

Abstract
Lenvatinib is a type I tyrosine kinase inhibitor exhibiting powerful antiangiogenic activity in cancer therapy. Displaying activity in multiple solid tumors, it has been approved in differentiated thyroid cancer, hepatocellular carcinoma, and renal cell carcinoma as single agent or in combination. In addition, lenvatinib has shown promise in several other tumor types including medullary, anaplastic thyroid, adenoid cystic, and endometrial cancer. Exploring synergy between angiogenic and immune checkpoint inhibitors, the lenvatinib/pembrolizumab combination is poised to become the next pair of active drugs in endometrial, lung, and gastrointestinal malignancies. Despite robust activity, the drug can be difficult to tolerate. Optimization of dose and biomarkers for prediction of efficacy and toxicities will be of great help.
Implications for Practice
Readers will be presented with an update on U.S. Food and Drug Administration approval of lenvatinib and suggestions for off‐label use in thyroid cancer and adenoid cystic carcinomas. They will become familiarized with the common side effects, frequency, and predicators of response. In addition, they will learn that different strengths of lenvatinib are prescribed and why. Finally, readers are pointed to the latest efforts to combine lenvatinib and pembrolizumab, as well as to unresolved issues such as long‐term side effects/toxicities of this drug.
Keywords: Lenvatinib, Cancer, Angiogenesis inhibitor, Pembrolizumab
Short abstract
This article reviews the discovery, development, clinical testing, and regulatory approval of the tyrosine kinase inhibitor lenvatinib (E7080) and highlights future potential in the management of solid tumors.
Introduction
Tyrosine kinases (TKs) are key regulators of cell function and constitute a large and functionally diverse group of family members sharing a conserved kinase domain. By adding a phosphate group from adenosine triphosphate (ATP) to the tyrosine residue on their substrate(s), TKs change the substrates activity, localization, and interaction with many other proteins 1, 2. They are intimately involved in numerous cellular processes such as angiogenesis and cell cycle control 3, 4. TKs are critical components of the cell signaling pathway, and their activities are strictly regulated. Aberrant activity of TKs has been linked to many disease states, including cancer; therefore, tyrosine kinase inhibitors (TKIs) are perhaps best studied in cancer. Starting from the success of imatinib for the treatment of chronic myeloid leukemia, TKIs are successively tested and introduced into the treatment of patients with cancer with remarkable improvement of cancer outcome 5, 6. Here we review the discovery, development, clinical testing, and regulatory approval of the TKI lenvatinib (also known as E7080) with affinity to multiple receptor TKs (RTK). We also review the hurdles to compliance and highlight its future potential in the management of other solid tumors that are not currently approved.
Lenvatinib was initially reported in 2008 as a multitargeted RTK inhibitor capable of inhibiting multiple kinases in nanomole concentration (half maximal inhibiting concentration (IC50) 4–100 nM) 7. In animal experiments, lenvatinib potently inhibited angiogenesis, causing tumors to regress in mouse model. Further testing in a breast cancer model revealed it suppressed breast cancer metastasis to the lymph nodes and to the lungs by targeting vascular endothelial growth factor receptor (VEGFR) 3 during angiogenesis and lymphangiogenesis 8. Activities were also shown in an orthotopic malignant mesothelioma mouse model, which is known to respond to angiogenesis inhibitor. Lenvatinib significantly prolonged the survival of mice injected with three mesothelioma cell lines 9 and mice carrying sarcoma xenograft 10. Lenvatinib was further developed as an orally available TKI in solid tumor (see below).
Lenvatinib functions as an inhibitor to multiple RTKs. The IC50s to VEGF1–3 kinases assayed ranged from 2.3 to 4.7 nM, fibroblast growth factor receptor (FGFR) 1–4, 27–61 nM, platelet‐derived growth factor (PDGFR) α, β 29–39 Nm, and v‐Kit Hardy‐Zuckerman 4 feline sarcoma viral oncogene homolog (KIT) 85 nM. In contrast, lenvatinib did not inhibit epidermal growth factor receptor (EGFR; IC50 6,500 nM) or other kinases such as Src or cyclin‐dependent kinase 4 (all >10,000 nM) 7, 11. In a mouse model of EGFR wild‐type lung cancer, lenvatinib suppressed endothelial and lymph epithelial growth and decreased the numbers of lung nodules and metastatic foci 12. Using a mouse model of human osteosarcoma, it was shown that lenvatinib did not have a big impact on tumor cell proliferation. However, it had profound effects on tumor cell migration and invasion, likely through suppressing FGFR and PDGFR because PDGFRβ‐negative cell line was not affected by the drug. Knockdown of PDGFRβ had also inhibited tumor cell migration 13. A kinetic interaction analysis of lenvatinib with VEGFR2 (KDR) and x‐ray analysis of the crystal structure of VEGFR2‐lenvatinib complexes showed a rapid association constant and relatively slow dissociation rate. Lenvatinib binds at its ATP mimetic moiety to the ATP binding site and neighboring region of VEGFR2 in a unique way different from several other known VEGFR2 TKIs such as sorafenib and sunitinib 14. Lenvatinib is therefore a type I TKI 15.
Lenvatinib is rapidly absorbed and typically reaches its maximal concentration between 1 and 4 hours in healthy volunteers or patients with solid tumors after oral ingestion 16, 17. The absorption followed a first‐ and zero‐order kinetics not affected by elevated pH in the stomach. The absorption in patients with solid tumors followed a dose‐dependent linear pharmacokinetic with no drug accumulation after once‐daily doses (maximum concentration after multiple doses were the same as those after a single dose) 18. The relative bioavailability in capsule form was 90%. Population pharmacokinetics analysis of 779 subjects with preserved liver function who received between 3.2 and 32 mg of oral lenvatinib once‐daily dose confirmed a three‐compartment model of linear elimination. The population mean value for lenvatinib apparent clearance as a function of bioavailability (CL/F) was 6.51 Liter/hour and was independent of dose or time. None of the factors including dose, sex, race, age, performance, renal function, or thyroid function (by thyroid‐stimulating hormone) had any significant effects on CL/F. However, after the same dose, patients with lower body weight had higher steady‐state concentration and 90% prediction intervals compared with those with a higher body weight, and those who were getting CYP3A4 inhibitors also had slightly higher steady‐state concentration. Although lower albumin and high alkaline phosphatase increases drug exposure, none of them were clinically relevant to justify dose adjustment. To test how liver function affects pharmacokinetics of lenvatinib, patients with mild to moderate or severe liver impairment were compared with patients with normal hepatic function. Although mild to moderate liver function impairment had no clinically meaningful effects on lenvatinib pharmacokinetics, patients with severe liver dysfunction had area under the curve (AUC) increased to 170% and T1/2 increase from 23 hours to 37 hours. It was recommended that patients with severe liver impairment start lenvatinib at 14 mg instead of 24 mg 19. After a dose of 24 mg of C14 labeled lenvatinib, radioactivity peaked after 1.6 hours; lenvatinib remained mostly intact at the peak concentration and was 700 times higher than metabolites. After 10 days, recovery was 64% ± 11% in the feces and 25% ± 18% in the urine (89% recovery of administered dose). Lenvatinib is dosed differently in different cancer, according to body weight or whether it is in combination with another agent (see below for dosing considerations) 20.
Lenvatinib Use and Approval in Solid Tumors
Differentiated Thyroid Cancer
Initially, in a phase II trial enrolling 58 patients, lenvatinib showed robust activity in advanced, radiation‐refractory differentiated thyroid cancer (RR‐DTC; papillary, follicular, or Hurthle cell). The overall response rate (ORR) was 50%, lower in patients who had received one line of VEGFR inhibitor and higher in those who were naïve (41% vs. 54%). Median progression‐free survival (PFS) was 12.6 months and median duration of response was 12.7 months. The toxicities were manageable 21.
