

Abstract
Chromosome 7q (Ch.7q) is clonally amplified in colorectal cancer (CRC). We aimed to identify oncogenes on Ch.7q that are overexpressed through DNA copy number amplification and determine the biological and clinical significance of these oncogenes in CRC. We identified general transcription factor 2I repeat domain‐containing protein 1 (GTF2IRD1) as a potential oncogene using a CRC dataset from The Cancer Genome Atlas with a bioinformatics approach. We measured the expression of GTF2IRD1 in 98 patients with CRC using immunohistochemistry and RT‐quantitative PCR (RT‐qPCR). The biological effects of GTF2IRD1 expression were explored by gene set enrichment analysis (GSEA). Next, we undertook in vitro cell proliferation and cell cycle assays using siGTF2IRD1‐transfected CRC cells. We further investigated the oncogenic mechanisms through which GTF2IRD1 promoted CRC progression. Finally, we assessed the clinical significance of GTF2IRD1 expression by RT‐qPCR. GTF2IRD1 was overexpressed in tumor cells and liver metastatic lesions. The GSEA revealed a positive correlation between GTF2IRD1 expression and cell cycle progression‐related genes. GTF2IRD1 knockdown inhibited cell proliferation and induced cell cycle arrest in Smad4‐mutated CRC. GTF2IRD1 downregulated the expression of the gene encoding transforming growth factor β receptor 2 (TGFβR2), a tumor‐suppressor gene in Smad4‐mutated CRC. On multivariate analysis, high GTF2IRD1 expression was an independent poor prognostic factor. Clinicopathological analysis showed that GTF2IRD1 expression was positively correlated with liver metastasis. In conclusion, GTF2IRD1 promoted CRC progression by downregulating TGFβR2 and could be a prognostic biomarker on Ch.7q in CRC. GTF2IRD1 could also be a novel oncogene in CRC.
Keywords: cell cycle, colorectal cancer, GTF2IRD1, oncogene, TGFβR2
GTF2IRD1 is identified as a driver gene on chromosome 7q of colorectal cancer (CRC). GTF2IRD1 promotes cell cycle progression by downregulation of TGFβR2, and GTF2IRD1 could be a novel oncogene in Smad4‐mutated CRC.
1. INTRODUCTION
Colorectal cancer (CRC) is a leading cause of tumor‐associated morbidity and mortality worldwide, and its incidence continues to rise.1, 2 Despite recent advances in therapeutic approaches, including chemotherapy and molecular targeted therapy, relapse is frequently found among patients with CRC, particularly for those with advanced disease.3
Intratumor heterogeneity (ITH), defined as molecular and cellular heterogeneity within a single tumor, is thought to cause the development of resistance against chemotherapy and leads to therapeutic failure because the presence of distinct subpopulations of cells with different sensitivities to chemotherapeutic drugs increases the risk of resistance and recurrence.4 Therefore, identification of oncogenes expressed in all tumor cells within a single tumor is essential for the discovery of promising therapeutic targets despite ITH.
Amplification of chromosome 7 is frequently found in CRC and colorectal adenoma tissue.5, 6, 7 Moreover, we showed that amplification of the long arm of chromosome 7 (Ch.7q) exists in all regions of a single tumor by undertaking multiregional copy number analysis of tumor tissues in CRC,8, 9 suggesting that amplification of Ch.7q is a fundamental and predominant event and that this region harbors oncogenes that affect the tumorigenesis of CRC.
We recently established a screening system using a bioinformatics approach with public datasets to identify candidate oncogenes in CRC. Using this system, we identified phosphoserine phosphatase (PSPH) and eIF5‐mimic protein 1 (5MP1) as potential oncogenes on Ch.7p in CRC.10, 11 Furthermore, we found that their expression was significantly associated with cell cycle progression genes and was an independent poor prognostic factor in patients with CRC. This system enables us to comprehensively search for oncogenes in CRC.
In this study, we aimed to identify novel potential oncogenes on Ch.7q in CRC using this screening system and to clarify the biological and clinical significance of the identified oncogenes in CRC.
2. MATERIALS AND METHODS
2.1. Selection of candidate genes
We obtained RNA sequencing data and DNA copy number data from 615 patients with CRC from The Cancer Genome Atlas (TCGA) from the Broad Institute’s Firehose (http://gdac.broadinstitute.org/runs/stddata__2015_08_21/data/COADREAD/20150821/). The RNA sequencing data also included expression profiles from 51 paired normal colon samples. Using these data, we extracted candidate genes from 819 genes on Ch.7q that showed positive correlations between DNA copy numbers and mRNA expression levels (cut‐off correlation coefficient, 0.4) (criteria 1) and showed overexpression in tumor tissues compared with normal tissues (more than 2‐fold change) (criteria 2).
2.2. Cell culture
Human CRC cell lines (SW620, COLO205, COLO320, DLD1, HCT116, and LoVo) were purchased from the cell bank at RIKEN BioResource Center (Tsukuba). SW620, COLO205, and DLD1 cells were maintained in RPMI‐1640. COLO320 and HCT116 cells were maintained in DMEM. LoVo cells were maintained in Ham’s F12 medium. All media contained 10% FBS with 100 U/mL penicillin and 100 U/mL streptomycin sulfate. All CRC cells were cultured in a humidified 5% CO2 incubator at 37°C.
2.3. Total RNA extraction and RT‐quantitative PCR
Total RNA from tissues and cell lines was extracted using a modified acid guanidinium thiocyanate‐phenol‐chloroform extraction (AGPC) method with Isogen (Nippon Gene). Reverse transcription was carried out using 8 µg total RNA with M‐MLV reverse transcriptase (Invitrogen), according to the manufacturer’s instructions. Quantitative PCR (qPCR) was undertaken using LightCycler FastStart DNA Master SYBR Green I (Roche Diagnostics) as previously described.12 The expression levels of general transcription factor 2I repeat domain‐containing protein 1 (GTF2IRD1) and transforming growth factor β receptor 2 (TGFβR2) mRNA were normalized to GAPDH mRNA as an internal control. Gene expression was presented as the values relative to the expression level of the cDNA from Human Universal Reference Total RNA (Clontech). The primer sequences for qPCR were as follows: GTF2IRD1, forward 5′‐GTGCCAGCCAAAGACAGCAG‐3′ and reverse 5′‐TGGCCATTGCACGAGTGAGA‐3′; TGFβR2, forward 5′‐TGGACCCTACTCTGTCTGTGGA‐3′ and reverse 5′‐CCCACTGCATTACAGCGAGAT‐3′; p21, forward 5′‐GCGACTGTGATGCGCTAATG‐3′ and reverse 5′‐
GAAGGTAGAGCTTGGGCAGG‐3′; and GAPDH, forward, 5′‐TTGGTATCGTGGAAGGACTCTA‐3′ and reverse, 5′‐TGTCATATTTGGCAGGTT‐3′.
