

Abstract
Minimally invasive transcatheter embolization is a common non-surgical procedure in interventional radiology used for the deliberate occlusion of blood vessels for the treatment of diseased or injured vasculature. A wide variety of embolic agents including metallic coils, calibrated microspheres and liquids are available for clinical practice. Additionally, advances in biomaterials such as shape memory foams, biodegradable polymer and in situ gelling solutions have led to the development of novel pre-clinical embolic agents. The aim of this review is to provide a comprehensive overview of current and emerging technologies in endovascular embolization with respect to devices, materials, mechanisms, and design guidelines. Limitations and challenges in embolic materials are also discussed to promote advancement in the field.
Keywords: embolic agents, minimally invasive approach, catheter-based delivery, vascular embolization, interventional radiology
ToC Figure
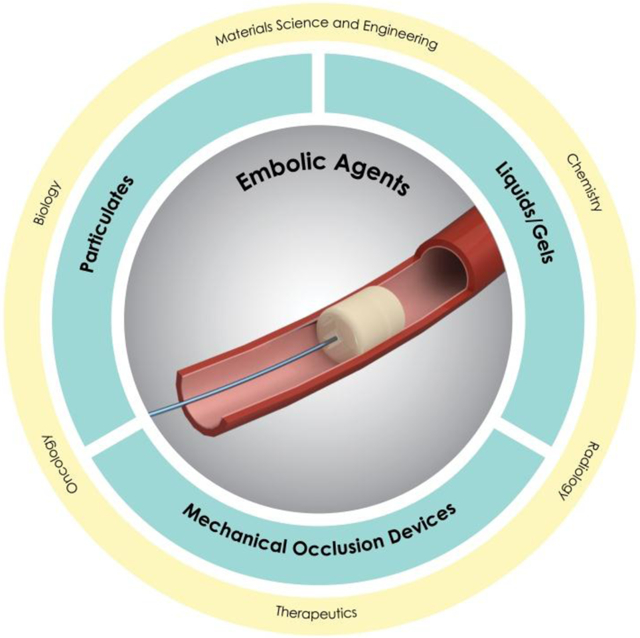
Advances in biotechnology and material science have facilitated the development of embolic agents for vascular embolization. In this review, both clinical and pre-clinical embolic agents including mechanical occlusion devices, particulates and liquids are discussed. The challenges and future insights in this field are also highlighted. An integrated and multidisciplinary approach may offer opportunities to revolutionize the next generation of embolic agents.
1. Introduction
Minimally invasive interventions are the cornerstone of modern medicine.[1] Over the past few decades there has been a gradual shift from complex open surgery to minimally invasive, image-guided interventions that have fewer complications, often better outcomes and lower cost. Blood vessels constitute a common route of entry into the body for many minimally invasive procedures and, through an endovascular approach, virtually any organ can be accessed. One frequent indication for targeting a lesion from an endovascular approach is to decrease blood flow to that part of the body--a procedure known as embolization.[2] Embolization refers to the deliberate occlusion of blood vessels with intravascular deposition of embolic agents from catheters for therapeutic purposes.[3] Therapeutic endovascular embolization is indicated in a variety of clinical scenarios. It can be used to treat acute potentially fatal bleeds to more chronic processes including aneurysms, vascular malformations, and in targeted oncologic applications such as the embolization of blood vessels supplying a tumor (Figure 1A).
Figure 1.

A) Schematic demonstrating embolization of (a) aneurysms; (b) AVMs; (c)malignant tumors (e.g., hepatocellular carcinoma, HCC) and (d) benign tumors (e.g., uterine fibroids). Figure components adapted and reproduced with permission.[12] Copyright 2000, Elsevier; 2014, BMJ Publishing Group Ltd.; 2015 Springer Nature; 2013, Elsevier; and 2016, Springer Nature. B) Summary of clinical and pre-clinical embolic agents. PVA: polyvinyl alcohol; TGMS: trisacryl gelatin microspheres; DEB: drug eluting bead; SM: sulfamethzaine; β-GP: β-glycerophosphate; Sal: polycationic salmine sulfate; IP6: polyanionic sodium inositol hexaphosphate; nBCA: N-bnutyl-2-cyanoacrylate; PPODA: poly(propylene glycol) diacrylate; QT: pentaerythritol tetrakis 3-mercaptopropionate.
The first documented embolization was performed in 1904 by Robert Dawbain, who embolized head and neck lesions of patients by preoperative injection of melted paraffin-petrolatum into external carotid arteries to reduce tumor blood flow.[4] In 1930, Barney Brooks first described vessel occlusion and proposed that the only satisfactory method to treat arteriovenous malformation (AVM) was to close the fistula’s opening.[5] Later, a thin strip of muscle was used to successfully embolize a carotid–cavernous fistula.[6] These and other pioneering studies created innovative directions using both natural and synthetic agents for embolization. In 1964, Charles Dotter, a radiologist from Oregon Health and Science University, performed the first percutaneous transluminal angioplasty for the treatment of atherosclerotic arterial stenosis, marking a breakthrough in the new era of catheter based interventions for diagnosis and treatment.[7] He also pioneered balloon catheterization and coronary angiography (X-ray imaging for vessel visualization), percutaneous arterial stenting, and is credited with developing an entirely new medical specialty, interventional radiology.[7b]
In the early years, muscle fragments, blood clots and stainless steel pellets were used as embolic agents.[8] Since the 1970s, however, improvements in imaging and catheter technology have significantly advanced embolization modalities.[9] In response to the failure of natural analogues for embolization, researchers and clinicians have used advances in materials to create new embolic agents to address specific clinical applications. Examples of these materials include metallic coils, calibrated microspheres and bioglue.[10] All current embolic materials have their advantages and shortcomings. Although no “one-size-fits-all” material exists for embolization, the general properties of an ideal embolic material include 1) biocompatibility, 2) ease of delivery, 3) resistance to fragmentation/migration, 4) biodegradability (depending on application), 5) visibility and trackability on common imaging modalities such as fluoroscopy, magnetic resonance imaging (MRI), computed tomography (CT) or ultrasound, and 6) the ability to serve as a therapeutic (e.g. anticancer drug) delivery vehicle.
A principal challenge in developing embolic agents is the versatility of embolization as an intervention. Clinical scenarios can call for the occlusion of blood vessels ranging in size from arteries (up to 1–2 cm diameter) to capillaries (5–10 μm diameter), as either a temporary or permanent intervention.[3a] Furthermore, the material must be deployable via catheter, yet also maintain structural integrity to withstand arterial blood pressure.[10–11] Therefore, the efficacy of an embolic agent is dependent on its ability to adapt to unique clinical scenarios presented by each patient and on its ability to deliver effective treatment while minimizing collateral injury to adjacent structures.
Here, we address the clinical needs of embolization in minimally invasive therapy and review the features of current and emerging embolic agents. This review starts with addressing the clinical utility of endovascular embolization (Section 2), followed by a brief introduction to the imaging modalities used in embolization (Section 3). Next, the embolic agents are discussed and are categorized into three groups based on the size of blood vessels they can occlude and on their physical properties (Figure 1B). These include mechanical occlusion devices (e.g., large vessel embolization) in Section 4, particulates (e.g., small vessel embolization) in Section 5 and liquid/gel embolic agents in Section 6. We characterize the current state of embolization inspired by clinical problems from a perspective rooted in materials science and engineering. This review evaluates both clinical embolic agents and materials under development, in terms of their mechanical/physical properties, mechanisms, and physiochemical/biological characteristics. We also highlight limitations and future directions in this field, as presented in Section 7.
2. Clinical Utility of Endovascular Embolization
Embolization is performed in a wide variety of clinical scenarios.[13] Successful embolization occurs when hemostasis is achieved, as the aim of embolization is to prevent blood flow. Examples of pathologic processes treated with embolization include hemorrhagic lesions, aneurysms and pseudoaneurysms, AVMs, and solid tumors (Figure 1A).[9]
2.1. Hemorrhage
Hemorrhage, a potentially fatal condition during which blood escapes the circulatory system either internally or externally; it can arise from trauma, from an underlying medical condition, or often a combination of both. Hemorrhage can lead to serious complications including cardiogenic shock and death with mortality rates of 40%.[14] Additionally, trauma-related hemorrhage is the leading cause of death among those aged 1–44 years.[15] Since the circulatory system carries blood throughout the body, hemorrhage can occur nearly anywhere in the body; common locations include the head, gastrointestinal tract, lungs, urinary tract, limbs and reproductive organs.[14a]
Prior to the development of endovascular techniques, the only treatment option for patients with hemorrhage was open surgery. However, in the contemporary practice of medicine, transarterial embolization (TAE) has supplanted surgery in some cases as the treatment of choice, given its potential for rapid and effective hemorrhage control using a minimally invasive approach.[16] In the context of embolization for gastrointestinal bleeding, the overall procedural success rates have been reported to be 93%−100% with clinical success rates of 51–88%.[17] Patients with coagulation disorders or who are on anti-coagulation therapy are more likely to present with hemorrhage, as their ability to form a clot is compromised.[18] For these coagulopathic patients, an ideal embolic agent is an intrinsic plug rather than one that leverages the coagulation cascade of the body to achieve hemostasis.
The aim for embolization in hemorrhage is to selectively reduce arterial perfusion pressure and blood extravasation from the bleeding vessels while simultaneously maintaining adequate collateral blood flow in order to minimize ischemic injury to normal tissues.[19] Successful embolization typically introduces an instantaneous mechanical effect which reduces blood pressure distal to the embolization site in order to minimize blood loss.[20] Subsequently, a hemostatic clot may develop to further prevent bleeding and facilitate the healing of injured vessel walls.
2.2. Aneurysms
An aneurysm is a focal dilatation of an artery due to weakening or injury to the vessel wall (Figure 1A(a)).[12a, 21] Aneurysms may occur in the cerebral circulation and in peripheral arteries.[22] Depending upon their size and location, aneurysms are prone to rupture with catastrophic bleeding. Intracranial aneurysm rupture affects approximately 30,000 people in the United States every year and can lead to severe disability or death.[23] The walls of cerebral arteries lack an external elastic lamina, a structural layer present in peripheral arterial system that reduces their compliance and results in increased susceptibility to aneurysm formation.[23b] Additional risk factors for intracranial aneurysm formation include hypertension and atherosclerosis.
Endovascular approaches including embolization have become a mainstay in treating patients with brain aneurysms given the risks associated with open surgical repair (e.g. craniotomy and clipping).[24] Every effort is made to treat aneurysms at risk for rupture in an elective setting, with TAE forming the cornerstone of treatment.[25] The ultimate goal of embolization is to fill the sac, excluding it from the circulation and thus reducing the risk of sac dilation and rupture.[20]
The development of soft steerable microcatheters and coils has allowed for safer and less traumatic intra-arterial access to aneurysms.[26] Factors determining the effectiveness of coil embolization of aneurysms have been studied in vitro, as well as through in silico methods involving finite element modeling and computational fluid dynamics.[27] These studies are useful to guide innovations in coil design, embolic materials and devices for aneurysm treatment. Additionally, the material (e.g. coils and gels) must be manufactured with a high degree of precision over a wide range of available sizes, as non-target embolization downstream can have extremely deleterious effects such as ischemia and infarction.[3a] Visibility of embolic agents on imaging modalities is also important for monitoring the real-time deployment of the material, as well as for assessing the outcomes of the intervention during follow up imaging.
2.3. Arteriovenous Malformation (AVM)
AVM is an abnormal connection between an artery and vein that bypasses the normal capillary circulation, often resulting in a tangle of vessels called a nidus (Figure 1A(b)).[12b, 12c] The malformation results in a high pressure shunting of arterial blood directly to the venous circulation, placing excessive stress on the venous wall.[12b, 28] Overstressed veins may enlarge, stretch, and eventually rupture. While AVMs can happen anywhere in the body (e.g., brain, spine, liver, pelvis and lung), they are associated with the highest morbidity and mortality when they occur in the central nervous system.[28] AVMs can also lead to localized hypoxia since the oxygen-enriched blood in the artery bypasses the capillary circulation.[29] Clinically, AVMs are treated by microsurgery, embolization and radiosurgery.[12b]
Acute bleeding from an AVM often requires urgent embolization to prevent fatal complications. Typical embolic agents for AVM management include coils, particulates and liquids, alone or in combination.[3a] The goal is to embolize upstream or within the arteriovenous shunt, thereby returning venous pressure to normal.[20] In particular, liquid embolic materials permit a vascular area to be homogenously filled and limit the risk of recanalization (i.e., a secondary reopening of the embolized area). [30] Liquid embolics that have been used in AVM embolization include absolute alcohol, N-butyl cyanoacrylate glues (e.g. TruFill®, Cordis, Miami Lakes, FL, approved by Food and Drug Administration, in 2000 for cerebral AVM embolization) and Onyx® liquid embolic system (Micro Therapeutics, Inc., Irvine, CA).[31]
The primary challenge in embolizing an AVM stems from the difficulty involved with adequately penetrating the dense, tortuous and low resistance nidus.[32] Proximal occlusion leads to the development of collateral vessels that continue supplying the nidus bed, promoting angiogenesis.[32] Therefore, blockage of the tortuous vessels in the nidus, in addition to the feeding arteries, is essential for successful embolization. To treat AVMs effectively, embolic materials should be radiopaque, deployable by microcatheters and durable to prevent recanalization and recurrence.[33]
2.4. Tumor Embolization (Malignant and Benign)
Although the conventional approach to local control of solid malignant tumors is surgical re, tumors can be difficult to remove due to their location, size or patient comorbidities. TAE has emerged as an effective tool fsectionor the treatment of inoperable hypervascular tumors (Figure 1A(c)).[12d] In this procedure, angiography and subsequent embolization is used to preferentially block a tumors blood supply relative to adjacent healthy parenchyma (Figure 2). This approach aims to “starve” the lesion by depriving it of oxygen and nutrients, thereby triggering ischemic injury and necrosis, resulting in reduction of tumor size. Generally, embolization deliberately devascularizes a tumor by blocking various levels of its blood supply (e.g. feeding arteries, peritumoral network, or intratumoral vessels).[20, 34] TAE can also be used on resectable tumors preoperatively to reduce intraoperative bleeding—a significant concern during surgery for highly vascular tumors.[35]
Figure 2.

(A-E) Catheter-based endovascular approaches to the treatment of cancer (A-E and F-G) and benign conditions (H-J and K-N). A) Digital subtraction angiography (DSA) from a catheter inside the celiac artery shows a liver tumor blush in the liver (black arrow). B) The same lesion is demonstrated in a contrast enhanced liver MRI measuring more than 6 cm in diameter. C) Following high dose segmentectomy radioembolization, the lesion is completely ablated allowing the patient to receive potentially curative surgical resection in D) with a 6 month follow-up CT imaging E) showing absence of any malignancy. F) and G) demonstrates MRI images before and after TACE embolization of a 4 cm liver tumor; white arrow in G) shows complete ablation of tumor. H) DSA from the prostate artery in a patient benign prostatic hyperplasia (BPH); corresponding Dyna-CT at the time of procedure is shown in I) Using particulate embolization, this prostate artery was completely blocked in J) (white arrow). (K-N) demonstrates particulate embolization of fibroids in uterus K). DSA of the uterine artery in L) and M) demonstrates the large fibroids; these were successfully embolized and follow-up MRI in N) shows successful treatment of the fibroids.
Hepatocellular carcinoma (HCC) is the most common malignant primary tumor of the liver and third most common cause of cancer related death worldwide.[36] The best potentially curative options are ablation, surgical resection or transplantation (e.g., Figure 2A–E).[37] However, at the time of diagnosis, 85% of patients are considered ineligible for curative treatments, resulting in poor prognoses and median survival times of less than a year.[38] Liver tumors are unique in that they receive the majority of their blood supply from the hepatic artery, while normal liver parenchyma receives the majority of its blood supply from the portal vein.[39] Therefore, TAE of the hepatic arteries or their segments can be used to starve a tumor of its blood supply; in 1974, the first use of TAE for HCC was reported.[40] Since this time, the role TAE plays in HCC treatment and the technique itself has evolved.
TAE has evolved over time to include more advanced techniques such as transarterial chemoembolization (TACE), which achieves ischemia via embolics while simultaneously delivering therapeutics such as chemotherapy (Figure 2F–G). TACE allows for the targeted delivery of chemotherapy to the tumor while reducing both the systemic circulation of these cytotoxic drugs and their associated toxicities. Conventional TACE includes administration of a chemotherapeutic agent (e.g., doxorubicin) emulsified in an oily suspension (e.g., lipiodol), followed by gelatin sponge embolization. The aim is to deliver therapeutics locally and confine the drug within the lesion for highest local drug concentration.[41] Alternatively, chemotherapeutics can be impregnated onto microspheres for use in a technique known as drug eluting bead TACE (DEB-TACE).[42] In addition, microspheres can be loaded with radioactive isotopes such as yttrium-90 (Y90) for use in radioembolization, which is a contemporary alternative to TACE that has demonstrated improved efficacy specifically when used as a bridge to surgery or transplantation (Figure 2A–E). [39, 43]
Embolization has also demonstrated efficacy for the treatment of benign tumors. Uterine fibroids are the most common benign tumors of the uterus and are also the most common benign tumors of women of reproductive age (Figure 1A(d)).[12e, 44] Since fibroids receive blood from branches of uterine arteries,[45] uterine artery embolization (UAE) has been used for uterine fibroid management (Figure 2K–N). The goal is to block blood flow to the uterus thereby shrinking the fibroids and reducing uterine bleeding. The first UAE was carried out successfully in 1995 with non-spherical particulates.[46] Compared to conventional hysterectomy and myomectomy, UAE represents a safe, effective and minimally invasive alternative for fibroid management[47] with the added benefits of reduced symptomatology and uterus preservation.[48] Recently, embolization of the prostate artery has also been described for the treatment of benign prostatic hyperplasia (Figure 2H–J). It has demonstrated similar efficacy to transurethral resection of the prostate with fewer complications or adverse side effects.[49] Prostate artery embolization (PAE) using microspheres has been FDA approved and is also approved by the United Kingdom’s National Healthcare System.
In summary, minimally invasive endovascular embolization has demonstrated utility for a variety of highly prevalent medical conditions. When compared to surgery, endovascular embolization has lower complication rates, avoids general anesthesia and can often be performed on an outpatient basis, reducing overall costs.
3. Imaging Modalities Used in Embolization Procedures
Vascular imaging plays an integral role in embolization procedures by providing a roadmap for catheter and wire navigation, guiding procedural execution, and confirming successful embolization. Visualization of embolic materials on imaging is essential as it enables the operator to determine the level of vessel occlusion, ensuring targeted embolization. Various imaging modalities are also used for procedural planning, procedural execution and follow up.
Imaging modalities used in embolization include radiation-based X-ray fluoroscopy, ultrasound, computed tomography (CT) and less commonly magnetic resonance imaging (MRI). Although no single imaging system meets all clinical needs, each system is well suited for specific treatment stages. Almost all embolization procedures are performed under live visualization using fluoroscopy which provides angiography to visualize blood vessels. Current fluoroscopy technology allows for real-time, high resolution imaging of radio-opaque devices and iodinated contrast agents. Intra-procedural cone beam CT or CT angiography can be used as an adjunct to fluoroscopy to determine optimal positioning of the catheter for selective tumor treatment and to evaluate tumor coverage after the injection of an embolic agent.[50]
Follow up imaging may include CT or MRI. CT is commonly used following embolization since it is widely available, rapid and robust. Disadvantages of CT include ionizing radiation, streak artifacts that limit visualization of the embolized region and relatively low soft-tissue contrast.[51] To differentiate vasculature and tissues, radiopaque contrast agents are usually administered and increase the attenuation of the tissues of interest. For CT, iodinated contrast agents are FDA approved and widely used.[52]
When compared to CT, MRI offers superior soft-tissue contrast.[53] New MRI sequences also allow for high-resolution vascular imaging without the need for intravenous gadolinium-based contrast agents. This is particularly advantageous for patients with compromised renal function or allergies to iodinated or gadolinium-based contrasts.[54] Despite these advantages, the lack of MRI-compatible interventional tools limits this modality’s utility for intraprocedural image guidance.
Doppler ultrasound is a non-invasive imaging modality that is widely used for vascular imaging. This technology allows for visualization of the vessel lumen and superficial structures while simultaneously providing hemodynamic information. It is limited, however, by depth of penetration of ultrasound signal, resolution and susceptibility to imaging artifacts that preclude the use of ultrasound for deep vessel imaging.[55] Ultrasound is widely used in interventional radiology, whether for gaining arterial or venous access or for guiding percutaneous thrombin injection for the treatment of common femoral artery pseudoaneurysms, which are often iatrogenically induced.[56]
The visibility of embolic materials is of paramount importance during embolization procedures, as well as for long term monitoring. The multimodal visibility of embolic agents is especially appealing since it offers flexibility for modality selection while providing both qualitative and quantitative intensity information for in vivo evaluation. Additionally, the tracking of drug diffusion and distribution in tissue may be desirable for chemoembolization assessment.
4. Mechanical Occlusion Devices
A variety of mechanical occlusion devices have been used clinically for embolization including coils, plugs, stents and balloons.[31, 57] Coils are most commonly used for the treatment of aneurysms and hemorrhage control. Often times, multiple coils are used and may be combined with other devices or agents. In this section, the evolution of coil materials and coil induced tissue reactions are discussed. Newly developed polymeric coils and shape memory foams are also briefly reviewed.
4.1. Coils
Coil embolization has been widely used to mechanically occlude aneurysms[58] or to block bleeding vessels.[58–59] During the procedure, a catheter is steered through the blood stream to reach the target vessel. Once positioned, the coil is pushed through the catheter using a delivery system to wedge it against the vessel lumen or the aneurysm sac, leading to embolization.[60]
Among the many properties of coils, biocompatibility is a key factor.[61] For long-term implantation, the coil materials should be bio-inert to avoid provoking a local or systemic adverse host response. Coils should also be malleable for ease of manufacturing, adequately pliable for catheter delivery and visible under imaging modalities. Another critical property of coils is surface thrombogenicity, which governs fibrin formation and platelet adhesion.[62]
Coils primarily consist of bio-inert metallic cores such as stainless steel, platinum or nitinol and can also contain a coating (Figure 3A). Theoretically, the core should possess relatively high longitudinal strength but low radial forces to prevent excessive stress on the vessel wall. These dense metals are also intrinsically radiopaque, allowing for fluoroscopically-guided, targeted embolizations. The mechanism of metallic coil embolization is primarily mechanical occlusion with low thrombogenic properties. For improved thrombus formation, coils with bioactive coatings have been developed.
Figure 3.

Illustrations of A) a AZUR® framing coil (coil OD of 0.014–0.015 inches or 0.022 inches); B) a polymer-fibered (either PLGA or nylon) Concerto™ 3D detachable coil system (available diameter ranging between 2mm and 18 mm with length spanning from 2 cm to 40 cm); C) a polymer-fibered (either PLGA or nylon) Concerto™ HELIX detachable coil system (available diameter ranging between 2mm and 20 mm with length spanning from 4 cm to 50 cm); and D) a AZUR® HydroCoil, which combined a platinum coil and an expandable hydrogel polymer (pre expansion coil OD at 0.014–0.017 inch (or 0.024–0.030 inch) and post expansion OD of 0.034 inch (or 0.048 inch)) (Courtesy of Terumo and Medtronic). OD represents outer diameter.
4.1.1. Bare Metallic Coils
Stainless Steel Coils:
Stainless steel has long been a dominant force in the field of biomaterials since it is bio-inert, corrosion resistant and has excellent mechanical properties. In 1975, the first documented stainless steel coil was introduced by Gianturco and colleagues.[16a] The team delivered the coils through 6 French (F) and 7F catheters into canines and patients for permanent arterial embolization.[16a] Stainless steel coils are radiopaque, though to a lesser extent than newer metallic alloys, such as platinum.[63] These coils are relatively stiff due to their high radial strength which can make dense packing a challenge. Stainless steel coils are primarily used for large vessel occlusion, as opposed to coils made of more flexible materials (e.g., platinum) that allow for smaller vessel occlusion. Stainless steel coils have demonstrated efficacy in the treatment of arteriovenous fistulas, hypernephromas, hypersplenism and bleeding from pelvic neoplasms.[64]
Platinum Coils:
For improved packing density, softer platinum coils have been introduced and are currently among the most widely used coils in contemporary clinical practice. Platinum coils are more malleable, radiopaque and softer than stainless steel coils. They can also be delivered through microcatheters (e.g., 2.8F). Due to their compliance, platinum coils conform into the shapes of often-irregular aneurysms upon release. Since they do not possess as high radial force like stainless steel coils, platinum coils can be packed tightly without exerting excess pressure on the lumen of aneurysm sac. Furthermore, unlike stainless steel, platinum is not prone to electrolysis and is three to four times more thrombogenic than stainless steel.[21] However, streak artifacts caused by platinum coils can lead to impaired evaluation of the embolized segments.
Nitinol Coils:
Shape memory nitinol is a relatively new coil core material, despite its extensive use in the stent industry.[65] It is an intermetallic alloy composed of almost equal amounts of nickel and titanium, and has the ability to restore its shape after deformation (i.e. self-expandable). Nitinol has a lower elastic modulus and greater strength compared to stainless steel.[66] It is 10 to 20 times more flexible than stainless steel, indicating excellent torqueability. Nitinol also has excellent biocompatibility and is more corrosion resistant when compared to stainless steel, given the presence of a formed passive titanium-oxide layer. This layer shields the toxic nickel component from leaking into its surroundings, protecting the bulk material from corrosion. Furthermore, it has been shown that nitinol reinforced stainless steel coils increase intravascular stability due to higher radial forces when compared to standard stainless steel coils, all without compromising coil handling.[67] Nitinol has also been incorporated as a secondary coil inside of platinum coils with larger diameters (Penumbra Coil 400™, Penumbra Inc., Alameda, CA) to provide structural support and stiffness, allowing for greater packing density.[68]
The unique properties of nitinol alloys including shape memory and superelasticity are due to their inherent phase transformation.[69] At high temperatures, nitinol exhibits a stable and non-transformable austenite phase. In contrast, at low temperatures, nitinol is in a martensitic phase, demonstrating a uniquely twinning structure with less symmetry. This characteristic allows martensitic nitinol to be sheared approximately ten times the strain of common alloys by gradually increasing force, without exhibiting permanent deformation.[69] The austenitic-martensitic transformation occurs either by lowering the temperature or by applying a stress. Thus, nitinol alloys can be deformed at a lower temperature and return to their original shape when activated by body heat.[69] Once the coil is in the body, nitinol restores its austenitic state and remains stable. These properties from thermoelastic martensitic transformations (e.g., shape memory effects) help promote flexibility and torqueability of nitinol based coils.
Furthermore, the stress-strain behavior of nitinol exhibits hysteresis (energy storage) after unloading, similar to certain biological materials (e.g., human hair, bone and tendon). This suggests that upon contact, the strain mismatch between nitinol and biological materials is minimized and therefore leads to faster healing and less trauma. The superelasticity of nitinol, therefore, allows deformation energy storage to accommodate imposed stresses and pressure.
4.1.2. Bioactive Coils
Bio-inert bare metallic coils stop blood flow mechanically and are dependent upon the patient’s individual ability to form a clot, which is triggered by coil-induced flow dynamics changes.[70]. Thus, the formed thrombus on the surface of bare metallic coil is usually thin and may vary significantly on different areas of the coil, between coils and from patient to patient.[71] Moreover, the newly developed thrombus mainly consists of erythrocytes and thin fibrin fibers, and therefore may not withstand arterial blood pressure. Additionally, physiologic thrombus breakdown can lead to coil compaction and migration which can lead to recurrence of bleeding.[72] Surface modifications to existing metallic coils can improve thrombogenicity which has led to the development of a new class of coils - bioactive coils.
Fibered Coils:
Fibered coils represent one class of the bioactive coils (Figure 3B and 3C). In 1975, Gianturco and his team used cotton-tailed coils for small artery embolization and wool coils for larger vessel embolization in canine models.[16a] Cotton-tailed coils consisted of 3 mm of steel tubing and 5 mm cotton threads. Woolen coils were composed of 3 cm long wool strands weaved onto a 5 cm long steel coil. It took approximately 10 minutes for a clot to form on woolen coils. Intense foreign body reaction and chronic inflammation was observed with wool coils.[73] Alternatively, coils with attached synthetic Dacron (polyester), Ivalon (polyvinyl alcohol) and silk fibers demonstrated reduced inflammation while maintaining platelet aggregation properties.[73–74] Nylon fiber coated platinum coils improved the occlusion rate compared to that of bare platinum coil from 85% to 96%.[71]
Compared to bare metallic coils, fibered coils are generally more thrombogenic and further stabilize the thrombus as a result of fiber dispersion within the clot[71]. Because the cellular and fibrin adhesions on fibered coils effectively increase with larger coil diameters, reduced thrombus dislodgement from the aneurysm has been reported.[71] However, in a retrospective study of 474 aneurysms, the recurrence rate was found to be comparable between bare platinum coils (27.8%) and platinum coils with nylon fiber (24%). Additionally, coil compaction rates were similar (27.8% for bare coils and 24% for fibered coils).[71]
Though fibers can effectively increase thrombogenicity, friction comes into play if the fibers are longer than 2–3 cm.[62] Enhanced friction makes pushing fibered coils through the catheter extremely difficult, especially when traversing tortuous vessels. Additionally, too rapid of thrombus formation associated with fibers is undesirable due to the high risk of thrombus dislodging from either the coil or within aneurysm during coil delivery and placement.[75]
Polymer Coated Coils:
Biodegradable poly(lactic-co-glycolic acid) (PLGA) or poly(glycolic acid) (PGA) polymers have been used as surface coatings on platinum coils to promote tissue response and thrombus formation.[76] The use of PLGA copolymer coated platinum coils was reported to accelerate aneurysm healing with intensified neck neointimal formation and pronounced fibrosis compared to bare platinum coils.[77] However, PLGA coated coils do not necessarily exhibit a sustained benefit over bare platinum coils.[78] For example, FDA approved Matrix® and Matrix2™ coils (Boston Scientific, Marlborough, MA, platinum coils with PLGA coating) showed disappointing clinical outcomes with elevated recanalization rates.[79] It is possible that PLGA degrades before thrombus formation, leading to coil compaction, insufficient volume filling and recanalization.[23a] PLGA coated coils were also technically more difficult to deliver through the catheter because of increased stiffness and friction against the catheter wall.[80] Additionally, a higher rate of headache and fever after embolization of intracranial aneurysm was reported with the use of PGA coils compared to bare platinum coils; this may be attributed to inflammation associated with PGA coating.[81]
In another example, polyurethane coated platinum coils demonstrated tunable thrombogenicity.[62] This was achieved by altering hard and soft segments or the ratio between the two components that produce polyurethane. In a rabbit model, bare platinum coils led to 40% embolization within 30 minutes in aortas, whereas thrombogenic polyurethane coated polymers led to 100% occlusion in 30 minutes. SEM images of retrieved coils after implantation further confirmed that thrombus coverage correlated with the degree of polyurethane thrombogenicity; the most thrombogenic polyurethane coated platinum coil was covered with the heaviest amount of thrombus.
The surface structure of polyurethane is complex. The thermodynamic immiscibility of the alternating hard and soft segments in polyurethane leads to its microphase separated structure,[82] creating a local microenvironment with spatially distributed chemical, physiological and mechanical properties due to different segment characteristics.[83] Major factors that influence the structure include chemical composition of the segments, the polarity of soft segments, the ratio between the hard/soft segment, as well as the environment. Therefore, the blood interface may respond differently to fibrin recruitment, platelet aggregation and cell migration.[83] Additionally, the urethane group content and the type chain extender also affect polyurethane characteristics. For instance, sulfonic acid groups,[84] amino acid[85] and peptide[86] based chain extenders can be incorporated to tailor the surface thrombogenicity and physical properties to selectively attract cellular elements for desired thrombus formation.
Protein Coated Coils:
Fibroblast invasion and thrombus formation are critical steps in aneurysm healing.[87] Some key proteins involved in the healing and recanalization process include monocyte chemoattractant protein-1 (MCP-1),[88] osteopontin (OPN), interleukin-10 (IL-10) and matrix metallopeptidase 9 (MMP-9).[87]
One of the notable proteins with pronounced elevated expression in densely packed and well-healed aneurysms is OPN.[89] It is a cell-adhesion molecule that mediates a variety of physiological and pathological activities such as chemotaxis and wound healing. In addition, its anti-apoptotic property promotes cell adhesion and migration to mediate vascular smooth muscle cell differentiation.[90] Chen et al. found that OPN modified PLGA/platinum coils markedly increased tissue growth in a rat carotid aneurysm model in 7 days compared to PLGA/platinum coils.[87] The tissue ingrowth of OPN modified PLGA/platinum coils was over twice that of PLGA/platinum coils post treatment at 7, 20 and 60 days, which highlighted the potential of using OPN as an effective protein coating to accelerate healing of aneurysms. In addition, IL-10 was also shown to facilitate tissue ingrowth but at a later stage (60 days). Conversely, MMP-9/PLGA did not promote, but rather inhibited tissue ingrowth compared to PGLA coils.[87] It is interesting to note that MMP-9 induces aneurysm recanalization while also promoting neointima formation.[90]
Additionally, the combination of fibrinogen and vascular smooth muscle cells may promote neointima generation at the aneurysm neck.[91] Specifically, human vascular endothelial growth factor coated platinum coil is beneficial in promoting endothelialization, clot organization and tissue/coil integration to hasten aneurysm occlusion.[92] Thrombogenic/antithrombolytic and fibroblast molecule coated coils have also been investigated.[93]
4.1.3. Hydrogel-Coated Coils
Recanalization after coil embolization often occurs due to coil compaction and at least 20–25% packing density is recommended to prevent recanalization. To address this issue, hybrid hydrogel-coated platinum coils (e.g. HydroCoil Embolic System, MicroVention, Inc., Aliso Viejo, CA) have been developed to improve packing density and achieve larger filling volumes when compared to Matrix and platinum coils (Figure 3D).[94] Upon contact with blood, polymer chains disentangle. Hydrogel-coated coils can expand up to five times their original diameters[79a, 95] and undergo full expansion within 20 minutes.[72] They appose the vessel wall to prevent dislodgement[60] without exerting excess pressure on the vessel wall itself, since they expand to the volume that is available. Furthermore, hydrogel-coated coils are especially useful in high flow vessels which are often difficult to embolize due to increased risk of coil migration.
Hydrogel-coated coils appear to exhibit reduced recanalization and recurrence rates in large aneurysms when compared to bare platinum coils at early stages.[25, 96] Additionally, in a pulmonary AVM study, no recanalization was reported for 57 patients treated with hydrogel-coated platinum coils over a median follow up time of 19 months.[97] This may be attributed to the increased packing density associated with hydrogel-coated coils compared to an equivalent amount of bare platinum coils. It is also possible that hydrogel gives rise to more tissue reaction and organization due to elastic lamina damage and vascular smooth muscle cell proliferation, which was observed in a rat sidewall aneurysm model.[98]
However, some studies suggest that current hydrogel-coated coils do not show sustained benefit over bare platinum coils in aneurysm treatment.[78, 99] Furthermore, delayed aseptic meningitis and hydrocephalus development have been reported as potential side effects associated with hydrogel-coated coils.[72, 96, 100] Additionally, relatively fast deployment of hydrogel-coated coil is required to prevent its pre-expansion inside of the catheter.
4.1.4. Coil-Tissue Interactions
Mechanism of Bare Platinum Coil Embolization:
The combined coil mass and subsequent induced thrombosis initiates lumen obliteration due to hemodynamic changes and intrinsic blood clotting pathways. Eventually, the blood clot becomes fibrotic and obliterates the vessel wall.[72, 101] Although the type of coil metal influences short term thrombus formation, the long term occlusive behavior depends on the formation of fibrous tissue.[102]
Histologic studies of treated human aneurysms illustrate the progression of acute thrombus formation to fibrosis following bare platinum coil embolization.[90] Generally within the first week, thin fibrin layer covers the coil and disorganized thrombus is observed around the coil.[103] Fibroblasts and macrophages begin to invade the thrombus. A thin but incomplete layer of fibrin then presents at the aneurysm neck. One to two weeks after coil embolization, there is an increase in the amount of fibrin surrounding the coil. This is accompanied by myofibroblast invasion and a thin fibrin layer at the aneurysm neck is observed.[104] More rigorous myofibroblast invasion persists up to one month after the embolization. During this period, the coil is covered by fibrin, and more organized thrombus starts to form. Additionally, neointima formation is observed at the aneurysm neck.[105] Afterwards, the inflammatory activity decreases within the sac due to apoptosis of myofibroblasts and macrophages, and endothelialization increases at the neck of aneurysm. After three months, vascularized connective tissue initiates within the sac, together with markedly increased fibrin around the coil and maturing of the endothelialization layer at the neck. Generally, at one year following the procedure, vascular fibrous connective tissue fills the sac. The aneurysm neck is covered with a layer of mature fibrous tissue and the coil is completely sealed within the sac.[90, 93a, 103]
Mechanical Properties of The Coil-Clot Complex:
An understanding of the underlying mechanical mechanism during coil embolization and mechanical properties of coil-clot complexes facilitates the prevention of aneurysm recurrence. It has been reported that reduced aneurysm wall stress is associated with increased stiffness of the filling material.[106]
Uniaxial compression tests were performed on coil-clot complexes[107] and clot stiffness was characterized by clot modulus (Young’s modulus).[107] After 3 hours of clot formation, bare platinum coils increased clot stiffness almost eight times that of clot alone, while HydroCoil did not have a significant impact on clot modulus (Figure 4A).[107] After 3 days, the stiffness of the bare platinum coil-clot complex reduced to the same order of the HydroCoil-clot system, whereas the sources which contributed to the stiffness of complex were totally different. In particular, the coil modulus determines the stiffness of the coil-clot complex while fibrin network contributes to the clot modulus. The fibrin structure governs fibrinolytic and viscoelastic properties of mesh,[108] with individual fibrin fiber modulus on the order of megapascals.[109] In addition, the implantation of platinum coils did not alter fibrin characteristics (e.g., fibrin diameter and density) compared to fibrin characteristics found in clots alone.
Figure 4.

