

Abstract
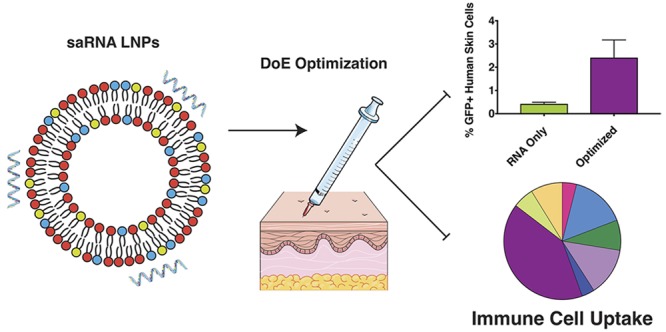
Messenger RNA (mRNA) is a promising tool for biotherapeutics, and self-amplifying mRNA (saRNA) is particularly advantageous, because it results in abundant protein expression and production is easily scalable. While mRNA therapeutics have been shown to be highly effective in small animals, the outcomes do not scale linearly when these formulations are translated to dose-escalation studies in humans. Here, we utilize a design of experiments (DoE) approach to optimize the formulation of saRNA lipid nanoparticles in human skin explants. We first observed that luciferase expression from saRNA peaked after 11 days in human skin. Using DoE inputs of complexing lipid identity, lipid nanoparticle dose, lipid concentration, particle concentration, and ratio of zwitterionic to cationic lipids, we optimized the saRNA-induced luciferase expression in skin explants. Lipid identity and lipid concentration were found to be significant parameters in the DoE model, and the optimized formulation resulted in ∼7-fold increase in luciferase expression, relative to initial 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP) formulation. Using flow cytometry, we observed that optimized formulations delivered the saRNA to ∼2% of the resident cells in the human skin explants. Although immune cells comprise only 7% of the total population of cells in skin, immune cells were found to express ∼50% of the RNA. This study demonstrates the powerful combination of using a DoE approach paired with clinically relevant human skin explants to optimize nucleic acid formulations. We expect that this system will be useful for optimizing both formulation and molecular designs of clinically translational nucleic acid vaccines and therapeutics.
Keywords: lipid nanoparticle, RNA, design of experiments, human skin, self-amplifying, nucleic acid, ex vivo
Messenger RNA (mRNA) has emerged as a versatile and advantageous tool for both vaccine and protein replacement therapeutics. RNA has several beneficial features over DNA and protein therapeutics; mRNA is noninfectious and nonintegrating, and the degradation of mRNA by normal cellular processes can be modulated through modifications and delivery vehicles.1−4 mRNA is the minimal genetic vector, and, thus, antivector immunity is avoided, even after repeated administration. Furthermore, mRNA is known to have adjuvanting properties induced by activation of innate sensing mechanisms, such as toll-like receptors (TLRs) and cytosolic pattern recognition receptors.5,6 Recently, self-amplifying RNA (saRNA) has been investigated as the next-generation approach for mRNA therapeutics. saRNA vectors are derived from the alphavirus genome7 and self-replicate upon delivery into cytoplasm, resulting in abundant protein expression and thus minimizing the required doses of RNA.8−10
Because mRNA therapeutics are negatively charged and not readily taken up into cells, they must be formulated with a delivery vehicle in order to enable efficient cellular uptake and expression. Previous formulations have included liposomes,11,12 polyplexes,13,14 and emulsions,15,16 typically with a cationic lipid or polymer used to complex/condense the RNA. Preclinical formulations are generally optimized in vitro or in vivo in small animal models, such as mice or rats, in order to assess the effectiveness prior to human clinical trials. Kauffman et al. used an innovative in vivo design of experiments (DoE) approach to optimize the expression of liposome-formulated erythropoietin (EPO)-encoding mRNA, which showed 7-fold greater protein expression.17 However, when the same formulation was used to deliver siRNA, no enhancement was observed, emphasizing the importance of optimizing each formulation based on the platform and indication.
While mRNA therapeutics have shown promising results in vivo in many small animal models, the transition to humans is poor. For example, Bahl et al. observed remarkable hemagglutinin (HA) inhibition titers in mice (100–1000), ferrets (∼10 000), and nonhuman primates (∼10 000) for HA proteins H10N8- and H7N9-encoding mRNA formulated in lipid nanoparticles (LNPs).18 However, in the corresponding first-in-human, escalating-dose phase I clinical trial, the HAI titer was merely <100. The reason for this inconsistency is unclear; perhaps it is because both the molecular and formulation components of mRNA therapeutics are optimized in models that are somewhat irrelevant to humans. While small animal models will likely always have a role in preclinical studies, we hypothesize that inherent differences in human innate sensing and tissue architecture pose a barrier to the translation of RNA therapeutics from the laboratory to the clinic. Because of these potential differences between humans and small animal models, we sought to optimize the formulated delivery of saRNA in a relevant human tissue. van den Berg et al. previously optimized a tattooed DNA vaccine in human skin explants;19 however, to our knowledge, this approach has not been previously applied to optimization of mRNA formulations.
Here, we present the optimization of LNP formulations of saRNA in human skin explants, using a DoE approach to maximize protein expression. We first characterized the temporal kinetics of firefly luciferase (fLuc) in human skin explants. Four complexing lipids were chosen, because of their previous use in nucleic acid formulations, including C12–200 (ionizable), dimethyldioctadecylammonium bromide [(DDA), cationic], 1,2-dioleoyl-3-trimethylammonium-propane [(DOTAP), cationic] and cephalin (zwitterionic). We used input parameters of complexing lipid, LNP dose, lipid concentration, particle concentration, and ratio of cationic to zwitterionic lipid, and a response variable of luciferase expression in human skin explants. Upon completion of the DoE, we used flow cytometry to confirm whether the LNP formulations enhanced saRNA delivery to the cells using GFP expression as a proxy. Finally, we characterized which resident cell types of the human skin explants were taking up and expressing saRNA.
Results and Discussion
A depiction of the lipid nanoparticle composition and complexing lipids is shown in Scheme 1. saRNA was adsorbed to the outside of LNPs, using a variety of complexing lipids, including ionizable (C12–200), cationic (DDA, DOTAP), and zwitterionic (cephalin) lipids. We used a DoE approach to optimize the luciferase-encoding saRNA delivery into human skin explants by varying different aspects of the formulation, including the complexing lipid, lipid concentration, particle concentration, and the ratio of zwitterionic to cationic lipid. We then assessed whether the enhanced luciferase expression was due to the quantity of cellular uptake, or enhanced expression in individual cells, and then characterized the identity of the skin cells that were expressing saRNA.
