

Abstract
Risankizumab has demonstrated efficacy in phase III trials in patients with moderate‐to‐severe plaque psoriasis. The exposure–response relationships for risankizumab efficacy (Psoriasis Area and Severity Index (PASI)75, PASI90, PASI100, and static Physician's Global Assessment (sPGA)0/1) at week 16 (N = 1,732) and week 52 (N = 598) as well as safety (incidence of any adverse event, serious adverse event, infection and infestation, or serious infection) over up to 52 weeks were characterized using the data from risankizumab phase II and III clinical trials in patients with moderate‐to‐severe plaque psoriasis. Impact of clinically relevant covariates was evaluated. Risankizumab phase III regimen (150 mg subcutaneously at weeks 0, 4, and every 12 weeks thereafter) achieved the plateau of the exposure–efficacy relationships with model‐estimated PASI90 and sPGA0/1 response probabilities of 77%, and 87%, respectively, at week 16 and 85%, and 88%, respectively, at week 52. There was no apparent relationship between risankizumab plasma exposure and the evaluated safety variables.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Risankizumab is a humanized immunoglobulin G1 monoclonal antibody that selectively blocks the activation of the interleukin (IL)‐23‐mediated signaling pathway and the release of proinflammatory cytokines.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ This study characterized the relationships between risankizumab plasma exposures and its efficacy (measured by Psoriasis Area and Severity Index (PASI)75, PASI90, PASI100, and static Physician's Global Assessment (sPGA)0/1) and safety (incidence of any adverse event, serious adverse event, infection and infestation, or serious infection) using the data from risankizumab phase II and III clinical trials in moderate‐to‐severe chronic plaque psoriasis patients.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ The analyses presented demonstrate that risankizumab phase III regimen (150 mg subcutaneous (SC) dose at weeks 0, 4, and every 12 weeks thereafter) maximized efficacy in moderate‐to‐severe chronic plaque psoriasis with no apparent relationship between risankizumab exposure and evaluated safety variables.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ The presented analyses supported adequacy of the proposed clinical regimen of risankizumab in patients with moderate‐to‐severe chronic plaque psoriasis.
Plaque psoriasis is a chronic, immune‐mediated, inflammatory, systemic disease associated epidermal thickening of the skin with prevalence estimated to be between 1% and 4% in the Western countries.1, 2 Symptoms of plaque psoriasis typically include painful, itchy, well demarcated, erythematous plaques covered with a silver scale that can negatively affect psychological well‐being and quality‐of‐life. Furthermore, patients with psoriasis are more likely to develop several comorbidities, including psoriatic arthritis, cardiovascular diseases, inflammatory bowel disease, and depression.3
Interleukin (IL)‐23 plays a pivotal role in the pathogenesis of several autoimmune disorders, including psoriasis.4 IL‐23 stimulates the proliferation and differentiation of T‐helper‐17 cells, which in turn are major sources of proinflammatory cytokines, including IL‐17.5 A growing body of evidence suggests that dysregulated IL‐23/IL‐17 responses contribute to the chronic inflammation underlying the pathophysiology of psoriasis. Several therapies targeting either IL‐23 or IL‐17 have shown efficacy in treating such disease conditions.6, 7, 8
Risankizumab is a humanized immunoglobulin G1 (IgG1) monoclonal antibody that selectively binds with high affinity to the p19 subunit of IL‐23,9 thereby blocking the activation of the IL‐23‐mediated signaling pathway and the release of proinflammatory cytokines. In a phase II study in patients with moderate‐to‐severe plaque psoriasis, risankizumab showed superior efficacy and rapid and durable skin clearance compared with ustekinumab.9 Risankizumab 150 mg dose administered subcutaneously (SC) at week 0, week 4, and every 12 weeks (q12w) thereafter was evaluated in four phase III clinical trials in patients with moderate‐to‐severe chronic plaque psoriasis (UltIMMa‐1 (NCT02684370),10 UltIMMa‐2 (NCT02684357),10 IMMvent (NCT02694523),11 and IMMhance (NCT02672852)9). The coprimary endpoints in these phase III trials were PASI90 response (i.e., proportion of patients who achieved at least 90% reduction in baseline Psoriasis Area and Severity Index (PASI) score) and sPGA (static Physician's Global Assessment) response of clear or almost clear skin (sPGA0/1) at week 16. Additional key efficacy endpoints were PASI90 and sPGA0/1 responses at week 52 and PASI75 and PASI100 responses (representing 75% and 100% reduction in baseline PASI score, respectively) at week 16 and week 52. The results from these phase III trials showed that risankizumab achieved significantly greater (P < 0.001) rates of skin responses (84–88% of patients with clear or almost clear skin) than ustekinumab (62–63% of patients), adalimumab (60% of patients) or placebo (7%) after 16 weeks of treatment.10, 11, 12
The objectives of the analyses presented herein were to characterize the relationships between risankizumab plasma exposure and key efficacy response variables, such as PASI75, PASI90, PASI100, and sPGA0/1 using data from one phase II and four phase III clinical trials in patients with moderate‐to‐severe chronic plaque psoriasis, and to assess the effect of patient‐specific covariates on the exposure–response relationships to support dosing recommendation of risankizumab in different patient subpopulations. In addition, the relationships between risankizumab plasma exposure and key safety variables, including any adverse events, serious adverse events, infections and infestations, and serious infections were also explored to evaluate the recommended dose of risankizumab in patients with moderate‐to‐severe chronic plaque psoriasis.
Results
Participant demographics and baseline characteristics
Data from a total of 1,732 patients were included in the week 16 exposure–efficacy analyses. Subsets of these patients had data available at week 52 for inclusion in the week 52 exposure–efficacy analyses (N = 598) and the exposure–safety analyses over the first 16 weeks (N = 1,606) and up to the first 52 weeks (N = 1,590) as described in the methods section. Demographics and baseline characteristics of the patients included in each analysis are summarized in Table 1. The majority of the psoriasis patients included in the analyses were males with mean age and bodyweight of 47 years and 90 kg, respectively. Approximately 11–15% of patients were of Asian race, while majority of the patients were white or other races. The mean baseline PASI score was ~20 and all patients had baseline sPGA score of at least 3. There were no substantial differences in characteristics among the populations for week 16 or week 52 efficacy or safety analyses.