A) Box plots of Young’s modulus of coil-clot complex. SEM images of B) HydroCoil–clot complex and C) bare platinum coil–clot complex, Figure components adapted and reproduced with permission.[107] Copyright 2014, BMJ Publishing Group Ltd..
As opposed to the observed mature fibrin network on the surface of platinum coil, no fibrin is observed on the surface of retrieved HydroCoils (Figure 4B and 4C). The lack of fibrin formation associated with HydroCoils has also been observed elsewhere.[110] The long-term occlusion by HydroCoil, therefore, does not depend on host response to the coil and thrombus formation, but rather the occlusion of the aneurysm.[111] In particular, HydroCoils fill a larger volume fraction (over four folds of magnitude) of the aneurysm compared to platinum coils alone.[110] A substantially improved volumetric packing of the aneurysm lumen is observed clinically compared to standard platinum coils (74% versus 32%).[112] The stable occlusion may be the reason behind the low recanalization rate and recurrence rate following hydrogel coated platinum coil embolization.[113]
4.1.5. Shape Memory Polymeric Coils
Recent advancements in shape memory polymers highlight their potential in biomedical applications. Shape memory polymers have been actively explored in the fields of stents, surgical sutures, scaffolds and tissue engineering.[114] External stimulation (e.g., from heat or light) can trigger the shape memory effect. Therefore, a large bulky device can be delivered in a temporarily compressed form through a minimally invasive approach and can then be expanded into its permanent shape.[114b, 115]
While embolic coils are primarily considered for permanent occlusion, a bioabsorbable water-induced shape memory plug was recently introduced for temporary vascular occlusion.[116] The plug consisted of a PLGA core doped with radiopaque materials (e.g., barium sulfate, tantalum and bismuth oxychloride) and a crosslinked poly(ethylene glycol) diacrylate (PEGDA) hydrogel coating for enhanced vessel occlusion.
The shape memory behavior of the plug was attributed to phase transformation between the crystalline and amorphous states of PLGA and PEGDA polymers in response to both physiological solution infusion and thermal activation. At an elevated temperature (70°C), both polymers were in their amorphous phases and could be deformed into temporary shape. The temporary configuration could then be fixed at a temperature below PEG glass transition temperature due to the immobilization of the PEG crystalline phase. The initial rigid gel stripe facilitated catheter delivery. When infused with aqueous solution at 37°C, the system became fully amorphous. The PEGDA coating expanded for improved occlusion in conjunction with the activated PEG crystalline phase. Specifically, buckling initiated in the PLGA core, which gradually overcame the resistance from the PEGDA coating, and the stripe eventually recovered its programmed coil shape.
The composite polymeric plug was examined in various arteries in a rabbit model and exhibited a complete occlusion time ranging between 38 seconds and 2 minutes. No vascular rupture was observed and significant enlargement of the vessels was clear at the embolization sites due to swollen PEGDA hydrogel. Additionally, the gel composite degraded in vivo (70% mass reduction in 10 weeks) primarily due to hydrolysis. Specifically, the PEGDA hydrogel coating encapsulated the degraded PLGA core and subsequent downstream migration, which was a clinical concern. [116]
A long lingering problem associated with smart hydrogels is their low modulus and strength which severely limits their biological applications.[117] Since the vast majority of the temperature-triggered hydrogel coils are too soft with moduli far below those of metallic coils, they deform easily within the catheter, making delivery difficult. Additionally, they may not return to their programmed shape in vivo. Recently, a high strength, high modulus and temperature-responsive radiopaque PAN-PAAm-PEG3kDMA hydrogel microcoil was developed for permanent embolization (Figure 5A–5C).[118] PAN-PAAm-PEG3kDMA gel, composed of acrylonitrile (AN), acrylamide (AAm, for bonding) and PEG3kDMA (synthesized from PEG3k and methacryloyl chloride) was synthesized through a combined copolymerization and phase conversion method. BaSO4 was blended to render the hydrogel coil radiopaque.
Figure 5.

A) Schematic fabrication process of radiopaque hydrogel coils. B) The temperature triggered response of shape memory hydrogel coil. C) The shape memory induced embolism by the hydrogel coil assessed at 37 °C. D) Schematic of the transarterial embolization process. E) Angiograms of radiopaque coil delivery (yellow dots outlined contour of delivered hydrogel coils) into the renal artery. F) Angiographic images obtained at 4, 8, and 12 weeks after embolization with red circles denotes microcoil position. G) Gross appearance of the embolized and normal kidneys at 4, 8, and 12 weeks, respectively, after procedure. (Scale bars: 2 cm). Figure components adapted and reproduced with permission.[118] Copyright 2018, John Wiley & Sons Inc..
The smart gel network consisted of dual physical crosslinking construction with strong hydrophobic dipole-dipole pairings of polyacrylonitrile and weak hydrophilic hydrogen bonding of polyacrylamide. The former interaction stabilized the latter and enhanced the hydrogel’s mechanical properties due to formed hydrophobic crystalline microdomains[119] that inhibited hydrogel swelling and facilitated stability at elevated temperature.[120] The reinforced hydrophobic aggregation, a result of physical crosslinking, offered a pronounced network strengthening of the gel which possessed a Young’s modulus of approximately 16 MPa at room temperature. The rigidity of the material helped the coil to be delivered smoothly through the catheter. While most hydrogels are brittle with a fracture energy on the order of ~10 J/m2,[117a] PAN-PAAm-PEG3kDMA gel was tough (fracture energy ~16 kJ/m2) and also highly stretchable (breaking elongation ~ 950%). Its fracture energy was higher than that of natural rubber (~10 kJ/m2). The superior mechanical properties allowed this microcoil to maintain stable occlusion in vivo.[117a, 121]
The embolization effect of shape memory PAN-PAAm-PEG3kDMA microcoil was also examined in a porcine renal artery model. The coil was delivered in the form of a rigid hydrogel strip via a 4F catheter under the protection of cooled saline (Figure 5D). When released into the blood vessels, the thermo-responsive strip softened and twisted into a coil in less than 10 seconds (Figure 5E). The microcoil remained stable in blood vessels with no evidence of recanalization 12 weeks after surgery, as confirmed by angiographic images (Figure 5F). The embolized kidneys became atrophic with histology revealing coagulation necrosis in the affected tissue, suggesting the therapeutic efficacy of the microcoil (Figure 5G).
4.1.6. Challenges in Coil Embolization
Challenges in coil embolization mainly include nontargeted embolization and coil migration. Non-target embolization usually results from coil misplacement or coil-vessel size mismatch. If the coils are too small, they may migrate into the systemic circulation. Systemic migration rates of up to 3% have been reported.[122] If the coils are too big for an aneurysm, they may extrude into the parent vessel and lead to thrombosis of non-target vessels. Due to excessive radial force, the oversized coil can also elongate within the sac and cause aneurysm rupture.
Recanalization is also a complication commonly observed after aneurysm embolization and may necessitate additional procedures. Reasons for recanalization include aneurysm regrowth, coil compaction, unstable thrombus formation and lack of neointima formation at the aneurysm neck.[90] There is a strong correlation between aneurysm volume, packing density and recanalization rate. Residual volume, determined by the difference of the volume of the inserted coils and the aneurysm volume (e.g., packing density), has been identified as a major risk factor in recanalization.[90] Furthermore, computational modeling between coil and blood flow interaction has suggested that densely and uniformly packed coils would reduce permeability, thereby promoting clotting and thrombosis formation.[123]
4.1.7. Design Guidelines for Coils
The physical properties (e.g. stiffness) of coils can be referenced to those of springs.[61, 124] The spring constant is usually related to the material modulus, number of turns per unit distance, the pitch of the coil, and stock wire diameter.[61, 124] Internal materials placed within the primary wind such as bioactive coatings and fibers also mediate coil stiffness.[125] Additionally, coils must be radiopaque for visualization during both treatment and follow-up imaging.
Endovascular coils need to be easily deployable through the catheter, without binding inside of catheter due to inappropriate rigidity or excess wall friction. After release, it is critical that coils are able to withstand blood pressure and restore to their desired shape without collapsing. In the case of aneurysm treatment, the coils should maintain adequate packing density without exerting excessive force on the aneurysm wall. Excellent mechanical strength and low stress relaxation behavior are characteristics that are critical to successful aneurysm occlusion in the long term.[115]
Finally, coils aid to provide either mechanical (e.g., hydrogel-coated coils) or biological support (e.g., bioactive coils) and these are two generally adapted approaches that manufacturers use in current coil design. Permanent occlusion and coil surface activation are desired to promote endothelium formation at the aneurysm neck, which prevents recanalization and re-bleeding of an aneurysm. Though each of above-mentioned characteristics and functionalities has been studied extensively, existing coils fail to employ multiple functionalities, necessitating further exploration to optimize coil design.
4.2. Shape Memory Polymer (SMP) Foam
Shape memory materials have emerged as an important class of stimuli-responsive materials in medicine for minimally invasive procedures.[126] Due to the inherent risks that are associated with metallic based coils including inflammation and recanalization, shape memory polymer (SMP) foam based occlusion devices have been proposed as an alternative.[127] They have many favorable properties that make them suitable as aneurysm filling materials.[128] Primary advantages include tunable pore sizes that can be tailored for optimal cellular infiltration and stable tissue integration within the foam.[129] Biocompatibility, mechanical property and contrast visualization enhancement has been characterized for SMP foams.[130] For example, less inflammatory activities have been found with SMP foam.[131] Additionally, a simulated aneurysm model demonstrated that typical hemodynamic forces do not hinder the shape recovery process. SMP foams can be rendered radiopaque with doped tantalum or tungsten.[131–132] They have also been coated onto the coils to facilitate delivery and packing performance in aneurysm treatment.[133]
In one study, polyurethane based SMP foam was crimped over a nitinol wire for transcatheter delivery (Figures 6A).[128] When released into the body, body temperature and body fluid absorption activated foam expansion. The foam expanded ten times its crimped diameter to fill a larger volume within 20 minutes (Figure 6B and 6C).[128] In vitro blood perfusion assay suggested that SMP foam stopped blood flow completely in 270 seconds and the foam was covered by dense fibrin rather than interspersed fibrin at 30 seconds of blood perfusion (Figure 6D). Furthermore, the porous structure of SMP foam provided a scaffold for cell growth and thrombus formation leading to ultimate tissue regeneration.[128] The degree of foam porosity (e.g., 0.5 mm, 1 mm and 1.5 mm of pore diameter) can be tailored by varying physical blowing agents (for foaming).[134] A 200–800 μm pore diameter has been reported to be effective for connective tissue regeneration in porcine side wall aneurysm occlusion.[131] The surface chemical properties of SMP foam can also be tuned (e.g., by plasma sterilization).[135] Moreover, the foam’s intrinsic echogenicity allow SMP foams to be imaged by ultrasound during both the delivery and follow-up processes. This could significantly reduce the patient’s radiation exposure from fluoroscopy.
Figure 6.

SMP device A) before and B) after expansion. C) Schematics demonstrating temporary shape of SMP foam device for catheter delivery into aneurysm, intermediate stage of SMP during expansion and fully recovered foam for aneurysm filling. D) Histology of SPM samples perfused with blood in vitro, with perfusion time of 30 seconds in a-c and 270 seconds in d-f. Samples a&d, b&e, and c&f represent proximal, middle and distal regimes, respectively. Pinkish red stained for erythrocytes and purple stained for fibrin and leukocytes. E) Gross examination of the implanted SMP foams in vein pouch aneurysm model thirty-minute, thirty-day and ninety-day after embolization, respectively. Figure components adapted and reproduced with permission.[128, 139] Copyright 2016, Elsevier and 2013, John Wiley & Sons, Inc.
The radial force and pressure exerted on the vessel wall by any occlusion device is important to identify in order to prevent rupture. The aneurysm wall strength is in the range of 700–5000 kPa[136] and Konig et al. reported an average burst pressure of human saphenous veins to be 213 ± 177 kPa.[137] For the polyurethane based SMP device, even when the polymer was expanded to over 50% of the size of the target vessel, the radial force exerted by the SMP was less than 0.5 N, which was significantly below the threshold for autologous vein rupture (107 N).
The polyurethane based SMP foam device also demonstrated encouraging in vivo results. The SMP foams resulted in complete occlusion of various porcine hind limb vessels in an average of 90 seconds .[138] More importantly, partial healing was observed at one month post SMP foam embolization in a porcine aneurysm model. Three months after the procedure, a complete endothelial layer presented across the aneurysm neck with minimal inflammatory response, indicating almost complete healing (Figure 6E).[139] Moreover, at 180 days, a 89–93% reduction in cross-sectional area of the neck was reported for SMP foam treated porcine vein pouch aneurysms, whereas coil treated aneurysms only demonstrated an 18–34% reduction.[140] These results suggested that SMP foam treated aneurysms exhibited superior healing at advanced stages compared to bare metal coil treated aneurysms.[140]
Compared to the large clot formation seen in conventional metallic coils, the porous polymeric foam allows for the generation and connection of numerous small clots, resulting in a faster coagulation and healing process. The porosity also permits ingrowth of granulation tissue and neointima formation to seal the aneurysm neck. These features effectively reduce the chances of recanalization.[129] Compared to shape memory alloy, SMP foams possesses the advantages of lower density, higher inelastic recovery (after plastic deformation) and lower cost. Furthermore, the mechanical properties and transition temperature of SMP foam can be tailored over a wide range with minimal adjustments to chemical structure and composition.[129] In addition to its biocompatibility and non-mutagenicity, polyurethane can also serve as drug carrier and hemostatic agent.[141] These studies highlight the potential of SMP foams as viable mechanical occlusion devices for aneurysm treatment with long term occlusion efficacy and benefits.
5. Particulates
Particulates were the first embolic agents to be developed and are currently the mostly commonly used agent due to their versatile functionality.[1, 142] They can be either irregularly shaped or calibrated, natural or synthetic, and permanent or bio-degradable. Although non-calibrated particulates have historically been widely used and are still used clinically today (e.g. gelatin pledgets), the in vivo travel trajectory and occlusion level are usually unpredictable. Therefore, calibrated microspheres are often preferred due to their controllable size distribution and spherical shape, which may improve embolization outcomes. There is a limit on how small particulates can be when used for embolization. It has been reported that particles with a mean diameter of 9 μm can lead to pulmonary emboli in rabbits.[143] Overall, 40 μm is an accepted lower limit for particulate embolic agents.[144]
This section begins with natural and polymeric non-spherical particles, followed by a discussion on calibrated microspheres and recently developed multi-functional beads.
5.1. Autologous Clots and Tissues
Naturally autologous embolics include blood clot and muscle tissues. Different types of blood clots have been used for embolization: unmodified clots, heated clots (to retard clot lysis), aminocaproic acid modified clots (to resist fibrinolysis), oxidized cellulose modified blood, thrombin modified blood, as well as aged blood with sprayed tantalum powder for radiopacity.[145] Blood clots are temporary embolic agents that can last for a few days; their lysis and fragmentation can lead to distal migration.[16a]
Conversely, autologous tissues are considered permanent embolic agents, though recanalization may occur after 14–18 days.[145] Muscle and subcutaneous tissue are taken from patients just before the procedure.[145] Autologous tissues are then cut into small pieces to form a “meatball” and suspended into saline for injection.
Both autologous clots and tissues can be individualized for each patient and both are non-toxic.[145] The main advantage is their low cost and inherent biocompatibility. However, autologous embolic agents are rarely used in current clinical practice, given rapid advancements in particle embolization technology.[16a]
5.2. Polyvinyl Alcohol (PVA) Particulates
Polyvinyl alcohol (PVA) particles have been used since the 1970s.[146] They are prepared by mechanical fragmentation of the PVA polymer block followed by sieving to separate particles with different size ranges.[147] However, PVA particulates tend to aggregate because of surface charges and due to surface hydrophobicity, they also aggregate in physiologic solutions. This can lead to unintended occlusion of proximal larger vessels,[148], which is often disadvantageous when more distal embolization is desired. The main challenges of PVA particulates are associated with their irregular shape and lack of size precision, since their dimension does not necessarily correlate with the diameter of occluded vessels. These shortcomings give rise to unpredictable embolization behavior and even blockage of the delivery catheter.[149]
Non-calibrated PVA particulates cause immediate mechanical occlusion of blood vessels, followed by thrombus formation. The sequence of histologic events following PVA particle embolization can be summarized into three stages: 1) within the first few hours or days, thrombus forms around the trapped particulates; 2) in a few weeks, inflammation processes starts, and vessel remodeling initiates; 3) in a few months, recanalization may occur due to particle migration.[20] PVA particle is considered a permanent occlusion agent. Its long-term biocompatibility, tissue effects and bio-inertness are well established in humans, though recanalization may occur through old thrombi.[150]
5.3. Gelatin Embolics
Gelatin-based systems are biodegradable embolic agents that are currently available on the market for temporary embolization. Gelatin foams (e.g., Gelfoam®, Pfizer, New York, NY) are prepared from purified porcine skin gelatin (a non-antigenic carbohydrate) and have been widely used in endovascular procedures since 1964.[151] Gelatin foams are available in different configurations, such as sponge and sterile sheets, which can be cut into pledgets (1–2 mm). Other standard uses are gelatin powders, which range in size between 40 and 60 μm, and gelatin microspheres. Gelatin foams serve as hemostatic embolic agents; they can be opacified through mixing with contrast medium (e.g., iodinated contrast) to form a slurry for injection. Clinically, gelatin-based systems are used as pre-operational embolic agents to reduce subsequent blood loss during surgery. They have also demonstrated promising clinical outcomes in the treatment of HCC,[152] bone malignancies,[152b, 153] uterine fibroids (both mid-term and long-term)[154] and massive arterial bleeding.[155]
Gelatin powders lead to distal vessel obstruction and thrombus formation both within and around the emboli.[58, 145] They mechanically pack a lumen with smaller diameter to promote blood clotting. Their hemostatic ability is almost identical to that of fibrin when directly adhering to the bleeding surface. The coagulation time is reduced from 9.5 minutes to 6.2 minutes when gelatin powder is added to a whole-blood sample.[155a] Furthermore, the porous structure of gelatin foam serves as a scaffold to promote cell adhesion and tissue regeneration. When placed in soft tissues, it usually biodegrades completely within 4–6 weeks, although gelatin microspheres with a degradation period of 5 days have been designed for clinical trials.[152b] Gelatin embolization is considered to be temporary because of its enzymatic degradation. The degradable nature of gelatin matrix has been considered advantageous for some applications such as the embolization of internal iliac arteries and occluding hepatic arteries in chemoembolization.[156] However, because its degradation profile is unpredictable, gelatin-based embolization may lead to early recovery of blood flow or can lead to permanent occlusion.[52]
5.4. Calibrated Microspheres
Unpredictable embolization resulting from irregular shaped particles has led to the development of calibrated microspheres. Accurate particle size is crucial for localized targeted embolization since delivery of microspheres is driven by blood flow and their accumulation in vivo is size-dependent. For example, in tumor embolization, a deep, distal microvascular embolization is often desired. This requires consistently sized, small microparticles (45–150 μm) to penetrate the vascular bed. Once distal embolization is complete, more proximal vessels may be embolized with larger particles (150–250 μm). This strategy of tailored embolization results in the greatest degree of tumor necrosis.[58] Additionally, for preoperative treatment of meningiomas, relatively small particles (45–150 μm) are most effective, whereas uterine fibroids are mostly managed with larger particles (up to 900 μm).[157] Moreover, multiple therapeutics (e.g., gene and cell therapies) can be encapsulated into microspheres to induce cancer cell apoptosis and limit angiogenesis.[144, 158]
5.4.1. Trisacryl Gelatin Microspheres (TGMS)
The first commercially available calibrated microsphere embolic agents were trisacryl gelatin microspheres (TGMS) (i.e., Embosphere® Microspheres, Merit Medical, South Jordan, UT, licensed in the US in 2000). They are hydrophilic, compressible, and non-biodegradable. TGMS consists of a trisacryl matrix with embedded gelatin.[159] Trisacryl polymer has long been used as base material in chromatography column manufacturing for protein filtration due to its low porosity and biocompatibility.[159–160] The impregnated gelatin allows for cell growth and spreading.[159] TGMS can be synthesized from a reversed emulsion process, followed by wet-sieving for size selection.[159, 160b] TGMS obtained from this methodology appear spherical, exhibit smooth contour and are homogenously distributed. They are soft and adaptable for delivery using microcatheters with no tendency of clogging or forming aggregates.
Embosphere is commercially available with a wide range of size distribution, including 40–120 μm, 50–100 μm, 100–300 μm, 300–500 μm, 500–700 μm, 700–900 μm and 900–1200 μm. Marked inflammatory responses (e.g., giant cell response after 4 weeks) have been observed with smaller particles (100–300 μm) compared to larger microspheres (300–500 μm), due to small particles’ deeper penetration and their allogeneic overcoat.[161] Simple coloration has been incorporated to enhance visualization of TGMS during injection; EmboGold® Microspheres (Merit Medical, South Jordan, UT) are commercially available as TGMS doped with 2% gold.
Clinical evidence indicates that Embosphere microspheres lead to deeper penetration in meningioma embolization when compared to PVA particles, with less intraoperative blood loss.[148] Interestingly, on histology, Embosphere microspheres do not cluster but form chains in smaller vessels.[148] PVA particles tend to clump, block wider blood vessels and are associated with more intense necrosis compared to TGMS embolization.[162] No direct correlation was found between sizes of PVA particles and occluded vessel diameters. In contrast, there was a strong correlation between the sizes of calibrated TGMS and the diameter of occluded vessels. Similar results were also obtained when comparing PVA particles to Embosphere microspheres in UAE.[163]
5.4.2. PVA Microspheres
Calibrated PVA microspheres overcome some of the aforementioned disadvantages associated with PVA particles.[164] For example, Bead Block® (Biocompatibles UK Ltd) (sizes ranges: 100–300 μm, 300–500 μm, 500–700 μm, 700–900 μm and 900–1200 μm) is a type of PVA microsphere (PVA crosslinked with acrylic polymer) that is commercially available for embolization. Additionally, LC Bead® (Biocompatibles UK Ltd) and LC Bead LUMI™ (with intrinsic radiopacity) (Biocompatibles UK Ltd) have also been cleared by FDA for the embolization of hypervascularized tumors and AVMs.
5.5. Temporary Microspherical Embolic Agents
Biodegradable embolic materials are selected when temporary, non-permanent embolization is desired, often in cases in which re-intervention at a later date may be planned or when non-target embolization to normal issue is unavoidable.[165] Clinical indication for temporary embolization may include trauma, postpartum hemorrhage and gastrointestinal bleeding.[166] The typical aim of temporary embolization is to stop blood flow, allowing hemostasis and healing of injured blood vessels. As the temporary embolization agent gradually degrades, the blood vessels recanalize at a later stage and restore the normal blood flow.[166]
A variety of degradable embolic particles/microspheres have been developed.[167] In addition to fundamental microsphere properties (calibrated sizes, compressibility and resiliency), biodegradable embolic agents should also encompass the following design considerations: occlusion period, fate of the degraded fragment, intrinsic radiopacity and therapeutic loading capability.
5.5.1. Chitin and Chitosan Derived Microspheres
Chitin, a (β−1,4) polymer chain of N-acetyl-D-glucosamine, is the second most abundant natural biopolymer in the world after cellulose.[168] Due to its limited solubility in common solvents, chitin’s derivative, chitosan (poly(β−1,4-D-glucosamine) (by chitin N-deacetylation), is more commonly used in biomedical and pharmaceutical applications.[169] Both chitin and chitosan can be extracted from the crust of crabs and shrimps, and are highly biocompatible, biodegradable and antigenic. They are anti-oxidant, anti-inflammatory, thermosensitive and can serve as drug carriers for chemoembolization.[168–170] Additionally, chitosan has a hydrophilic surface to promote cell adhesion, proliferation and differentiation, without evoking intense foreign body reaction in vivo.[171] It degrades slowly into harmless gluocosamine that can be absorbed completely by the body.
The embolic effect and degradation profile of chitin and chitosan particles were assessed in the renal arteries of rabbits.[172] Compared to similarly sized PVA particles, chitin and chitosan particles (150–250 μm) were found to pack blood vessels more compactly, with less frequent capillary vessels formation within the embolized arteries, suggesting less chances of recanalization. Giant cells were observed 1–2 weeks after embolization (before particle degradation occurs) and continued to be present until week 32, which was after complete chitosan absorption at 24 weeks. Histologic examinations revealed deformed and fragmented emboli, some of which were gradually engulfed by giant cells via phagocytosis.[172] These observations suggest that the degradation of chitin and chitosan particle is likely governed by enzymatic hydrolysis, a major degradation route of carbohydrates.
The level of deacetylation and amount of amino groups contributed to biocompatibility, physicochemical characteristics and degradation profile of chitosan microspheres.[173] Uniformly sized chitosan microspheres (CM) (132, 260 and 429 μm) were prepared by the water-in-oil (W/O) emulsification technique and aceylated chitosan microspheres (ACM) were obtained by aceylating CMS with anhydride acetic.[173] ACMs have reduced bovine serum albumin adsorption compared to CMs; in addition, ACMs and CMs exhibit hemolysis rates of less than 1.2% (non-hemolytic) and 3.3% (slightly hemolytic), respectively, suggesting that ACMs are more hemocompatible than CMs. In aqueous solution, amino groups on chitosan are protonated in to −NH3+. Therefore, less positively charged ACMs can reduce the attraction of negatively charged red blood cell glycoproteins, thereby preventing cell membrane rupture and reducing the amount of subsequently released hemoglobin. At constant temperature, ACMs deemed to maintain a stable swelling ratio, which was not significantly affected by pH value as CMs were. Hence, ACMs may be more stable (e.g. less swelling-induced pressure on vessel walls) in response to in vivo environmental changes.
The in vitro and in vivo biocompatibility and degradation profile were also assessed for both CMs and ACMs.[174] ACMs underwent faster lysozyme degradation (40% residual mass at 8 weeks) compared to CMs (60% residual mass at 8 weeks) under identical in vitro conditions. Their implantation in rat gluteal regions did not lead to noticeable adverse effects. Histologic sections revealed gradual degradation of CMs and ACMs over a period of 24 weeks. Cell infiltration was observed at early stages (at both 1 week and 8 weeks), followed by granulation tissue deposition (16 weeks) with connective tissue replacing the microspheres completely beginning at 24 weeks. The embolic efficacy of ACMs was examined in a rabbit ear model, which exhibited complete occlusion and ischemic necrosis 15 days after ACM administration.[174]
Chitosan’s poor long-term stability and restricted shelf-life remain significant challenge that hinders its clinical applicability. Both intrinsic and external factors that influence chitosan stability have been identified. Intrinsic parameters include purity, molecular weight, polydispersity, deacetylation and water content. External factors consist of environmental factors (e.g., humidity and temperature) and processing factors (e.g., acidic dissolution, sterilization, heating and lyophilization).[169] The addition of stabilizer, nonionic polymers, as well as physical and chemical crosslinkers are strategies that have been employed to effectively enhance the physiochemical stability of chitosan. Proper storage condition is also critical to optimize its performance.[169]
5.5.2. Chitosan/Carboxymethyl Cellulose Microspheres
Bioresorbable chitosan (CN)/carboxymethyl cellulose (CMC) microspheres have been made via the reverse emulsion process and their degradation profile depends on the composition and degree of oxidation of CMC.[175]. The two naturally-derived, highly biocompatible materials form hydrogel beads through in situ intermolecular crosslinking via a Schiff base reaction between amino groups on chitosan and aldehyde groups on oxidized CMC.[171, 175] The CMC oxidation level, therefore, directly affects cross-linking density. This methodology avoids small molecular cross-linkers for reduced toxicity. The particles are able to deform as much as 33% of their original size and pass through a catheter with a smaller diameter without structural compromise.[171] Their highly porous internal structures are also beneficial for therapeutic delivery.[175–176]
In vitro studies demonstrate that CN/CMC microspheres crack and break into small pieces in lysozyme with tunable degradation rate at 37°C (ranging from 15 days to over 4 weeks, depending on oxidation level). The degradation mechanism is based on hydrolysis due to cleavage of Schiff bases in an aqueous environment.[171, 175] In particular, highly cross-linked (e.g., high oxidation level of CMC) microspheres exhibit a lower degradation rate compared to microspheres with low cross-linking densities.
In vivo occlusion level, degradability and biocompatibility of CN/CMC microspheres were investigated in a rabbit kidney model. Successful renal embolization was observed 73 days after the procedure. Mild tissue reaction occurred, possibly due to ischemia and degraded byproduct, while no vessel wall destruction was observed. Partial degradation was observed on day 7 and no particles were found at day 73, whereas proximal vessels were patent.[175] Moreover, no systemic complications (e.g. necrosis and neutrophilic inflammation) were reported at either 6 or 12 months after CN/CMC particles delivery.[177] Pathological findings suggested mild local tissue responses at both time points, and new vessel formation was observed in treated kidneys at 6 months. Biodegradable CN/CMC microspheres demonstrated no adverse long-term effects, highlighting their potential as safe agents for arterial embolization.
5.5.3. Hydrogel Microspheres with Hydroxylamine Containing Crosslinker
A class of degradable hydrogel microspheres were synthesized by homopolymerizing a variety of acrylic monomers and hydroxylamine containing crosslinker, namely N,N’-(dimethacryloyloxy)adipamide (C6NCL), for temporary embolization.[166] Some of the used monomers include 2-hydroxyetheyl acrylate (HEA), acrylamide (AAm) and acrylic acid (AA). The in vitro degradation period of formulated microspheres varied between 6 hours to 31 days (at 37°C, pH=7.4).[166] The degradation profiles were tunable by the type of monomer, crosslinkers and pH values (of both physiological and storage solutions). Faster degradation kinetics are usually achieved by reduced cross-linking density; however, this can compromise the microsphere mechanical properties (e.g. breakage of particles due to fragility). Instead, the new hydrolytically degradable agent, C6NCL, was composed of two degradation sites that offered relatively faster degradation rates while maintaining the microspheres with desired mechanical properties.[166] Among all the candidates, HEA microspheres, with sizes between 300 and 500 μm, were selected for further investigation. They exhibited in vitro degradation time of 22 days and no swelling after three months of storage.
The occlusion efficacy of HEA microspheres was examined in both canine renal arteries (up to 3 weeks) and rabbit median auricular artery (up to 2 weeks).[166] Biodegradable Human Serum Albumin (HSA) microspheres and permanent EmboGold microspheres with same sizes were also used for comparison. After renal embolization in canine model, small infarcts were visible in the kidney with temporary embolic agents (both HEA and HAS microspheres), whereas more pronounced abnormalities and moderate inflammatory reactions were seen in kidney treated with permanent microspheres. The degradable microspheres were found to be encapsulated at different stages of degradation and phagocytosis. HSA microsphere embolized arterial branches recanalized in 1 week while no recanalization was observed for the permanent EmboGold microsphere treated vessels. For HEA microsphere embolized renal arteries, the occlusion persisted for 1 week and blood flow reestablished at 2–3 weeks, which is similar to the degradation time of that to gelatin sponge.[178] This is a reasonable degradation period that provides sufficient time to achieve tumor ischemia, vessel repair and embolization.[179]
5.5.4. Microspheres with PLGA-PEG-PLGA Crosslinker
Another hydrolysable cross-linker, PLGA-PEG-PLGA, was used in synthesizing poly(ethylene glycol) methacrylate (PEGMA) microspheres through a suspension polymerization process.[179] The degradation of PEGMA is based on the hydrolysis of the ester bond in the crosslinker. Its decomposition rate is mainly determined by the molecular weight of polymethacrylate chains, size of lacoyl/glycolidyl segments, crosslinking density and hydophilicity.[179]
The subcutaneous implantation of PEGMA microspheres in rabbits demonstrated that they degraded to an advanced stage in two days; PEGMA microspheres degraded completely in seven days, which was before the onset of a chronic inflammatory response. Such rapid degradation and body elimination occurred because the molecular weight of polymethacrylate in formulated microspheres was below the threshold of 50kg/mol to be filtered out by kidney.[179–180] Mild inflammatory reaction, without lymphocytes or giant cells, was observed: neutrophils were found 2 days after implantation and microphages appeared after 7 days, corresponding to normal acute and progression phases of foreign body reactions.
5.5.5. Hydrophobic Linear Polymers Based Microspheres
In addition to hydrolysable cross-linker, hydrophobic linear polymers represent another class of hydrolytically degradable polymer material (e.g., PLGA, plyanhydridge, and polyesters), which can decompose into oligomers or monomers.[181] As an example, PLGA degrades into lactic acid and glycolic acid by hydrolysis of ester linkage.[182] However, PLGA microspheres are typically dense and non-compressible; they are not flexible enough to pass through a catheter with smaller inner diameter.[183] Although PLGA microspheres are commercially available (e.g., Occlusin® 500 Embolization Microspheres, IMBiotechnologies, Ltd., Edmonton, AB, Canada) and have been used for embolization, their decomposition is usually slow and gradual (i.e., on the order of months).[183b, 184]
In a sheep UAE model, 150–212 μm OCL 503 (type I bovine fibrillar collagen-coated PLGA microspheres) was observed with significant shape deformation due to partial degradation at 1 month, with fibrous connective tissue between and surrounding the particles. They degraded completely in 6 months, whereas the arteries remained fully occluded 6 months after the procedure due to fibrous connective tissue; however, recanalization was observed at 12 months. Additionally, OCL 503 was shown to rapidly attract platelets enhancing its stability of formed clots through platelet-platelet interaction and cross-linking between the microspheres. Though no complications were observed over the study period, long-term clinical benefits associated with OCL 503 microspheres cannot be identified and a more rapidly biodegradable microsphere may be favorable when repeated embolization is necessary.[183b]
5.6. Imageable Microspheres
Currently most available microsphere embolic agents are radiolucent and, therefore, are usually mixed with intravenous contrast agents for fluoroscopic imaging. However, the body rapidly clears contrast media, precluding their applications in long-term monitoring.[185]. Simple mixing of contrast agent (usually heavy metal or heavy metal salts) with polymer can cause leakage of the radiopaque agent due to the lack of primary bond formation. This not only deteriorates the physiomechanical properties of polymer matrix, but can also lead to systemic toxicity or complications such as non-target embolization. Moreover, detection of loosely mixed contrast media does not necessarily correlate with the actual location of microspheres and whether repeated embolization is necessary is also hard to assess.[144, 186] Therefore, intrinsically radiopaque microspheres are needed, as they would reduce the need for multiple injections of contrast agents, thereby reducing allergic complications and potential nephrotoxicity.
The most commonly incorporated radiodense agents are organoiodine compounds.[52] Ionic contrast agents (e.g., ioxithalamate) have been found to cause transient endothelial cell injury, while non-ionic contrast agents (e.g., iomeprol) do not lead to detectable endothelial damage.[187] Compared to nonionic contrast agents, ionic contrast media have greater biological effects and high osmolality, which may cause osmotic dilution and renal toxicity.[52] Besides iodinated contrast agents, radio-opacifiers, including barium sulfate,[188] silver iodide,[189] tantalum[190] and zinc-silicate,[191] have also been impregnated into microspheres to render them radiopaque to monitor embolization and long-term occlusion level. Additionally, multimodal visibility on fluoroscopy, MRI, CT and ultrasound are worth exploring for the development of multimodal microspheres as the next generation of embolic microspheres.
5.6.1. Iodinated Microspheres
There are two major approaches to introduce intrinsic radiopacity to microspheres through primary bonding: a) polymerization of radiopaque monomers and b) attachment of the radiopaque moiety onto the host polymer structure.[192] Iodinated benzyl based starting materials are widely used in either approach due to synthetic flexibility and high iodine content (Figure 7A).[193]
Figure 7.