Scheme 1. Schematic of Lipid Nanoparticle (LNP) Formulations Used for DoE Analysis: (a) Lipid Nanoparticles Containing a Complexing Lipid, DOPE, and Cholesterol; and (b) Complexing Lipids Used in the DoE Library.

saRNA Luciferase Expression Kinetics in Human Skin Explants
While the kinetic profile of saRNA luciferase expression in mice has been well-characterized,9 we first sought to determine the peak expression of saRNA in human skin explants. A single tissue explant was treated with three separate injections, given simultaneously, containing a dose of 10 μg of fLuc saRNA complexed with DOTAP LNPs at a ratio of total lipid to RNA of 4:1 (w/w) and imaged over the course of 21 days (Figure 1). Quantification of the luciferase activity reveals that a signal is visible after only 24 h (∼40 000 p/s), peaks at day 11 (∼235 000 p/s), but persists for at least 21 days (Figure 1b). Tissue culture and imaging was discontinued after 21 days, because of a visible decline in tissue viability (see Figure S3 in the Supporting Information). As evidenced in Figure 1a, the bleb caused by intradermal (ID) injection of the formulation was not confined to the initial injection space, but rather spreads out across the tissue, which had a volume of ∼3 cm2. Thus, for future explant experiments, the tissue was cut into smaller volumes (1 cm2) in order to maintain the intended number of replicates. Furthermore, luciferase quantification for the DoE samples was performed only at 10 days, because this timing was determined to be best-suited to distinguish differences in formulation delivery. van den Berg et al. found that luciferase expression peaked in human skin 24 h after delivery via DNA tattooing, and was depleted by 72 h,19 likely because of high turnover of the epidermis or gene silencing.20 We hypothesized that the smaller volume of our tissue explants improved the viability, and the amplifying nature of saRNA yielded a delayed maximum signal. The observed kinetics of saRNA-induced fLuc expression in human skin is similar to the profile observed after intramuscular (IM) injection in mice, wherein the peak expression occurs between 7 and 14 days.9 The prolonged viability of human skin explants in these experiments enabled us to optimize LNP formulations for saRNA delivery.
Figure 1.

Firefly luciferase expression in human skin explants over the course of 21 days after ID injection of three separate, simultaneous injections of 10 μg of saRNA with a mass ratio of lipid to RNA of 4:1 (w/w): (a) time course of ex vivo imaging of luciferase expressed by replicon RNA delivered with the initial formulation of DOTAP LNPs; and (b) quantification of luciferase expression, expressed as the mean total flux (p/s) ± standard deviation for n = 3.
Effects of Complexing Lipid Identity and Lipid-to-RNA Ratio on Luciferase Expression
We first optimized the complexing lipid identity and the ratio of total lipid to RNA on saRNA-induced luciferase expression in human skin explants. We chose a range of complexing lipids to include in the DoE, since they had previously been used in liposomal nucleic acid formulations, including C12–200,17,21 cephalin,22 DDA,23,24 and DOTAP.25 As shown in Figure 2a, cephalin LNPs were found to have the highest luciferase expression (∼40 000 p/s). C12–200, DDA and DOTAP LNPs had peak luciferase expression of ∼20 000 p/s (Figure 2b). These experiments yielded lower luciferase expression than those presented in Figure 1, because of a lower dose of saRNA and decreased surface area of the skin explant utilized for the DoE. Interestingly, C12–200, cephalin, and DOTAP LNPs were found to have increasing luciferase expression with increasing ratio of lipids to RNA, but DDA had similar luciferase expression levels for all three tested ratios (1:1, 4:1, 18:1 (w/w)). We hypothesize that this occurs because DDA complexes the saRNA more efficiently than the other lipids, and thus the DDA LNPs were not saturated with saRNA at a ratio of 18:1 (w/w). C12–200 and DOTAP have previously been used in siRNA21,26 and mRNA17,22,27 delivery, but these lipids have never been formulated in similar LNPs and systematically compared. While both DOTAP and DDA have quaternary, cationic amines, DDA has only two methyl groups and DOTAP has three, which potentially accounts for the more-efficient complexation of DDA LNPs to the saRNA. Zwitterionic lipids such as cephalin are typically used as helper lipids to stabilize liposomes,28,29 in addition to a cationic lipid that complexes the nucleic acid; however, it has been previously observed that increasing the helper lipid composition of the liposome enhances liposome fusion efficiency.30 We were interested to evaluate the role of cephalin as a complexing lipid, because it represents a very different class of molecule with a different headgroup and tail saturation. We hypothesize that the primary amine on cephalin is able to directly complex the saRNA, and the comparatively higher molar composition of the cephalin LNPs enhance the fusion of these particles to the human skin cells, resulting in higher luciferase expression. Particle size and charge was not included as an input into the DoE, since this was variable between the formulations (Figure S1 in the Supporting Information); however, the impact of these characteristics on protein expression warrants future studies. Based on these observations, we used a ratio of total lipid to RNA of 18:1 (w/w) for further experimentation and completion of the DoE.
Figure 2.

Firefly luciferase expression in human skin explants injected intradermally with LNP formulations with varying lipid identity and LNP dose containing 2 μg of saRNA with medium particle concentration (108 particles/mL) at varying ratios of lipid to RNA (w/w): (a) ex vivo imaging of explants after 11 days and (b) quantification of luciferase imagine expressed as the mean total flux (p/s) ± standard deviation for n = 5.
Effects of Lipid and Particle Concentration on Luciferase Expression
Because we observed that increasing the total ratio of lipids to RNA generally increased the luciferase expression in human skin explants, we then sought to determine whether increasing either the particle or lipid concentration would further enhance luciferase expression. We prepared batches of LNPs with high lipid and particle concentration (H[L]/H[P]) or high lipid and low particle concentration (H[L]/L[P]) for each of the complexing lipids. The high and low particle concentrations were defined as 109 and 107 particles/mL, respectively, while the high lipid concentration was defined as 7.5 mg/mL. Using a ratio of total lipids to RNA of 90:1 (w/w), human skin explants were injected ID and quantified for luciferase expression (Figure 3a). We observed that, for cephalin, DDA, and DOTAP LNPs, the H[L]/H[P] formulation had enhanced luciferase expression, compared to the H[L]/L[P] formulation (Figure 3b), although there was no difference in the C12–200 formulations. The H[L]/H[P] cephalin LNPs had the highest luciferase expression (∼25 000 p/s), followed by the cationic H[L]/H[P] DDA and DOTAP LNPs (∼20 000 p/s) and finally the ionizable C12–200 LNPs (∼10 000 p/s). Despite this trend, the luciferase expression of the cephalin LNPs with low lipid concentration and medium particle concentration (Figure 2) was greater than any of the H[L]/H[P] or H[L]/L[P] formulations, emphasizing that, for the formulations of LNPs, a lower ratio of lipid to RNA is more optimal. We observed slight aggregation of H[L]/H[P] LNPs upon addition of saRNA, and we postulate that this phenomenon is due to exceeding the critical concentration of RNA in the particles, wherein the abrupt addition of RNA to a more highly concentrated particle environment causes aggregation. This observation is similar to previous formulations with polyplexes and saRNA, wherein increasing the ratio of saRNA to cationic polymer results in an aggregation of particles, but this has not been previously reported for LNP formulations of saRNA.31 These experiments confirm the optimized LNP formulation to be low lipid and medium particle concentrations.
