

Abstract
Mutations in the X‐linked MECP2 gene are responsible for Rett syndrome (RTT), a severe neurological disorder for which there is no treatment. Several studies have linked the loss of MeCP2 function to alterations of brain‐derived neurotrophic factor (BDNF) levels, but non‐specific overexpression of BDNF only partially improves the phenotype of Mecp2‐deficient mice. We and others have previously shown that huntingtin (HTT) scaffolds molecular motor complexes, transports BDNF‐containing vesicles, and is under‐expressed in Mecp2 knockout brains. Here, we demonstrate that promoting HTT phosphorylation at Ser421, either by a phospho‐mimetic mutation or inhibition of the phosphatase calcineurin, restores endogenous BDNF axonal transport in vitro in the corticostriatal pathway, increases striatal BDNF availability and synaptic connectivity in vivo, and improves the phenotype and the survival of Mecp2 knockout mice—even though treatments were initiated only after the mice had already developed symptoms. Stimulation of endogenous cellular pathways may thus be a promising approach for the treatment of RTT patients.
Keywords: Mecp2, Rett, BDNF, huntingtin, axonal transport
Subject Categories: Genetics, Gene Therapy & Genetic Disease; Neuroscience; Chemical Biology
BDNF is one of the most prominent deregulated factors in Rett syndrome, a severe neurodevelopmental disorder. This study reveals that huntingtin phosphorylation stimulates BDNF axonal transport and improves many features in a Rett mouse model, suggesting a possible therapeutic approach.
Introduction
MeCP2 (Methyl‐CpG‐binding protein 2) is one of the most abundant proteins in the brain, yet the precise nature of its activities remains controversial. It was originally discovered as a DNA methylation‐dependent transcriptional repressor (Meehan et al, 1992), but has also been shown to play various roles in chromatin compaction, global gene expression, alternative splicing, and miRNA processing (Young et al, 2005; Chahrour et al, 2008; Skene et al, 2010; Cheng et al, 2014). Interest in this protein rose sharply after it was discovered that mutations in MECP2 cause Rett syndrome (RTT), a severe developmental disorder (Amir et al, 1999; Lyst & Bird, 2015). Females with RTT begin life apparently healthy, but between 6 and 18 months of age they undergo regression of early milestones, with deterioration of motor skills, eye contact, speech, and motor control; they then develop a range of neurological symptoms, including anxiety, respiratory dysrhythmias, and seizures (Lyst & Bird, 2015; Katz et al, 2016). Whereas loss of MeCP2 function leads to RTT, duplication or triplication of the locus leads to intellectual disability, autistic features, and motor dysfunction, as observed in males with MECP2 duplication syndrome (Van Esch, 2012).
Disease‐causing mutations in MECP2 alter the expression of thousands of genes (Chahrour et al, 2008). Among these, brain‐derived neurotrophic factor (BDNF), a neuronal modulator that plays a key role in neuronal survival, development, and plasticity (Cheng et al, 2011), is one of the best studied (Chen et al, 2003, 2015; Chang et al, 2006; Sampathkumar et al, 2016). BDNF appears to be involved in the appearance and progression of the RTT phenotype in mice: Mecp2 knockout mice (KO) present lower BDNF levels, and conditional BDNF deletion in Mecp2 KO mice accelerates the onset of RTT‐like symptoms (Chang et al, 2006). Conversely, conditional BDNF overexpression in the brain of Mecp2 knockout mice leads to an improvement of certain locomotor and electrophysiological deficits (Chang et al, 2006). Although it is possible that the incomplete rescue reflects that MeCP2 has much broader regulatory effects than BDNF, it is possible that BDNF overexpression fails to restore synaptic and neuronal function because it does not target the appropriate neurons. In support of this hypothesis, recent evidence suggests that MeCP2 deficiency leads to the disruption of cell‐autonomous and autocrine BDNF signaling in excitatory glutamatergic neurons, and that increasing BDNF levels in diseased neurons restores their growth and ability to form synapses (Sampathkumar et al, 2016).
We have previously shown that BDNF homeostasis and transport involve huntingtin (HTT) and HTT‐associated protein 1 (Hap1) (Roux et al, 2012; Saudou & Humbert, 2016). Levels of both these proteins are lower than normal in excitatory cortical neurons deficient for Mecp2 (Roux et al, 2012). Here, we tested whether activating HTT, by a genetic or pharmacological approach, could improve BDNF homeostasis in Mecp2‐deficient neuronal circuits and in Mecp2 KO mice.
Results
BDNF transport is slowed in Mecp2‐deficient axons
To examine BDNF transport in Mecp2‐deficient neurons, we took advantage of the recent development and validation of microfluidic devices to reconstitute a neuronal network and monitor intracellular dynamics (Taylor et al, 2010; Virlogeux et al, 2018). We focused here on the corticostriatal network, which is altered in RTT and Mecp2 KO mice (Roux et al, 2012; Xu et al, 2014). The device consists of a presynaptic compartment containing cortical neurons, a postsynaptic chamber containing striatal neurons, and a synaptic chamber containing corticostriatal contacts. Cortical neurons plated in the first compartment extend axons that connect to striatal dendrites within the synaptic compartment (Fig 1A), thus creating an oriented corticostriatal network on‐a‐chip (Virlogeux et al, 2018). This network reproduces the physiological network as we previously showed that cortical neurons from embryonic day (E)15.5 are enriched in CTIP2/TBR1 neurons that correspond to the deepest layers of the cortex that send axons to the striatum. Also, most striatal neurons correspond to enkephalin‐positive neurons that are the output‐projecting neurons of the striatum (Virlogeux et al, 2018). Cortical neurons were transduced with mCherry‐tagged BDNF lentivirus (BDNF‐mCherry), and we used spinning disk confocal videomicroscopy to record the dynamics of BDNF‐mCherry containing vesicles within microchannels in the axons (Fig 1A and Movie EV1). The dynamics of vesicles are in agreement with our recent studies using this technology (Moutaux et al, 2018; Virlogeux et al, 2018).
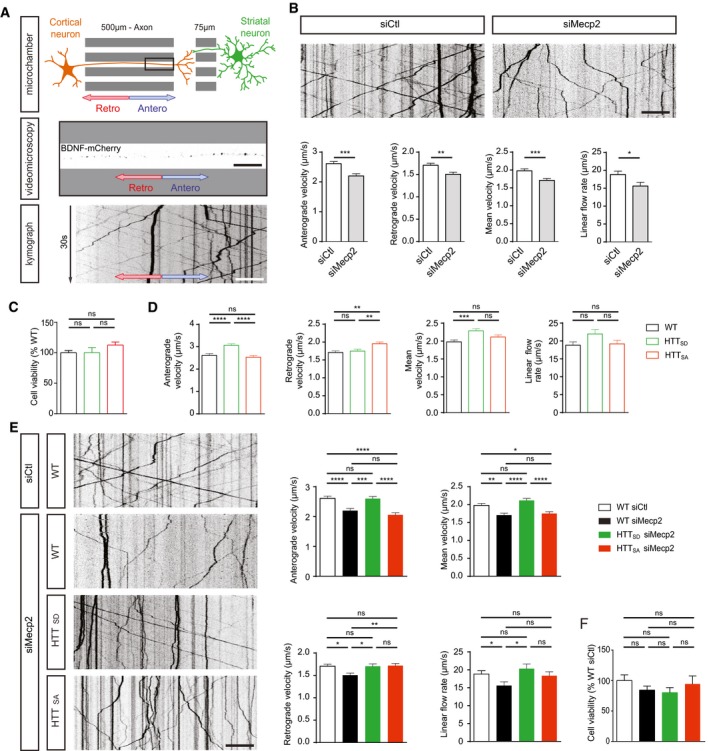
Figure 1. Huntingtin phosphorylation rescues BDNF transport in Mecp2‐deficient axons.
-
AMicrochamber that allows the isolation of axons within a corticostriatal network and live‐cell imaging of axonal BDNF‐mCherry transduced into mouse primary cortical neurons.
-
BRepresentative kymographs and quantification of anterograde, retrograde, mean velocity, and linear flow rate of BDNF‐mCherry‐containing vesicles in cortical neurons transfected with siMecp2 (n = 753 vesicles/81 axons) or siControl (siCtl) (n = 894 vesicles/94 axons), inducing significant silencing of Mecp2 (3 independent experiments; unpaired t‐test).
-
CQuantification of anterograde, retrograde, mean velocity, and linear flow rate of BDNF‐mCherry trafficking into cortical neurons obtained from WT (n = 753 vesicles/81 axons), HTTSA (n = 812 vesicles/81 axons), or HTTSD (n = 787 vesicles/82 axons) homozygous knock‐in mice in which S421 of HTT was replaced by an alanine (HTT S421A/S421A or HTTSA), mimicking the absence of phosphorylation, or by an aspartic acid (HTT S421D/S421D or HTTSD), mimicking constitutive phosphorylation (3 independent experiments; one‐way ANOVA test with Tukey's multiple comparisons).
-
DCell viability in WT (n = 3), HTTSA (n = 1), or HTTSD (n = 2) neurons measured by MTT assay. No differences were observed between the different groups (three independent experiments; one‐way ANOVA Kruskal–Wallis test, P = 0.5701)
-
EKymographs and quantification of anterograde, retrograde, mean velocity, and linear flow rate of BDNF‐mCherry axonal trafficking into WT (n = 894 vesicles/94 axons), HTTSA (n = 812 vesicles/82 axons), or HTTSD (n = 787 vesicles/94 axons) cortical neurons transfected with siCtl or siMecp2, which significantly silenced Mecp2 (three independent experiments; one‐way ANOVA with Tukey's multiple comparisons).
-
FCell viability in WT, HTTSA, or HTTSD neurons transfected with siCtl or siMecp2 measured by MTT assay (WT siCtl: n = 3; WT siMecp2: n = 3; HTTSD siMecp2: n = 3; HTTSA siMecp2: n = 2). No differences were observed between the different groups (three independent experiments; one‐way ANOVA Kruskal–Wallis test, P = 0.5701).
We transfected neurons with Mecp2 siRNA, which reduced Mecp2 protein levels by 63% compared to WT (Fig EV1A). This reduced the mean velocity and overall linear flow of BDNF vesicles reaching the corticostriatal contacts (Fig 1B). The number of moving vesicles was unchanged (Fig EV1B).