A) (a) 2,4,5 triiodinated benzyl nucleus in commercial contrast media; (b)-(d) examples of iodinated vinylic monomers used for microsphere synthesis; and (e) 2,3,5, triiodinated compound for conjugation. B) (a) Schematics of microfluidics technique to fabricate alginate microspheres with in situ synthesized BaSO4 nanoparticles; (b) optical image of hydrated BaSO4/ALG microsphere and SEM image of a dried particle; and (c) BaSO4/ALG microsphere solution. C) Digital radiography of tantalum nanoparticle loaded calcium alginate particle (a-e) and blank calcium alginate particle (f-j) immediately, 1 week, 2 weeks, 3 weeks and 4 weeks of rabbit kidney after embolization. Figure components adapted and reproduced with permission.[193, 199, 202] Copyright 2016, Elsevier; 2015, American Chemical Society and 2018 Ivyspring International Publisher.
For example, the triiodobenzyl moiety can be linked to PVA-AMPS microspheres (sulfonate-modified acrylamide-polyvinylalcohol) via hydroxyl groups presented on the surface.[193] These microspheres, with sizes between 70 and 150 μm, were infused for embolization into hepatic lobar arteries in a porcine model. Subsequent CT scans confirmed no loss of the bead signal over a 90-day period. This suggested successful covalent attachment of iodine moieties onto the microsphere. Pathologic studies indicated initial inflammation, as expected in a typical foreign body response, whereas no chronic inflammation was seen. The microspheres were shown to be biocompatible by passing standard ISO10993 biocompatibility tests.[193] A pilot pig study suggested that a minimum of 20 wt% iodine was required to distinguish individual particles. However, the determination of whether to visualize single or clusters of microspheres depends on the application, and the radiodensity is also device specific.[157a]
5.6.2. Nano-on-Micro Systems
Another commonly used approach to render embolic microspheres visible on imaging modalities is to introduce contrast agents in the form of nanoparticles, which can be incorporated into the microparticle matrix. This methodology allows the microspheres to be visualized under several imaging modalities, including fluoroscopy, CT and MRI. The advantage of MRI is its high sensitivity in soft tissue evaluation and non-invasiveness, which does not expose patients to ionizing radiation. On the other hand, CT can provide additional diagnostic information, e.g. pulmonary reflux of embolic material.[194]
Iron Oxide Nanoparticle Based Microspheres:
Iron oxide nanoparticles have received attention as potential MRI contrast agents, due to their magnetic properties and biocompatibility.[195] Several formulations of iron oxide nanoparticles have been approved by the FDA for clinical trials. These include Feraheme® (vascular imaging), Feridex I.V.® (liver and spleen imaging), Gastromark® (bowel imaging) and Combidex® (lymph node imaging).[195b]
Chitosan microspheres (100–900 μm) loaded with superparamangetic iron oxide nanoparticles (SPIONs) (10–15 nm) have been synthesized for both embolization and MRI visualization.[196] The nano-on-micro formulation was made by ionotropic gelation and PEG leaching, in order to distribute SPIONs uniformly within the chitosan microspheres.[197] The addition of SPION particles did not alter the morphology, or the size of chitosan microspheres. Though Young’s modulus increased five times (477kPa versus 95kPa), the SPION chitosan microspheres were still sufficiently deformable. Freeze dried SPION-loaded chitosan microspheres contain approximately 18 wt% of SPION and a maximum of 11.8 mg/mL SPION content was reported in wet condition. Effective blockage of the right renal artery of rabbits was observed for up to 18 weeks after embolization, with concomitant decrease in the size of the embolized kidney and hypertrophy of the contralateral kidney. The particles remained visible on T2-weighted images after 18 weeks.
In another study, SPION impregnated poly(acrylic acid) microspheres were developed via an inverse suspension polymerization method.[198] Due to the addition of SPION particles during the polymerization process, the Young’s modulus of SPION-poly(acrylic acid) microspheres (75 kPa) was almost half that of plain poly(acrylic acid) microspheres (146 kPa), as a result of decreased polymerization degree. The SPIONs content in this formulation was shown to be 18.3 ± 2.0 mmol Fe, which was detectable in mice by a clinical 3.0T MRI scanner. Furthermore, embolization with SPION loaded poly(acrylic acid) microspheres led to local tissue necrosis in rabbit kidney embolization. Their MRI signals were found to be detectable 7 days post embolization. In clinical practice, the degree of contrast on MRI depends on the strength of the magnetic coil, as well as size, surface charges, presence of coatings and saturation magnetization of SPIONs.
Barium Sulfate Nanoparticle Based Microspheres:
A one-step droplet microfluidic technique was used to fabricate alginate microspheres containing in situ synthesized BaSO4 nanoparticles (Figure 7B).[199] The monodispersed droplets were generated as a result of competition between viscous and capillary forces. Alginate matrix provided a porous structure for even dispersion of BaSO4 nanoparticles. The sizes of microspheres could be tailored by varying the concentration of and flow rate between the dispersed and continuous phases. The final particle size was 250 μm and the diameter in the dried state was 60 μm (Figure 7B).
Whereas a concentration of 5 wt% BaSO4 loaded alginate microspheres (ALG) is sufficiently visible, higher BaSO4 content (e.g., 9 wt%) leads to microspheres brittleness and consequent rupture.[199] The minimum BaSO4 content of in microspheres to meet the visibility criteria is approximately 5 wt% for CT and 10 wt% for fluoroscopy. [200] Furthermore, BaSO4/ALG microspheres are visible fluoroscopically up to 1.5 hours after kidney embolization in rabbits, and continue to be detectable with CT after 14 days. Their embolic efficacy and performance are comparable to commercial calcium alginate microspheres based on pathologic studies. Their intrinsic radiopacity is beneficial for tracking and follow-up assessment of the fate of the embolic agents in vivo.
Tantalum Nanoparticle Based Microspheres:
Tantalum is another radiodense material to render microsphere radiopaque.[201] A one-step electrospraying method can be used to produce monodisperse and radiopaque tantalum nanoparticle-loaded calcium alginate particles.[202] Tantalum suspended sodium alginate mixture is sprayed into CaCl2 solution and solidified. Tantalum nanoparticles are homogenously embedded in calcium alginate matrix. Microsphere morphology and radiopacity is controlled by spinneret voltage, spinneret size, Ca2+ concentration and tantalum nanoparticle concentration. Optimized tantalum loaded calcium alginate microspheres have a diameter of 330 μm and 10% (w/v) tantalum concentration. In renal embolization of rabbits, radiolucent calcium alginate microspheres (without tantalum) were used to embolize the kidney; these were not visible fluoroscopically (Figure 7C). However, Tantalum loaded calcium alginate microspheres demonstrated successful embolization, which were visible in both fluoroscopy (i.e., digital radiography) and CT scans 4 weeks after the procedure, indicating their potential for real-time imaging and long-term assessment.
5.6.3. Microspheres with Multimodal Visibility
Modifications have been made to Embozene™ Microspheres (polyphosphazene-coated polymethylmethacrylate, Boston Scientific, Marlborough, MA) to achieve multimodal imaging with fluoroscopy, CT and MRI by impregnating iodine, barium sulfate and iron oxide through precipitation.[203] Pronounced change in particle visibility, as quantitatively indicated by signal to noise ratio, after porcine liver embolization was observed on CT (up to 200% increase), as well as on T1- and T2-weighted MRI (up to 80% decrease). Control particles (without contrast agents) had a 15% intensity change and were not visible. These signal measurements quantitatively evaluated the visibility of contrast agent modified Embozene microspheres and the multimodal visibility of these particles may offer improved monitoring on both CT and MRI for embolotherapy.
Nano-on-micro microspheres comprised of iodinated monomer 2-methyacryloyloxyethyl (2,3,5-triiodobenzoate) (MAOETIB) and magnetic nanoparticles were synthesized as dual imaging modalities for CT and MRI.[204] P(MAOETIB-GMA) microsphere core (diameter of 40–200 μm) was first prepared by suspension copolymerization of MAOETIB and glycidyl methacrylate (GMA). GMA was used during the emulsion process to create a hydrophilic surface, allowing subsequent nucleation and growth of γ-Fe2O3 nanoparticles to form a thin layer on surface of P(MAOETIB-GMA) microspheres. Hence, while the microsphere core is visible by fluoroscopy and CT due to the presence of iodine, the external iron oxide shell makes the particle visible in MRI. The bulk of microsphere contained 56.9 wt% iodine and the surface iodine content was 0.6 wt%. γ-Fe2O3/P(MAOETIB-GMA) microspheres were successfully used to embolize a rat kidney.[204] Signal changes were observed by both fluoroscopy and T2-weighted MRI. Additionally, the size of the signal change (70 μm) appeared to be correlated with single particle diameter.
Another type of magnetic nanoparticles, Fe3O4 nanoparticles, were impregnated into P(MAOETIB-GMA) microspheres following homopolymerization.[194] Nanoparticle dispersion was achieved by adding rabbit albumin to relieve the aggregation due to surface charges. Rabbit renal arteries were successfully embolized by Fe3O4/P(MAOETIB-GMA) microspheres, as confirmed by histologic studies, revealing particles located at various sites inside of the arteries with associated thrombus.[194] The microspheres were visible under three imaging modalities, including CT (hyperdense and punctuate areas), T2* and T2-weighted MRI (hypointense and darkening confluent areas) and fluoroscopy (focal, small hyperdense areas). Larger particles containing more iodine would likely perform better for fluoroscopic and CT imaging, but not necessarily for MRI, since the relative amount of iron oxide decreases as a result of the reduced surface-to-volume ratio of larger particles. Hence, the balance between the contrast compound within the core and surface should to be further tailored for optimum results.[194]
5.7. Drug Eluting Beads (DEBs)
Localized drug delivery and controlled drug release can improve therapeutic efficacy and safety. Targeted drug carriers are critical in cancer therapy although localizing therapeutic agents to a specific area remains challenging. For instance, doxorubicin, a common anticancer drug, is intrinsically toxic. Because it is rapidly cleared, it must be administered to patients with multiple injections, leading to cardiotoxicity. Conventional TACE delivers a mixture of chemotherapeutics (e.g., doxorubicin), alone or with cisplatin and mitomycin C, and lipiodol (as a contrast agent) into the targeted organ (e.g., liver), followed by embolization with bland agents (e.g., gelatin sponge) (Figure 8).[205] Radioactive beads (Figure 8B) are also used extensively for brachytherapy to induce local cell death [206]. However, radioembolization is beyond the topic of current review.
Figure 8.

Schematics of A) arterial blood supply to HCC and B) mechanisms of bland embolization, conventional chemoembolization, DEB chemoembolization, and radioembolization. Pharmacokinetic assay measured during 7 days in patients treated with DEB-TACE and conventional TACE. Doxorubicin level in serum at different time points posted C) DEB-TACE treatment, and D) conventional TACE procedure. Figure components adapted and reproduced with permission.[205, 208] Copyright 2013, Elsevier and 2007, Elsevier.
In the early 2000s, a variety of DEBs were developed.[192b, 207] The aim of DEBs is to deliver drugs over a longer period of time directly to the tumor while reducing systemic toxicity (Figure 8C and 8D).[208] Ideally, DEBs should have the following characteristics: visibility, targetability and capability of delivering multiple therapeutic modalities, as well as maintaining effective drug concentrations for prolonged periods of time. These characteristics should strongly increase the efficiency of the drug while limiting complications. The matrix of the drug carrier is critical as it controls the general release profile,[144] while the drug pharmacokinetics are dominated by the physiology of particular embolized tissues.[209] In addition to the cytotoxic effects of the drug, DEBs retard tumoral growth through ischemic cell death via feeding vessel embolization.[210] Combining DEB-TACE and anti-angiogenic therapy (e.g., sorafenib) is well tolerated and has demonstrated favorable disease control rate in phase II trials for patients with unresectable HCC.[211]
Overall, therapeutic drugs are either encapsulated during microsphere fabrication procedures (e.g., emulsion and precipitation processes) or they can be loaded onto the microsphere surface via ion exchange or swelling. In particular, ion exchange is the major drug capture mechanism for commercially available, non-degradable beads. This method does not compromise drug bioactivity.[209a, 212] Additionally, the drug release profile can be modulated by microsphere microstructure (e.g., porosity).[186] In the following sections, drug encapsulation mechanisms, release kinetics for DEB applications, as well as corresponding pharmacokinetics are discussed.
5.7.1. Ion Exchange Mechanism
PVA microspheres containing sulfonate groups (e.g., DC Bead®, Biocompatibles UK Ltd, UK) or sodium acrylate groups (e.g., HepaSphere™ Microspheres, Merit Medical, South Jordan, UT) are commercially available embolic agents to encapsulate drugs for TACE in treating HCC.[213] In particular, DC beads are made of sulfonate-modified PVA hydrogel, which contains negatively charged 2-acrylamido-2-methylpropanesulphonate sodium (AMPS) units throughout its structure to bind to positively charged drug. Specifically, cationic drug molecules (e.g. protonated doxorubicin) interact with the sulfonic acid group (R-SO3-)[181b] on AMPS for both loading and release through ion exchange mechanism. Up to 99% of doxorubicin uptake occurs between 30 minutes and 8 hours, depending on bead size.[214]
The recommended doxorubicin loading level is 25 mg/mL in hydrated DC beads.[152a, 215] The high drug-loading efficacy allows drugs to remain in the tumor for at least three months, promoting apoptosis and cell death. Due to the targeted and sustained nature of drug release, DC beads deliver 11.5-fold more doxorubicin to a tumor than conventional TACE, as seen in a rabbit liver model.[216] Furthermore, clinical studies demonstrated improved 6-month response (disease control, objective response and complete response) in patients treated with DEB-TACE compared to conventional TACE.[217] DEBs have also demonstrated effectiveness in the treatment both primary and metastatic liver cancer. The liver is the most common metastatic site for colorectal cancer, and hepatic metastases occur in over 50% of colorectal cancer patients.[218] Additionally, irinotecan-loaded DC beads were shown to be an effective and well-tolerated therapy in patients with unresectable colorectal liver metastasis.[219] These studies suggested that DEBs offer a new level of therapeutic consistency and patient tolerability.[220]
By activating a portion of hydroxyl groups on DC beads with 1,1’-carbonyldiimidazol (CDI), radiodense species can be attached to render the microspheres radiopaque.[213] These chemically modifiable and reactive hydroxyl groups can then be used for attachment of positively charged drugs (e.g., doxorubicin, ironotecan and topotecan).[213] The final formulated beads can contain an average of 45.6% iodine and exhibited doxorubicin loading capacity of 40–80 mg/mL, which is comparable to the maximum theoretical loading capacity of DC bead of 45 mg/mL.[215]
However, for commercially available DC beads (although not FDA-approved for use in USA) with negative surface charges, only cationic drugs (e.g. doxorubicin, irinotecan[221] and sunitinib[222]) can be loaded. Therefore, their release kinetics are primarily governed by the ionic environment. Additionally, the nonbiodegradable nature of the beads could also lead to late stage inflammatory responses due to persistent occlusion.[223]
5.7.2. Swelling Mechanism
Superabsorbent HepaSphere Microsphere, comprised of polyvinyl alcohol and sodium acrylate copolymer, is a newly developed embolic material for controlled drug release.[224] As the name “superabsorbent” suggests, these microspheres absorb up to 64 times of their volume at dry state.[225] This swelling behavior allows massive loading of aqueous solutions(e.g., contrast agent, drug solution and serum). With their negatively charged surfaces, these particles are ideal for encapsulating positively charged cancer drugs (e.g., doxorubicin and paclitaxel) in TACE. Currently, HepaSphere is not approved for use or sale within the US, although clinical trials have been carried out in other countries for TACE in HCC with promising results.[226]
In another example, ketoprofen was loaded into N-(Tris(hydroxymethyl)methyl)–acrylamide gelatin beads through swelling mechanism for pain relief after UAE.[48] The plain microspheres were synthesized through inverse suspension polymerization, followed by drying. The dried particles (sized range between 150 and 1430 μm) were then soaked in concentrated drug solution for ketoprofen loading. It was found that the drying method (lyophilization versus anhydrous ethanol wash) affected ketoprofen loading efficacy; lyophilized particles provided an overall higher loading capacity (79–122 mg/mL) compared to ethanol dried particles (61–106 mg/mL). This was due to increased porosity as a result of volume expansion and water sublimation during the lyophilizing process. Since ketoprofen cannot be loaded during polymerization reaction due to its insolubility in either water or paraffin oil, the dry-and-soak method appears to be feasible for ketoprofen encapsulation.[48]
5.7.3. Emulsification/Precipitation Mechanism
Well-dispersed adriamycin hydrochloride (ADM) loaded alginate-chitosan microcapsules (mean diameter of 77 μm) can be prepared by an emulsification-gelation process.[227] The two biodegradable and nontoxic components are polyelectrolyte polymers containing opposite charges. The positively charged amino groups on chitosan interact with negatively charged carboxylic acid groups in alginate to form microcapsules electrostatically.[228] Embolization using plain alginate-chitosan microspheres was tested in a rabbit kidney model.[227] The particles caused permanent coagulation necrosis of the kidney and ischemic necrosis in renal tubular cells by 4 weeks. The particles were identified in renal tubules up to 8 weeks after procedure.[227]
The maximum reported ADM encapsulation efficiency was 84% (with higher drug/carrier ratio), possibly because of enhanced electrostatic interactions between ADM and sodium alginate. Additionally, the interaction between alginates and chitosan contributed to the formation of a complex polyelectrolyte layer on microsphere surface.[229] This complex polyelectrolyte membrane became thick and tight with elevated chitosan concentration, which increased encapsulation efficiency accordingly.[227] In vitro ADM release profile was strongly dependent on the medium pH, but less so on chitosan concentration. For instance, ADM was more likely to dissolve in acidic solutions. As the microcapsule shrank in low pH environment, ADM was rapidly released by dissolution and diffusion via H+ ion exchange. [227]
Using alginate-chitosan as matrix, norcantharidin (NCTD) loaded microspheres have also been prepared via an emulsification-gelation method, and examined in a VX2 rabbit liver cancer model.[230] Sustained release of NCTD of over 20 days (with accumulated release up to 90%) was obtained in both small (60–120 μm) and large microspheres (120–200 μm); large microspheres exhibited more extended release, and small microspheres significantly slowed the tumor growth rate. Histologic results revealed the presence of small infarcts and necrosis in embolized livers with no metastasis, which occurred in the control liver. The tumor tissue around the beads entrapped within in tumor vessels appeared to be necrotic without giant cells. The profound necrotizing effect at tumor margins, where the tumor cells are most vigorous, suggested that these microspheres have high therapeutic efficacy.[230]
Microfluidics is an effective emulsion technique to produce monodisperse (uniformly sized) microspheres for embolization.[156, 207c] Polysaccharide alginate microspheres (~25 μm) consisting of both gold nanorods and magnetic clusters for dual MRI/CT imaging, and 6-methyxyethylamino numonafide (MEAN) can be loaded for chemoembolization, as demonstrated in a rat model of HCC.[207c] In this study, FDA approved biodegradable polysaccharide alginate polymers were used as the matrix due to their biocompatibility and mild gelation at room temperature, which is critical to the microfluidic gelation process. An oil mixture was used as continuous phase, and the aqueous phase consisted MEAN-magnetic clusters, gold nanorods, and alginate solution. Gold nanorods (33×8.6 nm) were synthesized with seed mediated method as CT imaging component. The magnetic clusters (~62 nm), synthesized via high temperature hydrolysis reaction, allowed for imaging on MRI. After shearing from microfluidic channel, the aqueous phase contacted the CaCl2 solution, solidified and formed alginate based microspheres via cross-linking gelation.
The presence of iron oxide nanocluster influences the MEAN release profile. With MEAN loaded magnetic clusters, MEAN release lasted for 2 weeks, with burst release of 47% by day 1. However, without iron oxide clusters, 90% MEAN released within a single day. This indicates that iron oxide nanoclusters effectively slow release rates down, due to porous structure generated among iron oxide clusters. The porosity also altered local field gradients and disturbed hydrogen atoms locally, which gave rise to strong T2 contrast (Figure 9A and 9B). After microsphere infusion, the tumor edges were enhanced (with negative contrast to noise ratio) in MRI compared to before infusion (Figure 9C). In addition, both MRI and CT have been used to monitor and track microsphere embolization (Figure 9D). The particles were found to preferentially accumulate at HCC tumor margins, rather than in normal hepatic tissues, confirming their super-selectivity.
Figure 9.

T2-weighted MRI A) pre- and B) post- transcatheter intra-arterial infusion of nanocomposite microspheres consisting of gold nanorods and iron oxide coatings in a rat HCC model (yellow circle indicates tumor location), showing enhanced tumor edge contrast. C) contrast to noise ratio (CNR) of tumor margin before and after microspheres embolization. D) Whole body coronal CT (left panel) and three dimensional maximum intensity projection (right panel) image after nanocomposite microspheres embolization, with circles highlighting regions with enriched signal. Figure components adapted and reproduced with permission.[207c] Copyright 2016, Nature Publishing Group.
5.7.4. Biodegradable DEBs
For biodegradable DEBs, drug release mechanisms are primarily governed by microsphere swelling and polymer degradation, and the release profile can be triggered by heat or enzymatic hydrolysis.[144, 209a] Choi et al. developed an embolic microsphere consisting of sorafenib, 2,3,5-triiodobenzoic acid (TIBA, for CT imaging) and a PLGA matrix, with diameter between 24.8 and 28.5 μm, for HCC embolization.[184a] The imageable drug-loaded microsphere was fabricated by oil-in-water (O/W) emulsification, with up to 58% sorafenib encapsulation efficiency and 5.11% (w/w) sorafenib content. The sizes were chosen to match the internal diameter of the terminal arterioles (10–50 μm) and capillaries (8–10 μm) in rat liver. The microspheres were delivered through intra-arterial administration and the iodine content (average of 23.15% (w/w)) appeared to be adequate for CT visualization. In biological fluids, PLGA matrix degraded gradually into lactic acid and glycolic acid via hydrolysis, which directly contributed to the sustained SOF release.
Biodegradable poly(D,L-lactic acid) (PDLLA) microspheres were loaded with either cisplatin or sorafenib or both drugs through an emulsion solvent evaporation method to produce particles with final diameter ranging between 200 and 400 μm (~60% yield).[223, 231] Drug loading capacity in cisplatin, sorafenib and cisplatin+sorafenib microspheres was 12.4%, 15.7% and 4.8%+7.3%, respectively. Dual drug-loaded microspheres are highly porous due to solvent extraction during microsphere formation, compared to dense PDLLA, cisplatin-PDLLA and sorafenib-PDLLA microspheres.[231] The porosity resulted faster in vitro release rates during the first 14 days compared to single drug-loaded particles. The particle morphology, drug distribution and release kinetics were all controlled by nature of polymer, polymer concentration, type of solvent, temperature, stirring speed, and pH of the environment.[232]
Degradation of PDLLA microspheres in aqueous solution typically involves three steps: 1) water absorption; 2) hydrolysis of ester bonds and 3) polymer erosion and bulk degradation.[223, 231] As a result of PDLLA degradation process, the drug is usually associated with tri-phasic release profile: 1) a burst-release phase (initial release from microsphere surface and diffusion through water-filled pores), 2) a slow release phase (diffusion through polymer matrix); and 3) a faster release phase (onset of erosion).[223, 231] All types of microspheres were shown to be intact in vitro after three months. Therefore, drug release during the first two weeks was more likely to be dominated by surface release, rather than bulk release due to polymer degradation.
Cisplatin is a powerful cytotoxic and cytostatic agent used to treat a variety of solid tumors.[233] It binds to DNA to facilitate apoptosis and cell death. Sorafenib is a multi-kinase inhibitor with anti-angiogenic and anti-proliferative properties for liver and kidney cancers.[234] It inhibits receptor kinases to block angiogenesis, tumor growth, and subsequent metastasis.[234b] In nude mice, cisplatin-loaded microspheres have no significant effect on tumor growth compared to plain PDLLA microspheres, whereas dual drug loaded (cisplatin+sorafenib) PDLLA microspheres are the most effective, inducing tumor shrinkage at day 28, demonstrating a combined cytostatic and antiangiogenic effect on solid tumors. The dual agent may destroy tumor cells and suppress tumor vessel growth to effectively inhibit tumors in situ.
5.7.5. Drug Concentration and Distribution in Tissues
Drug concentration and distribution in serum and tissue at different times after HCC chemoembolization have been characterized in rats,[184b] rabbits,[216, 222, 235] pigs[236] and humans[208, 226a, 237] for pharmacokinetic and safety evaluation. Furthermore, the drug release profile of DEBs has also been investigated. For example, small radiopaque DEBs (70–150 μm) travel more distally compared to large radiopaque DEBs (100–300 μm) in a porcine liver model (Figure 10A), whereas no substantial difference in doxorubicin distribution or penetration from the DEB surfaces was observed between these two groups (Figure 10B and 10C).[236c]
Figure 10.

A) MicroCT of normal porcine liver embolized with small and large DEBs. Doxorubicin concentration and penetration depth from the bead surface of B) small (70–150 μm) and C) large (100–300 μm) radiopaque DEBs in porcine liver over one week. Figure components adapted and reproduced with permission.[236c] Copyright 2012, Elsevier.
In patient liver explants, doxorubicin was detected in the adjacent tissue at 8 hours, 9–14 days and 32–36 days after DEB-TACE, with overall reduced amount of doxorubicin with increased time after embolization (Figure 11A and 11B).[237a] Concentration also decreased with increasing distance to bead surface, e.g., from 8.45 μM at the bead surface to 3.55 μM at a distance of 600 μm away from bead edge 8 hours post procedure (Figure 11A). In addition, for the liver explanted 8 hours after embolization, histology revealed the distribution of DEB both inside and outside the tumor, with an average penetration depth of 3.8 mm inside of the lesion and 4.9 mm away from the tumor edge (Figure 11C). As time elapsed from embolization, there was no significant change in tissue response around the beads. For 9–14 day liver explants, inflammatory-fibrotic tissue was observed in half of cases, with 37% cases showing coagulative necrosis around the bead, 8% cases of viable liver parenchyma and 2% cases of viable tumor. In 32–36 day liver explants, 40% of the beads were surrounded by necrosis, while the remaining beads were covered by inflammatory-fibrotic tissue.[237a] This study demonstrated the efficacy of DEBs, which offered controlled drug delivery for over one month and diffused over 1 mm from the surface, with local drug concentration above the cytotoxic threshold. Moreover, clinical trials demonstrate better safety and efficacy of using DEB-TACE for HCC, compared to conventional TACE with doxorubicin-lipiodol emulsion (Figure 8),[208, 238] with lower complication rates, decreased toxicity and increased tumor response.[239] DEB-TACE may also be effective in treating liver metastases.[240]
Figure 11.

A) Doxorubicin distribution around and away from the bead and B) doxorubicin concentration in patient liver explants at 8 hours, 9–14 days, and 32–36 days after embolization. C) Distribution of beads in peritumoral and intratumoral regimes in the patient transplanted at 8 hours. Figure components adapted and reproduced with permission.[237a] Copyright 2011, Elsevier.
For cancer treatment, in addition to chemotherapy, efforts have also made in gene therapy by incorporating peptides or genes into microspheres to modify DNA replication in cancer cells. As described, these microspheres would be delivered by catheters directly to arteries that supply the tumor. Combined gene therapy, chemotherapy and possibly immunotherapy may be considered in future embolic microsphere designs.
5.8. Other Notable Bio-Functionalized Microspheres
5.8.1. Thrombin-Functionalized Microspheres
Additional surface biofunctionalization, such as immobilizing the coagulation factor thrombin on microspheres, can be used to trigger blood coagulation. Intrinsically radiopaque acrylic microspheres were first synthesized from iodinated monomer via suspension polymerization.[241] The addition of methacrylic acid allowed further surface conjugation. Thrombin was then covalently bound to the surface through EDC/NHS (EDC: 1-ethyl-3-(−3-dimethylaminopropyl) carbodiimide hydrochloride; NHS: N-Hydroxysuccinimide) reaction, aiming to facilitate thrombus formation.[241] Both optical imaging and SEM confirmed the aggregation of thrombin-loaded particles by thrombus formation, compared to dispersed non-functionalized microspheres. Thrombin coated particles were able to induce clotting of blood within 6–8 minutes while non-coated microspheres required 12–14 minutes (Figure 12). Though excessive thrombin may form thrombus too rapidly and cause premature clotting, the surface density of attached thrombin can be tuned to achieve a desired clotting time frame.
Figure 12.

Comparison between thrombin-loaded and non-thrombin coupled iodinated acrylic microspheres in platelet rich plasma at various time points. Thrombin-loaded microspheres aggregate within 30 seconds while control particles (without thrombin) do not show obvious aggregation after 20 minutes. Figure components adapted and reproduce with permission.[157a] Copyright 2007, Elsevier.
Thrombin was also encapsulated into alginate calcium microspheres (TACM) through an electrostatic droplet technique, without the need of either high temperatures or cross-linking agents.[242] This approach allowed for the production of compressible and monodispersed microspheres with average diameter of 350 μm. Super-selective hepatic artery embolization was conducted in a canine model and was successful in all animals during 90 day observation period. No systemic inflammatory reactions were induced. The hemostatic mechanism of TACMs combined with mechanical occlusion of blood vessels and procoagulant activities induced by thrombin release from TACMs could reinforce particle/clot complexes.[242] TACMs were simple to fabricate, hemocompatible, biocompatible, and exhibited long-term hemostasis, revealing their potential as new hemostatic embolic material for interventional applications.
5.8.2. PEGylated Microspheres
The mononuclear phagocytic system (MPS) is known to effectively digest large particles through phagocytosis and PEGylation is a widely used technique to shield the microspheres from MPS uptake.[243] The protective PEG coating results in a prolonged blood circulation of the particles, thereby improving efficacy of delivery. Moreover, PEG is widely used in biomedical applications, such as drug delivery and cell encapsulation, due to its excellent biocompatibility, chain flexibility and hydrophilicity for reduced cell adhesion and protein adsorption.[179, 244]
Madani et al. demonstrated that PEG-coated trisacryl microspheres lowered the complement system activation rate by a third, compared to non-coated counterparts.[243] The polymer contained positively charged surface and tertiary amine groups, which enabled efficient and reproducible loading of the negatively charged anti-inflammatory drug, indomethacin, through an ion-exchange mechanism. Indomethacin loaded to PEG-coated trisacryl microspheres was found to suppress complement activation, revealing their promising biological performance. Moreover, PEGylated DEBs appeared to be efficient and safe in HCC treatment.[245] Although PEG coatings have been widely accepted to be non-immunogenic and non-antigenic, some studies have demonstrated that pre-existing anti-PEG antibodies exist in some healthy individuals.[246] These antibodies may lead to rapid blood clearance and reduced efficacy of PEGylated therapeutics.
5.9. Physical and Mechanical Properties of Microsphere Embolic Agents
Despite similar morphologies (e.g., spherical shape and calibrated sizes) of microspherical embolic agents, their physical (e.g., surface tension, charges, hydrophobicity) and mechanical (e.g., stiffness, fatigue resistance, fracture strength) properties vary due to differences in their chemical composition and manufacturing processes, which in turn influence microsphere/tissue interactions and clinical outcomes.[161, 247]
The deformability of microspheres is one of the major determinants of embolization performance. Ideally, the particles should deform and pass through the catheter, especially when the catheter is in a torturous vessel, and then recover their shape upon release. During an embolization procedure, microspheres are localized within the targeted region by catheter infusion, followed by size-dependent accumulation in blood vessels near the organ. Theoretical and computational models have been carried out to predict fluid-driven trajectories, and the effect of particle physical properties (e.g., size and density) on transport extent.[248] An average of 9%−18% deformation in diameter of microsphere agents in both sheep uterus and kidney models was reported.[249]
Typical physical/mechanical characterization of microspheres include size distribution, compressibility, suspension evaluation (i.e., time to achieve suspension and time maintaining suspension) and catheter deliverability.[165] Failure stress also needs to be assessed; fragmentation of microspheres under pressure could lead to non-target embolization and cause tissue necrosis.[212] In addition, fracture behavior is important to characterize for any new embolic microspheres. This is because during injection both compression and shear forces are imposed on the microsphere, which may cause fracture before reaching its maximum deformation limit. [171]
Compression tests are typically performed to evaluate embolic microsphere deformability by assessing their elasticity[224] and viscoelasticity[250]. Since testing the compressibility of a single microsphere is usually challenging, compressing a monolayer of particles is more assessable. The mechanical properties of four types of commercially available microspheres (Embosphere, EmboGold, Bead Block and Contour SE) of sizes 700–900 μm were evaluated with a monolayer compression test.[165] The compression force of Embosphere (TGMS), Embogold (TGMS), Bead Block (PVA crosslinked with acrylic) all fell into the similar range of 21–27.5 kPa while the compression force for Contour SE (PVA) was substantially lower (5 kPa).[165] Contour SE microspheres were easily squeezed by simple finger compressions; they collapse, become flat and do not recover their shape until several minutes later. This is primarily due to their internal porosity, which can be tuned by the ratio between PVA and gelling agent.[251] In terms of occlusion level, highly compressible Contour SE would have higher potential to travel more distally compared to less porous particles in similar size range. This phenomenon has been confirmed in both sheep uterus and kidney models, with higher degree of deformation associated with Contour SE compared to Embosphere of same size.[162, 249]
Hidaka et al. systematically performed compression tests on a single microsphere with a micromanipulator[224] and an additional relaxation test on particle monolayer with a texture analyzer in physiological saline.[250] In the single particle compression test (Figure 13A), Young’s modulus was obtained by fitting the Hertz contact model to linear elastic range of the loading curve (up to 25% deformation). The TG-ms (Embosphere, 700–900 μm) exhibit a Young’s modulus of 19 ± 5 kPa, significantly higher than that (9 ± 2 kPa) of superabsorbent Hepasphere (300–350 μm).[224] Histological evaluation revealed Hepasphere’s excellent vessel wall adaptability, with no intravascular cells identified between the particles and arterial endothelium.[252] From monolayer compression testing, Young’s moduli of TG-ms (Embosphere, 900–1200 μm), acrylamino polyvinyl alcohol microspheres (APVA-ms) (Bead Block, 900–1200μm) and polyphosphazene-coated polymethylmethacrylate microspheres (PP-PMMA-ms) (Embozene, 900 μm) were 40 ± 5 kPa, 19 ± 4 kPa and 14 ± 2 kPa, respectively (Figure 13B).[250] In embolized porcine kidneys, Hepasphere and Bead Block tended to travel distally, compared to Embosphere and Contour microspheres in the similar size ranges.[252]
Figure 13.