Table 1.
Demographic and baseline characteristics of the patients included in the exposure–efficacy and exposure–safety analyses at weeks 16 and 52
| Characteristic | Exposure–efficacy | Exposure–safety | ||
|---|---|---|---|---|
| Week 16 (N = 1,732) | Week 52 (N = 598) | Week 16 (N = 1,606) | Week 52 (N = 1,590) | |
| Treatment | ||||
| 18 mg SC, N (%) | 43 (2.5) | — | — | — |
| 90 mg SC, N (%) | 41 (2.4) | — | — | — |
| 150 mg SC, N (%) | 1,306 (75.4) | 598 (100) | 1,306 (81) | 1,306 (82) |
| 180 mg SC, N (%) | 42 (2.4) | — | — | — |
| Placebo, N (%) | 300 (17.3) | — | 300 (19) | 284 (18) |
| Age (years) | ||||
| Mean (SD) | 47.5 (13.6) | 47.2 (13.6) | 47.6 (13.6) | 47.5 (13.6) |
| Body weight (kg) | ||||
| Mean (SD) | 90.4 (22.2) | 90.0 (22.4) | 90.4 (22.5) | 90.4 (22.5) |
| Body mass index (kg/m2) | ||||
| Mean (SD) | 30.6 (7.02) | 30.5 (7.00) | 30.6 (7.12) | 30.6 (7.14) |
| Sex | ||||
| Male, N (%) | 1,209 (70) | 415 (69) | 1,127 (70) | 1,111 (70) |
| Female, N (%) | 523 (30) | 183 (31) | 479 (30) | 479 (30) |
| Race | ||||
| Non‐Asian, (White and others), N (%) | 1,464 (85) | 487 (81) | 1,340 (83) | 1,327 (83) |
| Asian, N (%) | 268 (15) | 111 (19) | 266 (17) | 263 (17) |
| Region | ||||
| Asia, N (%) | 179 (10) | 65 (11) | 179 (11) | 177 (11) |
| Others, N (%) | 1,553 (90) | 533 (89) | 1,427 (89) | 1,413 (89) |
| Psoriatic arthritis | ||||
| Yes, N (%) | 460 (27) | 159 (27) | — | — |
| No, N (%) | 1,272 (73) | 439 (73) | — | — |
| C‐reactive protein high‐sensitivity (mg/L) | ||||
| Mean (SD) | 6.1 (10.2) | 5.8 (8.47) | — | — |
| Baseline PASI score | ||||
| Mean (SD) | 20.1 (7.70) | 20.6 (7.75) | — | — |
| Baseline sPGA scorea (3 or 4) | ||||
| sPGA 3, N (%) | 1,369 (79%) | 484 (81%) | — | — |
| sPGA 4, N (%) | 360 (21%) | 114 (19%) | — | — |
| Prior therapy | ||||
| Naive, N (%) | 434 (25%) | 122 (20%) | — | — |
| Non‐biologic systemic therapy, N (%) | 768 (44%) | 296 (50%) | — | — |
| Non‐TNF antagonist biologic therapy, N (%) | 481 (28%) | 129 (22%) | — | — |
| TNF antagonist, N (%) | 411 (24%) | 134 (22%) | — | — |
SC, subcutaneous; sPGA, static Physician's Global Assessment; TNF, tumor necrosis factor.
Baseline sPGA score of 5 was reported for three subjects in data sets representing week 16 efficacy analyses.
Exposure–efficacy analyses
The graphical exposure–efficacy relationships in the phase II trial demonstrated that maximum PASI90, PASI100, and sPGA0/1 responses at week 16 were observed at plasma exposures that correspond to risankizumab average plasma concentration (C avg) of 4–7.9 μg/mL (Figure 1). These graphical analyses supported that the phase III dosing regimen of risankizumab with median estimated C avg value of 7.3 μg/mL would maximize these efficacy responses in psoriasis patients.
Figure 1.
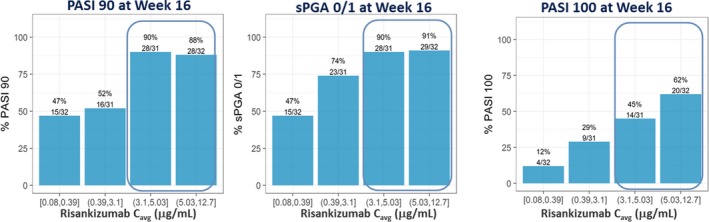
Relationships between risankizumab average plasma concentration (C avg) and observed PASI90, PASI100, and sPGA0/1 responses at week 16 in the phase II trial. Note: C avg represents average concentration from week 0 to 16. Median of C avg (μg/mL): quartile 1 = 0.24 μg/mL, quartile 2 = 1.97 μg/mL, quartile 3 = 3.97 μg/mL, and quartile 4 = 7.88 μg/mL. PASI, Psoriasis Area and Severity Index; sPGA, static Physician's Global Assessment. [Colour figure can be viewed at http://wileyonlinelibrary.com]
The model‐based exposure–response relationships between risankizumab plasma C avg values and efficacy endpoints, PASI75, PASI90, PASI100, and sPGA0/1 responses at week 16 and week 52 were well described by Emax (maximum effect) relationships. The parameter estimates of the regression analyses are shown in Table 2. The risankizumab EC50 (concentration producing 50% of maximum effect) values were significantly lower than the estimated median C avg values (7.3 μg/mL over weeks 0–16 and 5.4 μg/mL over weeks 40–52) for the risankizumab phase III clinical regimen. Based on the exposure–response analyses, at the median risankizumab C avg value for the clinical regimen, the estimated probabilities for PASI75, PASI90, PASI100, and sPGA0/1 responses were 92%, 77%, 49%, and 87%, respectively, at week 16 and 95%, 85%, 61%, and 88%, respectively, at week 52. These probabilities were at the estimated plateau of exposure–response relationship for each efficacy endpoint (Figure 2).