2.4. Protein extraction
For total protein extraction, cells were lysed in lysis buffer (25 mmol/L Tris‐HCl [pH 7.5], 150 mmol/L NaCl, 0.2 mmol/L EDTA, 0.1% NP‐40, 5% glycerol, and proteinase inhibitor cocktail).
2.5. Immunohistochemical analysis
Immunohistochemistry for GTF2IRD1 in CRC cases with liver metastasis was carried out on formalin‐fixed, paraffin‐embedded tissues as previously described.13 The primary Ab against GTF2IRD was used at a dilution of 1:100. Rabbit polyclonal Abs to GTF2IRD were purchased from Sigma‐Aldrich. Tumor histology was independently reviewed by an experienced research pathologist (T.T.) at Kyushu University.
2.6. Immunoblotting analysis
Immunoblotting analysis was carried out as previously described.14 Briefly, equal amounts of protein (35 μg) were electrophoresed on 4%‐20% Tris‐glycine gels and then electroblotted onto Immobilon‐P Transfer Membranes (Merck Millipore) at 70 V for 4 hours at room temperature. Nonspecific binding sites were blocked with blocking buffer (TBS and 0.1% Tween‐20 with 5% nonfat milk powder) for 1 hour at room temperature, and the blots were incubated with specific primary Abs in blocking buffer (anti‐GTF2IRD1, anti‐TGFβR2, and anti‐phospho‐cyclin‐dependent kinase [CDK] 2 [Tyr15] Abs at a 1:250 dilution; anti‐phospho‐Smad2, anti‐Smad2, anti‐p21, anti‐phospho retinoblastoma (pRb), anti‐bone morphogenetic protein receptor type 1B (BMPR1b), and anti‐β‐actin Abs at a 1:1000 dilution) at 4°C overnight. After washing, the blots were incubated with an appropriate secondary Ab conjugated with HRP for 1 hour at room temperature. After washing, the detection was undertaken using an ImageQuant LAS 4000 Mini system (GE Healthcare Japan). Rabbit polyclonal Abs targeting GTF2IRD1 were purchased from Atlas Antibodies. Rabbit polyclonal Abs targeting TGFβR2 and mouse mAbs to β‐actin were purchased from Santa Cruz Biotechnology. Rabbit polyclonal Abs targeting phospho‐Smad2, Smad2, p21, and pRb were purchased from Cell Signaling Technology. Rabbit polyclonal Abs targeting BMPR1b were purchased from GeneTex. Rabbit polyclonal Abs targeting phospho‐CDK2 (Tyr15) were purchased from Abcam. Protein concentrations were quantified using Bradford protein assays.
2.7. GTF2IRD1 siRNA transfection
GTF2IRD1‐specific siRNA (Silencer Predesigned siRNA: sense CAUCGUCCAUGACAAGUCATT and antisense UGACUUGUCAUGGACGAUGGA) and negative control siRNA (Silencer Negative Control 1 siRNA) were purchased from Ambion. Small interfering RNA oligonucleotides were transfected into SW620 or COLO205 cells using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s instructions.
2.8. Colony formation assay
Cells (1000 cells/well) were seeded in 6‐well plates. After incubation for 24 hours followed by siRNA transfection, the cells were cultured for an additional 14 days, and the colonies were stained using a Differential Quik Stain Kit (Sysmex) according to the manufacturer’s instructions. Visible colonies were photographed using a Chemiluminescence Imaging FUSION SOLO S (VILBER). Colony counts were determined using ImageJ software (NIH).
2.9. Cell proliferation assay
Colorectal cancer cell proliferation was assessed using an MTT assay kit (Roche Applied Science) as previously described.15 After incubation for 24 hours, followed by siRNA transfection, the cells were cultured for an additional 0‐5 days, and the absorbance of the samples was measured.
2.10. Cell cycle assay
Nocodazole (an inhibitor of tubulin assembly; 5 μg/mL) was added 48 hours after siRNA transfection, and the cells were incubated for an additional 16 hours. Cells were harvested, washed with PBS, and fixed in ice‐cold 70% ethanol at −20°C overnight. Samples were then washed with PBS and stained with propidium iodide containing RNase A for 20 minutes at 37°C. Cell cycle distribution was measured by flow cytometry (Sony).
2.11. In vitro invasion assay
Cell invasion capacities were assessed using the BD BioCoat Tumor Invasion System, 24 Multiwell (BD Bioscience) as previously described.15
2.12. Patients with CRC and collection of clinical samples
Primary CRC samples and paired normal tissues were obtained from 98 patients who underwent surgery at Kyushu University Beppu Hospital and affiliated hospitals from 1992 to 2007. All patients had a histological diagnosis of CRC and were closely followed at 3‐month intervals. The median follow‐up period was 3.0 years. All patients were treated in accordance with the Japanese Society of Cancer of the Colon and Rectum Guidelines for the Treatment of Colorectal Cancer. Written informed consent was obtained from all patients, and the Institutional Review Board of our university approved this study. Sample collection was carried out as previously described.14 Data on patient age, sex, histology, tumor depth of invasion, lymph node metastasis, lymphatic invasion, venous invasion, liver metastasis, and clinical stage were obtained from clinical and pathological records.
2.13. Cancer Cell Line Encyclopedia data analysis
We obtained normalized mRNA expression data and DNA copy number data from 58 available CRC cell lines from the Cancer Cell Line Encyclopedia (CCLE) dataset (http://www.broadinstitute.org/ccle/home). Candidate gene mRNA expression and DNA copy number data were extracted from this reference.
2.14. Gene set enrichment analysis
The correlations between GTF2IRD1 expression and previously annotated gene expression signatures were analyzed by applying gene set enrichment analysis (GSEA).16 We acquired CRC expression profiles from the NCBI’s Gene Expression Omnibus database (accession code GSE7963) and analyzed the expression profiles using GSEA. Gene sets of GTF2IRD1 targets were extracted from C2 curated gene sets in the Broad Institute database (http://www.broadinstitute.org/gsea/msigdb/collections.jsp).
2.15. The Cancer Genome Atlas data analysis
Paired RNA sequencing and survival data of 620 available patients with CRC were obtained from TCGA (http://cancergenome.nih.gov/). GTF2IRD1 mRNA expression, Smad4 mutation status, and survival data were extracted from this reference.