Interestingly, molecular profiling revealed that patients who carried a RAS (rat sarcoma virus) mutation (KRAS or NRAS), which were present in 35% of all patients, had 100% response rate and significantly improved PFS (80% at 14 months vs. 20% in wild‐type RAS group) 22.
Based on the above, the SELECT trial, a phase III study of lenvatinib 23, was conducted in the RR‐DTC population with radiographic progression within the prior 12 months. Patients received 24 mg once daily of lenvatinib or placebo in a 28‐day cycle at a 2:1 ratio in this trial enrolling a total of 392 (261:131) patients across 23 countries. Again, one line of prior anti‐VEGFR therapy was allowed. Crossover from placebo was also allowed. Enrollment was completed after 3 years. At the end, the study met its primary endpoint: PFS. The median PFS was 18.3 months in the lenvatinib group compared with 3.6 months in the placebo group (hazard ratio [HR] 0.21; 99% confidence interval [CI] 0.14–0.31; p < .001). The benefit was across all prespecified subgroups. Complete response (CR) plus partial response (PR) was 64.8% versus 1.5% in the placebo (p < .001). The median overall survival was not reached in either group. Toxicities were manageable but significant. Discontinuation of drug occurred in 37 patients (14.2%) in the lenvatinib and 3 (2.3%) in the placebo group. Treatment‐related adverse effects (AEs) occurred in more than 40% of the patients. They included hypertension (67.8%), diarrhea (59.4%) fatigue or asthenia (59%), decreased appetite (50.2%), decreased weight (46.4%), and nausea (41%). Of the 20 deaths that occurred during treatment, 6 were considered treatment related. Lenvatinib was subsequently approved by the U.S. Food and Drug Administration (FDA) for treatment of RR‐DTC in August 2015 24.
Medullary Thyroid Carcinoma
Lenvatinib inhibited rearranged during transfection (RET) kinase activity in early studies 7, 11. Lenvatinib was active inhibiting RET fusion protein driven tumor in animal models 25. Based on early indication of activity in thyroid cancer, lenvatinib was tested in a phase II trial enrolling 59 patients with advanced medullary thyroid carcinoma who were unresectable or metastatic in the prior 12 months 26. Prior use of anti‐VEGF was allowed. ORR of lenvatinib was 36%, all of which were partial. Disease control rate (DCR) was 80%. ORR was comparable between patients who received (35%) or did not receive (36%) anti‐VEGF therapy. Among the responders, median PFS was 9.0 months. Grade 3 and 4 (G3/4) AEs included diarrhea (14%), hypertension (7%), decreased appetite (7%), fatigue, and dysphagia. Although 67% of patients harbored a RET mutation (M918T or C634R), the presence of a RET mutation did not correlate with response. In contrast, lower levels of angiopoietin‐2 were associated with greater tumor reduction and prolonged PFS. Lenvatinib is not approved by the FDA.
Anaplastic Thyroid Carcinoma
Anaplastic thyroid carcinomas (ATCs) are rare compared with other thyroid cancers but chart a very aggressive course. These cancers are treated aggressively with surgery and chemoradiation. However, 1‐year survival is only 20%–40% 27, 28, 29. In 2018, the FDA granted approval for dabrafenib and trametinib combination in ATC with V600E mutation, which are present in 20%–50% of all ATCs 30, 31, 32, based on an open‐labeled trial in these patients. Of the 23 evaluable patients treated with the drug combo, PR and CR were 57% and 4%, respectively. Of the 14 patients who had response, 64% had no significant growth of tumor for 6 months or longer. Better treatment options are needed for this deadly cancer. Lenvatinib has been studied in ATCs and has been approved for use in patients with ATCs in Japan since 2015 33.
In one retrospective study of lenvatinib 34, three of the five unresectable patients who were given lenvatinib had PR. Time to progression (TTP) was 88 days. Four patients had hypothyroidism. Other AEs included hypertension, diarrhea, fatigue, and decreased appetite.
In another retrospective study 33, a total of 23 patients were enrolled. They were evaluated for overall survival (OS). Ten patients were able to receive surgical debulking first; the remaining 13 were not. All were then started on lenvatinib as life‐saving measures. ORR was 17.4% and DCR was 43.5%. Of the patients treated, 100% had AEs. Hypertension developed in 91.3%, and 39.1% had G3 or above toxicities. Ten patients had tumor fistula, and 9 of 23 discontinued drug because of G3 toxicities. OS was 166 days. Lenvatinib seemed active in ATC, and tumor fistula management was crucial for survival.
In a phase II, single‐arm, open‐labeled prospective study of lenvatinib in thyroid cancer, 17 patients with ATC were enrolled and received 24 mg once daily until disease progression or unacceptable toxicity. The median PFS was 7.4 months (95% CI 1.7–12.9). Median OS was 10.6 months (95% CI 3.8–19.8) with an ORR of 24% and DCR of 88%. Treatment‐emergent adverse events (TEAEs) were present in all participants. The most frequent TEAEs were decreased appetite (82%), hypertension (82%), fatigue (59%), and proteinuria (59%). One patient required withdrawal that was not related to study drug 35. In support of the lenvatinib efficacy in patients with ATCs, genetic analysis of 196 advanced ATCs by either Foundation One or Memorial Sloan Kettering‐IMPACT gene panels revealed the following: (a) ATCs have accumulated more gene mutations per tumor compared with DTCs owing to DNA mismatch repair defects and apolipoprotein B mRNA editing enzyme, catalytic polypeptide‐like cytidine deaminase activity in a subset of ATCs; and (b) amplification of receptor kinase KDR (VEGFR2), KIT, and PDGFRα, which are all targets of lenvatinib 36. In another report, 12 patients with ATCs treated with lenvatinib were analyzed for response and tumor expression of VEGFR2, a known target of lenvatinib. It was found that 4 of the 12 had PR, 3 had stable disease (SD), and 5 had progressive disease (PD). Interestingly, none of the responders or SD expressed VEGFR2 37.
Hepatocellular Carcinoma
In a phase I study of 20 patients with hepatocellular carcinoma (HCC) with liver function as Child‐Pugh A (CP‐A, score 5–6) and Child‐Pugh B (CP‐B, score 7–8), 12 mg of lenvatinib once daily was given to CP‐A and 8 mg to CP‐B; this resulted in PR in 3 patients and 14 patients with any degree of tumor shrinkage 38.
The first phase II trial of lenvatinib in HCC was conducted in Japan and South Korea 39. In this trial, lenvatinib 12 mg once daily in a 28‐day cycle was given to patients with HCC who did not qualify for local therapies 39. A total of 46 patients were enrolled. All but one was CP‐A. PR was seen in 37% of the patients. With SD seen in 41% of patients, the DCR was 78%. Median OS was 18.7 months and TTP was 7.4 months. The most common AEs were hypertension (76%), palmar‐plantar erythrodysesthesia syndrome (PPES; 65%), decreased appetite (61%), and proteinuria (61%). Seventy‐four percent was dose reduced with 22% drug discontinuation.
The pivotal trial REFLECT sought to compare lenvatinib with the then–standard of care agent sorafenib for unresectable HCC in a noninferiority design 40. Patients were deemed unresectable owing to either macroscopic portal vein invasion or extrahepatic spread or both. A total of 154 sites across 20 countries were involved, enrolling 954 eligible patients. Lenvatinib (n = 478) 12 mg (>60 kg of body weight) or 8 mg once daily (<60 kg) or sorafenib (n = 476) 400 mg twice a day were given to these patients in a 28‐day cycle. The study met its noninferiority goal with set margin of 1.08. Hazard ratio was 0.92 (CI 0.79–1.06). Median OS was 13.6 months in the lenvatinib arm and 12.3 months in the sorafenib arm. The REFLECT trial also demonstrated a statistically significant improvement in PFS with lenvatinib as compared with sorafenib. Median PFS was 7.3 months in the lenvatinib arm and 3.6 months in the sorafenib arm (HR 0.64; 95% CI 0.55–0.75; p < .001). Common AEs for lenvatinib included hypertension (42%), diarrhea (39%), decreased appetite (34%), and decreased body weight (31%). Commonly seen in patients receiving sorafenib were PPES (52%), diarrhea (46%), hypertension (30%), and decreased appetite (27%). In addition, lenvatinib demonstrated a significantly higher ORR of 24% compared with 9% for sorafenib (odds ratio 3.13; 95% CI 2.15–4.56; p < .001). Based on these data, the FDA approved lenvatinib use in the first‐line settings for unresectable HCC 41.