Table 2.
Population estimates and medians and 95% confidence intervals from the bootstrap evaluations (weeks 16 and 52)
| Parameter | Week 16 | Week 52 | ||
|---|---|---|---|---|
| Estimate | Bootstrap median (95% confidence interval) | Estimate | Bootstrap median (95% confidence interval) | |
| PASI75 | ||||
| Placebo response | 0.078 | 0.077 (0.049, 0.109) | — | — |
| Emax | 0.939 | 0.938 (0.915, 0.967) | 1a | — |
| EC50 (μg/mL) | 0.203 | 0.197 (0.081, 0.407) | 0.300 | 0.298 (0.214, 0.401) |
| Exponent for effect of baseline hs‐CRP on EC50 | 0.649 | 0.660 (0.323, 1.00) | — | — |
| PASI90 | ||||
| Placebo response | 0.031 | 0.030 (0.013, 0.053) | — | — |
| Emax | 0.812 | 0.812 (0.769, 0.872) | 1a | — |
| EC50 (μg/mL) | 0.441 | 0.434 (0.170, 0.990) | 0.956 | 0.958 (0.761, 1.20) |
| Exponent for effect of baseline hs‐CRP on EC50 | 0.616 | 0.618 (0.293, 0.952) | — | — |
| PASI100 | ||||
| Placebo response | 0.011 | 0.010 (0.001, 0.024) | — | — |
| Emax | 0.642 | 0.641 (0.541, 0.856) | 0.933 | 0.931 (0.718, 0.998) |
| EC50 (μg/mL) | — | — | 2.88 | 2.88 (1.04, 3.89) |
| EC50 for Asian race (μg/mL) | 9.65 | 9.80 (4.93, 18.8) | — | — |
| EC50 for Non‐Asian races (whites or other) (μg/mL) | 2.36 | 2.31 (0.974, 5.66) | — | — |
| Exponent for effect of baseline hs‐CRP on EC50 | 0.296 | 0.299 (0.109, 0.548) | — | — |
| sPGA0/1 | ||||
| Placebo response | 0.069 | 0.068 (0.042, 0.099) | — | — |
| Emax | 0.916 | 0.916 (0.882, 0.967) | 1a | — |
| EC50 (μg/mL) | 0.431 | 0.424 (0.194, 0.891) | 0.709 | 0.710 (0.565, 0.883) |
| Exponent for effect of baseline hs‐CRP on EC50 | 0.505 | 0.508 (0.211, 0.806) | — | — |
The effect of baseline hs‐CRP on EC50 was incorporated using power model by normalizing baseline hs‐CRP with median value in data set (2.94 mg/L). e.g., .
EC50, concentration producing 50% of maximum effect; Emax, maximum effect; hs‐CRP, high‐sensitivity C‐reactive protein; PASI, Psoriasis Area and Severity Index; sPGA, static Physician's Global Assessment.
The estimate was close to 1, fixing it to 1 allowed for estimation of its precision.
Figure 2.
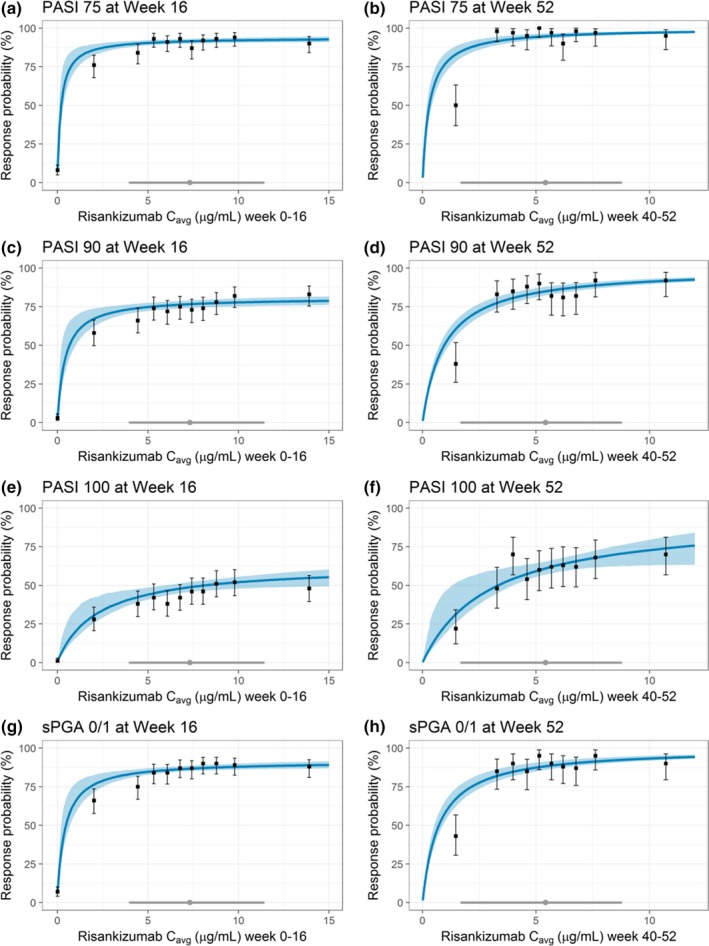
Observed and model‐estimated exposure–efficacy relationships for PASI and sPGA at weeks 16 and 52 using data from phase II and III trials. Black solid symbols with error bars: Observed PASI75 ((a) week 16 and (b) week 52), PASI90 ((c) week 16 and (d) week 52), PASI100 ((e) week 16 and (f) week 52), or sPGA0/1 ((g) week 16 and (h) week 52) response and corresponding 95% exact binomial confidence intervals within each C avg decile. Blue solid line and shaded area: MODEL‐estimated probability of response and 95% confidence interval. Gray solid symbol and line: Median C avg and 5th and 95th percentiles with 150 mg SC dose at weeks 0 and 4 and every 12 weeks thereafter. The model‐estimated probabilities for PASI and sPGA endpoints at week 16 are shown at the median value of baseline high‐sensitivity C‐reactive protein (hs‐CRP) for all endpoints and for non‐Asians for PASI100. C avg, average plasma concentration; PASI, Psoriasis Area and Severity Index; sPGA, static Physician's Global Assessment. [Colour figure can be viewed at http://wileyonlinelibrary.com]
Baseline high‐sensitivity C‐reactive protein (hs‐CRP) was a significant predictor for week 16 efficacy responses but had a minimal effect on efficacy with up to 5% lower PASI or sPGA0/1 responses estimated in patients with high baseline hs‐CRP values (patients at the 90th percentile in the data set (13.8 mg/L)) compared with those with median value (2.9 mg/L). Asian race was also a statistically significant covariate for risankizumab EC50 for only PASI100 response at week 16, and not for PASI75, PASI90, or sPGA0/1 response at week 16 or week 52 or PASI100 response at week 52. The EC50 estimate for PASI 100 at week 16 in Asians was higher compared with non‐Asians (Table 2) since the observed week 16 PASI100 response was lower for Asians, and the maximum week 16 PASI100 response parameter (Emax) was assumed to be the same across Asians and non‐Asians (an alternative model with Asian race