A) Compression testing on single TG-ms microsphere; Summary of B) modulus and C) relaxation half time of the TG-ms, APVA-ms, and PP-PMMA-ms obtained by compressing a monolayer of microspheres immersed in physiological saline. Figure components adapted and reproduced with permission.[224, 250] Copyright 2010, Elsevier and 2011, Elsevier.
Because most materials are not completely elastic, repeated compression and relaxation times are important to investigate for the recovery ability of embolic agents through stress relaxation tests, since the microspheres may undergo multiple compressions in the catheters and within blood vessels before reaching their destination.[212] Stress relaxation can be divided into two stages. During the initial stage, the resistance of microspheres decreases rapidly, due to the reduced hydraulic pressure resulting from water loss from the beads. This is followed by gradual decaying to an equilibrium state, where relaxation slows. The second stage, therefore, reveals the intrinsic viscoelastic characteristics of microspheres as a result of the deformation and rearrangement of polymer chains.[250] The relaxation half time, which characterizes the duration required for the force return to half of its initial value, for TG-ms, APVA-ms and PP-PMMA-ms are 52 ± 5 s, 60 ± 8 s and 31 ± 7 s, respectively (Figure 13C).[250] This suggests that Embozene returns to its equilibrium position faster than other types of particles. Embozene has considerably higher in vivo deformation than Embosphere, and travels more distally in renal and uterine vasculature in sheep.[253]
Less time required to distribute and the ability to remain in suspension in contrast agent are also important microsphere properties for clinical use. The four types of microspheres used in clinical practices (Embosphere, EmboGold, Contour SE and Bead Block) all exhibit times between 2 and 4 minutes to reach homogenous suspension in Isovue 300 contrast agent.[165] Particularly, Bead Block remains suspended for ~4.6 minutes, which is over twice as long as Contour SE (~2 minutes) and Embosphere (~2.5 minutes).[165] Particle density, surface properties and suspension solution all attribute to suspension time,[171] which need to be assessed on new particle agents to optimize microsphere/solution performance in a clinically relevant preparation time frame.
These results demonstrate the complexity of the physical and mechanical properties of microspherical embolic agents, despite similarities in their original morphological characteristics. Both elasticity and viscoelastic properties explain a microsphere’s flow behavior and its ability to adapt to the vessel wall, which is especially important to evaluate for DEB/vessel interaction in drug efficacy.[252] Furthermore, the addition of drugs alters the microsphere properties, including relaxation half time, suspension time and swelling behavior.[48, 212, 254] The accurate prediction between microsphere and blood flow would, therefore, facilitate efficient and safe use of particles to achieve desired occlusion, as well as assist agent selection to fit various therapeutic purposes.
6. Liquid/Gel Embolic Agents
There is a growing interest in liquid/gel embolic agents for peripheral interventions given their ability to penetrate into targets where catheters and coils cannot reach. Liquid/gel embolics block blood flow by forming permanent casts that do not depend on the patient’s coagulation system for thrombosis formation.[255]
For liquid/gel embolic agents, phase transformation of the material is typically required between administration (i.e., inside of the syringe and the catheter) and operating conditions (i.e., in the body). Low viscosity is desired to pass through the delivery system (e.g., syringe and catheter), while the formation of a more stiff and solid-like structure is required after exiting the catheter in order to achieve vessel casting. Such liquid- to solid-like (sol-gel) phase transition can be achieved via approaches categorized into physical crosslinking (e.g., precipitation and ionic cross-linking) and chemical crosslinking (e.g., polymerization).[256] In particular, the environmental difference between in vitro and in vivo conditions provides various triggering factors for certain physical mechanisms to take place, such as pH, ionic strength and temperature. Lastly, flow induced shear thinning effect (reduced viscosity with increasing shear rate) also enables the development of new embolic agents, regardless of environmental factors. A summary of liquid/gel materials in development is presented in Table 1.
Table 1.
Summary of liquid/gel embolic agents in development
| Liquid/Gel Embolic Agents | Mechanism | Special Delivery System | Radiopacity | Therapeutic Carrier | In vivo Embolization | Ref. |
|---|---|---|---|---|---|---|
| Iodine-containing cellulose mixed esters | Precipitation | No | Covalently bonded iodinated compounds | N/A | Sheep aorta | [257] |
| Iodobenzyl ethers of PVA | Precipitation | No | Covalently bonded iodinated compounds | N/A | N/A | [192a] |
| Yes | Iohexol | N/A | Rabbit pelvic vasculature and saccular aneurysm | [261] | ||
| BAB-type HEMA/NIPAAm triblock copolymers | Temperature triggered gelation | No | Iopromide | N/A | Porcine rete mirabile and kidney | [262] |
| N-isopropylacrylamide (NIPAAm)–N-propylacrylamide (NPAAm)–vinyl pyrrolidone (VP) terpolymers (PNINAVP) | Temperature triggered gelation | No | Iohexol | N/A | Porcine rete mirabile | [263] |
| No | Iohexol | N/A | Rabbit renal artery and VX2 liver tumor | [265] | ||
| P407-sodium alginate-HPMC-iodixanol hydrogel (PSHI-Ca2+) | Temperature triggered gelation | No | Iodixanol | N/A | Rabbit kidney and VX2 liver tumor | [266] |
| No | Iodixanol | N/A | Porcine splenic and gastric vessels | [270] | ||
| No | No | Doxorubicin, sorafenib | Rat McA-RH7777 liver tumor | [272] | ||
| No | Ethiodized oil or iohexol | Doxorubicin | Rabbit VX2 liver tumor | [275] | ||
| Poly(amino ester urethane) (PAEU) block copolymers | pH triggered gelation | No | Ethiodized oil | Doxorubicin | Rabbit VX2 liver cancer model | [276] |
| No | Iothalamate meglumine | N/A | Canine lateral wall aneurysm (temporary balloon protection) | [278] | ||
| Graphene-oxide (GO) enhanced generation five poly(amidoamine) dendrimers (PAMAM-5) hydrogel | Pre-activated chemical crosslinking | No | Iohexol | N/A | Rabbit subclavian arteries | [256] |
| Chitosan-doxycycline hydrogel | Temperature triggered gelation | No | Iodixanol | Doxycycline | Porcine renal artery | [279] |
| Carbazochrome-containing chitosan-PEG hydrogel (PCC) | Pre-activated chemical crosslinking | No | No | Carbazochrome | Rat kidney | [280] |
| Carboxymethyl chitosan and oxidized carboxymethyl cellulose based hydrogel | Pre-activated chemical crosslinking | No | Ioversol | Tetracycline, doxorubicin | Rabbit kidney | [176] |
| Dual-gelling poly(NIPAAm) based systems | Temperature triggered gelation and pre-activated chemical crosslinking | No | No | N/A | Porcine aneurysm (temporary balloon protection) | [281] |
| No | Docetaxel | Rabbit hepatic artery | [283] | |||
| Glyceryl monooleate (GMO) based liquid crystal | Water/Physiological fluid induced gelation | No | Tantalum powder | N/A | Rabbit renal artery | [284] |
| Polycationic salmine sulfate (Sal)-polyanionic sodium inositol hexaphosphate (IP6) coacervate (Sal-IP6) | Ionic strength difference (NaCl) induced gelation | No | Tantalum powder | N/A | Rabbit kidney | [285] |
| Polyphosphate based coacervates | Cation-induced phase separation | Yes | Sr, Ba | N/A | Rabbit auricular arteries | [286] |
| Gelatin and silicate nanoplatelets based shear thinning biomaterial (STB) | Flow-induced shear thinning | No | Iodixanol | N/A | Mouse femoral arteries; porcine lumbar artery, external iliac artery and forelimb veins | [10] |
In this section, both clinical and pre-clinical liquid/gel embolic agents are introduced, including the setting mechanism, settling time, change of mechanical properties upon phase transformation, therapeutic effects and embolic efficacy (if investigated) (Figure 1B). Finally, a brief design guideline of materials for liquid/gel agents is discussed.
6.1. Sclerosing Agents
Sclerosing agents include detergents, osmotic agents and chemical irritants that induce blood-vessel sclerosis. Ethanol is one of the most commonly used sclerosing agents for embolization.[20, 287] It leads to permanent endothelial cell injury, blood protein denaturation and also gives rise to vessel wall necrosis.[288] In AVM treatment, the maximum tolerated dose for absolute ethanol injection is 1 mL/kg to a maximum of 15 mL. Above this level, patients are at the risk for complications, such as respiratory depression, seizures and cardiac arrhythmias.[289] Ethanol is cost effective and accessible, though its high diffusivity limits its therapeutic window. A more viscous ethanol based emulsion (~0.55 Pa·s at 20°C) is commercially available as Ethibloc (Ethnor Laboratories/Ethicon, Norderstedt, Germany), which comprises maize protein zein, sodium diatrizoate, absolute ethanol and oleum papaverius.[20] It is a radiopaque agent for AVM treatment,[290] though its use has decreased in the last 10 years.[20]
6.2. Droplet Agents
Lipiodol (Ethiodol), in the form of ethyl esters of iodinated fatty acids from poppyseed oil, is an extensively used radiopaque liquid contrast agent. It contains 38 wt% iodine and has viscosity of 34–70 mPa·s at 20°C, slightly denser than water. Due to its rapid diffusivity, lipiodol penetrates the capillary bed evenly, and may serve as a capillary embolic agent to achieve capillary stasis.[291] However, due to blood scouring and tissue clearance, lipiodol is rapidly eliminated by the body and vascular recanalization occurs, lowering its embolic efficacy.[265]
6.3. In Situ Polymerization - nBCA
N-bnutyl-2-cyanoacrylate (nBCA) (e.g., TruFill), commonly known as “glue,” is a clear, free-flowing liquid used for embolization since the mid-1980s.[292] It is also widely used in wound healing since it provides an antimicrobial barrier with 99% efficacy for at least 72 hours.[11b] nBCA has relatively low viscosity of about 10 mPa·s.[293] It adheres to tissue by in situ polymerization upon contact with tissue fluid, an ionic and high pH environment, creating a glue effect. The polymerization process is anionic (induced by OH−) and exothermic (released heat is up to 80°C and can be very painful).[20] Due to its rapid polymerization upon contact of body fluid (in seconds),[294] nBCA may not adequately penetrate the tumor bed or reach distal vessels but is excellent for embolizing branches that are 500 μm or greater.
Clinically, nBCA has been mainly used to manage high-flow vascular malformations.[33] Its high thrombogenicity and excellent hemostatic properties require only a minimal injected volume for occlusion, thereby reducing embolization/fluoroscopy time and radiation exposure.[33, 295] The permanent embolic nature of nBCA is effective for permanent vascular embolization, although it is difficult to control. Hence, its administration requires more experienced operators as unpredictable polymerization may lead to non-target embolization, especially in high-flow regions. As an adhesive liquid casting agent, nBCA demonstrates adequate mechanical strength and homogeneity similar to tissue adhesives.[296] Proper dilution of nBCA and thorough flushing of the delivery catheter with nonionic solution (e.g., dextrose) prior to injection is necessary.[58] Additionally, its adhesive nature requires catheters to be withdrawn immediately after nBCA injection to avoid adhering to vessel walls. Since nBCA is radiolucent, lipiodol or tantalum microparticles are usually added for radiopacity and lipiodol has been shown to slow down the solidification time of nBCA.[255]
6.4. In Situ Precipitating Liquid Agents
6.4.1. EVOH/DMSO
In situ precipitation gelling solutions are composed of a polymer in a solvent. After the solvent is replaced by body fluids, the polymer precipitates and forms a cast.[20, 297] The first gelling solution consisted of ethylene vinyl alcohol (EVOH) polymer and dimethyl-sulphoxide (DMSO) solvent. It was developed in 1990 and revolutionized liquid embolic agents given its major advantage of non-adhesiveness.[298]
EVOH/DMSO forms a slightly viscous polymer suspension. When delivered inside the body, DMSO rapidly diffuses into the blood and EVOH precipitates. EVOH polymer hardens on the outer surface first, gradually gelling towards the core over several minutes. The viscosity of EVOH/DMSO liquid is adjustable by varying the concentration of EVOH, and its viscosity increases when more DMSO diffuses out. The tunable initial viscosity of the mixture is beneficial for tailoring occlusion level.[293] Since precipitation is due to the loss of solvent, not polymerization, its solidification time (10 minutes) is relatively longer than that of nBCA (few seconds),[299] allowing extended delivery in a controlled manner.[58] The slow solidification time allows EVOH to penetrate the tumor bed for percutaneous embolization. Similar to nBCA, EVOH completely obstructs blood vessels by mechanical casting, which is independent of thrombus formation.
The major limitation of the EVOH system is associated with the use of organic solvent. DMSO has exhibited both local and systemic cardiovascular toxicity. In addition, since DMSO is as a powerful solvent of plastic polymer, specialized and costly DMSO-compatible catheters are needed. It is important to note that rapid injection of DMSO containing gelling solutions induces vasospasm and can cause vascular toxicity.[300] Therefore, a slow injection pace (2–4 mL/min in the peripheral circulation and 0.6 mL/min in the cerebral circulation) is critical and required.[301] One alternative organic solvent to replace DMSO is n-methyl-2-pyrrolidone (NMP), which produces less vasospasm than DMSO when used as carrier for precipitating liquid embolics.[302] NMP-dissolved iodinated PVA liquid agent can effectively embolize aneurysms.[303] Another alternative is water-soluble liquid agent, such as hydrogels, which contain over 90% water and have are highly absorbent polymeric networks that exhibit the advantages of both liquid and solid embolic agents.[256] The gel composition can be tuned to achieve good injectability and strong mechanical properties for embolization, without complications associated with organic solvents.[304]
One commercially available product based on EVOH gelling solutions is Onyx (Onyx, Micro Therapeutics, Inc., Irvine, CA), mainly used in brain and pulmonary AVMs, as well as in pseudoaneurysms.[305] Onyx was FDA-approved for cerebral AVM treatment in 2005.[31] It was shown to block vessels down to 5 μm compared to 20 μm vessel occlusion achieved by nBCA, demonstrating Onyx’s ability for deeper penetration.[306] Onyx is also easier to manipulate compared to nBCA.[307] Both agents cause inflammatory reactions,[306] but Onyx gives rise to smaller inflammatory areas, increasing tolerability for patients.[308]
The radiolucency of EVOH/DMSO requires the addition of contrast agents (e.g., tantalum powders) for fluoroscopic visualization. However, upon rapid release of organic solvent, the originally entrapped contrast agent (e.g., tantalum powders) may precipitate in the catheter or slowly release to surrounding areas. Additionally, the long-term fate of leached contrast agent is also concerning, and multiple injections of contrast agents are often needed. Hence, the development of leach free liquid embolic agents represents major progress, as discussed in the next section.
6.4.2. PHIL™
Recently, a newly developed precipitating hydrophobic injectable liquid (PHIL™) (MicroVention, Aliso Viejo, CA, USA) with intrinsic radiopacity was introduced for endovascular use. PHIL consists of nonadhesive copolymer, polylactide-co-glycolide and polyhydroxyethylmethacrylate, in DMSO solvent.[307, 309] Similar to Onyx, PHIL diffuses into blood and precipitate into a cast. It solidifies from the outside to the inside within 5 minutes [307]. The major advantage of PHIL over Onyx is its intrinsic radiopacity due to chemically bound iodine component (triiodophenol), which produces considerably less artifact on CT compared to tantalum-based embolic materials.[310] Furthermore, PHIL is ready to use and does not require the additional 20 minute contrast agent mixing time as needed in Onyx preparation.[8]
The embolization characteristics of PHIL are similar to those of Onyx.[309a] Additionally, both PHIL and Onyx cause moderate disintegration and mild inflammation of embolized blood vessels during a 7-day period, due to both contained active substances and interactions with DMSO.[309a] However, the use of PHIL does not lead to angionecrosis, which has been observed after Onyx treatment of human cerebral AVMs.[309b] Additionally, PHIL has been shown to penetrate 2.9 μm blood vessels while Onyx may only penetrate 5 μm vessels.[309b] PHIL is clinically available for embolization in Europe and is used to treat intracranial dural arteriovenous fistulas and cerebral AVMs in patients.[307, 310–311] Its feasibility and effectiveness in aneurysm embolization were previously investigated in a canine model.[312]
6.4.3. Other Radiopaque In Situ Precipitating Liquid Embolic Agents
The incorporation of leach free contrast agents into liquid embolic agents has become a major area of study. In addition to PHIL, other iodinated in situ precipitating polymeric liquid embolic agents have also been developed. For example, Mottu et al. verified the effectiveness of iodinated cellulose based polymeric precipitating solution in cerebral malformations in a sheep model.[257] The degree of radiopacity was shown to be affected by the amount of iodine content and steric effects.[313]
Leach free radiopaque iodinated polymeric agents can be made by attaching iodobenzyl bromides covalently to PVA backbones via the Williamson etherification reaction.[192a] Its chemical stability, due to ether linkage, resists hydrolysis. However, their iodobenzoyl esters are prone to hydrolysis and consequent fragmentation. The newly formed iodinated polymer, which contains 40–70 wt% of iodine, can be prepared either in NMP or DMSO. Its viscosity generally increases with increasing molecular weight of PVA; however, all synthesized iodinated PVA liquids exhibit much lower viscosity than Onyx with the same polymer concentration. For instance, 50 wt% iodinated PVA solution has viscosity up to 1.8 Pa·s, which is difficult to inject; however, this value is still much lower than the viscosity of Onyx 500 (~2.25 Pa·s) with 20% EVOH. Suspension of PVA polymers with high molecular weight formulations form a cohesive mass through precipitation within 3 minutes, similar to the time frame for Onyx. With relatively low viscosity, the radiopaque iodinated PVA/DMSO/NMP liquid has the potential for AVM embolization with a tunable degree of occlusion by tailoring the polymer’s molecular weight, concentration, degree of radiopaque group substitution, and the type of organic solvent.[192a]
6.5. Ionically Crosslinked System - Calcium Alginate
Calcium alginate, a two-component biocompatible polymer system, demonstrates potential as an embolic agent due to its high mechanical strength in its solid state and low viscosity in its liquid form.[314] Calcium alginate gel (ALGEL) (Neural Intervention Technologies, Ann Arbor, MI) is a relatively new embolic agent developed primarily for neuroendovascular procedures.[31]
Alginates are a class of water-soluble salts derived from alginic acid, a natural polysaccharide gel found in brown seaweed. [314–315] They have been used for polymer films,[316] cell encapsulation,[317] wound dressings[318] and surgical sponges.[319] Alginate consists of blocks of mannuronic (M) and guluronic (G) acids in various configurations along a polymer chain (Figure 14A).[314] Molecular weight, M/G ratio, block length and block arrangement are the key factors that govern the elastic moduli and degradation rate of alginate as well as its resultant gels.[320] Particularly, the M/G ratio contributes to the structural and biocompatible characteristics of alginate. The purity of alginate also governs biocompatibility, since contaminants and proteins (phenols and endotoxins) lead to undesirable immune responses.[258, 321] Purified alginates can be compressed up to 40% without significant loss in elasticity.[258]
Figure 14.

A) Alginate structure and reaction mechanism with divalent calcium ions, transitioning from liquid to solid gel states. B) Strength and polymer yield percentage of the four alginates, with different compositions, at 40% and 60% compression levels. C) Concentration dependent viscosity of four types of alginate (at room temp. 28°C) versus alginate concentration. D) Flow rates at different catheter diameters for non-Newtonian alginate and Newtonian glycerin solution with similar initial viscosities, injected at a maximum pressure of 2100 kPa. The shear-thinning properties allow alginate flow at wider ranges compared to Newtonian fluid with same viscosity. Figure components adapted and reproduced with permission.[258] Copyright 2000, John Wiley & Sons Inc.
Water-soluble alginate can be ionically cross-linked with nontoxic divalent cation solution, such as calcium chloride (Figure 14A).[258] The calcium ions bind to the G-blocks of alginate strands and form a stable gel. G-blocks are the most important fragments in intermolecular cross-linking with divalent cations (Ca2+) at both acidic and neutral pH to form gel, whereas M-blocks only support acidic gel formation.[315] The addition of Ca2+ into alginate solution replaces the weak polar interactions between the polysaccharide chains with stronger cation-induced electrostatic interactions. For example, Ca2+ replaces the Na+ in sodium alginate and connects two polymer strands. The cross-linking by Ca2+ makes the gel significantly stronger but less elastic. The gelation kinetics and rheological properties are tunable by adjusting the calcium content, polymer concentration and gelation temperature.[322]
Becker et al. found that G/M of 66/34 provides the alginate gel, namely PHG, with optimum strength and injectability (Figure 14B).[258] PHG exhibits the best strength, highest polymer yield (high G-block composition increases polymer yield) (Figure 14B) and low viscosity (<250 mPa·s with concentration <3 wt%) of all composed gels (Figure 14C). PHG alginate could be compressed to 66 kPa, which is five times the cerebrovasular pressure (13 kPa).[258] A 38% reduction occurs in viscosity of 2.5 wt% PHG with increasing shear rate (158 mPa·s at low shear rate 10 s−1 versus 98 mPa·s at high shear rate 2000 s−1). A 15% decrease in viscosity at low shear rate (10 s−1) occurs (158 mPa·s at 28°C and 135 mPa·s at 37°C) when temperature is elevated from room temperature to body temperature.[258] Alginate concentration that is too high renders the solution too viscous to inject. Whereas the intrinsic shear thinning property of alginate mixture allowed it to be injected at a flow rate that was significantly higher than Newtonian fluid (i.e., viscosity remains constant, independent of shear stress, at constant temperature) with the same viscosity (Figure 14D).
The biocompatibility and embolization properties of calcium alginate were tested in rat kidney capsules and rabbit kidneys, respectively.[258] PHG alginate (2.5 wt%) (via venous injection) and CaCl2·2H2O (0.68M) (via arterial injection) were delivered from opposite directions (i.e., bi-directional injection) into the vascular bed of a rabbit kidney and reacted in situ. A stable polymeric plug formed, stopping all flow out of kidney and maintaining vascular occlusion during a one week study. Histologic analysis reveals that PHG gel is biocompatible and does not cause vessel wall injury.
The effectiveness of calcium alginate gel has also been examined in a porcine aneurysm model.[259] The gel entered the aneurysm in a strand form and then formed a cohesive mass.[323] Complete aneurysm filling was achieved (90%−100%) when the gel was used with temporary balloon protection across the aneurysm neck. This filling percentage was difficult to achieve with either coils or Onyx. One month following embolization, a minor bioactive response was observed, with fibrous tissue across the aneurysm neck excluding the sac from the parent vessel. Cerebral aneurysms completely healed within 90 days with no tissue necrosis or neointimal growth into the parent vessel. No migration or dislodgment of calcium alginate gel was observed three months after embolization. Additionally, evaluation of ALGEL for AVM treatment was performed in a porcine model for 1 week and 6 months respectively.[260] During 6-month long-term study, increased stability of ALGEL was observed and no tissue damage was seen.
The main advantage of the two-component calcium alginate system is its rapid in situ gelation when released from the catheter without depending on thrombosis formation for occlusion. However, separate delivery of alginate and calcium ions is required for the system, e.g., concentric/dual-lumen catheter, to prevent polymerization inside catheter. Thus, meticulous clinical experience is essential for this technique, limiting its use.[31, 323]
Lastly, a mixture of enzyme alginate lyase and ethylenediaminetetraacetic acid (EmboClear) was also developed to selectively dissolve calcium-alginate gel (EmboGel).[261] As opposed to ALGEL, EmboGel contains iohexol which renders the calcium-alginate gel radiopaque.[324] Following EmboGel administration to occlude pelvic vessels in a rabbit model, EmboClear solution was administrated, dissolving the gel within 1 minute.[261] The initially occluded arteries then regained patency. EmboClear has been used successfully to dissolve EmboGel in aneurysms and in the craniocervical circulation in rabbits.[261] Theoretically, the EmboGel/EmboClear system could reverse non-target embolization, which would represent an innovative safety feature.
6.6. Temperature-Induced In Situ Gelling Systems
Thermally responsive gels take advantage of temperature-induced material property changes between room and body temperatures for embolization.[262] The liquid embolic agent ideally possesses low enough viscosity during injection and forms a gel with sufficient strength in response to elevated temperature at the target site inside of the body to impede flow. For example, a temperature-sensitive N-isopropylacrylamide (NIPAAm)-based system has been extensively investigated for embolization (e.g., AVM).[262–263, 325] In this section, a few major classes of thermosensitive materials proposed for embolization are presented.
6.6.1. PNIPAM Nanogels
Nanogels are formed from polymer nanoparticles and the crosslinked, highly hydrophilic network has the ability to hold large amounts of water without being dissolved.[326] The unique properties and versatile utility of nanogels, such as their stimuli-responsive behavior and their ability to deliver small molecule therapeutics, even under harsh conditions, demonstrate their potential as the next generation of functional materials for biomedical applications. As a result of their spatial microtopological structure, nanogels exhibit more prominent shear-thinning properties compared to corresponding bulk polymers. Additionally, their porosity allows for a high drug loading capacity and sustained release, and they may serve as injectable bioactive scaffolds.[327]
Zhao et al. evaluated the potential of p(N-isopropylacrylamide-co-butyl methacrylate) (PNIPAM) based nanogel as a new embolic agent due to its temperature sensitivity.[265] Pure PNIPAM gels above 46 °C, which is above body temperature, whereas the addition of methylacrylate reduces this gelation temperature to 36.5°C. The newly formed nanogel, namely PIB, travels quickly inside of blood vessel due to good diffusivity. The subsequent vessel occlusion is achieved by the formation of a three dimensional hierarchical gel network due to hydrophobic interactions above phase transition temperature.
Iohexol has been blended into translucent PIB through physiosorption for fluoroscopic visualization and the radiopaque gel, namely PIB-I-6150, reduced the sol-gel transition during nanogel formation while maintaining a similar gelation trend of PIB.[265] Phase 1, 2 and 3 represent its swollen-gel phase, flowable-gel phase and shrunken-gel phase, respectively (Figure 15A and 15B). Figure 15B demonstrates that PIB-I-6150 possesses low enough viscosity in sol phase (storage modulus, G’< loss modulus, G”) for delivery and high strength (G’) in gel phase (G’>G”) at body temperature. The gel point (sol-gel transition point) can be evaluated from loss tangent, tanδ, a ratio between G” and G’ to measure the viscous component to elastic portion. The gelation time can be tailored by temperature change (ΔT) experienced by the gel and larger ΔT leads to the slower sol-gel transition kinetics. For instance, the gelation time for ΔT at 32°C, 13°C and 7°C was found to be 192s, 113s and 77s, respectively.
Figure 15.

Sol-gel transition behavior of temperature sensitive PIB-I-6150 A) phase diagram and B) storage modulus, loss modulus and loss tangent. C) Digital subtraction angiography (DSA) images of VX2 rabbit tumor embolization. (a-c) are DSA images of PIB-I-6150 treated group before embolization, 7 and 14 days post embolization, respectively. (d-f) are DSA images of Lipiodol treated group before embolization, 7 and 14 days post embolization, respectively. D) Schematics of Ivalon, Lipiodol and PIB-I-6150 embolization. Figure components adapted and reproduced with permission.[265] Copyright 2011, Jon Wiley & Sons Inc..
The embolic effect of radiopaque gel PIB-I-6150 was studied in rabbit renal arteries and compared to clinically used Ivalon (PVA) and lipidol (Figure 15C and 15D). [265] Flow recovery and collateral circulation occurred in renal artery treated with Ivalon after 35 days, while PIB-I-6150 remained stable, with no recanalization after 60 days. Ivalon’s incomplete embolization was attributed to disordered packing in vessels, whereas nanogel resisted blood scouring and tissue clearance. For tumor embolization, both lipiodol and PIB-I-6150 nanogel resulted in immediate vascular occlusion in VX2 liver tumors in rabbits. However, recanalization occurred 2 weeks after embolization with lipiodol, but not with PIB-I-6150 (Figure 15C). Histological analysis revealed better tumor necrosis, lower tumor growth rate, smaller tumor nests and no surviving tumor cells in PIB-I-6150-treated tumors, compared to lipiodol-embolized liver. These results suggest that nanogel successfully inhibits collateral circulation and vascular recanalization.
In a rabbit kidney model, the penetration depth of iohexol/PIB gels was shown to be dependent on injection rate.[264] Slowly injected (e.g., 0.05 mL/s) gel reached the segmental and interlobar arteries while rapidly injected (e.g., 0.15 mL/s) gel penetrated interlobular arteries, afferent arterioles and glomerular capillaries.[264] Flow conditions at the injection site (e.g., high flow AVM versus the terminal circulation of a main renal artery) also affected the distribution of PIB gels. Homogenous and persistent occlusion of rabbit kidney arteries was observed during a 3-month observation period. The durable embolization may be attributed to the addition of iohexol contrast agent, which forms hydrogen bonding with nanogel particles, strengthening the embolic’s structure.[328] No granulomas, neovascularization, recanalization or vessel wall destruction was observed,[264] contrary to findings with PVA particles[329] and nBCA.[330] The bio- and hemo-compatibility of PIB based nanogel suggests its utility for permanent and peripheral embolization.[265]
6.6.2. Poloxamer 407 and Alginate Based Composite Hydrogel
Thermosensitive liquid embolic hydrogel (PSHI) consists of poloxamer 407, sodium alginate, hydroxymethyl cellulose and iodixanel, and was synthesized for liver cancer therapy.[266] Poloxamer (or Pluronic) is a triblock copolymer of polyethylene oxidea–polypropylene oxideb–polyethylene oxidea. PSHI flows at 4°C and forms into a gel when introduced into the body.[331]
The exact sol-gel transition temperature of PSHI depends on the concentration of poloxamer, which remains a monomer in liquid and self-assembles into micelles for gelation.[332] The sol-gel transition time of PSHI varies from 30 s to 140 s, depending on the concentration of components, the addition of contrast agent (e.g., 23% v/v iodixanel) and temperature differences. Additionally, the storage modulus and viscosity of PSHI are influenced by the concentration of poloxamer 407 and are strongly affected by temperature. For 18% poloxamer solution, the storage modulus at 25°C and 37°C was 0.08 Pa and 10MPa, while the viscosity at these two temperatures was ~ 0.2 Pa·s (25°C) and 3000 Pa·s (37°C), respectively.
Though PSHI gel is stiffer at higher temperatures, it undergoes significant erosion (completely dissolution after 250 minutes).[266] To address this challenge, Ca2+ was introduced into PSHI and formed PSHI-Ca2+ gel with improved strength. Its weight remained unchanged for 5 hours, suggesting Ca2+ hinders the erosion of PSHI in the aqueous environment. This may be achieved by diffusion of Ca2+ through the Poloxamer 407 micelle for the formation of a stable three-dimensional network.
PSHI-Ca2+ gel occluded normal rabbit blood vessel for 7 days, whereas PSHI led to recanalization 30 minutes after embolization. Furthermore, complete occlusion was achieved with PSHI-Ca2+ with concomitant disappearance of tumor (VX2 liver cancer). Tumor cell apoptosis and necrosis were observed histologically. Conversely, both saline- and PSHI-treated groups had no significant differences before and after embolization, with tumor still persisting in both cases. This study suggests the embolic efficacy of PSHI-Ca2+ gel, whereas its injection procedure remains to be optimized, as indicated by the authors.[266]
6.6.3. Chitosan-Based Thermosensitive Materials
Chitosan/ β-glycerophosphate (β-GP) solution was first introduced in 2000 as a new class of material that underwent sol-gel transition at body temperature.[333] Pure chitosan solution is highly pH sensitive and precipitates once the pH exceeds 6.2, a major limitation to its biological application. Glycerophosphate, a weak base, is naturally found in the body and its intravenous administration is FDA-approved.[267] The addition of β-GP adjusted purely pH dependent chitosan solution to become a temperature-controlled and pH-dependent mixture.[334] For example, chitosan/β-GP remains a liquid in the physiological pH range (6.8–7.2) at low temperatures and transitions into a gel when trigged by body heat[333].
Unlike divalent anions (e.g., Ca2+) that induce ionic cross-linking for the gelation of chitosan, β-GP affects chitosan purely with secondary interactions. These include electrostatic interactions between the ammonium groups of chitosan and the phosphate groups in β-GP, hydrogen bonding associated with chitosan interchains, and chitosan-chitosan hydrophobic interactions, which is believed to play a major role in the gelation process.[333] Specifically, the glycerol (polyol) part of β-GP modulates thermo-induced hydration or dehydration of chitosan chains due to its influence on water.[333] The polyol components create hydrogen bonds to prevent chitosan chains from forming gels at low temperature. While this protection mechanism is removed upon temperature elevation, chitosan chains are left to interact with each other through stronger hydrophobic bonding and give rise to gelation.[335] Chemical composition, size and structure (e.g., attachment between polyol and phosphate moiety) of the gelling agents all play roles in the gelation process. Additionally, greater deacetylation degree in chitosan leads to faster gelation, which may be due to increased cross-link density between ammonium groups of chitosan and phosphate groups of β-GP.[335b]
The liquid to gel transition of chitosan/β-GP at body temperature makes it a potential embolic material.[267, 270] In one study, a liquid composed of 2% chitosan and 56% β-GP was prepared at a volume ratio of 7:1 and tantalum powders were doped as contrast agent. This liquid agent embolized and completely occluded rabbit renal arteries with no recanalization during an 8-week observation period. The occlusion led to the infarction of the kidney, which appeared pale and substantially smaller than the control kidney, without inflammation. Since the gelation time was approximately 2 minutes for this particular chitosan/β-GP system, the injection window was controlled between 1.8–2 minutes to prevent gel wash away. No catheter blockage or non-target embolization occurred.[267] In another study, a radiopaque system consisted of chitosan, β-GP and Visipaque demonstrated successful acute embolization of splenic and gastric vessels in pigs.[270]
Chitosan/β-GP mixture was also used for embolization of cerebral AVM in a porcine model.[268] The chitosan (80.4 kDa) used in the study had a viscosity of 130 mPa·s and 94% degree of deacetylation. The optimum ratio between 2% chitosan and 8% β-GP was selected to be 1:1. The mixture had a final pH value of 7.28, a modulus of up to 14 kPa, and an average gelation time of 2 minutes at 37°C. Tantalum powders or Omnipaque (GE healthcare, Chicago, IL) were blended to achieve radiopacity. Histological analysis confirmed that the basicranial rete mirabile (i.e., an AVM model) vascular cavity was filled with gel, and the tunica intima and muscle layer remained clear and intact. No vessel wall inflammation or necrosis was observed.[268]
The chitosan/β-GP system may also be used as an injectable drug carrier for embolization.[336] It has been shown that the addition of β-GP in chitosan reduced the initial burst release of chlorpheniramine maleate; chitosan alone was associated with a 53% release compared to 15% release in the chitosan/β-GP system within 4 hours.[335b] SEM micrographs demonstrated an interconnected pore structure of chitosan/β-GP system with pore diameters between 5 and 10 μm.[335b] The “open-cell” structure was dependent on the ratio between chitosan and β-GP, which also influenced the gelation temperature, swelling kinetics and degradation rate.[337] The highly porous structure suggested that chitosan/β-GP system allowed the delivery of large molecules. In one study, sodium tetradecyl sulfate (STS) was combined with radiopaque (iopamidol) chitosan/β-GP to produce a seclerosing gel for endoleak repair after aneurysm treatment.[269]
It is important to note that pure chitosan solution solidifies even without the presence of β-GP as a result of buffer release into the surroundings. However, this spontaneous process is usually too slow to form highly organized structures and will lead to burst release of therapeutics.[335b] Hence, the addition of β-GP into chitosan is essential for the efficient formation of an organized hierarchical porous structure for embolization.[335b]
In addition to β-GP-induced physical gelation of chitosan, other gelling agents may be used to provide chitosan based gel adequate strength to impede physiological pressure.[338] For example, sodium bicarbonate coupled with phosphate buffer has been used as gelling agent and mixed with chitosan.[279] By varying gelling agent composition, the physical/mechanical properties of the final gel was optimized with initial G’ of ~2233 Pa at body temperature. The thermosensitive gel was loaded with doxycycline, a MMP inhibitor, as a sclerosing embolic agent for aneurysm repair. Doxycycline inhibited MMP progression by combining to zinc, a factor required for MMP function in aneurysm recurrence.[90, 339] Embolization of the porcine renal artery with this formulated doxycycline/chitosan gel exhibited short term occlusion success of 86%, without gel migration or catheter blockage.[279]
6.6.4. Silk-Elastinlike Protein Polymer (SELP)
Synthetically designed silk-elastin-like protein polymer (SELP) was investigated as a novel formulation for TACE in HCC.[271, 340] SELP sequential block copolymers consist of two natural materials, silk and elastin proteins, where silk provides the strength and elastin provides flexibility.[272b] In particular, the crystalline structure in silk domains, formed via long-range physical interactions between polymer chains, governs hydrogel stiffness.[271] Elastin blocks offer the water-solubility of the polymer and also separate silk components to create a porous structure.[341] When injected into the body, SELP gels rapidly due to irreversible thermally induced sol-gel transition without the need for chemical crosslinking.[342] The rate of gelation depends on polymer composition, concentration, temperature and presence of additive, which either hinders or accelerates hydrogen bond-mediated crystallization[342] The polymer is soluble in aqueous solution and can be loaded with drugs at room temperature (Figure 16A).
Figure 16.