2.16. Statistical analysis
For continuous variables, data are expressed as mean ± SD, and statistical analyses were carried out using Student’s t tests. The degree of linearity was estimated by Pearson’s correlation coefficient. Categorical variables were compared using χ2 tests or Fisher’s exact tests. Overall survival was estimated using the Kaplan‐Meier method, and survival curves were compared using log‐rank tests. Based on the levels of GTF2IRD1 mRNA expression in our dataset, cases were divided into 2 groups by the minimum P value approach, a comprehensive method to find the optimal risk separation cut‐off point in continuous gene expression measurement.17 Data analyses were undertaken using JMP 12 software (SAS Institute) and R software version 3.1.1 (The R Foundation for Statistical Computing).18 Clinicopathological factors and clinical stages were classified using the TNM system of classification.
3. RESULTS
3.1. GTF2IRD1 is a potential oncogene in CRC
We identified 8 genes that satisfied the criteria described above. Among the 8 genes, we focused on GTF2IRD1 (Figure 1A) because this gene has been reported to promote mammary tumor growth.19 GTF2IRD1 mRNA expression in tumor tissues was 4.57‐fold higher than that in normal tissues (P < .0005; Figure 1B). GTF2IRD1 mRNA expression and copy numbers were positively correlated in TCGA dataset (R = .48, P < .001; Figure 1C). Consistent with this result, there was a significant positive correlation between GTF2IRD1 mRNA expression and copy numbers in the CCLE dataset (R = .35, P < .05; Figure 1C). Next, we undertook immunohistochemical analysis to confirm GTF2IRD1 protein expression in CRC tumor cells from our hospital. GTF2IRD1 was stained more intensely in the nuclei and cytoplasm of CRC tumor cells than in those of normal colon cells (Figure 1D). Notably, GTF2IRD1 showed similar strong staining in tumor cells from liver metastatic lesions from the same patients (Figure 1D). Moreover, RT‐qPCR analysis also showed that GTF2IRD1 mRNA expression in tumor tissues was significantly higher than that in paired normal tissues (P < .0005; Figure 1B). Indeed, GTF2IRD1 mRNA expression levels in tumor tissues were higher than those in normal tissues in 91.3% of 98 patients with CRC. In addition, GSEA revealed a positive correlation between GTF2IRD1 mRNA expression and the expression of a gene set involved in cell cycle progression (P < .05; Figure 1E). These findings suggested that GTF2IRD1 was a novel oncogene in CRC.
Figure 1.
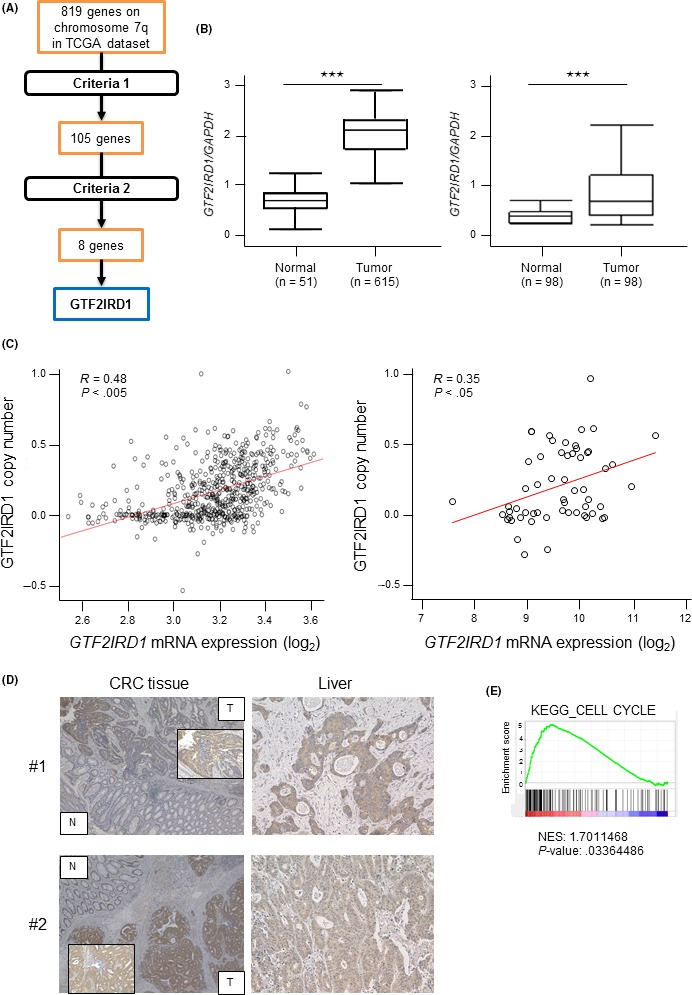
Identification of candidate oncogenes on chromosome 7q in colorectal cancer (CRC). A, Schematic diagram of the strategy for candidate oncogene selection. Criteria 1: Positive correlations between DNA copy numbers and mRNA expression levels (cut‐off correlation coefficient, 0.4). Criteria 2: Overexpressed in tumor tissues compared with normal tissues (>2‐fold change). B, Left, GTF2IRD1 mRNA expression between 615 CRC tissues and 51 normal colon tissues in The Cancer Genome Atlas (TCGA) dataset. Right, GTF2IRD1 mRNA expression in 98 CRC tissues and paired normal colon tissues in our dataset by RT‐quantitative PCR. ***P < .0005. C, Left, Correlation between GTF2IRD1 copy number and GTF2IRD1 mRNA expression in TCGA dataset. Right, Correlation between GTF2IRD1 copy number and GTF2IRD1 mRNA expression in the Cancer Cell Line Encyclopedia dataset. R, Pearson’s correlation coefficient. D, Immunohistochemical staining for GTF2IRD1 in normal colon and tumor tissues (left), and liver metastatic lesion tissues (right) in the same patients. Original magnification, 40× and 200×. E, Gene set enrichment analysis of the expression of GTF2IRD1 and cell cycle‐related genes using reference gene sets in the CRC dataset. KEGG, Kyoto Encyclopedia of Genes and Genomes; N, normal tissue; NES, Normalized Enrichment Score; T, tumor tissue
3.2. GTF2IRD1 promotes proliferation of CRC cells
The results of GSEA motivated us to investigate whether GTF2IRD1 regulated cell cycle progression and consequent tumor proliferation. Accordingly, RT‐qPCR analysis was undertaken to quantify GTF2IRD1 mRNA expression in several CRC cell lines. Endogenous GTF2IRD1 mRNA expression was higher in SW620 and COLO205 cells (Figure 2A). Therefore, SW620 and COLO205 cells were selected for subsequent experiments. To examine the biological roles of GTF2IRD1 in CRC, we carried out knockdown experiments using siRNA. siGTF2IRD1 induced significant downregulation of GTF2IRD1 mRNA expression in SW620 and COLO205 cells (Figure 2B). Immunoblotting analysis confirmed a substantial decrease in GTF2IRD1 protein in siGTF2IRD1‐transfected SW620 and COLO205 cells (Figure 2B).