Renal Cell Carcinoma
In a phase I study 42, lenvatinib showed encouraging activity in kidney cancer. Median PFS was more than 500 days. Another phase I study also revealed response in kidney cancer 43. Subsequently, a phase Ib study showed that lenvatinib at 12 mg or 18 mg (maximum tolerated dose [MTD]) in combination with 5 mg of everolimus given to unresectable or metastatic renal cell carcinoma (RCC) yielded a best of response of 83% (n = 18), PR 33% and SD 50% 44. A randomized, phase II, open‐label, multicenter trial 45 was performed at 37 centers in five countries, enrolling patients with advanced clear‐cell RCC. Patients who had received treatment with a VEGF‐targeted therapy and progressed on or within 9 months were included. Patients were randomized in a 1:1:1 ratio to receive lenvatinib (24 mg/day), everolimus (10 mg/day), or lenvatinib and everolimus (18 mg/day and 5 mg/day) in 28‐day cycles until disease progression or unacceptable toxicity. The primary objective was PFS in the intention‐to‐treat population. In a little over 15 months, 153 patients were allocated to receive lenvatinib plus everolimus (n = 51), single‐agent lenvatinib (n = 52), or single‐agent everolimus (n = 50). Lenvatinib plus everolimus significantly prolonged PFS over everolimus alone (median 14.6 months vs. 5.5 months; HR 0.40; 95% CI 0.24–0.68; p = .0005). However, lenvatinib plus everolimus was not significantly different from lenvatinib alone (PFS 7.4 months; HR 0.66; 95% CI 0.30–1.10; p = .12). Single‐agent lenvatinib significantly prolonged PFS compared with everolimus alone (HR 0.61; 95% CI 0.38–0.98; p = .048). G3 and 4 events were less common in patients allocated single‐agent everolimus (50%) compared with those assigned to lenvatinib alone (79%) or lenvatinib plus everolimus (71%). The most common G3/4 treatment‐emergent adverse event in patients allocated to lenvatinib plus everolimus was diarrhea (20%); in those assigned single‐agent lenvatinib, it was proteinuria (19%); and in those assigned single‐agent everolimus, it was anemia (12%). Two deaths occurred during treatment, one cerebral hemorrhage in the lenvatinib plus everolimus group and one myocardial infarction with single‐agent lenvatinib, both deemed related to the study drug. Based on this trial, the FDA approved the combination of 18 mg of lenvatinib and 5 mg of everolimus once daily as a second‐line treatment for patients with clear‐cell RCC who have progressed after one previous VEGF‐targeted therapy 46.
Adenoid Cystic Carcinoma
In a phase II study reported recently 47, 33 patients with recurrent and metastatic adenoid cystic carcinoma with radiographic progression or symptoms were treated with 24 mg of lenvatinib once daily. The primary endpoint for the study was ORR, and the secondary endpoints were PFS, toxicity, Myb expression, and genomic alteration. Of the 32 patients evaluable, 5 had PR (15.6%), 24 had SD (75%), 2 were discontinued (3.1%) and 1 had PD (1.5%). Median PFS was 17.5 months. In all, 23 patients needed dose modification and 18 patients were discontinued. The most common G3/4 toxicities were hypertension (n = 9, 28.1%) and oral pain (n = 3, 9.4%). There were three G4 toxicities: one myocardial infarction, one intracranial hemorrhage, and one posterior reversible encephalopathy syndrome. This study met its primary endpoint, and lenvatinib showed activity in these patients with side effects comparable to previous studies. Currently, lenvatinib is not approved for this indication.
Dosing Considerations
Lenvatinib tolerability and toxicity were tested alone or in combination in different tumor types in multiple phase I trials. In patients with toxicities, dose modification ensues.
The first phase I trial in 27 patients 48 was performed in solid tumors in a 2‐weeks‐on, 1‐week‐off schedule. Starting at 0.5 mg b.i.d., the dose went up to 13, 16, and 20 mg b.i.d. During cycle 1, no G3 or 4 toxicities were observed in patients receiving up to 13 mg b.i.d. Dose‐limiting toxicities (DLTs) appeared when patients were given higher doses. They included G3 increase in aspartate aminotransferase/alanine aminotransferase in one patient at 16 mg b.i.d. and G3 platelet count decrease in two patients at 20 mg b.i.d. It was decided that lenvatinib up to 13 mg b.i.d. in a 2‐weeks‐on and 1‐week‐off schedule would likely have a manageable toxicity profile from this study.
Another phase I study was conducted in 87 patients. Lenvatinib dose was slowly escalated from 0.2 mg to 32 mg once daily in a 28‐day cycle. Pharmacokinetics analyses were performed at day 1, 8, 15, 22, and 28. DLT was determined to be G3 proteinuria, and the MTD was 25 mg once daily 42. Hence, the current recommended dose of lenvatinib is 24 mg daily when used alone in patients with solid tumors with preserved liver function.
A multicenter, open‐label, phase I, dose‐escalation study 38 was performed in patients with HCC refractory to standard therapy. They are stratified by hepatic function as CP‐A (score, 5–6) and CP‐B (score, 7–8). Lenvatinib was administered continually once daily for 4‐week cycles. MTD was defined as the maximum dose associated with ≤1 DLT within the first cycle among six patients. Of the 20 patients enrolled (9 in CP‐A and 11 in CP‐B), the MTD was 12 and 8 mg once daily in CP‐A and CP‐B, respectively. DLTs included proteinuria, hepatic encephalopathy, and hyperbilirubinemia. The most common G3 toxicities included hypertension in CP‐A and hyperbilirubinemia in CP‐B. PRs were observed in 3 patients and tumor shrinkage occurred in 14 patients. Lenvatinib (12 mg once daily) was determined to be the dose that showed preliminary efficacy with manageable toxicity and was the recommended dose for patients with HCC with liver function as CP‐A.
In a dose‐finding study based on population pharmacodynamics and exposure–response analysis in patients with HCC with CP‐A, it was found that there was an increase in AUC as body weight decreased in subjects with HCC. An exposure–response relationship was observed, with higher lenvatinib AUC and lower body weight resulting in earlier drug withdrawal or dose reduction. The best cutoff values for body weight and lenvatinib AUC were 57.8 kg and 2,430 ng·h/mL, respectively, to predict the group at high risk for early drug withdrawal or dose reduction. Therefore, 12 mg and 8 mg once‐daily starting doses for patients ≥60 kg and < 60 kg, respectively, were recommended for patients with HCC CP‐A 49.
To find the safe dose for RCC, a phase Ib trial combining lenvatinib and everolimus was performed to treat the disease. Starting from lenvatinib 12 mg once daily (n = 7) in a three‐plus‐three fashion, lenvatinib went up to 18 mg (n = 11) and 24 mg (n = 2) in combination with 5 mg of everolimus, both given once a day. It was determined that the MTD was 18 mg of lenvatinib and 5 mg of everolimus a day 44.