A) Schematics of drug loaded SELP liquid embolic agent. B) Illustration of SELP-815 K structure. In vitro drug release profiles from 12% SELP-815 K loaded with doxorubicin and sorafenib powders. Relative release rates of C) Doxorubicin and D) Sorafenib from single drug loaded gels (either 25 mg/mL or 50 mg/mL) or from dual drug loaded gel at 25 mg/mL loading (per drug). Statistical significance between single drug loaded versus dual drug loaded gels (*); single drug loaded 25 mg/mL versus single drug loaded 50 mg/mL gels (+);single drug loaded 50 mg/mL gels and the dual drug loaded gels (•). Statistical significance is reported as p < 0.05, highly significant as p < 0.01 and very highly significant as p < 0.001. Figure components adapted and reproduced with permission.[271, 272b] Copyright 2016, American Chemical Society and 2015, Elsevier.
The mechanical and physiochemical properties of SELP, such as stiffness, swelling ratio and drug-loading capacity, were tunable by varying the ratio and sequence between two components, as well as externally applied shear processing.[343] The shearing caused drastic increases in gelation kinetics and stiffness. This may have been due to the breakage of weak intramolecular bonds, followed by realignment of polymer strands to form stronger intermolecular hydrogen bonds between silk units that made up major SELP networking.[343]
After optimization, one final formulation, namely sheared 12 w/w SELP-815K, was selected for further in vitro and in vivo testing due to its excellent physical properties. SELP-815K was genetically engineered with eight silk and fifteen elastin units and a lysine (K) modified elastin (Figure 16B). It had initial viscosity of less than 150 mPa·s, gelation time within 5 minutes and final gel stiffness over 100 kPa. Injected sheared 12 w/w SELP-815K successfully occluded in vitro microfluidic devices mimicking flow conditions in a vasculature system. In vivo hard stasis was observed within 5 minutes after injection of 0.8–0.9 mL SELP into the hepatic artery of rabbits and stable embolus was achieved 10 minutes after injection. SELP-815 gels reached arterioles with no non-target embolization in draining veins.[271] Its long-term embolic persistence and stability remain to be examined.
Doxorubicin and sorafenib can be loaded into SELP-815K gel through physical mixing for potential chemoembolization of HCC.[272b] Drug loaded SELP has demonstrated a total drug loading level of 25 mg/mL, which is comparable to the clinically specified loading level of doxorubicin-loaded DEBs.[215, 272b] Theoretically, combining both drugs would be beneficial as they have complimentary mechanisms of action: doxorubicin induces cytotoxicity and intercalates DNA to prevent DNA replication, whereas sorafenib blocks tumor angiogenesis. Although sorafenib cannot be delivered by DC beads because the drug is not sufficiently soluble and also not positively charged, it can be loaded on SELP and a 50 mg/mL loading has been achieved. Additionally, drug formulation (base form versus salt form, and powdered form versus solvent dissolved suspension) influences mixture viscosity, which is also temperature-dependent.[272b, 344]
Whereas the in vitro release profile of doxorubicin is similar in single or dual drug systems, sorafenib has a higher release rate in dual compared to single drug systems (Figure 16C and 16D).[272b] Additionally, doxorubicin (in both single and dual drug systems) exhibits a burst release (up to 18%) by day 1, with an overall release of 30% by day 30. For sorafenib, a sustained and relative linear release profile has been observed in single and dual drug systems up to 20 days, with cumulative release of 16% and 28%, respectively. In vitro characterization reveals the ability of the SELP system to achieve a therapeutically effective drug concentration from a clinical standpoint for a minimum of 14 days and 30 days, for single and dual systems, respectively. The enhanced release profile of sorafenib may be due to relatively greater hydrophilicity of doxorubicin, which facilitates water diffusion into the gel.[272b] The prolonged release of drug is primarily attributed to the hydrophobic domains within SELP that shield the drug from aqueous solution. Preliminary study in a rat liver cancer model has been carried out to demonstrate synergetic effects favoring dual-drug, compared to single-drug delivery systems.[272a]
6.7. pH-Sensitive Gelling Systems
pH-sensitive materials undergo sol-gel transition due to pH differences between the injection and operating conditions. This pH difference changes the charges on polymer chains to activate consecutive swelling and drug release.[345] pH-triggered materials may serve as embolic agents since they can be delivered in liquid form and gel at the targeted site in response to pH shifts. Whereas temperature-responsive embolic agents may pre-maturely gel within long catheters placed inside of a vessel, the on-demand gelation of pH sensitive hydrogel, which only occurs once the material has emerged from the catheter, may avoid catheter blockage.
Sulfamethzaine (SM)-based pH-sensitive hydrogels have been proposed as injectable liquid embolic agents for vascular embolization and chemoembolization.[273–275] A variety of SM containing polymeric hydrogels have been formed. These include a copolymer comprising poly(ethylene glycol) and poly(urethane sulfide sulfamethazine) (PEG-PUSSM),[274] a block copolymer system composed of triblock poly(ɛ-caprolactone-co-lactide)-b-poly(ethyleneglycol)-b-poly(ɛ-caprolactone-co-lactide) (PCLA-PEG-PCLA) and SM(PCLA-PUSSM),[275] and polycaprolactone(PCL)-PEG-SM block copolymers.[273]
As an anionic pH-sensitive moiety, SM transitions from an ionized state at high pH to a deionized state at physiological pH. This feature is responsible for the gel’s sol-gel transition behavior.[274] For example, in the PEG-PUSSM system, decreased pH values deionize SM and induce PUSSM to transition from a hydrophilic to hydrophobic state, leading to the formation of micelles that ultimately interconnect into a three-dimensional hydrogel network for occlusion.[274] Therefore, SM-based hydrogels undergo phase transition from a liquid state at high pH (e.g., 8.5 during injection and within the catheter) to a gel state at physiological pH (e.g., 7.4) or a relatively acidic tumor pH (e.g., 6.5–7.2). For example, for 25 wt% PCL-PEG-SM solution, the viscosity has been measured to be less than 1 Pa·s at injection conditions (i.e., room temperature and pH 8.0) and ~3900 Pa·s at operating conditions (i.e., 37°C and pH 7.4).[273] Storage moduli before and after activation were not reported.
The stability and degradability of SM based gel have also been investigated in vivo.[273–275] Specifically, PEG-PUSSM was found to have 74% of its original weight 12 weeks after subcutaneous implantation into rats. Its slow degradation profile may be attributed to erosion or cleavage of urethane bonds. Faster degradation was reported for PCL-PUSSM system (e.g., loss of 50% mass at 4 weeks). The long term embolization stability for both systems has been examined in rabbit renal and liver models. In vivo gelation of PEG-PUSSM was observed to occur within 30 minutes and was observed within 15 minutes for PCL-PUSSM system.
The chemoembolization potential of doxorubicin-loaded PCLA-PUSSM and doxorubicin-loaded PCL-PEG-SM was investigated both in vitro and in a VX2 rabbit liver cancer model. Sustained release of doxorubicin over four weeks was observed in vitro in both systems. Released doxorubicin exhibited a bioactivity level (with respect to inhibiting hepatic cancer cell proliferation) comparable to that of free doxorubicin.[273, 275] This suggests that both systems could be used for sustained delivery of active anti-cancer drugs. Moreover, intraarterially injected doxorubicin-loaded PCLA-PUSSM hydrogel led to a significant decrease (~48%) of tumor volume at 2 weeks, suggesting its chemoembolization effect.[275]
In addition to the anionic SM based systems, biodegradable cationic poly(amino ester urethane) (PAEU) block copolymers also exhibit pH-triggered sol-gel transition and have been investigated as potential liquid chemoembolic agents.[276] In contrast to the SM-based system, PAEU-based hydrogels undergo sol-gel transition with increased pH (e.g., sol state at pH 6.5 and gel phase at pH 7.4). For example, the viscosity increased from 2.7 to 1901.3 Pa·s when the pH shifted from 6.6 to 7.4. The gelation kinetics are affected by PAEU fraction, molecular weight and concentration of copolymer. The critical gelation pH is influenced by the addition of contrast agent (e.g., lipiodol), which introduces hydrophobicity to the system. The in vivo occlusion level and stability of PAEU gel was investigated subcutaneously and also in hepatic tumor artery, and were directly correlated to the completeness of gelation, which ultimately governs gel strength.[275–276]
6.8. Pre-Activated Chemically Crosslinked Systems
In addition to on-site polymerization due to change in solvent states (e.g., protein and enzymes) for chemically induced sol-gel transition (e.g., nBCA), gelation can also be initiated chemically beforehand and then delivered to the targeted site within a precise window. The predictability of curing time is critical because the material may breakdown if gelation is not complete, leading to migration or potential micro-emboli downstream, which can have harmful consequences (e.g., stroke).[346] Conversely, delivery that is too delayed can lead to pre-mature gelation of the liquid/gel in the catheter , resulting in catheter blockage. Next, embolic systems that depend on pre-activated chemical crosslinking are presented.
6.8.1. PPODA-QT System
The PPODA-QT polymeric system is composed of liquid organic monomers poly(propylene glycol) diacrylate (PPODA) and pentaerythritol tetrakis 3-mercaptopropionate (QT), which are mixed and reacted in a basic environment (NaOH).[347] The diffusion of OH− groups from aqueous droplets into organic phases initiates the reaction.[348] The thiol groups on QT undergo deprotonation in basic aqueous solutions and then interact with the acrylate group on PPODA. The material gradually gels through cross-linking via Michael-type addition to form a network in a time-dependent manner.[346] Before the polymer becomes fully cross-linked, the mixture is still deliverable through the catheter. Therefore, there is a critical window for delivery during which the solution has low enough viscosity.
Higher pH, increased pre-mixing time and presence of surfactant all lead to faster reaction kinetics of PPODA-QT. Furthermore, the type of contrast agent also alters gelation kinetics as a result of interactions between the contrast medium and PPODA-QT. Faster gelation occurs in the Conray™ (Mallinckrodt, St Louis, MO) system (e.g., ~11.1 minutes with 0.5-minute pre-mix) compared to the Omnipaque system (e.g., 28.5 minutes with 0.5-minute pre-mix).[347] In addition, the Conray gels swell nearly 60% in volume, whereas Omnipaque gels swell only 30–40% after 10 months.[348] Uncontrollable and unpredictable swelling is undesirable after embolization as it is possible to overstress weakened blood vessels. Both types of gels have remain intact after 10 months, with a minimum Young’s modulus of 0.5 MPa.[348] Conray gels have exhibited lower initial in vitro cytotoxicity, which may encourage neointimal tissue growth to overcome its limitation in swelling. Furthermore, the addition of iodobenzoyl poly(ethylene glycol) diacrylate (IPEGA) renders PPODA-QT intrinsically radiopaque.[297]
The biocompatibility and deliverability of PPODA-QT was examined in lateral wall carotid artery aneurysms in a porcine model under balloon protection across the neck.[277] For this small-scale study, the gel was formulated with a relatively long gelation time of 10–15 minutes in order to offer a flexible delivery window. Incomplete initial filling appeared to provide the best outcome, with accelerated neointimal tissue growth and progressive occlusion 1 month after embolization. 100% material filling appeared to overstress the aneurysm, but still facilitated neointimal tissue growth.
Furthermore, a 6 month pilot study was carried out in a canine lateral wall aneurysm model with temporary balloon occlusion.[278] Three groups were tested 1) PPODA-QT only; 2) placement of coil followed by PPODA-QT; and 3) coil only. Group 1 led to complete obliteration with no signs of recanalization or material compaction; a protective continuous tissue layer formed over the polymer. Group 2 exhibited complete occlusion at 6 months, though the radiographically dense coil caused difficulty in visualizing the PPODA-QT during delivery. Group 3 failed to induce a neointimal layer and instead developed nonuniform and flimsy tissue. This study suggests the biocompatibility of PPODA-QT system as well as its long-term stability for potential aneurysm treatment. The water-born system eliminates the use of organic solvents.[348] One concern of the PPODA-QT system is potential leakage of unreacted QT, a very toxic monomer.[349]
6.8.2. Graphene-oxide Enhanced Hydrogel
A graphene-oxide (GO) enhanced generation five poly(amidoamine) dendrimers (PAMAM-5) hydrogel demonstrates satisfactory mechanical strength for arterial occlusion.[256] Briefly, the gel is prepared by first mixing PAMAM-5 and sodium hydroxide (NaOH) with GO, followed by adding fresh glucono-delta-lactone (GDL). The gelation mechanism results from the reaction between GO and NaOH. The slowly released H+ from GDL during its hydrolytic process activates carboxyl group on GO. Therefore, the gel has to be freshly mixed prior to administration since addition of GDL initiates the gelation process.
The addition of GO improves the mechanical performance of PAMAM-5 hydrogels, while its potential toxicity is still under evaluation. Its mechanical properties depend on system pH, which is governed by the NaOH/GDL ratio. By varying this ratio, acceptable storage moduli for embolization application is achieved and is 2050Pa. Subcutaneous implantation of GO/PAMAM-5 hydrogel into rats induces a foreign body response, mainly on the fifth day after injection. Due to its nonbiodegradable nature, the gel induces granulomas and forms a clear border with surrounding tissues. No infections have been observed four weeks after injection.
The embolic strength of GO/PAMAM-5/Iohexol mixture was assessed in the subclavian artery of rabbits. The gel was injected rapidly within 5 minutes after addition of GDL. No complications or artifacts were observed during two-week observation period. Granulomas containing inflammatory cells and macrophages were observed in the artery and fibroblasts were found around vascular emboli, indicating recanalization was not possible. Active fibrosis and thrombus blocked arteries completely. Therefore, the GO/PAMAM-5 hydrogel not only occluded the vessel but also irritated the lumen, stimulating thrombus formation. The highly thrombogenic nature of GO induces thrombus formation by platelet aggregation at a scale comparable to that of thrombin.[350] The main challenge associated with this technique is its limited operating window. As the authors mentioned, although premix time for this gel has been shortened from 20 minutes to 2 minutes, prolonged injection time may lead to hydrogel clogging inside of the microcatheter.
6.8.3. Carbazochrome-Containing Chitosan-PEG Hydrogel
A dibenzaldehyde-terminated poly(ethylene-glycol) (DF-PEG) gelator has been shown to interact with glycol-chitosan and carbazochrome through Schiff-base cross-linking, and a self-healable drug-loaded hydrogel (PCC) starts to form in 200 seconds.[280] 10 wt% PCC exhibits a storage modulus of 5.7 kPa and its embolization efficacy was demonstrated in a rat kidney model. The self-healing property of PCC is established by uncoupling and recoupling imine linkage in Schiff-based bonds. This characteristic offers tunable hemostatic release in conjunction with occlusion.
6.9. Tandem Thermal Gelling Chemical Cross-Linking System
A simultaneous physically and chemically crosslinked NIPAAm-based system has been developed and demonstrated increased strength and reduced creep (i.e., time dependent permanent deformation under elevated temperatures and stresses, which are lower than the material’s yield strength) in embolization applications.[351] Physical gels formed by secondary interaction are usually soft and easy to inject, though their viscoelastic properties are not suitable for AVM treatment given their tendency to undergo considerable creep [352] in response to the constant stress exerted by blood pressure.[353] Conversely, because chemically cross-linked gels are stronger and less viscoelastic due to covalent bonds, they can be difficult to inject and may undergo swelling at body temperature.[354] Therefore, a combined physical and chemical cross-linking mechanism provides a unique approach to balance injectability at room temperature and high strength/stability at body temperature.
The tandem system is based on poly(NIPAAm), a temperature-sensitive moiety with low critical solution temperature (LCST) at approximately 32°C.[354] Below LCST, it is soluble in aqueous solutions with an unfolded conformation. It undergoes sol-gel transition at sufficient polymer concentration above LCST due to chain entanglement (physical crosslinking). Chemical crosslinking has been achieved through a Michael-type addition between vinyl groups present on either acrylate[355] or vinylsulfone functionalized[351] poly(NIPAAm) system (produced by free radical polymerization), and a thiol compound (such as cysteamine[281] or QT[354]). Some of the investigated poly(NIPAAm) systems include poly(NIPAAm-co-cysteamine) (NC), poly(NIPAAm-cysteamine-co-vinylsulfone) (NCVS) and poly(NIPAAm-co-HEMA-acrylate) (NHA), with LCST at 30°C, 34°C, and 29°C, respectively.[351] The conjugation of hydrophobic monomers decreases the LCST while hydrophilic monomers increase the LCST.[351]
Gelation time determines the delivery window for the tandem agent. Pre-mixing time, pH, buffer strength, number and type of thiol and acrylates, and the molecular weights of polymers all affect poly(NIPAAm) gelation kinetics. [349, 354] For instance, NC-NHA gels within 22 to 37 minutes depending on pre-mixing duration, whereas NC-NCVS gels immediately regardless of pre-mixing time. This may be due to both higher cross-linking density and higher reactivity of vinylsulfone groups on NCVS compared to NHA. Furthermore, NC-NHA has superior mechanical strength, with G’ measuring 10 kPa, compared to that of its pure chemical gel with G’ of 1 kPa. Deliverability and embolization efficacy for both NC-NHA and NC-NCVS were examined in a porcine aneurysm model for 1 hour, as proof of concept.[281]
Compared to exclusive physical gels, the tandem system exhibits low-frequency load bearing, an indicator of reduced creep compliance and long-term stability confirmed by rheological studies, owing to its chemical crosslinking.[354] Importantly, the swelling ratios (i.e., a representation of percentage weight increase compared to initial formulation) for both NC-NCVS and NC-NHA systems remain stable for 1.5 years in vitro. Lastly, the compatibility of the NIPAAm-based polymeric system (poly(NIPAAm-co-CysOEt) and NC-NHA) have been examined after subcutaneous implantation in rats at 1 and 4 weeks.[354] Minimal chronic inflammation was observed, without toxic systematic reactions.
The class of NIPAAm-based tandem systems offers advantages over purely physical or chemical gels. The physical cross-linking enables smooth delivery and filling of the targeted vasculature, which can be tortuous and irregular (e.g., AVMs and aneurysms). Gradual curing from chemical crosslinking further strengthens the mixture and maintains long-term occlusion. System parameters, including delivery window and final storage modulus, may be further optimized for appropriate use in vivo.
6.10. Other Notable Classes of In Situ Gelling Systems
6.10.1. Liquid Crystals
Liquid crystals are polar amphiphilic cohesive materials that can be reconfigured upon external stimuli (e.g., light, electrical field and pH values).[356] They self-assemble in water by aggregation of amphiphilic molecules, which are beneficial for enhanced drug delivery.[282, 356b, 357] A cubic phase forms from polar amphiphilic material in aqueous environments. This cubic phase is of great importance in biological and medical applications since it can hold hydrophilic, amphiphilic, and hydrophobic substances. Indeed most anti-cancer drugs are hydrophobic.[282, 358] The cubic phase precursor (e.g., DMSO and NMP) can be prepared as a liquid for injection.[282] The precursor then gels in situ to cubic phase by water absorption and forms a cast. This phase transformation is helpful for embolization and may overcome some of the hurdles of current embolic agents.
Liquid crystal was discovered in 1888.[359] However, its utility in embolic materials has not been investigated until recently. In one study, glyceryl monooleate (GMO) based liquid crystalline material has been proposed for embolization.[284] GMO precursors (GMO-ethanol-water system), with viscosity less than 30 mPa·s, were formed to facilitate microcatheter delivery. Upon contact with blood, GMO-based liquid crystals transitioned from low viscous injectable liquid into a highly viscous cubic phase gel that forms vessel casts in less than 15 seconds. Superselective occlusion of rabbit renal artery was confirmed 5 weeks after GMO embolization and biodegradation was observed. For this system, the catheter needs to be flushed with ethanol thoroughly before and after GMO precursor delivery to avoid catheter blockage due to phase transformation upon aqueous exposure.
Recently, Qin et al. investigated phytantrol based liquid crystals as a potential embolic liquid agent for delivering hydroxycamptothecin (HCPT), an antiangiogenic inhibitor for HCC.[282] Although HCPT has been used clinically, its poor stability and solubility hinder loading capability while its burst release limits its clinical application. [360] As such, liquid crystal may serve as a novel and effective drug vehicle for HCPT delivery since the cubic phase can hold drugs with different polarities, protect a drugs bioactivity, and achieve a high drug loading rate, in addition to facilitating its sustained release. The phytantrol-water-ethanol system has been shown to embolize rabbit hepatic artery with phytantriol cubic phase.[283]
NMP-based precursor (loaded with HCPT) passes through a 2.8F microcatheter with no difficulty and forms into a bulky hydrogel when exposed to excess water at 37°C in vitro. The precursor solution is able to contain up to 20 mg/mL (2% w/w) HCPT. However, precipitates form after 30 minutes of standing when HCPT concentration exceeds 0.5% w/w. Therefore, 0.5% w/w HCPT loaded precursor was selected for further study and demonstrated a viscosity of less than 200 mPa·s at 37°C.[361] The overall gelation time was on the order of 40 seconds despite precursor formulations and the formed gel maintained its integrity in the flow of saline at 0.3 mL/s.
In vitro, HCPT exhibits a rapid release (up to 15%) during the first day, followed by sustained release for over 30 days with a cumulative release of up to 90%. The initial rapid release may be due to cubic gel formation which expels some of the HCPT (initially dissolved in NMP) when NMP diffuses out. Since HCPT has poor solubility in water, only small amounts of drug are released at the beginning, while the majority of the drug remains crystallized and encapsulated within the gel. From a clinical perspective, the initial release of HCPT induces an adequate drug concentration to inhibit topoisomerase-I for cancer cell apoptosis. The sustained and extended release of HCPT inhibits DNA replication that is essential for cancer cell growth, while also creating an antiangiogenic effect.[362] Its in vivo performance and therapeutic efficacy remains to be tested.
6.10.2. Coacervates
Complex coacervation is an associative, liquid-liquid phase separation in response to the change of thermodynamic states (i.e., driven by entropy) caused by electrostatic interaction of oppositely charged micro-ions.[363] Upon aqueous colloidal solution separation, one phase contains little colloid and the other colloid rich phase is the coacervate.[364] These hierarchically structured materials possess self-healing properties due to their non-covalent nature of molecular interactions.[365] Their structural recovery ability could be exploited to create an embolic agent that has low viscosity for injection and returns to gel phase when recovered.[363] As an important class of self-assembling materials, coacervates’ dynamic behaviors are poorly understood. Rheology studies have been conducted to understand coacervate dynamics, such as their structure, self-assembly and intermolecular interactions.[366]
Sal-IP6:
This liquid embolic agent was inspired by the undersea adhesiveness of marine sandcastle worms.[285, 367] The adhesiveness of underwater glues arises from electrostatically condensed, oppositely charged polyelectrolytes, which are complex coacervates.[368] Mimicking the chemistry of these undersea fluid adhesives, Jones et al. developed phase-separated embolic coacervates (Sal-IP6) from polyelectrolytes, which consisted of polycationic salmine sulfate (Sal) and polyanionic sodium inositol hexaphosphate (IP6) (Figure 17). Both Sal and IP6 are FDA-approved.[285]
Figure 17.

A) Illustration of fabrication process of injectable radiopaque Sal-IP6 embolic coacervate. B) The fluid rapidly solidifies into a stable gel under physiological saline. C) Fluoroscope image and D) Post-mortem dorsal three dimensional image of a rabbit kidney 90 min after arterial embolization with Sal-IP6. E) Morphological changes of Sal-IP6 as a function of NaCl concentration. Figure components adapted and reproduced with permission.[285] Copyright 2016, John Wiley & Sons Inc..
The sol-gel phase shift is attributed to the morphological change of Sal-IP6 in response to the presence of NaCl, which has different concentrations between prepared stock solution and physiological conditions (Figure 17A, 17B and 17E). Specifically, Sal-IP6 transitions from a clear, homogenous fluid at high ionic strength (e.g., 1200 mM NaCl) with a viscosity ~1 Pa·s, to a stiff gel formulation at physiological ionic strength (e.g., 150 mM NaCl, similar to that in plasma) with a viscosity of ~39.7 Pa·s (Figure 17E). Therefore, the change of viscosity of Sal-IP6 was triggered on site as a result of NaCl ionic strength differences. For fluoroscopic visualization, tantalum microparticles were added into Sal-IP6, leading to increased viscosity (e.g., 1.1 Pa·s to 56.8 Pa·s from 1200 mM to 150 mM NaCl) and reverse shear thinning behavior. The injection pressure (maximum 4.3 MPa) has also been shown to be within of the range of that normally handled by endovascular radiologists.[285]
In vivo studies have demonstrated that Sal-IP6 penetrates the entire arterial vasculature of a rabbit kidney, precluding the venous sides of the renal cortex or ducts (Figure 17C and 17D).[285] Up to 4 cm of total distance was reached by the coacervate. No adverse tissue reactions were observed. Since the solidification mechanism of Sal-IP6 depends on the diffusion of NaCl out of the material, no catheter blockage occurred. Therefore, Sal-IP6, an ionic dependent embolic coacervate, can be potentially delivered discontinuously in a controllable manner without the danger of gluing the catheter or use of organic solvent, which are the major disadvantages of nBCA and EVOH/DMSO.
Polyphosphate Based Coacervates:
Radiopaque coacervates have been developed by chelating polyphosphate (PP) and divalent cations (e.g., Sr2+, Ba2+ and Ca2+) as temporary liquid embolic agents.[286] The cation-induced phase separation is governed by both electrostatic and hydrophobic interactions.[369].[364a]
The physical appearance and material kinetics of PP coacervates depend on PP concentration and degree of polymerization (Dp).[286] Viscosity of low Dp is much lower compared to that at high Dp [286]. The incorporation of divalent cation reduces the viscosity (maximum of 67 Pa·s) compared to unloaded NaPP solution (over 2000 Pa·s), which is favorable for injection. This is because the chelation process reduces the amount of negative charges in PP. Pre-loaded Ba and Sr ions have been shown to reduce cell toxicity to a greater degree than Ca2+ because the greater stability of Ba and Sr reduces their ion release. Rheological studies show that impregnated Ba and Sr ions enhance elasticity and viscosity more than Ca2+ only counterparts.[370]
The addition of heavy metal Sr and Ba into PP significantly improves its radiopacity (up to 3 folds compared to PP coacervates).[286] The Ba/P molar ratio contributes more to the contrast than Sr/P does, due to higher atomic number of Ba. Successful occlusion and radiopacity was documented in PP embolized rabbit auricular arteries after 15 days. Higher levels of macrophages were observed with PP based coacervate implants compared to gelatin sponge. The higher degree of complex foreign body response may be due to local drops in pH from PP dissolution, though no gross toxicity was observed. Similar to calcium alginate system, a concentric delivery device was deployed in this work to deliver ions (e.g., CaCl2) and PP in separate paths before getting into contact on site. Though in both in vitro and in vivo studies, the embolic mass was noted to fragment, suggesting the need for further improvement of the embolic’s cohesiveness to avoid errant dislodgement.
Coacervates represent an intriguing class of materials as they can be loaded with cations for intrinsic radiopacity, hydrolyzed into simple byproducts (e.g., phosphate salts), and carry drugs.[364a] Drug encapsulation has been reported with coacervates while the addition of drug could lower the system storage modulus significantly.[371] Coacervate-drug systems are usually complicated as temperature, pH, and presence of protein can all affect release kinetics.[371] Studies also show that coacervates enhance platelet adhesion and reduce clotting time by promoting thrombin activation and consequent fibrin generation, highlighting their hemostatic abilities.[364a]
6.11. Shear Thinning Hemostatic Hydrogels
Shear thinning hydrogels have received great attention in minimally invasive delivery since they are injectable, self-healable, and capable of delivering cells and growth factors.[372] The shear thinning property is ideal to facilitate injection during transcatheter delivery due to reduced viscosity at increased strain rate and can be characterized by rheology.[373]
A shear thinning biomaterial (STB), consisting of gelatin and silicate nanoplatelets, has been developed as an hemostatic embolic agent and attempts to take advantage of the strong electrostatic interactions between these two components (Figure 18A).[10] Highly charged synthetic bioactive silicate nanoplatelets have negative charges on their surfaces and positive charges along the edges.[374] Due to anisotropic charge distribution, the positively charged gelatin interacts with silicate nanoplatelets. They self-assemble, dynamically form and break upon external stimuli and exhibit shear thinning behavior.[375] Additionally, nanoplatelets induce coagulation by concentrating clotting factors and the procoagulant response is dependent on their surface properties and structure.[376] As a result of the intrinsic coagulation of gelatin and silicate nanoplatelets, STB exhibits hemostasis behavior and was original discovered as a hemostasis agent.[375] Its in vitro clotting time in anticoagulated whole blood is between 3 and 5 minutes, similar to that of coils.
Figure 18.

A) Schematic illustration of STB fabrication. STB is extruded from the catheter tip. B) Storage moduli (G′) of 6% (w/v) STBs after repeated cycles of low and high strain. C) A representative injection force curve to deliver STB through the catheter. Laser Doppler microperfusion imaging in mouse showing D) hindlimb perfusion before STB injection and E) no perfusion in STB injected limb. F) Coronal CT study confirmed no pulmonary embolism occurred in porcine model during 24 days after STB embolization. G) Gross evaluation at 24 days shows STB (arrow) occluding the vein. H) Percentage of vessel remodeling (replacement of STB with connective tissue) over time. Figure components adapted and reproduced with permission.[10] Copyright 2016, The American Association for the Advancement of Science.
Compared to other in situ gelling systems that undergo sol-gel transition, STB remains in its natural gel structure both before and after injection. Its syringeability depends on the reduced viscosity at high shear rates during injection. Blood vessel occlusion is achieved by recovered STB strength upon the removal of shear stress after delivery, followed by thrombus formation (Figure 18B). The modulus and viscosity of STB can be tailored by varying the ratio between gelatin and silicate nanoplatelets.[10] Among all formulations, 6NC75 (1.5 wt% gelation, 4.5 wt% nanoclay and 94 wt% water) was selected as the optimal composition, which provided comfortable injection forces between 20 and 25 N (Figure 18C). The material exhibits fluid-like behavior within the catheter during injection. When stress is removed, it returns to its original gel formulation with storage modulus ~200 Pa for operating condition. The rheology and injectability of STB can be affected by the presence of salt in physiological environments.[377]
STB has demonstrated promising results in arterial embolization in both small and large animals.[10] When injected into the left external iliac artery of a pig, STB immediately ceases blood with no fragmentation or migration. This has important clinical implications; the iliac artery in pig has high flow rate at 100 cm/s, similar to that of human iliac arteries. These results suggest that STB renders stable and complete occlusion in high flow targets. Furthermore, its excellent stability to resist cyclic loading and frequent movement was confirmed in forelimb vein embolization in the pig (Figure 18D and 18E) Both CT and histological analysis revealed complete occlusion 24 days after embolization in a porcine model with no evidence of pulmonary embolism (Figure 18F and 18G).[10]
Moreover, STB occludes vessels mechanically without depending on thrombus formation, while both gelatin and silicate nanoplatelets facilitated coagulation to further stabilize STB in place to prevent dislodgement. Histological analysis revealed that macrophages surrounded STB locally, indicating ongoing degradation and clearance of residual STB inside of the embolized vessel. Continuous vessel remodeling in conjunction with STB degradation was observed in the lumen of occluded vessels (Figure 18H). Connective tissue was present at occlusion sites, further suggesting the unlikelihood of recanalization.[10] This work indicates that STB has the potential to serve a hemostatic embolic agent.
6.12. Characterization and Injectability of Liquid/Gel Embolic Agents
For liquid/gel embolic agents, their viscosity must be low enough at room temperature for smooth delivery, followed by an increase in viscosity and stiffness when introduced into the target site to avoid washout by blood flow. Therefore, the physical and mechanical properties of the material at the injection site (e.g., inside of the syringe at room temperature), within the catheter (e.g., during delivery at body temperature) and in the target environment (e.g., stasis within the body) must be carefully balanced.
A key physical and mechanical property of a liquid/gel embolic agent is its viscosity, which determines its feasibility for transcatheter delivery. Viscosity represents the material’s ability to resist deformation in response to applied stress and depends on the molecular weight of the polymer, the type of the solvent and agent concentration.[192a] It is also a measure of the response of the material to change of shear strain during injection. For injectable embolic material, viscosity less than 150 mPa·s is preferred for smooth transcatheter delivery.[272b]
The critical viscosity value depends on the targeted vessel diameter and blood pressure, and may be used to identify the safety range of liquid materials when used for embolization in different clinical applications. For instance, for AVM embolization and small capillaries, low viscosity in the 30–150 mPa·s range is optimal, whereas high viscosity (e.g., 400 mPa·s and above) is preferred to fill larger structures such as aneurysms.[192a] Less viscous liquids may pass the target site into the venous side while highly viscous materials may not reach the nidus but occlude the proximal arteries only, limiting the extent of transport.
Additional critical rheological parameters to characterize liquid/gel embolic agent include storage moduli and loss moduli before (liquid-like state) and after injection (solid-like state), since the material’s strength at body temperature determines the embolization efficacy in both the short and long term. In response to oscillatory shear, storage moduli (G’) measures the material’s ability to absorb energy, while loss moduli (G”) provides information on whether material undergo stress relaxation to dissipate energy. Overall, storage and loss moduli evaluate the elastic and viscous properties of a material, respectively. Most importantly, storage modulus measures material stiffness and is an indicator of gel network cross-linking.[272b] It describes the agent’s ability to cast blood vessels against flow and the capability to withstand blood pressure without breakage. It has been suggested that the initial G’ of the embolic agent needs to be higher than 800 Pa upon injection to resist pressure up to 200 mmHg.[270] Additionally, the final storage modulus of the gel should be more than 0.1 MPa for stable gel embolus formation against hepatic blood pressure.[272b] However, a wide range (i.e., from few hundreds of pascals to thousands of pascals) of final gel moduli have been reported with successful embolization, as presented. Furthermore, a material’s viscoelastic behavior (e.g., physically versus chemically cross-linked gels) reflects its fatigue resistance, an important characteristic of the embolic material subjected to endovascular oscillation pressures.[258, 275] The formulation of embolic materials is, therefore, critical to be tuned for optimized embolic performance.
The critical gelation time also needs to be defined and correlated to both delivery time and procedure duration to avoid catheter blockage due to pre-gelation. In particular, the sol-gel transition time governs the delivery window (for pre-activated mixture) and the onset of effective occlusion (for on-site gelation). It has been reported that the ideal sol-gel transition time in arterial vessels is less than 5 minutes to prevent venous washout.[272b] Alternatively, the use of photocurable polymers may obtain rapid increase in strength by on site curing.[378] It is important to note that for temperature responsive liquid/gel agents, the material property may be affected due to long catheter that has been pre-heated to body temperature before actual delivery. Therefore, too rapid temperature triggered gelation may cause catheter blockage. As a result, assessment of deliverability should always be carried out in clinically relevant catheters, not needles.
Though rheology properties influence the injectability of materials, injection force determines whether the agents are suitable for clinical use and is the key parameter that is directly related physicians’ experience. Injection force is usually measured by compression test using either force sensors or mechanical testing machines, e.g., pushing down the plunger.[10, 373, 379] A typical force-time or force-displacement plot includes plunger-stopper break loose force (i.e., the force to initiate plunger movement) and injection force (also known as dynamic glide force which is the force to sustain the movement of syringe) (Figure 18C).[380] The break-loose force is critical, as in practice, this is the force that physician needs to overcome to initiate liquid dispersal.
In additional to the nature of the embolic agent, the injection force can be influenced by many factors, such as injection speed, environment (e.g., temperature, pH), types of syringe and catheter, and whether the injection is delivered to open air, liquid or tissues. Regardless, the system must be useable by any physician.[373, 379b, 381] An injection is typically performed with the thumb pushing on the plunger with ipsilateral index and middle fingers to stabilize flanks.[381a] At this position, the maximum force applied by a male is 95.4 N and is 64.1 N for females.[382] In vitro injection forces less than 50 N are considered injectable and forces between 51–100 N correspond to injectable but with some difficulty, and over 100 N indicates too difficult to inject.[379b] The injection force can be predicted from Hagen–Poiseuille law.[314, 383]
7. Outlook and Conclusions
The development of current and next generation embolic agents depends on the understanding of their physical, mechanical and physiochemical properties. Thorough material characterizations (e.g., mechanical and rheological testing) offer fundamental knowledge to predict occlusion level and embolic agent/tissue interactions. However, there are no standardized protocols to compare different products and agents (e.g., elasticity of calibrated microspheres). Additionally, the clinically relevant shelf life of newly developed embolic agents is overlooked. Both short- and long- term stability and biocompatibility should to be assessed. Furthermore, despite the development of multifunctional microspheres and on-demand gelling liquid embolic agent, their retrievability and reversibility have not been adequately addressed. The development of a reversible embolic mechanism would allow for material retrieval, which offers flexibility to benefit embolization practice.
Furthermore, material-specific cell/tissue interactions, which are known to influence biological performance, play an important role in any embolization application. The surface properties of the embolic agents, such as hydrophilicity and lipophilicity, directly affect cell adhesion, growth, migration and death. There is constant exchange between cells and materials via direct penetration, endocytosis, exocytosis and immune reactions.[256] Therefore, post embolization inflammatory reactions (e.g., macrophages, giant cells, and lymphocytes) should be assessed to quantitatively evaluate and compare tissue changes in response to different embolic agents.[161] One crucial property of embolic materials is thrombogenicity, which is largely determined by net electric charges. Even small amounts of voltage may damage endothelium, giving rise to non-specific thrombosis formation. Therefore, balanced surface charge is required for optimal thrombogenicity, since positively charged materials appear more thrombogenic but Hageman factor activation requires a negative charged surface.[384]
In order to determine the best embolic agent to use, one needs to at least ask the following questions: 1) is the vessel to be blocked large or small; 2) would the embolization be temporary or permanent; and 3) is the tissue supplied by the vessel to remain viable after the embolization?[9, 385] Regardless of composition, the smaller the agent, the greater the likelihood of organ ischemia.[142] The specific type of embolic material depends on the clinical condition, though one particular medical condition can be treated with different embolic materials.[386] In any case, embolic agents need to be biocompatible, cost effective and easily deployed. The ideal agent should be able to block an artery rapidly, fill 100% of the targeted vasculature, prevent recanalization, fragmentation and dislodgement, carry therapeutics, endow tunable embolic duration, render intrinsic visibility, leave limited trace artifacts, and possess clinically relevant shelf life. Table 2 summarized clinical used embolic agents discussed in this review.
Table 2.
Summary of clinically used embolic agents
| Embolic Agents | Description | Examples of Clinical Applications | Permanent? | Instant Hemostasis? | Effective with Coagulopathies? | Imaging Artifact-free? | Easy to Deliver? | Cost per procedure? |
|---|---|---|---|---|---|---|---|---|
| Coils |
|
Hemorrhage control and aneurysm embolization | Yes | No | No | No | No | $$-$$$$$ |
| Particulates |
|
Hypervascular tumor (i.e., TACE), uterine fibroids and prostate artery embolization | Yes | No | No | Yes | No | $$-$$$$ |
| Gelfoam Embolics |
|
Hemorrhage control and TACE | No | No | No | Yes | No | $ |
| Liquid Embolics |
|
AVM embolization | Yes | Yes | Yes | No | No | $$$$$ |
Unmet needs include controlled biodegradation and sustained drug release for embolization applications. While DEBs have been developed and clinically deployed, they cannot penetrate arteriocapillaries due to finite sizes. Though tremendous amounts of efforts have been devoted into biodegradable and drug loadable microspheres, systems designed and examined specifically for embolization are rather limited. Alternatively, liquid/gel embolic agents serve as a promising platform to penetrate deeply and encompass all branches inside of the tumor. However, they should never traverse into draining veins and their ability to carry therapeutics is limited.[272b] Drug release mechanism and profile depends on the hierarchical network of the gel, drug-drug interactions, drug-matrix interactions, drug surface charges, and drug hydrophobicity.[387] As a general design criterion for a gel-based drug system, inclusion of hydrophobic segments is commonly used to contain hydrophobic drugs and reduce porosity for controlled release.[388] Regardless, the development of therapeutic liquid/gel embolic agents is still limited (compared to DEBs) and remains to be explored.
Before closing, it is important to note that the investigation of embolic agent efficacy relies on accurate preclinical models. In theory, the pig makes an ideal large animal model because its fibrinolytic system closely simulates that of humans. Although canine models are also used widely as large animal models, the englobulin lysis time in dog is much shorter (40 min) than man (2–8 hours).[145] The porcine rete mirabile has widely been used as endovascular embolization model for AVM since it has a fine vascular network that resembles human arterioles with the same histological structures.[309a] Regarding aneurysm models, pigs have an aggressive healing response that is different from humans. Therefore, porcine aneurysms may seal rapidly with presence of thrombus. To address this issue, anticoagulation treatment should be considered. Also, thrombus formation should to be closely monitored at the aneurysm neck during healing process.[259] Furthermore, when examining recurrence after aneurysm embolization, an animal model that is prone to recanalization would be more appropriate, such as a canine bifurcation aneurysm model and the elastase-induced aneurysm in rabbits.[389] Lastly, to model liver tumor embolization, the VX2 tumor in rabbit is commonly used because of its rapid growth and similarity in blood supply to that of human HCC, though VX2 tumor is not of hepatic origin.[251] The liver is widely selected for embolization because of its dual blood supply. Its unique structure minimizes necrosis and fibrotic tissue change that may influence the infiltration of inflammatory cells.[161, 390]
Successful embolization relies on material selection, radiological imaging and the experience of the interventional radiologists. Thorough understanding of material properties, anatomy and meticulous surgical technique are required to investigate embolic efficacy of newly developed agents and minimize complications. Bridging engineering and patient-inspired problems provides an emerging paradigm to address limitations in clinically used embolic agents and highlights the design criteria for desired materials.[391] This fertile and multidisciplinary field requires integrated effort from all disciplines to revolutionize the next generation embolic materials.
Acknowledgements
We thank Dr. Aman Saini for manuscript editing, Dr. Zefu Zhang and Yash Pershad for helpful discussions and Xinyu Li for assistance with graphics.
Author Biographies and photographs
Dr. Jingjie Hu PhD is a postdoctoral research fellow at the Minimally Invasive Therapeutics Laboratory at Mayo Clinic working on the development and characterization of embolic agents for endovascular embolization. She received her B.S.E. from the University of Michigan, Ann Arbor and her Ph.D. from Princeton University in Mechanical Engineering. Her broad research interests include biomaterials, nanotechnology and mechanical properties of materials.

Dr. Rahmi Oklu MD, PhD, FSIR is a Professor, Chair and Consultant in the Division of Vascular & Interventional Radiology and the Director of the Minimally Invasive Therapeutics Laboratory at Mayo Clinic. He received his BS degree from Yale University, MD degree from Northwestern University and his PhD degree in Biochemistry from the University of Cambridge, UK. He completed his surgery and radiology training at Columbia University NY Presbyterian Hospital and his fellowship in Vascular & Interventional Radiology at the Massachusetts General Hospital, Harvard Medical School. His current research is focused on patient inspired engineering to develop medical devices.

Footnotes
Conflict of Interest
RO and AK have recently launched a joint start-up Obsidio, Inc. in the field of vascular embolization.
Contributor Information
Jingjie Hu, Division of Vascular & Interventional Radiology, Minimally Invasive Therapeutics Laboratory, Mayo Clinic, 13400 East Shea Blvd., Scottsdale, Arizona 85259, USA.
Hassan Albadawi, Division of Vascular & Interventional Radiology, Minimally Invasive Therapeutics Laboratory, Mayo Clinic, 13400 East Shea Blvd., Scottsdale, Arizona 85259, USA.
Rahmi Oklu, Division of Vascular & Interventional Radiology, Minimally Invasive Therapeutics Laboratory, Mayo Clinic, 13400 East Shea Blvd., Scottsdale, Arizona 85259, USA.
Brian W Chong, Departments of Radiology and Neurological Surgery, Mayo Clinic, 13400 East Shea Blvd., Scottsdale, Arizona 85259, USA.
Amy R. Deipolyi, Department of Interventional Radiology, Memorial Sloan Kettering Cancer Center, Weill Cornell Medical Center, 1275 York Avenue, New York, New York 10065, USA.
Rahul A. Sheth, Department of Interventional Radiology, The University of Texas MD Anderson Cancer Center, Houston, Texas 77054, USA
Ali Khademhosseini, Department of Bioengineering, Department of Radiological Sciences, Department of Chemical and Biomolecular Engineering, Center for Minimally Invasive Therapeutics, California Nanosystems Institute, University of California, 410 Westwood Plaza, Los Angeles, California 90095, USA.
References
- [1].Sheth RA, Sabir S, Krishnamurthy S, Avery RK, Zhang YS, Khademhosseini A, Oklu R, J. Funct. Biomater 2017, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Leyon JJ, Littlehales T, Rangarajan B, Hoey ET, Ganeshan A, Curr. Probl. Diagn. Radiol 2014, 43, 35. [DOI] [PubMed] [Google Scholar]
- [3].a) Transcatheter Embolization and Therapy 2010, 1; [Google Scholar]; b) Behravesh S, Yakes W, Gupta N, Naidu S, Chong BW, Khademhosseini A, Oklu R, Cardiovascular Diagnosis and Therapy 2016, 6, 557; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gunn AJ, Oklu R, J. Obes 2014, 2014, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Interventional Radiology, Williams and Wilkins, Baltimore: 1997; [Google Scholar]; b) Luessenhop AJ, Spence WT, JAMA, J. Am. Med. Assoc 1960, 172, 1153; [DOI] [PubMed] [Google Scholar]; c) Dawbarn RHM, J. Am. Med. Assoc 1904, 43, 0792. [Google Scholar]
- [5].Brooks B, South. Med. J 1930, 23, 100. [Google Scholar]
- [6].a) Vitek JJ, Smith MJ, J. Neurointerv. Surg 2009, 1, 108; [DOI] [PubMed] [Google Scholar]; b) Hamby WB, Gardner WJ, Arch. Surg 1933, 27, 676. [Google Scholar]
- [7].a) Dotter CT, Judkins MP, Circulation 1964, 30, 654; [DOI] [PubMed] [Google Scholar]; b) Payne MM, Tex. Heart Inst. J 2001, 28, 28. [PMC free article] [PubMed] [Google Scholar]
- [8].Poursaid A, Jensen MM, Huo E, Ghandehari H, J. Controlled Release 2016, 240, 414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lubarsky M, Ray CE, Funaki B, Seminars in interventional radiology 2009, 26, 352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Avery RK, Albadawi H, Akbari M, Zhang YS, Duggan MJ, Sahani DV, Olsen BD, Khademhosseini A, Oklu R, Sci. Transl. Med 2016, 8. [DOI] [PubMed] [Google Scholar]
- [11].a) Ginat DT, Saad WEA, Turba UC, Techniques in vascular and interventional radiology 2009, 12, 224; [DOI] [PubMed] [Google Scholar]; b) Hickman DA, Pawlowski CL, Sekhon UDS, Marks J, Sen Gupta A, Adv. Mater 2018, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Guglielmi G, Operative Techniques in Neurosurgery 2000, 3, 191; [Google Scholar]; b) Lawton MT, Rutledge WC, Kim H, Stapf C, Whitehead KJ, Li DY, Krings T, terBrugge K, Kondziolka D, Morgan MK, Moon K, Spetzler RF, Nature Reviews Disease Primers 2015, 1, 15008; [DOI] [PubMed] [Google Scholar]; c) Spiotta AM, James RF, Lowe SR, Vargas J, Turk AS, Chaudry MI, Bhalla T, Janjua RM, Delaney JJ, Quintero-Wolfe S, Turner RD, J. Neurointerv. Surg 2015, 7, 721; [DOI] [PubMed] [Google Scholar]; d) Idee JM, Guiu B, Critical Reviews in Oncology Hematology 2013, 88, 530; [DOI] [PubMed] [Google Scholar]; e) Stewart EA, Laughlin-Tommaso SK, Catherino WH, Lalitkumar S, Gupta D, Vollenhoven B, Nature Reviews Disease Primers 2016, 2. [DOI] [PubMed] [Google Scholar]
- [13].Jiang B, Paff M, Colby GP, Coon AL, Lin L-M, Stroke and vascular neurology 2016, 1, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) Irita K, Korean J. Anesthesiol 2011, 60, 151; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Froberg L, Helgstrand F, Clausen C, Steinmetz J, Eckardt H, J. Emerg. Trauma Shock 2016, 9, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Kauvar DS, Wade CE, Critical Care 2005, 9, 1; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Rossaint R, Bouillon B, Cerny V, Coats TJ, Duranteau J, Fernandez-Mondejar E, Hunt BJ, Komadina R, Nardi G, Neugebauer E, Ozier Y, Riddez L, Schultz A, Stahel PF, Vincent JL, Spahn DR, Critical Care 2010, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].a) Gianturco C, Anderson JH, Wallace S, Am. J. Roentgenol 1975, 124, 428; [DOI] [PubMed] [Google Scholar]; b) Eriksson LG, Ljungdahl M, Sundbom M, Nyman R, J. Vasc. Interv. Radiol 2008, 19, 1413; [DOI] [PubMed] [Google Scholar]; c) Weiss VJ, Lumsden AB, World J. Surg 1999, 23, 406. [DOI] [PubMed] [Google Scholar]
- [17].a) Gomes AS, Lois JF, McCoy RD, Am. J. Roentgenol 1986, 146, 1031; [DOI] [PubMed] [Google Scholar]; b) Loffroy R, Guiu B, D’Athis P, Mezzetta L, Gagnaire A, Jouve JL, Ortega-Deballon P, Cheynel N, Cercueil JP, Krausé D, Clin. Gastroenterol. Hepatol 2009, 7, 515; [DOI] [PubMed] [Google Scholar]; c) Poultsides GA, Kim CJ, Orlando R III, Peros G, Hallisey MJ, Vignati PV, Arch. Surg 2008, 143, 457; [DOI] [PubMed] [Google Scholar]; d) Moger T, Atle Bjørnbeth B, Lygren I, Kløw N-E, Scand. J. Gastroenterol 2008, 43, 217; [DOI] [PubMed] [Google Scholar]; e) Loffroy R, Guiu B, Cercueil J-P, Lepage C, Latournerie M, Hillon P, Rat P, Ricolfi F, Krausé D, J. Clin. Gastroenterol 2008, 42, 361; [DOI] [PubMed] [Google Scholar]; f) Širvinskas A, Smolskas E, Mikelis K, Brimienė V, Brimas G, Videosurgery and Other Miniinvasive Techniques 2017, 12, 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Shoeb M, Fang MC, Thromb J. Thrombolysis 2013, 35, 312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ramaswamy RS, Choi HW, Mouser HC, Narsinh KH, McCammack KC, Treesit T, Kinney TB, World journal of radiology 2014, 6, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Loffroy R, Guiu B, Cercueil JP, Krause D, Curr. Vasc. Pharmacol 2009, 7, 250. [DOI] [PubMed] [Google Scholar]
- [21].Guglielmi G, Vinuela F, Sepetka I, Macellari V, J. Neurosurg 1991, 75, 1. [DOI] [PubMed] [Google Scholar]
- [22].Bonneville F, Sourour N, Biondi A, Neuroimaging Clin. N. Am 2006, 16, 371. [DOI] [PubMed] [Google Scholar]
- [23].a) Rodriguez JN, Hwang WJ, Horn J, Landsman TL, Boyle A, Wierzbicki MA, Hasan SM, Follmer D, Bryant J, Small W, Maitland DJ, J. Biomed. Mater. Res. A 2015, 103, 1577; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Stehbens WE, Neurol. Res 1990, 12, 29. [DOI] [PubMed] [Google Scholar]
- [24].Molyneux AJ, Kerr RSC, Yu LM, Clarke M, Sneade M, Yarnold JA, Sandercock P, Grp IC, Lancet 2005, 366, 809. [DOI] [PubMed] [Google Scholar]
- [25].Brilstra EH, Rinkel GJE, van der Graaf Y, van Rooij WJJ, Algra A, Stroke 1999, 30, 470. [DOI] [PubMed] [Google Scholar]
- [26].Guglielmi G, Interv. Neuroradiol 2007, 13, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].a) Nair P, Chong BW, Indahlastari A, Lindsay J, DeJeu D, Parthasarathy V, Ryan J, Babiker H, Workman C, Gonzalez LF, Frakes D, JBiom 2016, 49, 2118; [DOI] [PubMed] [Google Scholar]; b) Nair P, Chong BW, Indahlastari A, Ryan J, Workman C, Haithem Babiker M, Yadollahi Farsani H, Baccin CE, Frakes D, J. Biomech. Eng 2016, 138, 021011. [DOI] [PubMed] [Google Scholar]
- [28].Friedlander RM, New Engl. J. Med 2007, 356, 2704. [DOI] [PubMed] [Google Scholar]
- [29].Ostergaard L, Engedal TS, Moreton F, Hansen MB, Wardlaw JM, Dalkara T, Markus HS, Muir KW, J. Cereb. Blood Flow Metab 2016, 36, 302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Brassel F, Meila D, Clin. Neuroradiol 2015, 25, 333. [DOI] [PubMed] [Google Scholar]
- [31].Vaidya S, Tozer KR, Chen J, Seminars in interventional radiology 2008, 25, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Nassiri N, Cirillo-Penn NC, Thomas J, J. Vasc. Surg 2015, 62, 1667. [DOI] [PubMed] [Google Scholar]
- [33].Rosen RJ, Contractor S, Seminars in interventional radiology 2004, 21, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Deipolyi AR, Zhang YS, Khademhosseini A, Naidu S, Borad M, Sahin B, Mathur AK, Oklu R, J. Clin. Med 2017, 6, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ladner TR, He L, Lakomkin N, Davis BJ, Cheng JS, Devin CJ, Mocco J, J. Neurointerv. Surg 2016, 8, 210. [DOI] [PubMed] [Google Scholar]
- [36].Ghouri Y, Mian I, Rowe J, J. Carcinog. 2017, 16, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kanwal F, Befeler A, Chari RS, Marrero J, Kahn J, Afdhal N, Morgan T, Roberts L, Mohanty SR, Schwartz J, VanThiel D, Li J, Zeringue A, Di Bisceglie A, Aliment. Pharmacol. Ther 2012, 36, 257. [DOI] [PubMed] [Google Scholar]
- [38].a) Roxburgh P, Evans TRJ, Adv. Ther 2008, 25, 1089; [DOI] [PubMed] [Google Scholar]; b) Nordenstedt H, White DL, El-Serag HB, Dig. Liver Dis 2010, 42, S206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lencioni R, Crocetti L, Radiology 2012, 262, 43. [DOI] [PubMed] [Google Scholar]
- [40].Doyon D, Mouzon A, Jourde AM, Regensberg C, Frileux C, Ann. Radiol. (Paris) 1974, 17, 593. [PubMed] [Google Scholar]
- [41].a) Nishikawa H, Kita R, Kimura T, Osaki Y, Anticancer Res. 2014, 34, 6877; [PubMed] [Google Scholar]; b) Osuga K, Maeda N, Higashihara H, Hori S, Nakazawa T, Tanaka K, Nakamura M, Kishimoto K, Ono Y, Tomiyama N, Int. J. Clin. Oncol 2012, 17, 306. [DOI] [PubMed] [Google Scholar]
- [42].Deipolyi AR, Oklu R, Al-Ansari S, Zhu AX, Goyal L, Ganguli S, J. Vasc. Interv. Radiol 2015, 26, 516. [DOI] [PubMed] [Google Scholar]
- [43].a) Bester L, Meteling B, Boshell D, Chua TC, Morris DL, J. Med. Imaging Radiat. Oncol 2014, 58, 341; [DOI] [PubMed] [Google Scholar]; b) Sheth RA, Patel MS, Koottappillil B, Shah JA, Oklu R, Mueller P, Vagefi PA, Ganguli S, J. Vasc. Interv. Radiol 2015, 26, 1761. [DOI] [PubMed] [Google Scholar]
- [44].Wallach EE, Vlahos NF, Obstet. Gynecol 2004, 104, 393. [DOI] [PubMed] [Google Scholar]
- [45].Saralidze K, Koole LH, Knetsch MLW, Materials 2010, 3, 3537. [Google Scholar]
- [46].Ravina JH, Herbreteau D, Ciraruvigneron N, Bouret JM, Houdart E, Aymard A, Merland JJ, Lancet 1995, 346, 671. [DOI] [PubMed] [Google Scholar]
- [47].Mara M, Kubinova K, Int. J. Women’s Health 2014, 6, 623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Zhou C, Cui DC, Zhang Y, Yuan HY, Fan TY, J. Mater. Sci.: Mater. Med 2012, 23, 409. [DOI] [PubMed] [Google Scholar]
- [49].a) Pisco JM, Bilhim T, Pinheiro LC, Fernandes L, Pereira J, Costa NV, Duarte M, Oliveira AG, J. Vasc. Interv. Radiol 2016, 27, 1115; [DOI] [PubMed] [Google Scholar]; b) Malling B, Røder MA, Brasso K, Forman J, Taudorf M, Lönn L, Eur. Radiol 2019, 29, 287. [DOI] [PubMed] [Google Scholar]
- [50].a) Orth RC, Wallace MJ, Kuo MD, J. Vasc. Interv. Radiol 2008, 19, 814; [DOI] [PubMed] [Google Scholar]; b) Tacher V, Radaelli A, Lin M, Geschwind J-F, Radiology 2015, 274, 320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Choi JY, Lee JM, Sirlin CB, Radiology 2014, 273, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Tian L, Lu LF, Feng J, Melancon MP, Acta Pharm. Sin. B 2018, 8, 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Frydrychowicz A, Lubner MG, Brown JJ, Merkle EM, Nagle SK, Rofsky NM, Reeder SB, J. Magn. Reson. Imaging 2012, 35, 492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].a) Hodnett PA, Koktzoglou I, Davarpanah AH, Scanlon TG, Collins JD, Sheehan JJ, Dunkle EE, Gupta N, Carr JC, Edelman RR, Radiology 2011, 260, 282; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Carr JC, JACC Cardiovasc. Imaging 2017, 10, 1125. [DOI] [PubMed] [Google Scholar]
- [55].Evangelista A, Flachskampf FA, Erbel R, Antonini-Canterin F, Vlachopoulos C, Rocchi G, Sicari R, Nihoyannopoulos P, Zamorano J, European Assoc E, Eur. J. Echocardiogr 2010, 11, 645. [DOI] [PubMed] [Google Scholar]
- [56].Sizemore G, Ayubi F, Clark B, Kellicut D, Journal of Vascular Diagnostics and Interventions 2018, 2018:6, 1. [Google Scholar]
- [57].Zhu Y, Zhang H, Zhang Y, Wu H, Wei L, Zhou G, Zhang Y, Deng L, Cheng Y, Li M, Santos HA, Cui W, Adv. Mater 2018, 0, 1805452. [DOI] [PubMed] [Google Scholar]
- [58].Lazzaro MA, Badruddin A, Zaidat OO, Darkhabani Z, Pandya DJ, Lynch JR, Front. Neurol 2011, 2, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Galal MO, J. Interv. Cardiol 2003, 16, 157. [DOI] [PubMed] [Google Scholar]
- [60].Tisnado J, Beachley MC, Cho SR, Amendola M, Am. J. Roentgenol 1979, 133, 324. [DOI] [PubMed] [Google Scholar]
- [61].White JB, Ken CGM, Cloft HJ, Kallmes DF, Am. J. Neuroradiol 2008, 29, 1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Ahuja AA, Hergenrother RW, Strother CM, Rappe AA, Cooper SL, Graves VB, Am. J. Neuroradiol 1993, 14, 794. [PMC free article] [PubMed] [Google Scholar]
- [63].Slesnick TC, Schreier J, Soriano BD, Kutty S, Nutting AC, Kim DW, Powell AJ, Valente AM, Pediatr. Cardiol 2016, 37, 62. [DOI] [PubMed] [Google Scholar]
- [64].a) Wallace S, Gianturco C, Anderson JH, Goldstein HM, Davis LJ, Bree RL, Am. J. Roentgenol 1976, 127, 381; [DOI] [PubMed] [Google Scholar]; b) Zollikofer C, Castanedazuniga WR, Galliani C, Rysavy JA, Tadavarthy M, Formanek A, Amplatz K, Radiology 1981, 138, 229. [DOI] [PubMed] [Google Scholar]
- [65].Duerig T, Pelton A, Stockel D, Mater. Sci. Eng., A 1999, 273, 149. [Google Scholar]
- [66].Thompson SA, Int. Endod. J 2001, 33, 297. [DOI] [PubMed] [Google Scholar]
- [67].Konya A, Maxin M, Wright KC, J. Vasc. Interv. Radiol 2001, 12, 869. [DOI] [PubMed] [Google Scholar]
- [68].Mascitelli JR, Polykarpou MF, Patel AA, Kamath AA, Moyle H, Patel AB, J. Neurointerv. Surg 2013, 5, 573. [DOI] [PubMed] [Google Scholar]
- [69].Waters NE, Br. J. Orthod 1992, 19, 319. [DOI] [PubMed] [Google Scholar]
- [70].Lanzino G, Kanaan Y, Perrini P, Dayoub H, Fraser K, Neurosurgery 2005, 57, 449. [DOI] [PubMed] [Google Scholar]
- [71].Liebig T, Henkes H, Fischer S, Weber W, Miloslavski E, Mariushi W, Brew S, Kuhne D, Interv. Neuroradiol 2004, 10, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Reinges MHT, Krings T, Drexler AY, Ludolph A, Sellhaus B, Bovi M, Geibprasert S, Agid R, Scherer K, Hans FJ, Interv. Neuroradiol 2010, 16, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Barth KH, Strandberg JD, Kaufman SL, White RI, Am. J. Roentgenol 1978, 131, 455. [DOI] [PubMed] [Google Scholar]
- [74].White RI Jr., Pollak JS, Vascular Embolotherapy, Vol 1: General Principles, Chest, Abdomen, and Great Vessels 2006, 35. [Google Scholar]
- [75].Song JK, Niimi Y, Fernandez PM, Brisman JL, Buciuc R, Kupersmith MJ, Berenstein A, Am. J. Neuroradiol 2004, 25, 1147. [PMC free article] [PubMed] [Google Scholar]
- [76].Fiorella D, Albuquerque FC, McDougall CG, Neurosurgery 2006, 58, 51. [DOI] [PubMed] [Google Scholar]
- [77].Murayama Y, Vinuela F, Tateshima S, Song JK, Gonzalez NR, Wallace MP, J. Neurosurg 2001, 94, 454. [DOI] [PubMed] [Google Scholar]
- [78].Vance A, Welch BG, Neurol. Res 2014, 36, 356. [DOI] [PubMed] [Google Scholar]
- [79].a) Ding YH, Dai DY, Lewis DA, Cloft HJ, Kallmes DF, Am. J. Neuroradiol 2005, 26, 1757; [PMC free article] [PubMed] [Google Scholar]; b) Ansari SA, Dueweke EJ, Kanaan Y, Chaudhary N, Gandhi D, Thompson BG, Gemmete JJ, J. Neurointerv. Surg 2011, 3, 324. [DOI] [PubMed] [Google Scholar]
- [80].a) Rivet DJ, Moran CJ, Mazumdar A, Pilgram TK, Derdeyn CP, Cross DT, Am. J. Neuroradiol 2007, 28, 1736; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Taschner CA, Leclerc X, Rachdi H, Barros AM, Pruvo JP, Stroke 2005, 36, 2176. [DOI] [PubMed] [Google Scholar]
- [81].Takigawa T, Matsumaru Y, Nakai Y, Nakamura K, Hayakawa M, Tsuruta W, Matsumura A, Headache 2012, 52, 312. [DOI] [PubMed] [Google Scholar]
- [82].Grasel TG, Cooper SL, Biomaterials 1986, 7, 315. [DOI] [PubMed] [Google Scholar]
- [83].Xu LC, Bauer JW, Siedlecki CA, Colloids Surf., B 2014, 124, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Okkema AZ, Visser SA, Cooper SL, J. Biomed. Mater. Res 1991, 25, 1371. [DOI] [PubMed] [Google Scholar]
- [85].Skarja GA, Woodhouse KA, J. Biomater. Sci., Polym. Ed 1998, 9, 271. [DOI] [PubMed] [Google Scholar]
- [86].Parrag IC, Woodhouse KA, J. Biomater. Sci., Polym. Ed 2010, 21, 843. [DOI] [PubMed] [Google Scholar]
- [87].Chen JY, Yang LJ, Chen Y, Zhang GS, Fan ZN, J. Neurol. Sci 2016, 360, 13. [DOI] [PubMed] [Google Scholar]
- [88].a) Aoki T, Kataoka H, Ishibashi R, Nozaki K, Egashira K, Hashimoto N, Stroke 2009, 40, 942; [DOI] [PubMed] [Google Scholar]; b) Hoh BL, Hosaka K, Downes DP, Nowicki KW, Fernandez CE, Batich CD, Scott EW, Circulation 2011, 124, 2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].a) Kadirvel R, Ding YH, Dai DY, Lewis DA, Kallmes DF, Radiology 2010, 257, 418; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kadirvel R, Ding YH, Dai DY, Lewis DA, Kallmes DF, Neurosurgery 2010, 66, 578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Brinjikji W, Kallmes DF, Kadirvel R, Am. J. Neuroradiol 2015, 36, 1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Raymond J, Desfaits AC, Roy D, Stroke 1999, 30, 1657. [DOI] [PubMed] [Google Scholar]
- [92].Abrahams JM, Forman MS, Grady MS, Diamond SL, Am. J. Neuroradiol 2001, 22, 1410. [PMC free article] [PubMed] [Google Scholar]
- [93].a) Bavinzski G, Richling B, Binder BR, Gruber A, Talazoglu V, Dietrich W, Schwendtenwein I, Plenk H, Minim. Invasive Neurosurg 1999, 42, 167; [DOI] [PubMed] [Google Scholar]; b) Marx WF, Cloft HJ, Helm GA, Short JG, Do HM, Jensen ME, Kallmes DF, Am. J. Neuroradiol 2001, 22, 323; [PMC free article] [PubMed] [Google Scholar]; c) Dai D, Ding YH, Danielson MA, Kadirvel R, Helm GA, Lewis DA, Cloft HJ, Kallmes DF, Am. J. Neuroradiol 2008, 29, 739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Cruise GM, Shum JC, Plenk H, JMCh 2007, 17, 3965. [Google Scholar]
- [95].Fanning NF, Berentei Z, Brennan PR, Thornton J, Neuroradiology 2007, 49, 139. [DOI] [PubMed] [Google Scholar]
- [96].Berenstein A, Song JK, Niimi Y, Namba K, Heran NS, Brisman JL, Nahoum MC, Madrid M, Langer DJ, Kupersmith MJ, Am. J. Neuroradiol 2006, 27, 1834. [PMC free article] [PubMed] [Google Scholar]
- [97].Shimohira M, Kawai T, Hashizume T, Muto M, Kitase M, Shibamoto Y, Cardiovasc. Intervent. Radiol 2018, 41, 848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Zhang C, Chaudhary N, Gemmete JJ, Thompson BG, Xi GH, Pandey AS, J. Neurointerv. Surg 2014, 6, 480. [DOI] [PubMed] [Google Scholar]
- [99].Raymond J, Klink R, Chagnon M, Barnwell SL, Evans AJ, Mocco J, Hoh BH, Turk AS, Turner RD, Desal H, Fiorella D, Bracard S, Weill A, Guilbert F, Lanthier S, Fox AJ, Darsaut TE, White PM, Roy D, Am. J. Neuroradiol 2017, 38, 432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Marchan EM, Sekula RF, Ku A, Williams R, O’Neill BR, Wilberger JE, Quigley MR, J. Neurosurg 2008, 109, 186. [DOI] [PubMed] [Google Scholar]
- [101].Kulcsar Z, Houdart E, Bonafe A, Parker G, Millar J, Goddard AJP, Renowden S, Gal G, Turowski B, Mitchell K, Gray F, Rodriguez M, van den Berg R, Gruber A, Desal H, Wanke I, Rufenacht DA, Am. J. Neuroradiol 2011, 32, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Byrne JV, Hope JKA, Hubbard N, Morris JH, Am. J. Neuroradiol 1997, 18, 29. [PMC free article] [PubMed] [Google Scholar]
- [103].Bavinzski G, Talazoglu V, Killer M, Richling B, Gruber A, Gross CE, Plenk H, J. Neurosurg 1999, 91, 284. [DOI] [PubMed] [Google Scholar]
- [104].Groden C, Hagel C, Delling G, Zeumer H, Am. J. Neuroradiol 2003, 24, 579. [PMC free article] [PubMed] [Google Scholar]
- [105].a) Horowitz MB, Purdy PD, Burns D, Bellotto D, Am. J. Neuroradiol 1997, 18, 688; [PMC free article] [PubMed] [Google Scholar]; b) Shimizu S, Kurata A, Takano M, Takagi H, Yamazaki H, Miyasaka Y, Fujii K, Am. J. Neuroradiol 1999, 20, 546. [PMC free article] [PubMed] [Google Scholar]
- [106].RAMACHANDRAN R, MS University of Cincinnati, 2008. [Google Scholar]
- [107].Haworth KJ, Weidner CR, Abruzzo TA, Shearn JT, Holland CK, J. Neurointerv. Surg 2015, 7, 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Weisel JW, J. Thromb. Haemost 2007, 5, 116. [DOI] [PubMed] [Google Scholar]
- [109].Collet JP, Shuman H, Ledger RE, Lee ST, Weisel JW, Proc. Natl. Acad. Sci. U. S. A 2005, 102, 9133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Killer M, Plenk H, Minnich B, Al-Schameri R, Lametschwantner A, Richling B, Minim. Invasive Neurosurg 2009, 52, 170. [DOI] [PubMed] [Google Scholar]
- [111].Kallmes DF, Fujiwara NH, Am. J. Neuroradiol 2002, 23, 1580. [PMC free article] [PubMed] [Google Scholar]
- [112].Cloft HJ, Kallmes DF, Am. J. Neuroradiol 2004, 25, 60. [PMC free article] [PubMed] [Google Scholar]
- [113].Serafin Z, Di Leo G, Palys A, Nowaczewska M, Beuth W, Sardanelli F, Eur. J. Radiol 2015, 84, 1954. [DOI] [PubMed] [Google Scholar]
- [114].a) El Feninat F, Laroche G, Fiset M, Mantovani D, Adv. Eng. Mater 2002, 4, 91; [Google Scholar]; b) Lendlein A, Langer R, Sci 2002, 296, 1673. [DOI] [PubMed] [Google Scholar]
- [115].Kunkel R, Laurence D, Wang J, Robinson D, Scherrer J, Wu Y, Bohnstedt B, Chien A, Liu Y, Lee C-H, J. Mech. Behav. Biomed. Mater 2018, 88, 422. [DOI] [PubMed] [Google Scholar]
- [116].Wong YS, Salvekar AV, Da Zhuang K, Liu H, Birch WR, Tay KH, Huang WM, Venkatraman SS, Biomaterials 2016, 102, 98. [DOI] [PubMed] [Google Scholar]
- [117].a) Sun JY, Zhao XH, Illeperuma WRK, Chaudhuri O, Oh KH, Mooney DJ, Vlassak JJ, Suo ZG, Nature 2012, 489, 133; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Haque MA, Kurokawa T, Gong JP, Poly 2012, 53, 1805. [Google Scholar]
- [118].Zhang YY, Gao HJ, Wang H, Xu ZY, Chen XW, Liu B, Shi Y, Lu Y, Wen LF, Li Y, Li ZS, Men YF, Feng XQ, Liu WG, Adv. Funct. Mater 2018, 28. [Google Scholar]
- [119].Zhang YY, Li YM, Liu WG, Adv. Funct. Mater 2015, 25, 471. [Google Scholar]
- [120].Wang W, Zhang YY, Liu WG, Prog. Polym. Sci 2017, 71, 1. [Google Scholar]
- [121].Lake GJ, Rubber Chem. Technol 1995, 68, 435. [Google Scholar]
- [122].Chuang VP, Wallace S, Gianturco C, Soo CS, Am. J. Roentgenol 1981, 137, 809. [DOI] [PubMed] [Google Scholar]
- [123].a) Chueh JY, Vedantham S, Wakhloo AK, Carniato SL, Puri AS, Bzura C, Coffin S, Bogdanov AA, Gounis MJ, J. Neurointerv. Surg 2015, 7, 676; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Morales HG, Larrabide I, Geers AJ, Aguilar ML, Frangi AF, JBiom 2013, 46, 2158; [DOI] [PubMed] [Google Scholar]; c) Cha KS, Balaras E, Lieber BB, Sadasivan C, Wakhloo AK, J. Biomech. Eng 2007, 129, 873. [DOI] [PubMed] [Google Scholar]
- [124].Jou LD, Am. J. Neuroradiol 2010, 31, E42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Eddleman CS, Welch BG, Vance AZ, Rickert KL, White JA, Pride GL, Purdy PD, J. Neurointerv. Surg 2013, 5, 104. [DOI] [PubMed] [Google Scholar]
- [126].a) Behl M, Razzaq MY, Lendlein A, Adv. Mater 2010, 22, 3388; [DOI] [PubMed] [Google Scholar]; b) Serrano MC, Carbajal L, Ameer GA, Adv. Mater 2011, 23, 2211. [DOI] [PubMed] [Google Scholar]
- [127].Small W, Buckley PR, Wilson TS, Benett WJ, Hartman J, Saloner D, Maitland DJ, ITBE 2007, 54, 1157. [DOI] [PubMed] [Google Scholar]
- [128].Landsman TL, Bush RL, Glowczwski A, Horn J, Jessen SL, Ungchusri E, Diguette K, Smith HR, Hasan SM, Nash D, Clubb FJ, Maitland DJ, J. Mech. Behav. Biomed. Mater 2016, 63, 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Metcalfe A, Desfaits AC, Salazkin I, Yahia L, Sokolowski WM, Raymond J, Biomaterials 2003, 24, 491. [DOI] [PubMed] [Google Scholar]
- [130].Peterson GI, Dobrynin AV, Becker ML, Adv. Healthcare Mater 2017, 6. [DOI] [PubMed] [Google Scholar]
- [131].Rodriguez JN, Yu YJ, Miller MW, Wilson TS, Hartman J, Clubb FJ, Gentry B, Maitland DJ, Ann. Biomed. Eng 2012, 40, 883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].a) Hampikian JM, Heaton BC, Tong FC, Zhang ZQ, Wong CP, Mater. Sci. Eng., C: Biomimetic Supramol. Syst 2006, 26, 1373; [Google Scholar]; b) Hwang W, Singhal P, Miller MW, Maitland DJ, Journal of Medical Devices-Transactions of the Asme 2013, 7. [Google Scholar]
- [133].a) Boyle AJ, Wierzbicki MA, Herting S, Weems AC, Nathan A, Hwang W, Maitland DJ, Med. Eng. Phys 2017, 49, 56; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nash LD, Monroe MBB, Ding YH, Ezell KP, Boyle AJ, Kadirvel R, Kallmes DF, Maitland DJ, Polymers 2017, 9; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Boyle AJ, Landsman TL, Wierzbicki MA, Nash LD, Hwang W, Miller MW, Tuzun E, Hasan SM, Maitland DJ, J. Biomed. Mater. Res., Part B 2016, 104, 1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Singhal P, Rodriguez JN, Small W, Eagleston S, de Water JV, Maitland DJ, Wilson TS, J. Polym. Sci., Part B: Polym. Phys 2012, 50, 724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].De Nardo L, Alberti R, Cigada A, Yahia L, Tanzi MC, Fare S, Acta Biomater 2009, 5, 1508. [DOI] [PubMed] [Google Scholar]
- [136].MacDonald DJ, Finlay HM, Canham PB, Ann. Biomed. Eng 2000, 28, 533. [DOI] [PubMed] [Google Scholar]
- [137].Konig G, McAllister TN, Dusserre N, Garrido SA, Iyican C, Marini A, Fiorillo A, Avila H, Wystrychowski W, Zagalski K, Maruszewski M, Jones AL, Cierpka L, de la Fuente LM, L’Heureux N, Biomaterials 2009, 30, 1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Rodriguez JN, Miller MW, Boyle A, Horn J, Yang CK, Wilson TS, Ortega JM, Small W, Nash L, Skoog H, Maitland DJ, J. Mech. Behav. Biomed. Mater 2014, 40, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Rodriguez JN, Clubb FJ, Wilson TS, Miller MW, Fossum TW, Hartman J, Tuzun E, Singhal P, Maitland DJ, J. Biomed. Mater. Res. A 2014, 102, 1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Horn J, Hwang W, Jessen SL, Keller BK, Miller MW, Tuzun E, Hartman J, Clubb FJ, Maitland DJ, J. Biomed. Mater. Res., Part B 2017, 105, 1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].a) Kim BK, Lee SY, Xu M, Poly 1996, 37, 5781; [Google Scholar]; b) Cherng JY, Hou TY, Shih MF, Talsma H, Hennink WE, Int. J. Pharm 2013, 450, 145. [DOI] [PubMed] [Google Scholar]
- [142].Lubarsky M, Ray C, Funaki B, Seminars in interventional radiology 2010, 27, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].van Es OFRJJ, Dullens HFJ, Bernsen MR, Bosman F, Hennink WE, Slootweg PJ, Lab. Anim 1999, 33, 175. [DOI] [PubMed] [Google Scholar]
- [144].Bannerman D, Wan WK, Expert Opin. Drug Delivery 2016, 13, 1289. [DOI] [PubMed] [Google Scholar]
- [145].Kunstlinger F, Brunelle F, Chaumont P, Doyon D, Am. J. Roentgenol 1981, 136, 151. [DOI] [PubMed] [Google Scholar]
- [146].a) Tadavarthy SM, Moller JH, Amplatz K, Am. J. Roentgenol 1975, 125, 609; [DOI] [PubMed] [Google Scholar]; b) Porstmann W, Wierny L, Warnke H, Gerstberger G, Romaniuk PA, Radiol. Clin. North Am 1971, 9, 203. [PubMed] [Google Scholar]
- [147].Herrera M, Rysavy J, Kotula F, Rusnak B, Castanedazuniga WR, Amplatz K, Radiology 1982, 144, 638. [DOI] [PubMed] [Google Scholar]
- [148].Bendszus M, Klein R, Burger R, Warmuth-Metz M, Hofmann E, Solymosi L, Am. J. Neuroradiol 2000, 21, 255. [PMC free article] [PubMed] [Google Scholar]
- [149].Horton JA, Marano GD, Kerber CW, Jenkins JJ, Davis S, Am. J. Neuroradiol 1983, 4, 143. [PMC free article] [PubMed] [Google Scholar]
- [150].Davidson GS, Terbrugge KG, Am. J. Neuroradiol 1995, 16, 843. [PMC free article] [PubMed] [Google Scholar]
- [151].SPEAKMAN TJ, J. Neurosurg 1964, 21, 303. [DOI] [PubMed] [Google Scholar]
- [152].a) Wang YXJ, De Baere T, Idee JM, Ballet S, Chin. J. Cancer Res 2015, 27, 96; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nitta N, Ohta S, Tanaka T, Takazakura R, Nagatani Y, Kono N, Sonoda A, Seko A, Furukawa A, Takahashi M, Murata K, Tabata Y, Eur. J. Radiol 2008, 67, 536. [DOI] [PubMed] [Google Scholar]
- [153].Owen RJT, Radiol. Clin. North Am 2008, 46, 535. [DOI] [PubMed] [Google Scholar]
- [154].a) Katsumori T, Nakajima K, Mihara T, Tokuhiro M, Am. J. Roentgenol 2002, 178, 135; [DOI] [PubMed] [Google Scholar]; b) Katsumori T, Kasahara T, Akazawa K, Am. J. Roentgenol 2006, 186, 848. [DOI] [PubMed] [Google Scholar]
- [155].a) Miyayama S, Yamakado K, Anai H, Abo D, Minami T, Takaki H, Kodama T, Yamanaka T, Nishiofuku H, Morimoto K, Soyama T, Hasegawa Y, Nakamura K, Yamanishi T, Sato M, Nakajima Y, Jpn. J. Radiol 2014, 32, 242; [DOI] [PubMed] [Google Scholar]; b) Rosch J, Keller FS, Kozak B, Niles N, Dotter CT, Radiology 1984, 151, 365. [DOI] [PubMed] [Google Scholar]
- [156].Kim B, Han SW, Choi SE, Yim D, Kim JH, Wyss HM, Kim JW, Biomacromolecules 2018, 19, 386. [DOI] [PubMed] [Google Scholar]
- [157].a) van Hooy-Corstjens CSJ, Saralidze K, Knetsch MLW, Emans PJ, de Haan MW, Magusin PCMM, Mezari B, Koole LH, Biomacromolecules 2008, 9, 84; [DOI] [PubMed] [Google Scholar]; b) Siskin GP, Englander M, Stainken BF, Ahn J, Dowling K, Dolen EG, Am. J. Roentgenol 2000, 175, 767. [DOI] [PubMed] [Google Scholar]
- [158].Alvarez MM, Aizenberg J, Analoui M, Andrews AM, Bisker G, Boyden ES, Kamm RD, Karp JM, Mooney DJ, Oklu R, Peer D, Stolzoff M, Strano MS, Trujillo-de Santiago G, Webster TJ, Weiss PS, Khademhosseini A, ACS Nano 2017, 11, 5195. [DOI] [PubMed] [Google Scholar]
- [159].Laurent A, Beaujeux R, Wassef M, Rufenacht D, Boschetti E, Merland JJ, Am. J. Neuroradiol 1996, 17, 533. [PMC free article] [PubMed] [Google Scholar]
- [160].a) Obrenovitch A, Maintier C, Sene C, Boschetti E, Monsigny M, Biol. Cell 1982, 46, 249; [Google Scholar]; b) Brown E, Racois A, Boschetti E, Corgier M, JCh 1978, 150, 101. [Google Scholar]
- [161].Stampfl U, Stampfl S, Bellemann N, Sommer CM, Lopez-Benitez R, Thierjung H, Radeleff B, Berger I, Richter GM, Cardiovasc. Intervent. Radiol 2009, 32, 303. [DOI] [PubMed] [Google Scholar]
- [162].Laurent A, Techniques in vascular and interventional radiology 2007, 10, 248. [DOI] [PubMed] [Google Scholar]
- [163].a) Weichert W, Denkert C, Gauruder-Burmester A, Kurzeja R, Amm B, Dietel M, Kroencke TJ, Am. J. Surg. Pathol 2005, 29, 955; [DOI] [PubMed] [Google Scholar]; b) Laurent A, Wassef M, Namur J, Martal J, Labarre D, Pelage JP, Fertil. Steril 2009, 91, 884; [DOI] [PubMed] [Google Scholar]; c) Walker WJ, S. McDowell J, Am. J. Obstet. Gynecol 2006, 195, 1266; [DOI] [PubMed] [Google Scholar]; d) Laurent A, Pelage JP, Wassef M, Martal J, Fertil. Steril 2008, 89, 1371; [DOI] [PubMed] [Google Scholar]; e) Pelage JP, Laurent A, Wassef M, Bonneau M, Germain D, Rymer R, Flaud P, Martal J, Merland JJ, Radiology 2002, 224, 436. [DOI] [PubMed] [Google Scholar]
- [164].Derdeyn CP, Moran CJ, Cross DT, Dietrich HH, Dacey RG, Am. J. Neuroradiol 1995, 16, 1335. [PMC free article] [PubMed] [Google Scholar]
- [165].Lewis AL, Adams C, Busby W, Jones SA, Wolfenden LC, Leppard SW, Palmer RR, Small S, J. Mater. Sci.: Mater. Med 2006, 17, 1193. [DOI] [PubMed] [Google Scholar]
- [166].Schwarz A, Zhang HM, Metcalfe A, Salazkin I, Raymond J, Biomaterials 2004, 25, 5209. [DOI] [PubMed] [Google Scholar]
- [167].Doucet J, Kiri L, O’Connell K, Kehoe S, Lewandowski RJ, Liu DM, Abraham RJ, Boyd D, J. Funct. Biomater 2018, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Yadav P, Yadav H, Shah VG, Shah G, Dhaka G, J. Clin. Diagn. Res 2015, 9, ZE21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Szymanska E, Winnicka K, Mar. Drugs 2015, 13, 1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Park JM, Lee SY, Lee GH, Chung EY, Chang KM, Kwak BK, Kuh HJ, Lee J, J. Microencapsul. 2012, 29, 695. [DOI] [PubMed] [Google Scholar]
- [171].Weng LH, Le HC, Talaie R, Golzarian J, J. Vasc. Interv. Radiol 2011, 22, 1464. [DOI] [PubMed] [Google Scholar]
- [172].Kwak BK, Shim HJ, Han SM, Park ES, Radiology 2005, 236, 151. [DOI] [PubMed] [Google Scholar]
- [173].Zhou X, Kong M, Cheng XJ, Li JJ, Li J, Chen XG, Colloids Surf., B 2014, 123, 387. [DOI] [PubMed] [Google Scholar]
- [174].Zhou X, Kong M, Cheng XJ, Feng C, Li J, Li JJ, Chen XG, Carbohydr. Polym 2014, 113, 304. [DOI] [PubMed] [Google Scholar]
- [175].Weng LH, Rostamzadeh P, Nooryshokry N, Le HC, Golzarian J, Acta Biomater 2013, 9, 6823. [DOI] [PubMed] [Google Scholar]
- [176].Weng LH, Rostambeigi N, Zantek ND, Rostamzadeh P, Bravo M, Carey J, Golzarian J, Acta Biomater. 2013, 9, 8182. [DOI] [PubMed] [Google Scholar]
- [177].Weng LH, Seelig D, Souresrafil O, Cardiovasc. Intervent. Radiol 2018, 41, 951. [DOI] [PubMed] [Google Scholar]
- [178].Tyler T, Rosenbaum HD, Invest. Radiol 1978, 13, 71. [DOI] [PubMed] [Google Scholar]
- [179].Louguet S, Verret V, Bedouet L, Servais E, Pascale F, Wassef M, Labarre D, Laurent A, Moine L, Acta Biomater. 2014, 10, 1194. [DOI] [PubMed] [Google Scholar]
- [180].a) Yamaoka T, Tabata Y, Ikada Y, J. Pharm. Sci 1995, 84, 349; [DOI] [PubMed] [Google Scholar]; b) Fox ME, Szoka FC, Frechet JMJ, Acc. Chem. Res 2009, 42, 1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].a) Okada M, Prog. Polym. Sci 2002, 27, 87; [Google Scholar]; b) Chiang PF, Peng CL, Shih YH, Cho YH, Yu CS, Kuo YM, Shieh MJ, Luo TY, ACS Biomater. Sci. Eng 2018, 4, 3425. [DOI] [PubMed] [Google Scholar]
- [182].Makadia HK, Siegel SJ, Polymers 2011, 3, 1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].a) Grandfils C, Flandroy P, Nihant N, Barbette S, Jerome R, Teyssie P, Thibaut A, J. Biomed. Mater. Res 1992, 26, 467; [DOI] [PubMed] [Google Scholar]; b) Owen RJ, Nation PN, Polakowski R, Biliske JA, Tiege PB, Griffith IJ, Cardiovasc. Intervent. Radiol 2012, 35, 636. [DOI] [PubMed] [Google Scholar]
- [184].a) Choi JW, Park JH, Cho HR, Chung JW, Kim DD, Kim HC, Cho HJ, Sci. Rep 2017, 7; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Choi JW, Park JH, Baek SY, Kim DD, Kim HC, Cho HJ, Colloids Surf., B 2015, 132, 305. [DOI] [PubMed] [Google Scholar]
- [185].Liu Y, Liu J, Ai K, Yuan Q, Lu L, Contrast Media Mol. Imaging 2014, 9, 26. [DOI] [PubMed] [Google Scholar]
- [186].Sang L, Luo DD, Wei ZY, Qi M, Mater. Sci. Eng., C 2017, 75, 1389. [DOI] [PubMed] [Google Scholar]
- [187].Gabelmann A, Haberstroh J, Weyrich G, Acta Radiol. 2001, 42, 422. [PubMed] [Google Scholar]
- [188].Thanoo BC, Sunny MC, Jayakrishnan A, J. Appl. Biomater 1991, 2, 67. [DOI] [PubMed] [Google Scholar]
- [189].Horak D, Cervinka M, Puza V, Biomaterials 1998, 19, 1303. [DOI] [PubMed] [Google Scholar]
- [190].Thanoo BC, Jayakrishnan A, J. Microencaps 1991, 8, 95. [DOI] [PubMed] [Google Scholar]
- [191].Hasan MS, Kehoe S, Boyd D, J. Biomater. Appl 2014, 29, 566. [DOI] [PubMed] [Google Scholar]
- [192].a) Agusti G, Jordan O, Andersen G, Doelker E, Chevalier Y, J. Appl. Polym. Sci 2015, 132; [Google Scholar]; b) Duran R, Sharma K, Dreher MR, Ashrafi K, Mirpour S, Lin MD, Schernthaner RE, Schlachter TR, Tacher V, Lewis AL, Willis S, den Hartog M, Radaelli A, Negussie AH, Wood BJ, Geschwind JFH, Theranostics 2016, 6, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Sharma KV, Bascal Z, Kilpatrick H, Ashrafi K, Willis SL, Dreher MR, Lewis AL, Biomaterials 2016, 103, 293. [DOI] [PubMed] [Google Scholar]
- [194].Bartling SH, Budjan J, Aviv H, Haneder S, Kraenzlin B, Michaely H, Margel S, Diehl S, Semmler W, Gretz N, Schonberg SO, Sadick M, Invest. Radiol 2011, 46, 178. [DOI] [PubMed] [Google Scholar]
- [195].a) Revia RA, Zhang M, Mater. Today 2016, 19, 157; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Stephen ZR, Kievit FM, Zhang M, Mater. Today 2011, 14, 330; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Chakravarty R, Valdovinos HF, Chen F, Lewis CM, Ellison PA, Luo H, Meyerand ME, Nickles RJ, Cai W, Adv. Mater 2014, 26, 5119; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Park JH, von Maltzahn G, Zhang L, Schwartz MP, Ruoslahti E, Bhatia SN, Sailor MJ, Adv. Mater 2008, 20, 1630; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Hu J, Obayemi JD, Malatesta K, Kosmrlj A, Soboyejo WO, Mater. Sci. Eng., C 2018, 88, 32; [DOI] [PubMed] [Google Scholar]; f) Obayemi JD, Hu J, Uzonwanne VO, Odusanya OS, Malatesta K, Anuku N, Soboyejo WO, J. Mech. Behav. Biomed. Mater 2017, 68, 276. [DOI] [PubMed] [Google Scholar]