Figure 3.

Firefly luciferase expression in human skin explants injected intradermally with LNP formulations with varying lipid and particle concentrations containing 2 μg of saRNA with a ratio of total lipid to RNA of 90:1 (w/w): (a) ex vivo imaging of explants after 11 days and (b) quantification of luciferase imagine expressed as the mean total flux (p/s) ± standard deviation for n = 5. High and low particle concentrations are defined as 109 and 107 particles/mL, respectively, while high lipid concentration is defined as 7.5 mg/mL. H[L]/H[P] denotes high lipid concentration and high particle concentration; H[L]/L[P] denotes high lipid concentration and low particle concentration.
Effects of Combining Zwitterionic and Cationic Complexing Lipids on Luciferase Expression
After observing the higher luciferase expression from LNPs with a single cationic or zwitterionic complexing lipid (Figure 2), we prepared formulations of combinations of cationic and zwitterionic lipids with varying ratios from 10:1 to 0.1:1 while maintaining the low lipid concentration and medium particle concentration. We observed that, generally, the DDA/cephalin LNPs had higher luciferase expression (∼25 000 p/s) than the DOTAP/cephalin LNPs (∼10 000 p/s) (Figure 4). These trends are similar to the formulations, wherein only a single complexing lipid was included (Figure 2). There was no added benefit to combining cationic and zwitterionic lipids into a single LNP. In addition, even the LNP formulations that were primarily cephalin (0.1:1) had lower luciferase expression than the cephalin alone, indicating that combining cationic and zwitterionic lipids is detrimental for saRNA delivery. We postulate that this could be because including cationic lipids alters the net charge of the particles, as shown in Figure S1, thus limiting the cephalin-aided fusion of the particles into cells, reducing uptake. However, this hypothesis warrants further studies to confirm whether this is the case, or if incorporating the cationic lipid complexes irreversibly to the saRNA, rendering it functionally useless within the cytoplasm.
Figure 4.

Firefly luciferase expression in human skin explants injected intradermally with LNP formulations with varying ratios of cationic and zwitterionic lipids containing 2 μg of saRNA with a total lipid to RNA ratio of 18:1 (w/w) and medium particle concentration (108 particles/mL): (a) ex vivo imaging of explants after 11 days and (b) quantification of luciferase imagine expressed as mean total flux (p/s) ± standard deviation for n = 5.
Design of Experiments (DoE) Analysis
The DoE approach allows for exploration and characterization of the three-dimensional formulation space in order to identify optimal parameters for saRNA formulation. This approach paired with human skin explants is a clinically relevant way to optimize formulations, as tissue is readily available to facilitate the large number of samples required for an exhaustive full factorial DoE, and ID injections are a clinically viable route of administration for RNA vaccines.32,33 Using input parameters of complexing lipid identity (C12–200, cephalin, DDA, DOTAP), ratio of total lipid to RNA, lipid concentration, particle concentration, and ratio of cationic to zwitterionic lipid and luciferase expression as the response, we used standard least-squares effect screening to construct a model of which input parameters significantly impact luciferase expression in human skin explants (Figure 5). Our model had a correlation coefficient (R2) of 0.55, with p = 0.0043 for the experimental versus predicted luciferase expression. We found that lipid identity and lipid concentration were the only significant factors. In particular, cephalin lipid identity had the strongest effect, with p = 0.0018. To further visualize the design space, we plotted the fold change of luciferase expression when compared to the original DOTAP LNPs administered at a total lipid-to-RNA ratio of 1:1 (w/w) (see Figure 6). A value of 1 indicates no enhancement of luciferase expression. Cephalin LNPs with a ratio of total lipids to RNA of 18:1 (w/w), low lipid concentration, and medium particle concentration yielded a 7-fold increase in luciferase expression over the original formulation. This observation emphasizes the importance of optimizing the formulation and evidence the practicality of a DoE approach. Previous approaches for DoE optimization of liposomal-formulated mRNA delivery found that the ratio of ionizable lipid to mRNA and the identity of the phospholipid were significant factors in enhancing delivery to the liver.17 They postulated that increasing the surface charge of the particles enhanced interaction with the cell membrane and resulted in increased particulate uptake.34 While the ratio of complexing lipid to saRNA was not statistically significant in this model, this is likely due to the DDA LNPs having similar luciferase expression at the tested doses. Cephalin is known to associate with lipid bilayers and cell membranes35 and, thus, may lightly complex to saRNA and facilitate membrane fusion of the LNPs. We showed that luciferase expression in human skin explants is influenced by the complexing lipid identity and the lipid concentration, and the DoE optimization performed in these experiments resulted in a 7-fold increase in protein expression.
Figure 5.
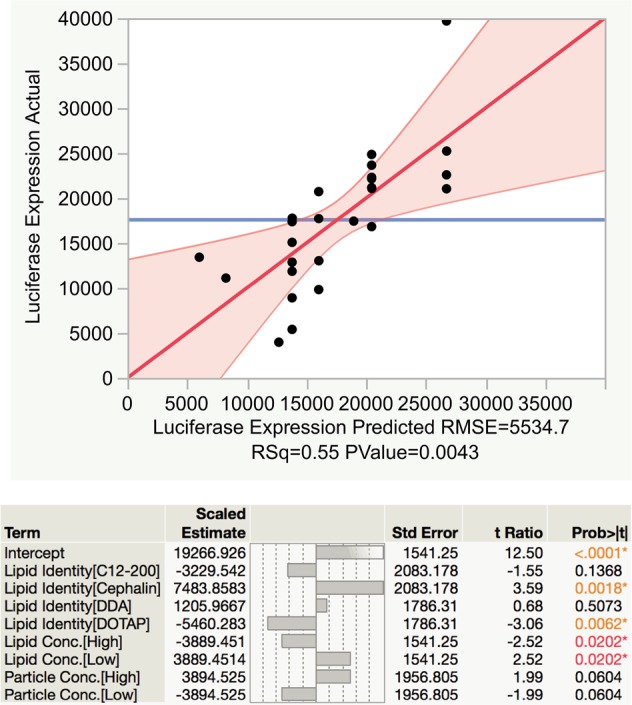
Standard least-squares effect screening DoE analysis of lipid nanoparticle formulation in human skin explants.