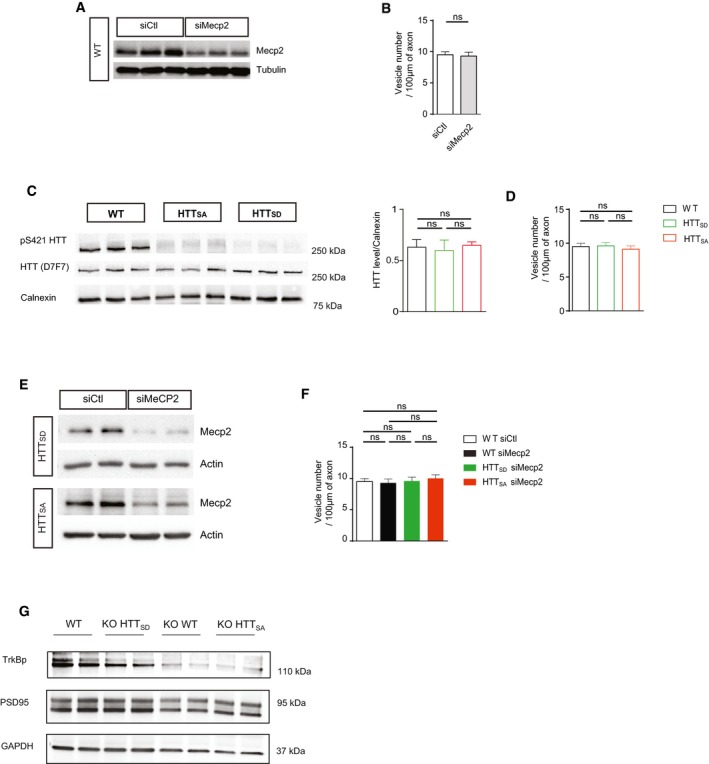
Figure EV1. Mecp2 levels and axonal vesicle number in cortical Mecp2‐silenced neurons.
-
AWestern blot analysis of DIV 5 cortical neurons transfected with siMecp2 or siControl (siCtl) to assess Mecp2 protein levels.
-
BWe quantified the number of BDNF‐mCherry‐containing vesicles trafficking within cortical axons transfected with siMecp2 or siControl (siCtl). Data are presented as the mean ± SEM of at least three independent experiments, with 94 siCtl and 81 siMecp2 axons and 894 siCtl and 753 siMecp2 vesicles (unpaired t‐test).
-
CBrain extracts from WT, WT HTTSD, and WT HTTSA mice were analyzed by Western blotting for total HTT and phospho‐HTT. Quantification of n = 3 samples per genotype did not reveal any significant differences on total HTT. The mutation of HTT S421 into alanine or aspartic acid abrogated the recognition of phospho‐HTT by the phospho‐specific antibody, further demonstrating the specificity of HTT phosphorylation at S421. Data are presented as means ± SEM, and one‐way ANOVA test followed by Tukey's post‐hoc analysis for multiple comparisons.
-
DWe quantified the number of BDNF‐mCherry‐containing vesicles trafficking within WT, HTTSA, or HTTSD cortical axons. Data are presented as the mean ± SEM of at least three independent experiments, with 80 axons and 700 vesicles per condition (one‐way ANOVA test followed by Tukey's post‐hoc analysis for multiple comparisons).
-
EWestern blot analysis of DIV 5 HTTSA or HTTSD cortical neurons transfected with siMecp2 or siControl (siCtl) to assess Mecp2 protein levels.
-
FQuantification of the number of BDNF‐mCherry‐containing vesicles trafficking within WT, HTTSA, or HTTSD cortical axons transfected with siMecp2 or siControl (siCtl). Data are presented as the mean ± SEM of at least three independent experiments, with WT (n = 753 vesicles/81 axons), HTTSA (n = 812 vesicles/81 axons), or HTTSD (n = 787 vesicles/82 axons) (one‐way ANOVA test followed by Tukey's post‐hoc analysis for multiple comparisons). ns = not significant.
-
GRepresentative Western blot analysis of PSD‐95 and phospho‐TrkB protein levels in striatum samples from WT, KO HTTSD, KO WT, and KO HTTSA mice.
Source data are available online for this figure.
HTT phosphorylation status influences anterograde and retrograde BDNF transport
Overexpression of phosphorylated HTT at S421 leads to increased outward transport in cells (Colin et al, 2008), but the role of endogenous HTT phosphorylation in axons remains unknown. We isolated cortical neurons from homozygous knock‐in mice in which S421 of HTT was replaced by an alanine (HTT S421A/S421A or HTTSA), mimicking the absence of phosphorylation, or by an aspartic acid (HTT S421D/S421D or HTTSD), mimicking constitutive phosphorylation (Thion et al, 2015). Mutating HTT at S421 abrogated the capacity of our phospho‐HTT specific antibody to recognize endogenous phosphorylated protein (Fig EV1C). Importantly, the S into A or into D mutations had no effect on HTT protein levels, as quantification showed no difference in HTT protein expression between WT, HTTSD, and HTTSA mice (Fig EV1C). Also, we found that the mutations had no effect on cell viability, as shown by the MTT assay (Fig 1C). We then reproduced a corticostriatal network by plating the neurons within microfluidic devices. Phospho‐mimetic HTT (HTTSD) significantly increased the speed of BDNF vesicles moving in the anterograde direction (Fig 1D, Movie EV1) without affecting the number of moving BDNF vesicles (Fig EV1D). In contrast, the absence of HTT phosphorylation (HTTSA) significantly increased the retrograde velocity of BDNF vesicles (Fig 1D). Thus, HTT phosphorylation status influences the velocity of BDNF vesicles in axons.
We next investigated whether HTT phosphorylation status influences the observed dysregulation of BDNF transport in Mecp2 siRNA‐transfected neurons (Figs 1E and EV1E and F). Phospho‐mimetic HTT (HTTSD) rescued both anterograde and retrograde transport of BDNF, along with the mean velocity of BDNF vesicles and linear flow (Fig 1E). Preventing HTT phosphorylation (HTTSA) restored only the retrograde velocity of BDNF and linear flow rate to control levels (Fig 1E). The overall effect of HTT phosphorylation on BDNF transport under normal or low‐Mecp2 conditions was not due to a change in the number of moving BDNF vesicles (Fig EV1D and F) or in cell viability, since we observed no toxicity in Mecp2 siRNA‐transfected HTTSD or HTTSA neurons compared to Mecp2 siRNA‐transfected WT neurons (Fig 1F). These results demonstrate that genetically promoting HTT phosphorylation at S421 rescues the transport of BDNF vesicles in projecting corticostriatal siMecp2 neurons.
Constitutive phosphorylation of HTT rescues corticostriatal BDNF transport and increases postsynaptic TrkB phosphorylation and markers of postsynaptic density in vivo
Mecp2 KO mice show altered corticostriatal connections and reduced BDNF levels in the striatum (Roux et al, 2012). We therefore assessed the impact of huntingtin phosphorylation on BDNF transport in vivo. We crossed Mecp2 KO mice with either HTTSA or HTTSD mice. The resulting double‐mutant male mice, deficient for the Mecp2 gene (KO) and homozygous for the S421A (HTT S421A/S421A) or S421D mutation (HTT S421D/S421D), will from hereon be referred to as KO/HTTSA and KO/HTTSD, respectively. Most of the BDNF protein located in the striatum comes from the cortex by anterograde transport within corticostriatal afferences (Altar et al, 1997). We therefore quantified the level of BDNF proteins using ELISA at the site of translation (the cortex) and the target site (the striatum) of 55‐day‐old WT mice, KO, KO/HTTSD, and KO/HTTSA mice (Fig 2A). The ratio of striatal and cortical BDNF is an indicator of the in vivo efficacy of BDNF axonal transport to the corticostriatal synapses. The ratio we observed in KO/HTTSD mice (1.57 ± 0.3) was equivalent to what we observed in WT mice (1.57 ± 0.6) and was significantly higher than that in Mecp2 KO mice (1.14 ± 0.1) or KO/HTTSA mice (1.1 ± 0.2). These results suggest that HTT phosphorylation rescues corticostriatal BDNF transport in vivo.
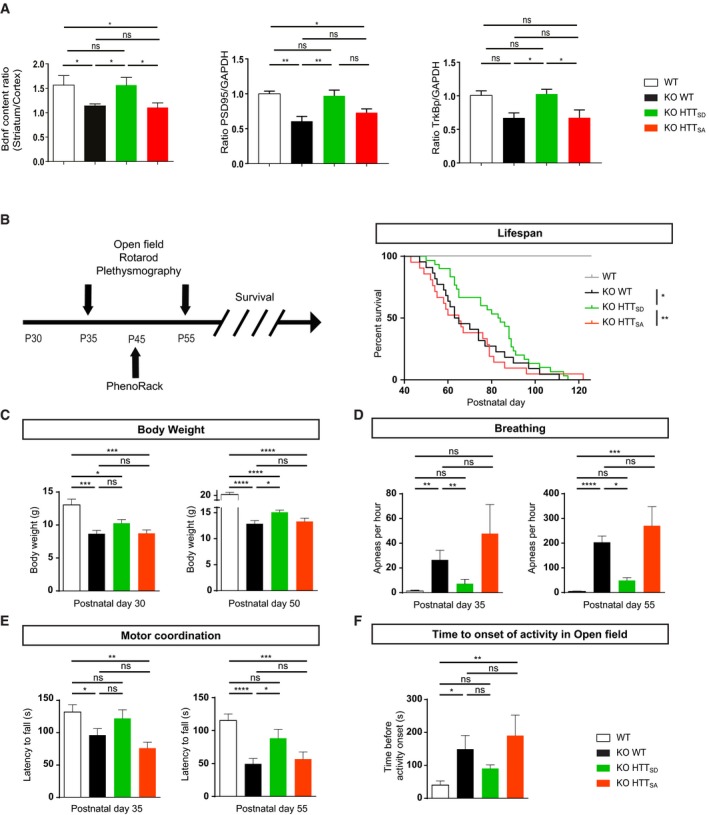
Figure 2. The S421D mutation improves BDNF corticostriatal transport in vivo, the motor and autonomic functions of Mecp2‐deficient mice and increases their lifespan.
-
ALeft: The ratio between the striatal and the cortical BDNF determined by quantifying the level of BDNF proteins at the cortex and the striatum of 55‐day‐old WT (n = 8), Mecp2 KO (n = 6), Mecp2 KO/HTTSD (mimicking the absence of phosphorylation) (n = 4), and Mecp2 KO/HTTSA mice (mimicking constitutive phosphorylation) (n = 5). Since striatal BDNF depends only on BDNF transport from the cortex, this ratio reflects BDNF transport through corticostriatal pathway (Mann–Whitney test). Right: Quantitative analysis of phospho‐TrkB protein level in striatum of KO WT mice and KO HTTSD mice by immunoblotting. The relative expression levels of phospho‐TrkB were normalized against GAPDH and are presented as the ratio (n = 6 mice per group) (Mann–Whitney test). Middle: Quantitative analysis of PSD‐95 protein level in striatum of Mecp2 KO/HTT WT mice and KO/HTTSD mice by immunoblotting. The relative expression levels of PSD‐95 were normalized against GAPDH and are presented as the ratio (n = 6 mice per group) (Mann–Whitney test).
