

Abstract
Bacterial infections continue to threaten humankind and the rapid spread of antibiotic resistant bacteria is alarming. Current antibiotics target essential bacterial processes and thereby apply a strong selective pressure on pathogenic and non-pathogenic bacteria alike. One alternative strategy is to block bacterial virulence systems that are essential for the ability to cause disease but not for general bacterial viability. We have previously show that the plant natural product (-)-hopeaphenol blocks the type III secretion system (T3SS) in the Gram-negative pathogens Yersinia pseudotuberculosis and Pseudomonas aeruginosa. (-)-Hopeaphenol is a resveratrol tetramer and in the present study we explore various resveratrol dimers, including partial structures of (-)-hopeaphenol, as T3SS inhibitors. To allow rapid and efficient assessment of T3SS inhibition in P. aeruginosa, we developed a new screening method by using a green fluorescent protein reporter under the control of the ExoS promoter. Using a panel of assays we showed that compounds with a benzofuran core structure i.e. viniferifuran, dehydroampelopsin B, anigopreissin A, dehydro-δ-viniferin and resveratrol-piceatannol hybrid displayed significant to moderate activities towards the T3SS in Y. pseudotuberculosis and P. aeruginosa.
Subject terms: Chemical biology, Drug discovery, Microbiology
Introduction
The discovery and introduction of antibiotics is recognized as one of the greatest advances in therapeutic medicine during the 20th century. The possibility to employ antibiotics to directly target essential processes enabled us to take the lead in the arms race with pathogenic bacteria that have plagued humans for centuries. However, we are on the verge of losing this advantage to the ever-evolving microbes that develop and spread antibiotic resistances with alarming speed. To be able to turn the tide, decisive action needs to be taken. It is important to reduce the use of broad-spectrum antibiotics and replace them with pathogen-selective compounds that, in contrast to broad acting agents, do not facilitate acquisition, maintenance and spread of resistance determinants by pathogens and non-virulent bystanders alike. One strategy is to develop therapeutic agents that act on major virulence systems in pathogenic bacteria and thus disarm them and assist the immune defense in clearing the pathogen. The advantage with this strategy is that only the pathogenic bacteria are affected and that the commercial flora is kept intact. In addition, virulence blocking agents are expected to act also on bacterial strains resistant to conventional antibiotics in current use.
One of the important virulence systems in Gram-negative bacteria is the type III secretion system (T3SS). The T3SS is a syringe-like apparatus composed of a basal body spanning the inner and outer membrane of the bacterium and a needle complex protruding from the bacterial surface. Upon host cell contact the system translocates toxins into the cytosol of the eukaryotic cell to induce cell injury, subvert the host defense and promote bacterial proliferation. Antibodies binding to proteins at the tip of the needle structure block T3SS function and promote passive protection in animal infection models1. In addition, mutation of the T3SS reduces infection of eukaryotic cells and mice2,3. The T3SS is conserved among a large number of Gram-negative pathogens and research over the last decades has demonstrated that the T3SS constitutes a validated target for the development of novel pathogen selective therapeutic agents4.
Yersinia spp. and Pseudomonas aeruginosa are two Gram-negative bacteria that harbor very homologous T3SS, but each causes very different diseases. Yesinia constitutes eleven species, of which Y. entercolitica, Y. pestis, and Y. pseudotuberculosis are pathogenic to humans5. Y. pestis, the causative agent of bubonic plague, is probably the most well-known of these since it was the pathogen responsible for the Black Death in the middle of the 14th century. Y. entercolitica and Y. pseudotuberculosis cause inflammation in the gastrointestinal tract of humans and spread through the fecal-oral route, usually from contaminated food or water. Y. pseudotuberculosis is an excellent model organism to study and explore the T3SS as target for therapeutic intervention. The opportunistic pathogen P. aeruginosa is the leading cause of hospital-acquired (nosocomial) infections as well as chronic infections in cystic fibrosis patients. In addition, it causes pneumonia and urinary tract, wound, burn, and bloodstream infections. P. aeruginosa is a “superbug” with a unique capacity to develop resistance6. This is due to a combination of intrinsic, acquired and adaptive resistance. The intrinsic resistance is due to a generally low outer membrane permeability, β-lactamase production and constitutive expression of efflux pumps. Acquired resistance results from horizontal gene transfer and mutations leading to reduced uptake, efflux pump overexpression, target mutations, and expression of antibiotic modifying enzymes such as extended-spectrum β-lactamases. Adaptive resistance is the result of triggering factors such as antibiotics, biocides, polyamines, pH, anaerobiosis, cations, and carbon sources, as well as social behavior in biofilm formation and swarming. These factors modulate expression of genes that lead to increased resistance. This has resulted in multi-drug resistant P. aeruginosa strains for which no effective antibiotic treatment is available; moreover, these strains are becoming more frequent.
(-)-Hopeaphenol, a dihydrobenzofuran based resveratrol tetramer, has been isolated from the leaves of the Papua New Guinean rainforest tree Anisoptera thurifera in gram quantities7. We recently established that this natural product has antibacterial activity towards Y. pseudotuberculosis, Chlamydia trachomatis and P. aeruginosa8. In Y. pseudotuberculosis (-)-hopeaphenol irreversibly blocks the T3SS by an unknown mechanism. (-)-Hopeaphenol can be isolated in substantial quantities from natural sources, but in order to establish structure-activity relationships (SARs) and explore the potential for further development, access to analogs is required. However, synthetic efforts toward (-)-hopeaphenol and derivatives have been challenging9,10 due to the complex core structure composed of multiple fused rings and the presence of a number of stereocenters. As a first step, we therefore turned our attention to simplified hopeaphenol-related structures and synthesized (dihydro)benzofuran resveratrol dimers, additional stilbenoid natural products and analogues including viniferifuran, ampelopsin A and B, resveratrol-piceatannol hybrid and anigopreissin A11,12. Moreover, while (-)-hopeaphenol and related compounds compromise the Lipinski rules of 513 and are at the border of “hard to optimize” structures beyond the rule of 514, the simplified structures of resveratrol dimers and analogues could be more amendable for further exploration.