Figure 6.

Comparison of fold change luciferase expression of tested LNP formulations normalized to original DOTAP formulation (blue bar). Values are expressed fold change total flux (p/s) ± standard deviation.
LNP Delivery and Expression of eGFP saRNA in Human Skin Cells
In order to confirm that our LNP formulations were enhancing the delivery of saRNA into human skin cells within the explant, we used saRNA encoding eGFP and flow cytometry to further investigate these observations. Our main question was whether the LNP formulations were increasing the total number of cells that the saRNA was being expressed in, or whether they were simply facilitating a higher copy number of saRNA per cell, which would result in increased eGFP intensity signal per cell. Based on the results from the DoE, we used each of the LNP formulations with individual complexing lipids, as well as the cephalin H[L]/H[P] and cephalin H[L]/L[P] LNPs in order to account for all the input variables. In keeping with the luciferase expression, we observed that cephalin LNPs resulted in the highest increase in GFP expression (Figure 7a). Furthermore, cephalin LNPs had the highest total number of cells (∼2.5%) expressing GFP (Figure 7b), which was significantly increased above RNA only, which has previously been shown to induce protein expression sans formulation.36 While the DDA and DOTAP LNPs had higher average percentages of GFP-positive cells, they were not statistically significantly more than RNA only, and there was no increase in the total number of GFP-positive cells with the C12–200, cephalin H[L]/H[P], or cephalin H[L]/L[P] formulations. The RNA only did result in increased GFP intensity above the background (Figure 7a), there is a slight shift in GFP intensity from each of the LNP formulations on a per cell basis, indicating that the LNPs all facilitate increased total amount of saRNA per cell, while the cephalin, DDA, and DOTAP LNPs also increased the total number of positive cells. While the cellular uptake has not been previously characterized for LNP formulations of saRNA, a previous study revealed a similar increase in the number of cells expressing GFP in human skin explants for a pDNA construct transfected into the cells using a tattoo device.19 It has not yet been defined whether a higher percentage of cells taking up and/or expressing saRNA enhances the immunogenicity of the formulation, and whether there is a balance between RNA expression and innate activation. These results confirm the luciferase DoE results and emphasize the potential for enhanced delivery by increasing the total number of cells affected by saRNA formulations.
Figure 7.

GFP expression in human skin cells after intradermal injection with LNP formulations, as determined using flow cytometry: (a) histogram of number of cells expressing GFP for each formulation, and (b) percentage of GFP-positive cells of total live cells for each sample. Bar represents the average ± standard deviation, with a significance of α = 0.05 indicated by an asterisk (*).
Identification of Cells Expressing eGFP in Human Skin Cells
Given the relatively low number of cells observed to be expressing GFP in the human skin explants, we then sought to identify which resident cells are present in the explants and which ones actually express the RNA (see Figure 8, as well as Figure S4 in the Supporting Information). We identified which cells were resident in human skin explants, using a flow cytometry panel capable of identifying epithelial cells (CD45-), fibroblasts (CD90+), NK cells (CD56+), leukocytes (CD45+), Langerhans cells (CD1a+), monocytes (CD14+), dendritic cells (CD11c+), and T cells (CD3+). We found that all of these cells were resident in human skin explants (Figure 8a), and that the majority of cells (93%) are epithelial cells (82.6%) and fibroblasts (10.4%). The immune cells comprised only 7% of the total skin population, which is composed of 0.45% NK cells, 1.33% leukocytes, 3.78% Langerhans cells, 0.24% monocytes, 0.79% dendritic cells, and 0.41% T cells, assuming that the flow cytometry preparation did not bias the presence of any cell types. Interestingly, however, the immune cells comprise a low percentage of the total resident skin cells; they were found be ∼50% of the GFP-expressing cell population (Figure 8b). This observation was true for both formulated and unformulated RNA, with the order of expression as follows: epithelial cells, dendritic cells, Langerhans cells, monocytes, leukocytes, fibroblasts, T cells, and NK cells. We did not observe differences in the cell-viability-tested LNP formulations (see Figure S5 in the Supporting Information). These results indicate that the resident skin immune cells preferentially take up and/or express saRNA, despite formulation. These findings agree strongly with a previous study, which observed that monocytes and dendritic cells were the main cell types that express mRNA after intramuscular or ID injection in rhesus macaques.37 While the results are consistent, future work is warranted to investigate whether uptake and expression into the plethora of epithelial cells and fibroblasts present in the skin can be enhanced by tailoring the formulation, and whether this would lead to enhanced overall protein expression. Furthermore, it remains to be defined whether preferential uptake by immune cells enhances the immunogenicity of saRNA vaccines and therapeutics, and is likely dependent on the indication of the injection. For example, it may be more beneficial to target immune cells in the context of saRNA vaccines for the prevention of infectious diseases, as opposed to protein replacement therapies.
Figure 8.

Identity of cells present in human skin explants and GFP+ cells after ID injection of LNP formulations, as determined by flow cytometry: (a) identity of cells in the population of cells extracted from human skin explants, and (b) identity of GFP-expressing skin cells from explants treated with LNP-formulated RNA. The blue sections of each of the small pie charts indicate the total percentage of immune cells in the GFP+ cell population.
Conclusions
Here, we optimize LNP formulations of saRNA in human skin explants using a DoE approach. Skin explants were cultured for up to 3 weeks and showed luciferase expression after 24 h, which peaked at 10 days. Of the tested input parameters of lipid identity, including cationic (DDA, DOTAP), ionizable (C12–200), and zwitterionic (cephalin) lipids, the ratio of total lipid to RNA, lipid concentration, particle concentration, and ratio of cationic to zwitterionic lipid, only the lipid identity and lipid concentration significantly affected the saRNA-induced luciferase expression in human skin. Despite general use as a “helper lipid” in LNP formulations, cephalin was found to be the most effective complexing lipid. The DoE enabled a 7-fold increase in luciferase expression, compared to the original formulation. Flow cytometry revealed that all of the formulations enhanced the eGFP expression in human skin cells and paralleled the enhanced delivery with cephalin, DDA, and DOTAP LNPs observed with luciferase imaging studies. Finally, while epithelial cells and fibroblasts were found to comprise the majority of the resident skin cell population, the immune cells were found to express more of the administered RNA, with respect to their proportion of the total cell population. This study demonstrates the powerful combination of using a DoE approach paired with clinically relevant human skin explants to optimize nucleic acid formulations. We expect that this system will be useful for optimizing both formulation and molecular designs of clinically translational nucleic acid vaccines and therapeutics.