Figure 2.
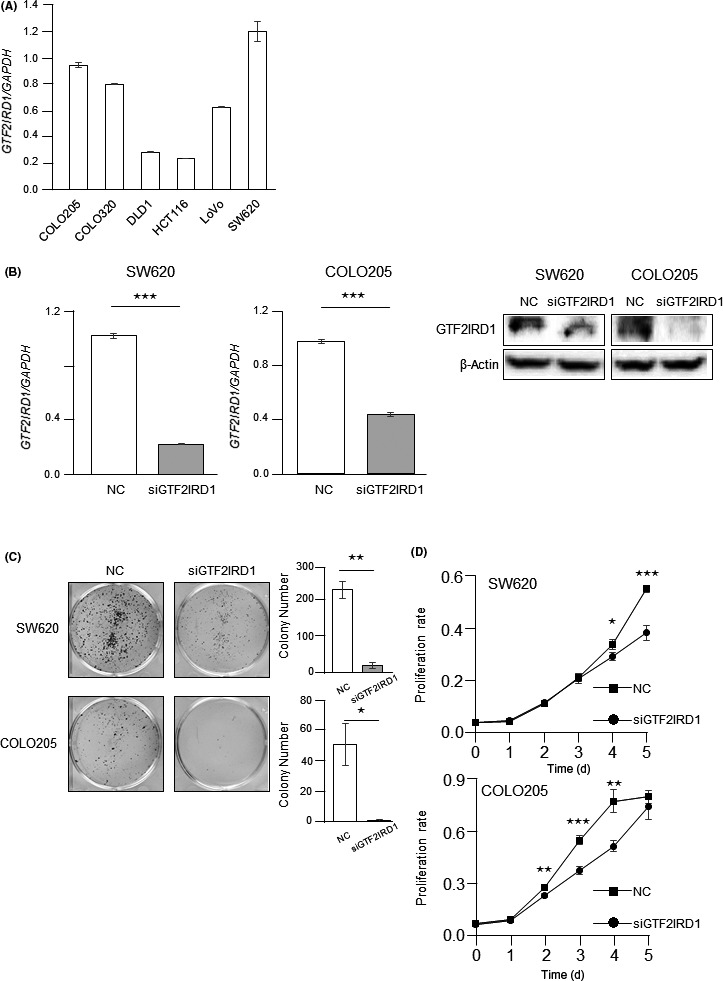
Effects of GTF2IRD1 knockdown on cell proliferation in colorectal cancer (CRC) cells. A, RT‐quantitative PCR (RT‐qPCR) analysis of GTF2IRD1 mRNA expression in 6 CRC cell lines. B, Left, RT‐qPCR for GTF2IRD1 mRNA expression in GTF2IRD1 siRNA‐transfected SW620 and COLO205 cells and control siRNA‐transfected cells. ***P < .0005. Right, Immunoblotting for total protein expression of GTF2IRD1 in GTF2IRD1 siRNA‐transfected SW620 and COLO205 cells and control siRNA‐transfected cells. C, Left, Colony formation assays. SW620 and COLO205 cells were cultured after GTF2IRD1 siRNA or control siRNA transfection for 14 d. Right, Total number of colonies. *P < .05, **P < .005. D, MTT proliferation assays. Proliferation rates of GTF2IRD1 siRNA‐transfected SW620 and COLO205 cells were compared with those of control siRNA‐transfected cells (NC). *P < .05, **P < .005, ***P < .0005
The effects of GTF2IRD1 on proliferation in CRC were examined by colony formation assays and MTT assays. GTF2IRD1 knockdown significantly reduced colony formation (Figure 2C). The MTT assays showed that GTF2IRD1 knockdown significantly inhibited cancer cell proliferation (Figure 2D). In contrast, GTF2IRD1 knockdown did not affect invasion potential (Figure S1).
3.3. GTF2IRD1 promoted cell cycle progression in CRC cells
Next, we undertook cell cycle analysis. GTF2IRD1 knockdown significantly decreased the S phase fraction (Figure 3A). Furthermore, there was a significant increase in the G1/S phase fraction in GTF2IRD1 knockdown cells in the presence of nocodazole, which prevents reentry of cells into G1 phase (Figure 3B). Immunoblotting analysis showed a substantial increase in phosphorylation of CDK2 at Tyr15 and decrease in phosphorylation of Rb, indicating G1/S arrest, in GTF2IRD1‐knockdown cells (Figure 3C). Taken together, these data indicated that GTF2IRD1 promoted cell proliferation by promoting cell cycle progression in CRC.
Figure 3.
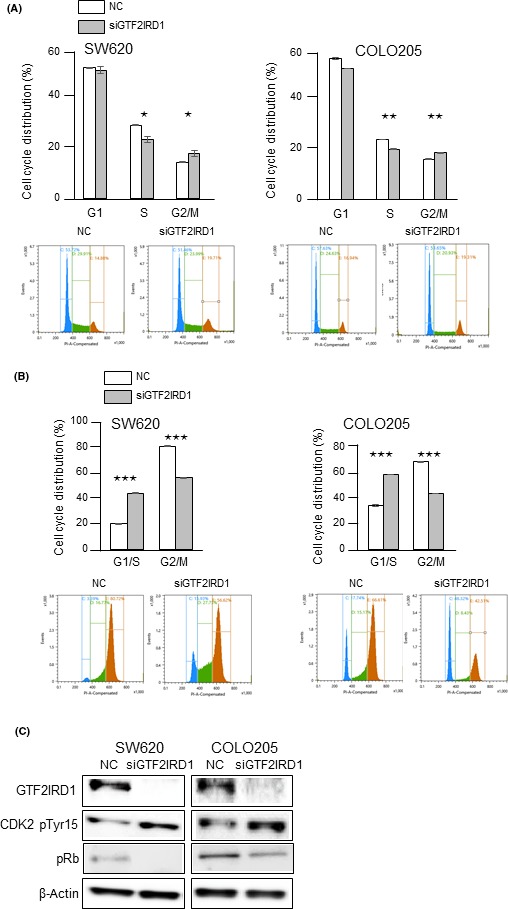
Effects of GTF2IRD1 knockdown on the cell cycle in colorectal cancer cells. A, Cell cycle distributions were analyzed by flow cytometry. *P < .05, **P < .005. B, Cell cycle distributions in the presence of nocodazole were analyzed by flow cytometry. ***P < .0005. C, Immunoblotting for phospho‐cyclin‐dependent kinase 2 (CDK2) (Tyr15) and pRb in GTF2IRD1 siRNA‐transfected SW620 and COLO205 cells and control siRNA‐transfected cells (NC)
3.4. GTF2IRD1 downregulates TGFβR2
Expression of TGFβR2, a well‐known tumor suppressor gene in CRC, is downregulated by GTF2IRD1 in mouse embryonic fibroblasts. 20, 21 We initially evaluated the expression of this gene using siGTF2IRD1‐transfected SW620 and COLO205 cells. TGFβR2 mRNA and protein expression levels were significantly higher in siGTF2IRD1‐transfected cells than in mock‐transfected cells (Figure 4A,B). To further test whether TGFβR2 was a downstream target of GTF2IRD1, we measured phospho‐Smad2 levels in GTF2IRD1‐knockdown cells. Immunoblotting analysis showed that phospho‐Smad2 levels were significantly higher in siGTF2IRD1‐transfected cells. Furthermore, there was a strong increase in p21 mRNA and protein expression (Figure 4A,B), which is upregulated by activation of TGFβ. These data indicated that GTF2IRD1 downregulated membrane TGFβR2 levels, resulting in reduced TGFβ activity.