as a covariate on week 16 PASI100 Emax was inferior to the model with race as a covariate on week 16 PASI100 EC50). With the 150 mg SC at weeks 0 and 4 and q12w thereafter risankizumab regimen, the estimated PASI100 responses at week 16 for Asians and non‐Asians were 30% and 48%, respectively. None of the other covariates evaluated, including demographic variables (age, sex, bodyweight, body mass index (BMI)), baseline disease characteristics (presence of psoriatic arthritis, PASI, and sPGA scores), prior treatment history, and immunogenicity status (antidrug antibodies (ADA) or neutralizing antibodies (NAb) positive vs. negative status) were statistically significant at either time point. Based on pharmacokinetic analyses, risankizumab exposures were correlated with bodyweight; Figure 3 further shows the lack of effect of bodyweight on efficacy based on risankizumab exposures in subjects with higher bodyweight. These results are consistent with observed efficacy data in phase III trials; the placebo‐adjusted PASI90 responses were 72.1% and 69.2%, and sPGA0/1 responses were 77.5% and 80.0% in patients with bodyweight ≤100 kg and >100 kg, respectively. Consistent with the results from exposure–response analyses, Figure S1 shows lack of an effect of ADA or NAb status on week 16 and week 52 efficacy responses based on subgroup analyses of efficacy.
Figure 3.
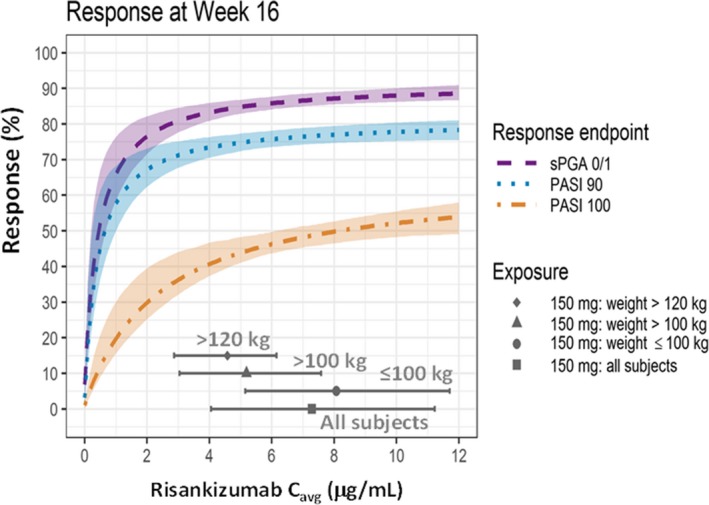
Estimated relationships for PASI90, PASI100, and sPGA0/1 responses at week 16 and average risankizumab plasma concentration (C avg) and illustration of achieving plateau of response across body weight categories with the clinical regimen in psoriasis patients. Dashed lines and shaded area: Model‐predicted probability of response, median (dashed lines) and 95% confidence interval. Gray solid symbols (median) and lines (5th and 95th percentiles) represents distribution of risankizumab C avg values with phase III clinical dosing regimen of risankizumab (i.e., 150 mg SC at weeks 0 and 4 and every 12 weeks thereafter). C avg, average plasma concentration; PASI, Psoriasis Area and Severity Index; sPGA, static Physician's Global Assessment. [Colour figure can be viewed at http://wileyonlinelibrary.com]
The observed and model‐estimated relationships between risankizumab C avg values and efficacy endpoints at week 16 and week 52 were in good agreement across the wide range of exposures associated with the doses in the range of 18–180 mg SC evaluated in phase II and phase III trials (Figure 2 and Figure S2 ). An apparent overprediction of week 52 efficacy response was observed in the first decile of C avg values, which was due to early discontinuation of some patients with low exposure values and imputation of these patients as nonresponders based on nonresponder imputation method. With the exclusion of these patients, the observed efficacy responses were comparable to the model‐estimated values (data not shown). The bootstrap analyses indicated that the exposure–response relationships were precisely estimated as indicated by the 95% confidence intervals, with negligible deviation from estimates based on the observed data set and successful convergence of all bootstrap replicates (Table 2).
Exposure–safety analyses
The exposure–response relationships between risankizumab plasma C avg values and the percentages of subjects who experienced any adverse event (AE), serious AE (SAE), infection and infestation, or serious infection over up to a 52‐week treatment period using pooled data from the phase III trials are presented in Figure 4. There was no apparent relationship between risankizumab exposure and any of the evaluated safety variables through 52 weeks of treatment. Consistent results were seen over the first 16‐week treatment period (Figure S3 ). This was further confirmed with logistic regression analyses that indicated that, for subjects treated with risankizumab, there was no statistically significant correlation between risankizumab plasma exposure and the evaluated safety variables at week 16 or week 52 (data not shown).