Methods
RNA Synthesis and Purification
Self-amplifying RNA derived from the Venezuelan Equine Encephalitis Virus (VEEV) encoding either firefly luciferase (fLuc) or enhanced green fluorescent protein (eGFP) was prepared using in vitro transcription. pDNA was transformed in Escherichia coli and cultured in 50 mL of LB with 1 mg/mL carbenicillin (Sigma–Aldrich, U.K.) and isolated using a Plasmid Plus Maxiprep kit (QIAGEN, U.K.). pDNA concentration and purity was measured on a NanoDrop One (ThermoFisher, U.K.) and then linearized using MluI for 2 h at 37 °C and heat inactivated at 80 °C for 20 min. Uncapped in vitro RNA transcripts were synthesized using 1 μg of linearized DNA template in a MEGAScript reaction (Promega, U.K.), according to the manufacturer’s protocol. Transcripts were then purified by overnight LiCl precipitation at −20 °C, pelleted by centrifugation at 14 000 rpm for 20 min, washed once with 70% EtOH, centrifuged at 14 000 rpm for 5 min, and then resuspended in UltraPure H2O. Purified transcripts were then capped using the ScriptCap m7G Capping System (CellScript, Madison, WI, USA) and ScriptCap 2′-O-Methyltransferase Kit (CellScript, Madison, WI, USA) simultaneously, according to the manufacturer’s protocol. Capped transcripts were then purified again by LiCl precipitation, resuspended in ultraPure H2O, and stored at −80 °C until use.
Production of Lipid Nanoparticles
Dimethyldioctadecylammonium bromide (DDA) (Sigma, U.K.), 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP) (Avanti Polar Lipids, Alabaster, AL, USA), and cephalin (soy phosphatidylethanolamine) (Avanti Polar Lipids, Alabaster, AL, USA) were used as received. C12–200 was synthesized by reacting 1 mol equiv of N1-(2-(4-(2-aminoethyl)piperazin-1-yl)ethyl)ethane-1,2-diamine (Enamine Ltd., Kyiv, Ukraine) with 7 mol equiv of 1,2-epoxydodecane (Sigma, U.K.) at 80 °C for 2.5 days, according to previous protocols.21 LNPs were prepared on a μEncapsulator 1 System (Dolomite Bio, Royston, U.K.). The lipid solution was prepared by dissolving lipids in 90% EtOH at a total concentration of 1.5 mg/mL, consisting of 35 mol % complexing lipid (C12–200, cephalin, DDA, or DOTAP), 49 mol % cholesterol (Sigma, U.K.) and 16 mol % 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) (Avanti Polar Lipids, Alabaster, AL, USA). For high lipid concentration particles, the total lipid concentration was increased to 7.5 mg/mL. One hundred microliters (100 μL) of the lipid was loaded into one side of the μEncapsulator reservoir, while the other side was loaded with 100 μL of citrate buffer (pH 3), and the solutions were then loaded into the corresponding pumps. A 50 μm fluorophilic chip with a T-junction and subsequent PBS dilution channel was used. LNPs were prepared using the following conditions: chip temperature, 70 °C; lipid solution pump pressure, 2000 Pa; citrate buffer pump pressure, 666 Pa; and PBS pump pressure, 2000 Pa. LNPs were purified by dialyzing against PBS in a 3500 MWCO dialysis cartridge (Thermo Fisher, U.K.) overnight. In these studies, high, medium, and low particle concentrations correspond to 109, 108, and 107 particles/mL, respectively, diluted in PBS. For combinations of cationic and zwitterionic lipids, the lipid solutions were prepared such that the total complexing lipid mole percentage was maintained at 35 mol % by varying the ratio of DDA/DOTAP to cephalin (10:1, 1:1, or 0.1:1).
Particle Characterization
LNPs were characterized for size, particle concentration, and surface charge prior to complexation with RNA (Figure S1). One hundred microliters (100 μL) of LNPs was diluted into 900 μL PBS (Sigma, U.K.) and equilibrated at room temperature prior to analysis. The particle size and concentration were characterized on a NanoSight LM10 (Malvern Instruments, U.K.) with NanoSight NTA 3.0 software (Malvern Instruments, U.K.) using an infusion rate of 20, a capture duration of 1 min, a gain of 2, and a camera level of 7. Processing parameters were kept constant for all samples. The surface charge of the LNPs was characterized on a Zetasizer Nano ZS (Malvern Instruments, U.K.) with Zetasizer 7.1 software (Malvern, U.K.) using 850 μL of diluted particles in a 1 mL cuvette and the following settings: material refractive index, 1.529; absorbance, 0.010; dispersant viscosity, 0.8820 cP; refractive index, 1.330; and dielectric constant, 79. Each sample was analyzed for up to 100 runs until the measurement stabilized.
Human Skin Explant Injection, Culture, and Imaging
Surgically resected specimens of human skin tissue were collected at Charing Cross Hospital, Imperial College London, U.K. All tissues were collected after receiving signed informed consent from all patients, under protocols approved by the Local Research Ethics Committee. The tissue was obtained from patients undergoing elective abdominoplasty or mastectomy surgeries. Tissue was refrigerated until its arrival in the laboratory, where it was cut into 1 cm2 section, and the subcutaneous layer of fat removed. Explants were incubated at 37 °C with 5% CO2 in Petri dishes with 10 mL of Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% FBS, 5 mg/mL l-glutamine, and 5 mg/mL penicillin/streptomycin (Thermo Fisher, U.K.). Media was replaced every 3 days, and explants were cultured for up to 21 days. Explants were injected intradermally using a Micro-Fine Demi 0.3 mL syringe (Becton Dickinson, U.K.) with 2 μg of RNA and 25 μL of LNPs in PBS, unless otherwise indicated. After 10 days, explants were inverted such that the epidermis was submerged in the media, and the media was supplemented with 30 ug/mL XenoLight RediJect D-Luciferin (PerkinElmer, U.K.). Samples were imaged with a In Vivo Imaging System (IVIS) FX Pro (Kodak Co., Rochester, NY, USA) equipped with Molecular Imaging Software Version 5.0 (Carestream Health, Rochester, NY, USA), for 60 min. Signal from each tissue explant was analyzed using Molecular Imaging software and expressed as total flux (p/s).
Flow Cytometry
For flow cytometry experiments, eGFP signal was analyzed after 3 days of culture. Skin was minced well with scissors and incubated in 3 mL DMEM supplemented with 1 mg/mL collagenase P (Sigma, U.K.) and 5 mg/mL Dispase II (Sigma, U.K.) for 4 h at 37 °C on a rotational shaker. Digests were then filtered through a 70 μm cell strainer and centrifuged at 1750 rpm for 5 min. Cells were then resuspended in 1 mL of FACS buffer (PBS + 2.5% fetal calf serum (FCS)) at a concentration of 107 cells/mL. One hundred microliters (100 μL) of cell suspension was added to a FACS tube and stained with fixable aqua live/dead cell stain (Thermo Fisher, U.K.) diluted 1:400 in FACS buffer for 20 min on ice. Cells were then washed with 2.5 mL of FACS buffer, centrifuged at 1750 rpm for 5 min, and stained with a panel of antibodies to identify each cell type, as described in Table S1 in the Supporting Information, for 30 min. Cells were then washed with 2.5 mL of FACS buffer, centrifuged at 1750 rpm for 5 min, and resuspended in 250 μL of PBS. Cells were fixed with 250 μL of 3% paraformaldehyde for a total final concentration of 1.5% and refrigerated until flow cytometry analysis. Samples were analyzed on a LSRFortessa (BD Biosciences, U.K.) with FACSDiva software (BD Biosciences, U.K.) with 30 000 acquired events. Gating strategy is shown in Figure S2 in the Supporting Information. GFP positive cells were quantified using FlowJo Version 10 (FlowJo LLC, Ashland, OR, USA).
Design of Experiment and Statistical Analysis
DoE analysis was performed in JMP, version 13.0, using a full factorial design with complexing lipid identity (C12–200, cephalin, DDA, DOTAP), lipid concentration (high, low), particle concentration (high, low), and ratio of cationic lipid to zwitterionic lipid (10:1, 1:1, 0.1:1) as input factors (Table 1), and luciferase expression in human skin explants after 10 days as the response. The data were analyzed using a fit model of standard least-squares for effect screening with the model effects designated as first- and second-order effects only. Nonsignificant effects were excluded from the model. Graphs were prepared in GraphPad Prism software, version 7.0. Flow cytometry statistical analysis was performed in Prism software, using a two-tailed t test with α = 0.05, which was used to indicate significance.
Table 1. Specifications of Design of Experiment Input Parameters.
| lipid identity | ratio of total lipid to RNA (w/w) | lipid concentration | particle concentration | ratio cationic to zwitterionic lipids(mol/mol) | ID # |
|---|---|---|---|---|---|
| C12–200 | 18:1 | medium | medium | – | 1 |
| 4:1 | medium | medium | – | 2 | |
| 1:1 | medium | medium | – | 3 | |
| 90:1 | high | high | 4 | ||
| 90:1 | high | low | 5 | ||
| cephalin | 18:1 | medium | medium | – | 6 |
| 4:1 | medium | medium | – | 7 | |
| 1:1 | medium | medium | – | 8 | |
| 90:1 | high | high | 9 | ||
| 90:1 | high | low | 10 | ||
| DDA | 18:1 | medium | medium | – | 11 |
| 4:1 | medium | medium | – | 12 | |
| 1:1 | medium | medium | – | 13 | |
| 18:1 | medium | medium | 10:1 | 14 | |
| 18:1 | medium | medium | 1:1 | 15 | |
| 0.1:1 | 16 | ||||
| 90:1 | high | high | 17 | ||
| 90:1 | high | low | 18 | ||
| DOTAP | 18:1 | high/low | high/low | – | 19 |
| 4:1 | low | low | – | 20 | |
| 1:1 | low | low | – | 21 | |
| 18:1 | low | low | 10:1 | 22 | |
| 18:1 | low | low | 1:1 | 23 | |
| 18:1 | low | low | 0.1:1 | 24 | |
| 90:1 | high | high | 25 | ||
| 90:1 | high | low | 26 |
Acknowledgments
We gratefully acknowledge the nursing team and patients at Charing Cross Hospital and Dormeur Investment Service Ltd. for providing funds to purchase equipment used in these studies.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.9b01774.
LNP characterization of particle size and surface charge (Figure S1); eGFP expression gating strategy (Figure S2); skin explant cell viability (Figure S3); identity of cells present in total population and GFP+ cells (Figure S4); cell viability of human skin explants after LNP-formulation saRNA injection (Figure S5); antibodies used for flow cytometry (Table S1) (PDF)
Author Contributions
A.K.B., P.F.M., and R.J.S. designed the experiments. A.K.B., B.I.Y., and P.F.M. performed the experiments and analyzed the data. A.K.B., B.I.Y., P.F.M., J.E.H., E.A.D., and R.J.S. wrote the manuscript. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Funding was provided by a Whitaker Post-Doctoral Fellowship (to A.K.B.), Innovate UK, the EPSRC Future Vaccines Manufacturing Research Hub at Imperial College, and the Imperial Confidence in Concept (ICiC) scheme (No. RSRO P75207).
The authors declare no competing financial interest.