Figure 4.
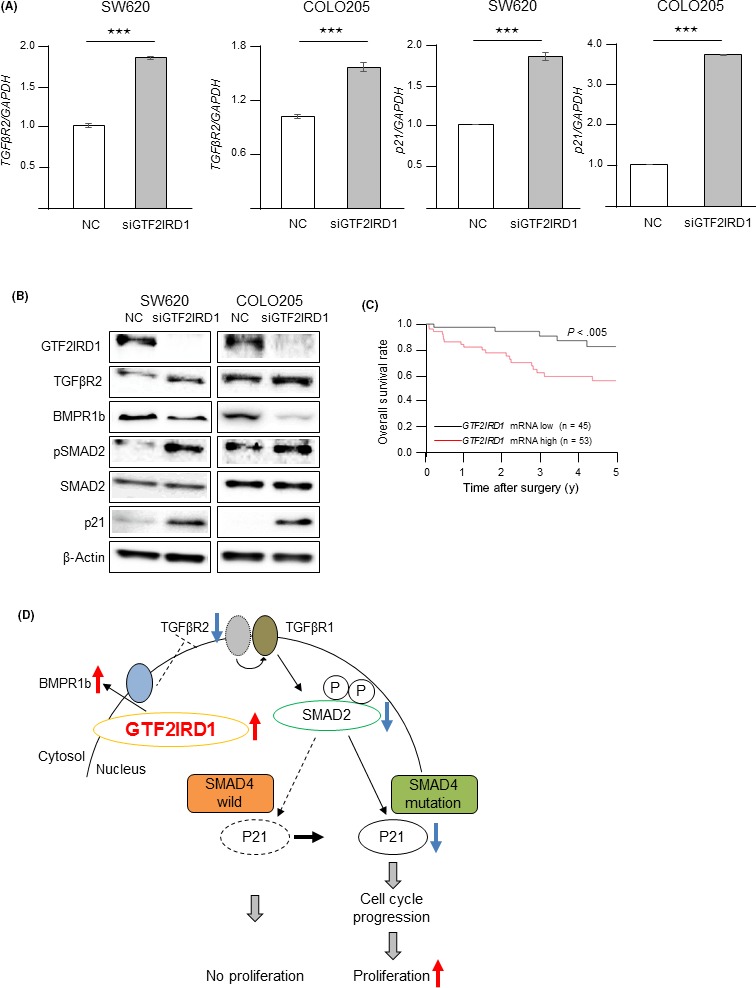
GTF2IRD1 downregulated the TGFβR2 gene in colorectal cancer (CRC) cells. A, RT‐quantitative PCR for TGFβR2 and p21 mRNA expression in GTF2IRD1 siRNA‐transfected SW620 and COLO205 cells and control siRNA‐transfected cells (NC). ***P < .0005. B, Immunoblotting for TGFβR2, BMPR1b, phospho‐SMAD2, SMAD2, and p21 protein expression in GTF2IRD1 siRNA‐transfected SW620 and COLO205 cells and control siRNA‐transfected cells. C, Overall survival rate in patients with CRC according to GTF2IRD1 mRNA expression in tumor tissues in our dataset. D, Schema indicating that overexpression of GTF2IRD1 promotes cell cycle progression and tumor proliferation
Bone morphogenetic protein (BMP) is a member of the TGFβ superfamily, and BMPR1b is a BMP signal receptor. It is well known that TGFβ and BMP signals play an opposing role in kidney disease22 and bone formation.23 These motivated us to assess the expression of BMPR1b by GTF2IRD1‐knockdown experiments. Levels of BMPR1b expression were significantly lower in siGTF2IRD1‐transfected cells than in mock‐transfected cells (Figure 4B). This result suggested that GTF2IRD1 upregulated membrane BMPR1b levels.
3.5. GTF2IRD1 promotes proliferation of Smad4‐mutated CRC cells
It is well known that the Smad signaling pathway is composed of the TGFβR2, TGFβR1, and Smad proteins. The cytoplasmic Smad2 and Smad3 proteins are phosphorylated by TGFβ signal through TGFβR2 and TGFβR1, which allows them to form a heteromeric complex with Smad4. This Smad complex is translocated into the nucleus and activates gene transcription, including p21. Our experimental data showed that GTF2IRD1 downregulated TGFβR2 expression, resulting in the downregulation of phospho‐Smad2 and p21 expression. However, SW620 and COLO205 harbor loss of function mutation of Smad4,24 indicating that the TGFβ signal pathway is inactivated in these cells. Thus, we undertook the cell proliferation assay and immunoblotting analysis using Smad4‐wild CRC cells, COLO320, whose endogenous GTF2IRD1 mRNA expression was near to SW620 and COLO205 cells (Figure 2A). Unexpectedly, GTF2IRD1 knockdown did not upregulate colony formation (Figure S2A). The MTT assays showed that GTF2IRD1 knockdown did not promote cancer cell proliferation (Figure S2B). The TGFβR2 and phospho‐Smad2 protein expression levels in Smad4‐wild cells was higher as well as Smad4‐mutated cells, but p21 protein expression levels were lower in siGTF2IRD1‐transfected WT cells than in mock‐transfected WT cells (Figure S2c), indicating that GTF2IRD1 downregulated TGFβR2 expression, resulting in reduced p21 expression in Smad4‐mutated CRC, but not in Smad4‐wild CRC.