-
BLeft: We investigated the behavior of Mecp2 KO mice at 35, 45, and 55 days of age and assessed their survival. Right: KO/HTTSD mice (n = 30) and WT mice (n = 17) had a significantly longer lifespan than KO (n = 22) or KO/HTTSA mice (n = 21) (Kaplan–Meier survival test).
-
CBody weight of 10 WT, 24 KO, 32 KO/HTTSD, and 24 KO/HTTSA mice at P30 and P50 (one‐way ANOVA with Tukey's comparison).
-
DFrequency of apnea of 9 WT, 10 KO, 14 KO/HTTSD, and 9 KO/HTTSA mice at P35 and P55 (Kruskal–Wallis test with Dunn's comparison).
-
EKO/HTTSD mice (n = 19) performed as well as WT (n = 17) at P35 on the accelerating rotarod test and continued to outperform Mecp2 KO (n = 16) and KO/HTTSA (n = 13) at P55 (one‐way ANOVA with Fisher's LSD test).
-
FTime before the onset of spontaneous locomotor activity of 17 WT, 20 KO, 19 KO/HTTSD, and 17 KO/HTTSA mice at P55. (Kruskal–Wallis test with Dunn's comparison).
We also found that improved corticostriatal BDNF transport in KO/HTTSD mice increased levels of TrkB phosphorylation at the postsynaptic level compared to KO WT (+36%, P < 0.05) and KO/HTTSA (+35%, P < 0.05), showing that the BDNF release is stimulated in vivo (Figs 2A and EV1G). As a consequence, the postsynaptic marker PSD‐95 is increased in KO/HTTSD striatum compared to KO/WT striatum (+37%, P < 0.01) (Figs 2A and EV1G). We conclude that promoting HTT phosphorylation stimulates striatal BDNF pathways and helps maintain corticostriatal synapse homeostasis in vivo.
Constitutive phosphorylation of HTT reduces the loss of body weight of Mecp2 KO mice and extends their lifespan
We next investigated whether manipulating HTT phosphorylation in vivo could have an effect on Mecp2 KO mouse symptoms. We first assessed the behavior of HTTSA and HTTSD homozygous mice using a modified SHIRPA primary screen (Appendix Table S1) and various behavioral assays (Fig EV2A–D) and found no significant differences between WT and HTTSA or HTTSD mice at 6 months in motor activity, strength, coordination, exploratory behavior, or body weight. Mecp2 KO mice carrying the S421D mutation (KO/HTTSD) had a longer lifespan than Mecp2 KO mice, though they still were subject to premature lethality, whereas KO/HTTSA mice showed no improvement over that of Mecp2 KO mice (Fig 2B). KO/HTTSD mice also had greater body weight than the Mecp2 KO mice, whereas the absence of phosphorylation in the KO/HTTSA mice had no effect on weight (Figs 2C and EV2E).
Figure EV2. Phenotypic characterization of WT, HTTSD, HTTSA (A, B, C, and D) mice and WT, KO/HTTSD, KO/HTTSA (E, F, G, and H) mice.
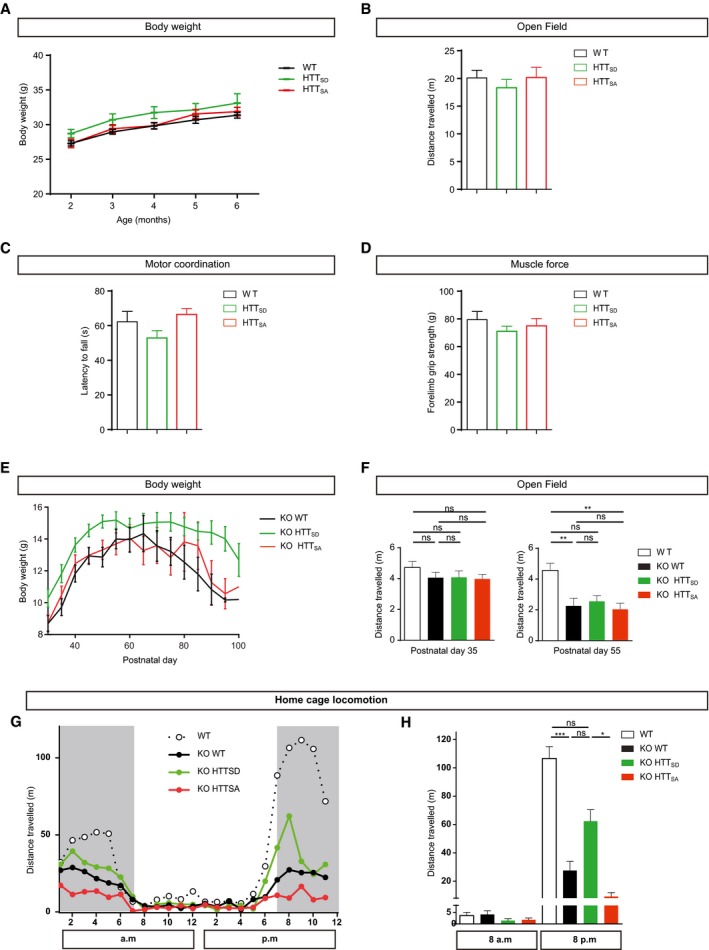
-
AThere were no significant differences in body weight between 6‐month‐old WT, HTTSD, and HTTSA mice (WT: n = 11; HTTSD: n = 6, and HTTSA: n = 10).
-
BNo significant differences in the distance travelled in the open‐field between 4‐month‐old WT, HTTSD, and HTTSA mice (WT: n = 10; HTTSD: n = 6 HTTSA: n = 11).
-
CNo differences between genotypes in the accelerating Rotarod test for assessing motor coordination of 6‐month‐old WT (n = 11), HTTSD (n = 11), and HTTSA mice (n = 12).
-
DNo differences in forelimb strength of 6‐month‐old WT (n = 11), HTTSD (n = 11), and HTTSA mice (n = 12) as assessed by the grip strength test.
-
ENo significant differences in body weight between KO/HTTSD (n = 32), KO/HTTSA (n = 24), and KO mice (n = 24) at different postnatal timepoints.
-
FDistance travelled during the open‐field test by WT (n = 17), KO (n = 20), KO/HTTSD (n = 19), and KO/HTTSA mice (n = 17) at P35 and P55. Results are expressed in meters.
-
G, HResults of 24‐h Phenorack monitoring to measure the spontaneous activity of 12 WT, 12 KO, 8 KO/HTTSD, and 7 KO/HTTSA mice at P45 (G). Distance travelled at 8 AM versus 8 PM (H).
Constitutive phosphorylation of HTT reduces apneas in Mecp2 KO mice
Breathing disturbances are prominent and deleterious in RTT patients (Kerr et al, 1997) and Mecp2 KO mice (Viemari et al, 2005). We found that apnea frequency increased with age in Mecp2 KO mice, from P35 to P55 (Fig 2D). The KO/HTTSD mice had significantly fewer apneas than Mecp2 KO mice at both time points (Fig 2D). The absence of HTT phosphorylation slightly worsened this phenotype.
These data suggest that promoting HTT phosphorylation in vivo improves respiration in Mecp2 KO mice.
Constitutive phosphorylation of HTT improves motor function of Mecp2 KO mice
We next investigated motor coordination on the accelerating rotarod test (Pratte et al, 2011). In agreement with previous studies, Mecp2 KO mice showed a progressive, significant decrease in the latency to fall relative to WT mice (Fig 2E). Promoting HTT phosphorylation delayed the appearance of motor incoordination until P55, but ablating HTT phosphorylation worsened motor coordination at both time points (Fig 2E). HTT phosphorylation had no effect on the overall exploration pattern of Mecp2 KO mice in the open‐field (OF) test (Fig EV2F), but the time before initiation of the first movement was significantly longer for the Mecp2 KO than WT mice (Fig 2F). The latency to explore the OF arena was further increased in KO/HTTSA mice, whereas it was reduced in the KO/HTTSD mice toward the values observed for WT mice (Fig 2F).
We next monitored circadian activity and found no deregulation in the circadian rhythm in the different genotypes during the day (Fig EV2G). There was, however, a striking suppression of spontaneous locomotion in the absence of Mecp2 during the dark phase (7:00 PM–7:00 AM) (Fig EV2G). Promoting HTT phosphorylation enhanced spontaneous night activity, increasing the distance travelled by the KO/HTTSD mice, although it did not reach the value of the WT mice (Fig EV2H). Conversely, the distance travelled by the KO/HTTSA mice was significantly less than that covered by the KO/HTTSD mice. Overall, these results show that promoting HTT phosphorylation improves sensorimotor coordination and locomotor activity in Mecp2 KO mice.
Inhibition of calcineurin by FK506 restores BDNF transport in Mecp2‐silenced axons
Since chronic phosphorylation rescues BDNF trafficking in vitro and improves symptoms in mice, we investigated whether pharmacological induction of HTT phosphorylation could be of therapeutic value in RTT. We previously reported that HTT phosphorylation can be inhibited by the protein phosphatase calcineurin (Pardo et al, 2006; Pineda et al, 2009). We therefore evaluated whether FK506, a calcineurin inhibitor, can restore BDNF transport in Mecp2‐silenced neurons by increasing HTT phosphorylation. Cortical neurons connected to striatal neurons within the microfluidic devices were transduced with the BDNF‐mCherry lentiviral vector and a siRNA targeting either a control sequence or that of Mecp2. Five days after plating, we incubated the microfluidic chambers for 1 h with 1 μM FK506 or vehicle and recorded BDNF axonal transport.
FK506 treatment increased HTT phosphorylation (Fig 3A) and rescued the reduced BDNF trafficking measured after Mecp2 silencing (Fig 3B and C, Movie EV2). Both mean anterograde and retrograde vesicle velocities were increased in siMecp2‐transfected neurons, with a significant overall effect on mean velocity and linear flow (Fig 3C). The number of moving vesicles did not change (Fig 3C). FK506‐induced calcineurin inhibition thus mitigates deficits of BDNF transport observed in Mecp2‐silenced neurons.
Figure 3. FK506 restores BDNF axonal transport in Mecp2‐silenced axons.
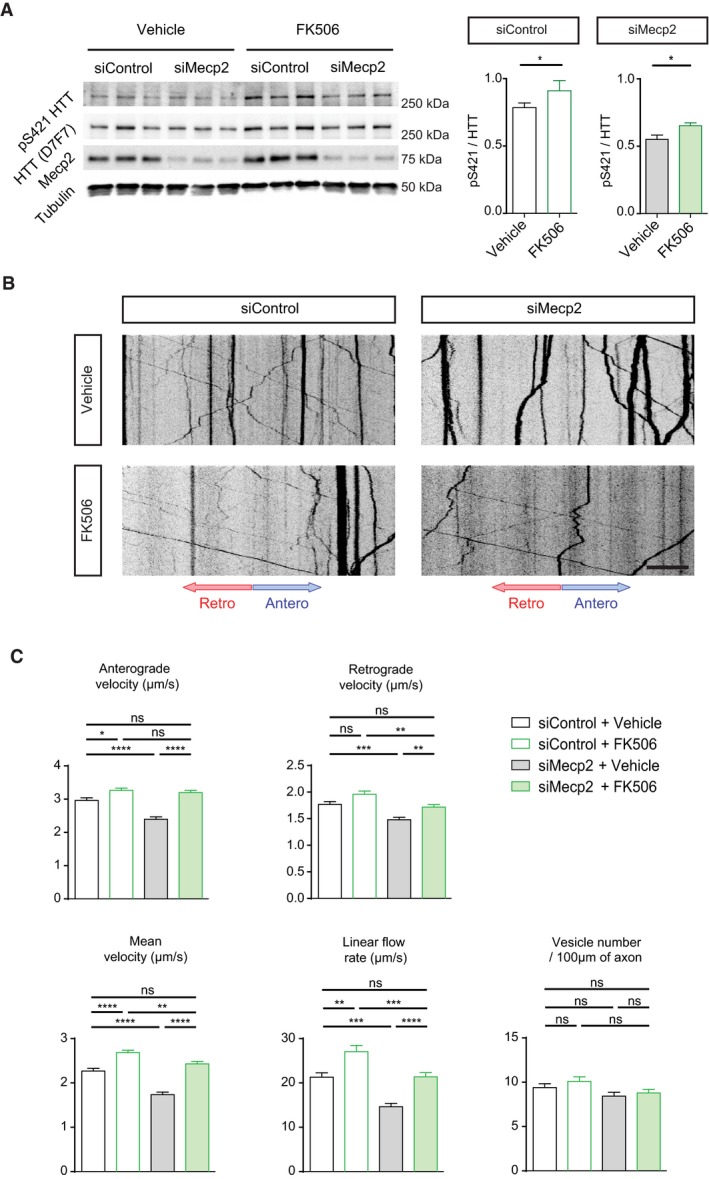
-
AWestern blot analysis of DIV 5 cortical neurons transfected with siMecp2 or siControl (siCtl) and treated with 1 µM FK506 or vehicle for 1 h, and quantification of pS421 HTT (n = 9 per group). Data are presented as means ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001, Mann–Whitney test.
-
BRepresentative kymographs showing axonal trafficking of BDNF‐mCherry‐containing vesicles in cortical neurons transfected with siMecp2 or siControl (siCtl) and treated with 1 µM FK506 or vehicle for 1 h. Scale bar = 20 µm.
-
CQuantification of anterograde, retrograde, mean velocity, linear flow rate, and number of BDNF‐mCherry‐containing vesicles from the data in (A) (siMecp2+Vehicle: n = 801 vesicles/95 axons; siMecp2+FK506: n = 1020 vesicles/116 axons; siCtl+Vehicle: n = 780 vesicles/83 axons; siCtl+FK506: n = 1,029 vesicles/102 axons). Data are presented as the mean ± SEM of at least three independent experiments (one‐way ANOVA with Tukey's comparison). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns = not significant.
Source data are available online for this figure.
FK506 increases HTT phosphorylation in Mecp2 KO mice
To determine whether calcineurin inhibition via FK506 treatment could improve Mecp2 KO mouse symptoms, we treated Mecp2 KO, WT and HTTSA mice with FK506 to induce HTT phosphorylation. Mice were first injected intraperitoneally with FK506 (5 mg/kg) and sacrificed 2 h after administration, as previously described (Pardo et al, 2006). We analyzed HTT phosphorylation at S421 by immunoblotting fresh whole‐brain protein extract (Figs 4A and EV3A and B). As previously described, administration of FK506 in WT mice increased HTT phosphorylation, by about 1.3‐fold (Pardo et al, 2006). In Mecp2 KO mice, FK506 doubled HTT phosphorylation relative to vehicle. FK506 had no effect on HTT phosphorylation in HTTSA mice (Fig EV3A and B).
Figure 4. Calcineurin inhibition by FK506 in Mecp2‐deficient mice improves motor and autonomic functions and extends lifespan through huntingtin‐dependent phosphorylation.
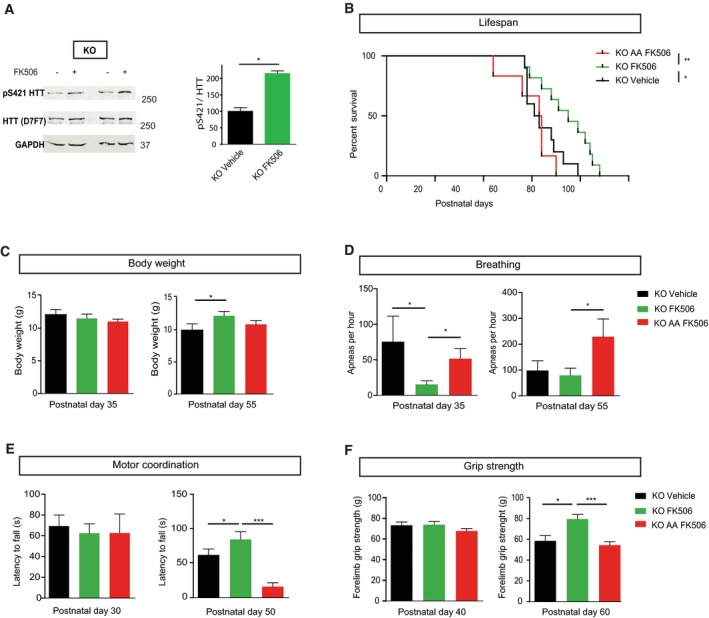
-
AWestern blot of HTT S421 phosphorylation. We treated 30‐day‐old Mecp2 KO mice intraperitoneally with FK506 (5 mg/kg) or vehicle and analyzed brain extracts for endogenous HTT phosphorylation by Western blotting, 2 h after administration, using an anti‐phospho‐HTT‐S421 specific antibody. The D7F7 antibody recognizes total HTT. The relative protein level of phospho‐HTT was normalized on total HTT protein level and is presented as the ratio (KO FK506 n = 4, KO Vehicle n = 4). Data are presented as means ± SEM, *P < 0.05, Mann–Whitney test).
-
B–FWe treated 30‐day‐old Mecp2 KO mice (n = 10) and KO/HTTSA mice (n = 10) with 5 mg/kg FK506 three times a week by intraperitoneal injection and assessed them in various behavioral tests. (B) FK506‐treated KO mice lived longer than vehicle‐treated KO mice and FK506‐treated KO HTTSA mice (Kaplan–Meier survival test). (C) Body weight of FK506‐treated KO, FK506‐treated KO HTTSA, and vehicle‐treated KO mice at P35 and P55 (Mann–Whitney test). (D) Frequency of apnea of FK506‐treated KO, FK506‐treated KO HTTSA, and vehicle‐treated KO mice at P35 and P55 (Mann–Whitney test). (E) Motor coordination of FK506‐treated KO, FK506‐treated KO HTTSA, and vehicle‐treated KO mice on the accelerating rotarod test at P30 and P50 (Mann–Whitney test). (F) Forelimb strength of FK506‐treated KO, FK506‐treated KO HTTSA, and vehicle‐treated KO mice assessed by the grip strength test at P40 and P60 (Mann–Whitney test).
Figure EV3. Acute and chronic FK506 treatment of WT, HTTSA, and Mecp2 KO mice.
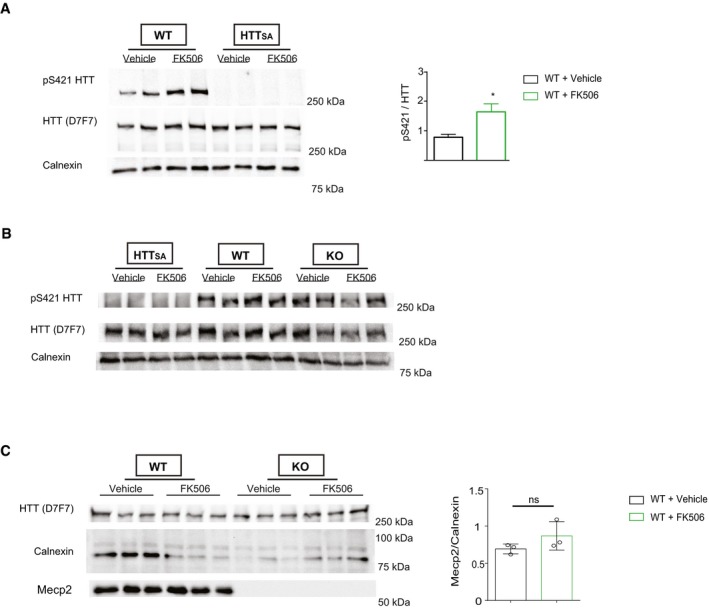
-
AWestern blot of HTT S421 phosphorylation. We treated WT and HTTSA 30‐day‐old mice intraperitoneally with FK506 (5 mg/kg) or vehicle and analyzed brain extracts for endogenous HTT phosphorylation by Western blotting, 2 h after administration (WT FK506 = 3, WT Vehicle = 6, HTTSA FK506 = 3, HTTSA Vehicle = 3). The relative protein level of phospho‐HTT was normalized on total HTT protein level and is presented as the ratio. Data are presented as the means ± SEM, *P < 0.05, Mann–Whitney test.
-
BWestern blot of HTT S421 phosphorylation. We treated WT, HTTSA, and Mecp2 KO 30‐day‐old mice intraperitoneally with FK506 (5 mg/kg) or vehicle chronically for 20 days and analyzed brain extracts for endogenous HTT phosphorylation by Western blotting, 2 h after the last administration (KO FK506 n = 2, KO Vehicle n = 2, WT FK506 = 2, WT Vehicle = 2, HTTSA FK506 = 2, HTTSA Vehicle = 2).
-
CWestern blot of HTT and Mecp2. We treated WT and Mecp2 KO 30‐day‐old mice intraperitoneally with FK506 (5 mg/kg) or vehicle chronically for 20 days (WT FK506 = 3, WT Vehicle = 3, HTTSA FK506 = 3, HTTSA Vehicle = 3). Data are presented as the means ± SEM, ns = not significant, Mann–Whitney test.
Source data are available online for this figure.
FK506 treatment improves respiration and motor function of Mecp2 KO mice and extends their lifespan via HTT phosphorylation
To determine whether FK506 could improve the Mecp2‐deficient phenotype, we treated Mecp2 KO mice by intraperitoneal FK506 injection (10 mg/kg), three times a week, starting at P30. We choose P30 as a starting point, since the onset of the Mecp2‐knockout phenotype is already apparent at this stage as breathing abnormalities, locomotor deficits, and body weight loss (Guy et al, 2001; Viemari et al, 2005; Matagne et al, 2017). FK506 treatment significantly increased the lifespan of Mecp2 KO mice (Fig 4B) and induced a significant increase in body weight by P55 (Fig 4C). The number of apneas was significantly lower in the FK506‐treated group than the vehicle group at P35 and remained lower at P55 (Fig 4D). FK506‐treated Mecp2 KO mice performed better on the accelerating rotarod at P50 (Fig 4E) and showed better forelimb muscle strength than the vehicle group (Fig 4F). We next investigated cell death in the striatum of these mice. In accord with previous reports (Reiss et al, 1993; Kishi & Macklis, 2004; Armstrong, 2005; Roux et al, 2007), we did not detect any cell death in Mecp2 KO mice by cleaved caspase‐3 immunodetection or by TUNEL assay (Fig EV4A and B). Importantly, we found that FK506 treatment did not induce any neuronal toxicity or cell death after 20 days of treatment (Fig EV4A and B).
Figure EV4. Absence of cytotoxicity and increased neuronal activity after chronic FK506 treatment.
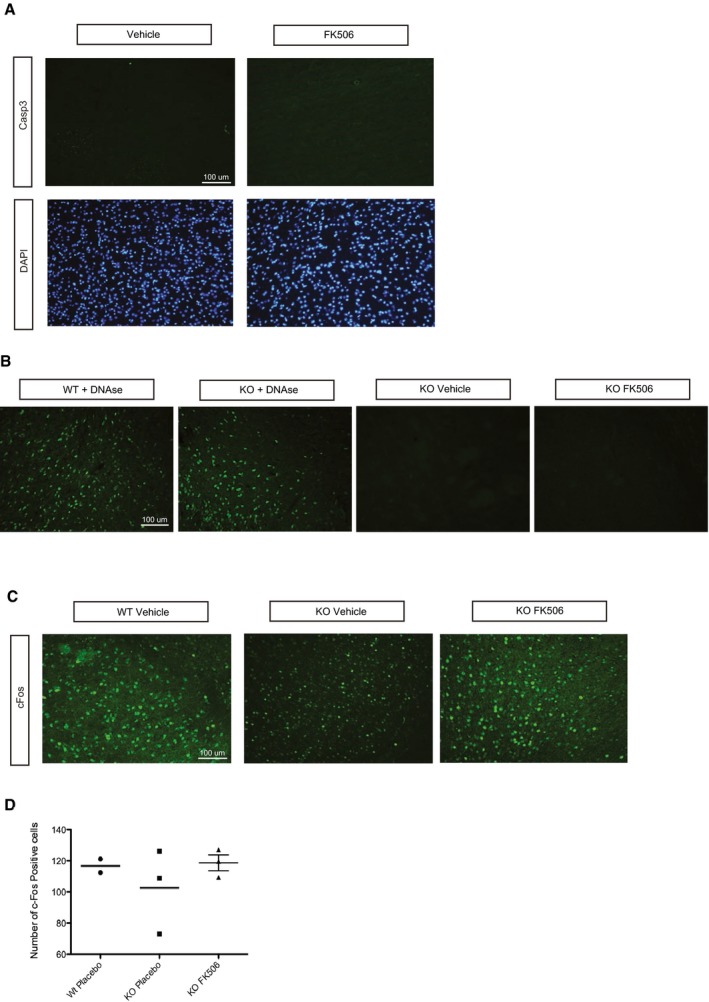
-
AFK506 did not increase the levels of cleaved caspase‐3, as shown by immunostaining. Representative images of Striatum labeled with Casp3 (green) and DAPI of 50‐day‐old Mecp2 KO mice treated with FK506 (right panel) or vehicle (left panel) for 20 days.
-
BAbsence of apoptosis in the dorsal striatum of Mecp2‐knockout mice treated chronically with FK505 or with Vehicle. TUNEL assay was used to detect potential apoptotic neurons in 50‐day‐old Mecp2‐deficient mice (n = 3 for each stage). Left panel: detection of cells containing fragmented DNA after desoxyribonuclease I treatment of a representative dorsal striatum section originating from one Mecp2‐knockout and one wild‐type animal. No labeled cells were visible after counting all striatum sections of 50‐day‐old Mecp2‐knockout mice.
-
CFK506 tends to increase the number of cFos‐positive neurons. Representative images of striatum labeled with cFos (green) of 50‐day‐old KO mice treated with FK506 (right panel) or vehicle (middle panel) for 20 days.
-
DVertical scatter plot representing the number of cFos positive neurons counted in the dorsal striatum of P50 Wt placebo (n = 2)‐, KO placebo (n = 3)‐, and KO FK506 (n = 3)‐treated mice in a picture of length 650 μm and width 445 μm. Each dot represents the mean of three successive sections.
We also verified the induction of HTT phosphorylation in Mecp2‐deficient mice after 20 days of FK506 chronic treatment. Mouse brain samples were analyzed 2 hrs after the last treatment. We did not detect any difference in Mecp2 protein levels or in HTT phosphorylation in wild‐type mice treated with FK506 (Fig EV3B and C). However, at this time point we did not detect elevated HTT phosphorylation in the samples treated with FK506 relative to DMSO‐treated animals, likely because repeated treatments desensitize the pathway. We observed a trend toward reduction in cFos staining in Mecp2‐deficient mice that was increased back to control levels in FK506‐treated Mecp2 KO mice, suggesting a positive treatment effect (Fig EV4C and D).
To establish that the beneficial effect of FK506 is mediated by HTT phosphorylation at S421, we treated KO/HTTSA mice by intraperitoneal FK506 injection as previously done (Fig 4). Interestingly, the lifespan of Mecp2 KO/HTTSA mice was significantly shorter than that of Mecp2 KO mice (Fig 4B). Moreover, FK506 treatment affected neither motor function nor apneas of KO HTTSA mice (Fig 4D–F). These results demonstrate that HTT phosphorylation at S421 is essential for the therapeutic effect of FK506.
Discussion
Here, we demonstrate that genetically or pharmacologically inducing HTT phosphorylation at S421 rescues BDNF vesicular transport in Mecp2‐silenced projecting corticostriatal neurons and improves several pathophysiological features of Mecp2 KO mice. The ability of FK506 as a proof‐of‐principle candidate to improve Mecp2 symptoms highlights the feasibility of pharmacological stimulation of HTT S421‐P in a mouse model of RTT. Importantly, our findings show that HTT phosphorylation can stimulate endogenous machinery to promote BDNF trafficking in the corticostriatal network.
There has been considerable interest in modulating BDNF expression and signaling as a treatment for RTT. Unfortunately, BDNF itself has very low blood–brain barrier (BBB) permeability, precluding its peripheral administration as a potential therapy. Several studies have used indirect stimulation of BDNF metabolism via fingolimod (Deogracias et al, 2012) or ampakine treatment (Ogier et al, 2007) to circumvent this limitation, but these pharmacological treatments only partially improved the phenotype of Mecp2 KO mice. Daily injection of IGF‐1, another neurotrophic factor that can cross the BBB and is known to induce Akt phosphorylation (Humbert et al, 2002), has been found to improve survival, locomotor activity, and respiratory rhythm in Mecp2 KO mice (Tropea et al, 2009). Finally, potential agonists of the TrkB receptor, such as 7,8‐dihydroxyflavone (Johnson et al, 2012) and LM22A‐4 (Schmid et al, 2012; Kron et al, 2014; Li et al, 2017), improved breathing patterns in Mecp2 KO mice. It is important to note, however, that the improvements observed in these different studies required that pharmacological treatments be initiated before the appearance of the first symptoms.
Given that RTT is not diagnosed until long after symptoms have begun, we searched for a more practical translational approach based on observed improvements in the RTT phenotype by constitutive phosphorylation of HTT. We selected FK506 because Pardo et al (2006) found that this drug increases phosphorylation of mutant HTT. Importantly, FK506 also induces HTT phosphorylation in Mecp2 KO mice. Our results showed that FK506 treatment, starting at an already‐symptomatic stage, improved the lifespan, motor strength and coordination, and exploratory behavior, and reduced the frequency of apneas in Mecp2 KO mice in a manner that requires HTT phosphorylation at S421. Thus, part of the beneficial effect of FK506 treatment is due to the stimulation of the HTT‐dependent transport of BDNF. It is possible that FK506 also modulates the trafficking of other cargo, such as mitochondria, which could contribute to the in vivo improvement we saw in the Mecp2 KO mice. Future studies investigating such additional effects would be of interest, as they would increase the therapeutic relevance of FK506 treatment for Mecp2 symptoms (Reddy et al, 2012).
We conclude that BDNF trafficking and supply are diminished in the absence of Mecp2 and can be effectively stimulated by promoting HTT phosphorylation. Our results are in accord with a recent study that demonstrated that BDNF acts cell‐autonomously in an autocrine loop, as wild‐type neurons were unable to rescue growth deficits of neighboring Mecp2‐deficient neurons (Sampathkumar et al, 2016). Thus, HTT phosphorylation may increase the bioavailability of BDNF at the synapse through autocrine and paracrine mechanisms in the brains of Mecp2 KO mice and likely represents a strategy of therapeutic interest versus a general, non‐synapse‐specific increase of BDNF levels in the brain.
Materials and Methods
Mouse breeding and genotyping
All mouse lines were on a C57BL/6J genetic background. The Mecp2tm1‐1Bird Mecp2‐deficient mice were obtained from the Jackson Laboratory and maintained on a C57BL/6 background by using C56BL/6J male breeders (also from the Jackson Laboratory). The HTT knock‐in mice were previously generated by inserting a point mutation in exon 9 (AGC>GAC, Ser>Asp called S421D or AGC>GCC, Ser>Ala called S421A) (Thion et al, 2015). To obtain double‐mutant mice (Mecp2 KO and S421A or S421D), heterozygous females (Mecp2 +/−) were crossed with homozygous S421D or S421A males.
Hemizygous mutant males (Mecp2 −/y also called Mecp2 KO) were generated by crossing heterozygous females (Mecp2 +/−) with C57BL/6 males. Genotyping was performed by routine PCR technique following a previously described protocol (Roux et al, 2012). Animals were housed under standard conditions of temperature (21 ± 2°C) and humidity (55 ± 5%), with food and water ad libitum in a 12:12 h day/night cycle.
Only male mice were used for all genotypes and experiments.
Experimental protocols were approved by the ethical committee of the Aix Marseille University and the French M.E.N.E.S.R. minister (Permit Number: 02910.02).
The experimental procedures were carried out in keeping with the European guidelines for the care and use of laboratory animals (EU directive 2010/63/EU), the guide for the care and use of the laboratory animals of the French national institute for science and health (INSERM). All experiments were made to minimize animal suffering. In order to reduce animal suffering, endpoints were fixed as weight loss limit (below 80% of maximum weight), obvious breathing defects, or severe injury. In these cases, animals were euthanized with an overdose of pentobarbital (100 mg/kg BW i.p., Ceva Santé Animale, La Ballastiere, France).
FK506 chronic in vivo treatment of Mecp2‐deficient mice (KO and KO/HTTSA)
Mecp2 KO mice were randomly assigned in groups. From P30, animals received an i.p. injection three times a week of either 10 mg/kg FK506 in 17% DMSO or vehicle alone (17% DMSO). The first group was used for behavioral testing and evaluating survival (see below). A second group was used to assess the chronic effects of FK506 treatment from P30 to P50 on cellular and molecular parameters.
Behavioral testing
All mice were weighed every 5 days and assessed for survival. At P35, P45, and P55, each animal underwent a set of behavioral tests to assess motor function, activity, and breathing pattern (Fig 2). All testing occurred during the light phase of the light–dark cycle (except for the PhenoRack monitoring). All the behavioral experiments were performed blinded to HTT genotype and treatment.
Open field
An arena made of clear perspex (38 × 30 cm), under controlled light conditions (300 lx), was used for the FK506 study. For the genetic study, mice were placed in a 1‐m‐diameter arena under controlled light conditions (300 lx). Activity was recorded using the Videotrack software (Viewpoint, Lyon, France) for 20 min. Activity and average velocity (cm/s) were determined from the total distance moved and activity duration. Vertical activity (rearing, leaning, and grooming) was noted by an experimenter blind of the genotype.
Rotarod
Sensorimotor abilities were assessed by the accelerating Rotarod apparatus (Panlab LE‐8200, Harvard Apparatus). Each trial started at 4 rpm and reached 40 rpm speed after 300 s. Mice underwent three trials, with 5‐min rest time in between. The trial ended when the mouse fell off the rod or after 300 s. Latency to fall (in second) was measured, and only, the best trial was recorded. In the case of mice clinging to the rod, the trial was stopped and the passive rotation was considered a failure in performance like falling (Brown et al, 2005).
Grip strength
A Bioseb grip strength meter (Panlab) was used to measure forelimb strength. Five measures for each mouse were taken, and means were calculated from the three best trials.
Whole‐body plethysmography
To assess apneas, mice were placed in a clear plexiglass chamber (200 ml) and allowed to breathe naturally under conscious and unrestrained conditions. After a ~30‐min adaptation, breathing was recorded: The spirogram was obtained by recording the pressure difference between the two chambers, and then, the signal was amplified, filtered and fed to an analog‐to‐digital converter (sampling frequency, 1 kHz), and finally analyzed by the Spike2 interface and software (v.5.04, Cambridge Electronic Design Ltd., Cambridge, UK). Apneas were defined by more than 1s without breathing, as previously described (Roux et al, 2007). Breathing cycles were divided into four groups according to their duration: hyperventilation (including cycles in the range 0–0.3 s range); ventilation (0.3–0.7 s); hypoventilation (0.7–1 s); and apneas (1–∞ s). The breathing variability was calculated as the mean standard variability. Breathing parameters were obtained from the analysis of quiet period of at least 100 consecutive cycles (Fig EV5).
Figure EV5. Breathing patterns in 55‐day‐old WT, KO Vehicle, KO FK506, KO/HTTSD, and KO/HTTSA mice.
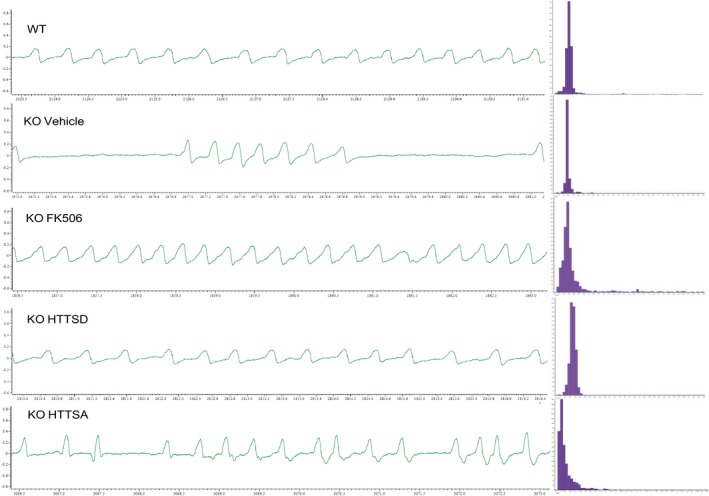
Left: Typical plethysmographic recordings of breathing (inspiration upward) performed in quiet, 55‐day‐old WT, KO Vehicle, KO FK506, KO/HTTSD, and KO/HTTSA mice. Right: Distribution of frequency values recorded in WT, KO Vehicle, KO FK506, KO/HTTSD, and KO/HTTSA mice at P55. Frequency histograms represent the number of occurrences (ordinate) of breathing frequency during 4 consecutive minutes separated in 0.05‐s time windows.
Home cage activity
To analyze spontaneous activity and circadian rhythm, mice were put in individual cages and monitored by the PhenoRack system (Viewpoint, Lyon, France). Mice locomotion was tracked by infrared light during 48 h, and after a 24‐h adaptation phase, only the last 24‐h activity was analyzed. The recorded data allowed us to analyze activity, distance (cm), and velocity (cm/sec).
BDNF immunoassay
P55 (n = 4 Wt; n = 4 Mecp2 KO; n = 4 KO/HTTSD; n = 5 KO/HTTSA) male mice were euthanized by cervical dislocation, and their brains were dissected out within the first 2 min post‐mortem. The cortex and the striatum were microdissected using a punching needle (0.5 mm in diameter). Briefly, brain area dissection was performed on cryostat brain sections with the help of a 5× magnifying lens, following their stereotaxic coordinates (Paxinos and Franklin, 2001). We dissected cortical and striatal samples only coming from the same slice in the same rostro‐caudal level. Tissue samples were freshly isolated and lysed in 200 μl of the extraction buffer (100 mM Tris‐pH 7.5, 125 mM NaCl, 0.1 mM EGTA, 0.1% Triton X‐100, Roche® protease inhibitors cocktail), sonicated, centrifugated. The supernatant was stored at −80°C until assay. Total protein concentration was determined by using the bicinchoninic acid (BCA) protein assay (Thermo Fisher scientific) and measured with a spectrophotometer (Glomax, Promega). The level of BDNF protein from tissue extracts was determined with the BDNF Emax® ImmunoAssay System (Promega) using the manufacturer's instruction. In the present study, we measured only free mature BDNF, and therefore, we proceeded directly to the ELISA protocol avoiding any acid treatment. In each assay, duplicate wells were assigned for each sample. A Victor 4 PerkinElmer microplate reader was used to measure signal intensity from the wells at 450 nm. A linear standard curve was generated with standard BDNF from 5.8 to 500 pg/ml. The total amount of BDNF per well was calculated based on the standard curve, and each sample value was within the linear range. The relative BDNF value was then calculated by normalizing the amount of BDNF against the total amount of protein input.
Western blotting
Adult (P55) male mice were euthanized by cervical dislocation, and their brains were dissected out within the first 2 min post‐mortem. Brains were dissected on ice, and proteins were extracted by sonication and isolated in a lysis buffer containing 50 mM Tris–HCl (pH = 7.5), 150 mM NaCl, 2 mM EGTA, 2 mM EDTA, 1% Triton X‐100, 10 nM betaglycerophosphate, 5 mM sodium pyrophosphate, 50 mM sodium fluoride, and HaltTM proteases and phosphatases inhibitor cocktail (Pierce Thermo Fisher). Proteins were extracted from neuronal culture with lysis buffer containing 4 mM HEPES, pH 7.4, 320 mM sucrose, and protease inhibitor cocktail (Roche). Protein concentrations were determined by the bicinchoninic acid method. After a denaturation step at 96°C for 5 min, proteins (100 μg) were separated on 4–20% SDS‐polyacrylamide gel (Life technology) and transferred onto a nitrocellulose membrane by electroblotting using the Trans‐Blot turbo transfer system (Bio‐Rad). The membrane was blocked with blocking buffer (Millipore, WBAVDFL01) for 1 h at room temperature. Primary antibodies for HTT [1:1,000, rabbit, clone D7F7, CST (Lunkes et al, 2002)], HTT‐pS421 [1:500. rabbit homemade previously described (Humbert et al, 2002)], Mecp2 (1:1,000, rabbit, CST), TRKB‐p816 (Millipore ABN1381), PSD‐95 (1:1,000, Neuromab), Tubulin (1:5,000, mouse, Sigma), GAPDH (1:5,000, rabbit, Sigma), Actin (1:10,000, mouse, Millipore), and Calnexin (1:1,000, rabbit, Sigma C4731) were diluted in the same solution and incubated overnight at 4°C. The membrane was incubated using appropriate HRP secondary antibodies (donkey anti‐rabbit 711‐035‐152 and donkey anti‐mouse 715‐035‐150; Jackson Immunoresearch Laboratory and Bio‐Rad ChemiDoc XRS System) or fluorescent secondary antibodies (IRDye 800 CW, IRDye 680 RD, LI‐COR, and LI‐COR Odyssey Imager). Quantitative analyses of signal intensity were performed using ImageJ software. For quantification, total (phosphoindependent) protein signals were used to normalize the phospho‐protein signal.
Immunohistochemistry
Mice were euthanized with pentobarbital and transcardially perfused with 0.01 M PBS followed by 4% paraformaldehyde (PFA) in phosphate buffer. Brains were removed, fixed in 4% PFA overnight at 4°C, and then cryopreserved in 20% sucrose for 3 days. Brains were then rapidly frozen and coronally sectioned into 50‐μm sections using a Leica VT1200s cryostat (Leica Biosystems). Sections were rehydrated in PBS three times for 10 min and blocked with 7% normal goat serum in PBS for 1 h and then incubated overnight at room temperature (RT) with anti‐cFos (1/200, rabbit, Abcam ab209794) or anti‐cleaved caspase‐3 (1/400, rabbit, Cell signaling) antibodies. The sections were then washed three times for 10 min each in PBS followed by incubation for 2 h with the following secondary antibodies: Alexa Fluor 488‐labeled goat anti‐rabbit at 1/400. After staining, the sections were washed three times, for 10 min each in PBS, and then incubated 10 min in DAPI. After a last 10‐min wash, sections were mounted in Immu‐mount (Thermo Scientific). The immunolabeled slices were digitized and recorded using an Apotome Axioimager 2 (Carl Zeiss) or a Zeiss Lumar stereomicroscope coupled to Axiocam digital camera (Axiovision 4.4, Carl Zeiss).
Videomicroscopy
Embryonic (E15.5) neuronal cultures were prepared as previously described (Liot et al, 2013). Ganglionic eminences and cortex were dissected, and dissociated cortical neurons were nucleofected with ON‐TARGET plus mouse Mecp2 siRNA or Non‐targeting siRNA 1 (Dharmacon) according to the protocol of Amaxa Nucleofection (Lonza). Then, neurons were plated into microchambers coated with poly‐D‐lysine (0.1 mg/ml) in the cortical and synaptic compartment or poly‐D‐lysine and laminin (10 μg/ml, Sigma) into the striatal compartment and cultured at 37°C in a 5% CO2 incubator for 5 days. After 24 h in culture, cortical neurons were transduced with lentivector coding for BDNF‐mCherry into presynaptic neuron chamber for axonal transport analysis as previously described (Virlogeux et al, 2018). Acquisitions were done on microgrooves, at the limit of the synaptic compartment, at 5 Hz for 30 s on inverted microscope (Axio Observer, Zeiss) coupled to a spinning disk confocal system (CSU‐W1‐T3; Yokogawa) connected to an electron‐multiplying CCD (charge‐coupled device) camera (ProEM+1024, Princeton Instrument) at 37°C and 5% CO2. Quantifications of vesicle velocity, linear flow rate, and vesicle number were done on 100 μm of axon using KymoTool Box ImageJ plug‐in as previously described (Zala et al, 2013; Virlogeux et al, 2018). Vesicle velocity corresponds to segmental anterograde or retrograde velocity. Directional net flux is the anterograde cumulative distance minus the retrograde cumulative distance. Regarding vesicle number, a vesicle is considered anterograde when the distance travelled by one vesicle is more anterograde than retrograde.
MTT assay
In a corticostriatal network, at DIV 11 or 12, presynaptic chamber was filled with MTT 1/10e from MTT solution at 5 mg/ml. After a 3.5‐h incubation at 37°C, MTT solution was carefully removed from the presynaptic chamber and MTT solvent was added (10% SDS, 1.2 mM HCl) for 30 min under agitation. Then, MTT solvent was removed from the microfluidics device and put in a well within a 96‐well plate. The absorbance was read at 490 nm on a microplate reader (PHERAstar FS, BMG labtech). Negative controls (dead cells) were treated with H2O2 at 100 mM for 48 h before the experiment.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) of apoptotic cells
Cell apoptosis was measured in vivo using the Click‐It™ Plus TUNEL Assay (Invitrogen) according to the manufacturer's protocol.
Statistical analysis
All analyses were performed using GraphPad Prism for Windows/MacOS (GraphPad Software, La Jolla, California, USA, http://www.graphpad.com). The results are reported as mean ± standard error of the mean (SEM). A P < 0.05 was considered to be statistically significant. For group comparisons, normality distribution of the datasets was tested before performing any statistical test by Shapiro's test. In case of non‐normal distribution, a non‐parametric test was performed. When ANOVA was used, Brown Forsythe's test verifies that the variance is homogenous. One‐way ANOVAs were performed as indicated with Dunnett's post‐hoc analysis for pairwise comparisons when normal distribution. Kruskal–Wallis test with Dunn's multiple comparison was performed on datasets without normal distribution. When appropriate, two groups were compared with an unpaired two‐way t‐test or Mann–Whitney test on datasets without normal distribution. The Kaplan–Meier log‐rank test was used for survival studies.
Author contributions
YE, JB, NP, Y‐SA, BD, LV, FS, and JCR designed experiments. YE, JB, NP, Y‐SA, LS, VM, CS, EB, and HV performed experiments. YE, JB, NP, Y‐SA, LS, VM, CS, HV, BD, LV, FS, EB, and J‐CR analyzed the data, and YE, JB, FS, and J‐CR wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
The paper explained.
Problem
Rett syndrome (RTT) is a severe neurodevelopmental disorder that becomes apparent in the second year of life, robbing girls of the motor, language, and social skills they acquired and replacing them with learning impairments, stereotypic behaviors, seizures, respiratory dysrhythmias, and a host of other symptoms. RTT is caused by mutations in the X‐linked gene MECP2 (Methyl‐CpG‐binding protein 2), which encodes the MeCP2 protein, an epigenetic factor that governs the expression of thousands of neuronal genes. Even if gene therapy becomes possible, the introduction of the MECP2 gene into a cell already expressing a wild‐type copy would cause a problem of MeCP2 dosage (MeCP2 duplication syndrome is an equally severe disorder). We therefore need viable approaches to therapy. Several studies have linked the loss of MeCP2 function to reductions in the levels of BDNF (brain‐derived neurotrophic factor) in the brain of RTT patients as well as in mouse models. BDNF is crucial to learning and memory, but transport of BDNF‐containing vesicles from the cortex to the striatum is abnormally reduced in Mecp2 knockout (KO) mouse neurons. Intriguingly, BDNF is transported within neurons by the Huntingtin protein (HTT), so named because mutations in HTT underlie Huntington's disease. In the present study, we tested whether promoting HTT's transport function by stimulating its phosphorylation at serine 421 (S421) might restore BDNF transport.
Results
We used genetic and pharmacological approaches to promote HTT phosphorylation at S421 in both Mecp2‐deficient neurons and Mecp2 knockout mice. We also evaluated the consequences of HTT S421 phosphorylation on BDNF axonal trafficking in projecting corticostriatal neurons using microfluidic devices that mimic the excitatory network. We found that promoting huntingtin phosphorylation restores the intracellular trafficking of BDNF in Mecp2‐silenced neurons, dramatically improves several key symptoms in Mecp2 knockout mice, and extends their lifespan.
Impact
Our data demonstrate that promoting BDNF transport in the appropriate neuronal circuits is more effective at restoring normal function in Mecp2‐deficient neurons than non‐specific BDNF overexpression. Stimulation of endogenous cellular pathways, such as HTT phosphorylation at S421, may provide a promising new approach for the treatment of RTT patients.
Supporting information
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Source Data for Expanded View
Review Process File
Source Data for Figure 3
Source Data for Figure 4
Acknowledgements
We thank S. Humbert for sharing the HTT S421A and HTT S421D mice and for advice; the GIN imaging facility (PIC‐GIN) for help with image acquisitions; G. Froment, D. Nègre, and C. Costa from the lentivirus production facility of SFR Biosciences (UMS3444/CNRS, US8/Inserm, ENS de Lyon, UCBL); and V. Brandt for critical reading of the manuscript. This work was supported by grants from Agence Nationale pour la Recherche (ANR‐2012‐BSV1‐0003‐03 ANTARES, J.C.R. & F.S., ANR‐14‐CE35‐0027‐01 PASSAGE, F.S.; ANR‐15‐JPWG‐0003‐05 EU Joint Program‐Neurodegenerative Disease (JPND) Research project CircProt (no. 643417), (F.S.), Fondation pour la Recherche Médicale (FRM, équipe labellisée, F.S.), Fondation Bettencourt Schueller (F.S.), Fédération pour la Recherche sur le Cerveau (F.S.), Inserm (J.C.R. & F.S.), Aix‐Marseille Université (J.C.R.), AFSR (J.C.R.), Institut d'etablissement d'AMU “Marseille Maladies Rares” (MarMaRa) (J.C.R), Promex Stiftung Für Die Forschung (JCR), Rettsyndrome.org (JCR), and NeuroCoG in the framework of the “Investissements d'avenir” program (ANR‐15‐IDEX‐02, F.S.). F.S. laboratory is member of the Grenoble Center of Excellence in Neurodegeneration (GREEN).
EMBO Mol Med (2020) 12: e10889
Contributor Information
Frédéric Saudou, Email: frederic.saudou@inserm.fr.
Jean‐Christophe Roux, Email: jean-christophe.roux@univ-amu.fr.