Figure 4.
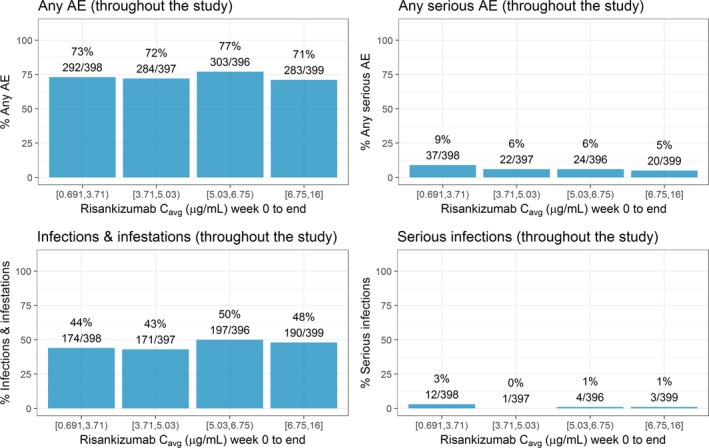
Exposure–response relationships between risankizumab plasma C avg and safety events of interest over the first 52 weeks treatment period. Note: Week 0 to end represents time period week 0–44 in IMMvent trial and week 0–52 in ultIMMa‐1, ultIMMa‐2, and IMMhance trials. C avg represents average concentration from week 0 to end. Median of C avg (μg/mL): quartile 1 = 2.98 μg/mL, quartile 2 = 4.25 μg/mL, quartile 3 = 5.87 μg/mL, quartile 4 = 7.93 μg/mL. AE, adverse event; C avg, average plasma concentration. [Colour figure can be viewed at http://wileyonlinelibrary.com]
Discussion
This manuscript describes the exposure–response analyses of risankizumab efficacy, as assessed by PASI75, PASI90, PASI100, and sPGA0/1 responses at week 16 and week 52, using data from one phase II and four phase III trials in patients with moderate‐to‐severe plaque psoriasis, as well as exploratory exposure–response analyses of risankizumab safety as assessed by key safety variables of interest including any AE, SAE, infection and infestation, and serious infection during the first 16 weeks and up to 52 weeks of treatment duration using data from the four phase III trials of risankizumab.
There were statistically significant relationships between risankizumab C avg and evaluated PASI and sPGA0/1 efficacy responses, both at week 16 and week 52, in subjects with plaque psoriasis. Nonlinear regression analyses based on Emax models adequately described the exposure–response relationships for the efficacy endpoints. These analyses showed that the PASI75, PASI90, PASI100, and sPGA0/1 responses both at week 16 and week 52 were at the plateau of exposure–response relationship following administration of the phase III clinical regimen, 150 mg SC at weeks 0 and 4 and q12w thereafter and are in good agreement with the observed responses in the phase III trials.
Comparison of observed vs. model‐estimated exposure–response relationships and bootstrap analyses indicated that models adequately described the exposure–response relationships. Statistically significant relationships were also noted when risankizumab plasma trough concentration (C trough at week 16 or week 52) was used instead of C avg. However, C avg was chosen as it provided a better correlation with efficacy endpoints, and contrary to C trough, C avg represents an integrated measure of exposure over a period of time (i.e., over the first 16 weeks or a dosing interval) and also takes into account any differences in dosing frequencies. It is noteworthy that the C avg range across the first 16 weeks (week 0–16, Figure 2 a,c,e,g) is higher than that at steady state (week 40–52 dosing interval, Figure 2 b,d,f,h) because of the higher risankizumab exposures early in treatment with the week 4 loading dose. In addition to the nonlinear regression, logistic‐regression analyses were also evaluated using Emax, sigmoid Emax, or linear function to describe the exposure–efficacy relationships; however, these were statistically inferior to nonlinear regression analyses and were not further evaluated.
In the analyses, baseline hs‐CRP values and race were statistically significant covariates for week 16 efficacy responses. High baseline hs‐CRP, representing high inflammatory burden, although statistically significant, did not result in a clinically meaningful impact on efficacy responses. In these analyses with the phase III clinical regimen of risankizumab, PASI100 response at week 16 for Asians was approximately 30% compared with 48% for non‐Asians. All other efficacy responses (PASI75, PASI90, and sPGA0/1) were comparable between Asian and non‐Asian patients. The lower PASI100 response at week 16 for Asian patients appears to be primarily driven by lower PASI100 response for Asian patients enrolled in Asian countries (27%); the Asian patients enrolled in the rest of the world had PASI100 response (38%) consistent with the overall phase III population (36% to 51%).10 The reason for the lower PASI100 response in Asian patients is unknown and could be an artifact of the lower sample size of Asian subjects in the trials. Neither baseline hs‐CRP nor race was a significant covariate for any efficacy responses at week 52. Other evaluated covariates, including demographic variables (body weight, BMI, age, and sex), baseline disease characteristics (baseline PASI/sPGA scores) and comorbidities (presence of psoriatic arthritis), immunogenicity (treatment‐emergent ADA or NAb), and prior therapies were also not statistically significant in the exposure–response analyses of PASI and sPGA0/1 responses at week 16 or week 52.
Although bodyweight was not a statistically significant covariate for any of the efficacy endpoints in these analyses, the population pharmacokinetic analysis estimated 30% lower risankizumab plasma exposures for patients >100 kg compared with ≤100 kg.13 Figure 3 shows that across the distribution of C avg values in heavier patients (>100 kg or even >120 kg) enrolled in phase III trials, the estimated coprimary efficacy endpoints (PASI90 and sPGA0/1 responses at week 16) as well as the PASI100 response at week 16 were at the plateau of exposure–response relationships, which indicates that the phase III clinical regimen of risankizumab is optimal to maximize efficacy regardless of bodyweight in the psoriasis patient population.
Immunogenicity against biologics can potentially affect the efficacy response. In this analysis, the ADA or NAb positive status was not a significant predictor for any of the evaluated efficacy responses at week 16 or week 52. This is consistent with the subgroup analysis of key efficacy endpoints, PASI90 and sPGA0/1 at week 16 or week 52, based on ADA and NAb status from phase III trials (summarized in Figure S1 ). Across all phase III trials of risankizumab, following administration of 150 mg SC dose at weeks 0 and 4 and q12w thereafter, the ADA positive and NAb positive incidence over 52‐week treatment duration were 24% and 14%, respectively. The majority of these ADA positive patients had lower titer values (ADA titer <128) with only 1.5% of the overall phase III population having high ADA titers (≥128). Based on the population pharmacokinetic analyses of risankizumab, risankizumab plasma exposures in ADA positive patients with titers <128 were comparable to ADA negative patients, while approximately 30% lower exposures were estimated in patients with high ADA titers (≥128) compared with ADA negative patients, which was also not clinically relevant for efficacy, consistent with the lack of the impact of 30% lower exposure on efficacy in patients with higher bodyweight as shown in Figure 3. Overall, these data support that immunogenicity to risankizumab is not clinically relevant for efficacy responses.
The phase III trials demonstrated that continued treatment with risankizuamb beyond 16 weeks results in incremental efficacy benefit, particularly for PASI100 response,10 which can be further appreciated from the data presented in Figure 2 and from the Emax estimate for PASI100 at week 52 compared with week 16 (Table 2).
Exploratory analyses of the key safety variables of interest indicated no apparent relationship between risankizumab exposure and any AE, SAE, infection and infestation, or serious infection over the first 16 weeks and up to 52 weeks of treatment duration using pooled data from all four phase III trials of risankizumab in patients with moderate to severe plaque psoriasis. These analyses indicate that the evaluated phase III clinical regimen of risankizumab is supported from a safety perspective as well.
In conclusion, the exposure–efficacy and exposure–safety analyses results indicated that the clinical regimen of risankizumab, 150 mg SC at weeks 0 and 4 and q12w thereafter, achieved the plateau of efficacy with no apparent relationship between risankizumab exposure and key safety events in patients with moderate to severe plaque psoriasis. Overall, these results support that the risankizumab 150 mg SC dose maximized efficacy in different patient subpopulations regardless of age, bodyweight, sex, baseline disease characteristics, or comorbidities, such as presence of psoriatic arthritis or immunogenicity status.
Methods
Data sources, patients, and study design
The studies were conducted in accordance with Good Clinical Practice guidelines and the ethical principles that have their origin in the Declaration of Helsinki. The protocols and informed consent forms were approved by the institutional review boards, and written informed consents were provided by all patients before any study‐related procedures were performed.
Efficacy data from five randomized, double‐blind trials in patients with moderate‐to‐severe chronic plaque psoriasis were included in the analyses: one phase II trial and four phase III trials (UltIMMa‐1, UltIMMa‐2, IMMhance, and IMMvent). Safety data from four phase III trials, noted above, were included in the analyses. In the phase II trial, patients received risankizumab 18 mg SC at week 0, or 90 mg at weeks 0, 4, and 16 or 180 mg SC at weeks 0 and 4 and 16. In phase III trials patients received risankizumab 150 mg SC at weeks 0 and 4 and q12w thereafter, while the patients who were randomized to receive placebo for the first 16 weeks received risankizumab 150 mg SC q12w, starting week 16.
Detailed description of study designs for these trials has been published10, 11 previously. Briefly, in the phase II trial, patients were randomized (1:1:1:1) to receive risankizumab 18 mg SC at week 0, or 90 mg at weeks 0, 4, and 16, or 180 mg SC at weeks 0 and 4 and 16, or ustekinumab as active comparator (45 mg or 90 mg weight‐based per label). Among the phase III trials, UltIMMa‐1 and UltIMMa‐2 trials were 52 weeks in duration with patients randomized (3:3:1) to receive risankizumab (150 mg SC at weeks 0 and 4 and q12w thereafter), placebo or ustekinumab (45 mg or 90 mg weight‐based per label) through week 40. The IMMhance trial was a withdrawal and retreatment study with 104 weeks in duration in which patients were randomized (4:1) at week 0 to receive risankizumab (150 mg SC at weeks 0 and 4 and q12w thereafter) or placebo. At week 28, patients initially randomized to risankizumab at week 0 were rerandomized to subsequent treatment, either continuing to receive risankizumab or switching to placebo, based on their sPGA response. The UltIMMa‐1, UltIMMa‐2, and IMMhance trials had a placebo‐controlled period during the first 16 weeks (week 0–16); patients who were randomized to receive placebo switched to risankizumab 150 mg SC q12w starting week 16. The IMMVent trial was 44 weeks in duration, with patients randomized (1:1) to receive risankizumab (150 mg SC at weeks 0 and 4 and q12w thereafter) or adalimumab (80 mg SC at week 0, then 40 mg every other week per label) through week 16. At week 16, patients who were randomized to the adalimumab treatment at week 0 were assigned to subsequent treatment, either adalimumab or risankizumab based on percent reduction in PASI scores, while those randomized to risankizumab at week 0 continued to receive risankizumab after week 16.
Efficacy, safety, and exposure assessments
For the exposure–efficacy analyses, the efficacy endpoints were PASI75, PASI90, PASI100, and sPGA0/1 responses (based on nonresponder imputation of missing data) at week 16 from the phase II and four phase III trials and at week 52 from two phase III trials (UltIMMa‐1, UltIMMa‐2).
For the exposure–efficacy analyses of PASI and sPGA responses at week 16 (placebo‐controlled period), responses following treatment with risankizumab dosing regimens of 18 mg SC at week 0, or 90 mg SC or 180 mg SC at weeks 0, 4, and 16 from the phase II trial and following treatment with placebo or risankizumab clinical regimen of 150 mg SC at weeks 0 and 4 and q12w thereafter from the phase III trials were included in the analyses. For the analyses of PASI and sPGA responses at week 52, responses from only those patients who received the 150 mg dose of risankizumab at weeks 0 and 4 and q12w thereafter in UltIMMa‐1 and UltIMMa‐2 trials were included in the analyses. Data from IMMVent and IMMhance trials were excluded from analyses of week 52 efficacy responses. This was primarily because the IMMVent trial was of shorter treatment duration with final efficacy assessment at week 44 and because in the IMMhance trial patients randomized to risankizumab clinical regimen were rerandomized to different treatments based on their week 28 efficacy response, which would bias the efficacy response at week 52 as only selected patients were evaluated at week 52 based on their week 28 efficacy response.
For the exposure–safety analyses, the safety variables of interest were any AE, SAE, AE within the MedDRA (Medical Dictionary for Regulatory Activities) system organ class of infection and infestation, or serious infections during the first 16 weeks (weeks 0–16) and up to 52 weeks of treatment duration (week 0–52 in UltIMMa‐1, UltIMMa‐2, and IMMhance or week 0–44 in IMMVent). For the safety variables across the first 16 weeks (placebo‐controlled period), safety data following treatment with the clinical regimen of risankizumab, 150 mg SC at weeks 0 and 4 and q12w thereafter, and placebo were included in the analyses. For the analyses of safety variables up to 52 weeks, subjects who received at least one dose of risankizumab, but no exposure of active control during the trials were included.
Serial blood samples were collected over a period of 44 to 104 weeks from patients enrolled in the phase II and phase III trials for the assessment of risankizumab pharmacokinetics, ADA, and NAb: 7‐14 blood samples for pharmacokinetic assessment and 5–10 blood samples for ADA and NAb assessment per patient. Risankizumab pharmacokinetic data combined across all phase I, II, and III trials in patients with plaque psoriasis were analyzed together using a population‐pharmacokinetic analyses approach with details previously described.13 For the exposure–response analyses, risankizumab plasma exposure metrics for individual patients in phase II and III trials were derived using the individual pharmacokinetic parameters based on the population‐pharmacokinetic analyses13 and the actual dosing history of each patient. The C avg across the first 16 weeks (week 0–16) was used for the analyses of week 16 efficacy endpoints or safety variables over the first 16‐week treatment period, and the C avg at steady state across the week 40–52 dosing interval was used for the analysis of week 52 efficacy endpoints. The C avg over up to a 52‐week treatment period in phase III trials were used for the analyses of safety variables over up to a 52‐week treatment period.
Exposure–efficacy analyses
Graphical exploration of exposure–efficacy relationship was conducted using data from the phase II trial. The percentage of patients who achieved PASI90, PASI100, and sPGA0/1 responses at week 16 in each risankizumab plasma C avg (over 16 weeks) quartile were shown graphically as quartile plots. The exposure–efficacy regression analyses between C avg and efficacy endpoints at week 16 and week 52 as well as assessment of the effect of relevant patient‐specific covariates on the exposure–efficacy relationships were conducted using data from the phase II and III trials. For these exposure–efficacy analyses, nonlinear regression models using an Emax function (Eq. 1) were used to estimate the probability of efficacy responses as a function of risankizumab C avg. The Emax relationship estimates the maximum probability of achieving an efficacy response (i.e., Emax) and EC50, which represents the C avg value at which the response is one‐half of estimated maximum.
| (1) |
where P(efficacy) represents the probability of an efficacy endpoint (e.g, PASI75), PL is the probability of occurrence of efficacy endpoint for subjects with risankizumab plasma exposure value of zero (i.e., placebo effect), and represents the C avg value for subject i.
The patient‐specific covariates evaluated in the analyses included demographic variables (e.g., age, sex, bodyweight, BMI, race (Asian vs. non‐Asian (white and others)), region (Asia vs. others)), baseline disease characteristics (presence of psoriatic arthritis (Yes/No), baseline hs‐CRP, PASI and sPGA scores), prior treatment history (i.e., naïve to prior psoriasis therapy or prior therapy with nonbiologic systemic therapy or prior treatment with tumor necrosis factor–antagonist or non‐tumor necrosis factor–antagonist biologic therapy) and immunogenicity (ADA positive vs. negative status or NAb positive vs. negative status). For the phase II study, prior treatment history was not available, and subjects were assumed to be naïve to psoriasis therapy for purposes of covariate evaluation. These covariates were tested on the Emax or EC50 values and included in the exposure–response relationship if found significant based on predefined statistical criteria (α = 0.01 for forward inclusion and α = 0.001 for backward elimination) using likelihood ratio test.
These analyses were performed using a nonlinear mixed‐effects modeling approach in NONMEM version 7.4 (ICON Development Solutions, Hanover, MD) compiled with the GNU Fortran compiler (Version 4.8.3). The NONMEM code for exposure–response analysis of PASI75, as a representative endpoint and the corresponding sample dataset (subset from full dataset), are included in the supplementary material (Data S1 ). The exposure–response relationships were evaluated graphically by comparing the model‐estimated probabilities of efficacy endpoints against the observed proportion of subjects achieving those endpoints. Bootstrap analyses with 1,000 replicates were conducted to evaluate the robustness of the exposure–response relationship and the precision of the regression estimates.14
Exposure–safety analyses
The relationships between risankizumab plasma exposures (C avg) and safety variables of interest during the first 16 weeks and up to 52 weeks (as defined earlier) were evaluated graphically. The percentage of subjects who experienced each safety event was determined among subjects in each risankizumab plasma C avg quartile and shown graphically as quartile plots, which were visually inspected for potential exposure–response relationships.