In this study we tested a set of resveratrol dimers and identified several compounds that block the T3SS in Y. pseudotuberculosis. In addition, we developed a new screening method for P. aeruginosa by using a green fluorescent protein reporter under the control of the ExoS promoter and confirmed activity against this pathogen as well. Fluorescence microscopy was subsequently used to show the interaction of the T3SS inhibitor viniferifuran with bacterial cells.
Results
In this study, we investigated the biological effects of selected natural benzofuran resveratrol dimers and analogues on the T3SS in comparison to (-)-hopeaphenol. These compounds are readily prepared by biomimetic methods or total synthesis and include ε-viniferin, ω-viniferin, ampelopsin B, ampelopsin A, viniferifuran, dehydroampelopsin B, δ-viniferin, dehydro-δ-viniferin, anigopreissin A and a resveratrol-piceatannol hybrid (Table 1, see Methods for details).
Table 1.
Activity against the T3SS and bacterial growth of Y. pseudotuberculosis (see Methods for details).
| Structure | No | Name | E-lux IC50 µM | YopH IC50 µM | Growth inhibition |
|---|---|---|---|---|---|
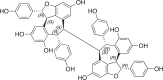 |
1 | (-)-Hopeaphenol | 10 | 7 | No |
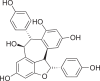 |
2 | (±)-Ampelopsin A | >50 | >50 | No |
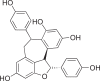 |
3 | (±)-Ampelopsin B | >50 | 40 | No |
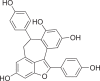 |
4 | (±)-Dehydro-ampelopsin B | 40 | 20 | No |
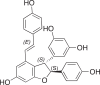 |
5 | (+)-ε-Viniferin | >50 | 15 | No |
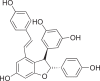 |
6 | (±)-ε-Viniferin | >50 | 30 | No |
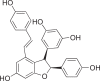 |
7 | (±)-Cis-ε-viniferin (Omega-viniferin) | >50 | 48 | No |
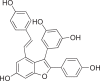 |
8 | Viniferifuran | 10 | 4 | No |
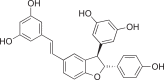 |
9 | (±)-δ-Viniferin | 29 | 15 | No |
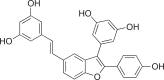 |
10 | Dehydro-δ-viniferin | 5 | 4 | ND* |
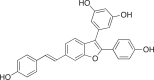 |
11 | Anigopreissin A | 12 | 6 | Yes |
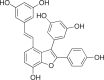 |
12 | Resveratrol-piceatannol hybrid | 15 | 8 | ND* |
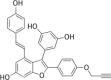 |
13 | Propagylated viniferifuran | 6 | 3 | ND* |
*Not determined.
Inhibition of yopE expression and YopH secretion
The compounds were tested for inhibition of the T3SS in the combined yopE-lux and YopH phosphatase assay for dose-dependent activity as described previously8. In addition, inhibition of bacterial growth was measured to allow identification of T3SS selective inhibitors with little or no effect on bacterial viability. The results are compiled in Table 1. The direct half of (-)-hopeaphenol (1) i.e. ampelopsin A (2) and B (3) as well as dehydroampelopsin B (4), which all contain a central 7-membered ring structure, showed no or modest inhibition of the T3SS. Similar data was obtained for the related opened form compounds ε-viniferin (5) and ω-viniferin (7) (IC50> 50 μM, yopE). The related benzofuran based viniferifuran (8), on the other hand, showed good inhibition of the T3SS (Fig. 1) with an IC50 of 10 μM (yopE) and 4 μM (YopH), without any effect on bacterial growth, i.e. the inhibition profile is similar to that of the original lead (-)-hopeaphenol. The fact that the inhibition profile of the resveratrol tetramer (-)-hopeaphenol is maintained by the resveratrol dimer viniferifuran is encouraging since it has a simplified structure that can be prepared in gram quantities in only four steps11. The resveratrol-piceatannol hybrid (10) that differs from viniferifuran in its hydroxylation pattern has a similar inhibition profile as viniferifuran in vitro. δ-Viniferin (9) with the stilbene substitution at position 6 displayed a moderate activity (IC50 = 29 μM, yopE) and the corresponding benzofuran analog dehydro-δ-viniferin (10) was found to be equally potent as viniferifuran. Anigopreissin A (11), with the stilbene group in position 7, inhibits the T3SS, but its detrimental effect on bacterial growth indicates that the inhibition is non-selective (Fig. S1, Table 1). All compounds with a benzofuran core structure i.e. viniferifuran, dehydroampelopsin B, anigopreissin A, dehydro-δ-viniferin and the resveratrol-piceatannol hybrid displayed significant to moderate activities (IC50 = 5–40 μM, yopE).
Figure 1.

Viniferifuran (8), dehydro-δ-viniferin (10), anigopreissin A (11) and resveratrol-piceatannol hybrid (12) show dose-dependent inhibition of yopE expression (a) and YopH secretion (b). The YPIII(pIB102E-lux) was induced for T3S and compounds were added at 1–100 µM. The graphs show the mean value (n = 3) +/−SD.
Viniferifuran is an irreversible inhibitor of T3SS
Viniferifuran was further tested for reversibility using a Western blot analysis as described previously8. Vinferifuran (8) and (-)-hopeaphenol (1) were added to the bacteria and after induction of T3SS, the compounds were washed away and the bacterial solution was divided into two, where one was treated again with compound under T3SS inducing conditions (-Ca2+) and one was used as a control by adding dimethyl sulfoxide (DMSO) under inducing conditions. Both compounds irreversibly block secretion of effector molecules, i.e. the T3SS could not be reactivated by washing (Fig. 2).
Figure 2.