Side Effects and Predictors of Response
In the phase III SELECT trial, serious TEAE related to treatment was 24% above the placebo arm with 2.3% fatal (Table 1). AEs characteristic of angiogenesis inhibitors included hypertension (68%), diarrhea (59%), PPES (32%), and proteinuria (31%). Stomatitis was reported in 36% of enrolled patients. Diarrhea, which typically appeared early in treatment and was managed by dose modification, was associated with improved OS according to analysis of the REFLECT trial 50. Another study found hypertension as TEAE predicted better PFS (18.8 months vs. 12.9 months; HR 0.59; 95% CI 0.39–0.88; p = .0085) compared with those who did not have hypertension 51. Better ORR (69% vs. 56%, not statistically significant) and better OS were also noted (not reached vs. 21.7 months; HR 0.43; 95% CI 0.27–0.69; p = .0003). Those patients who were older than 65 years had a higher percentage of G3 toxicity than those younger than 65 years (89% vs. 67%) 52.
Table 1.
Adverse events from the SELECT trial in differentiated thyroid carcinomaa
| Adverse event | Lenvatinib arm | Placebo arm | ||
|---|---|---|---|---|
| All grades, % | Grade ≥3, % | All grades, % | Grade ≥3, % | |
| Hypertension | 67.8 | 41.8 | 9.2 | 2.3 |
| Diarrhea | 59.4 | 8.0 | 8.4 | 0 |
| Fatigue | 59.0 | 9.2 | 27.5 | 2.3 |
| Decreased appetite | 50.2 | 5.4 | 11.5 | 0 |
| Decreased weight | 46.4 | 9.6 | 9.2 | 0 |
| Nausea | 41.0 | 2.3 | 13.7 | 0.8 |
| Stomatitis | 35.6 | 4.2 | 3.8 | 0 |
| PPES | 31.8 | 3.4 | 0.8 | 0 |
| Proteinuria | 31.0 | 10.0 | 1.5 | 0 |
| Vomiting | 28.4 | 1.9 | 6.1 | 0 |
| Headache | 27.6 | 2.7 | 6.1 | 0 |
| Dysphonia | 24.1 | 1.1 | 3.1 | 0 |
| Arthralgia | 18.0 | 0 | 0.8 | 0 |
| Dysgeusia | 16.9 | 0 | 1.5 | 0 |
| Rash | 16.1 | 0.4 | 1.5 | 0 |
| Constipation | 14.6 | 0.4 | 8.4 | 0 |
| Myalgia | 14.6 | 1.5 | 2.3 | 0 |
| Dry mouth | 13.8 | 0.4 | 3.8 | 0 |
| Upper abdominal pain | 13.0 | 0 | 3.8 | 0 |
| Abdominal pain | 11.5 | 0.4 | 0.8 | 0.8 |
| Peripheral edema | 11.1 | 0.4 | 0 | 0 |
| Alopecia | 11.1 | 0 | 3.8 | 0 |
| Dyspepsia | 10.0 | 0 | 0 | 0 |
| Oropharyngeal pain | 10.0 | 0.4 | 0.8 | 0 |
| Hypercalcemia | 6.9 | 2.7 | 0 | 0 |
| Pulmonary embolism | 2.7 | 2.7 | 1.5 | 1.5 |
| Fatal TEAE (TR, %) | 2.3 | 0 | ||
| Serious TEAE (TR, %) | 30.3 | 6.1 | ||
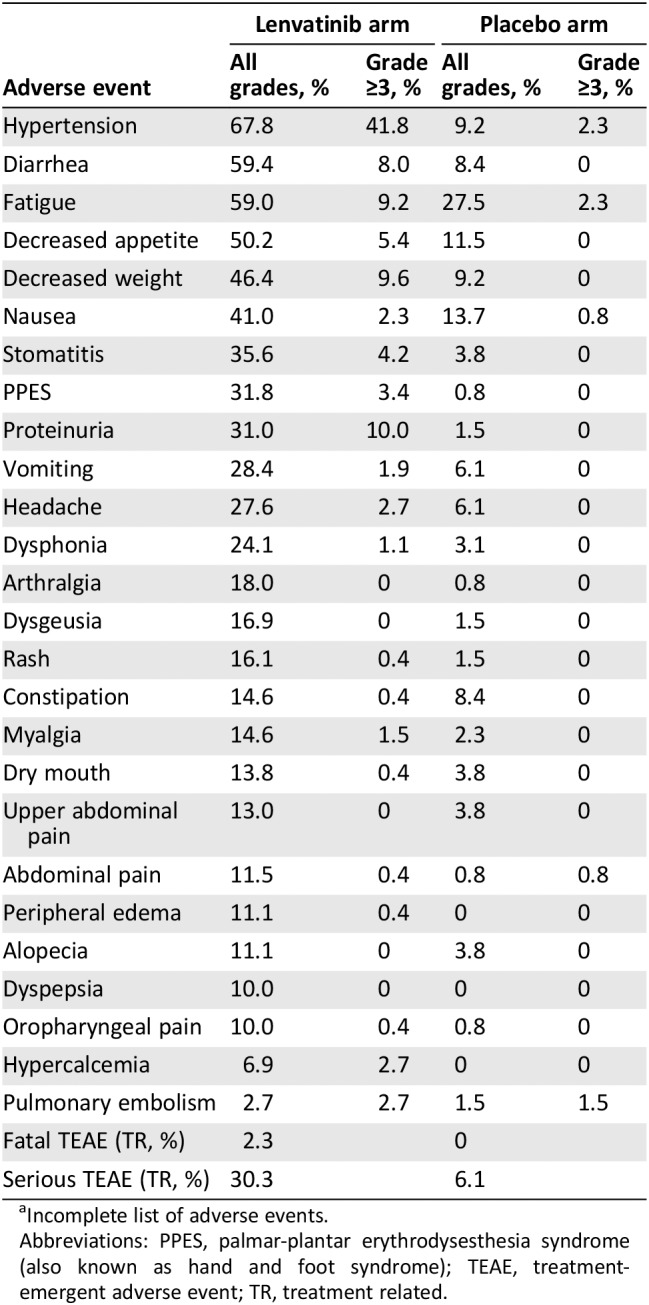
Incomplete list of adverse events.
Abbreviations: PPES, palmar‐plantar erythrodysesthesia syndrome (also known as hand and foot syndrome); TEAE, treatment‐emergent adverse event; TR, treatment related.
Compared with sorafenib, lenvatinib caused more hypothyroidism (16% vs. 2%) and proteinuria (25% vs. 11%) but fewer PPES or hand‐foot syndrome (27% vs. 52%) and alopecia (3% vs. 25%) in patients with HCC (Table 2) 40. AEs that worsened when lenvatinib was combined with temsirolimus (combination vs. lenvatinib vs. temsirolimus) included diarrhea (85% vs. 72% vs. 34%), fatigue (59% vs. 50% vs. 36%), vomiting (45% vs. 39% vs. 10%), hypercholesterolemia (33% vs. 12% vs. 16%), and peripheral edema (27% vs. 15% vs. 18%) in the RCC trial (Table 3) 45.
Table 2.