Supplementary Material
References
- Guan S.; Rosenecker J. Nanotechnologies in Delivery of mRNA Therapeutics Using Nonviral Vector-Based Delivery Systems. Gene Ther. 2017, 24, 133. 10.1038/gt.2017.5. [DOI] [PubMed] [Google Scholar]
- Karikó K.; Muramatsu H.; Welsh F. A.; Ludwig J.; Kato H.; Akira S.; Weissman D. Incorporation of Pseudouridine into mRNA Yields Superior Nonimmunogenic Vector with Increased Translational Capacity and Biological Stability. Mol. Ther. 2008, 16, 1833–1840. 10.1038/mt.2008.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thess A.; Grund S.; Mui B. L.; Hope M. J.; Baumhof P.; Fotin-Mleczek M.; Schlake T. Sequence-Engineered mRNA without Chemical Nucleoside Modifications Enables an Effective Protein Therapy in Large Animals. Mol. Ther. 2015, 23, 1456–1464. 10.1038/mt.2015.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffman K. J.; Webber M. J.; Anderson D. G. Materials for Non-Viral Intracellular Delivery of Messenger RNA Therapeutics. J. Controlled Release 2016, 240, 227–234. 10.1016/j.jconrel.2015.12.032. [DOI] [PubMed] [Google Scholar]
- Jensen S.; Thomsen A. R. Sensing of RNA Viruses: A Review of Innate Immune Receptors Involved in Recognizing RNA Virus Invasion. J. Virol. 2012, 86, 2900. 10.1128/JVI.05738-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T.; Akira S. The Role of Pattern-Recognition Receptors in Innate Immunity: Update on Toll-Like Receptors. Nat. Immunol. 2010, 11, 373. 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- Perri S.; Greer C. E.; Thudium K.; Doe B.; Legg H.; Liu H.; Romero R. E.; Tang Z.; Bin Q.; Dubensky T. W.; Vajdy M.; Otten G. R.; Polo J. M. An Alphavirus Replicon Particle Chimera Derived from Venezuelan Equine Encephalitis and Sindbis Viruses Is a Potent Gene-Based Vaccine Delivery Vector. J. Virol. 2003, 77, 10394–10403. 10.1128/JVI.77.19.10394-10403.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahal J. S.; Khan O. F.; Cooper C. L.; McPartlan J. S.; Tsosie J. K.; Tilley L. D.; Sidik S. M.; Lourido S.; Langer R.; Bavari S.; Ploegh H. L.; Anderson D. G. Dendrimer-RNA Nanoparticles Generate Protective Immunity against Lethal Ebola, H1N1 Influenza, and Toxoplasma Gondii Challenges with a Single Dose. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E4133–E4142. 10.1073/pnas.1600299113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geall A. J.; Verma A.; Otten G. R.; Shaw C. A.; Hekele A.; Banerjee K.; Cu Y.; Beard C. W.; Brito L. A.; Krucker T.; O’Hagan D. T.; Singh M.; Mason P. W.; Valiante N. M.; Dormitzer P. R.; Barnett S. W.; Rappuoli R.; Ulmer J. B.; Mandl C. W. Nonviral Delivery of Self-Amplifying RNA Vaccines. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 14604. 10.1073/pnas.1209367109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogers W. M.; Oostermeijer H.; Mooij P.; Koopman G.; Verschoor E. J.; Davis D.; Ulmer J. B.; Brito L. A.; Cu Y.; Banerjee K.; Otten G. R.; Burke B.; Dey A.; Heeney J. L.; Shen X.; Tomaras G. D.; Labranche C.; Montefiori D. C.; Liao H.-X.; Haynes B.; Geall A. J.; Barnett S. W. Potent Immune Responses in Rhesus Macaques Induced by Nonviral Delivery of a Self-Amplifying RNA Vaccine Expressing HIV Type 1 Envelope with a Cationic Nanoemulsion. J. Infect. Dis. 2015, 211, 947–955. 10.1093/infdis/jiu522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perche F.; Benvegnu T.; Berchel M.; Lebegue L.; Pichon C.; Jaffrès P.-A.; Midoux P. Enhancement of Dendritic Cells Transfection In Vivo and of Vaccination against B16F10 Melanoma with Mannosylated Histidylated Lipopolyplexes Loaded with Tumor Antigen Messenger RNA. Nanomedicine (N. Y., NY, U. S.) 2011, 7, 445–453. 10.1016/j.nano.2010.12.010. [DOI] [PubMed] [Google Scholar]
- Uchida S.; Kinoh H.; Ishii T.; Matsui A.; Tockary T. A.; Takeda K. M.; Uchida H.; Osada K.; Itaka K.; Kataoka K. Systemic Delivery of Messenger RNA for the Treatment of Pancreatic Cancer Using Polyplex Nanomicelles with a Cholesterol Moiety. Biomaterials 2016, 82, 221–228. 10.1016/j.biomaterials.2015.12.031. [DOI] [PubMed] [Google Scholar]
- Fotin-Mleczek M.; Zanzinger K.; Heidenreich R.; Lorenz C.; Kowalczyk A.; Kallen K.-J.; Huber S. M. mRNA-Based Vaccines Synergize with Radiation Therapy to Eradicate Established Tumors. Radiat. Oncol. 2014, 9, 180. 10.1186/1748-717X-9-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough K. C.; Bassi I.; Milona P.; Suter R.; Thomann-Harwood L.; Englezou P.; Démoulins T.; Ruggli N. Self-Replicating Replicon-RNA Delivery to Dendritic Cells by Chitosan-Nanoparticles for Translation In Vitro and In Vivo. Mol. Ther.--Nucleic Acids 2014, 3, e173 10.1038/mtna.2014.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekele A.; Bertholet S.; Archer J.; Gibson D. G.; Palladino G.; Brito L. A.; Otten G. R.; Brazzoli M.; Buccato S.; Bonci A.; Casini D.; Maione D.; Qi Z.-Q.; Gill J. E.; Caiazza N. C.; Urano J.; Hubby B.; Gao G. F.; Shu Y.; De Gregorio E.; Mandl C. W.; Mason P. W.; Settembre E. C.; Ulmer J. B.; Craig Venter J.; Dormitzer P. R.; Rappuoli R.; Geall A. J. Rapidly Produced SAM(®) Vaccine against H7N9 Influenza Is Immunogenic in Mice. Emerging Microbes Infect. 2013, 2, 1–7. 10.1038/emi.2013.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazzoli M.; Magini D.; Bonci A.; Buccato S.; Giovani C.; Kratzer R.; Zurli V.; Mangiavacchi S.; Casini D.; Brito L. M.; De Gregorio E.; Mason P. W.; Ulmer J. B.; Geall A. J.; Bertholet S. Induction of Broad-Based Immunity and Protective Efficacy by Self-Amplifying mRNA Vaccines Encoding Influenza Virus Hemagglutinin. J. Virol. 2016, 90, 332. 10.1128/JVI.01786-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffman K. J.; Dorkin J. R.; Yang J. H.; Heartlein M. W.; DeRosa F.; Mir F. F.; Fenton O. S.; Anderson D. G. Optimization of Lipid Nanoparticle Formulations for mRNA Delivery In Vivo with Fractional Factorial and Definitive Screening Designs. Nano Lett. 2015, 15, 7300–7306. 10.1021/acs.nanolett.5b02497. [DOI] [PubMed] [Google Scholar]