3.6. High expression of GTF2IRD1 mRNA predicts poor prognosis in CRC patients
Because our experimental data suggested that GTF2IRD1 was associated with tumor aggressiveness in CRC, we assessed the prognostic and clinical significance of GTF2IRD1 mRNA expression in CRC. First, we evaluated the survival rates according to GTF2IRD1 mRNA expression in patients with CRC. The overall survival rate in patients in the high GTF2IRD1 mRNA expression group was significantly lower than that in patients in the low expression group in our dataset (P < .0005; Figure 4C). In the univariate analysis, poor histology, higher T factor (≥SE), lymph node metastasis, lymphovascular invasion, liver metastasis, and high GTF2IRD1 mRNA expression were significantly associated with a lower overall survival rate (Table 1). The multivariate analysis indicated that liver metastasis (P < .05) and high GTF2IRD1 mRNA expression (P < .05) were independent poor prognostic factors in CRC (Table 1). Also, the survival rates according to Smad4 mutation status in patients with CRC were examined using TCGA dataset because our experimental data suggested that GTF2IRD1 showed malignant potential in Smad4‐mutated CRC. As expected, the overall survival rate in patients in the high GTF2IRD1 mRNA expression group was significantly lower than that in patients in the low expression group in Smad4‐mutated CRC cases (Figure S3A). In Smad4‐wild CRC cases, there was no significant difference in the overall survival rate between high and low GTF2IRD1 mRNA expression group (Figure S3B). These results imply that GTF2IRD1 acts as an oncogene in the Smad4‐mutated CRC case, but not in the Smad4‐wild CRC case.
Table 1.
Univariate and multivariate analysis of clinicopathological factors affecting overall survival rate in colorectal cancer patients
| Factor | Univariate analysis | Multivariate analysis | ||||
|---|---|---|---|---|---|---|
| RR | 95% CI | P value | RR | 95% CI | P value | |
| Age, >65/≤65 years | 1.853 | 0.843‐4.178 | .124 | – | – | – |
| Sex, male/female | 2.027 | 0.856‐5.568 | .112 | – | – | – |
| Histology grade: well, moderate/others | 0.188 | 0.071‐0.651 | .012* | 0.275 | 0.091‐1.059 | .059 |
| T factor, ≤SS/≥SE | 0.193 | 0.078‐0.438 | <.001* | 0.512 | 0.181‐1.389 | .189 |
| Lymph node metastasis, absent/present | 0.189 | 0.069‐0.449 | <.001* | 0.581 | 0.187‐1.669 | .314 |
| Lymphatic invasion, absent/present | 0.349 | 0.154‐0.768 | .009* | 0.633 | 0.235‐1.703 | .362 |
| Venous invasion, absent/present | 0.225 | 0.101‐0.534 | .001* | 0.392 | 0.144‐1.072 | .068 |
| Liver metastasis, absent/present | 0.091 | 0.041‐0.219 | <.001* | 0.229 | 0.074‐0.691 | .009* |
| GTF2IRD1 mRNA expression, low/high | 0.269 | 0.089‐0.664 | .003* | 0.336 | 0.103‐0.914 | .032* |
Abbreviations: CI, confidence interval; RR, relative risk; SE, serosa; SS, subserosa.
Statistically significant.
3.7. Clinicopathological significance of GTF2IRD1 mRNA expression in CRC
Next, we examined the association between GTF2IRD1 mRNA expression and clinicopathological factors in patients with CRC from our hospital (Table 2). The high GTF2IRD1 mRNA expression group (n = 53) had a higher frequency of liver metastasis (P < .05) compared with the low expression group (n = 45), suggesting that CRC cells with high expression of GTF2IRD1 should grow faster than with the low expression at metastatic regions, including liver, resulting in early clinical detection of metastasis of GTF2IRD1‐high expressed CRC cells because GTF2IRD1 can facilitate cell proliferation of CRC. There were no significant associations between GTF2IRD1 mRNA expression and age, sex, poor histology, depth of invasion, lymph node metastasis, lymphatic invasion, venous invasion, or clinical stage.
Table 2.
Correlation between GTF2IRD1 mRNA expression of tumor tissues and clinicopathological factors in colorectal cancer
| Factors | Low (n = 45) | High (n = 53) | P value |
|---|---|---|---|
| Number (%) | Number (%) | ||
| Age (years) | |||
| <65 | 16 (35.6) | 24 (45.3) | .329 |
| ≥65 | 29 (64.4) | 29 (54.7) | |
| Sex | |||
| Male | 32 (71.1) | 31 (58.5) | .194 |
| Female | 13 (28.9) | 22 (41.5) | |
| Histology | |||
| Well/moderate | 42 (93.3) | 49 (92.4) | .866 |
| Others | 3 (6.7) | 4 (7.6) | |
| Depth of invasion | |||
| ≤SS | 28 (62.2) | 32 (60.4) | .852 |
| ≥SE | 17 (37.8) | 21 (39.6) | |
| Lymph node metastasis | |||
| Absent | 29 (64.4) | 27 (50.9) | .178 |
| Present | 16 (35.6) | 26 (49.1) | |
| Lymphatic invasion | |||
| Absent | 30 (66.7) | 31 (58.5) | .405 |
| Present | 15 (33.3) | 22 (41.5) | |
| Venous invasion | |||
| Absent | 38 (84.4) | 43 (81.1) | .666 |
| Present | 7 (15.6) | 10 (18.9) | |
| Liver metastasis | |||
| Absent | 44 (97.8) | 44 (83.0) | .016* |
| Present | 1 (2.2) | 9 (17.0) | |
| UICC TNM stage | |||
| Ⅰ, Ⅱ | 27 (60.0) | 25 (47.2) | .205 |
| Ⅲ, Ⅳ | 18 (40.0) | 28 (52.8) | |
Abbreviations: SE, serosa; SS, subserosa.
Statistically significant.
4. DISCUSSION
In this study, we identified GTF2IRD1 as a driver gene on Ch.7q of CRC using our bioinformatics approach and determined the biological and clinical significance of GTF2IRD1 expression in CRC. To the best of our knowledge, this is the first study to provide evidence that GTF2IRD1 might act as a novel oncogene in CRC.
GTF2IRD1 is a member of the GTF2I gene family, which encodes a set of multifunctional transcription factors. GTF2IRD1 is located in the Williams‐Beuren syndrome critical region 7q11.23.25, 26 This syndrome is a genetic disorder associated with multiple systemic abnormalities, including craniofacial dysmorphology and several symptoms, such as hypertension and anxiety.27 Additional studies have reported that there is a positive correlation between the expression of GTF2IRD1 and that of cell cycle progression‐related genes and genes involved in the noncanonical Wnt‐calcium pathway, which is known to modulate migration.28, 29 Consistent with these data, our GSEA revealed that GTF2IRD1 expression in CRC was positively correlated with cell cycle‐related gene sets, and in vitro analysis showed that GTF2IRD1 promoted cell cycle progression and consequently accelerated cell proliferation.