Adverse events from the REFLECT trial in hepatocellular carcinoma
| Adverse event | Lenvatinib arm | Sorafenib arm | ||
|---|---|---|---|---|
| All grades, % | Grade ≥3, % | All grades, % | Grade ≥3, % | |
| PPES | 27 | 3 | 52 | 11 |
| Diarrhea | 39 | 4 | 46 | 4 |
| Hypertension | 42 | 23 | 30 | 14 |
| Decreased appetite | 34 | 5 | 27 | 1 |
| Decreased weight | 31 | 8 | 22 | 3 |
| Fatigue | 30 | 4 | 25 | 4 |
| Alopecia | 3 | 0 | 25 | 0 |
| Proteinuria | 25 | 6 | 11 | 2 |
| Dysphonia | 24 | 1 | 12 | 0 |
| Nausea | 20 | 1 | 14 | 1 |
| Abdominal pain | 17 | 2 | 18 | 3 |
| Decreased platelet | 18 | 5 | 12 | 3 |
| Increased AST | 14 | 5 | 17 | 8 |
| Hypothyroidism | 16 | 0 | 2 | 0 |
| Vomiting | 16 | 1 | 8 | 1 |
| Constipation | 16 | 1 | 11 | 0 |
| Rash | 10 | 0 | 16 | 1 |
| Increased bilirubin | 15 | 7 | 13 | 5 |
| TEAE (TR, %) | 57 | 49 | ||
| Serious TEAE (TR, %) | 18 | 10 | ||
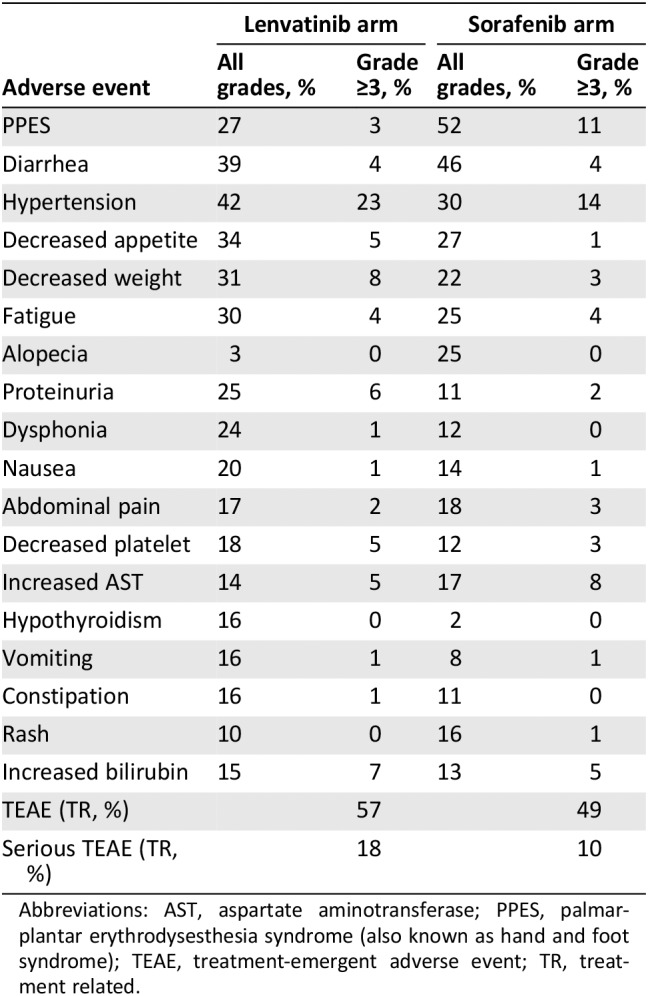
Abbreviations: AST, aspartate aminotransferase; PPES, palmar‐plantar erythrodysesthesia syndrome (also known as hand and foot syndrome); TEAE, treatment‐emergent adverse event; TR, treatment related.
Table 3.
Adverse events from the phase II study of lenvatinib plus everolimus for renal cell carcinoma
| Adverse event | Lenvatinib/temsirolimus arm | Lenvatinib arm | Temsirolimus arm | |||
|---|---|---|---|---|---|---|
| All grades, % | Grade ≥3, % | All grades, % | Grade ≥3, % | All grades, % | Grade ≥3, % | |
| Diarrhea | 85 | 20 | 72 | 12 | 34 | 2 |
| Decreased appetite | 51 | 6 | 58 | 4 | 18 | 0 |
| Fatigue/asthenia | 59 | 14 | 50 | 8 | 36 | 2 |
| Vomiting | 45 | 8 | 39 | 4 | 10 | 0 |
| Nausea | 41 | 6 | 52 | 8 | 16 | 0 |
| Cough | 37 | 0 | 17 | 2 | 30 | 0 |
| Hypercholesterolemia | 33 | 2 | 12 | 2 | 16 | 0 |
| Decreased weight | 31 | 2 | 48 | 6 | 8 | 0 |
| Stomatitis | 29 | 0 | 25 | 2 | 42 | 2 |
| Hypertriglyceridemia | 35 | 8 | 14 | 4 | 24 | 8 |
| Hypertension | 41 | 14 | 48 | 17 | 10 | 2 |
| Peripheral edema | 27 | 0 | 15 | 0 | 18 | 0 |
| Abdominal pain | 30 | 4 | 31 | 4 | 10 | 0 |
| Hypothyroidism | 24 | 0 | 37 | 2 | 2 | 0 |
| Arthralgia | 24 | 0 | 25 | 0 | 4 | 0 |
| Dyspnea | 24 | 2 | 21 | 2 | 22 | 8 |
| Dysphonia | 20 | 0 | 37 | 0 | 4 | 0 |
| Pyrexia | 22 | 2 | 10 | 0 | 10 | 2 |
| Epistaxis | 18 | 0 | 8 | 0 | 22 | 0 |
| Proteinuria | 22 | 4 | 31 | 19 | 14 | 2 |
| Rash | 18 | 0 | 17 | 0 | 22 | 0 |
| Hyperglycemia | 16 | 0 | 6 | 0 | 22 | 10 |
| Back pain | 20 | 4 | 21 | 0 | 14 | 0 |
| Headache | 18 | 2 | 25 | 4 | 10 | 2 |
| Insomnia | 18 | 2 | 14 | 0 | 2 | 0 |
| Any TEAE | 99 | 71 | 94 | 79 | 96 | 50 |
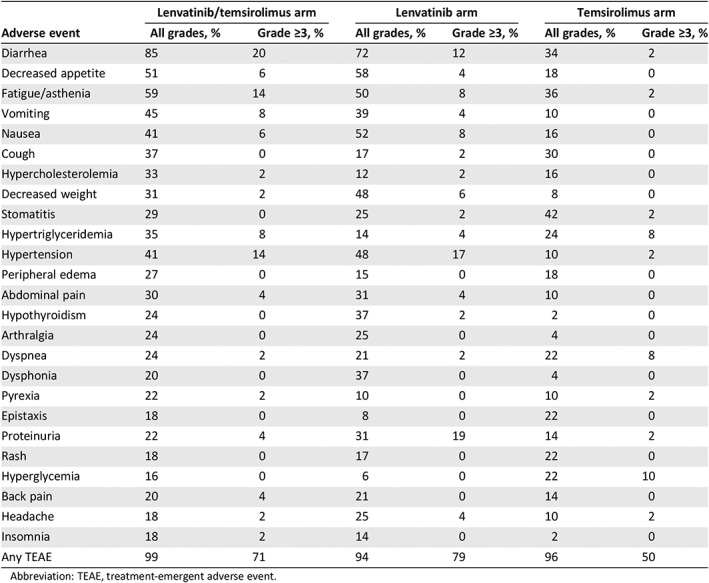
Abbreviation: TEAE, treatment‐emergent adverse event.
Antiangiogenesis and Checkpoint Blockade Combination
Antiangiogenic therapy is known to reprogram the immunosuppressive tumor microenvironment and enhances immunotherapy 53, 54. Angiogenic inhibitors do so through normalization of tumor vasculature and inhibition of immunosuppressive cells such as myeloid‐derived suppressor cells 55. This concept was explored in lung cancer and renal cell carcinoma with proof of concept.
In the IMpower150 trial enrolling patients with nonsquamous non‐small cell lung cancer appropriate for bevacizumab, the efficacies of carboplatin/paclitaxel/bevacizumab (BCP) with or without atezolizumab were compared. Atezolizumab clearly improved the efficacy of the BCP combination with PFS and OS benefits. Interestingly, the addition of atezolizumab improved efficacy over those patients who were traditionally resistant to BCP. Such patients either harbored EGFR mutation or had baseline liver metastases. BCP and atezolizumab, carboplatin, paclitaxel regimen did not differ in PFS or OS. Bevacizumab and atezolizumab had clear synergy 56. Atezolizumab was approved by the FDA on December 7, 2018, for this indication. Based on prior phase Ib data indicating synergy of angiogenesis inhibitor axitinib with pembrolizumab, the combination was compared with sunitinib in RCC in a phase III trial in the first‐line setting 57. After a median follow‐up of 12.8 months, 12‐month survival was better in the combination group than in the sunitinib group (89.9% vs. 78.3%; HR 0.53; 95% CI 0.38–0.74; p < .0001). There was also improved PFS (15.1 vs. 11.1 months; HR 0.69; 95% CI 0.57–0.84; p < .001) and ORR (59.3% vs. 35.7%; p < .001). G3 or above toxicities were similar in both groups. The FDA approved the indication on April 22, 2019. Avelumab added to axitinib outperformed sunitinib in the same patient population 58. FDA approved the combination on May 14, 2019.
