypertension, and cardiac hypertrophy. Hypertension. 2008;51:1345–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Montezano AC, Touyz RM. Oxidative stress, noxs, and hypertension: Experimental evidence and clinical controversies. Ann Med. 2012;44 Suppl 1:S2–16 [DOI] [PubMed] [Google Scholar]
- 30.Lim SS, Vos T, Flaxman ADea. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: A systematic analysis for the global burden of disease study 2010. Lancet (London, England). 2012;380:2224–2260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coffman TM. Under pressure: The search for the essential mechanisms of hypertension. Nat Med. 2011;17:1402–1409 [DOI] [PubMed] [Google Scholar]
- 32.Naik SH, Sathe P, Park HY, Metcalf D, Proietto AI, Dakic A, Carotta S, O’Keeffe M, Bahlo M, Papenfuss A, Kwak JY, Wu L, Shortman K. Development of plasmacytoid and conventional dendritic cell subtypes from single precursor cells derived in vitro and in vivo. Nat Immunol. 2007;8:1217–1226 [DOI] [PubMed] [Google Scholar]
- 33.Rudemiller N, Lund H, Jacob HJ, Geurts AM, Mattson DL, PhysGen Knockout P. Cd247 modulates blood pressure by altering t-lymphocyte infiltration in the kidney. Hypertension. 2014;63:559–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De Miguel C, Guo C, Lund H, Feng D, Mattson DL. Infiltrating t lymphocytes in the kidney increase oxidative stress and participate in the development of hypertension and renal disease. Am J Physiol Renal Physiol. 2011;300:F734–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saleh MA, McMaster WG, Wu J, Norlander AE, Funt SA, Thabet SR, Kirabo A, Xiao L, Chen W, Itani HA, Michell D, Huan T, Zhang Y, Takaki S, Titze J, Levy D, Harrison DG, Madhur MS. Lymphocyte adaptor protein lnk deficiency exacerbates hypertension and end-organ inflammation. The Journal of clinical investigation. 2015;125:1189–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mattson DL. Infiltrating immune cells in the kidney in salt-sensitive hypertension and renal injury. Am J Physiol Renal Physiol. 2014;307:F499–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang J, Rudemiller NP, Patel MB, Karlovich NS, Wu M, McDonough AA, Griffiths R, Sparks MA, Jeffs AD, Crowley SD. Interleukin-1 receptor activation potentiates salt reabsorption in angiotensin ii-induced hypertension via the nkcc2 co-transporter in the nephron. Cell Metab. 2016;23:360–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jia Z, Aoyagi T, Yang T. Mpges-1 protects against doca-salt hypertension via inhibition of oxidative stress or stimulation of no/cgmp. Hypertension. 2010;55:539–546 [DOI] [PubMed] [Google Scholar]
- 39.Jonsson S, Becirovic-Agic M, Isackson H, Tveitaras MK, Skogstrand T, Narfstrom F, Karlsen TV, Liden A, Leh S, Ericsson M, Nilsson SK, Reed RK, Hultstrom M. Angiotensin ii and salt-induced decompensation in balb/cj mice is aggravated by fluid retention related to low oxidative stress. Am J Physiol Renal Physiol. 2019;316:F914–F933 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.