Our clinical study showed that GTF2IRD1 was overexpressed due to copy number amplification at Ch.7q in CRC. The expression of GTF2IRD1 was positively associated with the malignant pathological phenotype. Furthermore, high expression of GTF2IRD1 was an independent poor prognostic factor in CRC. Huo et al revealed that high GTF2IRD1 expression correlated with poor overall survival in patients with breast cancer, lung cancer, and ovarian cancer.19 These clinical findings strongly supported that GTF2IRD1 is a tumor‐promoting oncogene in human cancers, including CRC. Moreover, high GTF2IRD1 expression could be a novel biomarker of poor prognosis in patients with CRC.
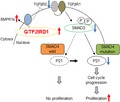
Next, we assessed the mechanisms through which GTF2IRD1 promoted CRC progression. We found that GTF2IRD1 downregulated TGFβR2 expression, resulting in reduced TGFβ activity in Smad4‐mutated CRC (Figure 4D). Our schema was strongly supported by the fact that suppression of TGFβ activity accelerated cell cycle progression and consequent cell proliferation.30, 31 Mutational inactivation of TGFβR2 occurs in approximately 30% of CRCs and promotes the formation of CRC by inhibiting the tumor‐suppressor activity of the TGFβ signaling pathway.21, 32 In a recent paper, GTF2IRD1 was reported to repress obesity‐associated adipose tissue fibrosis and improve systemic glucose homeostasis by suppressing the TGFβ signaling pathway.33
Loss of chromosome 18q21 including Smad4 has been detected in up to 60% of CRC, and Smad4 was reported to harbor loss of function mutation frequently in CRC,34, 35 suggesting that Smad4 is dysfunctional in the majority of CRC. Ijichi et al reported that p21 was upregulated through Smad2/3‐dependent transcriptional activation through a Smad4‐independent manner in Smad4‐null pancreatic cancer cells.36 This report suggested a novel mechanism of p21 regulation that Smad4 is not involved. In our study, p21 expression was downregulated in Smad4‐mutated CRC cells, but not in Smad4‐wild CRC cells, suggesting that GTF2IRD1 acts as an oncogene possibly in a Smad4‐independent manner in Smad4‐mutated CRC cases, but not in Smad4‐wild CRC cases. Survival analysis according to Smad4 mutation status supported our hypothesis described above. Our results and the reports suggested that downregulation of TGFβR2 could be an important pathway in Smad4‐mutated CRC formation and progression, but not in Smad4‐wild CRC, resulting in the difference of malignant phenotype between Smad4‐mutated and Smad4‐wild CRC. Further studies will be required to confirm this.
Recently, some reports have focused on the cross‐talk between TGFβ signal and the BMP signaling pathway in the progression of cancers. BMP7 attenuates TGFβ‐mediated tumor suppression in proliferation or invasion in prostate cancer or breast cancer respectively.37, 38 In our study, GTF2IRD1 downregulated TGFβR2, but upregulated BMPR1b expression in CRC cells, suggesting that the upregulation of BMPR1b by GTF2IRD1 might indirectly decrease TGFβR2 expression, resulting in suppression of TGFβ signal (Figure 4D).
Some limitations exist in this study. First, only COLO320 was available as Smad4‐wild CRC cells for in vitro analysis. Second, we could not identify the novel signal mediator regulating p21 instead of Smad4. Further in vitro and in vivo experiments are required to clarify these mechanisms.
In summary, our study identified GTF2IRD1 as a novel oncogene on Ch.7q and showed that GTF2IRD1 promoted cell cycle progression by downregulation of TGFβR2 in CRC. Furthermore, because overexpression of GTF2IRD1 due to copy number amplification of Ch.7q is considered to be a universal driver event present in all CRC cells, GTF2IRD1 could be a therapeutic target to overcome ITH in patients with CRC.
DISCLOSURE
No potential conflicts of interest were disclosed.
Supporting information
ACKNOWLEDGMENTS
This study used the super‐computing resource provided by the Human Genome Center, Institute of Medical Science, University of Tokyo (http://sc.hgc.jp/shirokane.html). We thank M. Oshiumi, M. Utou, K. Oda, M. Kasagi, S. Sakuma, N. Mishima, and T. Kawano for their excellent technical assistance. This work was supported in part by the following grants and foundations: Japan Society for the Promotion of Science (JSPS) Grant‐in‐Aid for Science Research (grant nos. JP16K07177, JP16K10543, JP16K19197, JP17K16454, JP17K16521, JP17K10593, and JP17K19608); OITA Cancer Research Foundation; Daiwa Securities Health Foundation; Grant‐in‐Aid for Scientific Research on Innovative Areas (grant no. 15H0912); Priority Issue on Post‐K computer (grant nos. hp170227, hp170227, and hp160219); JSPS KAKENHI (grant no. 15H05707); Eli Lilly Japan KK Grant; and the Japanese Foundation for Multidisciplinary Treatment of Cancer.