Funding
The studies analyzed were supported by Boehringer Ingelheim and AbbVie. Boehringer Ingelheim contributed to the study designs and data collection, and AbbVie contributed to the analysis and interpretation of the data and the writing, review, and approval of the manuscript. Boehringer Ingelheim contributed to the approval of the manuscript.
Conflicts of Interest
A.A.S., A.R.P., and A.A.O. are employees and shareholders of AbbVie. A.K. is a former employee of AbbVie and may hold AbbVie stocks.
Author Contributions
A.K., A.A.S., A.R.P., and A.A.O. wrote the manuscript, designed the research, and performed the research. A.A.S. analyzed the data.
Data Sharing Statement
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual and trial‐level data (analysis data sets), as well as other information (e.g., protocols and Clinical Study Reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. This clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html.
Supporting information
Figure S1. Lack of an effect of immunogenicity (ADA or NAb status) on week 16 and week 52 efficacy (PASI90 and sPGA0/1) responses. ADA, anti‐drug antibodies; NAb, neutralizing antibodies; PASI, Psoriasis Area and Severity Index; sPGA, static Physician's Global Assessment.
Figure S2. Graphical comparison of observed and model‐predicted probability of PASI and sPGA endpoints at weeks 16 and 52 based on analyses of the phase II and III trials. PASI, Psoriasis Area and Severity Index; sPGA, static Physician's Global Assessment.
Figure S3. Exposure–response relationships between risankizumab C avg and safety events of interest over the first 16 Weeks. C avg, average plasma concentration.
Data S1. NONMEM (ICON Development Solutions, Hanover, MD) code for PASI75 (week 16) model. PASI, Psoriasis Area and Severity Index.
Acknowledgments
The authors thank AbbVie employees Allison Kitten and Wesley Wayman for medical writing support.
References
- 1. Di Meglio, P. , Villanova, F. & Nestle, F.O. Psoriasis. Cold Spring Harb. Perspect Med. 4 (2014). 10.1101/cshperspect.a015354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Boehncke, W.H. & Schön, M.P. Psoriasis. Lancet 386, 983–994 (2015). [DOI] [PubMed] [Google Scholar]
- 3. Conrad, C. & Gilliet, M. Psoriasis: from pathogenesis to targeted therapies. Clin. Rev. Allergy Immunol. 54, 102–113 (2018). [DOI] [PubMed] [Google Scholar]
- 4. Haugh, I.M. , Preston, A.K. , Kivelevitch, D.N. & Menter, A.M. Risankizumab: an anti‐IL‐23 antibody for the treatment of psoriasis. Drug Des. Devel. Ther. 12, 3879–3883 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Al‐Salama, Z.T. & Scott, L.J. Guselkumab: a review in moderate to severe plaque psoriasis. Am. J. Clin. Dermatol. 19, 907–918 (2018). [DOI] [PubMed] [Google Scholar]
- 6. Stelara (ustekinumab) [US prescribing information] (Janssen Biotech, Horsham, PA, 2018). [Google Scholar]
- 7. Tremfya (guselkumab) [US prescribing information] (Janssen Biotech, Horsham, PA, 2017). [Google Scholar]
- 8. Reich, K. et al Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE 1 and reSURFACE 2): results from two randomised controlled, phase 3 trials. Lancet 390, 276–288 (2017). [DOI] [PubMed] [Google Scholar]
- 9. Papp, K.A. et al Risankizumab versus ustekinumab for moderate‐to‐severe plaque psoriasis. N. Engl. J. Med. 376, 1551–1560 (2017). [DOI] [PubMed] [Google Scholar]
- 10. Gordon, K.B. et al Efficacy and safety of risankizumab in moderate‐to‐severe plaque psoriasis (UltIMMa‐1 and UltIMMa‐2): results from two double‐blind, randomised, placebo‐controlled and ustekinumab‐controlled phase 3 trials. Lancet 392, 650–661 (2018). [DOI] [PubMed] [Google Scholar]
- 11. AbbVie . Press release: Risankizumab meets all co‐primary and ranked secondary endpoints, achieving significantly greater efficacy versus standard biologic therapies in three pivotal phase 3 psoriasis studies <https://news.abbvie.com/news/risankizumab-meets-all-co-primary-and-ranked-secondary-endpoints-achieving-significantly-greater-efficacy-versus-standard-biologic-therapies-in-three-pivotal-phase-3-psoriasis-studies.htm> (2017). Accessed June 4, 2018
- 12. AbbVie . Press release: Risankizumab meets all primary endpoints reporting positive results in fourth pivotal phase 3 psoriasis study <https://news.abbvie.com/news/risankizumab-meets-all-primary-endpoints-reporting-positive-results-in-fourth-pivotal-phase-3-psoriasis-study.htm> (2017). Accessed June 4, 2018.
- 13. Suleiman, A.A. , Minocha, M. , Khatri, A. , Pang, Y. & Othman, A.A. Population pharmacokinetics of risankizumab in healthy volunteers and subjects with moderate to severe plaque psoriasis: integrated analyses of phase I‐III clinical trials. Clin. Pharmacokinet. (2019). 10.1007/s40262-019-00759-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Efron, B. Bootstrap methods: another look at the jackknife. Ann. Stat. 7, 1–26 (1979). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Lack of an effect of immunogenicity (ADA or NAb status) on week 16 and week 52 efficacy (PASI90 and sPGA0/1) responses. ADA, anti‐drug antibodies; NAb, neutralizing antibodies; PASI, Psoriasis Area and Severity Index; sPGA, static Physician's Global Assessment.
Figure S2. Graphical comparison of observed and model‐predicted probability of PASI and sPGA endpoints at weeks 16 and 52 based on analyses of the phase II and III trials. PASI, Psoriasis Area and Severity Index; sPGA, static Physician's Global Assessment.
Figure S3. Exposure–response relationships between risankizumab C avg and safety events of interest over the first 16 Weeks. C avg, average plasma concentration.
Data S1. NONMEM (ICON Development Solutions, Hanover, MD) code for PASI75 (week 16) model. PASI, Psoriasis Area and Severity Index.