The effect of viniferifuran (a) and (-)-hopeaphenol (b) on Y. pseudotuberculosis is irreversible. Western blot analysis of the reversibility of viniferifuran and (-)-hopeaphenol treatment of Y. pseudotuberculosis. Lanes 1–4 show the secretion profile of bacteria pretreated with DMSO and lanes 5–8 bacteria pretreated with viniferifuran or (-)-hopeaphenol. After pretreatment the two cultures were divided into four, washed and two of them were diluted in lysogeny broth (LB) supplemented with Ca2+ (non-inducing conditions) and the other two were diluted in Ca2+ depleted LB (inducing conditions). (-)-Hopeaphenol, viniferifuran (40 µM final concentration) or DMSO were then added and subsequetly the protein secretion profiles were analysed.
Binding of viniferifuran to Yersinia pseudotuberculosis
Propagylation of viniferifuran with propargyl bromide resulted in three mono-propargylated compounds that could be separated by chromatography (Supplementary information). These fractions were tested for their biological activity and compound 13 was selected for further experiments due to its retained biological activity (Table 1) as well as the fact that it could be produced in the highest amounts of all of the mono-propargylated products. Y. pseudotuberculosis (YPIII (pIB102, pFU95gfp)), which constitutively expresses green fluorescent protein (GFP), was treated with compound 13 or DMSO followed by cupper catalyzed click chemistry of the propargyl functionality with the azide group of sulfo-cyanine azide. The bacteria were analyzed using confocal spinning disc microscopy (The CellASIC ONIX2 Microfluidic System, Merck). All bacteria display green fluorescence from the constitutively expressed GFP and the bacteria that has bound propagylated viniferifuran labeled with the azide fluorophore appear red. The results showed a clear binding of viniferifuran to the bacteria (Fig. 3b in top panel). Competition experiments showed that the binding could be outcompeted by adding five times more of free viniferifuran compared to the propargylated compound 13 (Fig. 3). Furthermore, the results showed that the compound binds directly to the bacteria without causing aggregation of the bacteria.
Figure 3.

Binding of viniferifuran to bacteria. Top panel shows the YPIII(pIB102, pFU95gfp) strain treated with 10 µM compound 13 followed click chemistry with sulfo-cyanine azide. Bottom panel shows bacteria treated with 10 µM compound 13 and 50 µM viniferifuran followed click chemistry with sulfo-cyanine azide. (a) GFP signal from the bacteria, (b) red signal, showing bacteria labeled with compound 13, (c) all bacteria visible with light microscopy, (d) merged pictures.
Inhibition of exoS expression in P. aeruginosa
The compounds were further tested for efficacy on the opportunistic pathogen P. aeruginosa. A reporter-gene was constructed where the promoter of the P. aeruginosa T3S-toxin, ExoS, was transcriptionally fused to the GFP reporter gene. The gene construct was then cloned into the high copy plasmid pUCP18, together with orf1, coding the chaperon of ExoS15. To be able to select for the plasmid, the gene coding for gentamicin resistant was inserted into the plasmid, creating plasmid pCS500 (Supporting information). The resulting plasmid was inserted into wild-type P. aeruginosa strain PAK and the T3SS activator mutant PAKexsA, resulting in strains that expresses the GFP when the ExoS promoter is activated. The T3SS is induced by incubating the bacteria in calcium depleted medium at 37 °C. The wild-type PAK and the PAKexsA strains with the reporter plasmid were grown under induced conditions and the compounds were added to the growth medium at different concentrations. After 16 h of incubation the GFP signal was measured in a plate reader (Biotek Synergy) and the inhibition of ExoS expression was calculated. The PAK(pCS500) control (no compound added) showed a high GFP level corresponding to a high expression of exoS, while the PAKexsA(pCS500) control strain showed a low level of GFP corresponding to a low level of exoS expression. The level of GFP seen by the PAKexsA(pCS500) strain was equal to a full inhibition of exoS by a fully active compound (Figure S3). Simultaneously, the inhibition of bacterial growth was measured to select for selective T3SS inhibitors. All compounds with a benzofuran core structure i.e. viniferifuran (8), dehydroampelopsin B (4), anigopreissin A (11), dehydro-δ-viniferin (10) and the resveratrol-piceatannol hybrid (12) exhibited significant activities by inhibiting expression of ExoS in a dose dependent manner, with IC50 values of approximately 8–30 µM (Fig. 4 and Table 2). However, compounds containing the central 7-membered ring structure displayed higher IC50 values, as exemplified by comparison of dehydroampelosin B (4, IC50 33 µM) and viniferifuran (8, IC50 11 µM) (Table 2). None of the compounds showed inhibition of bacterial growth and could therefore be considered as selective T3SS inhibitors in P. aeruginosa.
Figure 4.
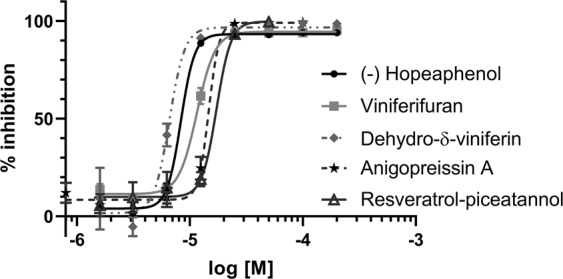
Inhibition of ExoS expression by (-)-hopeaphenol (1), viniferifuran (8), dehydro-δ-viniferin (10), anigopreissin A (11) and resveratrol-piceatannol hybrid (12). Wild-type P. aeruginosa PAK(pCS500) with GFP under the control of the exoS promoter was treated with different concentration of compounds under T3SS-inducing contitions. As controls the the untreated PAK(pCS500) (wt) and PAKexsA(pCS500) (exsA−) were used. The expression of exoS was calculated as percenteage of the untreated wild-type PAK(pCS500). Samples were run in triplicates and the experiment was repeated three times. The graph shows the mean value (n = 3) +/−SD.
Table 2.