Nambara S, Masuda T, Kobayashi Y, et al. GTF2IRD1 on chromosome 7 is a novel oncogene regulating the tumor‐suppressor gene TGFβR2 in colorectal cancer. Cancer Sci. 2020;111:343–355. 10.1111/cas.14248
REFERENCES
- 1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69‐90. [DOI] [PubMed] [Google Scholar]
- 2. Takahashi Y, Sugimachi K, Yamamoto K, et al. Japanese genome‐wide association study identifies a significant colorectal cancer susceptibility locus at chromosome 10p14. Cancer Sci. 2017;108:2239‐2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. DeSantis CE, Lin CC, Mariotto AB, et al. Cancer treatment and survivorship statistics, 2014. CA Cancer J Clin. 2014;64:252‐271. [DOI] [PubMed] [Google Scholar]
- 4. McGranahan N, Swanton C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell. 2015;27:15‐26. [DOI] [PubMed] [Google Scholar]
- 5. Douglas EJ, Fiegler H, Rowan A, et al. Array comparative genomic hybridization analysis of colorectal cancer cell lines and primary carcinomas. Cancer Res. 2004;64:4817‐4825. [DOI] [PubMed] [Google Scholar]
- 6. Herbergs J, Arends JW, Bongers EM, Ramaekers FC, Hopman AH. Clonal origin of trisomy for chromosome 7 in the epithelial compartment of colon neoplasia. Genes Chromosomes Cancer. 1996;16:106‐112. [DOI] [PubMed] [Google Scholar]
- 7. Zarzour P, Boelen L, Luciani F, et al. Single nucleotide polymorphism array profiling identifies distinct chromosomal aberration patterns across colorectal adenomas and carcinomas. Genes Chromosomes Cancer. 2015;54:303‐314. [DOI] [PubMed] [Google Scholar]
- 8. Uchi R, Takahashi Y, Niida A, et al. Integrated multiregional analysis proposing a new model of colorectal cancer evolution. PLoS Genet. 2016;12:e1005778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Saito T, Niida A, Uchi R, et al. A temporal shift of the evolutionary principle shaping intratumor heterogeneity in colorectal cancer. Nat Commun. 2018;9:2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sato K, Masuda T, Hu Q, et al. Phosphoserine phosphatase is a novel prognostic biomarker on chromosome 7 in colorectal cancer. Anticancer Res. 2017;37:2365‐2371. [DOI] [PubMed] [Google Scholar]
- 11. Sato K, Masuda T, Hu Q, et al. Novel oncogene 5MP1 reprograms c‐Myc translation initiation to drive malignant phenotypes in colorectal cancer. EBioMedicine. 2019;44:387‐402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Masuda TA, Inoue H, Nishida K, et al. Cyclin‐dependent kinase 1 gene expression is associated with poor prognosis in gastric carcinoma. Clinical Cancer Res. 2003;9:5693‐5698. [PubMed] [Google Scholar]
- 13. Nambara S, Iguchi T, Oki E, Tan P, Maehara Y, Mimori K. Overexpression of CXCR7 is a novel prognostic indicator in gastric cancer. Dig Surg. 2017;34:312‐318. [DOI] [PubMed] [Google Scholar]
- 14. Hirata H, Sugimachi K, Komatsu H, et al. Decreased expression of fructose‐1,6‐bisphosphatase associates with glucose metabolism and tumor progression in hepatocellular carcinoma. Cancer Res. 2016;76:3265‐3276. [DOI] [PubMed] [Google Scholar]
- 15. Kurashige J, Hasegawa T, Niida A, et al. Integrated molecular profiling of human gastric cancer identifies DDR2 as a potential regulator of peritoneal dissemination. Sci Rep. 2016;6:22371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545‐15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mizuno H, Kitada K, Nakai K, Sarai A. PrognoScan: a new database for meta‐analysis of the prognostic value of genes. BMC Med Genomics. 2009;2:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tsutsumi S, Saeki H, Nakashima Y, et al. Programmed death‐ligand 1 expression at tumor invasive front is associated with epithelial‐mesenchymal transition and poor prognosis in esophageal squamous cell carcinoma. Cancer Sci. 2017;108:1119‐1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huo Y, Su T, Cai Q, Macara IG. An in vivo gain‐of‐function screen identifies the Williams‐Beuren syndrome gene GTF2IRD1 as a mammary tumor promoter. Cell Rep. 2016;15:2089‐2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chimge NO, Mungunsukh O, Ruddle F, Bayarsaihan D. Expression profiling of BEN regulated genes in mouse embryonic fibroblasts. J Exp Zool B Mol Dev Evol. 2007;308:209‐224. [DOI] [PubMed] [Google Scholar]
- 21. Biswas S, Chytil A, Washington K, et al. Transforming growth factor beta receptor type II inactivation promotes the establishment and progression of colon cancer. Cancer Res. 2004;64:4687‐4692. [DOI] [PubMed] [Google Scholar]
- 22. Meng XM, Chung AC, Lan HY, et al. Role of the TGF‐beta/BMP‐7/Smad pathways in renal diseases. Clin Sci. 2013;124:243‐254. [DOI] [PubMed] [Google Scholar]
- 23. Wu M, Chen G, Li Y, et al. TGF‐β and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res. 2016;4:16009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Woodford‐Richens KL, Rowan AJ, Gorman P, et al. SMAD4 mutations in colorectal cancer probably occur before chromosomal instability, but after divergence of the microsatellite instability pathway. Proc Natl Acad Sci USA. 2001;14:9719‐9723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Franke Y, Peoples RJ, Francke U. Identification of GTF2IRD1, a putative transcription factor within the Williams‐Beuren syndrome deletion at 7q11.23. Cytogenet Cell Genet. 1999;86:296‐304. [DOI] [PubMed] [Google Scholar]
- 26. Tassabehji M, Hammond P, Karmiloff‐Smith A, et al. GTF2IRD1 in craniofacial development of humans and mice. Science. 2005;310:1184‐1187. [DOI] [PubMed] [Google Scholar]
- 27. Game X, Panicker J, Fowler CJ. Williams‐Beuren syndrome. N Engl J Med. 2010;362:1449. [DOI] [PubMed] [Google Scholar]
- 28. Bikle DD, Xie Z, Tu CL. Calcium regulation of keratinocyte differentiation. Expert Rev Endocrinol Metab. 2012;7:461‐472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Corley SM, Canales CP, Carmona‐Mora P, et al. RNA‐Seq analysis of Gtf2ird1 knockout epidermal tissue provides potential insights into molecular mechanisms underpinning Williams‐Beuren syndrome. BMC Genom. 2016;17:450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Massague J. TGFbeta in cancer. Cell. 2008;134:215‐230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Colak S, Ten Dijke P. Targeting TGF‐beta signaling in cancer. Trends Cancer. 2017;3:56‐71. [DOI] [PubMed] [Google Scholar]
- 32. Biswas S, Trobridge P, Romero‐Gallo J, et al. Mutational inactivation of TGFBR2 in microsatellite unstable colon cancer arises from the cooperation of genomic instability and the clonal outgrowth of transforming growth factor beta resistant cells. Genes Chromosomes Cancer. 2008;47:95‐106. [DOI] [PubMed] [Google Scholar]
- 33. Hasegawa Y, Ikeda K, Chen Y, et al. Repression of adipose tissue fibrosis through a PRDM16‐GTF2IRD1 complex improves systemic glucose homeostasis. Cell Metab. 2018;27:180‐194.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Thiagalingam S, Lengauer C, Leach FS, et al. Evaluation of candidate tumour suppressor genes on chromosome 18 in colorectal cancers. Nat Genet. 1996;13:343‐346. [DOI] [PubMed] [Google Scholar]
- 35. The Cancer Genome Atlas Network . Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330‐337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ijichi H, Otsuka M, Tateishi K, et al. Smad4‐independent regulation of p21/WAF1 by transforming growth factor‐beta. Oncogene. 2004;23:1043‐1051. [DOI] [PubMed] [Google Scholar]
- 37. Yang S, Zhong C, Frenkel B, et al. Diverse biological effect and Smad signaling of bone morphogenetic protein 7 in prostate tumor cells. Cancer Res. 2005;65:5769‐5777. [DOI] [PubMed] [Google Scholar]
- 38. Naber HP, Wiercinska E, Pardali E, et al. BMP‐7 inhibits TGF‐beta‐induced invasion of breast cancer cells through inhibition of integrin beta(3) expression. Cell Oncol. 2012;35:19‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials