

Abstract
Psoriasis is a common autoimmune disorder that affects the skin. Approximately 30% of individuals with psoriasis will develop inflammatory arthritis, often in the setting of human leukocyte antigen B27. Both forms of disease are thought to be the result of prolonged inflammation mediated by T lymphocytes, dendritic cells, and keratinocytes. While there are treatments aimed at immunomodulation, targeting T cell co-inhibitory receptors signaling pathways may provide therapeutic benefit. This review will discuss in detail four T cell co-inhibitory receptors and their potential application for the treatment of psoriasis and psoriatic arthritis.
Psoriasis (Ps) is a common autoimmune skin disorder affecting nearly 2% to 4% of the global population.1 Despite its broad dispersion in the global population, these conditions are more commonly associated with the American, Canadian, and European populations compared to African, African-American, and Asian populations.2,3 Men and women are affected with equal frequency. The disease may begin at any age. There are five different types of psoriasis: plaque, guttate, inverse, pustular, and erythrodermic.4 Plaque psoriasis, also known as psoriasis vulgaris, makes up about 90% of cases. It typically presents with plaques that have an erythematous base and a silvery surface. Areas of the body most commonly affected are the back of the forearms, shins, around the navel, and the scalp. Psoriasis can also cause changes to the nails, such as pitting or onycholysis, hyperkeratosis under the nails, and horizontal ridging. Being multifactorial, psoriasis has both genetic and environmental factors that trigger the onset of disease.1 Identical twins are three times more likely to be affected compared to non-identical twins. Certain environmental conditions, such as stress, can also trigger psoriasis.5 Psoriasis has many comorbidities associated with it. These include, but are not limited to, cardiovascular disease, inflammatory bowel disease, nonalcoholic fatty liver disease, and lymphoma.6,7 Current treatments include topical agents, phototherapy, non-biologic systemic agents (e.g., methotrexate, cyclosporine, and retinoids), and biologics (e.g., anti-TNF-α, anti-IL-12/23, and anti-IL-17 monoclonal antibodies).
About 30% of people with Ps develop psoriatic arthritis (PsA). PsA is classified as a seronegative spondyloarthropathy and occurs more commonly in patients with human leukocyte antigen (HLA) type B27. Psoriatic arthritis is characterized by asymmetrical oligoarthritis of the hands and wrists present in most cases as well as dactylitis, a sausage like swelling of the fingers. PsA patients can also develop sacroiliitis or spondylitis, which is present in a majority of cases. Another important clinical feature of PsA is enthesitis frequently affecting the Achilles tendon.
Ps and PsA are autoimmune disorders and according to current view arise as a consequence of the aberrant interplay between T cells, dendritic cells, and keratinocytes, giving rise to a self-perpetuating loop that amplifies and sustains inflammation in the skin and in the joints. In particular, myeloid cell secretion of IL-1, IL-23, and IL-12 activates IL-17-producing T cells (TH17) and TH1 cells, leading to the production of additional inflammatory cytokines, such as IL-17, IFN-γ, TNF-α, and IL-22. These cytokines mediate effects on keratinocytes, thus establishing the inflammatory cycle. Additional T cells subtypes, such as the autoreactive CD8 T cells, which were discovered in the 1990s, and the more recent γδ T cells, were also implicated in the disease pathogenesis.
Engagement of T cells requires two signals: antigen recognition through the T cell receptor (TCR) and major histocompatibility complex (MHC); and co-stimulatory or co-inhibitory signals. Chemokine receptors, CD28, CD4, and CD8 are well-known co-stimulatory receptors, while cytotoxic T-cell lymphocyte antigen-4 (CTLA-4), programmed-death receptor-1 (PD-1), T-cell immunoglobulin and mucin domain-3 (TIM-3), and leukocyte activation gene-3 (LAG-3) are co-inhibitory receptors. Several drug therapies utilized today involve antagonists of specific receptors in attempt to down-regulate the immune system. There have also been several suggestions to use agonists of the co-inhibitory receptors in an attempt to treat inflammation. This review will focus on four major co-inhibitory receptors and their respective backgrounds in light of their potential therapeutic application in autoimmunity.
Cytotoxic T Lymphocyte Antigen (CTLA)-4
CTLA-4 is a type I transmembrane protein that is part of the immunoglobulin superfamily. It is considered a homologue of CD28 that is capable of binding to both CD80 and CD86 expressed on antigen presenting cells (Fig. 1), but unlike CD28, CTLA-4 is functionally a co-inhibitory receptor.8 CTLA-4 has a cytoplasmic tail containing an YxxM motif that plays an important role in downstream signaling as well as in its subcellular localization.9 In resting cells, CTLA-4 is expressed predominately intracellularly in vesicles, the Golgi apparatus, endosomes, and lysosomes.9–11 CTLA-4 can be detected on the cell surface upon activation downstream of the TCR. Next, the YxxM motif is phosphorylated, and CTLA-4 is stabilized on the surface of T cells. Its surface expression is also based on calcium flux.12–15 When intracellular calcium levels increase, cell surface expression increases as well. When the YxxM motif is dephosphorylated, it can interact with the clathrin-associated adaptor protein AP-2, and CTLA-4 becomes endocytosed.12,13,16,17 While expression on effectors T cells must be induced by activation,11,18,19 CTLA-4 is constitutively expressed on the surface regulatory T cells (Treg).
Figure 1.
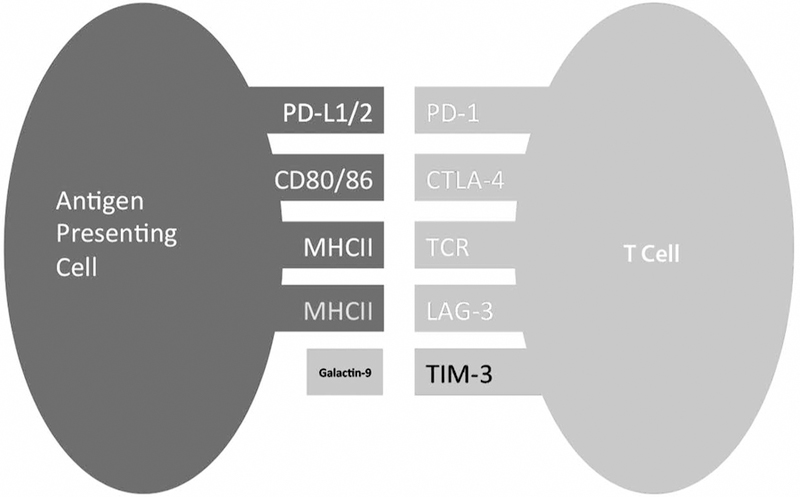
Inhibitory receptors expressed on T cell and their counter ligands.
The mechanism for CTLA-4-mediated cellular inhibition has not been well defined, but there are a few models for the means of its inhibition. Both CTLA-4 and CD28 can bind to the same ligands CD80 (B7–1) and CD86 (B7–2), but CTLA-4 binds at higher affinity.11 Thus, due to the competitive binding of B7 to CTLA-4, it can prevent the co-stimulatory signal that is provided by CD28.15,20–22 Following the engagement of either B7–1 or B7–2 ligands to the MYPPPY binding ecto-motif of CTLA-4, a decrease in T cell proliferation, cytokine production, and overall responsiveness ensue. Intrinsic models for inhibition involve the cytoplasmic tail of CTLA-4. At the molecular level, both proximal TCR signaling events and downstream signaling that are normally enhanced by CD28 and B7–1 or B7–2 are inhibited. Conceptually, there are two proposed modes for CTLA-4-mediated inhibition: threshold and attenuation.20 In the threshold model, CTLA-4 integration into the immunological synapse raises the threshold level for activation of T cells and by that controls the response in an antigen-dependent manner.23 In the attenuation model, CTLA-4 essentially reduces the signals delivered by CD28.
Several studies have reported involvement of the CTLA-4/CD28/B7 system in Ps and PsA pathogenesis (Table 1). One study performed by Summers and coworkers examined the expression of both CD80 and CD86 on synovial dendritic cells (DC) isolated from patients with PsA. Low levels of both CD80 and CD86 were found in the majority of patients. The conclusion of this study was that lack of expression of CD80 and CD86 on synovial DC might explain the altered cellular immune responses in these patients.24 Another prospect is that the low levels of CD80 and CD86 on these cells explain why CTLA-4 itself is not properly engaged to inhibit T cell signaling and functions.
Table 1.
Summary of Clinical Studies of Inhibitory Receptors
| Targeted Receptor and Focus of Study | Descriptive Findings | Study |
|---|---|---|
| CTLA-4 | ||
| Synovial dendritic cells | Expression and function of CD80 and CD86 costimulator molecules on synovial dendritic cells in chronic arthritis. | Summers K, et al., 1996 |
| T cells | HLA-DR and IL-2R identify persistently activated T cells in psoriasis vulgaris lesional skin: blood and skin comparisons by flow cytometry. | Ferenczi L, et al., 2000 |
| Genetic polymorphisms of CTLA-4 | CTLA-4 polymorphisms in Japanese patients with psoriasis vulgaris. | Tsunemi Y, et al., 2003 |
| Lack of associations of CTLA-4 and ICAM-1 polymorphisms with psoriasis in the Korean population. | Kim Y, et al., 2003 | |
| CTLA-4 gene polymorphisms and natural soluble CTLA-4 protein in psoriasis vulgaris. | Luszczek W, et al., 2006 | |
| Regulatory T cells (Tregs) | Dysfunctional blood and target tissue CD4+CD25high regulatory T cells in psoriasis: mechanism underlying unrestrained pathogenic effector T cell proliferation. | Sugiyama H, et al., 2005 |
| FoxP3 mRNA splice forms in synovial CD4+ T cells in rheumatoid arthritis and psoriatic arthritis. | Ryder L, et al., 2000 | |
| CTLA-4-Ig (Abatacept) | Abatacept in the treatment of patients with psoriatic arthritis: results of a 6-month, multicenter, randomized, double-blind, placebo-controlled, phase II trial. | Mease P, et al., 2011 |
| Inhibition of pro-inflammatory cytokine generation by CTLA-4-Ig in the skin and colon of mice adoptively transplanted with CD45Rhigh CD4+ T cells correlates with suppression of psoriasis and colitis. | Davenport CM, et al., 2002 | |
| Anti-CTLA-4 in cancer therapy | Ipilimumab therapy in patients with advanced melanoma and preexisting autoimmune disorders. | Johnson DB, et al., 2016 |
| 8-methoxypsoralen plus UVA photo chemotherapy (PUVA) | 8-methoxypsoralen plus ultraviolet A therapy acts via inhibition of the IL-23/Th17 axis and induction of Foxp3+ regulatory T cells involving CTLA-4 signaling in a psoriasis-like skin disorder. | Singh TP, et al., 2010 |
| PD-1 | ||
| PD-1 expression | PD-1 signaling in primary T cells. | Riley JL, 2009 |
| Expression of programmed death-1 in skin biopsies of benign inflammatory vs. lymphomatous erythroderma. | Cetinozman F, Jansen PM, Willemze R, 2014 | |
| Analysis of programmed death-1 in patients with psoriatic arthritis. | Peled M, et al., 2015 | |
| PD-1 blockade | PD-1 regulates Imiquimod-induced psoriasiform dermatitis through inhibition of IL-17A expression by innate gamma deltaLow T cells. | Imai Y, et al., 2015 |
| Programmed cell death ligand 1 alleviates psoriatic inflammation by suppressing IL-17A production from programmed cell death 1high T cells. | Kim JH, et al., 2016 | |
| Anti-PD-1 in cancer therapy (Nivolumab and Pembrolizumab) | Exacerbation of psoriasis during nivolumab therapy for metastatic melanoma. | Matsumura N, et al., 2016 |
| Severe psoriasis flare after anti-programmed death ligand 1 (PD-L1) therapy for metastatic non-small cell lung cancer (NSCLC). | Chia PL, John T, 2016 | |
| TIM-3 | ||
| Expression | Impaired expression of Tim-3 on Th17 and Th1 cells in psoriasis. | Kanai Y, et al., 2009 |
| Soluble galactin-9 | Stable form of galectin-9, a Tim-3 ligand, inhibits contact hypersensitivity and psoriatic reactions: a potent therapeutic tool for Th1- and/or Th17-mediated skin inflammation. | Niwa H, et al., 2009 |
| LAG-3 | ||
| No published studies | ||
To gain a better understanding of the surface markers expressed on T cells in patients with Ps, Ferenczi and colleagues studied epidermal T cells from skin lesions through flow cytometry. They found that most T cells isolated from Ps patients express CD80 constitutively, along with other activated T cell markers,25 but there were no significant differences in the levels of CD86 or CTLA-4 expression.
Considering that CTLA-4 could play an important role in the development of Ps vulgaris, Tsunemi and associates26 looked at polymorphisms in the CTLA-4 gene that could designate susceptibility of developing the disease. One hundred fifty-three unrelated Japanese patients were compared for single nucleotide polymorphisms (SNPs) in the 3’ UTR (318 C/T) and in the first exon (49 A/G), and compared them with 104 healthy control individuals. No significant differences were found between patients diagnosed with Ps vulgaris and the control individuals.26 A similar study by Kim and coworkers27 was performed on 137 individuals of Korean descent, compared to a control group of 191 individuals without Ps. The frequency of the CTLA-4 49 A/G variance was slightly higher in Ps patients versus the control group (54.7% vs. 45%), and the CTLA-4 49 G/G homozygous genotype was lower in Ps patients (45.3% vs. 55%).27 Another similar study by Luszczek and colleagues28 was performed on Caucasian patients with Ps vulgaris. They compared additional CTLA-4 SNPs (1147 C/T, 318 C/T, and 49 A/G) from 116 Caucasians diagnosed with Ps vulgaris, and 123 healthy blood donors. Again, no statistically significant difference was found among the two groups. Their study was taken a step further by testing serum for soluble CTLA-4 levels (sCTLA-4). They found that patients who developed Ps before the age of 40 years old had higher levels sCTLA-4. They hypothesized that elevated levels of sCTLA-4 could compete with activated T cells expressing CTLA-4 by blocking its ability to regulate T cell responses.28
As mentioned, CTLA-4 is constitutively expressed on Tregs. T cell activation plays a critical role in the development of Ps, but CD4+CD25+ Treg effector dysfunction could be another possible explanation for their uncontrolled phenotype. In a study by Sugiyama and associates,29 the group successfully revealed that Ps CD4+CD25+ Treg are dysfunctional when compared to cells isolated from normal individuals. Ps CD4+CD25+ Treg showed only 60.6% inhibition of CD4+CD25− Teff proliferation compared to 87.8% in normal individuals. An 8-fold increase in the ratio of Treg:Teff was required to achieve a 50% proliferation inhibition in CD4+CD25− Teff cells. Ps CD4+CD25− Teff also showed increased proliferative response with allogenic APCs compared to normal CD4+CD25− Teffcells. The investigators indicated that the source of the dysfunction could be associated with a proliferative functional deficit in the Ps Treg compartment.29
Ryder and coworkers30 suggested that Treg may be dysfunctional in PsA patients. They found that Foxp3 (Treg transcription factor) expression in CD4+ Treg was elevated in both synovial fluid and peripheral blood of PsA cells compared to healthy individuals. Interestingly, CTLA-4 mRNA expression was not increased in synovial fluid CD4+ Treg and was decreased in peripheral blood CD4+ Treg for arthritis patients. These findings may indicate that synovial fluid and peripheral blood Treg are induced locally or selectively recruited to the sites of inflammation in joints.30
Moving to interventional patient data, Abrams and coworkers performed phase I drug testing on Ps patients. They used previous knowledge that activated T cells plays a critical role in the development of Ps. Toward that end, they treated the patient with CTLA-4-Ig (abatacept), a fusion protein that contained an extracellular domain of human CTLA-4 and a fragment Fc portion of human IgG1. This soluble chimeric protein could bind B7 proteins on APC and effectively block co-stimulatory signals to CD28. They found that 46% of patients had a total of 50% or greater improvement in their disease activity indexes compared to the baseline upon drug treatment. The efficacy of the drug was further proved in another study published by the same group in the year 2000. During administration of the drug intravenously, they noticed a decrease in mature DCs and replacement with immature DCs, which could be due to a decreased co-stimulation provided by T cells that was blocked through the administration of the CTLA-4-Ig.31 CTLA-4-Ig has also been used to treat PsA. In a randomized double-blind study, 48% of PsA patients administered CTLA-4-Ig at 10 mg/kg for 6 months showed improvement in their clinical disease activity compared to only 19% in the placebo group. The study also showed that patients receiving CTLA-4-Ig at 10 mg/kg without prior anti-TNF agents achieved ACR20 of 56% compared to 31% for those previously treated with anti-TNF agents. However, when evaluating its effectiveness for treatment for Ps, the results were inconsistent.32,33
Another study utilizing CTLA-4-Ig by Davenport and colleagues34 exploited immunocompromised mice and transplanted them with CD45RBhi T cells and staphylococcal antigen. None of the mice treated with CTLA-4-Ig developed clinical or histological skin lesions, while all mice left untreated developed skin lesions. This study demonstrated that immunocompromised mice treated with CTLA-4-Ig were able to successfully inhibit the development of induced Ps.34
Anti-CTLA-4 based immune therapies have also been extensively used in cancer treatment. Administration of ipilimumab, a mouse anti-CTLA-4 blocking monoclonal antibody, has shown to have adverse side effects in the treatment of melanoma with patients who have baseline autoimmune disorders. A study by Johnson and associates35 showed that up to 27% of the patients who had previously been diagnosed with an autoimmune disorder showed recurrent manifestations of their symptoms, which included worsening plaques in psoriasis. This study concluded that that anti-immune suppressive therapy has the potential to exacerbate autoimmune disorders such as Ps.35
Another standard dermatological therapy for Ps is utilization of 8-methoxypsoralen plus UVA photochemotherapy (PUVA). A study by Singh and coworkers36 sought to understand the mechanisms involved in regulating Ps by using K5.hTGF-β1 transgenic mice that develop scaly erythema and skin lesions similar to those of humans with Ps. They found that administration prolonged the survival of mice with Ps-like skin alterations, as compared to similar results with humans that have pustular or erythrodermic Ps. Following PUVA treatment, there was an up-regulation of the number of CD4+CD25+ Treg. They were also capable of suppressing the proliferation of Teff cells (unlike normal Ps Treg cells whose suppressive activity is dysfunctional). To understand the suppressive mechanism of these Tregs, K5.hTGF-β1 mice undergoing PUVA treatment were administered anti-CTLA-4 monoclonal antibodies. Following this treatment, PUVA was unable to suppress disease in K5.hTGF-β1 mice, unlike the administration of the isotype control antibodies. While these results are based on mice models, the results were promising due to similarities in Ps development in both the K5.hTGF-β1 murine model and humans.36
In conclusion, the CTLA-4/CD28/B7 system has still yet to be explored further as a potential therapy for Ps or PsA. Despite other studies where polymorphisms of CTLA-4 have been linked to autoimmune disorders, such as systemic lupus erythematosus, rheumatoid arthritis, Grave’s disease, Hashimoto’s thyroiditis, and type I diabetes,37 this has not been the case for either Ps or PsA. While manipulating the pathways has been utilized efficiently by drugs, such as abatacept, this form of treatment takes advantage of its binding capabilities to B7–1 or B7–2 and out-competes CD28. PUVA treatment has also proven to be efficient in murine models, likely by helping improving the function of Treg. To further understand the mechanism of this system in Ps and PsA additional studies using both CTLA-4 agonist and antagonist are needed.
Programmed Cell Death (PD)-1
PD-1 is a co-inhibitory receptor found mainly on the surface of activated T cells, as well as on other hematopoietic cell types, such as B cells, macrophages, natural killer T cells (NKT), and certain types of dendritic cells (DC).38,39 Structurally, PD-1 contains an Ig variable-type (V-type) domain, a transmembrane domain, and a cytoplasmic domain. The cytoplasmic domain contains an immunoreceptor tyrosine-based inhibitory motif (ITIM) and an immunoreceptor tyrosine-based switch motif (ITSM).38,40–42 Phosphorylated ITSM binds to the phosphatase SHP-2 that in return acts on and deactivates key signaling proteins downstream of the TCR.
PD-1 is first seen on the surface of double negative (CD4− CD8−) T cells during their thymic development.40,43 In more mature cells, PD-1 is expressed on single positive CD4+ and CD8+ T cells upon activation in the periphery.39,40 PD-1 binds to either PD-L1 (B7-H1; CD274) or PD-L2 (B7-DC; CD273),42,44,45 (Fig. 1) and has a three-fold higher affinity for the later.40 PD-1 engagement by its ligands causes a decrease in T cell proliferation, cytokine production, cytolytic function, protein synthesis, and overall cell survival.40,46
The expression of PD-1 in Ps was first reported in 2014 when skin biopsies were collected from patients with erythrodermic Ps (Table 1). Half of the patients had up to 50% dermal PD-1 expressing T cells. While this study did not make any conclusions on the utilization of PD-1 expression as marker for Ps, it did show that PD-1 is expressed on activated epidermal T cells.47
A more recent study has shown that PD-1 can actually play an important role in regulating Ps disease activity. Imai and coworkers48 tested the effects PD-1 blockade, either by genetic knockout or monoclonal antibodies administration, on a murine imiquimod-induced psoriasis model. At moderate concentrations of imiquimod, there was enhanced psoriaform dermatitis in mice that had PD-1 blockade, suggesting that PD-1 might have a regulatory function in psoriasiform dermatitis. Interestingly, the lesions were characterized by increased neutrophilic infiltration, epidermal hyperplasia, and increased expression of TH17 cytokines.48 Another study by Kim and colleagues49 showed that PD-1 was over-expressed on T cells from an imiquimod-induced Ps model and on human T cells collected from Ps patients. Moreover, the same group reported that IL-17 T cells obtained from Ps patients had higher expression of PD-1 compared to that of normal individuals. Furthermore, imiquimod-induced murine Ps treated with PD-L1-Fc showed decreased inflammation,49 suggesting that PD-1 may exert an anti-inflammatory effect in this model.
Additionally, it has been observed that patients treated with anti-PD1 immunotherapy in the treatment of malignancy develop psoriatic lesions. This was first discovered in a patient with metastatic melanoma treated with nivolumab, who developed Ps plaques scattered over the trunk and extremities.50 Subsequently, this was also reported in a patient with metastatic non-small cell lung cancer undergoing treatment with pembrolizumab who developed diffuse erythematous and scaly plaque-like psoriasis lesions in his upper and lower limbs, trunk, and back.51
Our group has investigated the role of the potential role of PD-1 as a treatment modality for PsA. Due to PsA being a T cell mediated autoimmune disorder, we analyzed the expression levels of PD-1 in T cells isolated from patients with PsA and from healthy controls. PD-1 was expressed in 11% ± 2% of the CD3+ T cells isolated from the patients, compared to 1.2% ± 5% of the cells collected from the healthy population. Furthermore, the disease activity scores inversely correlated with PD-1 expression levels, suggesting insufficient PD-1 signaling might contribute to disease pathogenesis.52 Clearly, more data about PD-1 biology (e.g., polymorphism, signaling, and function) is absolutely needed prior to any attempt to engage the PD-1 receptor aimed at treatment of Ps and PsA.
T cell Immunoglobin and Mucin Domain-3 (TIM-3)
Previously known as a receptor for galectin-9 and phosphatidylserine, TIM-3 is a 33 Kd type I transmembrane protein. It is expressed on terminally differentiated TH1 cells, TH17 cells, DC cells, macrophages, natural killers cells, and some cancer cells.53–58 Like other inhibitory receptors, it mediated exhaustion TH1 responses by regulating the interaction with APCs. It consists of an IgV and mucin extracellular domains, a single transmembrane domain, and a cytoplasmic tail. The IgV domain is capable of binding phosphatidylserine molecules that are found on the surface of APCs.59
Differential functions are the results of interaction with different ligands. Some inhibitory effects are shown following engagement with galactin-9 (Fig. 1). One study showed that binding induces an intracellular calcium influx and cell death for TH1 cells60 while another study also suggested that Bat3 is released from the cytoplasmic tail of TIM-3 and activates the cell death pathway.61 Overall binding to galactin-9 leads to immune suppression of T cells, and following injection of galactin-9, there has been an upregulation of Treg cells.59,62–64
Due to its immunosuppressive capabilities, TIM-3 has been studied for potential therapeutic use in Ps (Table 1). It has been shown that TIM-3 is expressed in activated TH1 cells but is also expressed in murine TH17 cells.65 Considering that galactin-9 was found in skin lesions of Ps patients, Kanai and associates66 studied if TH1 and TH17 cells in blood expressed TIM-3, and whether its capabilities of immunosuppression were impaired. They found that Ps patients’ TH1 and TH17 cells had lower levels of TIM-3 expression compared to healthy patients. They also hypothesized that despite high levels of expression of galactin-9 in fibroblast cells of skin lesions, the lowered expression of TIM-3 could impair the function of TH1 and TH17 cells. The cause of the impairment was suggested to occur during cell differentiation or expansion of T cell clones in Ps patients.66
Another study by Niwa and coworkers67 sought to use a stable form of galactin-9 (sGal-9) that is resistant to proteolysis and to engage TH1 and TH17 cells to induce T cell apoptosis in IL-23 based Ps murine models. Following induction of disease, administration of sGal-9 reduced epidermal hyperplasia in the ear lobes of mice. They also found that administration of sGal-9 increased the numbers of Foxp3+CD25+CD4+ Treg. The sGal-9 resistance of proteolytic inactivation of sGal-9 properties make it a potential therapeutic tool to engage TH17 cells that can potentially alleviate autoimmune disorders, such as Ps.67
In conclusion, while research in TIM-3 biology and signaling is still ongoing, its ability to suppress the autoimmune responses of T cells has therapeutic potential for both Ps and PsA. TH17 cells are the main mediators of Ps and PsA, and the fact that they are capable of expressing TIM-3 is promising. By engaging this receptor and inducing apoptosis, we can potentially reduce T cell mediated inflammation. Possible side effects of reducing neutrophil recruitment during a bacterial infection may occur, since TH17 cells secrete IL-17, a potent neutrophil chemo-attractant. The preliminary studies mentioned above provide insight into utilizing this inhibitory receptor to our advantage, but further investigations still need to occur before a drug is developed for human trials.
Lymphocyte Activation Gene −3 (LAG-3)
LAG-3 is a co-inhibitory receptor that is expressed on the cell surface of hematopoietic cells.68 More specifically, it is expressed on the surface of B cells, NK cells, NKT cells, plasmacytoid DC, γδ T cells, activated T cells, and Tregs.69–76 It represented a homologue of CD4, and it is capable of binding to MHC class II molecules but with higher affinity.68 It is also expressed intracellularly in close vicinity to microtubule-organizing centers (MTOC).
LAG-3 has different regulatory roles in different cell types. Effector CD4 T cells are capable of expressing MHC class II molecules upon activation. The molecular mechanism of LAG-3 downstream signaling is unclear, but ligand engagement can inhibit T cell proliferation, cytokine production, and calcium influx.77,78 Furthermore, LAG-3 can outcompete CD4 for binding to MHC class II molecule for activation. LAG-3 has a unique cytoplasmic tail that contains three domains. One of those, the KIEELE motif, has been shown to have an important role in the suppressive capabilities of CD4 T cells. Naïve CD8 T cells also express LAG-3 but at very low levels. Following activation, LAG-3 is upregulated,73 but the exact biologic meaning of that is not clear.
Currently, the data is limited in regard to the therapeutic potential of LAG-3 in the treatment of Ps or PsA. Due to its immunosuppressive function, it may serve as an additional therapeutic target to consider.
Conclusions
Most of the targeted therapies for Ps and PsA have been geared toward immunosuppression; while utilization of the natural suppressive capabilities of the immune system have been rarely used or poorly understood (Table 1). CTLA-4 has been the most widely studied receptor for Ps and PsA treatment therapy but still requires further studies. Other agonists targeting the receptors PD-1, Tim-3, and LAG-3 have the potential to be powerful treatments for Ps and PsA as well. Another potential therapeutic option is to combine therapies that would utilize multiple co-inhibitory receptors to enhance the suppression of dysfunctional T cells. There is a fine balance between immunosuppression against autoimmune disorders and controlled regulation against malignancy. Again there needs to be more basic and clinical research geared toward the co-inhibitory receptors in order to understand their potential as treatment modalities against Ps and PsA.
Acknowledgments
Disclosure Statement
This work was supported by the Irma T. Hirschl Trust (A.M.), the Colton family (A.M.), the Rheumatology Research Foundation (A.M.), and NIH R01AI125640 (A.M.).
Contributor Information
Nehal Shah, Touro College of Osteopathic Medicine, and Department of Medicine, New York University School of Medicine, New York, New York.
Sabina Sandigursky, Department of Medicine, New York University School of Medicine, New York, New York.
Adam Mor, Department of Medicine, and Perlmutter Cancer Center, New York University School of Medicine, New York, New York.
References
- 1.Alexopoulos A, Chrousos GP. Stress-related skin disorders. Rev Endocr Metab Disord 2016. September;17(3):295–304. [DOI] [PubMed] [Google Scholar]
- 2.Harden JL, Krueger JG, Bowcock AM. The immunogenetics of psoriasis: a comprehensive review. J Autoimmun 2015. November;64:66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perera GK, Di Meglio P, Nestle FO. Psoriasis. Annu Rev Pathol 2012;7:385–422. [DOI] [PubMed] [Google Scholar]
- 4.Boehncke WH, Schon MP. Psoriasis. Lancet 2015. September 5;386(9997):983–94. [DOI] [PubMed] [Google Scholar]
- 5.Rampton DS. The influence of stress on the development and severity of immune-mediated diseases. J Rheumatol Suppl 2011. November;88:43–7. [DOI] [PubMed] [Google Scholar]
- 6.Ni C, Chiu MW. Psoriasis and comorbidities: links and risks. Clin Cosmet Investig Dermatol 2014. April 17;7:119–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Onumah N, Kircik LH. Psoriasis and its comorbidities. J Drugs Dermatol 2012. May;11(5 Suppl):s5–10. [PubMed] [Google Scholar]
- 8.Verhagen J, Sabatos CA, Wraith DC. The role of CTLA-4 in immune regulation. Immunol Lett 2008. January 15;115(1):73–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leung HT, Bradshaw J, Cleaveland JS, Linsley PS. Cytotoxic T lymphocyte-associated molecule-4, a high-avidity receptor for CD80 and CD86, contains an intracellular localization motif in its cytoplasmic tail. J Biol Chem 1995. October 20;270(42):25107–14. [DOI] [PubMed] [Google Scholar]
- 10.Valk E, Leung R, Kang H, et al. T cell receptor-interacting molecule acts as a chaperone to modulate surface expression of the CTLA-4 coreceptor. Immunity 2006. November;25(5):807–21. [DOI] [PubMed] [Google Scholar]
- 11.Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol Rev 2009. May;229(1):12–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shiratori T, Miyatake S, Ohno H, et al. Tyrosine phosphorylation controls internalization of CTLA-4 by regulating its interaction with clathrin-associated adaptor complex AP-2. Immunity 1997. May;6(5):583–9. [DOI] [PubMed] [Google Scholar]
- 13.Bradshaw JD, Lu P, Leytze G, et al. Interaction of the cytoplasmic tail of CTLA-4 (CD152) with a clathrin-associated protein is negatively regulated by tyrosine phosphorylation. Biochemistry 1997. December 16;36(50):15975–82. [DOI] [PubMed] [Google Scholar]
- 14.Linsley PS, Bradshaw J, Greene J, et al. Intracellular trafficking of CTLA-4 and focal localization towards sites of TCR engagement. Immunity 1996. June;4(6):535–43. [DOI] [PubMed] [Google Scholar]
- 15.Egen JG, Kuhns MS, Allison JP. CTLA-4: new insights into its biological function and use in tumor immunotherapy. Nat Immunol 2002. July;3(7):611–8. [DOI] [PubMed] [Google Scholar]
- 16.Chuang E, Alegre ML, Duckett CS, et al. Interaction of CTLA-4 with the clathrin-associated protein AP50 results in ligand-independent endocytosis that limits cell surface expression. J Immunol 1997. July 1;159(1):144–51. [PubMed] [Google Scholar]
- 17.Zhang Y, Allison JP. Interaction of CTLA-4 with AP50, a clathrin-coated pit adaptor protein. Proc Natl Acad Sci U S A 1997. August 19;94(17):9273–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perkins D, Wang Z, Donovan C, et al. Regulation of CTLA-4 expression during T cell activation. J Immunol 1996. July 1;156(11):4154–9. [PubMed] [Google Scholar]
- 19.Takahashi T, Tagami T, Yamazaki S, et al. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med 2000. July 17;192(2):303–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chambers CA, Kuhns MS, Egen JG, Allison JP. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu Rev Immunol 2001;19:565–94. [DOI] [PubMed] [Google Scholar]
- 21.Parry RV, Chemnitz JM, Frauwirth KA, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol 2005. November;25(21):9543–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol 2016. February;39(1):98–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Egen JG, Allison JP. Cytotoxic T lymphocyte antigen-4 accumulation in the immunological synapse is regulated by TCR signal strength. Immunity 2002. January;16(1):23–35. [DOI] [PubMed] [Google Scholar]
- 24.Summers KL, O’Donnell JL, Williams LA, Hart DN. Expression and function of CD80 and CD86 costimulator molecules on synovial dendritic cells in chronic arthritis. Arthritis Rheum 1996. August;39(8):1287–91. [DOI] [PubMed] [Google Scholar]
- 25.Ferenczi K, Burack L, Pope M, et al. CD69, HLA-DR and the IL-2R identify persistently activated T cells in psoriasis vulgaris lesional skin: blood and skin comparisons by flow cytometry. J Autoimmun 2000. February;14(1):63–78. [DOI] [PubMed] [Google Scholar]
- 26.Tsunemi Y, Saeki H, Kishimoto M, et al. Cytotoxic T lymphocyte antigen-4 gene (CTLA4) polymorphisms in Japanese patients with psoriasis vulgaris. J Dermatol Sci 2003. August;32(2):163–5. [DOI] [PubMed] [Google Scholar]
- 27.Kim YK, Pyo CW, Hur SS, et al. No associations of CTLA-4 and ICAM-1 polymorphisms with psoriasis in the Korean population. J Dermatol Sci 2003. October;33(1):75–7. [DOI] [PubMed] [Google Scholar]
- 28.Luszczek W, Kubicka W, Jasek M, et al. CTLA-4 gene poly-morphisms and natural soluble CTLA-4 protein in psoriasis vulgaris. Int J Immunogenet 2006. June;33(3):217–24. [DOI] [PubMed] [Google Scholar]
- 29.Sugiyama H, Gyulai R, Toichi E, et al. Dysfunctional blood and target tissue CD4+CD25high regulatory T cells in psoriasis: mechanism underlying unrestrained pathogenic effector T cell proliferation. J Immunol 2005. January 1;174(1):164–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ryder LR, Bartels EM, Woetmann A, et al. FoxP3 mRNA splice forms in synovial CD4+ T cells in rheumatoid arthritis and psoriatic arthritis. APMIS 2012. May;120(5):387–96. [DOI] [PubMed] [Google Scholar]
- 31.Abrams JR, Kelley SL, Hayes E, et al. Blockade of T lymphocyte costimulation with cytotoxic T lymphocyte-associated antigen 4-immunoglobulin (CTLA4Ig) reverses the cellular pathology of psoriatic plaques, including the activation of keratinocytes, dendritic cells, and endothelial cells. J Exp Med 2000. September 4;192(5):681–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huynh D, Kavanaugh A. Psoriatic arthritis: current therapy and future directions. Expert Opin Pharmacother 2013. September;14(13):1755–64. [DOI] [PubMed] [Google Scholar]
- 33.Mease P, Genovese MC, Gladstein G, et al. Abatacept in the treatment of patients with psoriatic arthritis: results of a six-month, multicenter, randomized, double-blind, placebo-controlled, phase II trial. Arthritis Rheum 2011. April;63(4):939–48. [DOI] [PubMed] [Google Scholar]
- 34.Davenport CM, McAdams HA, Kou J, et al. Inhibition of pro-inflammatory cytokine generation by CTLA4-Ig in the skin and colon of mice adoptively transplanted with CD45RBhi CD4+ T cells correlates with suppression of psoriasis and colitis. Int Immunopharmacol 2002. April;2(5):653–72. [DOI] [PubMed] [Google Scholar]
- 35.Johnson DB, Sullivan RJ, Ott PA, et al. Ipilimumab therapy in patients with advanced melanoma and preexisting autoimmune disorders. JAMA Oncol 2016. February;2(2):234–40. [DOI] [PubMed] [Google Scholar]
- 36.Singh TP, Schön MP, Wallbrecht K, et al. 8-methoxypsoralen plus ultraviolet A therapy acts via inhibition of the IL-23/Th17 axis and induction of Foxp3+ regulatory T cells involving CTLA4 signaling in a psoriasis-like skin disorder. J Immunol 2010. June 15;184(12):7257–67. [DOI] [PubMed] [Google Scholar]
- 37.Romo-Tena J, Gomez-Martin D, Alcocer-Varela J. CTLA-4 and autoimmunity: new insights into the dual regulator of tolerance. Autoimmun Rev 2013. October;12(12):1171–6. [DOI] [PubMed] [Google Scholar]
- 38.Gianchecchi E, Delfino DV, Fierabracci A. Recent insights into the role of the PD-1/PD-L1 pathway in immuno-logical tolerance and autoimmunity. Autoimmun Rev 2013. September;12(11):1091–100. [DOI] [PubMed] [Google Scholar]
- 39.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 2008;26:677–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev 2010. July;236:219–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang X, Schwartz JC, Guo X, et al. Structural and functional analysis of the costimulatory receptor programmed death-1. Immunity 2004. March;20(3):337–47. [DOI] [PubMed] [Google Scholar]
- 42.Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating auto-immunity and infection. Nat Immunol 2007. March;8(3):239–45. [DOI] [PubMed] [Google Scholar]
- 43.Nishimura H, Agata Y, Kawasaki A, et al. Developmentally regulated expression of the PD-1 protein on the surface of double-negative (CD4-CD8-) thymocytes. Int Immunol 1996. May;8(5):773–80. [DOI] [PubMed] [Google Scholar]
- 44.Okazaki T, Honjo T. PD-1 and PD-1 ligands: from discovery to clinical application. Int Immunol 2007. July;19(7):813–24. [DOI] [PubMed] [Google Scholar]
- 45.Nurieva RI, Liu X, Dong C. Yin-Yang of costimulation: crucial controls of immune tolerance and function. Immunol Rev 2009. May;229(1):88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Riley JL. PD-1 signaling in primary T cells. Immunol Rev 2009. May;229(1):114–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cetinozman F, Jansen PM, Willemze R. Expression of programmed death-1 in skin biopsies of benign inflammatory vs. lymphomatous erythroderma. Br J Dermatol 2014. September;171(3):499–504. [DOI] [PubMed] [Google Scholar]
- 48.Imai Y, Ayithan N, Wu X, et al. Cutting Edge: PD-1 regulates imiquimod-induced psoriasiform dermatitis through inhibition of IL-17A expression by innate gammadelta-low T cells. J Immunol 2015. July 15;195(2):421–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim JH, Choi YJ, Lee BH, et al. Programmed cell death ligand 1 alleviates psoriatic inflammation by suppressing IL-17A production from programmed cell death 1-high T cells. J Allergy Clin Immunol 2016. May;137(5):1466–76 e3. [DOI] [PubMed] [Google Scholar]
- 50.Matsumura N, Ohtsuka M, Kikuchi N, Yamamoto T. Exacerbation of psoriasis during nivolumab therapy for metastatic melanoma. Acta Derm Venereol 2016. February;96(2):259–60. [DOI] [PubMed] [Google Scholar]
- 51.Chia PL, John T. Severe psoriasis flare after anti-programmed death ligand 1 (PD-L1) therapy for metastatic non-small cell lung cancer (NSCLC). J Immunother 2016. June;39(5):202–4. [DOI] [PubMed] [Google Scholar]
- 52.Peled M, Strazza M, Azoulay-Alfaguter I, et al. Analysis of programmed death-1 in patients with psoriatic arthritis. Inflammation 2015. August;38(4):1573–9. [DOI] [PubMed] [Google Scholar]
- 53.Freeman GJ, Casasnovas JM, Umetsu DT, DeKruyff RH. TIM genes: a family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol Rev 2010. May;235(1):172–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cheng L, Ruan Z. Tim-3 and Tim-4 as the potential targets for antitumor therapy. Hum Vaccin Immunother 2015;11(10):2458–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liberal R, Grant CR, Holder BS, et al. The impaired immune regulation of autoimmune hepatitis is linked to a defective galectin-9/tim-3 pathway. Hepatology 2012. August;56(2):677–86. [DOI] [PubMed] [Google Scholar]
- 56.Wu W, Shi Y, Li S, et al. Blockade of Tim-3 signaling restores the virus-specific CD8(+) T-cell response in patients with chronic hepatitis B. Eur J Immunol 2012. May;42(5):1180–91. [DOI] [PubMed] [Google Scholar]
- 57.Anderson AC. Tim-3, a negative regulator of anti-tumor immunity. Curr Opin Immunol 2012. April;24(2):213–6. [DOI] [PubMed] [Google Scholar]
- 58.Han G, Chen G, Shen B, Li Y. Tim-3: an activation marker and activation limiter of innate immune cells. Front Immunol 2013. December 10;4:449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gorman JV, Colgan JD. Regulation of T cell responses by the receptor molecule Tim-3. Immunol Res 2014. August;59(1– 3):56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhu C, Anderson AC, Schubart A, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol 2005. December;6(12):1245–52. [DOI] [PubMed] [Google Scholar]
- 61.Rangachari M, Zhu C, Sakuishi K, et al. Bat3 promotes T cell responses and autoimmunity by repressing Tim-3-mediated cell death and exhaustion. Nat Med 2012. September;18(9):1394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Seki M, Oomizu S, Sakata KM, et al. Galectin-9 suppresses the generation of Th17, promotes the induction of regulatory T cells, and regulates experimental autoimmune arthritis. Clin Immunol 2008. April;127(1):78–88. [DOI] [PubMed] [Google Scholar]
- 63.Sehrawat S, Suryawanshi A, Hirashima M, Rouse BT. Role of Tim-3/galectin-9 inhibitory interaction in viral-induced immunopathology: shifting the balance toward regulators. J Immunol 2009. March 1;182(5):3191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mengshol JA, Golden-Mason L, Arikawa T, et al. A crucial role for Kupffer cell-derived galectin-9 in regulation of T cell immunity in hepatitis C infection. PLoS One 2010. March 4;5(3):e9504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hastings WD, Anderson DE, Kassam N, et al. TIM-3 is expressed on activated human CD4+ T cells and regulates Th1 and Th17 cytokines. Eur J Immunol 2009. September;39(9):2492–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kanai Y, Satoh T, Igawa K, Yokozeki H. Impaired expression of Tim-3 on Th17 and Th1 cells in psoriasis. Acta Derm Venereol 2012. July;92(4):367–71. [DOI] [PubMed] [Google Scholar]
- 67.Niwa H, Satoh T, Matsushima Y, et al. Stable form of galectin-9, a Tim-3 ligand, inhibits contact hypersensitivity and psoriatic reactions: a potent therapeutic tool for Th1- and/ or Th17-mediated skin inflammation. Clin Immunol 2009. August;132(2):184–94. [DOI] [PubMed] [Google Scholar]
- 68.Huard B, Mastrangeli R, Prigent P, et al. Characterization of the major histocompatibility complex class II binding site on LAG-3 protein. Proc Natl Acad Sci U S A 1997. May 27;94(11):5744–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Workman CJ, Wang Y, El Kasmi KC, et al. LAG-3 regulates plasmacytoid dendritic cell homeostasis. J Immunol 2009. February 15;182(4):1885–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kisielow M, Kisielow J, Capoferri-Sollami G, Karjalainen K. Expression of lymphocyte activation gene 3 (LAG-3) on B cells is induced by T cells. Eur J Immunol 2005. July;35(7):2081–8. [DOI] [PubMed] [Google Scholar]
- 71.Huard B, Tournier M, Triebel F. LAG-3 does not define a specific mode of natural killing in human. Immunol Lett 1998. April;61(2–3):109–12. [DOI] [PubMed] [Google Scholar]
- 72.Byun HJ, Jung WW, Lee DS, et al. Proliferation of activated CD1d-restricted NKT cells is down-modulated by lymphocyte activation gene-3 signaling via cell cycle arrest in S phase. Cell Biol Int 2007. March;31(3):257–62. [DOI] [PubMed] [Google Scholar]
- 73.Workman CJ, Rice DS, Dugger KJ, et al. Phenotypic analysis of the murine CD4-related glycoprotein, CD223 (LAG-3). Eur J Immunol 2002. August;32(8):2255–63. [DOI] [PubMed] [Google Scholar]
- 74.Huang CT, Workman CJ, Flies D, et al. Role of LAG-3 in regulatory T cells. Immunity 2004. October;21(4):503–13. [DOI] [PubMed] [Google Scholar]
- 75.Grosso JF, Kelleher CC, Harris TJ, et al. LAG-3 regulates CD8+ T cell accumulation and effector function in murine self- and tumor-tolerance systems. J Clin Invest 2007. November;117(11):3383–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sierro S, Romero P, Speiser DE. The CD4-like molecule LAG-3, biology and therapeutic applications. Expert Opin Ther Targets 2011. January;15(1):91–101. [DOI] [PubMed] [Google Scholar]
- 77.Macon-Lemaitre L, Triebel F. The negative regulatory function of the lymphocyte-activation gene-3 co-receptor (CD223) on human T cells. Immunology 2005. June;115(2):170–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: Co-inhibitory receptors with specialized functions in immune regulation. Immunity 2016. May 17;44(5):989–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]