Summery of efficacy and toxicity towards P. aeruginosa and eukaryotic cells (J774).
| No | Name | IC50 ExoS-gfp | Tocxicity P.a. | Toxicity* J774 | Efficacy Pa J774 |
|---|---|---|---|---|---|
| 1 | (-)-Hopeaphenol | 9 | No | No | Yes |
| 2 | (±)-Ampelopsin A | >200 | No | No | No |
| 3 | (±)-Ampelopsin B | 134 | No | No | No |
| 4 | (±)-Dehydro-ampelopsin B | 33 | No | No | No |
| 6 | (±)-ε-Viniferin | 89 | No | No | No |
| 8 | Viniferifuran | 11 | No | Tox | Moderate |
| 10 | Dehydro-δ-viniferin | 8 | No | Tox | Yes |
| 11 | Anigopreissin A | 15 | No | Some tox | No |
| 12 | Resveratrol-piceatannol hybrid | 14 | No | Some tox | Yes |
*For more information about toxicity on J774 cells, see Fig. S2.
Inhibition of T3S-mediated cytotoxicity of eukaryotic cells
The T3SS is important for P. aeruginosa infection of eukaryotic cells. A T3SS activator mutant or a mutant lacking the T3SS tip protein PcrV is not able to infect eukaryotic cells16,17. The J774 cells (ATCC) were infected with the wild-type PAK strain and the T3SS mutant with or without compounds. The T3SS mediated cytotoxic effect on the eukaryotic cells was measured by using a viability staining method, Uptiblue (Interchim), after 4, 5 and 6 h of infection. Simultaneously, uninfected cells were treated with compounds and toxicity towards eukaryotic cells was determined using the same assay. The cells were also studied by microscopy after 6 h to assess cell morphology. The results showed a clear dose-dependent rescue effect of (-)-hopeaphenol (1), with complete inhibition of cytotoxicity at 50 µM, which is in agreement with previous results8 (Fig. 5). Additionally, dehydro-δ-viniferin (10) and resveratrol-pinceatannol hybrid (12) showed a significant dose-dependent rescue effect on eukaryotic cells, while viniferifuran (8), (±)-ε-viniferin (5) and anigopreissin A (11) showed modest or no efficacy (Fig. 5). Viniferifuran (8) and dehydro-δ-viniferin (10) showed substantial toxicity towards eukaryotic cells at 100 µM, while some toxicity could be seen for the resveratrol-pinceatannol hybrid (12, Figure S2). Toxicity towards eukaryotic cells limits the possibility to study and accurately assess virulence blocking properties of these compounds.
Figure 5.

Effect of the (-)-hopeaphenol analogues on P. aeruginosa infection. J774 cells were infected with wild-type P. aeruginosa strain PAK and compounds were added and the T3SS dependent cell survival was analysed using a viability staining method. As controls uninfected cells, cells infected with the PAKpcrV-mutant and cells infected with the wild-type strain (PAK) were used. The survival was calculated as percenteage of the uninfected control. Samples were run in triplicates and the experiment were repeated three times. Viniferifuran (8) and dehydro-δ-viniferin (10) were very toxic at 100 µM, while some toxicity could be seen for resveratrol-pinceatannol hybrid (12). The graph shows the mean value (n = 3) +/−SD after 6 h of infection.
Discussion
Most antibiotics in clinical use today are either natural products of microbial origin or improved derivatives obtained by synthesis. Development of new antibiotics based on new synthetic scaffolds have proved to be challenging and this holds true also for identification of chemical entities that block virulence systems by mechanisms distinct from conventional antibiotics e.g. attenuation of T3SS in Gram-negative pathogens. A substantial number of synthetic T3SS inhibitors have been identified by predominantly phenotypic screening, and a selection of compounds have entered medicinal chemistry programs as summarized in recent review articles18,19. Several studies indicate that natural sources also have the potential to provide T3SS inhibitors for further exploration. Screening of marine invertebrate extracts led to the identification of the glycolipid caminoside A as an inhibitor of T3SS in enteropathogenic Eschericia coli (EPEC)20 and later caminocide B-D were also found to be T3SS inhibitors21. In addition to activity against T3SS in EPEC, the caminosides also display antimicrobial activity against vancomycin resistant Enterococcus and methicillin resistant Staphylococcus aureus, suggesting that these compounds act as general antibiotics rather than selective T3SS inhibitors20,21. In another study guadinimine A-F, isolated from a soil-derived Streptomyces broth, were found to block T3SS dependent hemolysis of erythrocytes by EPEC22,23. Aurodox is a known actinomycete derived antibiotic that inhibits protein biosynthesis by binding to bacterial elongation factor Tu22,23. Recently it was reported that aurodox inhibited EPEC T3SS-mediated hemolysis and protected mice infected with a lethal dose of Citrobacter rodentium24. The mechanism for its T3SS inhibition has however not been established. Pseudoceramines A-D and spermatinamine produced by the marine sponge Pseudoceratina spp. were identified as putative inhibitors of T3SS in Y. pseudotuberculosis, but the compounds also exhibited a detrimental effect on bacterial growth, indicating general antibacterial properties25. Phenolic compounds involved in plant defense signaling have been reported to block the T3SS in P. aeruginosa via the GacS-GacA two-component signal transduction system26. Cytosporone B, a fungal metabolite, and analogues selectively block the secretion of Salmonella pathogenicity island 1 (SPI-1)-associated effector proteins, without significant toxicity against bacteria27 and similar effects have been reported for prenylated plant flavonoids, of which licoflavonol proved to be the most promising compound28. It was recently reported that the T3SS of Y. pseudotuberculosis is effectively blocked by piericidin A1, a metabolite produced by a marine actinobacterium29. The compound was identified by screening of a library composed of marine-derived extracts and its virulence blocking activity is not attributed to general toxicity towards the bacteria.
Based on our finding that the plant derived complex resveratrol tetramer (-)-hopeaphenol (1) is an irreversible T3SS inhibitor we tested a series of resveratrol dimers that can be obtained by biomimetic or total synthesis. We identified the resveratrol dimer viniferifuran (8) and analogues as inhibitors of T3SS that target both Y. pseudotuberculosis and P. aeruginosa. Since we could observe inhibition of T3SS-toxins in both Y. pseudotuberculosis and P. aeruginosa it is not likely that the resveratrol dimers act on the toxins per se, since they are different between the bacterial species. The compounds rather act on a homologous function of the T3SS as the needle like structure or tip-proteins on the bacterial surface or some common regulatory system. Viniferifuran was found to irreversibly block the T3SS, indicating that (-)-hopeaphenol and viniferifuran might act by the same mechanism, although the chemical structures are substantially different. The efficacy of viniferifuran in cell infection experiments could not be accurately assessed due to its toxicity toward J774 cells.
Due to its ease of preparation, we still consider viniferifuran a promising scaffold amendable for medicinal chemistry to optimize its potency toward diverse Gram-negative pathogens and reduce its toxicity. The resveratrol-piceatannol hybrid (12) and the dehydro-δ-viniferin (10) showed better efficacy to rescue eukaryotic cells from infection, however they both show toxicity towards the eukaryotic cells at high concentrations, even though the toxicity of the resveratrol-piceatannol hybrid is minor compared to viniferifuran. (-)-Hopeaphenol is non-toxic toward both bacteria and eukaryotic cells and possibly this can be attributed to its size, which likely prevents cell permeability and suggest that (-)-hopeaphenol acts on the bacterial surface.
Incubation of Y. pseudotuberculosis with the monopropargylated viniferifuran derivative 13, followed by click chemistry using sulfo-cyanine azide and subsequent confocal spinning disc microscopy, suggested that viniferifuran might be located at the outer membrane of the bacteria but an intracellular localization cannot be excluded. Importantly, the pictures showed that the compound binds to the bacteria and the T3SS inhibition is not due to e.g. aggregation of bacteria.
Interestingly, all the most promising compounds in the present study share the unsaturated benzofuran scaffold as opposed to parent compound (-)-hopeaphenol that contains the related dihydrobenzofuran scaffold. Other compounds with the dihydrobenzofuran core e.g. ampelopsin A (2) and B (3) were essentially void of activity.
In conclusion, we have identified resveratrol dimers as novel inhibitors of T3SS in Y. pseudotuberculosis and P. aeruginosa. Compared to the parent compound, (-)-hopeaphenol, these compounds are amendable to medicinal chemistry and are thus useful as starting points to develop lead compounds and initiate drug discovery programs.
Methods
Compounds
(-)-Hopeaphenol (1) was obtained from extraction of Anisoptera thurifera leafs as described previously7 and (+)-ε-viniferin (5) was from Sigma Aldrich. (±)-ε-Viniferin (6), (±)-ω-viniferin (7), (±)-ampelopsin B (3), viniferifuran (8), (±)-dehydroampelopsin B (4) were prepared following a biomimetic or total synthetic procedures11,12. (±)-Ampelopsin A (2) was prepared from ε-viniferin following a biomimetic procedure30. Anigopreissin A (11) was isolated from the Australian plant Macropidia fuliginosa31 or prepared as reported previously11. Dehydro-δ-viniferin (10) and resveratrol-piceatannol hybrid (11) were prepared as described previously11.
yopE reporter gene and YopH phosphatase assays
The combined luciferase and phosphatase assay was performed essentially as previously described8. The Y. pseudotuberculosis serotype III strain YPIII(pIB102E-lux) (Table 3) was grown overnight in Luria broth (LB) supplemented with chloramphenicol, diluted to an OD600 of 0.08 in calcium depleted LB medium (addition of ethyleneglycol- bis(β-aminoethyl)-N,N,Nʹ,Nʹ-tetraacetic acid (EGTA) and MgCl2, at a final concentration of 5 mM and 20 mM, respectively) and incubated in 96-well plate 100 µl/well (Nunc flat bottom, white). The compounds were added to a final concentration between 1 and 100 µM. The plate was incubated for 1 h at 26 °C and 2 h at 37 °C, shaking. After incubation 10 µl of the bacterial suspension was transformed to a new 96-well plate (Nunc flat bottom, transparent) containing 90 µl of phosphatase mixture (25 mM p-nitro phenyl phosphatase, 40 mM 2-(N-morpholino) ethanesulfonic acid, pH 5.0, and 1.6 mM dithiotheitol in water). The plate was incubated for 15–20 min at 37 °C. 20 µl 1 M NaOH was added to stop the reaction and the absorbance, i.e. YopH secretion, was measured at 405 nm in a plate reader (Wallac Viktor2 1420 Multilabel counter). The remaining 90 µl of bacterial suspension in the white 96-well plate was used for measuring the luciferase activity, i.e. the yopE expression), by adding 50 µl of decanal in water (5 µl/500 ml) and the chemiluminescence was detected in a microplate reader (Wallac Viktor2 1420 Multilabel counter). The IC50 values were calculated using GraphPad Prism software.
Table 3.
Characteristics and references of bacterial strains and crusal plasmids used in this study.
| Strain or plasmid | Characteristics and genotype | Reference/source |
|---|---|---|
| Yersinia pseudotuberculosis | ||
| YPIII(pIB102) | yadA | 32 |
| YPIII(pIB102-Elux) | Luciferace reporter under the control of yopE promoter | 33 |
| YPIII(pIB102; pFU95) | Constitutive gfp expression | This paper |
| Pseudomonas aeruginosa | ||
| PAK | Wild-type | FOI* |
| PAKpcrV | pcrV | 17 |
| PAKexsA | exsA | 34 |
| PAK(pCS500) | gfp under the control of the exoS promoter | This paper |
| PAKexsA(pCS500) | exsA, gfp under the control of the exoS promoter | This paper |
| Relevant plasmids | ||
| pFU95 | Constitutive gfp expression | 35 |
| pTS103 | Source of Orf1 and exoS | 15 |
| pCS500 | gfp under the control of the exoS promoter, Gm resistant | This paper |
| pRIC-gfp | Source of gfp | FOI* |
| p34S-Gm | ApR GmR, source of GmR cassette | 36 |
*Swedish Defence Research Agency.
Inhibition of bacterial growth
For growth control, the Y. pseudotuberculosis serotype III strain YPIII(pIB102E-lux) (Table 3) was grown overnight in LB supplemented with chloramphenicol, diluted to an OD600 of 0.2 in LB medium with 2.5 mM CaCl2 and added into 96-well plate 100 µl/well (Nunc flat bottom, transparent). Compounds were then added and the plate was incubated at 37 °C for 24 h, shaking. The absorbance at OD600 was measured every hour for 8 h and then after 24 h in a microplate reader (Wallac Viktor2 1420 Multilabel counter).
Western blot analysis of supernatant samples of Y. pseudotuberculosis for studying reversibility
The experiment was performed essentially as described earlier8. A final concentration of 40 µM of the compounds were added to a 1:20 diluted overnight culture of YPIII(pIB102). Control cultures received only DMSO. The cultures were incubated at 26 °C for 30 min followed by 2 h at 37 °C to induce T3SS. The samples were then centrifuged and the pellet washed once with LB where after it was suspended in LB complemented with 20 mM MgCl2 and 5 mM EGTA for Ca2+ depletion (induction of T3SS) or 2.5 mM CaCl2. The samples were divided and 40 µM compound or DMSO was added followed by incubation for 45 min at 37 °C. The cultures supernatant was mixed with sample buffer and loaded on to a 12% sodium dodecyl sulfate (SDS) polyacrylamide gel and blotted onto a PVDF membrane. Polyclonal rabbit anti Yop-antiserum and polyclonal goat anti-rabbit immunoglobulin conjugated with horseradish peroxidase (HRP) (Dako Denmark) were used together with Millipore Immobilon Western chemiluminescent HRP substrate to determine Yop proteins in the supernatant. The chemiluminiscense signals were detected with a charge-coupled device (CCD) camera.
Visualizing viniferifuran binding to bacterial cells
The Y. pseudotuberculosis serotype III strain YPIII (pIB102; pFU95) (Table 3) was grown overnight in LB supplemented with carbenicillin. 100 µl YPIII (pIB102; pFU95) without compound in calcium depleted LB medium at an OD600 of 0.11, was mixed with propagylated viniferifuran (13) to a final concentration of 10 µM. To investigate selectivity of the compound viniferifuran (8) was added to a final concentration of 50 µM together with propagylated viniferifuran (13). The DMSO concentration was kept to 1% in all samples and bacteria treated only with 1% DMSO were used as control. The samples were incubated in 96-well plates for 1 h at 26 °C and 2 h in 37 °C with shaking. After incubation, the bacteria were diluted 1/5 in phosphate buffered saline (PBS) and 50 µl was loaded into a CellASIC ONIX B04A-03 Microfluidic Bacteria Plate of the CellASIC ONIX2 Microfluidic System (Merck), with a flow rate of 15 s at 4 psi twice to get them into the plate layers. After additional 5–10 times at 6 psi the bacteria reached the correct position (trap heights of 0.7 µm).
The click chemistry reaction mixture was made with a final concentration of CuSO4 (0.1 mM), tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA, 128 µM), sodium ascorbate (5 mM), sulfo-cyanine5 azide (100 µM, Lumiprobe, ex/em 646/662) in PBS. The plate was washed with for PBS 10 min at 4 psi, where after the click reaction mix was added for 5 min at 8 psi and 60 min at 4 psi. Finally, the plate was washed with PBS for 10 min at 8 psi and 20 min at 4 psi and the results were analyzed using a Confocal spinning disc microscope, far red 647 laser. The plate was incubated at 37 °C during the experiment.
Cloning and exoS expression assay
The pRIC-GFP plasmid was cut with PstI and EcoRI and the GFP fragment was purified and ligated into pTS103 plasmid15 cut with NsiI and EcoRI. The resulting plasmid was purified and cut with BamHI and EcoRI and ligated into the pUCP18 plasmid cut with BamHI and EcoRI. The resulting plasmid was cut with ScaI (blunt, cutting in the amp gene) and the gentamicin (Gm) gene was inserted by cleaving the p34S-Gm plasmid with Ecl136II (SstI) (blunt). The resulting plasmid, pCS500, was resistant to gentamicin and the gfp-gene is transcriptionally fused with the exoS promoter. pCS500 was transformed into the P. aeruginosa strain PAK and the T3SS activator mutant PAKexsA.
Overnight cultures of PAK(pCS500) and PAKexsA(pCS500) were diluted 1:30 in LB supplemented with 3 µg/ml Gm and incubated at 37 °C for 6 h on a rotary shaker. The cultures were then diluted to an OD600 of 0.0004 in calcium deleted LB supplemented with 3 µg/ml Gm and added into a flat-bottom 96-well transparent plate (Nunc). Compounds were added to give a final concentration of 1–100 µM. As a positive control 1% DMSO was added to the PAK(pCS500) strain and as a negative control 1% DMSO was added to the PAKexsA (pCS500). The fluorescence was measured after 16 h of incubation on a rotary shaker (ex/em, 488/509) in a plate reader (Biotek Synergy). The value for the wt PAK(pCS500) strain with no additives was set to 100% GFP expression (i.e. 100% ExoS expression). The IC50 values were calculated using GraphPad Prism software. Inhibition of bacterial growth was also measured after 16 h using the same plate by measurement of the absorbance at OD600. All experiments were carried out in triplicates.
Infection and toxicity assay
J774 cells (ATCC) was seeded to a concentration of 5 × 104 cells/well (100 µl/well) in 96-well plates and incubated 18 h prior to infection in Dulbecco’s modified eagle medium (DMEM, GlutaMAX (Glu), 3 µg/ml Gentamicin (Gm) + 10% FCS). Overnight cultures of PAK and PAKpcrV were diluted 1:10 in DMEM (Glu) medium and incubate at 37 °C, 250 rpm for 1 h. Meanwhile, the cells were wash once with PBS pH 7.2, where after 30 µl of compound solutions was added diluted in DMEM (Glu, 10% FCS). 1% DMSO was added to the control wells. 30 µl of the induced PAK bacteria solution, OD600 = 0.0008, was then added to the wells giving a final OD600 of 0.0004. PAKpcrV mutant, deficient for translocation of exoenzymes, was used to control that the effect on the cells was T3SS dependent. In a parallel plate, the compound solutions were added without bacteria to screen for toxicity. A viability staining method to investigate cell survival, after 2.5 h, 10 µl Uptiblue (Interchim, France) was added to each well, efficacy and toxicity of the compounds was measured 4, 5 and 6 h after infection (ex/em, 535/595) in a plate reader (Biotek Synergy). For efficacy, the compounds were tested at 50, 25, 12.5 and 6.25 µM in triplicates.
Acknowledgements
We would like to thank the Swedish Research Council for finical support, Irene Martinez Carrasco at Biochemical Imaging Centre Umeå (BICU), Umeå University for technical support with confocal spinning disc microscopy, Rémi Caraballo, Anders Lindgren and Laura Lekam Umeå University for technical assistance and Åke Forsberg FOI/Umeå University for the kind gifts of plasmids and bacterial strains.
Supplememtary information
Author contributions
Planning experiments and analyzing data; C.S., C.Z., D.D.V. and M.E. Performing experiments; C.S. (Figures 1, 4, 5, S2, S3 and cloning) and C.Z. (Figures 1, 2, 3 and S1). Compound synthesis; D.D.V. Providing compounds; R.B. and S.U. (anigopreissin A31), D.D.V. and M.E. (previously reported resveratrol dimers and analogues11). Writing the manuscript; C.S. and M.E. Critical reading the manuscript; C.S., C.Z., D.D.V., S.U. and M.E. Approving the manuscript; all authors.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-020-58872-0.
References
- 1.Baer M, et al. An engineered human antibody fab fragment specific for Pseudomonas aeruginosa PcrV antigen has potent antibacterial activity. Infect. Immun. 2009;77:1083–1090. doi: 10.1128/IAI.00815-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anantharajah A, et al. Inhibition of the Injectisome and Flagellar Type III Secretion Systems by INP1855 Impairs Pseudomonas aeruginosa Pathogenicity and Inflammasome Activation. J. Infect. Dis. 2016;214:1105–1116. doi: 10.1093/infdis/jiw295. [DOI] [PubMed] [Google Scholar]
- 3.Uusitalo P, et al. The salicylidene acylhydrazide INP0341 attenuates Pseudomonas aeruginosa virulence in vitro and in vivo. J. Antibiot. 2017;70:937–943. doi: 10.1038/ja.2017.64. [DOI] [PubMed] [Google Scholar]
- 4.Hauser AR. The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat. Rev. Microbiol. 2009;7:654–665. doi: 10.1038/nrmicro2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cornelis GR. The Yersinia Ysc-Yop ‘type III’ weaponry. Nat. Rev. Mol. Cell Biol. 2002;3:742–752. doi: 10.1038/nrm932. [DOI] [PubMed] [Google Scholar]
- 6.Breidenstein EB, de la Fuente-Nunez C, Hancock RE. Pseudomonas aeruginosa: all roads lead to resistance. Trends Microbiol. 2011;19:419–426. doi: 10.1016/j.tim.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 7.Davis RA, et al. Solving the supply of resveratrol tetramers from Papua New Guinean rainforest anisoptera species that inhibit bacterial type III secretion systems. J. Nat. Prod. 2014;77:2633–2640. doi: 10.1021/np500433z. [DOI] [PubMed] [Google Scholar]
- 8.Zetterstrom CE, et al. The resveratrol tetramer (-)-hopeaphenol inhibits type III secretion in the gram-negative pathogens Yersinia pseudotuberculosis and Pseudomonas aeruginosa. PLoS One. 2013;8:e81969. doi: 10.1371/journal.pone.0081969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keylor MH, et al. Synthesis of resveratrol tetramers via a stereoconvergent radical equilibrium. Sci. 2016;354:1260–1265. doi: 10.1126/science.aaj1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takaya Y, Yan KX, Terashima K, He YH, Niwa M. Biogenetic reactions on stilbenetetramers from Vitaceaeous plants. Tetrahedron. 2002;58:9265–9271. doi: 10.1016/S0040-4020(02)01191-2. [DOI] [Google Scholar]
- 11.Vo DD, Elofsson M. Total Synthesis of Viniferifuran, Resveratrol-Piceatannol Hybrid, Anigopreissin A and Analogues - Investigation of Demethylation Strategies. Adv. Synth. Catal. 2016;358:4085–4092. doi: 10.1002/adsc.201601089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lindgren, A. E. G., Oberg, C. T., Hillgren, J. M. & Elofsson, M. Total Synthesis of the Resveratrol Oligomers (+/−)-Ampelopsin B and (+/−)-E-Viniferin. Eur J Org Chem, 426–429 (2016).