- Bahl K.; Senn J. J.; Yuzhakov O.; Bulychev A.; Brito L. A.; Hassett K. J.; Laska M. E.; Smith M.; Almarsson Ö.; Thompson J.; Ribeiro A.; Watson M.; Zaks T.; Ciaramella G. Preclinical and Clinical Demonstration of Immunogenicity by mRNA Vaccines against H10N8 and H7N9 Influenza Viruses. Mol. Ther. 2017, 25, 1316–1327. 10.1016/j.ymthe.2017.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg J. H.; Nuijen B.; Beijnen J. H.; Vincent A.; van Tinteren H.; Kluge J.; Woerdeman L. A. E.; Hennink W. E.; Storm G.; Schumacher T. N.; Haanen J. B. A. G. Optimization of Intradermal Vaccination by DNA Tattooing in Human Skin. Hum. Gene Ther. 2009, 20, 181–189. 10.1089/hum.2008.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelfant S. Of Mice and Men” the Cell Cycle in Human Epidermis In Vivo. J. Invest. Dermatol. 1982, 78, 296–299. 10.1111/1523-1747.ep12507367. [DOI] [PubMed] [Google Scholar]
- Love K. T.; Mahon K. P.; Levins C. G.; Whitehead K. A.; Querbes W.; Dorkin J. R.; Qin J.; Cantley W.; Qin L. L.; Racie T.; Frank-Kamenetsky M.; Yip K. N.; Alvarez R.; Sah D. W. Y.; de Fougerolles A.; Fitzgerald K.; Koteliansky V.; Akinc A.; Langer R.; Anderson D. G. Lipid-Like Materials for Low-Dose, In Vivo Gene Silencing. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 1864–1869. 10.1073/pnas.0910603106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball R. L.; Hajj K. A.; Vizelman J.; Bajaj P.; Whitehead K. A. Lipid Nanoparticle Formulations for Enhanced Co-Delivery of siRNA and mRNA. Nano Lett. 2018, 18, 3814–3822. 10.1021/acs.nanolett.8b01101. [DOI] [PubMed] [Google Scholar]
- Henriksen-Lacey M.; Christensen D.; Bramwell V. W.; Lindenstrøm T.; Agger E. M.; Andersen P.; Perrie Y. Comparison of the Depot Effect and Immunogenicity of Liposomes Based on Dimethyldioctadecylammonium (DDA), 3β-[N-(N′,N′-Dimethylaminoethane)Carbomyl] Cholesterol (DC-Chol), and 1,2-Dioleoyl-3-Trimethylammonium Propane (DOTAP): Prolonged Liposome Retention Mediates Stronger Th1 Responses. Mol. Pharmaceutics 2011, 8, 153–161. 10.1021/mp100208f. [DOI] [PubMed] [Google Scholar]
- De Serrano L. O.; Burkhart D. J. Liposomal Vaccine Formulations as Prophylactic Agents: Design Considerations for Modern Vaccines. J. Nanobiotechnol. 2017, 15, 83. 10.1186/s12951-017-0319-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martino S.; di Girolamo I.; Tiribuzi R.; D’Angelo F.; Datti A.; Orlacchio A. Efficient siRNA Delivery by the Cationic Liposome DOTAP in Human Hematopoietic Stem Cells Differentiating into Dendritic Cells. J. Biomed. Biotechnol. 2009, 2009, 410260. 10.1155/2009/410260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneda M. M.; Sasaki Y.; Lanza G. M.; Milbrandt J.; Wickline S. A. Mechanisms of Nucleotide Trafficking During siRNA Delivery to Endothelial Cells Using Perfluorocarbon Nanoemulsions. Biomaterials 2010, 31, 3079–3086. 10.1016/j.biomaterials.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Li B.; Luo X.; Zhao W.; Jiang J.; Zhang C.; Gao M.; Chen X.; Dong Y. Biodegradable Amino-Ester Nanomaterials for Cas9 mRNA Delivery In Vitro and In Vivo. ACS Appl. Mater. Interfaces 2017, 9, 25481–25487. 10.1021/acsami.7b08163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B.-K.; Hwang G.-B.; Seu Y.-B.; Choi J.-S.; Jin K. S.; Doh K.-O. DOTAP/DOPE Ratio and Cell Type Determine Transfection Efficiency with DOTAP-Liposomes. Biochim. Biophys. Acta, Biomembr. 2015, 1848, 1996–2001. 10.1016/j.bbamem.2015.06.020. [DOI] [PubMed] [Google Scholar]
- Mével M.; Haudebourg T.; Colombani T.; Peuziat P.; Dallet L.; Chatin B.; Lambert O.; Berchel M.; Montier T.; Jaffrès P.-A.; Lehn P.; Pitard B. Important Role of Phosphoramido Linkage in Imidazole-Based Dioleyl Helper Lipids for Liposome Stability and Primary Cell Transfection. J. Gene Med. 2016, 18, 3–15. 10.1002/jgm.2869. [DOI] [PubMed] [Google Scholar]
- Cavalcanti R. R. M.; Lira R. B.; Riske K. A. Study of the Fusion Mechanism of Fusogenic Cationic Liposomes with Anionic Model Membranes. Biophys. J. 2018, 114, 606a. 10.1016/j.bpj.2017.11.3314. [DOI] [Google Scholar]
- Démoulins T.; Milona P.; Englezou P. C.; Ebensen T.; Schulze K.; Suter R.; Pichon C.; Midoux P.; Guzmán C. A.; Ruggli N.; McCullough K. C. Polyethylenimine-Based Polyplex Delivery of Self-Replicating RNA Vaccines. Nanomedicine (N. Y., NY, U. S.) 2016, 12, 711–722. 10.1016/j.nano.2015.11.001. [DOI] [PubMed] [Google Scholar]
- Pardi N.; Parkhouse K.; Kirkpatrick E.; McMahon M.; Zost S. J.; Mui B. L.; Tam Y. K.; Karikó K.; Barbosa C. J.; Madden T. D.; Hope M. J.; Krammer F.; Hensley S. E.; Weissman D. Nucleoside-Modified mRNA Immunization Elicits Influenza Virus Hemagglutinin Stalk-Specific Antibodies. Nat. Commun. 2018, 9, 3361. 10.1038/s41467-018-05482-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichmuth A. M.; Oberli M. A.; Jaklenec A.; Langer R.; Blankschtein D. mRNA Vaccine Delivery Using Lipid Nanoparticles. Ther. Delivery 2016, 7, 319–334. 10.4155/tde-2016-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albanese A.; Tang P. S.; Chan W. C. W. The Effect of Nanoparticle Size, Shape, and Surface Chemistry on Biological Systems. Annu. Rev. Biomed. Eng. 2012, 14, 1–16. 10.1146/annurev-bioeng-071811-150124. [DOI] [PubMed] [Google Scholar]
- Golestani R.; Pourfathollah A. A.; Moazzeni S. M. An Extreme Strategy for the Production of Hybridoma. Hybridoma 2009, 28, 139–144. 10.1089/hyb.2008.0076. [DOI] [PubMed] [Google Scholar]
- Phua K. K. L.; Leong K. W.; Nair S. K. Transfection Efficiency and Transgene Expression Kinetics of mRNA Delivered in Naked and Nanoparticle Format. J. Controlled Release 2013, 166, 227–233. 10.1016/j.jconrel.2012.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang F.; Lindgren G.; Lin A.; Thompson E. A.; Ols S.; Röhss J.; John S.; Hassett K.; Yuzhakov O.; Bahl K.; Brito L. A.; Salter H.; Ciaramella G.; Loré K. Efficient Targeting and Activation of Antigen-Presenting Cells In Vivo after Modified mRNA Vaccine Administration in Rhesus Macaques. Mol. Ther. 2017, 25, 2635–2647. 10.1016/j.ymthe.2017.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.