Axitinib was also given with pembrolizumab in another phase II clinical trial in patients with soft tissue sarcoma including alveolar soft part sarcoma (ASPS) 59. A total of 33 patients, including 12 with ASPS, were enrolled. With a median follow‐up of 14.7 months, 3‐month PFS for all evaluable patients was 65.6%; this was slightly higher for ASPS at 72.7%. AEs included hypertension (15%), nausea (6%), and seizure (6%). Serious adverse events (SAEs) occurred in 21% patients, including hypertriglyceridemia.
In a retrospective study of pembrolizumab added to 12 patients with ATC receiving lenvatinib, there were five PR (42%), four SD (33%), and three PD (25%). Median OS was 10.43 months with a 95% CI ranging from 6.02 to 14.83 months (5.4–40 months). Median OS from the time of adding pembrolizumab was 6.93, and median PFS was 2.96 months. AEs included hypertension, fatigue, and anemia 60.
Recently, a phase II study was also reported that evaluated the addition of lenvatinib to pembrolizumab in advanced and recurrent endometrial carcinoma 61. The study enrolled 54 patients, of whom 53 were evaluable. With a median follow‐up of 13.3 months, 21 of the 53 patients responded by week 24, ORR 39.6%. SAEs occurred in 16 of the 53 patients (30%), with one death. The most common AEs included hypertension (54%), fatigue (55%), diarrhea (51%), and hypothyroidism (47%). G3 hypertension was 34% and G3 diarrhea was 8%. Except for worsening hypothyroidism, no new AE was detected. This is clearly suggesting an active combination. A phase III trial is being planned.
Concept Under Exploration
Currently, lenvatinib is being investigated in a number of solid tumors including lung, gastrointestinal, hepatocellular, endometrial, urothelial, renal cell, and melanoma, in combination with pembrolizumab; head and neck with cetuximab; and soft tissue sarcoma with eribulin (Table 4). Most tumor types are known to respond to both angiogenesis and immune checkpoint inhibitors.
Table 4.
Concept under active exploration
| Drug(s) | n | Cancer type | Phase | Primary objective | NCT no. |
|---|---|---|---|---|---|
| Lenvatinib/pembrolizumab | 29 | Advanced gastric | II | ORR | 03609359 |
| Lenvatinib/TACE | 336 | HCC | III | OS | 03905967 |
| Lenvatinib/pembrolizumab | 24 | Gastroesophageal | IIa | OS | 03321630 |
| Lenvatinib/cetuximab | 16 | Squamous cell carcinoma (head/neck, skin) | I/Ib | MTD | 03524326 |
| Lenvatinib/pembrolizumab | 329 | Selected solid tumor | I/II | MTD/DLT/ORR | 02501096 |
| Lenvatinib/pembrolizumab | 60 | DTC | II | CR, ORR | 02973997 |
| Lenvatinib/everolimus vs. lenvatinib/pembrolizumab vs. sunitinib | 1,050 | RCC | III | PFS | 02811861 |
| Lenvatinib/pembrolizumab vs. chemotherapy | 780 | Endometrial | III | PFS, OS | 03517449 |
| Lenvatinib/eribulin | 30 | STS | Ib/II | ORR | 03526679 |
| Lenvatinib/pembrolizumab | 694 | Urothelial | III | PFS, OS | 03898180 |
| Lenvatinib/capecitabine/radiation | 28 | Rectal | I | MTD, DLT | 02935309 |
| Lenvatinib/pembrolizumab | 660 | Melanoma, first line | III | PFS, OS | 03820986 |
| Lenvatinib/pembrolizumab | 100 | Melanoma, second line | II | ORR | 03776136 |
| Lenvatinib/pembrolizumab or placebo | 750 | HCC | III | PFS, OS | 03713593 |
| Lenvatinib/pembrolizumab or chemotherapy | 720 | Advanced, recurrent endometrial | III | PFS, OS | 03884101 |
| Lenvatinib/everolimus | 40 | DTC | II | PFS | 03139747 |
| Lenvatinib/nivolumab | 50 | HCC | II | ORR, PR, CR | 03841201 |
| Lenvatinib ± carboplatin, pemetrexed/pembrolizumab | 726 | Nonsquamous, non‐small cell lung cancer | III | PFS, OS | 03829319 |
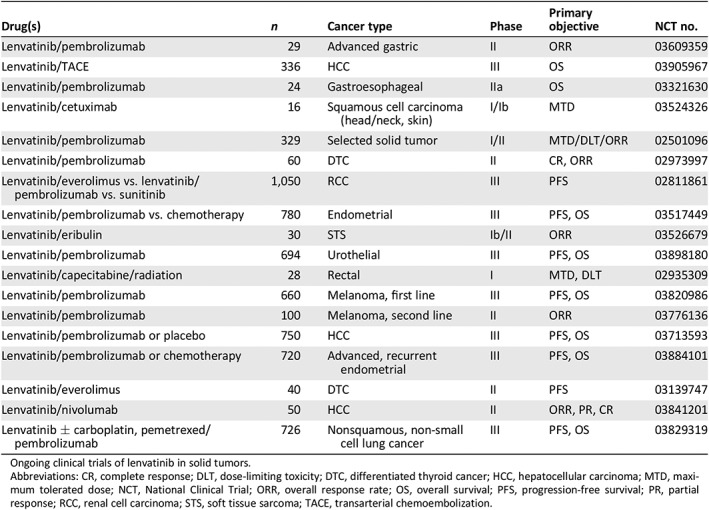
Ongoing clinical trials of lenvatinib in solid tumors.
Abbreviations: CR, complete response; DLT, dose‐limiting toxicity; DTC, differentiated thyroid cancer; HCC, hepatocellular carcinoma; MTD, maximum tolerated dose; NCT, National Clinical Trial; ORR, overall response rate; OS, overall survival; PFS, progression‐free survival; PR, partial response; RCC, renal cell carcinoma; STS, soft tissue sarcoma; TACE, transarterial chemoembolization.
Unresolved Issues
Through years of exploration, we clearly discovered a multitargeted TKI that is active in multiple solid tumor malignancies. However, it is also clear that treatment‐emergent adverse events are common and often cause dose interruption or treatment discontinuation 62. Dose interruption more than 10% had worse outcome than dose interruption less than 10% 63. Lower dose is being explored as to whether it will reduce toxicity but retain efficacy 64. In addition, biomarkers that predict efficacy and/or drug toxicity need to be further explored so that patients are not exposed to unnecessary risk. Finally, lenvatinib seems to be active for tumors grown in the brain 65, 66. Its role in controlling cancers metastasized to the brain should be explored further.
Author Contributions
Conception/design: Zhonglin Hao
Provision of study material or patients: Zhonglin Hao
Collection and/or assembly of data: Zhonglin Hao, Peng Wang
Data analysis and interpretation: Zhonglin Hao, Peng Wang
Manuscript writing: Zhonglin Hao, Peng Wang
Final approval of manuscript: Zhonglin Hao, Peng Wang
Disclosures
The authors indicated no financial relationships.
Acknowledgments
We thank Heather Russell‐Simmons for editorial assistance.
Disclosures of potential conflicts of interest may be found at the end of this article.