References
- Altar CA, Cai N, Bliven T, Juhasz M, Conner JM, Acheson AL, Lindsay RM, Wiegand SJ (1997) Anterograde transport of brain‐derived neurotrophic factor and its role in the brain. Nature 389: 856–860 [DOI] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY (1999) Rett syndrome is caused by mutations in X‐linked MECP2, encoding methyl‐CpG‐binding protein 2. Nat Genet 23: 185–188 [DOI] [PubMed] [Google Scholar]
- Armstrong DD (2005) Neuropathology of Rett syndrome. J Child Neurol 20: 747–753 [DOI] [PubMed] [Google Scholar]
- Brown SDM, Chambon P, de Angelis MH, Eumorphia Consortium (2005) EMPReSS: standardized phenotype screens for functional annotation of the mouse genome. Nat Genet 37: 1155 [DOI] [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong STC, Qin J, Zoghbi HY (2008) MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320: 1224–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Q, Khare G, Dani V, Nelson S, Jaenisch R (2006) The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron 49: 341–348 [DOI] [PubMed] [Google Scholar]
- Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, Jaenisch R, Greenberg ME (2003) Derepression of BDNF transcription involves calcium‐dependent phosphorylation of MeCP2. Science 302: 885–889 [DOI] [PubMed] [Google Scholar]
- Chen L, Chen K, Lavery LA, Baker SA, Shaw CA, Li W, Zoghbi HY (2015) MeCP2 binds to non‐CG methylated DNA as neurons mature, influencing transcription and the timing of onset for Rett syndrome. Proc Natl Acad Sci USA 112: 5509–5514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng P‐L, Song A‐H, Wong Y‐H, Wang S, Zhang X, Poo M‐M (2011) Self‐amplifying autocrine actions of BDNF in axon development. Proc Natl Acad Sci USA 108: 18430–18435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng T‐L, Wang Z, Liao Q, Zhu Y, Zhou W‐H, Xu W, Qiu Z (2014) MeCP2 suppresses nuclear microRNA processing and dendritic growth by regulating the DGCR8/Drosha complex. Dev Cell 28: 547–560 [DOI] [PubMed] [Google Scholar]
- Colin E, Zala D, Liot G, Rangone H, Borrell‐Pagès M, Li X‐J, Saudou F, Humbert S (2008) Huntingtin phosphorylation acts as a molecular switch for anterograde/retrograde transport in neurons. EMBO J 27: 2124–2134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deogracias R, Yazdani M, Dekkers MPJ, Guy J, Ionescu MCS, Vogt KE, Barde Y‐A (2012) Fingolimod, a sphingosine‐1 phosphate receptor modulator, increases BDNF levels and improves symptoms of a mouse model of Rett syndrome. Proc Natl Acad Sci USA 109: 14230–14235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Hendrich B, Holmes M, Martin JE, Bird A (2001) A mouse Mecp2‐null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet 27: 322–326 [DOI] [PubMed] [Google Scholar]
- Humbert S, Bryson EA, Cordelières FP, Connors NC, Datta SR, Finkbeiner S, Greenberg ME, Saudou F (2002) The IGF‐1/Akt pathway is neuroprotective in Huntington's disease and involves Huntingtin phosphorylation by Akt. Dev Cell 2: 831–837 [DOI] [PubMed] [Google Scholar]
- Johnson RA, Lam M, Punzo AM, Li H, Lin BR, Ye K, Mitchell GS, Chang Q (2012) 7,8‐dihydroxyflavone exhibits therapeutic efficacy in a mouse model of Rett syndrome. J Appl Physiol 112: 704–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz DM, Bird A, Coenraads M, Gray SJ, Menon DU, Philpot BD, Tarquinio DC (2016) Rett syndrome: crossing the threshold to clinical translation. Trends Neurosci 39: 100–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr AM, Armstrong DD, Prescott RJ, Doyle D, Kearney DL (1997) Rett syndrome: analysis of deaths in the British survey. Eur Child Adolesc Psychiatry 6(Suppl 1): 71–74 [PubMed] [Google Scholar]
- Kishi N, Macklis JD (2004) MECP2 is progressively expressed in post‐migratory neurons and is involved in neuronal maturation rather than cell fate decisions. Mol Cell Neurosci 27: 306–321 [DOI] [PubMed] [Google Scholar]
- Kron M, Lang M, Adams IT, Sceniak M, Longo F, Katz DM (2014) A BDNF loop‐domain mimetic acutely reverses spontaneous apneas and respiratory abnormalities during behavioral arousal in a mouse model of Rett syndrome. Dis Model Mech 7: 1047–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Bellot‐Saez A, Phillips ML, Yang T, Longo FM, Pozzo‐Miller L (2017) A small‐molecule TrkB ligand restores hippocampal synaptic plasticity and object location memory in Rett syndrome mice. Dis Model Mech 10: 837–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liot G, Zala D, Pla P, Mottet G, Piel M, Saudou F (2013) Mutant Huntingtin alters retrograde transport of TrkB receptors in striatal dendrites. J Neurosci 33: 6298–6309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunkes A, Lindenberg KS, Ben‐Haïem L, Weber C, Devys D, Landwehrmeyer GB, Mandel J‐L, Trottier Y (2002) Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol Cell 10: 259–269 [DOI] [PubMed] [Google Scholar]
- Lyst MJ, Bird A (2015) Rett syndrome: a complex disorder with simple roots. Nat Rev Genet 16: 261–275 [DOI] [PubMed] [Google Scholar]
- Matagne V, Ehinger Y, Saidi L, Borges‐Correia A, Barkats M, Bartoli M, Villard L, Roux J‐C (2017) A codon‐optimized Mecp2 transgene corrects breathing deficits and improves survival in a mouse model of Rett syndrome. Neurobiol Dis 99: 1–11 [DOI] [PubMed] [Google Scholar]
- Meehan RR, Lewis JD, Bird AP (1992) Characterization of MeCP2, a vertebrate DNA binding protein with affinity for methylated DNA. Nucleic Acids Res 20: 5085–5092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moutaux E, Christaller W, Scaramuzzino C, Genoux A, Charlot B, Cazorla M, Saudou F (2018) Neuronal network maturation differently affects secretory vesicles and mitochondria transport in axons. Sci Rep 8: 13429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogier M, Wang H, Hong E, Wang Q, Greenberg ME, Katz DM (2007) Brain‐derived neurotrophic factor expression and respiratory function improve after ampakine treatment in a mouse model of Rett syndrome. J Neurosci 27: 10912–10917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo R, Colin E, Régulier E, Aebischer P, Déglon N, Humbert S, Saudou F (2006) Inhibition of calcineurin by FK506 protects against polyglutamine‐huntingtin toxicity through an increase of huntingtin phosphorylation at S421. J Neurosci 26: 1635–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ (2001) The mouse brain in stereotaxic coordinates, 2nd edn San Diego: Academic Press; [Google Scholar]
- Pineda JR, Pardo R, Zala D, Yu H, Humbert S, Saudou F (2009) Genetic and pharmacological inhibition of calcineurin corrects the BDNF transport defect in Huntington's disease. Mol Brain 2: 33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratte M, Panayotis N, Ghata A, Villard L, Roux J‐C (2011) Progressive motor and respiratory metabolism deficits in post‐weaning Mecp2‐null male mice. Behav Brain Res 216: 313–320 [DOI] [PubMed] [Google Scholar]
- Reddy PH, Tripathi R, Troung Q, Tirumala K, Reddy TP, Anekonda V, Shirendeb UP, Calkins MJ, Reddy AP, Mao P et al (2012) Abnormal mitochondrial dynamics and synaptic degeneration as early events in Alzheimer's disease: implications to mitochondria‐targeted antioxidant therapeutics. Biochim Biophys Acta 1822: 639–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss AL, Faruque F, Naidu S, Abrams M, Beaty T, Bryan RN, Moser H (1993) Neuroanatomy of Rett syndrome: a volumetric imaging study. Ann Neurol 34: 227–234 [DOI] [PubMed] [Google Scholar]
- Roux J‐C, Dura E, Moncla A, Mancini J, Villard L (2007) Treatment with desipramine improves breathing and survival in a mouse model for Rett syndrome. Eur J Neurosci 25: 1915–1922 [DOI] [PubMed] [Google Scholar]
- Roux J‐C, Zala D, Panayotis N, Borges‐Correia A, Saudou F, Villard L (2012) Modification of Mecp2 dosage alters axonal transport through the Huntingtin/Hap1 pathway. Neurobiol Dis 45: 786–795 [DOI] [PubMed] [Google Scholar]
- Sampathkumar C, Wu Y‐J, Vadhvani M, Trimbuch T, Eickholt B, Rosenmund C (2016) Loss of MeCP2 disrupts cell autonomous and autocrine BDNF signaling in mouse glutamatergic neurons. Elife 5: e19374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saudou F, Humbert S (2016) The biology of huntingtin. Neuron 89: 910–926 [DOI] [PubMed] [Google Scholar]
- Schmid DA, Yang T, Ogier M, Adams I, Mirakhur Y, Wang Q, Massa SM, Longo FM, Katz DM (2012) A TrkB small molecule partial agonist rescues TrkB phosphorylation deficits and improves respiratory function in a mouse model of Rett syndrome. J Neurosci 32: 1803–1810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, Illingworth RS, Webb S, Kerr ARW, James KD, Turner DJ, Andrews R, Bird AP (2010) Neuronal MeCP2 is expressed at near histone‐octamer levels and globally alters the chromatin state. Mol Cell 37: 457–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM, Dieterich DC, Ito HT, Kim SA, Schuman EM (2010) Microfluidic local perfusion chambers for the visualization and manipulation of synapses. Neuron 66: 57–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thion MS, McGuire JR, Sousa CM, Fuhrmann L, Fitamant J, Leboucher S, Vacher S, du Montcel ST, Bièche I, Bernet A et al (2015) Unraveling the role of huntingtin in breast cancer metastasis. J Natl Cancer Inst 107: djv208 [DOI] [PubMed] [Google Scholar]
- Tropea D, Giacometti E, Wilson NR, Beard C, McCurry C, Fu DD, Flannery R, Jaenisch R, Sur M (2009) Partial reversal of Rett Syndrome‐like symptoms in MeCP2 mutant mice. Proc Natl Acad Sci USA 106: 2029–2034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Esch H (2012) MECP2 duplication syndrome. Mol Syndromol 2: 128–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viemari J‐C, Roux J‐C, Tryba AK, Saywell V, Burnet H, Peña F, Zanella S, Bévengut M, Barthelemy‐Requin M, Herzing LBK et al (2005) Mecp2 deficiency disrupts norepinephrine and respiratory systems in mice. J Neurosci 25: 11521–11530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virlogeux A, Moutaux E, Christaller W, Genoux A, Bruyère J, Fino E, Charlot B, Cazorla M, Saudou F (2018) Reconstituting corticostriatal network on‐a‐chip reveals the contribution of the presynaptic compartment to Huntington's disease. Cell Rep 22: 110–122 [DOI] [PubMed] [Google Scholar]
- Xu X, Kozikowski AP, Pozzo‐Miller L (2014) A selective histone deacetylase‐6 inhibitor improves BDNF trafficking in hippocampal neurons from Mecp2 knockout mice: implications for Rett syndrome. Front Cell Neurosci 8: 68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JI, Hong EP, Castle JC, Crespo‐Barreto J, Bowman AB, Rose MF, Kang D, Richman R, Johnson JM, Berget S et al (2005) Regulation of RNA splicing by the methylation‐dependent transcriptional repressor methyl‐CpG binding protein 2. Proc Natl Acad Sci USA 102: 17551–17558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zala D, Hinckelmann M‐V, Yu H, Lyra da Cunha MM, Liot G, Cordelières FP, Marco S, Saudou F (2013) Vesicular glycolysis provides on‐board energy for fast axonal transport. Cell 152: 479–491 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Source Data for Expanded View
Review Process File
Source Data for Figure 3
Source Data for Figure 4