- 13.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug. Deliv. Rev. 2001;46:3–26. doi: 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 14.Doak BC, Over B, Giordanetto F, Kihlberg J. Oral druggable space beyond the rule of 5: insights from drugs and clinical candidates. Chem. Biol. 2014;21:1115–1142. doi: 10.1016/j.chembiol.2014.08.013. [DOI] [PubMed] [Google Scholar]
- 15.Frithz-Lindsten E, Du Y, Rosqvist R, Forsberg A. Intracellular targeting of exoenzyme S of Pseudomonas aeruginosa via type III-dependent translocation induces phagocytosis resistance, cytotoxicity and disruption of actin microfilaments. Mol. Microbiol. 1997;25:1125–1139. doi: 10.1046/j.1365-2958.1997.5411905.x. [DOI] [PubMed] [Google Scholar]
- 16.Sundin C, Hallberg B, Forsberg A. ADP-ribosylation by exoenzyme T of Pseudomonas aeruginosa induces an irreversible effect on the host cell cytoskeleton in vivo. FEMS Microbiol. Lett. 2004;234:87–91. doi: 10.1111/j.1574-6968.2004.tb09517.x. [DOI] [PubMed] [Google Scholar]
- 17.Sundin C, Wolfgang MC, Lory S, Forsberg A, Frithz-Lindsten E. Type IV pili are not specifically required for contact dependent translocation of exoenzymes by Pseudomonas aeruginosa. Microb. pathogenesis. 2002;33:265–277. doi: 10.1006/mpat.2002.0534. [DOI] [PubMed] [Google Scholar]
- 18.Gu L, Zhou S, Zhu L, Liang C, Chen X. Small-Molecule Inhibitors of the Type III Secretion System. Molecules. 2015;20:17659–17674. doi: 10.3390/molecules200917659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marshall NC, Finlay BB. Targeting the type III secretion system to treat bacterial infections. Expert. Opin. Ther. Targets. 2014;18:137–152. doi: 10.1517/14728222.2014.855199. [DOI] [PubMed] [Google Scholar]
- 20.Linington RG, et al. Caminoside A, an antimicrobial glycolipid isolated from the marine sponge Caminus sphaeroconia. Org. Lett. 2002;4:4089–4092. doi: 10.1021/ol0268337. [DOI] [PubMed] [Google Scholar]
- 21.Linington RG, et al. Caminosides B-D, antimicrobial glycolipids isolated from the marine sponge Caminus sphaeroconia. J. Nat. Products. 2006;69:173–177. doi: 10.1021/np050192h. [DOI] [PubMed] [Google Scholar]
- 22.Iwatsuki M, et al. Guadinomines, Type III secretion system inhibitors, produced by Streptomyces sp. K01-0509. II: physico-chemical properties and structure elucidation. J. Antibiot. 2008;61:230–236. doi: 10.1038/ja.2008.33. [DOI] [PubMed] [Google Scholar]
- 23.Iwatsuki M, et al. Guadinomines, Type III secretion system inhibitors, produced by Streptomyces sp. K01-0509. I: taxonomy, fermentation, isolation and biological properties. J. Antibiot. 2008;61:222–229. doi: 10.1038/ja.2008.32. [DOI] [PubMed] [Google Scholar]
- 24.Kimura K, et al. A small-molecule inhibitor of the bacterial type III secretion system protects against in vivo infection with Citrobacter rodentium. J. Antibiot. 2011;64:197–203. doi: 10.1038/ja.2010.155. [DOI] [PubMed] [Google Scholar]
- 25.Yin S, et al. Pseudoceramines A-D, new antibacterial bromotyrosine alkaloids from the marine sponge Pseudoceratina sp. Org. Biomol. Chem. 2011;9:6755–6760. doi: 10.1039/c1ob05581j. [DOI] [PubMed] [Google Scholar]
- 26.Yamazaki A, et al. Derivatives of plant phenolic compound affect the type III secretion system of Pseudomonas aeruginosa via a GacS-GacA two-component signal transduction system. Antimicrob. Agents Chemother. 2012;56:36–43. doi: 10.1128/AAC.00732-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li J, et al. Cytosporone B, an inhibitor of the type III secretion system of Salmonella enterica serovar Typhimurium. Antimicrob. Agents Chemother. 2013;57:2191–2198. doi: 10.1128/AAC.02421-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guo ZX, et al. Licoflavonol is an inhibitor of the type three secretion system of Salmonella enterica serovar Typhimurium. Biochem. Bioph Res. Co. 2016;477:998–1004. doi: 10.1016/j.bbrc.2016.07.018. [DOI] [PubMed] [Google Scholar]
- 29.Duncan MC, et al. An NF-kappaB-based high-throughput screen identifies piericidins as inhibitors of the Yersinia pseudotuberculosis type III secretion system. Antimicrob. Agents Chemother. 2014;58:1118–1126. doi: 10.1128/AAC.02025-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takaya Y, Yan KX, Terashima K, Ito J, Niwa M. Chemical determination of the absolute structures of resveratrol dimers, ampelopsins A, B, D and F. Tetrahedron. 2002;58:7259–7265. doi: 10.1016/S0040-4020(02)00785-8. [DOI] [Google Scholar]
- 31.Brkljaca R, White JM, Urban S. Phytochemical Investigation of the Constituents Derived from the Australian Plant Macropidia fuliginosa. J. Nat. Products. 2015;78:1600–1608. doi: 10.1021/acs.jnatprod.5b00161. [DOI] [PubMed] [Google Scholar]
- 32.Bolin I, Wolf-Watz H. Molecular cloning of the temperature-inducible outer membrane protein 1 of Yersinia pseudotuberculosis. Infect. Immun. 1984;43:72–78. doi: 10.1128/IAI.43.1.72-78.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Forsberg A, Rosqvist R. In vivo expression of virulence genes of Yersinia pseudotuberculosis. Infect. Agents Dis. 1993;2:275–278. [PubMed] [Google Scholar]
- 34.Frank DW, Nair G, Schweizer HP. Construction and Characterization of Chromosomal Insertional Mutations of the Pseudomonas-Aeruginosa Exoenzyme-S Transregulatory Locus. Infect. Immun. 1994;62:554–563. doi: 10.1128/IAI.62.2.554-563.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Uliczka, F. et al. Monitoring of Gene Expression in Bacteria during Infections Using an Adaptable Set of Bioluminescent, Fluorescent and Colorigenic Fusion Vectors. Plos One6 (2011). [DOI] [PMC free article] [PubMed]
- 36.Dennis JJ, Zylstra GJ. Plasposons: modular self-cloning minitransposon derivatives for rapid genetic analysis of gram-negative bacterial genomes. Appl. Env. Microbiol. 1998;64:2710–2715. doi: 10.1128/AEM.64.7.2710-2715.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]