References
- 1. Cohen P. The regulation of protein function by multisite phosphorylation—A 25 year update. Trends Biochem Sci 2000;25:596–601. [DOI] [PubMed] [Google Scholar]
- 2. Tarrant MK, Cole PA. The chemical biology of protein phosphorylation. Annu Rev Biochem 2009;78:797–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mansour SJ, Matten WT, Hermann AS et al. Transformation of mammalian cells by constitutively active map kinase kinase. Science 1994;265:966–970. [DOI] [PubMed] [Google Scholar]
- 4. Malumbres M, Barbacid M. Cell cycle kinases in cancer. Curr Opin Genet Dev 2007;17:60–65. [DOI] [PubMed] [Google Scholar]
- 5. Fedorov O, Muller S, Knapp S. The (un)targeted cancer kinome. Nat Chem Biol 2010;6:166–169. [DOI] [PubMed] [Google Scholar]
- 6. Levitzki A. Protein kinase inhibitors as a therapeutic modality. Acc Chem Res 2003;36:462–469. [DOI] [PubMed] [Google Scholar]
- 7. Matsui J, Yamamoto Y, Funahashi Y et al. E7080, a novel inhibitor that targets multiple kinases, has potent antitumor activities against stem cell factor producing human small cell lung cancer H146, based on angiogenesis inhibition. Int J Cancer 2008;122:664–671. [DOI] [PubMed] [Google Scholar]
- 8. Matsui J, Funahashi Y, Uenaka T et al. Multi‐kinase inhibitor E7080 suppresses lymph node and lung metastases of human mammary breast tumor MDA‐MB‐231 via inhibition of vascular endothelial growth factor‐receptor (VEGF‐R) 2 and VEGF‐R3 kinase. Clin Cancer Res 2008;14:5459–5465. [DOI] [PubMed] [Google Scholar]
- 9. Ikuta K, Yano S, Trung VT et al. E7080, a multi‐tyrosine kinase inhibitor, suppresses the progression of malignant pleural mesothelioma with different proangiogenic cytokine production profiles. Clin Cancer Res 2009;15:7229–7237. [DOI] [PubMed] [Google Scholar]
- 10. Bruheim S, Kristian A, Uenaka T et al. Antitumour activity of oral E7080, a novel inhibitor of multiple tyrosine kinases, in human sarcoma xenografts. Int J Cancer 2011;129:742–750. [DOI] [PubMed] [Google Scholar]
- 11. Hussein Z, Mizuo H, Hayato S et al. Clinical pharmacokinetic and pharmacodynamic profile of lenvatinib, an orally active, small‐molecule, multitargeted tyrosine kinase inhibitor. Eur J Drug Metab Pharmacokinet 2017;42:903–914. [DOI] [PubMed] [Google Scholar]
- 12. Ogino H, Hanibuchi M, Kakiuchi S et al. E7080 suppresses hematogenous multiple organ metastases of lung cancer cells with nonmutated epidermal growth factor receptor. Mol Cancer Ther 2011;10:1218–1228. [DOI] [PubMed] [Google Scholar]
- 13. Glen H, Mason S, Patel H, et al. E7080, a multi‐targeted tyrosine kinase inhibitor suppresses tumor cell migration and invasion. BMC Cancer 2011;11:309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Okamoto K, Ikemori‐Kawada M, Jestel A et al. Distinct binding mode of multikinase inhibitor lenvatinib revealed by biochemical characterization. ACS Med Chem Lett 2015;6:89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lamba V and Ghosh I. New directions in targeting protein kinases: Focusing upon true allosteric and bivalent inhibitors. Curr Pharm Des 2012;18:2936‐2945. [DOI] [PubMed] [Google Scholar]
- 16. Shumaker R, Aluri J, Fan J, et al. Evaluation of the effects of formulation and food on the pharmacokinetics of lenvatinib (e7080) in healthy volunteers. Int J Clin Pharmacol Ther 2014;52:284‐291. [DOI] [PubMed] [Google Scholar]
- 17. Nakamichi S, Nokihara H, Yamamoto N, et al. A phase 1 study of lenvatinib, multiple receptor tyrosine kinase inhibitor, in japanese patients with advanced solid tumors. Cancer Chemother Pharmacol 2015;76:1153‐1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gupta A, Jarzab B, Capdevila J, et al. Population pharmacokinetic analysis of lenvatinib in healthy subjects and patients with cancer. Br J Clin Pharmacol 2016;81:1124‐1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shumaker R, Aluri J, Fan J et al. Influence of hepatic impairment on lenvatinib pharmacokinetics following single‐dose oral administration. J Clin Pharmacol 2015;55:317–327. [DOI] [PubMed] [Google Scholar]
- 20. Dubbelman AC, Rosing H, Nijenhuis C et al. Pharmacokinetics and excretion of (14)C‐lenvatinib in patients with advanced solid tumors or lymphomas. Invest New Drugs 2015;33:233–240. [DOI] [PubMed] [Google Scholar]
- 21. Cabanillas ME, Schlumberger M, Jarzab B et al. A phase 2 trial of lenvatinib (E7080) in advanced, progressive, radioiodine‐refractory, differentiated thyroid cancer: A clinical outcomes and biomarker assessment. Cancer 2015;121:2749–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stjepanovic N, Capdevila J. Multikinase inhibitors in the treatment of thyroid cancer: Specific role of lenvatinib. Biologics 2014;8:129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schlumberger M, Tahara M, Wirth LJ et al. Lenvatinib versus placebo in radioiodine‐refractory thyroid cancer. N Engl J Med 2015;372:621–630. [DOI] [PubMed] [Google Scholar]
- 24. Nair A, Lemery SJ, Yang J et al. FDA approval summary: Lenvatinib for progressive, radio‐iodine‐refractory differentiated thyroid cancer. Clin Cancer Res 2015;21:5205–5208. [DOI] [PubMed] [Google Scholar]
- 25. Okamoto K, Kodama K, Takase K et al. Antitumor activities of the targeted multi‐tyrosine kinase inhibitor lenvatinib (E7080) against RET gene fusion‐driven tumor models. Cancer Lett 2013;340:97–103. [DOI] [PubMed] [Google Scholar]
- 26. Schlumberger M, Jarzab B, Cabanillas ME et al. A phase II trial of the multitargeted tyrosine kinase inhibitor lenvatinib (E7080) in advanced medullary thyroid cancer. Clin Cancer Res 2016;22:44–53. [DOI] [PubMed] [Google Scholar]
- 27. Gilliland FD, Hunt WC, Morris DM et al. Prognostic factors for thyroid carcinoma. A population‐based study of 15,698 cases from the Surveillance, Epidemiology and End Results (SEER) program 1973‐1991. Cancer 1997;79:564–573. [DOI] [PubMed] [Google Scholar]
- 28. Rao SN, Zafereo M, Dadu R et al. Patterns of treatment failure in anaplastic thyroid carcinoma. Thyroid 2017;27:672–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kebebew E, Greenspan FS, Clark OH et al. Anaplastic thyroid carcinoma. Treatment outcome and prognostic factors. Cancer 2005;103:1330–1335. [DOI] [PubMed] [Google Scholar]
- 30. Subbiah V, Kreitman RJ, Wainberg ZA et al. Dabrafenib and trametinib treatment in patients with locally advanced or metastatic BRAF V600‐mutant anaplastic thyroid cancer. J Clin Oncol 2018;36:7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sandulache VC, Williams MD, Lai SY et al. Real‐time genomic characterization utilizing circulating cell‐free DNA in patients with anaplastic thyroid carcinoma. Thyroid 2017;27:81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Landa I, Ibrahimpasic T, Boucai L et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J Clin Invest 2016;126:1052–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Iwasaki H, Yamazaki H, Takasaki H et al. Lenvatinib as a novel treatment for anaplastic thyroid cancer: A retrospective study. Oncol Lett 2018;16:7271–7277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Koyama S, Miyake N, Fujiwara K et al. Lenvatinib for anaplastic thyroid cancer and lenvatinib‐induced thyroid dysfunction. Eur Thyroid J 2018;7:139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tahara M, Kiyota N, Yamazaki T et al. Lenvatinib for anaplastic thyroid cancer. Front Oncol 2017;7:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pozdeyev N, Gay LM, Sokol ES et al. Genetic analysis of 779 advanced differentiated and anaplastic thyroid cancers. Clin Cancer Res 2018;24:3059–3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yamazaki H, Yokose T, Hayashi H et al. Expression of vascular endothelial growth factor receptor 2 and clinical response to lenvatinib in patients with anaplastic thyroid cancer. Cancer Chemother Pharmacol 2018;82:649–654. [DOI] [PubMed] [Google Scholar]