- [196].a) Chung EY, Kim HM, Lee GH, Kwak BK, Jung JS, Kuh HJ, Lee J, Carbohydr. Polym 2012, 90, 1725; [DOI] [PubMed] [Google Scholar]; b) Kim EH, Ahn Y, Lee HS, J. Alloys Compd 2007, 434, 633. [Google Scholar]
- [197].Wang H, Qin XY, Li ZY, Guo LY, Zheng ZZ, Liu LS, Fan TY, Int. J. Pharm 2016, 511, 831. [DOI] [PubMed] [Google Scholar]
- [198].Li ZY, Qin XY, Guo LY, Wang H, Liu XX, Zheng ZZ, Guan HT, Song L, Zou YH, Fan TY, Int. J. Pharm 2017, 527, 31. [DOI] [PubMed] [Google Scholar]
- [199].Wang Q, Qian K, Liu SS, Yang YJ, Liang B, Zheng CS, Yang XL, Xu HB, Shen AQ, Biomacromolecules 2015, 16, 1240. [DOI] [PubMed] [Google Scholar]
- [200].Barnett BP, Arepally A, Stuber M, Arifin DR, Kraitchman DL, Bulte JWM, Nat. Protoc 2011, 6, 1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Gobin YP, Vinuela F, Vinters HV, Ji C, Chow K, Radiology 2000, 214, 113. [DOI] [PubMed] [Google Scholar]
- [202].Zeng J, Li L, Zhang HS, Li JY, Liu LL, Zhou GF, Du Q, Zheng CS, Yang XL, Theranostics 2018, 8, 4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Stampfl U, Sommer C-M, Bellemann N, Holzschuh M, Kueller A, Bluemnnel J, Gehrig T, Shevchenko M, Kenngott H, Kauczor H-U, Radeleff B, J. Vasc. Interv. Radiol 2012, 23, 1225. [DOI] [PubMed] [Google Scholar]
- [204].Hagit A, Soenke B, Johannes B, Shlomo M, Biomacromolecules 2010, 11, 1600. [DOI] [PubMed] [Google Scholar]
- [205].Salem R, Lewandowski RJ, Clin. Gastroenterol. Hepatol 2013, 11, 604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].a) Liu Q, Qian Y, Li P, Zhang S, Liu J, Sun X, Fulham M, Feng D, Huang G, Lu W, Song S, Theranostics 2018, 8, 785; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Atassi B, Bangash AK, Bahram A, Pizzi G, Lewandowski RJ, Ryu RK, Sato KT, Gates VL, Mulcahy MF, Kulik L, Miller F, Yaghmai V, Murth R, Larson A, Omary RA, Salem R, Radiographics 2008, 28, 81; [DOI] [PubMed] [Google Scholar]; c) Sangro B, Bilbao JI, Boan J, Martinez-Cuesta A, Benito A, Rodriguez J, Panizo A, Gil B, Inarrairaegui M, Herrero I, Quiroga J, Prieto J, Int. J. Radiat. Oncol., Biol., Phys 2006, 66, 792; [DOI] [PubMed] [Google Scholar]; d) Deipolyi AR, Iafrate AJ, Zhu AX, Ergul EA, Ganguli S, Oklu R, J. Vasc. Interv. Radiol 2014, 25, 1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].a) Choi JW, Cho HJ, Park JH, Baek SY, Chung JW, Kim DD, Kim HC, PLoS One 2014, 9; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kim DH, Chen J, Omary RA, Larson AC, Theranostics 2015, 5, 477; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kim DH, Li WG, Chen JN, Zhang ZL, Green RM, Huang S, Larson AC, Sci. Rep 2016, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Varela M, Real MI, Burrel M, Forner A, Sala M, Brunet M, Ayuso C, Castells L, Montana X, Llovet JM, Bruix J, J. Hepatol 2007, 46, 474. [DOI] [PubMed] [Google Scholar]
- [209].a) Fuchs K, Duran R, Denys A, Bize PE, Borchard G, Jordan O, J. Controlled Release 2017, 262, 127; [DOI] [PubMed] [Google Scholar]; b) Sheth RA, Hesketh R, Kong DS, Wicky S, Oklu R, J. Vasc. Interv. Radiol 2013, 24, 1201. [DOI] [PubMed] [Google Scholar]
- [210].Horak D, Svec F, Adamyan A, Titova M, Skuba N, Voronkova O, Trostenyuk N, Vishnevskii V, Gumargalieva K, Biomaterials 1992, 13, 361. [DOI] [PubMed] [Google Scholar]
- [211].Pawlik TM, Reyes DK, Cosgrove D, Kamel IR, Bhagat N, Geschwind JFH, J. Clin. Oncol 2011, 29, 3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Cui DC, Lu WL, Sa EA, Gu MJ, Lu XJ, Fan TY, Int. J. Pharm 2012, 436, 527. [DOI] [PubMed] [Google Scholar]
- [213].Negussie AH, Dreher MR, Johnson CG, Tang YQ, Lewis AL, Storm G, Sharma KV, Wood BJ, J. Mater. Sci.: Mater. Med 2015, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Ahnfelt E, Sjogren E, Hansson P, Lennernas H, J. Pharm. Sci 2016, 105, 3387. [DOI] [PubMed] [Google Scholar]
- [215].Lewis AL, Gonzalez MV, Lloyd AW, Hall B, Tang YQ, Willis SL, Leppard SW, Wolfenden LC, Palmer RR, Stratford PW, J. Vasc. Interv. Radiol 2006, 17, 335. [DOI] [PubMed] [Google Scholar]
- [216].Hong K, Khwaja A, Liapi E, Torbenson MS, Georgiades CS, Geschwind JFH, Clin. Cancer. Res 2006, 12, 2563. [DOI] [PubMed] [Google Scholar]
- [217].Lammer J, Malagari K, Vogl T, Pilleul F, Denys A, Watkinson A, Pitton M, Sergent G, Pfammatter T, Terraz S, Benhamou Y, Avajon Y, Gruenberger T, Pomoni M, Langenberger H, Schuchmann M, Dumortier J, Mueller C, Chevallier P, Lencioni R, Investigators PV, Cardiovasc. Intervent. Radiol 2010, 33, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].a) Augestad KM, Bakaki PM, Rose J, Crawshaw BP, Lindsetmo RO, Dorum LM, Koroukian SM, Delaney CP, Cancer Epidemiol 2015, 39, 734; [DOI] [PubMed] [Google Scholar]; b) Kemeny NE, Chou JF, Boucher TM, Capanu M, R. DeMatteo P, Jarnagin WR, Allen PJ, Fong YC, Cercek A, D’Angelica MI, J. Surg. Oncol 2016, 113, 477; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Adam R, Lucidi V, Bismuth H, Surg. Clin. North Am 2004, 84, 659. [DOI] [PubMed] [Google Scholar]
- [219].a) Martin RCG, Joshi J, Robbins K, Tomalty D, Bosnjakovik P, Derner M, Padr R, Rocek M, Scupchenko A, Tatum C, Ann. Surg. Oncol 2011, 18, 192; [DOI] [PubMed] [Google Scholar]; b) Martin RCG, Robbins K, Tomalty D, O’Hara R, Bosnjakovic P, Padr R, Rocek M, Slauf F, Scupchenko A, Tatum C, World J. Surg. Oncol 2009, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Golfieri R, Giampalma E, Renzulli M, Cioni R, Bargellini I, Bartolozzi C, Breatta AD, Gandini G, Nani R, Gasparini D, Cucchetti A, Bolondi L, Trevisani F, G. R. P. Precision Italia Study, Br. J. Cancer 2014, 111, 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Jordan O, Denys A, De Baere T, Boulens N, Doelker E, J. Vasc. Interv. Radiol 2010, 21, 1084. [DOI] [PubMed] [Google Scholar]
- [222].Fuchs K, Bize PE, Dormond O, Denys A, Doelker E, Borchard G, Jordan O, J. Vasc. Interv. Radiol 2014, 25, 379. [DOI] [PubMed] [Google Scholar]
- [223].Wang YJ, Molin DGM, Sevrin C, Grandfils C, van den Akker NMS, Gagliardi M, Knetsch ML, Delhaas T, Koole LH, Int. J. Pharm 2016, 503, 150. [DOI] [PubMed] [Google Scholar]
- [224].Hidaka K, Nakamura M, Osuga K, Miyazaki H, Wada S, J. Mech. Behav. Biomed. Mater 2010, 3, 497. [DOI] [PubMed] [Google Scholar]
- [225].Liapi E, Geschwind J-FH, Cardiovasc. Intervent. Radiol 2011, 34, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].a) Malagari K, Pomoni M, Moschouris H, Kelekis A, Charokopakis A, Bouma E, Spyridopoulos T, Chatziioannou A, Sotirchos V, Karampelas T, Tamvakopoulos C, Filippiadis D, Karagiannis E, Marinis A, Koskinas J, Kelekis DA, Cardiovasc. Intervent. Radiol 2014, 37, 165; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jiaqi Y, Hori S, Minamitani K, Hashimoto T, Yoshimura H, Nomura N, Ishida T, Fukuda H, Tomoda K, Nakamura H, Nihon Igaku Hoshasen Gakkai zasshi. Nippon acta radiologica 1996, 56, 19; [PubMed] [Google Scholar]; c) Sun J-H, Zhou G-H, Zhang Y-L, Nie C-H, Zhou T-Y, Ai J, Zhu T-Y, Wang W-L, Zheng S-S, Oncotarget 2017, 8, 5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Li S, Wang XT, Zhang XB, Yang RJ, Zhang HZ, Zhu LZ, Hou XP, J. Controlled Release 2002, 84, 87. [DOI] [PubMed] [Google Scholar]
- [228].Hamman JH, Mar. Drugs 2010, 8, 1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Sezer AD, Akbuga J, J. Microencaps 1999, 16, 195. [DOI] [PubMed] [Google Scholar]
- [230].Zhang G-Y, Zhou X-F, Zhou X-Y, Wen Q-Y, You B-G, Liu Y, Zhang X-N, Jin Y, J. Biomed. Mater. Res. A 2013, 101, 3192. [DOI] [PubMed] [Google Scholar]
- [231].Wang YJ, Benzina A, Molin DGM, van den Akker N, Gagliardi M, Koole LH, J. Biomater. Sci., Polym. Ed 2015, 26, 77. [DOI] [PubMed] [Google Scholar]
- [232].a) Odonnell PB, McGinity JW, Adv. Drug Del. Rev 1997, 28, 25; [DOI] [PubMed] [Google Scholar]; b) Yang YY, Chung TS, Ng NP, Biomaterials 2001, 22, 231. [DOI] [PubMed] [Google Scholar]
- [233].Dasari S, Tchounwou PB, Eur. J. Pharmacol 2014, 740, 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].a) Gotink KJ, Verheul HMW, Angiogenesis 2010, 13, 1; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pignochino Y, Grignani G, Cavalloni G, Motta M, Tapparo M, Bruno S, Bottos A, Gammaitoni L, Migliardi G, Camussi G, Alberghini M, Torchio B, Ferrari S, Bussolino F, Fagioli F, Picci P, Aglietta M, Mol. Cancer 2009, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].a) Zhang SS, Huang C, Li ZZ, Yang YJ, Bao TT, Chen HB, Zou YH, Song L, Drug Deliv 2017, 24, 1011; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang YW, Ao J, Liu Y, Qiao MX, Yang XL, Tang SX, Li C, Xu K, Tumor Biol 2014, 35, 10905. [DOI] [PubMed] [Google Scholar]
- [236].a) Namur J, Wassef M, Millot JM, Lewis AL, Manfait M, Laurent A, J. Vasc. Interv. Radiol 2010, 21, 259; [DOI] [PubMed] [Google Scholar]; b) Lewis AL, Taylor RR, Hall B, Gonzalez MV, Willis SL, Stratford PW, J. Vasc. Interv. Radiol 2006, 17, 1335; [DOI] [PubMed] [Google Scholar]; c) Dreher MR, Sharma KV, Woods DL, Reddy G, Tang YQ, Pritchard WF, Chiesa OA, Karanian JW, Esparza JA, Donahue D, Levy EB, Willis SL, Lewis AL, Wood BJ, J. Vasc. Interv. Radiol 2012, 23, 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].a) Namur J, Citron SJ, Sellers MT, Dupuis MH, Wassef M, Manfait M, Laurent A, J. Hepatol 2011, 55, 1332; [DOI] [PubMed] [Google Scholar]; b) Guiu B, Schmitt A, Reinhardt S, Fohlen A, Pohl T, Wendremaire M, Denys A, Blummel J, Boulin M, J. Vasc. Interv. Radiol 2015, 26, 262; [DOI] [PubMed] [Google Scholar]; c) Malagari K, Kiakidis T, Pomoni M, Moschouris H, Emmanouil E, Spiridopoulos T, Sotirchos V, Tandeles S, Koundouras D, Kelekis A, Filippiadis D, Charokopakis A, Bouma E, Chatziioannou A, Dourakis S, Koskinas J, Karampelas T, Tamvakopoulos K, Kelekis N, Kelekis D, Cardiovasc. Intervent. Radiol 2016, 39, 1379. [DOI] [PubMed] [Google Scholar]
- [238].a) Poon RTP, Tso WK, Pang RWC, Ng KKC, Woo R, Tai KS, Fan ST, Clin. Gastroenterol. Hepatol 2007, 5, 1100; [DOI] [PubMed] [Google Scholar]; b) Martin R, Geller D, Espat J, Kooby D, Sellars M, Goldstein R, Imagawa D, Scoggins C, Hepatogastroenterology 2012, 59, 255; [DOI] [PubMed] [Google Scholar]; c) Lencioni R, de Baere T, Burrel M, Caridi JG, Lammer J, Malagari K, Martin RCG, O’Grady E, Real MI, Vogl TJ, Watkinson A, Geschwind JFH, Cardiovasc. Intervent. Radiol 2012, 35, 980; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Wu BL, Zhou J, Ling GH, Zhu DY, Long QY, World J. Surg. Oncol 2018, 16; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Richter G, Radeleff B, Stroszczynski C, Pereira P, Helmberger T, Barakat M, Huppert P, Cardiovasc. Intervent. Radiol 2018, 41, 587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [239].a) Malagari K, Chatzimichael K, Alexopoulou E, Kelekis A, Hall B, Dourakis S, Delis S, Gouliamos A, Kelekis D, Cardiovasc. Intervent. Radiol 2008, 31, 269; [DOI] [PubMed] [Google Scholar]; b) van Malenstein H, Maleux G, Vandecaveye V, Heye S, Laleman W, J. van Pelt, Vaninbroukx J, Nevens F, Verslype C, Onkologie 2011, 34, 368; [DOI] [PubMed] [Google Scholar]; c) Nam HC, Jang B, Song MJ, World J. Gastroenterol 2016, 22, 8853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].de Baere T, Deschamps F, Teriitheau C, Rao P, Conengrapht K, Schlumberger M, Leboulleux S, Baudin E, Hechellhammer L, J. Vasc. Interv. Radiol 2008, 19, 855. [DOI] [PubMed] [Google Scholar]
- [241].Saralidze K, van Hooy-Corstjens CSJ, Koole LH, Knetsch MLW, Biomaterials 2007, 28, 2457. [DOI] [PubMed] [Google Scholar]
- [242].Xuan FQ, Rong JJ, Liang M, Zhang XW, Sun JY, Zhao LJ, Li Y, Liu D, Li F, Wang XZ, Han YL, BioMed Res. Int 2017, DOI: 10.1155/2017/1875258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [243].Madani F, Bessodes M, Lakrouf A, Vauthier C, Scherman D, Chaumeil JC, Biomaterials 2007, 28, 1198. [DOI] [PubMed] [Google Scholar]
- [244].a) Phelps EA, Enemchukwu NO, Fiore VF, Sy JC, Murthy N, Sulchek TA, Barker TH, García AJ, Adv. Mater 2012, 24, 64; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hu J, Youssefian S, Obayemi J, Malatesta K, Rahbar N, Soboyejo W, Acta Biomater 2018, 71, 363. [DOI] [PubMed] [Google Scholar]
- [245].Aliberti C, Carandina R, Sarti D, Mulazzani L, Pizzirani E, Guadagni S, Fiorentini G, Am. J. Roentgenol 2017, 209, 430. [DOI] [PubMed] [Google Scholar]
- [246].a) Yang Q, Lai SK, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol 2015, 7, 655; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang P, Sun F, Liu SJ, Jiang SY, J. Controlled Release 2016, 244, 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [247].Abramowitz SD, Israel GM, McCarthy SM, Pollak JS, White RI, Tal MG, Radiology 2009, 250, 482. [DOI] [PubMed] [Google Scholar]
- [248].a) Mukherjee D, Padilla J, Shadden SC, ThCFD 2016, 30, 23; [Google Scholar]; b) Fabbri D, Long Q, Das S, Pinelli M, Biomech. Model. Mechanobiol 2014, 13, 289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].a) Laurent A, Wassef M, Namur J, Ghegediban H, Pelage JP, Cardiovasc. Intervent. Radiol 2010, 33, 995; [DOI] [PubMed] [Google Scholar]; b) Laurent A, Wassef M, Saint Maurice JP, Namur J, Pelage JP, Seron A, Chapot R, Merland JJ, Invest. Radiol 2006, 41, 8. [DOI] [PubMed] [Google Scholar]
- [250].Hidaka K, Moine L, Collin G, Labarre D, Grossiord JL, Huang N, Osuga K, Wada S, Laurent A, J. Mech. Behav. Biomed. Mater 2011, 4, 2161. [DOI] [PubMed] [Google Scholar]
- [251].Lee KH, Liapi E, Ventura VP, Buijs M, Vossen JA, Vali M, Geschwind JFH, J. Vasc. Interv. Radiol 2008, 19, 1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Bilbao JI, de Luis E, García de Jalón JA, de Martino A, Lozano MD, de la Cuesta AM, Sangro B, J. Vasc. Interv. Radiol 2008, 19, 1625. [DOI] [PubMed] [Google Scholar]
- [253].Verret V, Ghegediban SH, Wassef M, Pelage JP, Golzarian J, Laurent A, J. Vasc. Interv. Radiol 2011, 22, 220. [DOI] [PubMed] [Google Scholar]
- [254].de Baere T, Plotkin S, Yu R, Sutter A, Wu Y, Cruise GM, J. Vasc. Interv. Radiol 2016, 27, 1425. [DOI] [PubMed] [Google Scholar]
- [255].Medsinge A, Zajko A, Orons P, Amesur N, Santos E, Am. J. Roentgenol 2014, 203, 699. [DOI] [PubMed] [Google Scholar]
- [256].Zhou F, Chen LM, An QZ, Chen L, Wen Y, Fang F, Zhu W, Yi T, Sci. Rep 2016, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [257].Mottu F, Rufenacht DA, Laurent A, Doelker E, Biomaterials 2002, 23, 121. [DOI] [PubMed] [Google Scholar]
- [258].Becker TA, Kipke DR, Brandon T, J. Biomed. Mater. Res 2001, 54, 76. [DOI] [PubMed] [Google Scholar]
- [259].Becker TA, Preul MC, Bichard WD, Kipke DR, McDougall CG, Neurosurgery 2007, 60, 1119. [DOI] [PubMed] [Google Scholar]
- [260].a) Kipke DR, Becker TA, Bichard WD, McDougall CG, Preul MC, Neurosurgery 2002, 51, 453; [PubMed] [Google Scholar]; b) Becker TA, Kipke DR, Preul MC, Bichard WD, C. McDougall G, presented at 25th Annual International Conference of the IEEE-Engineering-in-Medicine-and-Biology-Society, Cancun, MEXICO 2003. [Google Scholar]
- [261].Barnett BP, Gailloud P, J. Vasc. Interv. Radiol 2011, 22, 203. [DOI] [PubMed] [Google Scholar]
- [262].Dai FY, Tang L, Yang JH, Zhao XL, Liu WG, Chen G, Xiao FS, Feng XQ, J. Mater. Sci.: Mater. Med 2009, 20, 967. [DOI] [PubMed] [Google Scholar]
- [263].Li XW, Liu WG, Ye GX, Zhang BQ, Zhu DW, Yao KD, Liu ZQ, Sheng XZ, Biomaterials 2005, 26, 7002. [DOI] [PubMed] [Google Scholar]
- [264].Zhao H, Zheng C, Feng G, Zhao Y, Liang H, Wu H, Zhou G, Liang B, Wang Y, Xia X, Am. J. Neuroradiol 2013, 34, 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [265].Zhao YB, Zheng CS, Wang Q, Fang JL, Zhou GF, Zhao H, Yang YJ, Xu HB, Feng GS, Yang XL, Adv. Funct. Mater 2011, 21, 2035. [Google Scholar]
- [266].Huang LL, Shen M, Li RX, Zhang XY, Sun Y, Gao P, Fu H, Liu HQ, He Y, Du YQ, Cao J, Duan YR, Oncotarget 2016, 7, 73280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [267].Wang Y, Xu N, Luo Q, Li Y, Sun L, Wang H, Xu K, Wang B, Zhen Y, Interv. Neuroradiol 2011, 17, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [268].Ning X, Zhao C, Pang J, Ding Z, Wang Y, Xu K, Chen H, Li B, Luo Q, Exp. Ther. Med 2015, 10, 316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [269].Fatimi A, Chabrot P, Berrahmoune S, Coutu JM, Soulez G, Lerouge S, Acta Biomater 2012, 8, 2712. [DOI] [PubMed] [Google Scholar]
- [270].Fatimi A, Zehtabi F, Lerouge S, J. Biomed. Mater. Res., Part B 2016, 104, 1551. [DOI] [PubMed] [Google Scholar]
- [271].Poursaid A, Price R, Tiede A, Olson E, Huo E, McGill L, Ghandehari H, Cappello J, Biomaterials 2015, 57, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [272].a) Poursaid A, Doctor of Philosophy The University of Utah; 2016; [Google Scholar]; b) Poursaid A, Jensen MM, Nourbakhsh I, Weisenberger M, Hellgeth JW, Sampath S, Cappello J, Ghandehari H, Mol. Pharm 2016, 13, 2736. [DOI] [PubMed] [Google Scholar]
- [273].Lym JS, Nguyen QV, Ahn DW, Huynh CT, Jae HJ, Kim YI, Lee DS, Acta Biomater 2016, 41, 253. [DOI] [PubMed] [Google Scholar]
- [274].Nguyen QV, Lee MS, Lym JS, Kim YI, Jae HJ, Lee DS, J. Mater. Chem. B 2016, 4, 6524. [DOI] [PubMed] [Google Scholar]
- [275].Nguyen QV, Lym JS, Huynh CT, Kim BS, Jae HJ, Kim YI, Lee DS, Polym. Chem 2016, 7, 5805. [Google Scholar]
- [276].Huynh CT, Nguyen QV, Lym JS, Kim BS, Huynh DP, Jae HJ, Kim YI, Lee DS, RSC Adv 2016, 6, 47687. [Google Scholar]
- [277].Brennecka CR, Preul MC, Bichard WD, Vernon BL, World Neurosurg 2012, 78, 469. [DOI] [PubMed] [Google Scholar]
- [278].Brennecka CR, Preul MC, Becker TA, Vernon BL, J. Neurosurg 2013, 119, 228. [DOI] [PubMed] [Google Scholar]
- [279].Zehtabi F, Ispas-Szabo P, Djerir D, Sivakumaran L, Annabi B, Soulez G, Mateescu MA, Lerouge S, Acta Biomater 2017, 64, 94. [DOI] [PubMed] [Google Scholar]
- [280].Zhou X, Li Y, Chen S, Fu Y-N, Wang S, Li G, Tao L, Wei Y, Wang X, Liang JF, Colloids Surf., B 2018, 172, 601. [DOI] [PubMed] [Google Scholar]
- [281].Bearat HH, Preul MC, Vernon BL, J. Biomed. Mater. Res. A 2013, 101, 2515. [DOI] [PubMed] [Google Scholar]
- [282].Qin LZ, Mei LL, Shan ZY, Huang Y, Pan X, Li G, Gu YK, Wu CB, Drug Dev. Ind. Pharm 2016, 42, 307. [DOI] [PubMed] [Google Scholar]
- [283].Han K, Pan X, Chen M, Wang R, Xu Y, Feng M, Li G, Huang M, Wu C, Eur. J. Pharm. Sci 2010, 41, 692. [DOI] [PubMed] [Google Scholar]
- [284].Du LR, Lu XJ, Guan HT, Yang YJ, Gu MJ, Zheng ZZ, Lv TS, Yan ZG, Song L, Zou YH, Fu NQ, Qi XR, Fan TY, Int. J. Pharm 2014, 471, 285. [DOI] [PubMed] [Google Scholar]
- [285].Jones JP, Sima M, O’Hara RG, Stewart RJ, Adv. Healthcare Mater 2016, 5, 795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [286].Momeni A, Valliant EM, Brennan-Pierce EP, Shankar JJS, Abraham R, Colp P, Filiaggi MJ, Acta Biomater 2016, 32, 286. [DOI] [PubMed] [Google Scholar]
- [287].Albanese G, Kondo KL, Seminars in interventional radiology 2010, 27, 391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [288].Sampei K, Hashimoto N, Kazekawa K, Tsukahara T, Iwata H, Takaichi S, Neuroradiology 1996, 38, 291. [DOI] [PubMed] [Google Scholar]
- [289].Mason KP, Michna E, Zurakowski D, Koka BV, Burrows PE, Radiology 2000, 217, 127. [DOI] [PubMed] [Google Scholar]
- [290].a) Flandroy P, Pruvo JP, Cardiovasc. Intervent. Radiol 1994, 17, 222; [DOI] [PubMed] [Google Scholar]; b) Grzyska U, Westphal M, Zanella F, Freckmann N, Herrmann HD, Zeumer H, Surg. Neurol 1993, 40, 476. [DOI] [PubMed] [Google Scholar]
- [291].Konya A, Stephens LC, Wright KC, J. Vasc. Interv. Radiol 2010, 21, 1091. [DOI] [PubMed] [Google Scholar]
- [292].Tamatani S, Koike T, Ito Y, Tanaka R, Interv. Neuroradiol 2000, 6, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [293].Yamashita K, Taki W, Iwata H, Nakahara I, Nishi S, Sadato A, Matsumoto K, Kikuchi H, Am. J. Neuroradiol 1994, 15, 1103. [PMC free article] [PubMed] [Google Scholar]
- [294].Li YJ, Barthès-Biesel D, Salsac AV, J. Mech. Behav. Biomed. Mater 2017, 69, 307. [DOI] [PubMed] [Google Scholar]
- [295].O’Leary JA, J. Surg. Oncol 1971, 3, 117. [DOI] [PubMed] [Google Scholar]
- [296].Vakalopoulos KA, Wu ZQ, Kroese L, Kleinrensink GJ, Jeekel J, Vendamme R, Dodou D, Lange JF, Ann. Surg 2015, 261, 323. [DOI] [PubMed] [Google Scholar]
- [297].Blakely B, Lee BH, Riley C, McLemore R, Pathak CP, Vernon BL, J. Biomed. Mater. Res., Part B 2010, 93B, 9. [DOI] [PubMed] [Google Scholar]
- [298].Taki W, Yonekawa Y, Iwata H, Uno A, Yamashita K, Amemiya H, Am. J. Neuroradiol 1990, 11, 163. [PMC free article] [PubMed] [Google Scholar]
- [299].Brothers MF, Kaufmann JC, Fox AJ, Deveikis JP, AJNR Am. J. Neuroradiol 1989, 10, 777. [PMC free article] [PubMed] [Google Scholar]
- [300].Grasso RF, Cazzato RL, Luppi G, Faiella E, Del Vescovo R, Giurazza F, Borzomati D, Coppola R, Zobel BB, Cardiovasc. Intervent. Radiol 2012, 35, 958. [DOI] [PubMed] [Google Scholar]
- [301].Regine R, Palmieri F, De Siero M, Rescigno A, Sica V, Cantarela R, Villari V, Interventional Medicine and Applied Science 2015, 7, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [302].Dudeck O, Jordan O, Hoffmann KT, Okuducu AF, Tesmer K, Kreuzer-Nagy T, Rufenacht DA, Doelker E, Felix R, Am. J. Neuroradiol 2006, 27, 1900. [PMC free article] [PubMed] [Google Scholar]
- [303].Dudeck O, Jordan O, Hoffmann KT, Okuducu AF, Husmann I, Kreuzer-Nagy T, Tesmer K, Podrabsky P, Bruhn H, Hilborn J, Rufenacht DA, Doelker E, Felix R, Am. J. Neuroradiol 2006, 27, 1849. [PMC free article] [PubMed] [Google Scholar]
- [304].Ahmed EM, Journal of Advanced Research 2015, 6, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [305].Vanninen RL, Manninen I, Cardiovasc. Intervent. Radiol 2007, 30, 196. [DOI] [PubMed] [Google Scholar]
- [306].Natarajan SK, Born D, Ghodke B, Britz GW, Sekhar LN, J. Neurosurg 2009, 111, 105. [DOI] [PubMed] [Google Scholar]
- [307].Leyon JJ, Chavda S, Thomas A, Lamin S, J. Neurointerv. Surg 2016, 8, 596. [DOI] [PubMed] [Google Scholar]
- [308].Duffner F, Ritz R, Bornemann A, Freudenstein D, Wiendl H, Siekmann R, Clin. Neuropathol 2002, 21, 13. [PubMed] [Google Scholar]
- [309].a) Vollherbst DF, Otto R, von Deimling A, Pfaff J, Ulfert C, Kauczor HU, Bendszus M, Sommer CM, Mohlenbruch MA, J. Neurointerv. Surg 2018, 10, 268; [DOI] [PubMed] [Google Scholar]; b) Kocer N, Hanimoglu H, Batur S, Kandemirli SG, Kizilkilic O, Sanus Z, Oz B, Islak C, Kaynar MY, Diagn. Interv. Radiol 2016, 22, 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [310].Lamin S, Chew HS, Chavda S, Thomas A, Piano M, Quilici L, Pero G, Holtmannspolter M, Cronqvist ME, Casasco A, Guimaraens L, Paul L, Garcia AG, Aleu A, Chapot R, Am. J. Neuroradiol 2017, 38, 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [311].Samaniego EA, Kalousek V, Abdo G, Ortega-Gutierrez S, J. Neurointerv. Surg 2016, 8, 1253. [DOI] [PubMed] [Google Scholar]
- [312].Berenstein A, J. Neurointerv. Surg 2016, 8, 934. [DOI] [PubMed] [Google Scholar]
- [313].Wang W, Wei Z, Sang L, Wang Y, Zhang J, Bian Y, Li Y, Eur. Polym. J 2018, 108, 337. [Google Scholar]
- [314].Becker TA, Kipke DR, J. Biomed. Mater. Res 2002, 61, 533. [DOI] [PubMed] [Google Scholar]
- [315].Lee KY, Mooney DJ, Prog. Polym. Sci 2012, 37, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [316].a) Benavides S, Villalobos-Carvajal R, Reyes JE, J. Food Eng 2012, 110, 232; [Google Scholar]; b) Olivas GI, Barbosa-Canovas GV, LWT--Food Sci. Technol 2008, 41, 359. [Google Scholar]
- [317].a) Tan WH, Takeuchi S, Adv. Mater 2007, 19, 2696; [Google Scholar]; b) Sarker B, Rompf J, Silva R, Lang N, Detsch R, Kaschta J, Fabry B, Boccaccini AR, Int. J. Biol. Macromol 2015, 78, 72. [DOI] [PubMed] [Google Scholar]
- [318].Balakrishnan B, Mohanty M, Umashankar PR, Jayakrishnan A, Biomaterials 2005, 26, 6335. [DOI] [PubMed] [Google Scholar]
- [319].Dai M, Zheng XL, Xu X, Kong XY, Li XY, Guo G, Luo F, Zhao X, Wei YQ, Qian ZY, J. Biomed. Biotechnol 2009, DOI: 10.1155/2009/595126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [320].a) George M, Abraham TE, J. Controlled Release 2006, 114, 1; [DOI] [PubMed] [Google Scholar]; b) Kong HJ, Kaigler D, Kim K, Mooney DJ, Biomacromolecules 2004, 5, 1720. [DOI] [PubMed] [Google Scholar]
- [321].Dusseault J, Tam SK, Menard M, Polizu S, Jourdan G, Yahia L, Halle JP, J. Biomed. Mater. Res. A 2006, 76A, 243. [DOI] [PubMed] [Google Scholar]
- [322].a) Kuo CK, Ma PX, Biomaterials 2001, 22, 511; [DOI] [PubMed] [Google Scholar]; b) Larsen BE, Bjornstad J, Pettersen EO, Tonnesen HH, Melvik JE, BMC Biotechnol 2015, 15; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Tan RW, Niu XF, Gan SL, Feng QL, J. Mater. Sci.: Mater. Med 2009, 20, 1245. [DOI] [PubMed] [Google Scholar]
- [323].Soga Y, Preul MC, Furuse M, Becker T, McDougall CG, Neurosurgery 2004, 55, 1401. [DOI] [PubMed] [Google Scholar]
- [324].Barnett BP, Hughes AH, Lin S, Arepally A, Gailloud PH, J. Vasc. Interv. Radiol 2009, 20, 507. [DOI] [PubMed] [Google Scholar]
- [325].Tian F, Zhang BQ, Zhao XL, Ji WH, Li XW, Cheng GX, Yao KD, Polym. Int 2006, 55, 405. [Google Scholar]
- [326].Soni KS, Desale SS, Bronich TK, J. Controlled Release 2016, 240, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [327].Zhang Q, Chen XH, Geng SN, Wei LF, Miron RJ, Zhao YB, Zhang YF, J. Biomed. Mater. Res. A 2017, 105, 1175. [DOI] [PubMed] [Google Scholar]
- [328].Thoniyot P, Tan MJ, Karim AA, Young DJ, Loh XJ, Adv. Sci 2015, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [329].Germano IM, Davis RL, Wilson CB, Hieshima GB, J. Neurosurg 1992, 76, 607. [DOI] [PubMed] [Google Scholar]
- [330].Vinters HV, Lundie MJ, Kaufmann JCE, New Engl. J. Med 1986, 314, 477. [DOI] [PubMed] [Google Scholar]
- [331].a) San Norberto EM, Taylor JH, Carrera S, Vaquero C, J. Vasc. Surg 2012, 56, 1782; [DOI] [PubMed] [Google Scholar]; b) Boodhwani M, Feng J, Mieno S, Ramlawi B, Sodha N, Clements R, Sellke FW, Eur. J. Cardiothorac. Surg 2006, 29, 736; [DOI] [PubMed] [Google Scholar]; c) Raymond J, Metcalfe A, Salazkin I, Schwarz A, Biomaterials 2004, 25, 3983. [DOI] [PubMed] [Google Scholar]
- [332].Huang K, Lee BP, Ingram DR, Messersmith PB, Biomacromolecules 2002, 3, 397. [DOI] [PubMed] [Google Scholar]
- [333].Chenite A, Chaput C, Wang D, Combes C, Buschmann MD, Hoemann CD, Leroux JC, Atkinson BL, Binette F, Selmani A, Biomaterials 2000, 21, 2155. [DOI] [PubMed] [Google Scholar]
- [334].a) Khodaverdi E, Tafaghodi M, Ganji F, Abnoos K, Naghizadeh H, AAPS PharmSciTech 2012, 13, 460; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wu J, Wei W, Wang LY, Su ZG, Ma GH, Biomaterials 2007, 28, 2220; [DOI] [PubMed] [Google Scholar]; c) Croisier F, Jerome C, Eur. Polym. J 2013, 49, 780. [Google Scholar]
- [335].a) Supper S, Anton N, Seidel N, Riemenschnitter M, Schoch C, Vandamme T, Langmuir 2013, 29, 10229; [DOI] [PubMed] [Google Scholar]; b) Ruel-Gariepy E, Chenite A, Chaput C, Guirguis S, Leroux JC, Int. J. Pharm 2000, 203, 89. [DOI] [PubMed] [Google Scholar]
- [336].a) Hsiao MH, Larsson M, Larsson A, Evenbratt H, Chen YY, Liu DM, J. Controlled Release 2012, 161, 942; [DOI] [PubMed] [Google Scholar]; b) Kim GO, Kim N, Kim DY, Kwon JS, Min B-H, Molecules 2012, 17, 13704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [337].Dang QF, Yan JQ, Li JJ, Cheng XJ, Liu CS, Chen XG, Carbohydr. Polym 2011, 83, 171. [Google Scholar]
- [338].Liu L, Gao Q, Lu X, Zhou H, Asian Journal of Pharmaceutical Sciences 2016, 11, 673. [Google Scholar]
- [339].Shanmuganathan S, Shanumugasundaram N, Adhirajan N, Lakshmi TSR, Babu M, Carbohydr. Polym 2008, 73, 201. [Google Scholar]
- [340].Cappello J, Crissman J, Dorman M, Mikolajczak M, Textor G, Marquet M, Ferrari F, Biotechnol. Prog 1990, 6, 198. [DOI] [PubMed] [Google Scholar]
- [341].Dandu R, Von Cresce A, Briber R, Dowell P, Cappello J, Ghandehari H, Poly 2009, 50, 366. [Google Scholar]
- [342].Cappello J, Crissman JW, Crissman M, Ferrari FA, Textor G, Wallis O, Whitledge JR, Zhou X, Burman D, Aukerman L, Stedronsky ER, J. Controlled Release 1998, 53, 105. [DOI] [PubMed] [Google Scholar]
- [343].Price R, Poursaid A, Cappello J, Ghandehari H, J. Controlled Release 2014, 195, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [344].Fulop Z, Gref R, Loftsson T, Int. J. Pharm 2013, 454, 559. [DOI] [PubMed] [Google Scholar]
- [345].Rizwan M, Yahya R, Hassan A, Yar M, Azzahari AD, Selvanathan V, Sonsudin F, Abouloula CN, Polymers 2017, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [346].McLemore R, Preul MC, Vernon BL, J. Biomed. Mater. Res., Part B 2006, 79B, 398. [DOI] [PubMed] [Google Scholar]
- [347].Riley CM, McLemore R, Preul MC, Vernon BL, J. Biomed. Mater. Res., Part B 2011, 96B, 47. [DOI] [PubMed] [Google Scholar]
- [348].Brennecka CR, Preul MC, Vernon BL, J. Biomed. Mater. Res., Part B 2012, 100B, 1298. [DOI] [PubMed] [Google Scholar]
- [349].Lee BH, Beart HH, Cheng V, McLemore R, Robb SA, Cui ZW, Dovigi A, Vernon BL, J. Biomater. Sci., Polym. Ed 2013, 24, 1575. [DOI] [PubMed] [Google Scholar]
- [350].Singh SK, Singh MK, Nayak MK, Kumari S, Shrivastava S, Gracio JJA, Dash D, ACS Nano 2011, 5, 4987. [DOI] [PubMed] [Google Scholar]
- [351].Bearat HH, Lee BH, Vernon BL, Acta Biomater 2012, 8, 3629. [DOI] [PubMed] [Google Scholar]
- [352].Xin Q, in Diesel Engine System Design, DOI: 10.1533/9780857090836.1.113 (Ed: Xin Q), Woodhead Publishing; 2013, p. 113. [DOI] [Google Scholar]
- [353].Vernon B, Martinez A, J. Biomater. Sci., Polym. Ed 2005, 16, 1153. [DOI] [PubMed] [Google Scholar]
- [354].Lee BH, West B, McLemore R, Pauken C, Vernon BL, Biomacromolecules 2006, 7, 2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [355].a) Cheng V, Lee BH, Pauken C, Vernon BL, J. Appl. Polym. Sci 2007, 106, 1201; [Google Scholar]; b) Robb SA, Lee BH, McLemore R, Vernon BL, Biomacromolecules 2007, 8, 2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [356].a) Lowe AM, Abbott NL, Chem. Mater 2012, 24, 746; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Negrini R, Mezzenga R, Langmuir 2011, 27, 5296; [DOI] [PubMed] [Google Scholar]; c) Wang L, Urbas AM, Li Q, Adv. Mater 2018, DOI: 10.1002/adma.201801335e1801335. [DOI] [Google Scholar]
- [357].a) Lee DR, Park JS, Bae IH, Lee Y, Kim BM, Int. J. Nanomed 2016, 11, 853; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Guo CY, Wang J, Cao FL, Lee RJ, Zhai GX, Drug Discov. Today 2010, 15, 1032. [DOI] [PubMed] [Google Scholar]
- [358].Chen YL, Ma PY, Gui S, BioMed Res. Int 2014, DOI: 10.1155/2014/815981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [359].Mitov M, Chemphyschem 2014, 15, 1245. [DOI] [PubMed] [Google Scholar]
- [360].a) Shenderova A, Burke TG, Schwendeman SP, Pharm. Res 1997, 14, 1406; [DOI] [PubMed] [Google Scholar]; b) Wang A, Li S, BMC Biotechnol 2008, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [361].Richter G, Rassweiler J, Kauffmann GW, Wenz W, Crawford DB, Invest. Radiol 1984, 19, 36. [DOI] [PubMed] [Google Scholar]
- [362].Senapati S, Mahanta AK, Kumar S, Maiti P, Signal Transduction Targeted Ther 2018, 3, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [363].Liu YL, Winter HH, Perry SL, Adv. Colloid Interface Sci 2017, 239, 46. [DOI] [PubMed] [Google Scholar]
- [364].a) Momeni A, Filiaggi MJ, Langmuir 2014, 30, 5256; [DOI] [PubMed] [Google Scholar]; b) Shao H, Stewart RJ, Adv. Mater 2010, 22, 729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [365].Wang WN, Xu YS, Li A, Li T, Liu MM, von Klitzing R, Ober CK, Kayitmazer AB, Li L, Guo XH, RSC Adv 2015, 5, 66871. [Google Scholar]
- [366].Spruijt E, Stuart MAC, van der Gucht J, Macromolecules 2013, 46, 1633. [Google Scholar]
- [367].Stewart RJ, Wang CS, Shao H, Adv. Colloid Interface Sci 2011, 167, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [368].a) Kudlay A, Ermoshkin AV, de la Cruz MO, Macromolecules 2004, 37, 9231; [Google Scholar]; b) Hwang DS, Zeng HB, Srivastava A, Krogstad DV, Tirrell M, Israelachvili JN, Waite JH, Soft Matter 2010, 6, 3232; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kim S, Huang J, Lee Y, Dutta S, Yoo HY, Jung YM, Jho Y, Zeng HB, Hwang DS, Proc. Natl. Acad. Sci. U. S. A 2016, 113, E847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [369].Momeni A, Filiaggi MJ, Acta Biomater 2016, 41, 328. [DOI] [PubMed] [Google Scholar]
- [370].Momeni A, Filiaggi MJ, JRheo 2016, 60, 25. [Google Scholar]
- [371].Tiwari A, Bindal S, Bohidar HB, Biomacromolecules 2009, 10, 184. [DOI] [PubMed] [Google Scholar]
- [372].a) Rodell CB, Lee ME, Wang H, Takebayashi S, Takayama T, Kawamura T, Arkles JS, Dusaj NN, Dorsey SM, Witschey WRT, Pilla JJ, Gorman JH, Wenk JF, Burdick JA, Gorman RC, Circ.: Cardiovasc. Interventions 2016, 9; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Loebel C, Rodell CB, Chen MH, Burdick JA, Nat. Protoc 2017, 12, 1521; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Highley CB, Rodell CB, Burdick JA, Adv. Mater 2015, 27, 5075. [DOI] [PubMed] [Google Scholar]
- [373].Chen MH, Wang LL, Chung JJ, Kim YH, Atluri P, Burdick JA, ACS Biomater. Sci. Eng 2017, 3, 3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [374].Gaharwar AK, Mihaila SM, Swami A, Patel A, Sant S, Reis RL, Marques AP, Gomes ME, Khademhosseini A, Adv. Mater 2013, 25, 3329. [DOI] [PubMed] [Google Scholar]
- [375].Gaharwar AK, Avery RK, Assmann A, Paul A, McKinley GH, Khademhosseini A, Olsen BD, Acs Nano 2014, 8, 9833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [376].Baker SE, Sawvel AM, Zheng N, Stucky GD, Chem. Mater 2007, 19, 4390. [Google Scholar]
- [377].Sheikhi A, Afewerki S, Oklu R, Gaharwar AK, Khademhosseini A, Biomater. Sci 2018, 6, 2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [378].Zhu C, Ninh C, Bettinger CJ, Biomacromolecules 2014, 15, 3474. [DOI] [PubMed] [Google Scholar]
- [379].a) Burckbuchler V, Mekhloufi G, Giteau AP, Grossiord JL, Huille S, Agnely F, Eur. J. Pharm. Biopharm 2010, 76, 351; [DOI] [PubMed] [Google Scholar]; b) Rungseevijitprapa W, Bodmeier R, Eur. J. Pharm. Sci 2009, 36, 524. [DOI] [PubMed] [Google Scholar]
- [380].Cilurzo F, Selmin F, Minghetti P, Adami M, Bertoni E, Lauria S, Montanari L, AAPS PharmSciTech 2011, 12, 604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [381].a) Vo A, Doumit M, Rockwell G, J. Med. Eng 2016, 2016, 5162394; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chien YW, Przybyszewski P, Shami EG, J. Parenter. Sci. Technol 1981, 35, 281. [PubMed] [Google Scholar]
- [382].Astin AD, Master of Science Virginia Polytechnic Institute and State University, 1999. [Google Scholar]
- [383].a) Overcashier DE, Chan EK, Hsu CC, Am. Pharm. Rev 2006, 9, 77; [Google Scholar]; b) Allmendinger A, Fischer S, Huwyler B, Mahler HC, Schwarb E, Zarraga IE, Mueller R, Eur. J. Pharm. Biopharm 2014, 87, 318; [DOI] [PubMed] [Google Scholar]; c) Sutera SP, Skalak R, AnRFM 1993, 25, 1. [Google Scholar]
- [384].Vroman L, Bull. N. Y. Acad. Med 1972, 48, 302. [PMC free article] [PubMed] [Google Scholar]
- [385].Coldwell DM, Stokes KR, Yakes WF, Radiographics 1994, 14, 623. [DOI] [PubMed] [Google Scholar]
- [386].Favard N, Moulin M, Fauque P, Bertaut A, Favelier S, Estivalet L, Michel F, Cormier L, Sagot P, Loffroy R, Quantitative Imaging in Medicine and Surgery 2015, 5, 806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [387].Dinerman AA, Cappello J, El-Sayed M, Hoag SW, Ghandehari H, Macromol. Biosci 2010, 10, 1235. [DOI] [PubMed] [Google Scholar]
- [388].Hoare TR, Kohane DS, Poly 2008, 49, 1993. [Google Scholar]
- [389].Bouzeghrane F, Naggara O, Kallmes DF, Berenstein A, Raymond J, C. Int Consortium Neuroendovasc, Am. J. Neuroradiol 2010, 31, 418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [390].Stampfl S, Stampfl U, Rehnitz C, Schnabel P, Satzl S, Christoph P, Henn C, Thomas F, Kauffmann GW, Richter GM, Cardiovasc. Intervent. Radiol 2007, 30, 257. [DOI] [PubMed] [Google Scholar]
- [391].Oklu R, Khademhosseini A, Weiss PS, ACS Nano 2015, 9, 7733. [DOI] [PubMed] [Google Scholar]