- 38. Ikeda M, Okusaka T, Mitsunaga S et al. Safety and pharmacokinetics of lenvatinib in patients with advanced hepatocellular carcinoma. Clin Cancer Res 2016;22:1385–1394. [DOI] [PubMed] [Google Scholar]
- 39. Ikeda K, Kudo M, Kawazoe S et al. Phase 2 study of lenvatinib in patients with advanced hepatocellular carcinoma. J Gastroenterol 2017;52:512–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kudo M, Finn RS, Qin S et al. Lenvatinib versus sorafenib in first‐line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non‐inferiority trial. Lancet 2018;391:1163–1173. [DOI] [PubMed] [Google Scholar]
- 41. Personeni N, Pressiani T, Rimassa L. Lenvatinib for the treatment of unresectable hepatocellular carcinoma: Evidence to date. J Hepatocell Carcinoma 2019;6:31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Boss DS, Glen H, Beijnen JH et al. A phase I study of E7080, a multitargeted tyrosine kinase inhibitor, in patients with advanced solid tumours. Br J Cancer 2012;106:1598–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hong DS, Kurzrock R, Wheler JJ et al. Phase I dose‐escalation study of the multikinase inhibitor lenvatinib in patients with advanced solid tumors and in an expanded cohort of patients with melanoma. Clin Cancer Res 2015;21:4801–4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Molina AM, Hutson TE, Larkin J et al. A phase 1b clinical trial of the multi‐targeted tyrosine kinase inhibitor lenvatinib (E7080) in combination with everolimus for treatment of metastatic renal cell carcinoma (RCC). Cancer Chemother Pharmacol 2014;73:181–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Motzer RJ, Hutson TE, Glen H et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: A randomised, phase 2, open‐label, multicentre trial. Lancet Oncol 2015;16:1473–1482. [DOI] [PubMed] [Google Scholar]
- 46. FDA approves drug combo for kidney cancer . Cancer Discov 2016;6:687–688. [DOI] [PubMed] [Google Scholar]
- 47. Tchekmedyian V, Sherman EJ, Dunn L et al. Phase II study of lenvatinib in patients with progressive, recurrent or metastatic adenoid cystic carcinoma. J Clin Oncol 2019:JCO1801859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yamada K, Yamamoto N, Yamada Y et al. Phase I dose‐escalation study and biomarker analysis of E7080 in patients with advanced solid tumors. Clin Cancer Res 2011;17:2528–2537. [DOI] [PubMed] [Google Scholar]
- 49. Tamai T, Hayato S, Hojo S et al. Dose finding of lenvatinib in subjects with advanced hepatocellular carcinoma based on population pharmacokinetic and exposure‐response analyses. J Clin Pharmacol 2017;57:1138–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Haddad RI, Schlumberger M, Wirth LJ et al. Incidence and timing of common adverse events in lenvatinib‐treated patients from the select trial and their association with survival outcomes. Endocrine 2017;56:121–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wirth LJ, Tahara M, Robinson B et al. Treatment‐emergent hypertension and efficacy in the phase 3 study of (E7080) lenvatinib in differentiated cancer of the thyroid (SELECT). Cancer 2018;124:2365–2372. [DOI] [PubMed] [Google Scholar]
- 52. Brose MS, Worden FP, Newbold KL et al. Effect of age on the efficacy and safety of lenvatinib in radioiodine‐refractory differentiated thyroid cancer in the phase III SELECT trial. J Clin Oncol 2017;35:2692–2699. [DOI] [PubMed] [Google Scholar]
- 53. Huang Y, Yuan J, Righi E et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci U S A 2012;109:17561–17566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bose A, Taylor JL, Alber S et al. Sunitinib facilitates the activation and recruitment of therapeutic anti‐tumor immunity in concert with specific vaccination. Int J Cancer 2011;129:2158‐2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lemmon C, Sadek I, Hao Z. Angiogenesis and anti‐tumor immunity in the tumor microenvironment: Opportunities for synergism in intervention. J Nat Sci 2017;3:e460. [Google Scholar]
- 56. Reck M, Mok TSK, Nishio M et al. Atezolizumab plus bevacizumab and chemotherapy in non‐small‐cell lung cancer (IMpower150): Key subgroup analyses of patients with EGFR mutations or baseline liver metastases in a randomised, open‐label phase 3 trial. Lancet Respir Med 2019;7:387–401. [DOI] [PubMed] [Google Scholar]
- 57. Rini BI, Plimack ER, Stus V et al. Pembrolizumab plus axitinib versus sunitinib for advanced renal‐cell carcinoma. N Engl J Med 2019;380:1116–1127. [DOI] [PubMed] [Google Scholar]
- 58. Motzer RJ, Penkov K, Haanen J et al. Avelumab plus axitinib versus sunitinib for advanced renal‐cell carcinoma. N Engl J Med 2019;380:1103–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wilky BA, Trucco MM, Subhawong TK et al. Axitinib plus pembrolizumab in patients with advanced sarcomas including alveolar soft‐part sarcoma: A single‐centre, single‐arm, phase 2 trial. Lancet Oncol 2019;20:837–848. [DOI] [PubMed] [Google Scholar]
- 60. Iyer PC, Dadu R, Gule‐Monroe M et al. Salvage pembrolizumab added to kinase inhibitor therapy for the treatment of anaplastic thyroid carcinoma. J Immunother Cancer 2018;6:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Makker V, Rasco D, Vogelzang NJ et al. Lenvatinib plus pembrolizumab in patients with advanced endometrial cancer: An interim analysis of a multicentre, open‐label, single‐arm, phase 2 trial. Lancet Oncol 2019;20:711–718. [DOI] [PubMed] [Google Scholar]
- 62. Jasim S, Iniguez‐Ariza NM, Hilger CR et al. Optimizing lenvatinib therapy in patients with metastatic radioactive iodine‐resistant differentiated thyroid cancers. Endocr Pract 2017;23:1254–1261. [DOI] [PubMed] [Google Scholar]
- 63. Tahara M, Brose MS, Wirth LJ et al. Impact of dose interruption on the efficacy of lenvatinib in a phase 3 study in patients with radioiodine‐refractory differentiated thyroid cancer. Eur J Cancer 2019;106:61–68. [DOI] [PubMed] [Google Scholar]
- 64. Yamazaki H, Iwasaki H, Takasaki H et al. Efficacy and tolerability of initial low‐dose lenvatinib to treat differentiated thyroid cancer. Medicine (Baltimore) 2019;98:e14774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Arai N, Sasaki H, Tamura R et al. Unusual magnetic resonance imaging findings of a glioblastoma arising during treatment with lenvatinib for thyroid cancer. World Neurosurg 2017;107:1047.e1049–e1047.e1015. [DOI] [PubMed] [Google Scholar]
- 66. Wang R, Yamada T, Arai S et al. Distribution and activity of lenvatinib in brain tumor models of human anaplastic thyroid cancer cells in severe combined immune deficient mice. Mol Cancer Ther 2019;18:947–956. [DOI] [PubMed] [Google Scholar]